

Abstract
Autism and autism spectrum disorders (ASDs) are behaviorally defined, but the biochemical pathogenesis of the underlying disease process remains uncharacterized. Studies indicate that antioxidant status is diminished in autistic subjects, suggesting its pathology is associated with augmented production of oxidative species and/or compromised antioxidant metabolism. This suggests ASD may result from defects in the metabolism of cellular antioxidants which maintain intracellular redox status by quenching reactive oxygen species (ROS). Selenium-dependent enzymes (selenoenzymes) are important in maintaining intercellular reducing conditions, particularly in the brain. Selenoenzymes are a family of ~25 genetically unique proteins, several of which have roles in preventing and reversing oxidative damage in brain and endocrine tissues. Since the brain's high rate of oxygen consumption is accompanied by high ROS production, selenoenzyme activities are particularly important in this tissue. Because selenoenzymes can be irreversibly inhibited by many electrophiles, exposure to these organic and inorganic agents can diminish selenoenzyme-dependent antioxidant functions. This can impair brain development, particularly via the adverse influence of oxidative stress on epigenetic regulation. Here we review the physiological roles of selenoproteins in relation to potential biochemical mechanisms of ASD etiology and pathology.
1. Introduction
The causes of autism and autism spectrum disorder (collectively, ASD) remain unknown, in part because of complex behavioral phenotypes and the likelihood that multiple genetic and environmental factors contribute to its etiology [1–3]. In the absence of biochemical tests for ASD, the diagnosis is based solely on clinical assessment of behavioral criteria that define deficits in social interaction, impairments in verbal and nonverbal receptive/expression, speech, and hyperfocused repetitive behaviors. The pathophysiology of ASD is primarily expressed in the neurologic, immunologic, and gastrointestinal (GI) systems and affects four times as many boys as girls [4–6]. Regression, with loss of previously acquired skills, can also interrupt apparently normal development. Children with severe autism can exhibit mental retardation, and autistic children have an elevated rate of seizure disorders [7].
Although ASD was previously thought to be rare, the number of persons receiving treatment for ASD has increased substantially during the past several decades and continues to increase. A recent US government report estimated the prevalence of ASD increased by 78% from 2002 to 2008 [8]. In 2011-2012, a prevalence of 20 per 1000 was reported for school aged children [9]. However, a portion of ASD's increasing incidence may reflect changes in diagnostic practice and the broadening of diagnostic criteria [10]. Other studies indicate a diagnostic shift or substitution may also have contributed to the rise in diagnosis, whereby the increase in autism diagnoses corresponds with declines in the usage of other diagnostic categories [11, 12]. Based on a meta-analysis of ASD studies, McDonald and Paul [13] concluded that it does not seem possible to assess whether or how much of the observed increases in cumulative incidence are real, although the number of individuals identified as having ASD has increased dramatically.
Until reliably accurate differential diagnoses are achieved, it is difficult to attain the goal of defining the biochemical and physiological lesions that initiate and/or perpetuate the dysfunctions of autism. However, one pathological mechanism present in many children with ASD involves defects in the control of oxidative damage. Distinctions in the nature of these perturbations in redox control may provide insight for identifying biochemically defined patient subgroups that may respond to specific therapeutic interventions. This paper focuses on potential associations between genetic and acquired defects in control of oxidative damage, particularly those that impinge upon selenium- (Se-)dependent enzymes (selenoenzymes). Se physiology is a vital process in the brain and neuroendocrine system, [25, 26] and, because of the high reactivity and low abundance of Se in these tissues, its vulnerability to inhibition by a variety of toxicants is markedly enhanced. However, its potential role in the pathophysiology of ASD remains largely unexplored.
2. Oxidative Stress in ASD
Chemically reactive oxygen-derived products like peroxide radicals (∙O2 2−), hydrogen peroxide (H2O2), superoxide–anion (O2 −), singlet oxygen (1O2), and hydroxyl radicals (∙OH), are products of ongoing aerobic metabolism via mitochondrial oxidative phosphorylation [27]. If not intercepted and detoxified, reactive oxygen species (ROS) are capable of chemically damaging all forms of cellular macromolecules. To avoid these consequences, numerous ROS-detoxifying reactions enable cells to maintain redox equilibrium and metabolic homeostasis. Thus, oxidative stress is a condition where the level of ROS production exceeds antioxidant capacity.
Increased oxidative stress has been observed in children with ASD [28–30]. Blood collected from autistic children shows low concentrations of membrane polyunsaturated lipids, higher phospholipase A2, and loss of the normal asymmetry of membrane lipoproteins, which may indicate increased oxidative damage [13, 31]. Levels of endogenous and exogenous antioxidant capacity are commonly reduced in ASD. Glutathione (GSH) is the primary intracellular antioxidant, and the ratio of its reduced (GSH) and oxidized (GSSG) forms (GSH/GSSG) provides a useful index of redox status. As shown in Table 1, numerous studies have reported significantly lower plasma levels of GSH and, in some cases, lower GSH/GSSG levels. Low GSH levels are associated with oxidative stress, increased inflammation, impaired immune response, and a decreased ability to detoxify environmental contaminants. Autistic children have been reported to be increasingly susceptible to recurrent infections, neuroinflammation, gastroinflammation, and impaired antioxidant and detoxification capacity. Diminished glutathione peroxidase (GPx), superoxide dismutase, and catalase enzyme activities have been associated with ASD, as well as low cysteine, Se, zinc (Zn), and Vitamins C, E, and A [32], although these associations are not consistently observed.
Table 1.
Glutathione and oxidative stress in autism.
| Authors (Reference) | Control n | Autistic n | GSH status | Additional related findings |
|---|---|---|---|---|
| James et al. [14] | 33 | 20 | 46%↓ | ↓GSH/GSSG, ↓SAM/SAH, ↓cysteine |
| James et al. [15] | 73 | 80 | 32%↓ | ↓GSH/GSSG, ↓SAM/SAH, ↓cysteine |
| D. A. Geier and M. R. Geier [16] | Lab-based normal values | 10 | 36%↓ | ↓Cysteine |
| Adams et al. [17] | 55 | 43 | 21%↓ | ↓SAM, ↓cysteine, ↓vitamin E, ↑FIGLU |
| Paşca et al. [18] | 13 | 15 | 33%↓ | ↓Cysteine |
| Pastural et al. [19] | 12 | 15 | 35%↓ | ↓Cysteine |
| Al-Gadani et al. [20] | 30 | 30 | 27%↓ | ↑Lipid peroxides, ↓vitamin E, ↓SOD, ↓GPx |
| Melnyk et al. [21] | 40 | 40 | 29%↓ | ↓GSH/GSSG, ↓SAM/SAH, ↓cysteine, ↓DNA methylation |
| James et al. [22] | 42 | 40 | 28%↓ | ↓GSH/GSSG, ↓SAM/SAH, ↓cysteine |
| Geier et al. [23] | 120 | 28 | 24%↓ | ↓Cysteine |
| Geier et al. [24] | Lab-based normal values | 28 | 24%↓ | ↑GSSG, ↓cysteine, ↓taurine, ↓sulfate |
Accumulation of oxidized glutathione (GS-SG) in plasma is a strong indication of intracellular oxidative stress, as cells export the GS-SG to maintain redox equilibrium. James and coworkers [14] were first to report that plasma levels of cysteine, GSH, and the GSH/GS-SG ratio were significantly decreased in autistic children. In that study, total GSH levels were decreased, and GS-SG was increased, resulting in a threefold reduction in the redox ratio of GSH/GS-SG. Cysteine, the rate-limiting amino acid for GSH synthesis, was significantly decreased relative to controls in over 65% of the autistic children tested. The finding of lower-plasma GSH has since been replicated by other research groups [17–20, 33], suggesting this is a prevalent feature of ASD. A recent meta-analysis finds that children with ASD have decreased blood GSH (27%), GPx activity (18%), and methionine (13%) and increased concentrations of GS-SG (45%) relative to nonautistic children [34]. In addition, levels of NADPH and NADH, which reflect redox status and help maintain GSH in its reduced state, were found to be significantly lower in autistic children [32]. Several studies have reported a decrease in the level of GSH in postmortem brain samples from ASD subjects, associated with a decrease in the GSH/GS-SG ratio and increased levels of oxidative stress biomarkers [35, 36]. In addition, activities of several GSH-related enzymes, including the selenoenzyme GPx, were lower in cerebellums of ASD subjects [37].
The folate and Vitamin B12-dependent enzyme methionine synthase (MS) is inhibited under oxidative stress conditions, resulting in a decrease in all methylation reactions, including DNA methylation [38]. The basis for MS inhibition is oxidation of its Vitamin B12 (cobalamin) cofactor, which is considered the most readily oxidized biomolecule, making it an ideal sensor of cellular redox status [39, 40]. Lower MS activity inhibits methylation by lowering the ratio of the methyl donor S-adenosylmethionine (SAM) to the methylation inhibitor S-adenosylhomocysteine (SAH), exerting a global dampening effect on >200 methylation reactions. Arguably, the most important among these are methylation of DNA and histones, which combine to exert epigenetic regulation over gene expression. As noted above, a decrease in SAM/SAH has been documented in plasma of ASD subjects in association with a decrease in GSH/GS-SG, reflecting the reciprocal relationship between oxidative stress and methylation. In neuronal cells methionine synthase activity is stimulated by growth factors and dopamine but inhibited by neurodevelopmental toxins, including mercury (Hg) [41]. Methionine synthase mRNA levels are significantly decreased in postmortem brains of autistic subjects, indicative of a deficit in methylation capacity secondary to oxidative stress [42].
Elevated urinary levels of 8-isoprostane-F2α (8-iso-PGF2α) and malondialdehyde (MDA), oxidative stress biomarkers, have also been noted in children with autism [28, 30]. A bimodal distribution of 8-iso-PGF2α was reported, with the majority of autistic subjects showing moderate increases in isoprostane levels, while a smaller group of autistic children showed dramatic increases. Levels of urinary 8-hydroxy-2-deoxyguanosine (8-OHdG), a major product of DNA oxidation, were also measured but did not reach statistical significance, although they indicated a trend toward increasing concentrations in children with autism [30]. No significant correlations were noted between the levels of these biomarkers and vitamin intake, dietary supplements, medicine, medical disorders, or history of regression. Therefore, these results suggest that lipid peroxidation is increased in autistic children and that certain autistic children have much greater oxidative stress than others.
3. Selenium-Dependent Antioxidant Metabolism and ASD
Selenoproteins are essential for brain development, redox control, and preventing and reversing oxidative damage in the brain and neuroendocrine tissues (Figure 1, Table 2). Therefore, control of intracellular oxidative tone and findings of increased oxidative damage in children with ASD may be indicative of disruptions of selenoenzyme activities. The molecular forms of Se most common in foods are the amino acids selenocysteine (Sec) and selenomethionine (SeMet), although traces of water-soluble inorganic forms (e.g., selenate and selenite) can also be present in food and drinking water. For both the organic and inorganic Se forms, biochemical utilization in selenoenzymes is initiated through the common intermediate hydrogen selenide (H2Se). Therefore, all ingested (and endogenous) forms of Se must be degraded to inorganic selenide before Se can be used for synthesis of Sec, the physiological active form of Se. Although proteins with SeMet contain Se, they are not considered selenoproteins because SeMet is nonspecifically incorporated into proteins as if it were Met. The nonspecific insertion of SeMet or Met is directed by AUG codons and no significant distinctions in the biochemical functions have been observed. This is in contrast to Sec, which is the catalytically active primary amino acid present in all selenoproteins [43] and is responsible for the principal functions of these enzymes (Table 2). In contrast to other amino acids, Sec is not recycled for reincorporation into new proteins but is, instead, degraded to release inorganic Se which can be utilized for resynthesis of Sec.
Figure 1.
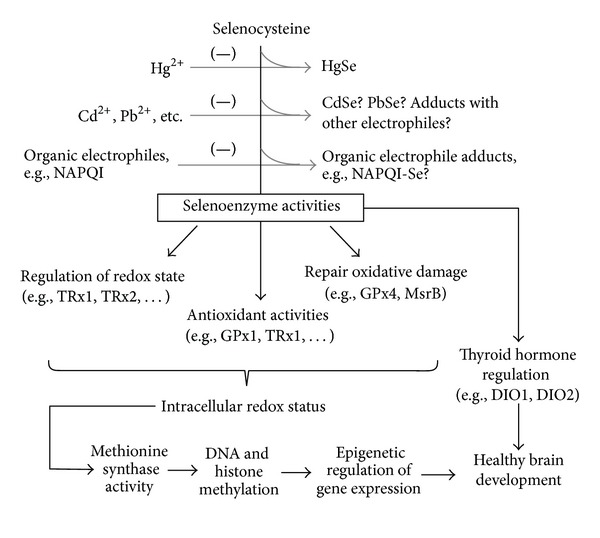
Selenoprotein synthesis and activities are sensitive to elemental and organic electrophiles. High exposures to soft electrophiles may additively impair redox regulation and thyroid hormone production, disrupting epigenetic regulation and normal brain development.
Table 2.
| Selenoprotein | Functions | References |
|---|---|---|
| GPx1 | Detoxifies peroxides in aqueous compartment of cellular cytosol | [58] |
| GPx2 | Expressed in cytosol of liver and tissues of the digestive system | [59] |
| GPx3 | Synthesized primarily by kidney; secreted into plasma for transport to other tissues | [60] |
| GPx4 | Prevents and reverses oxidative damage to lipids in brain and other tissues | [61] |
| TRx1 | Reduces T(SH)2, vitamin C, polyphenols, and other substrates to regulate intercellular redox state | [62–64] |
| TRx2 | Located in mitochondria and controls and regulates redox state | [63, 64] |
| TRx3 | Reduces mitochondrial glutathione disulfide, abundant in testes | [63, 64] |
| MsrB1 | Restores oxidatively damaged methionine (R-sulfoxides) to native configuration | [64] |
| DIO1 | Converts T4 (thyroxine) prohormone into T3 (active thyroid hormone) | [65] |
| DIO2 | Regulates thyroid hormone status, activating as well as inactivating T3 | [65] |
| DIO3 | Activates thyroid hormone in brain, placenta, important in fetal development | [65] |
| SPS2 | Creates the Se-phosphate precursor required for synthesis of all selenoproteins | [64] |
| SelM | Notably high expression levels in the brain, possible thiol-disulfide oxidoreductase | [64, 66] |
| SelN | Interacts with ryanodine receptor, mutations result in congenital muscular dystrophy | [64] |
| SelP | Transports Se in plasma (10 Sec/molecule) and delivers Se to brain and endocrine tissues | [64] |
| SelW | Expressed in a variety of tissues and may regulate redox state of 14-3-3 proteins | [64, 66, 67] |
| Sel15 | Oxidoreductase that may assist in disulfide formation and protein folding | [64] |
While Cys, the analogous sulfur amino acid, is inserted at UGU/UGC codons, the insertion of Sec is in response to UGA which is otherwise the “opal” stop codon for other proteins [44–46]. The selenoprotein mRNAs include a distinct Sec insertion sequence (SECIS) stem-loop structure in their untranslated 3′ region of the mRNA. This is recognized by specific SECIS binding proteins which function together with several transacting factors as well as a unique tRNA with an anticodon complementary to UGA to initiate de novo Sec synthesis. The tRNA is aminoacylated with serine prior to biosynthesis of Sec which is inserted into the protein's primary structure. In mRNA of most selenoproteins, the UGA of the Sec insertion codon is followed by a second or terminal UGA, which is then read as the stop codon.
Since the discovery of these genetically unique proteins, their enzyme activities have become increasingly well defined. Associations between compromised Se and ASD have been reported including low Se levels in red blood cells [47]. Since blood Se is less prone to contamination and more indicative of tissue selenoenzyme activities, it is considered to be a more reliable index than hair. Indeed, hair Se is variously reported as being increased [48], decreased [49], or unchanged [50] in subjects with ASD.
When considering the developmental roles of selenoproteins, as well as their involvement in redox control and protection from oxidative stress, the potential for selenoenzyme dysregulation in relation to the pathologies associated with ASD appears worthy of investigation. The three main families of characterized selenoproteins—iodothyronine deiodinases (DIO), thioredoxin reductases (TRx), and GPx—have critical roles in thyroid function, fetal development, hormone metabolism, and oxidative stress detoxification, particularly in endocrine and brain tissues. Although ~25 selenoproteins are known, only members of the major selenoenzyme/selenoprotein families with potential relevance to autism etiology and pathology are discussed below.
3.1. Roles of Selenoenzymes in Thyroid Hormone Regulation
Selenoenzymes regulate thyroid synthesis and metabolic functions contributing to thyroid hormone biosynthesis, antioxidant defense, redox control of thyrocytes, and thyroid hormone metabolism. Thyroid hormones have important roles in regulating many key biochemical reactions, especially protein synthesis and enzymatic activity, accompanied by an increase in basal metabolic rate. Thyroid hormone regulates several processes that are associated with brain differentiation, including dendrite and axon growth, synaptogenesis, neuronal migration, and myelination [51]. Disruption of thyroid hormone production during early child development leads to permanent deficiency in intelligence and sensorimotor functions [52], and it is hypothesized that Se deficiency may be responsible for the initiation of autoimmune thyroid disorders [53]. Interestingly, thyroid hormone increases plasma Se levels, as well as the levels of selenoenzymes such as DIO [54]. This relationship is logical, since increased metabolic activity places a higher demand on antioxidant resources.
The central nervous system is very sensitive to thyroid hormone supply during growth and development. The selenoprotein family of DIOs (DIO1, 2, and 3) is involved in the formation and regulation of the active thyroid hormone, triiodothyronine (T3). More than 80% of T3 in the brain is derived from intracellular deiodination of T4 by DIO2 [55, 56]. Since circulating T3 does not readily gain access to intracellular nuclear receptors [57], DIO2 provides an important regulatory function in the brain and central nervous system (CNS). During early childhood, T3 bound to nuclear receptors is entirely dependent on its local production from T4 via this selenoprotein.
3.2. Roles of TRx in Redox Regulation
The three distinct forms of TRx—TRx1, TRx2, and TRx3 (collectively, TRx)—are important in controlling redox state within major compartments of the cell. While TRx1 (cytosolic) and TRx2 (mitochondrial) restore oxidized thioredoxin (T[S-S]) to its reduced (T[SH]2) form and are responsible for reducing a variety of other essential antioxidant molecules [63] including Vitamin C [62]. Thioredoxin is a ubiquitous 12 kDa protein that employs vicinal cysteines (CXXC motif) and becomes oxidized to intramolecular disulfides T(S-S) during reduction of other molecules (Figure 2). Its action is essential for countering oxidative damage in the cytosol of aerobic organisms from bacteria to humans [68]. Since T(SH)2 is a central regulator of cellular redox status that is required for the redox-regulated function of transcription factors and hormonally regulated nuclear receptors, it is critical in DNA production, gene expression, cell survival, and embryogenesis. Thus TRx maintains T(SH)2 levels to enable basic processes and regulate multiple metabolic events. The antioxidant functions of TRx occur because they directly facilitate reduction of oxidized proteins through cysteine-thiol-disulfide exchange, forming an oxidized disulfide T(S-S) in the process. TRx is also directly involved in prevention and repair of damage caused by H2O2-based oxidative stress. Because intracellular reduction of selenite is required for de novo synthesis of Sec selenoproteins, TRx clearly has a central role in all Se physiology. It is assumed that Se's pivotal role in TRx explains why targeted disruption of the TRx1 [69] and T(SH)2 [70] genes are embryonically lethal.
Figure 2.
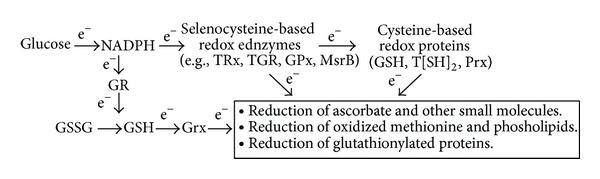
Selenoenzymes are central to providing antioxidant electrons to accomplish reduction of molecules in a number of biochemical processes. NADPH = nicotinamide adenine dinucleotide phosphate; GR = glutathione reductase; T(SH)2 = reduced thioredoxin; GSH = reduced glutathione; TRx = thioredoxin reductase; GSSG = oxidized glutathione; TGR = thioredoxin-glutathione reductase; GPx = glutathione peroxidase; MsrB = methionine sulfoxide reductase; Prx = peroxiredoxin; Grx = glutaredoxin; e− = electron. Levels of plasma GSH, erythrocyte NADH, and NADPH are notably reduced (P < 0.001) in children with autism [16, 32].
Additionally, investigations have shown that TRx1 synthesized without its penultimate Sec is an apoptosis initiator (GRIM-12) [71]. The only difference between the truncated GRIM-12 and full-length TRx1 is the absence of the final two amino acids, Sec and glycine. Truncation occurs when the codon UGA is interpreted as a stop codon instead of a signal for Sec insertion during times of Se deficiency or when its Sec is selectively derivatized. The Sec-deficient form of TRx1, GRIM-12, is a notably powerful apoptosis initiator that rapidly induces cell death [71].
3.3. Roles of GPx in Redox Regulation
GPx (five genetically distinct forms, GPx1–4 and GPx6) are selenoenzymes involved in antioxidant defense and redox regulation and modulation. GPx provide protection against oxidative damage and aid in the maintenance of membrane integrity by using GSH as a cofactor to catalyze reduction of hydrogen peroxide, forming oxidized glutathione (GS-SG) in the process. Thyroid hormone synthesis requires a continuous production of high concentrations of H2O2, which appears to be its rate-limiting step [72–74]. Therefore, since the thyrocyte is continually exposed to potentially toxic concentrations of H2O2 and lipid hydroperoxides, appropriate antioxidant defense systems are essential to control excess oxidative stress. Three of the five GPx are expressed in thyrocytes and thyroid tissue [75–77]. Studies indicate a distinct regulation of expression, secretion, and function of these selenoproteins for controlling thyrocyte growth, differentiation, and function [76–83]. When Se intake is adequate, the intracellular GPx and TRx systems protect the thyrocyte from peroxides; however, in Se deficiency, the thyrocyte's apoptotic response to H2O2 is increased [84]. Furthermore, in iodine deficiency, where hyperstimulation of the thyroid-stimulating hormone (TSH) receptor signals increased H2O2 production, GPx production is also stimulated, thus upregulating antioxidant protection. By virtue of its ability to increase basal metabolism, thyroid hormone increases oxygen utilization, thereby increasing the demand for antioxidant.
GPx4 reduces hydroperoxides of membrane phospholipid fatty acids and has particular relevance for autism. Along with TRx1, TRx2, DIO2, DIO3, and selenoprotein P (SelP), GPx4 is considered an essential selenoprotein whose levels are preserved in brain and endocrine tissues during Se deficiency. Suppression of neuronal GPx4 expression resulted in a selective loss of parvalbumin-expressing GABAergic interneurons [85] that are essential for dopamine-dependent regulation during attention [86] and has been linked to attention deficit hyperactivity disorder (ADHD) [87, 88]. Earlier studies also showed that expression of these interneurons was inhibited when glutathione synthesis was impaired, indicating a critical role for redox status in establishing the capacity for attention [89, 90].
3.4. Selenoenzyme Metabolism and Physiology
In addition to the TRx and GPx selenoenzyme families, further selenoproteins have recently been implicated in processes known to be involved in neurodegenerative diseases, including protein folding, degradation of misfolded membrane proteins, and control of cellular calcium homoeostasis [91]. Cerebral Se deficiency is associated with neurological disorders such as seizures and ataxia [92, 93], consistent with a restriction in the development of inhibitory interneurons. Knockout of selenoprotein synthesis in neurons specifically interfered with development of parvalbumin-expressing GABAergic interneurons, and knockout of GPx4 produced a similar deficit, indicating that these neurons have a particular requirement for Se [85]. Thus impaired selenoprotein synthesis or loss of their activities could contribute to the neurocognitive dysfunction and seizure activity in ASD.
The cerebral cortex, hippocampus, cerebellum, and olfactory bulb express the highest numbers of selenoproteins [94]. The brain and endocrine tissues are preferentially supplied with Se, predominantly through directed distribution and cellular uptake of SelP. Although other selenoproteins uniformly incorporate only a single Sec per molecule, SelP uniquely contains 10 Sec per molecule. Studies with SelP-deficient mice indicate that moderate reductions of brain Se content will impair brain function [95, 96].
Not only is SelP important for Sec transport, but it also appears to have a vital role in neurogenesis. A study of its brain distribution found a remarkably higher concentration in ependymal cells, which are found at the ventricle surface [97]. Ependymal cells are a source of neural stem cells which are produced upon asymmetric cell division and give rise to neuronal, astrocyte and oligodendrocyte lineages in the subependymal region [98]. Neurotrophic growth factors stimulate mitosis of these precursor cells [99] and provide an important source of postnatal neurons. SelP is taken up by neurons via apolipoprotein E receptor 2 (ApoER2), which is localized to synapses, and ApoER2 knockout mice show a decrease in synapse density as well as a decrease in the number of dendritic spines [100, 101].
All brain selenoenzymes are affected by loss of Se in the absence of SelP, but the DIOs, TRxs, and GPxs are the selenoproteins considered to have the most critical roles in the brain and endocrine system. Therefore, loss or compromise of their functions [96] would have dramatic effects on maturation of the neuroendocrine system. The unusual capacity of the brain and endocrine tissues (e.g., pituitary, testes) to retain their selenoenzyme activities during prolonged or even multigenerational deficiency states [43] indicates that their redox regulation effects are important in these tissues. The low GSH level of neurons (~0.2 mM) provides a unique opportunity for redox signaling as a mechanism for epigenetic control. Neurotrophic factors stimulate cysteine uptake and increase both GSH/GS-SG and SAM/SAH in association with a significant increase in DNA methylation [102]. However, Se-dependent redox regulation is more vulnerable to soft electrophiles, positively charged chemical species which bind to selenoproteins with exceptionally high affinity, as discussed below.
4. Genetic Influences and Metabolic Disturbances in ASD
Observations from family studies suggest that ASD has a strong genetic component [103–105], although the failure to identify genetic factors affecting more than a small proportion of ASD cases suggests that multiple etiologies may be responsible for the pathologies and neurobehavioral features of the disorder [106–117]. Moreover, genetic factors may increase the probability of oxidative damage and diminish the body's ability to detoxify ROS and free radicals. Interactions between genetic and environmental factors may potentiate increased oxidative stress in autistic children.
Genetic risk of autism may be related to a differential sensitivity to environmental factors. Using a strict definition of autism, a recent study found a 58% concordance rate for monozygotic male twins and 60% for females and 21% and 27% for male and female dizygotic twins, respectively [118]. Using the broader definition of ASD, monozygotic concordance increased to 77% and 50% for males and females, while dizygotic concordance was 31% and 36%, respectively. These rates are substantially lower than earlier estimates, and the authors concluded that environmental factors are more important than genetic factors, although genetic factors clearly play an important role. Moreover, no individual genetic cause of autism has been identified to account for more than 1%-2% of cases and, with the exception of Rett syndrome, there is no current evidence that ASD is linked to any specific genetic or nongenetic disorder. However, there is evidence suggesting that epigenetic factors and exposures to environmental modifiers may contribute to variable expression of autism-related traits [42]. Variants of major effect genes and numerous common variants with smaller effect genes have been identified in individuals with ASD and related conditions. These genetic variances are providing insights to common pathways and metabolic disturbances affected in ASD, particularly genes involved in oxidative stress and detoxification pathways [15, 119, 120].
Polymorphisms of genes involved in glutathione metabolism, including genes for GPx and glutathione S-transferase (GST), have been reportedly associated with ASD. GPx1 is the predominant and most abundant isoenzyme of GPx and plays an integral role in reducing oxidative stress by catalyzing the reduction of potentially harmful peroxides. Ming et al. [121] found significant disequilibrium in the overall transmission of a sequence polymorphism of GPx1 in ASD. Williams et al. [122] showed that the GSTP1-313A allele may be acting as a teratogenic allele, contributing to the phenotype of the affected child. GST proteins conjugate and detoxify products of oxidative stress and conjugate toxins that produce oxidative stress. By assessing genotypes of mothers and maternal grandparents, it was shown that the GSTP1A haplotype was significantly more frequently transmitted to mothers of individuals with ASD, suggesting that it may be acting in mothers during pregnancy to contribute to the phenotype of autism during fetal development [122].
James et al. [15] examined the frequency of several single nucleotide polymorphisms (SNPs) capable of affecting redox and methylation pathways in autistic subjects. They found significant differences in allele frequencies for the reduced folate carrier (RFC 80G>A), transcobalamin II (TCN2 776 G>C), methylenetetrahydrofolate reductase (MTHFR) 677 C>T and 1298 (A>C), catechol-O-methyltransferase (COMT 472 G>A), and GST M1 between autistic and control cohorts. These differences were associated with abnormal metabolite levels, suggesting that individuals with genetic vulnerability affecting redox and methylation capacity may be linked to a higher risk for autism. Any deficit in the function of selenoproteins could synergize with these genetic risk factors.
DNA copy number variants (CNVs) represent a major category of genetic risk for ASD and are implicated in approximately 10% of cases [123, 124]. Several of the genes likely affected by homozygous deletions are regulated by neuronal activity, and the expression of these genes can change in response to neuronal stimulation. Synapses mature partially as a function of experience-dependent neuronal activity, so disruption of those genes by mutation or copy number variation may alter the process of synaptic development. DNA methylation status is associated with the occurrence of CNVs [125], raising the possibility that impaired methylation capacity could contribute to increased CNVs in ASD.
5. Epigenetic Disturbances in ASD
Epigenetic regulation utilizes covalent modifications such as DNA methylation and the addition/removal of various chemical moieties to histone tails (collectively known as epigenetic marks) to provide stable, transgenerational changes in gene expression without alteration of the underlying nucleotide sequence [126, 127]. Epigenetic marks are dynamic and highly sensitive to cellular changes [128]. Thus, normal physiologic changes in the cellular environment, such as levels of growth factors, hormones, and neurotransmitters, as well as xenobiotic exposure, can translate into modifications in gene expression mediated by epigenetic regulation. Xenobiotic exposures affecting epigenetic status can, therefore, not only produce lifelong consequences, but their effects can be transmitted through germline cells to affect multiple succeeding generations [126, 128].
As noted above, MS exerts powerful control over all methylation reactions via its influence over the SAM/SAH ratio, and MS inhibition by oxidative stress will cause both a decrease in SAM and an increase in SAH, while reducing conditions will have the opposite effect. A twofold increase in MS activity induced by IGF-1 is associated with a twofold increase in global DNA methylation, while inhibition of MS activity by ethanol is associated with a large decrease [41]. Thus, xenobiotics affecting redox status can exert an epigenetic influence.
Beyond its direct epigenetic regulation of gene transcription, DNA methylation also regulates the activity of repetitive transposable elements dispersed throughout the human genome. Transposable elements comprise about 45% of the genome, and their earlier description as “junk DNA” has recently been revised in recognition of their ability to modulate gene transcription, mRNA splicing, micro-RNA formation, and other processes [129]. Reflecting their viral origin, retrotransposons such as the LINE-1 (long interspersed nuclear element-1) family are suppressed by methylation but can replicate and transpose to new locations, especially during early development and especially when methylation is suppressed. Based upon its impressive quantitative contribution to the genome, methylation of LINE-1 has been used as a surrogate for global DNA methylation [130], and factors regulating MS activity affect LINE-1 methylation [131]. LINE-1 retrotransposition is reported to occur at a higher rate in brain than to other tissues [132], and a higher rate was observed in Rett syndrome subjects carrying mutations in the methylated DNA binding protein MeCP2 [130]. Although more studies are needed to clarify their specific contribution, transposable elements such as LINE-1 are poised to provide a global genomic influence during development, so agents affecting their methylation state are likely to disrupt this process.
Oxidative stress and decreased methylation capacity are common in autism and abnormal epigenetic regulation may link the metabolic abnormalities to disruptions in brain development. Other comorbid features of autism, such as autoimmunity and gastrointestinal (GI) dysfunction [133, 134], may reflect similar manifestations of abnormal epigenetic regulation. A genome-wide comparison of DNA methylation in monozygotic twins discordant for autism found numerous differentially methylated regions associated with ASD, and the extent of these differences were correlated with severity of autistic trait scores [135].
From conception to maturation, human development is a highly orchestrated expression of epigenetic regulation, so it is not surprising that genetic and environmental factors adversely affecting oxidative tone and methylation status can contribute to developmental disorders. The exceptionally dynamic redox-dependent epigenetic regulation in the brain increases its vulnerability to neurodevelopmental disorders. Autism is a prominent feature of Rett, Angelman, Prader-Willi, and Fragile-X syndromes, each of which has been linked to interruption of methylation-dependent regulation [136–138]. Therefore, environmental exposures affecting redox and methylation status could reasonably result in neurodevelopment disorders such as ASD.
6. ASD in relation to Exposures to Potentially Neurotoxic Agents
Certain soft electrophiles are known to be neurotoxic at high exposures, presumably due to their effects on sulfur metabolism. Although these electrophilic species are highly interactive with cellular nucleophiles such as thiols, the Se of Sec is by far the strongest intracellular nucleophile. Therefore, selenoproteins are very vulnerable to enzyme inhibition by binding to neurotoxic electrophiles. Their toxic concentrations are generally miniscule in relation to sulfur, toxic levels generally equal or exceed the normal tissue concentrations of Se. Therefore, because of their high reactivity and low molar abundance, selenoenzymes are highly vulnerable to selective inhibition by high concentrations of electrophiles such as Hg. Soft electrophiles such as Hg have larger ionic radii and a more dispersed surface charge, making them more reactive with soft nucleophiles such as the Se of Sec at the active sites of enzymes.
Only certain electrophiles are notably neurotoxic. Those that bind and potentially sequester Se would cooperatively diminish the biological availability of Se for performance of its necessary physiological roles (Figure 1). For example, a multitude of electrophilic agents are naturally present in food in small amounts and the Se-sequestering effects of each would usually be minor. However, their additive effects on selenoenzyme synthesis and function could be detrimental in individuals with compromised Se status or metabolism.
Likewise, additional exposures to electrophiles are encountered in the form of environmental contaminants, such as toxic metals, pesticides, herbicides, and others. In addition, soft electrophiles are present as the active ingredients in many pharmaceuticals and food preservatives, while still others are produced during the degradation of these products. Therefore, instead of examining relationships between ASD incidence and exposures to individual agents, it may be more informative to examine ASD incidence in relation to aggregate exposures to these soft electrophiles and/or their effect on selenoenzyme activity in vulnerable individuals.
6.1. Thio-/Selenoreactive Elements
High exposures to soft electrophiles have the potential to incapacitate various sulfur- and Se-dependent metabolic processes, thus disrupting many redox regulatory mechanisms that are required for healthy cell growth and function, particularly in brain and endocrine tissues. Increased selenium status is known to counteract the adverse effects of elevated exposures to neurotoxic electrophiles such as Hg, cadmium (Cd), lead (Pb), and vanadium (V) [139]. These electrophilic elements may all be capable of selective, irreversible inhibition of selenoenzyme activities similar to the mechanism of Hg toxicity [140, 141]. However, relationships between Se status and the neurotoxic effects of these other elements have not been adequately examined, and additional mechanisms of toxicity have been recognized for some of these elements [142]. Because it is rich in cysteine, hair has often been used to provide a reflection of circulating amounts of thio-reactive electrophiles present in exposed individuals. The concentrations of Hg, As, Cd, or Pb in hair do not indicate consistent relationships with ASD incidence; however, some studies report unusually low concentrations in hair samples and suggest that individuals with ASD have diminished abilities to eliminate toxic metals [143, 144]. This is consistent with the finding that lower Hg concentrations are present in the hair of young (<6 y) children with ASD [144–146] although Majewska et al. [146] found older ASD children have higher hair Hg. Although serum was studied instead of RBC's or whole blood, a large study of Hg in relation to autism [147] reported finding no significant differences between nonautistic and ASD children. This is supported by the finding that no distinctions in expression levels of four genes that are known to respond to metal exposures were noted between ASD and typical children [148]. However, Stamova et al. [149] found a distinctive correlation between gene expression and blood Hg levels in boys with autism suggesting it is associated with a different pattern of gene transcription in response to Hg exposure. The ability of both Hg and Se to exert epigenetic effects was recently demonstrated in embryonic stem cells [150]. Several studies have reported potent toxic effects of methylmercury on neural stem cell differentiation and survival [151, 152], indicating its potential capacity for altering gene expression during development.
To prevent bacterial contamination in multiple-dose vials, thimerosal, the ethylmercury derivative of thiosalicylic acid, has been used as a preservative in various medical products, including vaccines. As autism rates increased, Bernard et al. [153] suggested that vaccine-derived Hg might be a contributing cause, a highly controversial proposal [154–156]. As a result, thimerosal was removed from all pediatric vaccines, except for some influenza vaccines, in the United States starting in 2001, but the incidence of autism continued to rise [157], furthering the doubts that vaccine-derived Hg exposures contributes to autism incidence. While a number of epidemiological studies do not indicate an association between thimerosal exposure and ASD [158, 159], possible associations between developmental disorders with Hg-containing vaccines [157] and delayed or even transgenerational influence of epigenetic changes have been suggested [160]. Such genetic or epigenetic defects of the antioxidant enzyme system could cooperatively interact with other environmental electrophiles and make vulnerable individuals more sensitive to exposure levels that would otherwise be harmless. Since deficits in selenoenzyme synthesis or function can increase the potential for oxidative stress and epigenetic dysregulation, sensitivity to neurotoxins, such as Hg and similar soft electrophiles, may differ among individuals. In this regard, there have been several reports of decreased GPx activity in autism [161, 162] and selective transmission of GPx-1 allelic variants [121]. Interestingly, Paşca et al. [120] found an inverse correlation between homocysteine and GPx activity in autistic subjects, indicating an association between low GPx activity and impaired methylation. Therefore, Castañeda et al. suggested that the removal of thimerosal from vaccines might not be immediately reflected as a reversal of epigenetic effects, especially if they involved effects on germline cells [163]. Notably, lower levels of DNA methylation increase novel insertions of transposable elements and increase the frequency of CNVs in germline cells [164–166], which are also elevated in autism. Therefore, the effects of elemental electrophiles on selenometabolism may be a contributing factor to ASD etiology and/or progression in vulnerable individuals.
6.2. Thio-/Selenoreactive Organic Electrophiles
Organic molecules such as acrylamide, acrolein, and diethyldithiocarbamate are structurally diverse, but these electrophilic species all share the potential to chemically react with strong nucleophiles [167]. Just as for inorganic electrophiles, the most likely target will be the most nucleophilic moieties of enzymes, such as Sec or thiols of Cys residues. Acetaminophen, the most commonly used analgesic and antipyretic drug in much of the world, is associated with toxic effects at high exposures. Excessive intake leads to impaired sulfur metabolism and life-threatening hepatotoxicity, involving depletion of GSH [168]. A survey of parents reported a higher frequency of acetaminophen use after the MMR (measles-mumps-rubella) vaccine for autistic children than for unaffected children [169], leading to the suggestion that use of acetaminophen might be causally linked to an increase in autism rates [170].
Interestingly, the major acetaminophen-binding protein in the liver is Se binding protein-2 (SeBP2) [171, 172]. Both SeBP1 and SeBP2 bind Se, but not in the Sec form characteristic of the genetically encoded selenoproteins. Increased expression of SeBP2 is associated with increased susceptibility to acetaminophen cytotoxicity [173]. In view of the male predominance of autism, it is interesting to note that SeBP2 levels are higher in males [174] and their vulnerability to acetaminophen hepatotoxicity is also greater. Males also display a decreased capacity to restore their GSH levels to normal [175] following high acetaminophen exposures.
N-acetylcysteine protects against acetaminophen-induced hepatotoxicity by maintaining or restoring hepatic concentrations of GSH [176]. Glutathione is required to inactivate N-acetyl-p-benzoquinone imine (NAPQI), a metabolic product of acetaminophen breakdown that is thought to be the proximal cause of hepatotoxicity following acetaminophen overdoses. When excessive quantities of NAPQI are formed, the primary metabolic (glucuronide and sulfate conjugation) pathways apparently become saturated. N-acetylcysteine is thought to counteract toxicity by either reducing NAPQI to the parent compound or providing sulfhydryl for conjugation of this metabolite [176]. If supplementation with a sulfhydryl-containing compound such as N-acetylcysteine can directly inactivate NAPQI, supplementation with Se to restore selenoenzyme status may be an important adjunct therapy to restore healthy redox status in the affected tissues. The significance of the liver in providing Se for delivery to the brain suggests that compromised Se availability in the liver temporarily induced by exposures to NAPQI and/or other electrophiles could diminish the amount of Se the brain receives. Exposures to agents which could cause either prolonged or excessive diminishments in the supply of Se to neuroendocrine tissues may therefore be important factors to consider in relation to ASD.
7. Summary and Conclusions
The nosology of ASD is complicated by the difficulties to differentiate the syndrome into subsets with similar symptoms and distinct etiologies. Children that share the diagnosis of ASD represent more than one distinct pathophysiological condition. Recognizing and distinguishing between groups with separate etiologies require identification of objective laboratory indices that clinicians can use for diagnosis and to monitor progression and treatment effects. The objective of this review is to discuss metabolic defects that may contribute to the onset and pathology of ASD, particularly in relation to Se physiology.
Existing evidence indicates children with ASD have disruptions in GSH metabolism, and that impaired selenoenzyme and thiol metabolic pathways may be involved. These disruptions could occur as the result of multiple exposures to elemental and organic electrophiles (Figure 3) with exacerbations related to congenital vulnerabilities. Therefore, genetic predisposing factors for ASD may be exacerbated secondary to high aggregate exposures to electrophiles acquired through environmental, pharmaceutical, or foodborne routes. Currently, no specific causal agents or conditions have been recognized in ASD and not all children with potentially predisposing congenital defects or environmental exposures acquire ASD. This may indicate that the contributing effects of causal agents have low potency or that more than one predisposing factor is required. Therefore, a confluence of determinants of individual vulnerabilities and sufficiently high exposures to potential causal agents may be required to result in pathological effects. Likewise, the mechanisms of action may additively or synergistically accentuate risks to individuals with underlying genetic, epigenetic, or nutritional susceptibilities.
Figure 3.
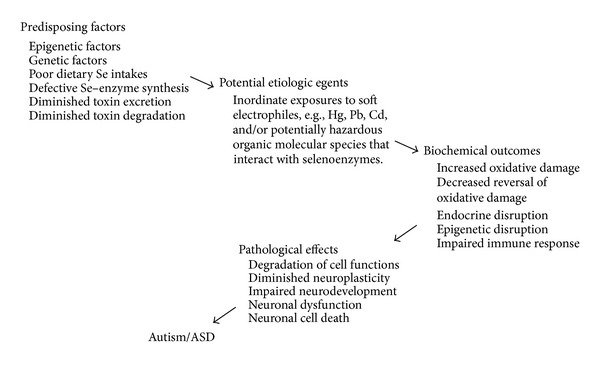
Depiction of potential etiologic contributors to disruptions of selenoenzyme physiology that may lead to disruptions of redox control and pathological consequences of autism and ASD. The factors and agents depicted are not all necessarily involved, but increases in predisposing factors along with additive contributions of increased exposures to thio- and selenoreactive electrophiles would be expected to increase the likelihood of progression to pathology.
Based upon the mechanisms outlined above, interventions designed to diminish oxidative damage and support methylation capacity could improve the health of individuals afflicted with ASD, particularly those with inadequate antioxidant defenses. In such individuals, dietary interventions may offer low-risk approaches with potential for significant improvements in neurodevelopmental outcomes. Although the etiology and pathology of ASD remain poorly resolved, research suggests that afflicted children may benefit from treatments designed to improve their antioxidant capacity.
Acknowledgment
Preparation of this paper was supported by funding from the Autism Research Institute. The sponsors had no influence on the contents of this work or the decision to submit this paper for publication.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends in Neurosciences. 2006;29(7):349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 2.Zahir FR, Brown CJ. Epigenetic impacts on neurodevelopment: pathophysiological mechanisms and genetic modes of action. Pediatric Research. 2011;69(5):92R–100R. doi: 10.1203/PDR.0b013e318213565e. [DOI] [PubMed] [Google Scholar]
- 3.Herbert MR. SHANK3, the synapse, and autism. The New England Journal of Medicine. 2011;365(2):173–175. doi: 10.1056/NEJMcibr1104261. [DOI] [PubMed] [Google Scholar]
- 4.Fombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. The Journal of Clinical Psychiatry. 2005;66(supplement 10):3–8. [PubMed] [Google Scholar]
- 5.Pessah IN, Seegal RF, Lein PJ, et al. Immunologic and neurodevelopmental susceptibilities of autism. NeuroToxicology. 2008;29(3):532–545. doi: 10.1016/j.neuro.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bauman ML. Medical comorbidities in autism: challenges to diagnosis and treatment. Neurotherapeutics. 2010;7(3):320–327. doi: 10.1016/j.nurt.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spence SJ, Schneider MT. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatric Research. 2009;65(6):599–606. doi: 10.1203/01.pdr.0000352115.41382.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators. Prevalence of autism spectrum disorders—autism and developmental disabilities monitoring network, 14 sites, United States, 2008. Morbidity and Mortality Weekly Report: Surveillance Summaries. 2012;61(3):1–19. [PubMed] [Google Scholar]
- 9.Blumberg SJ, Bramlett MD, Kogan MD, Schieve LA, Jones JR, Lu MC. Changes in prevalence of parent-reported autism spectrum disorder in school-aged U.S. children: 2007 to 2011-2012. National Health Statistics Reports. 2013;(65):1–7. [PubMed] [Google Scholar]
- 10.Taylor B. Vaccines and the changing epidemiology of autism. Child. 2006;32(5):511–519. doi: 10.1111/j.1365-2214.2006.00655.x. [DOI] [PubMed] [Google Scholar]
- 11.Shattuck PT. The contribution of diagnostic substitution to the growing administrative prevalence of autism in US special education. Pediatrics. 2006;117(4):1028–1037. doi: 10.1542/peds.2005-1516. [DOI] [PubMed] [Google Scholar]
- 12.Jick H, Kaye JA. Epidemiology and possible causes of autism. Pharmacotherapy. 2003;23(12):1524–1530. doi: 10.1592/phco.23.15.1524.31955. [DOI] [PubMed] [Google Scholar]
- 13.McDonald ME, Paul JF. Timing of increased autistic disorder cumulative incidence. Environmental Science and Technology. 2010;44(6):2112–2118. doi: 10.1021/es902057k. [DOI] [PubMed] [Google Scholar]
- 14.James SJ, Cutler P, Melnyk S, et al. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. The American Journal of Clinical Nutrition. 2004;80(6):1611–1617. doi: 10.1093/ajcn/80.6.1611. [DOI] [PubMed] [Google Scholar]
- 15.James SJ, Melnyk S, Jernigan S, et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. The American Journal of Medical Genetics B. 2006;141(8):947–956. doi: 10.1002/ajmg.b.30366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geier DA, Geier MR. A clinical and laboratory evaluation of methionine cycle-transsulfuration and androgen pathway markers in children with autistic disorders. Hormone Research. 2006;66(4):182–188. doi: 10.1159/000094467. [DOI] [PubMed] [Google Scholar]
- 17.Adams JB, Baral M, Geis E, et al. The severity of autism is associated with toxic metal body burden and red blood cell glutathione levels. Journal of Toxicology. 2009;2009:7 pages. doi: 10.1155/2009/532640.532640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paşca SP, Dronca E, Kaucsár T, et al. One carbon metabolism disturbances and the C677T MTHFR gene polymorphism in children with autism spectrum disorders. Journal of Cellular and Molecular Medicine. 2009;13(10):4229–4238. doi: 10.1111/j.1582-4934.2008.00463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pastural É, Ritchie S, Lu Y, et al. Novel plasma phospholipid biomarkers of autism: mitochondrial dysfunction as a putative causative mechanism. Prostaglandins Leukotrienes and Essential Fatty Acids. 2009;81(4):253–264. doi: 10.1016/j.plefa.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Al-Gadani Y, El-Ansary A, Attas O, Al-Ayadhi L. Metabolic biomarkers related to oxidative stress and antioxidant status in Saudi autistic children. Clinical Biochemistry. 2009;42(10-11):1032–1040. doi: 10.1016/j.clinbiochem.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Melnyk S, Fuchs GJ, Schulz E, et al. Metabolic imbalance associated with methylation dysregulation and oxidative damage in children with autism. Journal of Autism and Developmental Disorders. 2012;42(3):367–377. doi: 10.1007/s10803-011-1260-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.James SJ, Melnyk S, Fuchs G, et al. Efficacy of methylcobalamin and folinic acid treatment on glutathione redox status in children with autism. The American Journal of Clinical Nutrition. 2009;89(1):425–430. doi: 10.3945/ajcn.2008.26615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geier DA, Kern JK, Garver CR, et al. Biomarkers of environmental toxicity and susceptibility in autism. Journal of the Neurological Sciences. 2009;280(1):101–108. doi: 10.1016/j.jns.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 24.Geier DA, Kern JK, Garver CR, Adams JB, Audhya T, Geier MR. A prospective study of transsulfuration biomarkers in autistic disorders. Neurochemical Research. 2009;34(2):386–393. doi: 10.1007/s11064-008-9782-x. [DOI] [PubMed] [Google Scholar]
- 25.Chen J, Berry MJ. Selenium and selenoproteins in the brain and brain diseases. Journal of Neurochemistry. 2003;86(1):1–12. doi: 10.1046/j.1471-4159.2003.01854.x. [DOI] [PubMed] [Google Scholar]
- 26.Schweizer U, Bräuer AU, Köhrle J, Nitsch R, Savaskan NE. Selenium and brain function: a poorly recognized liaison. Brain Research Reviews. 2004;45(3):164–178. doi: 10.1016/j.brainresrev.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 27.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Biology and Medicine. 2010;48(6):749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chauhan A, Chauhan V, Brown WT, Cohen I. Oxidative stress in autism: increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin—the antioxidant proteins. Life Sciences. 2004;75(21):2539–2549. doi: 10.1016/j.lfs.2004.04.038. [DOI] [PubMed] [Google Scholar]
- 29.Zoroglu SS, Armutcu F, Ozen S, et al. Increased oxidative stress and altered activities of erythrocyte free radical scavenging enzymes in autism. European Archives of Psychiatry and Clinical Neuroscience. 2004;254(3):143–147. doi: 10.1007/s00406-004-0456-7. [DOI] [PubMed] [Google Scholar]
- 30.Ming X, Stein TP, Brimacombe M, Johnson WG, Lambert GH, Wagner GC. Increased excretion of a lipid peroxidation biomarker in autism. Prostaglandins Leukotrienes and Essential Fatty Acids. 2005;73(5):379–384. doi: 10.1016/j.plefa.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Sacco R, Curatolo P, Manzi B, et al. Principal pathogenetic components and biological endophenotypes in autism spectrum disorders. Autism Research. 2010;3(5):237–252. doi: 10.1002/aur.151. [DOI] [PubMed] [Google Scholar]
- 32.Adams JB, Audhya T, McDonough-Means S, et al. Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutrition and Metabolism. 2011;8, article 34 doi: 10.1186/1743-7075-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geier DA, Geier MR. A case series of children with apparent mercury toxic encephalopathies manifesting with clinical symptoms of regressive autistic disorders. Journal of Toxicology and Environmental Health A. 2007;70(10):837–851. doi: 10.1080/15287390701212141. [DOI] [PubMed] [Google Scholar]
- 34.Frustaci A, Neri M, Cesario A, et al. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radical Biology and Medicine. 2012;52(10):2128–2141. doi: 10.1016/j.freeradbiomed.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 35.Chauhan A, Audhya T, Chauhan V. Brain region-specific glutathione redox imbalance in autism. Neurochemical Research. 2012;37(8):1681–1689. doi: 10.1007/s11064-012-0775-4. [DOI] [PubMed] [Google Scholar]
- 36.Rose S, Melnyk S, Pavliv O, et al. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Translational Psychiatry. 2012;2(article e134) doi: 10.1038/tp.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu F, Chauhan V, Chauhan A. Impaired synthesis and antioxidant defense of glutathione in the cerebellum of autistic subjects: alterations in the activities and protein expression of glutathione-related enzymes. Free Radical Biology and Medicine. 2013;65:488–496. doi: 10.1016/j.freeradbiomed.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 38.Deth R, Muratore C, Benzecry J, Power-Charnitsky V-A, Waly M. How environmental and genetic factors combine to cause autism: a redox/methylation hypothesis. NeuroToxicology. 2008;29(1):190–201. doi: 10.1016/j.neuro.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 39.Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry. 2000;39(42):13005–13011. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- 40.Bandarian V, Ludwig ML, Matthews RG. Factors modulating conformational equilibria in large modular proteins: a case study with cobalamin-dependent methionine synthase. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8156–8163. doi: 10.1073/pnas.1133218100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waly M, Olteanu H, Banerjee R, et al. Activation of methionine synthase by insulin-like growth factor-1 and dopamine: a target for neurodevelopmental toxins and thimerosal. Molecular Psychiatry. 2004;9(4):358–370. doi: 10.1038/sj.mp.4001476. [DOI] [PubMed] [Google Scholar]
- 42.Muratore CR, Hodgson NW, Trivedi MS, et al. Age-dependent decrease and alternative splicing of methionine synthase mRNA in human cerebral cortex and an accelerated decrease in autism. PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0056927.e56927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kyriakopoulos A, Behne D. Selenium-containing proteins in mammals and other forms of life. Reviews of Physiology Biochemistry and Pharmacology. 2002;145:1–46. doi: 10.1007/BFb0116430. [DOI] [PubMed] [Google Scholar]
- 44.Gladyshev VN, Martin-Romero FJ, Xu X-M, et al. Molecular biology of selenium and its role in cancer, AIDS and other human diseases. Recent Research Developments in Biochemistry. 1999;1:145–167. [Google Scholar]
- 45.Low SC, Berry MJ. Knowing when not to stop: selenocysteine incorporation in eukaryotes. Trends in Biochemical Sciences. 1996;21(6):203–208. [PubMed] [Google Scholar]
- 46.Gromer S, Eubel JK, Lee BL, Jacob J. Human selenoproteins at a glance. Cellular and Molecular Life Sciences. 2005;62(21):2414–2437. doi: 10.1007/s00018-005-5143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jory J, McGinnis WR. Red-cell trace minerals in children with autism. The American Journal of Biochemistry and Biotechnology. 2008;4(2):101–104. [Google Scholar]
- 48.Lubkowska A, Sobieraj W. Concentrations of magnesium, calcium, iron, selenium, zinc and copper in the hair of autistic children. Trace Elements and Electrolytes. 2009;26(2):72–77. [Google Scholar]
- 49.Priya MDL, Geetha A. Level of trace elements (copper, zinc, magnesium and selenium) and toxic elements (lead and mercury) in the hair and nail of children with autism. Biological Trace Element Research. 2011;142(2):148–158. doi: 10.1007/s12011-010-8766-2. [DOI] [PubMed] [Google Scholar]
- 50.de Palma G, Catalani S, Franco A, Brighenti M, Apostoli P. Lack of correlation between metallic elements analyzed in hair by ICP-MS and autism. Journal of Autism and Developmental Disorders. 2012;42(3):342–353. doi: 10.1007/s10803-011-1245-6. [DOI] [PubMed] [Google Scholar]
- 51.Bernal J. Thyroid hormone receptors in brain development and function. Nature Reviews Endocrinology. 2007;3(3):249–259. doi: 10.1038/ncpendmet0424. [DOI] [PubMed] [Google Scholar]
- 52.Amara IB, Fetoui H, Guermazi F, Zeghal N. Dietary selenium addition improves cerebrum and cerebellum impairments induced by methimazole in suckling rats. International Journal of Developmental Neuroscience. 2009;27(7):719–726. doi: 10.1016/j.ijdevneu.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 53.Kohrle J, Gartner R. Selenium and thyroid. Best Practice & Research Clinical Endocrinology & Metabolism. 2009;23(6):815–827. doi: 10.1016/j.beem.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 54.Mittag J, Behrends T, Hoefig CS, Vennström B, Schomburg L. Thyroid hormones regulate selenoprotein expression and selenium status in mice. PLoS ONE. 2010;5(9) doi: 10.1371/journal.pone.0012931.e12931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crantz FR, Silva JE, Larsen PR. An analysis of the sources and quantity of 3,5,3′-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology. 1982;110(2):367–375. doi: 10.1210/endo-110-2-367. [DOI] [PubMed] [Google Scholar]
- 56.Courtin F, Chantoux F, Francon J. Thyroid hormone metabolism by glial cells in primary culture. Molecular and Cellular Endocrinology. 1986;48(2-3):167–178. doi: 10.1016/0303-7207(86)90039-0. [DOI] [PubMed] [Google Scholar]
- 57.Safran M, Farwell AP, Leonard JL. Evidence that type II 5′-deiodinase is not a selenoprotein. The Journal of Biological Chemistry. 1991;266(21):13477–13480. [PubMed] [Google Scholar]
- 58.Lei XG, Cheng W-H, McClung JP. Metabolic regulation and function of glutathione peroxidase-1. Annual Review of Nutrition. 2007;27:41–61. doi: 10.1146/annurev.nutr.27.061406.093716. [DOI] [PubMed] [Google Scholar]
- 59.Wingler K, Böcher M, Flohé L, Kollmus H, Brigelius-Flohé R. mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. European Journal of Biochemistry. 1999;259(1-2):149–157. doi: 10.1046/j.1432-1327.1999.00012.x. [DOI] [PubMed] [Google Scholar]
- 60.Lee YS, Kim AY, Choi JW, et al. Dysregulation of adipose glutathione peroxidase 3 in obesity contributes to local and systemic oxidative stress. Molecular Endocrinology. 2008;22(9):2176–2189. doi: 10.1210/me.2008-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conrad M, Schneider M, Seiler A, Bornkamm GW. Physiological role of phospholipid hydroperoxide glutathione peroxidase in mammals. Biological Chemistry. 2007;388(10):1019–1025. doi: 10.1515/BC.2007.130. [DOI] [PubMed] [Google Scholar]
- 62.Linster CL, van Schaftingen E. Vitamin C: biosynthesis, recycling and degradation in mammals. The FEBS Journal. 2007;274(1):1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 63.Arnér ESJ. Focus on mammalian thioredoxin reductases—important selenoproteins with versatile functions. Biochimica et Biophysica Acta. 2009;1790(6):495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 64.Reeves MA, Hoffmann PR. The human selenoproteome: recent insights into functions and regulation. Cellular and Molecular Life Sciences. 2009;66(15):2457–2478. doi: 10.1007/s00018-009-0032-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocrine Reviews. 2002;23(1):38–89. doi: 10.1210/edrv.23.1.0455. [DOI] [PubMed] [Google Scholar]
- 66.Reeves MA, Bellinger FP, Berry MJ. The neuroprotective functions of selenoprotein M and its role in cytosolic calcium regulation. Antioxidants and Redox Signaling. 2010;12(7):809–818. doi: 10.1089/ars.2009.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dikiy A, Novoselov SV, Fomenko DE, et al. SelT, SelW, SelH, and Rdx 12: genomics and molecular insights into the functions of selenoproteins of a novel thioredoxin-like family. Biochemistry. 2007;46(23):6871–6882. doi: 10.1021/bi602462q. [DOI] [PubMed] [Google Scholar]
- 68.Nordberg J, Arnér ESJ. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radical Biology and Medicine. 2001;31(11):1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 69.Jakupoglu C, Przemeck GKH, Schneider M, et al. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Molecular and Cellular Biology. 2005;25(5):1980–1988. doi: 10.1128/MCB.25.5.1980-1988.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsui M, Oshima M, Oshima H, et al. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Developmental Biology. 1996;178(1):179–185. doi: 10.1006/dbio.1996.0208. [DOI] [PubMed] [Google Scholar]
- 71.Anestål K, Arnér ESJ. Rapid induction of cell death by selenium-compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocysteine. The Journal of Biological Chemistry. 2003;278(18):15966–15972. doi: 10.1074/jbc.M210733200. [DOI] [PubMed] [Google Scholar]
- 72.Corvilain B, van Sande J, Laurent E, Dumont JE. The H2O2-generating system modulates protein iodination and the activity of the pentose phosphate pathway in dog thyroid. Endocrinology. 1991;128(2):779–785. doi: 10.1210/endo-128-2-779. [DOI] [PubMed] [Google Scholar]
- 73.Corvilain B, Laurent E, Lecomte M, van sande J, Dumont JE. Role of the cyclic adenosine 3′,5′-monophosphate and the phosphatidylinositol-Ca 2 + cascades in mediating the effects of thyrotropin and iodide on hormone synthesis and secretion in human thyroid slices. The Journal of Clinical Endocrinology and Metabolism. 1994;79(1):152–159. doi: 10.1210/jcem.79.1.8027219. [DOI] [PubMed] [Google Scholar]
- 74.Björkman U, Ekholm R. Generation of H2O2 in isolated porcine thyroid follicles. Endocrinology. 1984;115(1):392–398. doi: 10.1210/endo-115-1-392. [DOI] [PubMed] [Google Scholar]
- 75.Villette S, Bermano G, Arthur JR, Hesketh JE. Thyroid stimulating hormone and selenium supply interact to regulate selenoenzyme gene expression in thyroid cells (FRTL-5) in culture. FEBS Letters. 1998;438(1-2):81–84. doi: 10.1016/s0014-5793(98)01280-0. [DOI] [PubMed] [Google Scholar]
- 76.Zagrodzki P, Nicol F, McCoy MA, et al. Iodine deficiency in cattle: compensatory changes in thyroidal selenoenzymes. Research in Veterinary Science. 1998;64(3):209–211. doi: 10.1016/s0034-5288(98)90127-8. [DOI] [PubMed] [Google Scholar]
- 77.Howie AF, Walker SW, Akesson B, Arthur JR, Beckett GJ. Thyroidal extracellular glutathione peroxidase: a potential regulator of thyroid-hormone synthesis. Biochemical Journal. 1995;308(3):713–717. doi: 10.1042/bj3080713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Howie AF, Arthur JR, Nicol F, Walker SW, Beech SG, Beckett GJ. Identification of a 57-kilodalton selenoprotein in human thyrocytes as thioredoxin reductase and evidence that its expression is regulated through the calcium-phosphoinositol signaling pathway. The Journal of Clinical Endocrinology and Metabolism. 1998;83(6):2052–2058. doi: 10.1210/jcem.83.6.4875. [DOI] [PubMed] [Google Scholar]
- 79.Oertel M, Hesch RO, Kohrle J. Expression of iodothyronine deiodinase in cultured thyroid cells. Experimental and Clinical Endocrinology. 1991;97(2-3):182–186. doi: 10.1055/s-0029-1211061. [DOI] [PubMed] [Google Scholar]
- 80.Schreck R, Schnieders F, Schmutzler C, Kohrle J. Retinoids stimulate type I iodothyronine 5′-deiodinase activity in human follicular thyroid carcinoma cell lines. The Journal of Clinical Endocrinology and Metabolism. 1994;79(3):791–798. doi: 10.1210/jcem.79.3.8077363. [DOI] [PubMed] [Google Scholar]
- 81.Beech SG, Walker SW, Dorrance AM, et al. The role of thyroidal type-I iodothyronine deiodinase in tri-iodothyronine production by human and sheep thyrocytes in primary culture. The Journal of Endocrinology. 1993;136(3):361–370. doi: 10.1677/joe.0.1360361. [DOI] [PubMed] [Google Scholar]
- 82.Beech SG, Walker SW, Beckett GJ, Arthur JR, Nicol F, Lee D. Effect of selenium depletion on thyroidal type-I iodothyronine deiodinase activity in isolated human thyrocytes and rat thyroid and liver. Analyst. 1995;120(3):827–831. doi: 10.1039/an9952000827. [DOI] [PubMed] [Google Scholar]
- 83.Bermano G, Nicol F, Dyer JA, et al. Tissue-specific regulation of selenoenzyme gene expression during selenium deficiency in rats. Biochemical Journal. 1995;311(2):425–430. doi: 10.1042/bj3110425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Demelash A, Karlsson J-O, Nilsson M, Björkman U. Selenium has a protective role in caspase-3-dependent apoptosis induced by H2O2 in primary cultured pig thyrocytes. European Journal of Endocrinology. 2004;150(6):841–849. doi: 10.1530/eje.0.1500841. [DOI] [PubMed] [Google Scholar]
- 85.Wirth EK, Conrad M, Winterer J, et al. Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. The FASEB Journal. 2010;24(3):844–852. doi: 10.1096/fj.09-143974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nature Reviews Neuroscience. 2007;8(1):45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- 87.de Almeida J, Mengod G. D2 and D4 dopamine receptor mRNA distribution in pyramidal neurons and GABAergic subpopulations in monkey prefrontal cortex: implications for schizophrenia treatment. Neuroscience. 2010;170(4):1133–1139. doi: 10.1016/j.neuroscience.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 88.Swanson JM, Kinsbourne M, Nigg J, et al. Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychology Review. 2007;17(1):39–59. doi: 10.1007/s11065-007-9019-9. [DOI] [PubMed] [Google Scholar]
- 89.Cabungcal J-H, Nicolas D, Kraftsik R, Cuénod M, Do KQ, Hornung J-P. Glutathione deficit during development induces anomalies in the rat anterior cingulate GABAergic neurons: relevance to schizophrenia. Neurobiology of Disease. 2006;22(3):624–637. doi: 10.1016/j.nbd.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 90.Steullet P, Cabungcal J-H, Kulak A, et al. Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. The Journal of Neuroscience. 2010;30(7):2547–2558. doi: 10.1523/JNEUROSCI.3857-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shchedrina VA, Zhang Y, Labunskyy VM, Hatfield DL, Gladyshev VN. Structure—function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antioxidants and Redox Signaling. 2010;12(7):839–849. doi: 10.1089/ars.2009.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schomburg L, Schweizer U, Holtmann B, Flohé L, Sendtner M, Köhrle J. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochemical Journal. 2003;370(2):397–402. doi: 10.1042/BJ20021853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hill KE, Zhou J, McMahan WJ, et al. Deletion of selenoprotein P alters distribution of selenium in the mouse. The Journal of Biological Chemistry. 2003;278(16):13640–13646. doi: 10.1074/jbc.M300755200. [DOI] [PubMed] [Google Scholar]
- 94.Zhang Y, Zhou Y, Schweizer U, et al. Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. The Journal of Biological Chemistry. 2008;283(4):2427–2438. doi: 10.1074/jbc.M707951200. [DOI] [PubMed] [Google Scholar]
- 95.Hill KE, Zhou J, McMahan WJ, Motley AK, Burk RF. Neurological dysfunction occurs in mice with targeted deletion of the selenoprotein P gene. The Journal of Nutrition. 2004;134(1):157–161. doi: 10.1093/jn/134.1.157. [DOI] [PubMed] [Google Scholar]
- 96.Sunde RA, Raines AM, Barnes KM, Evenson JK. Selenium status highly regulates selenoprotein mRNA levels for only a subset of the selenoproteins in the selenoproteome. Bioscience Reports. 2009;29(5):329–338. doi: 10.1042/BSR20080146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Scharpf M, Schweizer U, Arzberger T, Roggendorf W, Schomburg L, Köhrle J. Neuronal and ependymal expression of selenoprotein P in the human brain. Journal of Neural Transmission. 2007;114(7):877–884. doi: 10.1007/s00702-006-0617-0. [DOI] [PubMed] [Google Scholar]
- 98.Kazanis I, Ffrench-Constant C. The number of stem cells in the subependymal zone of the adult rodent brain is correlated with the number of ependymal cells and not with the volume of the niche. Stem Cells and Development. 2012;21(7):1090–1096. doi: 10.1089/scd.2011.0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pérez-Martín M, Cifuentes M, Grondona JM, et al. IGF-I stimulates neurogenesis in the hypothalamus of adult rats. European Journal of Neuroscience. 2010;31(9):1533–1548. doi: 10.1111/j.1460-9568.2010.07220.x. [DOI] [PubMed] [Google Scholar]
- 100.Olson GE, Winfrey VP, NagDas SK, Hill KE, Burk RF. Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. The Journal of Biological Chemistry. 2007;282(16):12290–12297. doi: 10.1074/jbc.M611403200. [DOI] [PubMed] [Google Scholar]
- 101.Burk RF, Hill KE, Olson GE, et al. Deletion of apolipoprotein E receptor-2 in mice lowers brain selenium and causes severe neurological dysfunction and death when a low-selenium diet is fed. The Journal of Neuroscience. 2007;27(23):6207–6211. doi: 10.1523/JNEUROSCI.1153-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hodgson N, Trivedi M, Muratore C, Li S, Deth R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. Journal of Alzheimer's Disease. 2013;36:197–209. doi: 10.3233/JAD-130101. [DOI] [PubMed] [Google Scholar]
- 103.Bailey A, Le Couteur A, Gottesman I, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychological Medicine. 1995;25(1):63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 104.Hurley RSE, Losh M, Parlier M, Reznick JS, Piven J. The broad autism phenotype questionnaire. Journal of Autism and Developmental Disorders. 2007;37(9):1679–1690. doi: 10.1007/s10803-006-0299-3. [DOI] [PubMed] [Google Scholar]
- 105.Constantino JN, Todd RD. Intergenerational transmission of subthreshold autistic traits in the general population. Biological Psychiatry. 2005;57(6):655–660. doi: 10.1016/j.biopsych.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 106.Jamain S, Quach H, Betancur C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nature Genetics. 2003;34(1):27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Durand CM, Betancur C, Boeckers TM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nature Genetics. 2007;39(1):25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. The American Journal of Human Genetics. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weiss LA, Arking DE, Gene Discovery Project of Johns Hopkins & the Autism Consortium, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461(7265):802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. The New England Journal of Medicine. 2008;358(7):667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 112.Kumar RA, Karamohamed S, Sudi J, et al. Recurrent 16p11.2 microdeletions in autism. Human Molecular Genetics. 2008;17(4):628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 113.Wang K, Zhang H, Ma D, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459(7246):528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fernandez BA, Roberts W, Chung B, et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. Journal of Medical Genetics. 2010;47(3):195–203. doi: 10.1136/jmg.2009.069369. [DOI] [PubMed] [Google Scholar]
- 115.Glessner JT, Wang K, Cai G, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–572. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism. Nature. 2010;466(7304):368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Devlin B, Melhem N, Roeder K. Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain Research. 2011;1380:78–84. doi: 10.1016/j.brainres.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Archives of General Psychiatry. 2011;68(11):1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Serajee FJ, Nabi R, Zhong H, Huq AHMM. Polymorphisms in xenobiotic metabolism genes and autism. Journal of Child Neurology. 2004;19(6):413–417. doi: 10.1177/088307380401900603. [DOI] [PubMed] [Google Scholar]
- 120.Paşca SP, Nemeş B, Vlase L, et al. High levels of homocysteine and low serum paraoxonase 1 arylesterase activity in children with autism. Life Sciences. 2006;78(19):2244–2248. doi: 10.1016/j.lfs.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 121.Ming X, Johnson WG, Stenroos ES, Mars A, Lambert GH, Buyske S. Genetic variant of glutathione peroxidase 1 in autism. Brain and Development. 2010;32(2):105–109. doi: 10.1016/j.braindev.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 122.Williams TA, Mars AE, Buyske SG, et al. Risk of autistic disorder in affected offspring of mothers with a glutathione S-transferase P1 haplotype. Archives of Pediatrics and Adolescent Medicine. 2007;161(4):356–361. doi: 10.1001/archpedi.161.4.356. [DOI] [PubMed] [Google Scholar]
- 123.Merikangas AK, Corvin AP, Gallagher L. Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends in Genetics. 2009;25(12):536–544. doi: 10.1016/j.tig.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 124.Guerrero-Bosagna C, Settles M, Lucker B, Skinner MK. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS ONE. 2010;5(9) doi: 10.1371/journal.pone.0013100.e13100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Andrews J, Kennette W, Pilon J, et al. Multi-platform whole-genome microarray analyses refine the epigenetic signature of breast cancer metastasis with gene expression and copy number. PLoS ONE. 2010;5(1) doi: 10.1371/journal.pone.0008665.e8665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McCarrey JR. The epigenome as a target for heritable environmental disruptions of cellular function. Molecular and Cellular Endocrinology. 2012;354(1-2):9–15. doi: 10.1016/j.mce.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 127.Murgatroyd C, Spengler D. Epigenetics of early child development. Frontiers in Psychiatry. 2011;2(article 16) doi: 10.3389/fpsyt.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Skinner MK, Manikkam M, Guerrero-Bosagna C. Epigenetic transgenerational actions of endocrine disruptors. Reproductive Toxicology. 2011;31(3):337–343. doi: 10.1016/j.reprotox.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annual Review of Genomics and Human Genetics. 2011;12:187–215. doi: 10.1146/annurev-genom-082509-141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Muotri AR, Marchetto MCN, Coufal NG, et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010;468(7322):443–446. doi: 10.1038/nature09544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wernimont SM, Clark AG, Stover PJ, et al. Folate network genetic variation, plasma homocysteine, and global genomic methylation content: a genetic association study. BMC Medical Genetics. 2011;12, article 150 doi: 10.1186/1471-2350-12-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Singer T, McConnell MJ, Marchetto MCN, Coufal NG, Gage FH. LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends in Neurosciences. 2010;33(8):345–354. doi: 10.1016/j.tins.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Buie T, Campbell DB, Fuchs GJ, III, et al. Evaluation, diagnosis, and treatment of gastrointestinal disorders in individuals with ASDs: a consensus report. Pediatrics. 2010;125(1, supplement):S1–S18. doi: 10.1542/peds.2009-1878C. [DOI] [PubMed] [Google Scholar]
- 134.Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Molecular Psychiatry. 2012;17(4):389–401. doi: 10.1038/mp.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wong CCY, Meaburn EL, Ronald A, et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Molecular Psychiatry. 2013 doi: 10.1038/mp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jiang Y-H, Tsai T-F, Bressler J, Beaudet AL. Imprinting in Angelman and Prader-Willi syndromes. Current Opinion in Genetics and Development. 1998;8(3):334–342. doi: 10.1016/s0959-437x(98)80091-9. [DOI] [PubMed] [Google Scholar]
- 137.Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nature Reviews Genetics. 2006;7(6):415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- 138.Dolzhanskaya N, Merz G, Aletta JM, Denman RB. Methylation regulates the intracellular protein-protein and protein-RNA interactions of FMRP. Journal of Cell Science. 2006;119(9):1933–1946. doi: 10.1242/jcs.02882. [DOI] [PubMed] [Google Scholar]
- 139.Whanger PD. Selenium and the brain: a review. Nutritional Neuroscience. 2001;4(2):81–97. doi: 10.1080/1028415x.2001.11747353. [DOI] [PubMed] [Google Scholar]
- 140.Ralston NVC, Raymond LJ. Dietary selenium’s protective effects against methylmercury toxicity. Toxicology. 2010;278(1):112–123. doi: 10.1016/j.tox.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 141.Carvalho CML, Chew E-H, Hashemy SI, Lu J, Holmgren A. Inhibition of the human thioredoxin system: a molecular mechanism of mercury toxicity. The Journal of Biological Chemistry. 2008;283(18):11913–11923. doi: 10.1074/jbc.M710133200. [DOI] [PubMed] [Google Scholar]
- 142.Neal AP, Guilarte TR. Mechanisms of lead and manganese neurotoxicity. Toxicology Research. 2013;(2):99–114. doi: 10.1039/C2TX20064C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kern JK, Grannemann BD, Trivedi MH, Adams JB. Sulfhydryl-reactive metals in autism. Journal of Toxicology and Environmental Health A. 2007;70(8):715–721. [Google Scholar]
- 144.Obrenovich ME, Shamberger RJ, Lonsdale D. Altered heavy metals and transketolase found in autistic spectrum disorder. Biological Trace Element Research. 2011;144(1–3):475–486. doi: 10.1007/s12011-011-9146-2. [DOI] [PubMed] [Google Scholar]
- 145.Holmes AS, Blaxill MF, Haley BE. Reduced levels of mercury in first baby haircuts of autistic children. International Journal of Toxicology. 2003;22(4):277–285. doi: 10.1080/10915810305120. [DOI] [PubMed] [Google Scholar]
- 146.Majewska MD, Urbanowicz E, Rok-Bujko P, Namysłowska I, Mierzejewski P. Age-dependent lower or higher levels of hair mercury in autistic children than in healthy controls. Acta Neurobiologiae Experimentalis. 2010;70(2):196–208. doi: 10.55782/ane-2010-1791. [DOI] [PubMed] [Google Scholar]
- 147.Hertz-Picciotto I, Green PG, Delwiche L, Hansen R, Walker C, Pessah IN. Blood mercury concentrations in CHARGE study children with and without autism. Environmental Health Perspectives. 2010;118(1):161–166. doi: 10.1289/ehp.0900736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Owens SE, Summar ML, Ryckman KK, et al. Lack of association between autism and four heavy metal regulatory genes. NeuroToxicology. 2011;32(6):769–775. doi: 10.1016/j.neuro.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stamova B, Green PG, Tian Y, et al. Correlations between gene expression and mercury levels in blood of boys with and without autism. Neurotoxicity Research. 2011;19(1):31–48. doi: 10.1007/s12640-009-9137-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Arai Y, Ohgane J, Yagi S, et al. Epigenetic assessment of environmental chemicals detected in maternal peripheral and cord blood samples. The Journal of Reproduction and Development. 2011;57(4):507–517. doi: 10.1262/jrd.11-034a. [DOI] [PubMed] [Google Scholar]
- 151.Stummann TC, Hareng L, Bremer S. Hazard assessment of methylmercury toxicity to neuronal induction in embryogenesis using human embryonic stem cells. Toxicology. 2009;257(3):117–126. doi: 10.1016/j.tox.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 152.Tamm C, Duckworth J, Hermanson O, Ceccatelli S. High susceptibility of neural stem cells to methylmercury toxicity: effects on cell survival and neuronal differentiation. Journal of Neurochemistry. 2006;97(1):69–78. doi: 10.1111/j.1471-4159.2006.03718.x. [DOI] [PubMed] [Google Scholar]
- 153.Bernard S, Enayati A, Redwood L, Roger H, Binstock T. Autism: a novel form of mercury poisoning. Medical Hypotheses. 2001;56(4):462–471. doi: 10.1054/mehy.2000.1281. [DOI] [PubMed] [Google Scholar]
- 154.Dórea JG. Making sense of epidemiological studies of young children exposed to thimerosal in vaccines. Clinica Chimica Acta. 2010;411(21-22):1580–1586. doi: 10.1016/j.cca.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 155.Geier DA, Hooker BS, Kern JK, King PG, Sykes LK, Geier MR. A two-phase study evaluating the relationship between Thimerosal-containing vaccine administration and the risk for an autism spectrum disorder diagnosis in the United States. Translational Neurodegeneration. 2013;2(1, article 25) doi: 10.1186/2047-9158-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gallagher CM, Goodman MS. Hepatitis B vaccination of male neonates and autism diagnosis, NHIS 1997–2002. Journal of Toxicology and Environmental Health A. 2010;73(24):1665–1677. doi: 10.1080/15287394.2010.519317. [DOI] [PubMed] [Google Scholar]
- 157.Schechter R, Grether JK. Continuing increases in autism reported to California’s developmental services system: mercury in retrograde. Archives of General Psychiatry. 2008;65(1):19–24. doi: 10.1001/archgenpsychiatry.2007.1. [DOI] [PubMed] [Google Scholar]
- 158.Schultz ST, Klonoff-Cohen HS, Wingard DL, Akshoomoff NA, Macera CA, Ming J. Acetaminophen (paracetamol) use, measles-mumps-rubella vaccination, and autistic disorder: the results of a parent survey. Autism. 2008;12(3):293–307. doi: 10.1177/1362361307089518. [DOI] [PubMed] [Google Scholar]
- 159.Price CS, Thompson WW, Goodson B, et al. Prenatal and infant exposure to thimerosal from vaccines and immunoglobulins and risk of autism. Pediatrics. 2010;126(4):656–664. doi: 10.1542/peds.2010-0309. [DOI] [PubMed] [Google Scholar]
- 160.Walker DM, Gore AC. Transgenerational neuroendocrine disruption of reproduction. Nature Reviews Endocrinology. 2011;7(4):197–207. doi: 10.1038/nrendo.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yorbik O, Sayal A, Akay C, Akbiyik DI, Sohmen T. Investigation of antioxidant enzymes in children with autistic disorder. Prostaglandins Leukotrienes and Essential Fatty Acids. 2002;67(5):341–343. doi: 10.1054/plef.2002.0439. [DOI] [PubMed] [Google Scholar]
- 162.Meguid NA, Dardir AA, Abdel-Raouf ER, Hashish A. Evaluation of oxidative stress in autism: defective antioxidant enzymes and increased lipid peroxidation. Biological Trace Element Research. 2011;143(1):58–65. doi: 10.1007/s12011-010-8840-9. [DOI] [PubMed] [Google Scholar]
- 163.Castañeda J, Genzor P, Bortvin A. PiRNAs, transposon silencing, and germline genome integrity. Mutation Research. 2011;714(1-2):95–104. doi: 10.1016/j.mrfmmm.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 164.Gokcumen O, Babb PL, Iskow RC, et al. Refinement of primate copy number variation hotspots identifies candidate genomic regions evolving under positive selection. Genome Biology. 2011;12(5, article R52) doi: 10.1186/gb-2011-12-5-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li J, Harris RA, Cheung SW, et al. Genomic hypomethylation in the human germline associates with selective structural mutability in the human genome. PLoS Genetics. 2012;8(5) doi: 10.1371/journal.pgen.1002692.e1002692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Iskow RC, McCabe MT, Mills RE, et al. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141(7):1253–1261. doi: 10.1016/j.cell.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.LoPachin RM, Barber DS. Synaptic cysteine sulfhydryl groups as targets of electrophilic neurotoxicants. Toxicological Sciences. 2006;94(2):240–255. doi: 10.1093/toxsci/kfl066. [DOI] [PubMed] [Google Scholar]
- 168.Chun LJ, Tong MJ, Busuttil RW, Hiatt JR. Acetaminophen hepatotoxicity and acute liver failure. Journal of Clinical Gastroenterology. 2009;43(4):342–349. doi: 10.1097/MCG.0b013e31818a3854. [DOI] [PubMed] [Google Scholar]
- 169.Schultz ST. Does thimerosal or other mercury exposure increase the risk for autism? A review of current literature. Acta Neurobiologiae Experimentalis. 2010;70(2):187–195. doi: 10.55782/ane-2010-1790. [DOI] [PubMed] [Google Scholar]
- 170.Good P. Did acetaminophen provoke the autism epidemic? Alternative Medicine Review. 2009;14(4):364–372. [PubMed] [Google Scholar]
- 171.Pumford NR, Martin BM, Hinson JA. A metabolite of acetaminophen covalently binds to the 56 kDa selenium binding protein. Biochemical and Biophysical Research Communications. 1992;182(3):1348–1355. doi: 10.1016/0006-291x(92)91881-p. [DOI] [PubMed] [Google Scholar]
- 172.Hoivik DJ, Manautou JE, Tveit A, Mankowski DC, Khairallah EA, Cohen SD. Evidence suggesting the 58-kDa acetaminophen binding protein is a preferential target for acetaminophen electrophile. Fundamental and Applied Toxicology. 1996;32(1):79–86. doi: 10.1006/faat.1996.0109. [DOI] [PubMed] [Google Scholar]
- 173.Ishida T, Abe M, Oguri K, Yamada H. Enhancement of acetaminophen cytotoxicity in selenium-binding protein-overexpressed COS-1 cells. Drug Metabolism and Pharmacokinetics. 2004;19(4):290–296. doi: 10.2133/dmpk.19.290. [DOI] [PubMed] [Google Scholar]
- 174.Mattow J, Demuth I, Haeselbarth G, Jungblut PR, Klose J. Selenium-binding protein 2, the major hepatic target for acetaminophen, shows sex differences in protein abundance. Electrophoresis. 2006;27(8):1683–1691. doi: 10.1002/elps.200500703. [DOI] [PubMed] [Google Scholar]
- 175.Masubuchi Y, Nakayama J, Watanabe Y. Sex difference in susceptibility to acetaminophen hepatotoxicity is reversed by buthionine sulfoximine. Toxicology. 2011;287(1–3):54–60. doi: 10.1016/j.tox.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 176.Heard KJ. Acetylcysteine for acetaminophen poisoning. The New England Journal of Medicine. 2008;359(3):285–292. doi: 10.1056/NEJMct0708278. [DOI] [PMC free article] [PubMed] [Google Scholar]