

Abstract
Background and Purpose
The NO redox sibling nitroxyl (HNO) elicits soluble guanylyl cyclase (sGC)-dependent vasodilatation. HNO has high reactivity with thiols, which is attributed with HNO-enhanced left ventricular (LV) function. Here, we tested the hypothesis that the concomitant vasodilatation and inotropic actions induced by a HNO donor, Angeli's salt (sodium trioxodinitrate), were sGC-dependent and sGC-independent respectively.
Experimental Approach
Haemodynamic responses to Angeli's salt (10 pmol–10 μmol), alone and in the presence of scavengers of HNO (L-cysteine, 4 mM) or of NO [hydroxocobalamin (HXC), 100 μM] or a selective inhibitor of sGC [1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), 10 μM], a CGRP receptor antagonist (CGRP8–37, 0.1 μM) or a blocker of voltage-dependent potassium channels [4-aminopyridine (4-AP), 1 mM] were determined in isolated hearts from male rats.
Key Results
Angeli's salt elicited concomitant, dose-dependent increases in coronary flow and LV systolic and diastolic function. Both L-cysteine and ODQ shifted (but did not abolish) the dose–response curve of each of these effects to the right, implying contributions from HNO and sGC in both the vasodilator and inotropic actions. In contrast, neither HXC, CGRP8–37 nor 4-AP affected these actions.
Conclusions and Implications
Both vasodilator and inotropic actions of the HNO donor Angeli's salt were mediated in part by sGC-dependent mechanisms, representing the first evidence that sGC contributes to the inotropic and lusitropic action of HNO in the intact heart. Thus, HNO acutely enhances LV contraction and relaxation, while concomitantly unloading the heart, potentially beneficial actions in failing hearts.
Keywords: cardiac relaxation, cGMP, nitroxyl, vasodilatation, ventricular function
Introduction
Nitroxyl (HNO) is the one-electron reduced and protonated redox sibling of NO. Its therapeutic potential was first suggested when the effects of the anti-alcoholism drug, cyanamide, were found to be attributable to the release of HNO (Nagasawa et al., 1990). HNO is a transient species, readily undergoing dimerization to form hyponitrous acid with subsequent decomposition into nitrous acid and water (Dumond and King, 2011). Therefore, HNO donors are utilized in pharmacological studies, often with the prototypical HNO donor, sodium trioxodinitrate (Na2N2O3) or Angeli's salt (Miranda et al., 2005a). In recent years, HNO has emerged as a novel regulator of cardiovascular function, with vasoprotective (vasodilator, anti-aggregatory) and cardioprotective (i.e. positive inotrope, anti-hypertrophic) properties (Irvine et al., 2008; Bullen et al., 2011; Tocchetti et al., 2011; Lin et al., 2012). Interestingly, HNO serves as a positive cardiac inotrope and is protective in an experimental model of heart failure (Paolocci et al., 2001; 2003,), an action not shared by NO. HNO also exhibits antihypertrophic actions in the myocardium, an effect mediated via inhibition of NADPH oxidase-derived superoxide generation (Lin et al., 2012) and attenuation of the activity of a pro-hypertrophic signalling pathway, p38 MAPK (Wanstall et al., 2001; Favaloro and Kemp-Harper, 2009; Lin et al., 2012). As such, recent interest in the therapeutic potential of HNO has focused on cardiovascular disorders, such as vascular dysfunction, cardiac dysfunction, cardiac remodelling and heart failure (Irvine et al., 2007; 2008; Ritchie et al., 2009; El-Armouche et al., 2010; Bullen et al., 2011; Ding et al., 2011; Yuill et al., 2011; Lin et al., 2012).
In contrast to NO, HNO possesses several unique pharmacological properties. Firstly, HNO is resistant to scavenging by the reactive oxygen species (ROS), superoxide (levels of which are commonly elevated in cardiovascular pathologies), whereas NO is highly reactive with superoxide, forming a second ROS, peroxynitrite (Miranda et al., 2002). In addition, tolerance does not develop to vasodilator actions of HNO , a favourable difference from traditional clinically used nitrovasodilators (Irvine et al., 2007; 2011,). HNO reacts readily with metal centres of proteins such as iron-containing haem in oxymyoglobin and soluble guanylyl cyclase (sGC; nomenclature follows Alexander et al., 2013a, and in contrast to NO, preferentially targets ferric (Fe3+) rather than ferrous (Fe2+) haem groups and thus may activate these proteins when their iron is in the oxidized state (Miranda et al., 2003). Furthermore, HNO (but not NO) is highly thiolphilic, directly targeting thiol-containing proteins. Such an action of HNO underlies many of its unique properties in the CVS (Fukuto and Carrington, 2011). Indeed, the interaction of HNO with cysteine residues on Ca2+-cycling proteins, that is ryanodine receptors (RyR) and the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) on the sarcoplasmic reticulum of cardiomyocytes leads to enhanced cardiac contractility (Fukuto and Carrington, 2011; Tocchetti et al., 2011). The therapeutic advantages of HNO over NO are likely to be more obvious in settings where NO are exposed to significant levels of ROS which would limit the bioavailability of NO but not of HNO (Irvine et al., 2008; Ritchie et al., 2009; Bullen et al., 2011), and/or where specific HNO interactions with key cysteine residues confers protection, as with SERCA, a property not shared by NO (Fukuto and Carrington, 2011; Tocchetti et al., 2011). It is anticipated that HNO donors would thus be comparable with NO donors in other settings such as via inhalation for pulmonary hypertension (De Witt et al., 2001). However, the distinct pharmacological profile of HNO suggests that it offers favourable therapeutic advantages over its free radical sibling, NO, in vascular dysfunction, cardiac dysfunction, cardiac remodelling and heart failure.
NO predominantly utilizes sGC/cGMP to mediate vasodilatation and suppression of cardiomyocyte hypertrophy. In contrast, HNO has been shown to signal via both sGC-dependent and-independent pathways in the vasculature and myocardium. The mechanism of vasodilator actions of the HNO donor, Angeli's salt are largely sGC-dependent (Fukuto et al., 1992; Ellis et al., 2000; Irvine et al., 2003; 2007,; Favaloro and Kemp-Harper, 2007; 2009,), with a smaller contribution from K+ channels (Kv and KATP; nomenclature follows Alexander et al., 2013b) and calcitonin gene-related peptide (CGRP) evident in the resistance (Irvine et al., 2003; Favaloro and Kemp-Harper, 2007) and coronary vasculature (Favaloro and Kemp-Harper, 2007) respectively. These vasodilator properties are evident in both large (e.g. aorta) as well as smaller vessels such as in rodent-isolated thoracic aorta, rodent-isolated mesenteric arteries or isolated hearts in vitro (Ellis et al., 2000; Wanstall et al., 2001; Irvine et al., 2003; Favaloro and Kemp-Harper, 2007). The antihypertrophic actions of HNO donors in isolated cardiomyocytes are similarly cGMP dependent (Lin et al., 2012), whereas the superoxide-suppressing actions have been variably reported as cGMP dependent (Lin et al., 2012) or cGMP independent (Bullen et al., 2011), in cardiomyocytes and arteries respectively. In contrast, the acute enhancement of cardiac contractility elicited by HNO donors in the intact heart have been regarded as cGMP-independent, as no detectable changes in plasma cGMP content were observed in vivo (Paolocci et al., 2003). These studies in the intact heart have not however investigated HNO actions on cardiac contractility in the presence of cGMP inhibition. Of note, cardiac contractility is acutely enhanced by HNO donors in failing and normal hearts to an equivalent extent (Paolocci et al., 2001; 2003,).
The vasodilator and cardiac inotropic effects of HNO donors have been commonly attributed to cGMP-dependent and-independent mechanisms respectively. The concomitant effects of an HNO donor on vascular and cardiac function, and the net mechanism(s) of these actions, however, remain unresolved. The objective of the present study was to thus test the hypothesis that the concomitant vasodilator and inotropic actions induced by the HNO donor, Angeli's salt, are sGC-dependent and sGC-independent, respectively, in the rat isolated heart.
Methods
This investigation complies with the National Health and Medical Research Council of Australia code of practice for the care and use of animals for scientific purposes. All the procedures involved in this project were approved by The Alfred Medical Research Educational Precinct (AMREP) Animal Ethics Committee. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 53 animals were used in the experiments described here.
Hearts isolated from male Sprague-Dawley rats (350–450 g) n = 53 under ketamine-xylazine anaesthesia (100 and 12 mg·kg−1 i.p., respectively) were Langendorff perfused with Krebs buffer (pH 7.4, composition in mM: NaCl 118, KCl 4.7, MgSO4.7H2O 1.18, KH2PO4 1.2, EDTA 0.5, CaCl2 1.75, NaHCO3 25.0 and d-glucose 11, bubbled with 95% O2 and 5% CO2 at 37°C) under constant pressure, using the ADInstruments Langendorff System® (ADInstruments Pty, Ltd., Bella Vista, Australia). The STH Pump Controller (ADInstruments Pty, Ltd.) continuously detected coronary flow, in addition to maintaining a constant perfusion pressure (set to achieve coronary flow at baseline of 10 mL·min−1). A fluid-filled balloon was positioned in the left ventricle (LV) for continuous monitoring of LV systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), LV developed pressure (LVDP) and the first derivatives of LV pressure (LV ± dP/dt). The ADInstruments PowerLab data acquisition system acquired these variables, as well as coronary perfusion pressure, coronary flow and heart rate, throughout the protocol.
After 30 min equilibration, the thromboxane A2 mimetic U46619 (9,11-dideoxy-9α,11α-methanoepoxy PG F2α, 3 μM) was continuously infused into the aorta via a syringe infusion pump (0.1–2.5 mL·min−1), via a port just above the aortic cannula, to preconstrict the coronary vasculature to give a ∼50% reduction in baseline coronary flow-rate (i.e. from ∼10 to ∼5 mL·min−1). A single bolus dose of NaOH (10 mM, vehicle for Angeli's salt) was then administered to the heart via an injection port just above the aortic cannula, followed by a serial dose–response curve to Angeli's salt (10 pmol–10 μmol), constructed by administering bolus doses of the HNO donor to the heart via a second injection port just above the aortic cannula, in increasing doses 1 min apart. All parameters of contractile function had returned to baseline levels achieved with U46619 preconstriction. For coronary flow, this had either returned to baseline levels or had stabilized to a plateau, prior to the addition of the next bolus dose of Angeli's salt. In a parallel series of experiments, hearts were administered serial bolus doses of the equivalent volume of 10 mM NaOH, as a vehicle control.
Subsequent experiments were performed to examine the mechanism of the haemodynamic effects of Angel's salt in the intact heart, in which dose–response curves to Angeli's salt were performed in the presence of various selective pharmacological inhibitors, added to the reservoir of Krebs perfusion buffer. The relative contribution of HNO and NO to the actions of Angeli's salt was investigated in the presence of the HNO scavenger L-cysteine (4 mM), the NO scavenger hydroxocobalamin (HXC, 0.1 mM) or the thiol DTT (100 μM). Parallel experiments utilized the sGC inhibitor, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 10 μM), the CGRP receptor antagonist CGRP8–37 (0.1 μM), or the Kv channel blocker 4-aminopyridine (4-AP, 1 mM) to further examine the mechanisms of Angeli's salt actions. For comparison, dose–response curves to the pure NO donor diethylamine-NONOate (DEA-NO) were also performed.
Data analysis
Changes in all haemodynamic variables induced by each vasodilator dose were measured as the change (Δ) in each response relative to that elicited by the vehicle control (10 mM NaOH for Angeli's salt). All results were expressed as group mean ± SEM, with the number of independent experiments denoted as ‘n’. Data analysis was performed using Graphpad Prism® (version 5.0, La Jolla, CA, USA). Vasorelaxant responses were fitted to a sigmoidal logistic equation, to derive the pEC50 (vasodilator dose eliciting 50% maximal response, expressed as –log mol) and Rmax (maximal vasodilator response). The coefficient of variation, R2, for vasodilator responses was consistently >0.8 in all hearts studied. Dose–response curves to Angeli's salt in the absence and presence of each pharmacological inhibitor were compared on two-way anova, with the Bonferroni post hoc test. Baseline haemodynamic variables and the pEC50 and Rmax for Angeli's salt in the absence and presence of various inhibitors, were analysed using one-way anova with Dunnett's post hoc test for multiple comparisons. In all cases, P < 0.05 was considered statistically significant.
Materials
Angeli's salt, U46619, ODQ and DEA-NO were obtained from Cayman Chemical Company (Ann Arbor, MI, USA). All other reagents were purchased from Sigma Aldrich (St. Louis, MO, USA).
All stock and working solutions of Angeli's salt or DEA-NO were prepared fresh daily in 10 mM NaOH, and kept on ice until required. Aliquots of U46619 (1 mM in 100% ethanol) were stored at −20°C, and were further diluted on the day of use in Krebs buffer. Stock solutions of ODQ were prepared fresh daily (1 mM in 100% ethanol) with further dilution in Krebs buffer. Aliquots of CGRP8–37 (0.1 mM in distilled water) were stored at −20°C, with subsequent dilution in Krebs buffer on the day of use. L-cysteine, HXC, 4-AP and isoprenaline solutions were all prepared in Krebs buffer.
Results
Angeli's salt elicits HNO/sGC-dependent vasodilator actions in the whole heart
The baseline characteristics of all buffer-perfused rat hearts used in this study, at the end of equilibration, prior to commencement of any interventions, are shown in Table 1993. Haemodynamic variables after the commencement of infusion with pharmacological inhibitors and U46619 preconstriction are also shown. Baseline coronary flow prior to commencement of any interventions, as well as that immediately following U46619 preconstriction, was generally comparable across all experimental groups. A representative recording of all haemodynamic parameters on construction of a dose–response curve to Angeli's salt is shown in Figure 1. In the presence of U46619 preconstriction, the HNO donor, Angeli's salt (10 pmol–10 μmol) elicited a dose-dependent vasodilatation, with pEC50 (–log mol) of 8.55 ± 0.24 and Rmax (mL·min−1) of 5.14 ± 0.69 (Table 1993, Figure 2A). Significant increases in coronary flow were evident with doses of Angeli's salt ≥ 10 nmol. The selective HNO scavenger L-cysteine (4 mM, n = 6) caused a rightward shift in the dose–response curve of the vasodilator actions of Angeli's salt, with significant reductions in both the pEC50 and Rmax. In contrast, the selective NO scavenger HXC (100 μM, n = 5) not only failed to blunt the vasodilator effect of Angeli's salt, but actually tended to enhance the vasorelaxant effect of Angeli's salt (Figure 2A). The thiol DTT (100 μM, n = 5) did not affect the dose–response curve for Angeli's salt.
Figure 1.
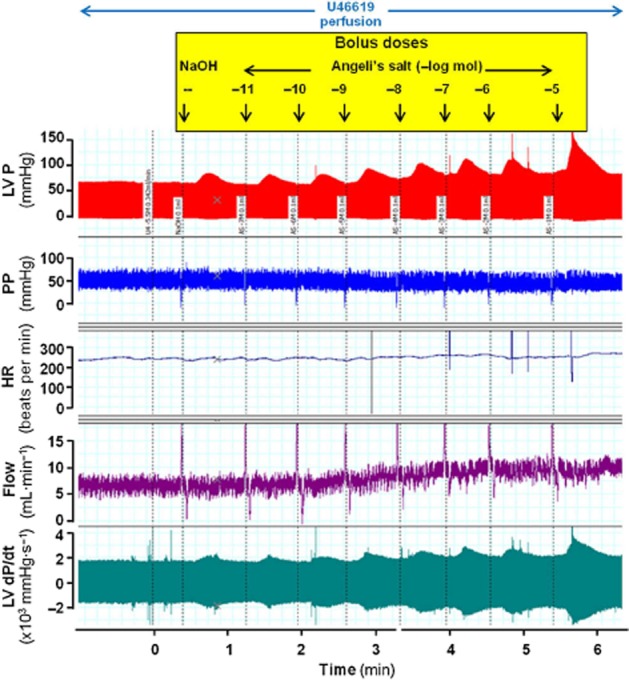
Representative dose–response curve to Angeli's salt, showing effects on LV pressure (LVP), perfusion pressure (PP), heart rate (HR), coronary flow and LV dP/dt.
Figure 2.
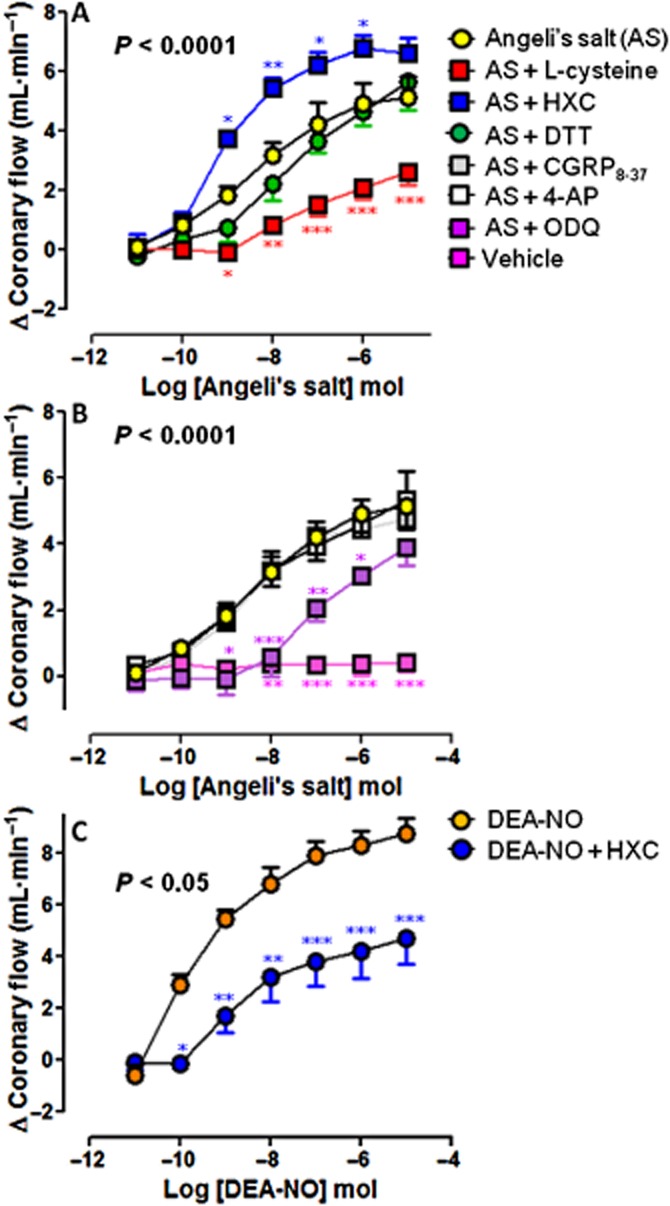
Dose–response curves to Angeli's salt (AS) (n = 8) on coronary flow in the absence and presence of (A) the HNO scavenger L-cysteine (4 mM, n = 6), the NO scavenger HXC (100 μM, n = 5) or the reducing agent DTT (100 μM, n = 5); and (B) the sGC inhibitor ODQ (10 μM, n = 6), the CGRP receptor antagonist CGRP8–37 (0.1 μM, n = 5) and the Kv channel inhibitor 4-AP (1 mM, n = 5). Serial bolus doses of 10 mM NaOH vehicle are shown for comparison (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 versus AS; two-way anova with Bonferroni post hoc test for multiple comparisons. (C) The dose–response curves to DEA-NO (n = 5) on coronary flow in the absence and presence of HXC (100 μM, n = 5) are shown for comparison.
Table 1.
Characteristics of all hearts in each experimental group, at each of baseline (at the end of the equilibration period), after pretreatment with each pharmacological inhibitor alone, and then the subsequent commencement of U46619 infusion (prior to the addition of AS or DEA-NO, shown as mean ± SEM)
| Experimental group | Timepoint | Haemodynamic Variable Prior to Vasodilator Dose–response Curve | n | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Coronary flow (mL·min−1) | Perfusion pressure (mmHg) | Heart rate (beats·min−1) | LVSP (mmHg) | LVDP (mmHg) | LVEDP (mmHg) | LV+dP/dt (mmHg·s−1) | LV-dP/dt (mmHg·s−1) | |||
| AS | Baseline | 10.6 ± 0.4 | 44.8 ± 1.3 | 242 ± 21 | 75.6 ± 6.2 | 76.7 ± 7.0 | −1.1 ± 1.8 | 1963 ± 94 | −1855 ± 101 | 8 |
| U46619 | 5.7 ± 0.5 | 51.1 ± 2.2 | 217 ± 18 | 55.9 ± 6.9 | 56.4 ± 8.4 | 0.6 ± 2.0 | 1721 ± 165 | −1617 ± 185 | ||
| AS + L-cysteine | Baseline | 10.2 ± 1.0 | 43.0 ± 1.6 | 268 ± 19 | 57.8 ± 6.0 | 52.6 ± 6.0* | 5.2 ± 1.3* | 1687 ± 105 | −1435 ± 83 | 6 |
| L-cysteine | 11.7 ± 1.1 | 42.3 ± 3.5 | 201 ± 23 | 57.5 ± 7.3 | 52.1 ± 6.6 | 5.4 ± 1.2 | 1635 ± 149 | −1366 ± 120 | ||
| U46619 | 7.2 ± 0.8 | 49.2 ± 3.5 | 187 ± 18 | 51.8 ± 5.9 | 47.1 ± 6.1 | 4.7 ± 0.8 | 1533 ± 98 | −1280 ± 82 | ||
| AS + HXC | Baseline | 10.5 ± 0.4 | 45.2 ± 1.5 | 296 ± 21 | 53.3 ± 5.0* | 55.2 ± 3.9* | −2.0 ± 1.4 | 1949 ± 153 | −1232 ± 131** | 5 |
| HXC | 9.1 ± 0.5 | 46.4 ± 2.0 | 280 ± 14 | 56.9 ± 7.9 | 61.0 ± 6.2 | −4.1 ± 2.0 | 2138 ± 211 | −1267 ± 58 | ||
| U46619 | 5.7 ± 0.5 | 48.7 ± 1.5 | 269 ± 11 | 41.7 ± 8.5 | 44.7 ± 7.9 | −3.0 ± 1.8 | 1588 ± 242 | −968 ± 96 | ||
| AS + DTT | Baseline | 11.2 ± 0.4 | 50.5 ± 0.6* | 291 ± 25 | 55.1 ± 1.3* | 50.7 ± 1.4* | 4.1 ± 1.3 | 1687 ± 218 | −1150 ± 81** | 5 |
| DTT | 16.5 ± 1.2** | 50.0 ± 2.1 | 299 ± 9 | 52.5 ± 4.1 | 52.3 ± 4.8 | 0.2 ± 2.0 | 1818 ± 193 | −1211 ± 92 | ||
| U46619 | 8.8 ± 0.5* | 54.9 ± 1.7 | 318 ± 19** | 42.4 ± 3.4 | 41.5 ± 3.6 | 0.9 ± 1.6 | 1530 ± 156 | 1123 ± 118 | ||
| AS + ODQ | Baseline | 10.2 ± 0.7 | 41.6 ± 0.9 | 294 ± 18 | 54.1 ± 4.5* | 51.0 ± 3.0* | 3.2 ± 1.2 | 1832 ± 177 | −1578 ± 180 | 6 |
| ODQ | 10.4 ± 0.6 | 42.6 ± 0.9 | 244 ± 20 | 69.5 ± 6.3 | 67.3 ± 5.4* | 2.2 ± 1.7 | 2047 ± 186 | −1812 ± 211 | ||
| U46619 | 5.5 ± 0.5 | 47.4 ± 1.8 | 208 ± 22 | 55.8 ± 9.5 | 52.6 ± 9.0 | 3.1 ± 1.1 | 1636 ± 192 | −1372 ± 222 | ||
| AS + CGRP8–37 | Baseline | 10.1 ± 0.3 | 41.4 ± 0.3 | 239 ± 16 | 64.8 ± 3.8 | 64.7 ± 2.9 | 0.1 ± 1.4 | 1674 ± 66 | −1320 ± 21 | 5 |
| CGRP8–37 | 10.1 ± 0.2 | 43.2 ± 1.6 | 255 ± 16 | 69.6 ± 3.4 | 71.6 ± 3.0 | −2.0 ± 1.5 | 1758 ± 116 | −1505 ± 99 | ||
| U46619 | 5.3 ± 0.2 | 49.9 ± 1.9 | 233 ± 14 | 53.7 ± 1.3 | 54.6 ± 1.5 | −0.8 ± 1.1 | 1481 ± 82 | −1201 ± 71 | ||
| AS + 4-AP | Baseline | 10.1 ± 0.8 | 46.1 ± 1.6 | 295 ± 19 | 53.5 ± 3.6* | 53.7 ± 3.4* | −0.2 ± 1.5 | 1718 ± 43 | −1500 ± 89 | 5 |
| 4-AP | 8.4 ± 1.5 | 52.0 ± 2.1 | 241 ± 9* | 72.7 ± 13.9 | 76.1 ± 15.2 | −3.4 ± 2.1 | 2324 ± 348 | −2079 ± 314 | ||
| U46619 | 4.5 ± 0.9 | 56.3 ± 2.2 | 226 ± 20 | 47.9 ± 11.5 | 47.9 ± 12.4 | −0.0 ± 1.8 | 1549 ± 319 | −1362 ± 245 | ||
| DEA-NO | Baseline | 10.4 ± 0.4 | 51.9 ± 3.1 | 254 ± 12 | 63.9 ± 6.7 | 58.6 ± 8.1 | 5.3 ± 2.6 | 1956 ± 248 | −1109 ± 57 | 5 |
| U46619 | 5.6 ± 0.3 | 57.9 ± 3.2 | 267 ± 10 | 51.5 ± 4.5 | 47.2 ± 6.2 | 4.3 ± 2.6 | 1637 ± 177 | −978 ± 67 | ||
| DEA-NO + HXC | Baseline | 10.6 ± 0.3 | 47.8 ± 2.2 | 271 ± 12 | 54.3 ± 2.4 | 55.5 ± 1.6 | −1.2 ± 3.4 | 1834 ± 66 | −1203 ± 92 | 5 |
| HXC | 10.8 ± 1.4 | 46.8 ± 2.3 | 257 ± 10 | 48.0 ± 7.6 | 50.5 ± 7.1 | −2.5 ± 3.2 | 1705 ± 193 | −1079 ± 63 | ||
| U46619 | 7.0 ± 1.2 | 50.5 ± 1.8 | 250 ± 11 | 44.7 ± 5.6 | 46.8 ± 3.9 | −2.1 ± 2.7 | 1589 ± 117 | −1080 ± 49 | ||
P < 0.05
P < 0.01 versus the analogous time point in hearts allocated to treatment with AS alone ;one-way anova (Dunnett's post hoc test).
AS, Angeli's salt; DEA-NO, diethylamine-NONOate; HXC, hydroxocobalamin; LVDP, left ventricle diastolic pressure; LVEDP, left ventricle end-diastolic pressure; LVSP, left ventricle systolic pressure; ODQ, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one.
Table 2.
Sensitivity (pEC50) and maximal relaxation response (Rmax) for the dose–response curves to AS and DEA-NO on coronary flow, in the absence and presence of selective inhibitors
| Experimental group | pEC50 (-log mol) | Rmax (mL·min−1) | n |
|---|---|---|---|
| AS | 8.55 ± 0.24 | 5.14 ± 0.69 | 8 |
| AS + L-cysteine | 7.53 ± 0.18** | 2.62 ± 0.44* | 6 |
| AS + HXC | 9.12 ± 0.12 | 6.85 ± 0.47 | 5 |
| AS + DTT | 7.85 ± 0.40 | 5.65 ± 0.93 | 5 |
| AS + ODQ | 7.36 ± 0.29** | 3.88 ± 0.52 | 6 |
| AS + CGRP8–37 | 8.49 ± 0.26 | 4.76 ± 0.52 | 5 |
| AS + 4-AP | 8.40 ± 0.30 | 5.36 ± 0.85 | 5 |
| DEA-NO | 9.60 ± 0.18 | 8.82 ± 0.61 | 5 |
| DEA-NO + HXC | 8.56 ± 0.19## | 4.77 ± 1.01## | 5 |
P < 0.05
P < 0.01 versus AS alone and
P < 0.01 versus DEA-NO alone.
AS, Angeli's salt; DEA-NO, diethylamine-NONOate; HXC, hydroxocobalamin; LVDP, left ventricle diastolic pressure; LVEDP, left ventricle end-diastolic pressure; LVSP, left ventricle systolic pressure; ODQ, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
As shown in Figure 2B, the selective sGC inhibitor, ODQ (10 μM, n = 6) also caused a rightward shift in the dose–response curve of the vasodilator actions of Angeli's salt, with significant reduction in the pEC50 (Figure 2B). The Rmax to Angeli's salt was not significantly affected by ODQ (Table 1993). Both the selective CGRP receptor antagonist CGRP8–37 (0.1 μM, n = 5) and the Kv channel inhibitor 4-AP (1 mM, n = 5) failed to affect the vasodilator actions of Angeli's salt (Figure 2B). Furthermore, serial bolus doses of 10 mM NaOH vehicle failed to elicit significant haemodynamic response (Figure 2B). As shown in Table 1993, neither L-cysteine, HXC alone nor other pharmacological inhibitors had any significant effect on basal vascular function, although DTT tended to enhance coronary flow and heart rate. For comparison, the NO donor DEA-NO (10 pmol–10 μmol) elicited a dose-dependent vasodilatation which was also shifted rightwards by HXC (both n = 5, Figure 2C and Table 1993).
Relative contribution of HNO/sGC (but not NO) to the inotropic effects of Angeli's salt
The vasorelaxant effect of Angeli's salt was accompanied by concomitant dose-dependent enhancement of myocardial inotropic function. Significant increases in LVSP (Figure 3A), LVDP (Figure 4A) and LV+dP/dt (Figure 5A), parameters of cardiac contractile function, were evident from ≥10 nmol Angeli's salt. Both L-cysteine and DTT (but not HXC) markedly blunted the effects of Angeli's salt on each of LVSP (Figure 3A), LVDP (Figure 4A) and LV+dP/dt (Figure 5A). Maximal increases in parameters of cardiac contractility induced by Angeli's salt were suppressed by ∼60% in the presence of L-cysteine. Angeli's salt also tended to increase heart rate at the highest dose studied (by 59 ± 7 beats per min), this was unaffected by either L-cysteine or HXC. Further, there was no evidence of arrhythmic events observed at any time. Inhibition of sGC with ODQ also markedly blunted (but did not abolish) the positive inotropic effect of Angeli's salt, on each of LVSP (Figure 3B), LVDP (Figure 4B) and LV+dP/dt (Figure 5B), by ∼50%. In contrast, inhibition of CGRP receptors or Kv channels failed to suppress the positive inotropic actions of Angeli's salt. Interestingly, the LV+dP/dt response tended to be exaggerated by 4-AP. For comparison, the NO donor DEA-NO elicited comparatively modest increases in LVSP (Figure 3C), LVDP (Figure 4C) and LV+dP/dt (Figure 5C), evident at higher doses of DEA-NO, which were insensitive to HXC (both n = 5). None of these inhibitors alone (L-cysteine, DTT, HXC, ODQ, CGRP8–37 and 4-AP) affected these parameters of contractile function prior to the construction of the dose–response curve to Angeli's salt, as shown in Table 1993).
Figure 3.
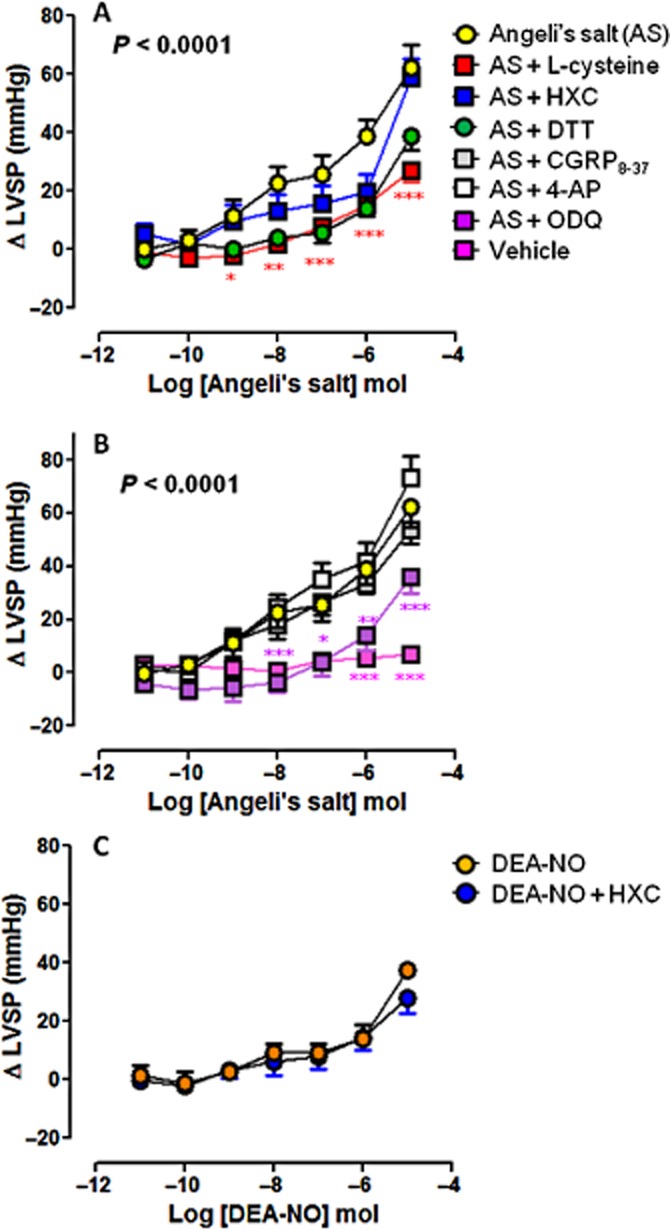
Dose–response curves to Angeli's salt (AS) (n = 8) on LVSP in the absence and presence of (A) L-cysteine (n = 6), HXC (n = 5) or DTT (n = 5); and (B) ODQ (n = 6), CGRP8–37 (n = 5) and 4-AP (n = 5). Serial bolus doses of 10 mM NaOH vehicle are shown for comparison (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 versus AS; two-way anova with Bonferroni post hoc test for multiple comparisons. (C) The dose–response curves to DEA-NO (n = 5) on LVSP in the absence and presence of HXC (100 μM, n = 5) are shown for comparison.
Figure 4.
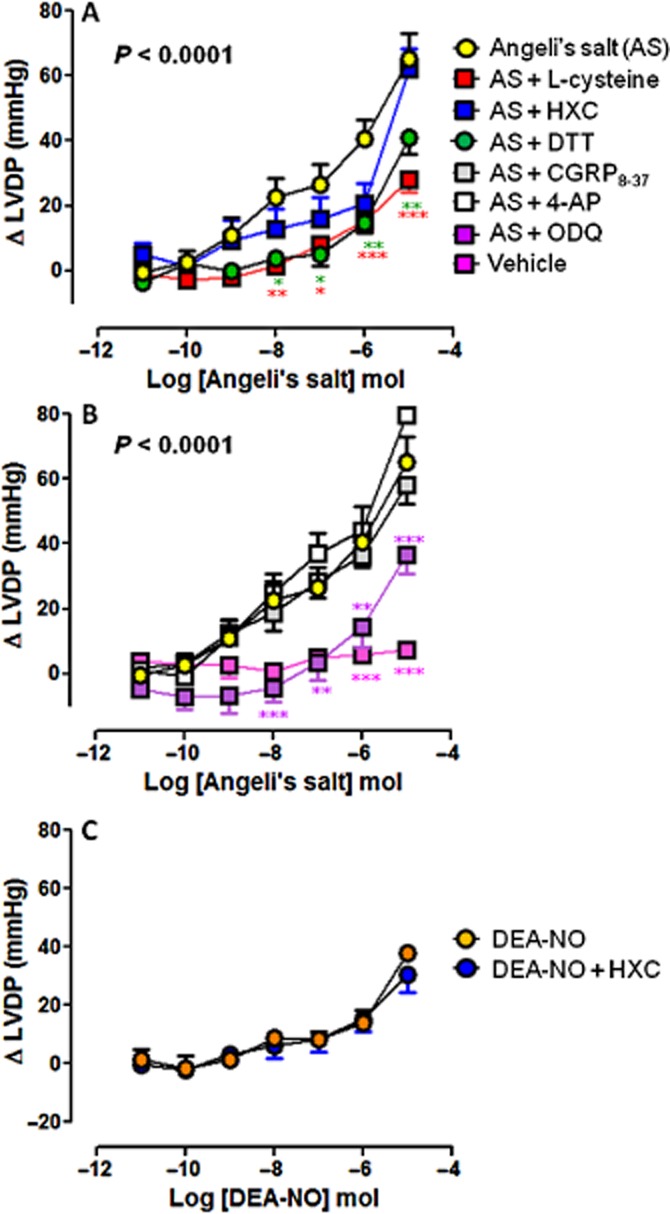
Dose–response curves to Angeli's salt (AS) (n = 8) on LVDP in the absence and presence of (A) L-cysteine (n = 6), HXC (n = 5) or DTT (n = 5); and (B) ODQ (n = 6), CGRP8–37 (n = 5) and 4-AP (n = 5). Serial bolus doses of 10 mM NaOH vehicle are shown for comparison (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 versus AS two-way anova with Bonferroni post-hoc test for multiple comparisons. (C) The dose–response curves to DEA-NO (n = 5) on LVDP in the absence and presence of HXC (100 μM, n = 5) are shown for comparison.
Figure 5.
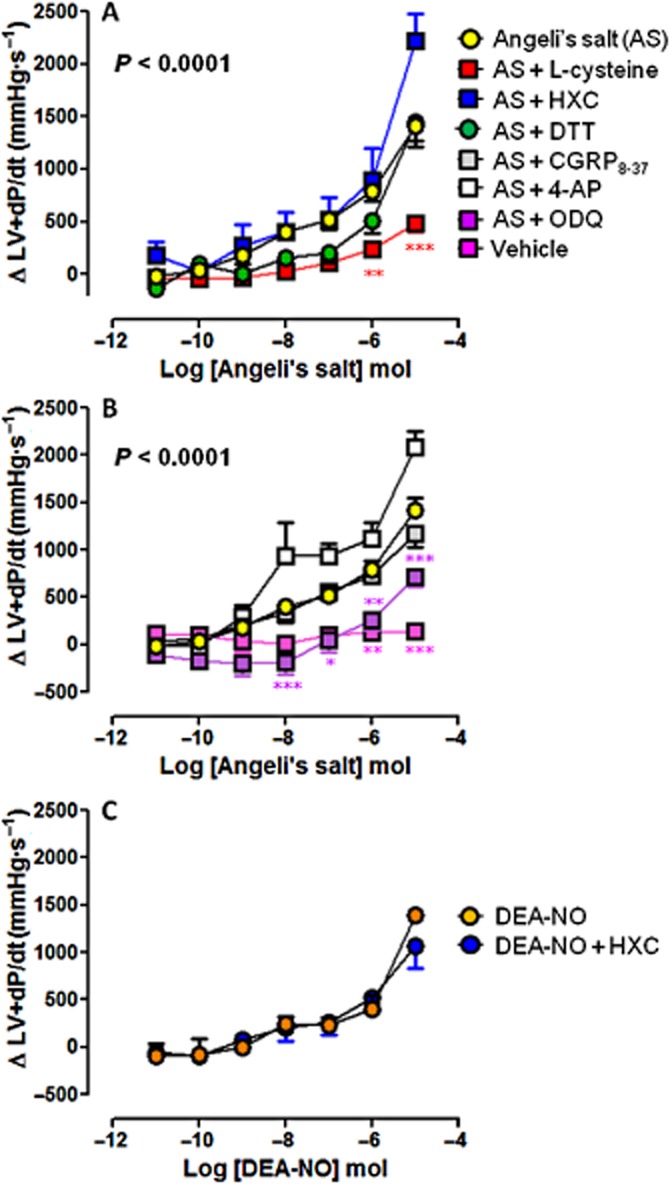
Dose–response curves to Angeli's salt (AS) (n = 8) on LV+dP/dt in the absence and presence of (A) L-cysteine (n = 6), HXC (n = 5) or DTT (n = 5); and (B) ODQ (n = 6), CGRP8–37 (n = 5) and 4-AP (n = 5). Serial bolus doses of 10 mM NaOH vehicle are shown for comparison (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 versus AS; two-way anova with Bonferroni post hoc test for multiple comparisons. (C) The dose–response curves to DEA-NO (n = 5) on LV+dP/dt in the absence and presence of HXC (100 μM, n = 5) are shown for comparison.
Contribution of HNO/sGC to the effects of Angeli's salt on cardiac relaxation
Angeli's salt elicited dose-dependent enhancement of myocardial lusitropic function, with progressive reduction in LVEDP (Figure 6) and potentiation of LV-dP/dt (Figure 7). These actions were blunted by L-cysteine, DTT and ODQ (Angeli's salt enhancement of LV-dP/dt was particularly sensitive to these inhibitors), but not by HXC or 4-AP (both of which tended to enhance the Angeli's salt effect). CGRP8–37 was without effect on the cardiac relaxation response to Angeli's salt (Figures 6 and 7). None of these inhibitors alone (L-cysteine, DTT, HXC, ODQ, CGRP8–37 and 4-AP) affected these parameters of cardiac relaxation alone, prior to the construction of the dose–response curve to Angeli's salt (Table 1993).
Figure 6.
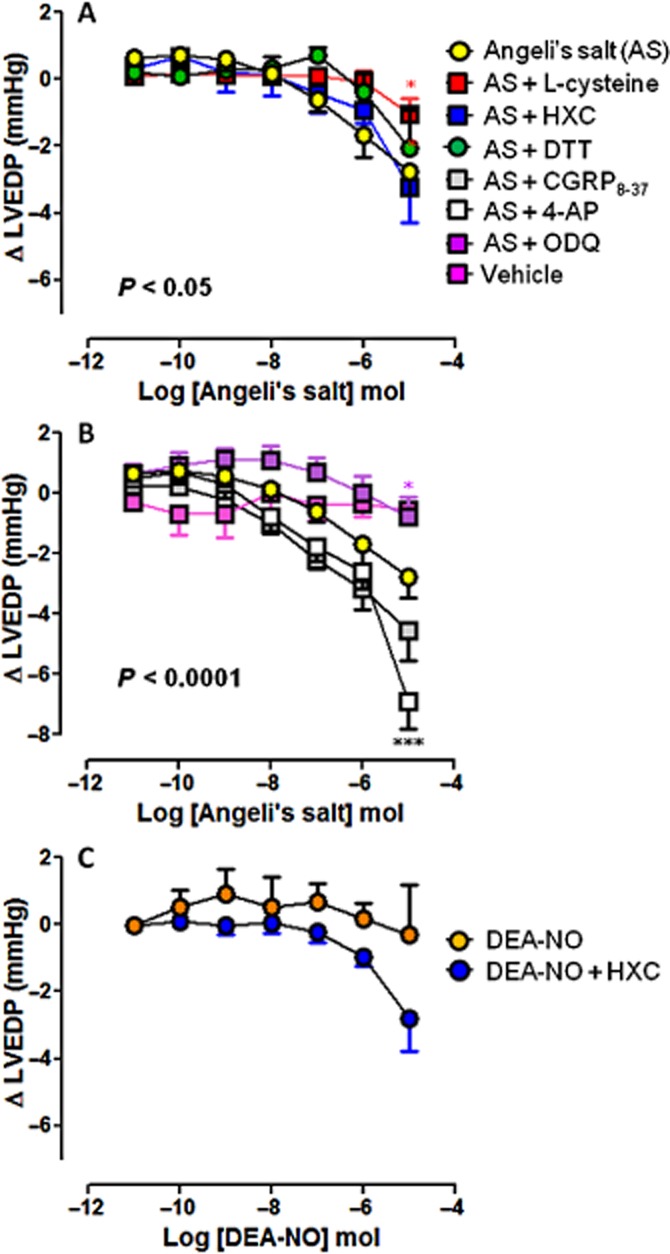
Dose–response curves to Angeli's salt (AS) (n = 8) on LVEDP in the absence and presence of (A) L-cysteine (n = 6), HXC (n = 5) or DTT (n = 5); and (B) ODQ (n = 6), CGRP8–37 (n = 5) and 4-AP (n = 5). Serial bolus doses of 10 mM NaOH vehicle are shown for comparison (n = 3). *P < 0.05, ***P < 0.001 versus AS on two-way anova with Bonferroni post hoc test for multiple comparisons. (C) The dose–response curves to DEA-NO (n = 5) on LVEDP in the absence and presence of HXC (100 μM, n = 5) are shown for comparison.
Figure 7.
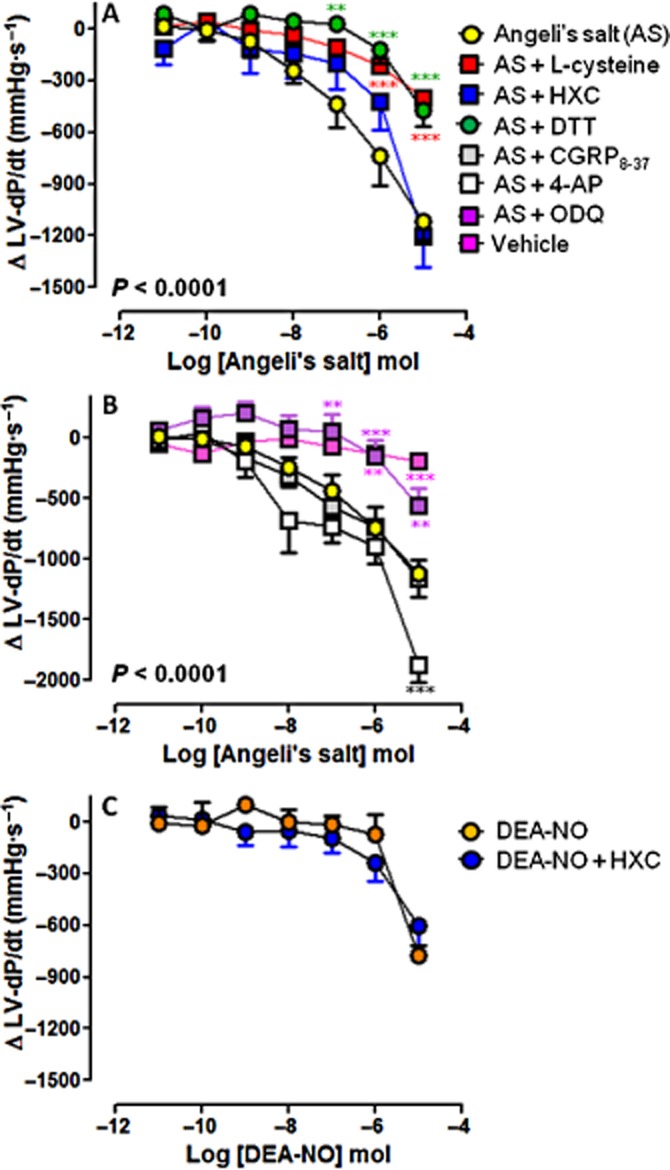
Dose–response curves to Angeli's salt (AS) (n = 8) on LV)-dP/dt in the absence and presence of (A) L-cysteine (n = 6), HXC) (n = 5) or DTT (n = 5); and (B) ODQ) (n = 6), CGRP8–37 (n = 5) and 4-AP) (n = 5). Serial bolus doses of 10 mM NaOH vehicle are shown for comparison (n = 3). **P < 0.01, ***P < 0.001 versus AS on two-way anova with Bonferroni post hoc test for multiple comparisons. (C) The dose–response curves to DEA-NO) (n = 5) on LV-dP/dt in the absence and presence of HXC (100 μM, n = 5) are shown for comparison.
Discussion
The key findings of the present study are that the HNO donor, Angeli's salt, elicits concomitant coronary vasodilator, inotropic and lusitropic actions in the intact rat heart, all of which are mediated by L-cysteine-sensitive, HNO-dependent mechanisms, with a significant contribution mediated via sGC. There appeared to be no role for extracellular oxidation of HNO to NO, or for CGRP receptors or Kv channels in the haemodynamic responses to Angeli's salt. These results are the first evidence that sGC may contribute, at least in part, to the inotropic and/or lusitropic action of HNO in the intact heart.
Our observations here that Angeli's salt induces HNO/sGC-mediated, dose-dependent vasodilatation in the intact rat heart are consistent with previous reports in isolated large conduit and smaller resistance-like vessels in vitro (Irvine et al., 2003; Favaloro and Kemp-Harper, 2009), as well as in the intact heart studied under conditions of constant flow ex vivo (Favaloro and Kemp-Harper, 2007). Although coronary vascular tone under basal, physiological conditions is largely regulated by Kv channels (Leblanc et al., 1994; Shimizu et al., 2000), we observed no role for Kv signalling in the vasodilator response to Angeli's salt in the rat coronary vasculature, consistent with previous observations (Irvine et al., 2003; Favaloro and Kemp-Harper, 2007). In contrast, the vasorelaxant actions of Angeli's salt are mediated, in part, via Kv channels in the mesenteric circulation (Irvine et al., 2003; Favaloro and Kemp-Harper, 2009), perhaps because of regional differences in K+ channel subtype distribution. Although KATP channels may also play a role in coronary vasodilatation in response to Angeli's salt (Favaloro and Kemp-Harper, 2007), this was not investigated in the present study.
Previous studies have suggested a potential contribution of CGRP to the coronary vasodilator response to Angeli's salt, as described in the isolated rat heart studied under constant flow conditions ex vivo (Favaloro and Kemp-Harper, 2007), but not to the peripheral arterial or venous vasorelaxation, as reported in a canine model in vivo (Paolocci et al., 2001). Although we detected no contribution of CGRP-dependent signalling to the vasodilator actions of Angeli's salt in the isolated rat heart studied under constant pressure conditions ex vivo, the reason for this discrepancy remains unresolved. Angeli's salt co-releases both HNO and nitrite at physiological pH (Miranda et al., 2005a), HNO rather than nitrite is likely to mediate the vasodilator responses observed here. Firstly, the HNO-selective scavenger, L-cysteine, markedly impaired these responses, and secondly, nitrite has almost negligible dilator activity in the rat coronary vasculature, with 15 000-fold less potency than Angeli's salt (Irvine et al., 2003; Favaloro and Kemp-Harper, 2007). Given that a residual, modest Angeli's salt-induced vasodilatation remains in the presence of L-cysteine, we cannot rule out the possibility of oxidation of HNO to NO under our experimental conditions. However, the inability of the NO-selective scavenger HXC to blunt the vasodilator response to Angeli's salt suggests this is unlikely, at least in the extracellular milieu. Intriguingly, this vasodilator response was actually augmented in the presence of HXC; whether this reflects a loss of endogenous NO and thus an increased responsiveness of sGC to stimulation by HNO was however not determined.
The positive cardiac inotropic and lusitropic actions of HNO donors are well established, both in the intact heart in vivo, as well as in isolated cardiomyocytes and trabeculae in vitro (Paolocci et al., 2001; Tocchetti et al., 2007; Kohr et al., 2010). We now confirm that the prototypical HNO donor, Angeli's salt, potently enhances both cardiac contraction and relaxation in the intact rat heart ex vivo. These actions were markedly attenuated by both L-cysteine and DTT, specifically implicating HNO. The positive inotropic and dilator effects of Angeli's salt are not likely to be mediated by co-release of nitrite, as this has no appreciable effect on cardiomyocyte contractility (Kohr et al., 2010). Early reports describing the positive inotropic actions implicated the neuropeptide CGRP at least in part in this mechanism of action, based on sensitivity to the CGRP receptor antagonist, CGRP8–37 (Paolocci et al., 2001; receptor nomenclature follows Alexander et al., 2013c). CGRP itself elicits positive inotropic and lusitropic effects via activation of cAMP/PKA/L-type Ca2+ channel signalling (Huang et al., 1999). These actions are however dependent on β-adrenoceptor signalling (Katori et al., 2005), in contrast to those of HNO, which are β-adrenoceptor-independent (Paolocci et al., 2003). Our results here are consistent with the absence of a role for CGRP in the inotropic and lusitropic actions of Angeli's salt.
As the myocardial effects of Angeli's salt are all evident even at relatively low doses (e.g. from 10 nmol), concomitant with doses required to elicit vasodilatation, this raises the possibility that these myocardial effects are a secondary effect to vasorelaxation, in accordance with the Gregg effect (Westerhof et al., 2006). However, the vasodilator response plateaus at ∼1 μmol, whereas the enhancement of LV contractility and relaxation induced by Angeli's salt progress further with increasing doses of the HNO donor, Angeli's salt. Given that previous reports suggest that the vasodilator actions of Angeli's salt are evident at markedly lower concentrations (e.g. 0.1 μM) than required for effects on cardiomyocyte function (e.g. 500 μM) (Favaloro and Kemp-Harper, 2007; Tocchetti et al., 2007), it remains likely that Angeli's salt-mediated vasodilatation occurs at lower concentrations while the contractile effect of Angeli's salt occurs only at higher concentrations.
The cardiac inotropic and lusitropic effects of HNO donors have been traditionally attributed to cGMP-independent mechanisms, through a thiol-mediated interaction with the sarcoplasmic reticulum Ca2+-handling proteins, RyR and SERCA (Tocchetti et al., 2007; Kohr et al., 2010). These previous reports concluded that the myocardial actions of HNO were cGMP-independent on the basis of an absence of detectable increases in plasma cGMP in vivo (Paolocci et al., 2001), as well as a perceived lack of sensitivity to ODQ (Tocchetti et al., 2007). Of note, the only previous investigation of the role for cGMP in the cardiac inotropic and lusitropic effects of HNO donors utilized isolated cardiomyocytes rather than the intact heart, and the concentration of HNO donor (1 mM) far exceeded that used for ODQ (10 μM) (Tocchetti et al., 2007). ODQ is considered an oxidizer (rather than a competitive inhibitor) of sGC, which irreversibly inhibits the enzyme. There is however one report that suprapharmacological concentrations of Angeli's salt (1 mM) may still be able to stimulate any residual sGC still in its reduced state (Zeller et al., 2009). In the present study, the effects of HNO on LV contractility and relaxation were determined in the intact heart, concomitantly with its vasorelaxant effects. Administration of ODQ under these conditions significantly attenuated (but did not abolish) the LV inotropic and lusitropic effects of Angeli's salt, suggesting for the first time that HNO may mediate a part of these actions via sGC/cGMP-dependent signalling.
Although the effects of both NO and sGC on cardiac contractile function have been previously examined in a broad range of scenarios, no consensus has yet been reached, with negative inotropic (Balligand et al., 1993; Brady et al., 1993; Grocott-Mason et al., 1994; Weyrich et al., 1994; Mohan et al., 1995; Kojda et al., 1996; Sandirasegarane and Diamond, 1999; Muller-Strahl et al., 2000; Gonzalez et al., 2008; Cawley et al., 2011; Derici et al., 2012), positive inotropic (Klabunde and Ritger, 1991; Smith et al., 1991; Kojda et al., 1995; 1996; 1997,,; Sarkar et al., 2000; Layland et al., 2002; Langer et al., 2003) or no change observed (Ritchie et al., 2006; 2009,). Indeed, the relationship between NO/sGC and myocardial force may be differentially modulated by concentration, whereby smaller increases in NO/sGC levels elicit positive inotropic effects either secondary to phosphodiesterase-3 inhibition (elevating cAMP), while high concentrations elicit a cGMP-mediated negative inotropic effect, perhaps secondary to formation of S-nitrosothiols on key cardiomyocyte Ca2+-handling proteins such as RyR, SERCA and phospholamban (Smith et al., 1991; Kojda et al., 1996; 1997,; Zahradnikova et al., 1997; Paolocci et al., 2000; Layland et al., 2002; Langer et al., 2003; Gonzalez et al., 2007; 2008,; Rastaldo et al., 2007; Wang et al., 2008; Ziolo, 2008). It is also likely that distinct cardiomyocyte pools of cGMP also contribute to this lack of consensus with respect to the nature of any possible effect of NO/sGC on inotropic mechanisms, as has been suggested for natriuretic peptide receptors (Qvigstad et al., 2010). There is however consensus with respect to cardiac relaxation, which is enhanced by NO (Paulus et al., 1994; Carnicer et al., 2013). In our study DEA-NO (which releases two NO molecules per molecule of DEA-NO) did tend to enhance systolic function, but this was more modest than that achieved by the equivalent concentration of Angeli's salt (despite it only releasing a single HNO molecule per molecule of Angeli's salt). We have previously demonstrated that HNO donors such as Angeli's salt and IPA-NO do not increase cardiomyocyte cAMP or CGRP content (Lin et al., 2012; Irvine et al., 2013).
In our hands, the thiols L-cysteine and DTT were similarly effective at blunting the Angeli's salt enhancement of inotropic and lusitropic function at the concentrations used (4 vs. 0.1 mM). In contrast, only L-cysteine (and not DTT) blunted the vasodilatation response. L-cysteine is conventionally used as an HNO scavenger (Tocchetti et al., 2011), blocking both Angeli's salt-induced coronary vasodilator and positive inotropic actions by removing available HNO. HNO is considered to enhance cardiac contractility and relaxation by inducing a reversible oxidation of key thiol residues on specific cardiomyocyte Ca2+ cycling/sensitization proteins (e.g. RyR and SERCA), without altering net thiol redox status (i.e. GSH:GSSG ratio; for review see Fukuto and Carrington, 2011; Tocchetti et al., 2011). Our findings with both thiols are perhaps consistent then with the Angeli's salt-induced vasodilatation dependent on HNO and sGC (but not proteins implicated in Ca2+ cycling/sensitization), whereas its enhancement of cardiac contractility and relaxation may be mediated at least in part by both sGC-dependent and sGC-independent mechanisms (such as HNO-mediated oxidation of RyR and SERCA).
The thiol modification induced by HNO is quite distinct to that induced by NO. NO leads to S-nitrosation via an indirect action, as it is initially oxidized to nitrous anhydride, which then reacts with protein thiol groups to form protein-SNO (Lima et al., 2010; Heinrich et al., 2013). In contrast, the interaction of HNO with thiols is direct and thus extremely rapid (Jackson et al., 2009), first generating the intermediate, N-hydroxysulphenamide, which can then either be irreversibly arranged to form N-hydroxysulphenamide, or alternatively can reversibly interact with an additional thiol, to form a disulphide and hydroxylamine. The predominant thiol modification induced by HNO is thus considered formation of a sulphinamide or disulphide, rather than S-nitrosation (Fukuto and Carrington, 2011). As Angeli's salt only releases NO at a very acidic pH (Miranda et al., 2005b), together with our finding that the coronary vasodilator action of Angeli's salt was not diminished in the presence of the NO scavenger HXC, it is highly unlikely that Angeli's salt will form S-NO in the presence of thiols such as L-cysteine. Thus, in contrast to NO donors, Angeli's salt dose-dependent enhancement of cardiac contractility and relaxation is unlikely to result from S-nitrosation of Ca2+-handling proteins.
In conclusion, the HNO donor Angeli's salt elicits dose-dependent enhancement of LV systolic and diastolic function, concomitant with vasodilatation, in the intact rat heart. These effects are all L-cysteine-sensitive and mediated by HNO, with contributions from both sGC-dependent and s-GC-independent mechanisms. No role for CGRP, NO or Kv in Angeli's salt cardiac effects was evident. HNO thus acutely modulates both LV contractile function and LV relaxation, while concomitantly unloading the heart. These properties, in combination with the powerful antihypertrophic and superoxide-suppressing actions we have previously demonstrated, may favour HNO donors as a potential strategy for managing heart failure (alone or in addition to standard care).
Acknowledgments
This work was supported by the National Health and Medical Research Council (NHMRC) of Australia (ID472642) and in part by the Victorian Government's Operational Infrastructure Support Program. K.Y.C. was supported by a Baker IDI Heart & Diabetes Institute Bright Sparks PhD stipend. R.H.R is the recipient of a NHMRC Senior Research Fellowship (ID472673) and B.K.K-H by a NHMRC Project Grant (ID606556).
Glossary
- 4-AP
4-aminopyridine
- CGRP
calcitonin gene-related peptide
- HNO
nitroxyl
- HXC
hydroxocobalamin
- LV ± dP/dt
first derivatives of LV pressure
- LV
left ventricle
- LVDP
LV developed pressure
- LVEDP
LV end-diastolic pressure
- LVSP
LV systolic pressure
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- RyR
ryanodine receptor
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- sGC
soluble guanylyl cyclase
- U46619
9,11-dideoxy-9α,11α-methanoepoxy PG F2α
Conflict of interest
The authors have no conflicts of interest to declare.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013a;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013c;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balligand JL, Kelly RA, Marsden PA, Smith TW, Michel T. Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc Natl Acad Sci U S A. 1993;90:347–351. doi: 10.1073/pnas.90.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady AJ, Warren JB, Poole-Wilson PA, Williams TJ, Harding SE. Nitric oxide attenuates cardiac myocyte contraction. Am J Physiol. 1993;265(1 Pt 2):H176–H182. doi: 10.1152/ajpheart.1993.265.1.H176. [DOI] [PubMed] [Google Scholar]
- Bullen ML, Miller AA, Dharmarajah J, Drummond GR, Sobey CG, Kemp-Harper BK. Vasorelaxant and antiaggregatory actions of the nitroxyl donor isopropylamine NONOate are maintained in hypercholesterolemia. Am J Physiol Heart Circ Physiol. 2011;301:H1405–H1414. doi: 10.1152/ajpheart.00489.2011. [DOI] [PubMed] [Google Scholar]
- Carnicer R, Crabtree MJ, Sivakumaran V, Casadei B, Kass DA. Nitric oxide synthases in heart failure. Antioxid Redox Signal. 2013;18:1078–1099. doi: 10.1089/ars.2012.4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawley SM, Kolodziej S, Ichinose F, Brouckaert P, Buys ES, Bloch KD. sGCα1 mediates the negative inotropic effects of NO in cardiac myocytes independent of changes in calcium handling. Am J Physiol Heart Circ Physiol. 2011;301:H157–H163. doi: 10.1152/ajpheart.01273.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Witt BJ, Marrone JR, Kaye AD, Keefer LK, Kadowitz PJ. Comparison of responses to novel nitric oxide donors in the feline pulmonary vascular bed. Eur J Pharmacol. 2001;430:311–315. doi: 10.1016/s0014-2999(01)01289-4. [DOI] [PubMed] [Google Scholar]
- Derici K, Samsar U, Demirel-Yilmaz E. Nitric oxide effects depend on different mechanisms in different regions of the rat heart. Heart Vessels. 2012;27:89–97. doi: 10.1007/s00380-011-0116-6. [DOI] [PubMed] [Google Scholar]
- Ding W, Li Z, Shen X, Martin J, King SB, Sivakumaran V, et al. Reversal of isoflurane-induced depression of myocardial contraction by a novel myofilament Ca2+ sensitizing agent, nitroxyl (HNO) J Pharmacol Exp Ther. 2011;339:825–831. doi: 10.1124/jpet.111.185272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumond JF, King SB. The chemistry of nitroxyl-releasing compounds. Antioxid Redox Signal. 2011;14:1637–1648. doi: 10.1089/ars.2010.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Armouche A, Wahab A, Wittkopper K, Schulze T, Bottcher F, Pohlmann L, et al. The new HNO donor, 1-nitrosocyclohexyl acetate, increases contractile force in normal and beta-adrenergically desensitized ventricular myocytes. Biochem Biophys Res Commun. 2010;402:340–344. doi: 10.1016/j.bbrc.2010.10.030. [DOI] [PubMed] [Google Scholar]
- Ellis A, Li CG, Rand MJ. Differential actions of L-cysteine on responses to nitric oxide, nitroxyl anions and EDRF in the rat aorta. Br J Pharmacol. 2000;129:315–322. doi: 10.1038/sj.bjp.0703058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaloro JL, Kemp-Harper BK. The nitroxyl anion (HNO) is a potent dilator of rat coronary vasculature. Cardiovasc Res. 2007;73:587–596. doi: 10.1016/j.cardiores.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Favaloro JL, Kemp-Harper BK. Redox variants of NO (NO and HNO) elicit vasorelaxation of resistance arteries via distinct mechanisms. Am J Physiol Heart Circ Physiol. 2009;296:H1274–H1280. doi: 10.1152/ajpheart.00008.2009. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Carrington SJ. HNO signaling mechanisms. Antioxid Redox Signal. 2011;14:1649–1657. doi: 10.1089/ars.2010.3855. [DOI] [PubMed] [Google Scholar]
- Fukuto JM, Chiang K, Hszieh R, Wong P, Chaudhuri G. The pharmacological activity of nitroxyl – a potent vasodilator with activity similar to nitric oxide and/or endothelium-derived relaxing factor. J Pharmacol Exp Ther. 1992;263:546–551. [PubMed] [Google Scholar]
- Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A. 2007;104:20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez DR, Fernandez IC, Ordenes PP, Treuer AV, Eller G, Boric MP. Differential role of S-nitrosylation and the NO-cGMP-PKG pathway in cardiac contractility. Nitric Oxide. 2008;18:157–167. doi: 10.1016/j.niox.2007.09.086. [DOI] [PubMed] [Google Scholar]
- Grocott-Mason R, Fort S, Lewis MJ, Shah AM. Myocardial relaxant effect of exogenous nitric oxide in isolated ejecting hearts. Am J Physiol. 1994;266(5 Pt 2):H1699–H1705. doi: 10.1152/ajpheart.1994.266.5.H1699. [DOI] [PubMed] [Google Scholar]
- Heinrich TA, Silva da RS, Miranda KM, Switzer CH, Wink DA, Fukuto JM. Biological nitric oxide signalling: chemistry and terminology. Br J Pharmacol. 2013;169:1417–1429. doi: 10.1111/bph.12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MH, Knight PR, Izzo JL. Ca2+-induced Ca2+ release involved in positive inotropic effect mediated by CGRP in ventricular myocytes. Am J Physiol. 1999;276(1 Pt 2):R259–R264. doi: 10.1152/ajpregu.1999.276.1.R259. [DOI] [PubMed] [Google Scholar]
- Irvine JC, Favaloro JL, Kemp-Harper BK. NO– activates soluble guanylate cyclase and Kv channels to vasodilate resistance arteries. Hypertension. 2003;41:1301–1307. doi: 10.1161/01.HYP.0000072010.54901.DE. [DOI] [PubMed] [Google Scholar]
- Irvine JC, Favaloro JL, Widdop RE, Kemp-Harper BK. Nitroxyl anion donor, Angeli's salt, does not develop tolerance in rat isolated aortae. Hypertension. 2007;49:885–892. doi: 10.1161/01.HYP.0000259328.04159.90. [DOI] [PubMed] [Google Scholar]
- Irvine JC, Kemp-Harper BK, Widdop RE. Chronic administration of the HNO donor Angeli's salt does not lead to tolerance, cross-tolerance, or endothelial dysfunction: comparison with GTN and DEA/NO. Antioxid Redox Signal. 2011;14:1615–1624. doi: 10.1089/ars.2010.3269. [DOI] [PubMed] [Google Scholar]
- Irvine JC, Cao N, Gossain S, Alexander AE, Love JE, Qin CX, et al. HNO/cGMP-dependent antihypertrophic actions of isopropylamine-NONOate in neonatal rat cardiomyocytes: potential therapeutic advantages of HNO over NO. Am J Physiol Heart Circ Physiol. 2013;305:H365–H377. doi: 10.1152/ajpheart.00495.2012. [DOI] [PubMed] [Google Scholar]
- Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp-Harper BK. Nitroxyl (HNO): The Cinderalla of the nitric oxide story. Trends Pharmacol Sci. 2008;29:601–608. doi: 10.1016/j.tips.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Jackson MI, Han TH, Serbulea L, Dutton A, Ford E, Miranda KM, et al. Kinetic feasibility of nitroxyl reduction by physiological reductants and biological implications. Free Radic Biol Med. 2009;47:1130–1139. doi: 10.1016/j.freeradbiomed.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katori T, Hoover DB, Ardell JL, Helm RH, Belardi DF, Tocchetti CG, et al. Calcitonin gene-related peptide in vivo positive inotropy is attributable to regional sympatho-stimulation and is blunted in congestive heart failure. Circ Res. 2005;96:234–243. doi: 10.1161/01.RES.0000152969.42117.ca. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klabunde RE, Ritger RC. NG-monomethyl-l-arginine (NMA) restores arterial blood pressure but reduces cardiac output in a canine model of endotoxic shock. Biochem Biophys Res Commun. 1991;178:1135–1140. doi: 10.1016/0006-291x(91)91010-a. [DOI] [PubMed] [Google Scholar]
- Kohr MJ, Kaludercic N, Tocchetti CG, Dong Gao W, Kass DA, Janssen PM, et al. Nitroxyl enhances myocyte Ca2+ transients by exclusively targeting SR Ca2+-cycling. Front Biosci. 2010;2:614–626. doi: 10.2741/e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojda G, Brixius K, Kottenberg K, Nix P, Schluter KD, Piper HM, et al. The new NO donor SPM3672 increases cGMP and improves contraction in rat cardiomyocytes and isolated heart. Eur J Pharmacol. 1995;284:315–319. doi: 10.1016/0014-2999(95)00448-t. [DOI] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K, Nix P, Schluter KD, Piper HM, Noack E. Low increase in cGMP induced by organic nitrates and nitrovasodilators improves contractile response of rat ventricular myocytes. Circ Res. 1996;78:91–101. doi: 10.1161/01.res.78.1.91. [DOI] [PubMed] [Google Scholar]
- Kojda G, Kottenberg K, Noack E. Inhibition of nitric oxide synthase and soluble guanylate cyclase induces cardiodepressive effects in normal rat hearts. Eur J Pharmacol. 1997;334:181–190. doi: 10.1016/s0014-2999(97)01168-0. [DOI] [PubMed] [Google Scholar]
- Langer M, Luttecke D, Schluter KD. Mechanism of the positive contractile effect of nitric oxide on rat ventricular cardiomyocytes with positive force/frequency relationship. Pflugers Arch. 2003;447:289–297. doi: 10.1007/s00424-003-1187-8. [DOI] [PubMed] [Google Scholar]
- Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540(Pt 2):457–467. doi: 10.1113/jphysiol.2001.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc N, Wan X, Leung PM. Physiological role of Ca2+-activated and voltage-dependent K+ currents in rabbit coronary myocytes. Am J Physiol. 1994;266(6 Pt 1):C1523–C1537. doi: 10.1152/ajpcell.1994.266.6.C1523. [DOI] [PubMed] [Google Scholar]
- Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EQ, Irvine JC, Cao AH, Alexander AE, Love JE, Patel R, et al. Nitroxyl (HNO) stimulates soluble guanylyl cyclase to suppress cardiomyocyte hypertrophy and superoxide generation. PLoS ONE. 2012;7:e34892. doi: 10.1371/journal.pone.0034892. doi: 10.1371/journal.pone.003489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda KM, Yamada K, Espey MG, Thomas DD, DeGraff W, Mitchell JB, et al. Further evidence for distinct reactive intermediates from nitroxyl and peroxynitrite: effects of buffer composition on the chemistry of Angeli's salt and synthetic peroxynitrite. Arch Biochem Biophys. 2002;401:134–144. doi: 10.1016/S0003-9861(02)00031-0. [DOI] [PubMed] [Google Scholar]
- Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, et al. A biochemical rationale for the discrete behavior of nitroxyl and nitric oxide in the cardiovascular system. Proc Natl Acad Sci U S A. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda KM, Dutton AS, Ridnour LA, Foreman CA, Ford E, Paolocci N, et al. Mechanism of aerobic decomposition of Angeli's salt (sodium trioxodinitrate) at physiological pH. J Am Chem Soc. 2005a;127:722–731. doi: 10.1021/ja045480z. [DOI] [PubMed] [Google Scholar]
- Miranda KM, Nagasawa HT, Toscano JP. Donors of HNO. Curr Top Med Chem. 2005b;5:649–664. doi: 10.2174/1568026054679290. [DOI] [PubMed] [Google Scholar]
- Mohan P, Sys SU, Brutsaert DL. Positive inotropic effect of nitric oxide in myocardium. Int J Cardiol. 1995;50:233–237. doi: 10.1016/0167-5273(95)02382-7. [DOI] [PubMed] [Google Scholar]
- Muller-Strahl G, Kottenberg K, Zimmer HG, Noack E, Kojda G. Inhibition of nitric oxide synthase augments the positive inotropic effect of nitric oxide donors in the rat heart. J Physiol. 2000;522(Pt 2):311–320. doi: 10.1111/j.1469-7793.2000.00311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa HT, DeMaster EG, Redfern B, Shirota FN, Goon DJ. Evidence for nitroxyl in the catalase-mediated bioactivation of the alcohol deterrent agent cyanamide. J Med Chem. 1990;33:3120–3122. doi: 10.1021/jm00174a001. [DOI] [PubMed] [Google Scholar]
- Paolocci N, Ekelund UE, Isoda T, Ozaki M, Vandegaer K, Georgakopoulos D, et al. cGMP-independent inotropic effects of nitric oxide and peroxynitrite donors: potential role for nitrosylation. Am J Physiol Heart Circ Physiol. 2000;279:H1982–H1988. doi: 10.1152/ajpheart.2000.279.4.H1982. [DOI] [PubMed] [Google Scholar]
- Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, et al. Nitroxyl anion exerts redox-sensitive positive cardiac inotropy in vivo by calcitonin gene-related peptide signaling. Proc Natl Acad Sci U S A. 2001;98:10463–10468. doi: 10.1073/pnas.181191198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolocci N, Katori T, Champion HC, St John ME, Miranda KM, Fukuto JM, et al. Positive inotropic and lusitropic effects of HNO/NO in failing hearts: independence from beta-adrenergic signaling. Proc Natl Acad Sci U S A. 2003;100:5537–5542. doi: 10.1073/pnas.0937302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus WJ, Vantrimpont PJ, Shah AM. Acute effects of nitric oxide on left ventricular relaxation and diastolic distensibility in humans. Assessment by bicoronary sodium nitroprusside infusion. Circulation. 1994;89:2070–2078. doi: 10.1161/01.cir.89.5.2070. [DOI] [PubMed] [Google Scholar]
- Qvigstad E, Moltzau LR, Aronsen JM, Nguyen CHT, Hougen K, Sjaastad I, et al. Natriuretic peptides increase β1-adrenoceptor signalling in failing hearts through phosphodiesterase 3 inhibition. Cardiovasc Res. 2010;85:763–772. doi: 10.1093/cvr/cvp364. [DOI] [PubMed] [Google Scholar]
- Rastaldo R, Pagliaro P, Cappello S, Penna C, Mancardi D, Westerhof N, et al. Nitric oxide and cardiac function. Life Sci. 2007;81:779–793. doi: 10.1016/j.lfs.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Ritchie RH, Zeitz CJ, Wuttke RD, Hii JT, Horowitz JD. Attenuation of the negative inotropic effects of metoprolol at short cycle lengths in humans: comparison with sotalol and verapamil. J Am Coll Cardiol. 2006;48:1234–1241. doi: 10.1016/j.jacc.2006.04.092. [DOI] [PubMed] [Google Scholar]
- Ritchie RH, Irvine JC, Rosenkranz AC, Patel R, Wendt IR, Horowitz JD, et al. Exploiting cGMP-based therapies for the prevention of left ventricular hypertrophy: NO* and beyond. Pharmacol Ther. 2009;124:279–300. doi: 10.1016/j.pharmthera.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Sandirasegarane L, Diamond J. The nitric oxide donors, SNAP and DEA/NO, exert a negative inotropic effect in rat cardiomyocytes which is independent of cyclic GMP elevation. J Mol Cell Cardiol. 1999;31:799–808. doi: 10.1006/jmcc.1998.0919. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Vallance P, Amirmansour C, Harding SE. Positive inotropic effects of NO donors in isolated guinea-pig and human cardiomyocytes independent of NO species and cyclic nucleotides. Cardiovasc Res. 2000;48:430–439. doi: 10.1016/s0008-6363(00)00202-9. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Yokoshiki H, Sperelakis N, Paul RJ. Role of voltage-dependent and Ca2+-activated K+ channels on the regulation of isometric force in porcine coronary artery. J Vasc Res. 2000;37:16–25. doi: 10.1159/000025709. [DOI] [PubMed] [Google Scholar]
- Smith JA, Shah AM, Lewis MJ. Factors released from endocardium of the ferret and pig modulate myocardial contraction. J Physiol. 1991;439:1–14. doi: 10.1113/jphysiol.1991.sp018653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocchetti CG, Wang W, Froehlich JP, Huke S, Aon MA, Wilson GM, et al. Nitroxyl improves cellular heart function by directly enhancing cardiac sarcoplasmic reticulum Ca2+ cycling. Circ Res. 2007;100:96–104. doi: 10.1161/01.RES.0000253904.53601.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocchetti CG, Stanley BA, Murray CI, Sivakumaran V, Donzelli S, Mancardi D, et al. Playing with cardiac ‘redox switches’: the ‘HNO way’ to modulate cardiac function. Antioxid Redox Signal. 2011;14:1687–1698. doi: 10.1089/ars.2010.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kohr MJ, Traynham CJ, Wheeler DG, Janssen PM, Ziolo MT. Neuronal nitric oxide synthase signaling within cardiac myocytes targets phospholamban. Am J Physiol Cell Physiol. 2008;294:C1566–C1575. doi: 10.1152/ajpcell.00367.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanstall JC, Jeffery TK, Gambino A, Lovren F, Triggle CR. Vascular smooth muscle relaxation mediated by nitric oxide donors: a comparison with acetylcholine, nitric oxide and nitroxyl ion. Br J Pharmacol. 2001;134:463–472. doi: 10.1038/sj.bjp.0704269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhof N, Boer C, Lamberts RR, Sipkema P. Cross-talk between cardiac muscle and coronary vasculature. Physiol Rev. 2006;86:1263–1308. doi: 10.1152/physrev.00029.2005. [DOI] [PubMed] [Google Scholar]
- Weyrich AS, Ma XL, Buerke M, Murohara T, Armstead VE, Lefer AM, et al. Physiological concentrations of nitric oxide do not elicit an acute negative inotropic effect in unstimulated cardiac muscle. Circ Res. 1994;75:692–700. doi: 10.1161/01.res.75.4.692. [DOI] [PubMed] [Google Scholar]
- Yuill KH, Yarova P, Kemp-Harper BK, Garland CJ, Dora KA. A novel role for HNO in local and spreading vasodilatation in rat mesenteric resistance arteries. Antioxid Redox Signal. 2011;14:1625–1635. doi: 10.1089/ars.2010.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradnikova A, Minarovic I, Venema RC, Meszaros LG. Inactivation of the cardiac ryanodine receptor calcium release channel by nitric oxide. Cell Calcium. 1997;22:447–454. doi: 10.1016/s0143-4160(97)90072-5. [DOI] [PubMed] [Google Scholar]
- Zeller A, Wenzl MV, Beretta M, Stessel H, Russwurm M, Koesling D, et al. Mechanisms underlying activation of soluble guanylate cyclase by the nitroxyl donor Angeli's salt. Mol Pharmacol. 2009;76:1115–1122. doi: 10.1124/mol.109.059915. [DOI] [PubMed] [Google Scholar]
- Ziolo MT. The fork in the nitric oxide road: cyclic GMP or nitrosylation? Nitric Oxide. 2008;18:153–156. doi: 10.1016/j.niox.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]