

Abstract
DNA methylation plays critical roles in the nervous system and has been traditionally considered to be restricted to CpG dinucleotides in metazoan genomes. Here we show that the single-base resolution DNA methylome from adult mouse dentate neurons consists of both CpG (~75%) and CpH (~25%) methylation (H = A/C/T). Neuronal CpH methylation is conserved in human brains, enriched in low CpG-density regions, depleted at protein-DNA interaction sites, and anti-correlated with gene expression. Functionally, both mCpGs and mCpHs can repress transcription in vitro and are recognized by MeCP2 in neurons in vivo. Unlike most CpG methylation, CpH methylation is established de novo during neuronal maturation and requires DNMT3A for active maintenance in post-mitotic neurons. These characteristics of CpH methylation suggest a significantly expanded proportion of the neuronal genome under cytosine methylation regulation and provide a new foundation for understanding the role of this key epigenetic modification in the nervous system.
INTRODUCTION
Accumulating evidence suggests critical roles of epigenetic mechanisms, including both histone and DNA modifications, in neuronal plasticity, neurogenesis, and neurological and psychiatric disorders1-8. Cytosine methylation is the predominant covalent modification of eukaryotic genomic DNA and regulates transcription in a highly cell type- and genomic context-dependent manner9, 10. In animals, DNA methylation is established and maintained by a conserved family of DNA methyltransferases (DNMTs)9, and can be removed in both passive and active manners11. The functions of DNA methylation, especially transcriptional repression, are in part mediated by a family of methylated DNA binding proteins (MBPs)12. Mutations in methyl-CpG binding protein 2 (MeCP2), a well-characterized MBP that is highly expressed in mature neurons, lead to deficits in neural development and neuronal functions and is causally linked to Rett syndrome, a severe neurodevelopmental disorder in humans13, 14.
In metazoan genomes, cytosine methylation is thought to be largely restricted to the CpG dinucleotide, which facilitates mitotic transmission of the methylation pattern15, 16. Interestingly, both the maintenance DNMT (DNMT1) and de novo DNMTs (DNMT3A and DNMT3B) have been shown to methylate non-CpG cytosines in vitro17, 18. Previous studies have shown that CpH methylation is present in cultured pluripotent stem cells, including embryonic stem cells (ESCs), induced pluripotent stem cells19-24, as well as in the mouse germ line25-27, but absent in most somatic tissues19, 23. Several recent profiling studies have shown the presence of CpH methylation in the adult mouse cortex28, 29 and human brains29, 30, which consist of mixtures of many neural subtypes. These observations raise important questions: Does CpH methylation play any roles in transcriptional regulation in mammalian cells, and if so, what protein readers may recognize CpH methylation? In addition, little is known about the enzymatic mechanisms that establish and maintain CpH methylation in neurons.
Here, we generated the single-base-resolution neuronal DNA methylation profile of the adult mouse dentate gyrus and characterized the genomic distribution of CpH methylation. We further demonstrated that CpH methylation is conserved in human brains in orthologous genes. Using a plasmid reporter system, we showed that CpH methylation could cause transcriptional repression in mouse neurons. Importantly, MeCP2 bound to mCpH both in vitro and in neurons in vivo. In addition, we found that CpH methylation was established postnatally during neuronal maturation and required DNMT3A for its active maintenance in neurons in vivo.
RESULTS
Single-base-resolution neuronal DNA methylome
To systematically characterize the in vivo neuronal methylome, we purified genomic DNA from a relatively homogeneous population of granule neurons from the adult mouse dentate gyrus31-33 and performed whole-genome bisulfite sequencing (Bisulfite-Seq) for two biological replicates. We obtained a total of ~ 43 Gb sequences (~ 1.5 billion 2x100 bp paired-end reads mapped; ~ 16x coverage per strand) that were uniquely mapped to the in silico bisulfite-converted mouse genome with no mismatch. To identify significantly methylated cytosines (mCs) genome-wide, we used a stringent binomial distribution-based filter to eliminate false positives from incomplete bisulfite conversion and sequencing errors. Our analysis pipeline confirmed the previous finding that CpH methylation is present in human ESCs, but not in fibroblasts20 (data not shown). Importantly, our analysis also revealed that ~ 25% of all mC loci in the adult mouse dentate neuronal genome were mCpHs (Fig. 1a), which consisted of ~ 4% mCHGs and ~ 21% mCHHs, with mCHGs being underrepresented (p < 10−15, χ2 test). Global CpG and CpH methylation levels were similar among autosomes, whereas sex chromosomes exhibited the lowest levels of CpH methylation (Supplementary Fig. 1). Methylation levels of individual mCpHs and mCpGs between two biological replicates were highly correlated (Supplementary Fig. 2).
Figure 1. Pervasive CpH methylation in the in vivo DNA methylome of adult dentate granule neurons.

(a) Composition of all mC loci in the genome of adult mouse dentate granule neurons. (b) Genomic DNA samples from adult dentate granule neurons were digested at methylated CmC motifs by FspEI. Sensitivity to FspEI digestion was measured by qPCR using primers flanking the predicted digestion sites (Supplementary Table 1a). Values represent mean ± s.e.m. (n = 3). Bisulfite-Seq results for each region are indicated by grey bars. (c) Four CpH-methylated loci were further examined by Sanger bisulfite sequencing in independent adult mouse dentate gyrus and spleen samples and FACS-sorted NeuN+ neuronal nuclei. Each row represents one DNA clone. Each column represents one mC site. Unmethylated and methylated cytosines are represented by open and filled boxes, respectively. CpG and CpH methylation are color-coded (black, CpG; red, CpH). Corresponding Bisulfite-Seq results are shown on the top panel.
We next selected a group of loci with high levels of CpH methylation for more detailed analyses. Bisulfite conversion-independent measurements using a methylation-dependent restriction enzyme FspEI, which selectively digests CmC motifs34, confirmed the presence of mCpH in the adult dentate gyrus (Fig. 1b and Supplementary Table 1a). Sanger bisulfite sequencing in independent samples also confirmed the presence of CpH methylation at selected loci in the adult mouse dentate gyrus (Fig. 1c and Supplementary Table 1b). CpH methylation at these loci was virtually absent in mouse spleen, whereas methylation of the examined CpG loci was largely conserved (Fig. 1c). In contrast to CpG methylation, CpH methylation was apparently heterogeneous among different alleles from a homogenous population of neurons, although it did not clearly segregate into hypermethylated and hypomethylated alleles (Fig. 1c). To exclude the potential contribution from non-neuronal cells, such as neural progenitors, astrocytes and oligodendrocytes, we analyzed the genomic DNA of FACS-purified NeuN+ neuronal nuclei from the adult dentate gyrus33 and observed similar levels of CpG and CpH methylation (Fig. 1c). Therefore, our study comprehensively and reliably identified a large number of mCpHs in the adult mouse dentate neuronal DNA methylome in vivo.
Conservation of CpH methylation in adult human brains
To examine whether neuronal CpH methylation identified in mouse genomic DNA was conserved in other mammals, we performed Sanger bisulfite sequencing in the orthologous genomic regions using adult human brain and spleen DNA samples (Supplementary Table 1c). Despite the fact that most of these regions exhibited different CpG patterns, highly reproducible levels of CpH and CpG methylation were observed in all orthologous regions examined using adult human brain genomic DNA from different individuals, but not spleen DNA (Fig. 2a, b), suggesting the evolutionary conservation of neuronal CpH methylation.
Figure 2. Conserved CpH methylation in orthogolous regions of the human brain DNA.

(a) Sanger bisulfite sequencing results of orthologous regions in the adult human brain and spleen genomic DNA. (b) Consistent levels of CpH methylation in multiple adult human cortical genomic DNA samples (Ctx1–3). (c) Reduced representation bisulfite sequencing (RRBS) data generated by the ENCODE project30 were analyzed using the mitochondrial CpH methylation rate (~1%) as the background probability. Percentage of mCpG/mCpH was corrected by the FDR estimated by a binomial distribution. (d) Quantification of numbers of mCpHs for each of the 15,417 one-to-one orthologous gene pairs between mouse and human (Ensembl annotations). A gene was considered CpH-methylated if two or more CpHs were ≥25% methylated (P value is indicated, χ2 test)
To extend this finding to the genome scale, we analyzed the reduced representation bisulfite sequencing (RRBS) data generated by the ENCODE consortium30 and observed much higher global levels of CpH methylation in the human brain and placenta than in other somatic tissues (Fig. 2c). Finally, we focused on genes with clear one-to-one orthologs in human and mouse (15,417 ortholog pairs in total), and quantified the degree of overlap of CpH-methylated genes (genes that contained ≥ 2 mCpHs with ≥ 25% methylation levels) in human and mouse brains. Although RRBS did not provide genome-wide high coverage and therefore fewer genes were identified as being CpH-methylated in the human brain than in the mouse brain, a majority (~ 83%) of the human CpH-methylated genes had their orthologs also CpH-methylated in the mouse brain (Fig. 2d). Together, these results indicated that CpH methylation not only widely existed in mammalian neurons, but also marked conserved sets of genes in both mice and humans.
Genomic features of neuronal CpH methylation
To characterize the genome-wide distribution of CpH methylation in adult dentate neurons, we focused on high-coverage (≥ 20x) cytosines for detailed bioinformatic analyses. Unlike individual mCpGs, which exhibited a bimodal distribution of methylation levels, intermediate methylation levels (mostly ~ 25%) were observed in most mCHGs and mCHHs (Fig. 3a), suggesting that only a fraction of all alleles were methylated at the steady state. Although most mCHGs and mCHHs were adjacent to highly methylated CpGs (Supplementary Fig. 3a), a chromosome-wide view of three classes of cytosine methylation (CpG, CHG and CHH) showed that the correlation between CHG and CHH methylation was much higher than that between CpG and CpH methylation (Fig. 3c and Supplementary Fig. 3), mostly due to the regions with high levels of CpG methylation levels but low levels of CpH methylation. Motif analysis further identified a prominent CAC preference for CpH methylation in neurons (Fig. 3b). The preference for A at the +1 position would predict asymmetric methylation patterns on two DNA strands. Indeed, while genome-wide CpG methylation on two strands was highly correlated, the correlations were much weaker for CHG and CHH methylation (Supplementary Fig. 4a). Also predicted by the CpA preference, for each mCHG (mostly CAG), the C at the +2 position in the opposite strand (mostly CTG) was rarely methylated (Supplementary Fig. 4b).
Figure 3. Genomic features of the neuronal CpH methylation.

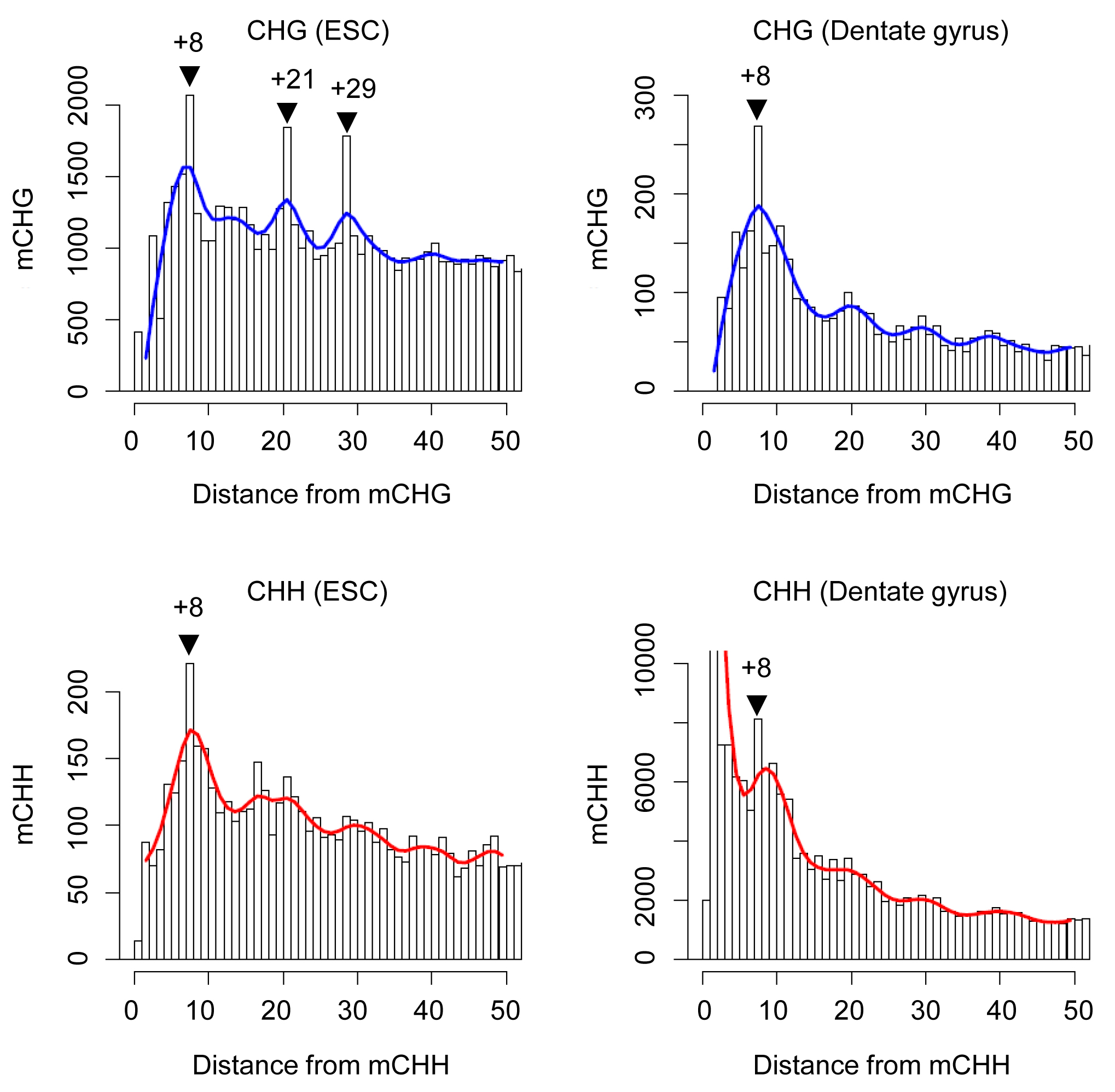
(a) Distributions of methylation levels of mCpG (black), mCHG (blue) and mCHH (red) in the genome of adult mouse dentate granule neurons. (b) Motif analysis of all mCpHs, with mCs being in position 0. (c) A chromosome-wide view of three types of mCs on chromosome 14 (chr14). Methylation levels were moving-averaged by 100 kb windows. Refseq transcript annotation is shown at the bottom. (d) Spacing analysis of adjacent mCpHs. The cubic spline smoothing curve is shown. Please see comparison between adult dentate granule neurons and embryonic stem cells in Supplementary Fig. 5. (e) Relationship between mCpG/mCpH occurrence and their local CpG densities.
In contrast to the distinct spacing patterns of mCHG and mCHH found in ESCs23, the same analysis showed a similar 8 bp spacing for both mCHGs and mCHHs in neurons (Fig. 3d and Supplementary Fig. 5), suggesting a common mechanism regulating CHG and CHH methylation in neurons. In addition, we observed a periodicity of ~ 180 bp for neuronal mCpHs (Fig. 3d), supporting previous findings on the relationship between DNA methylation and nucleosome positioning35. Importantly, mCpHs preferentially resided in low-CpG density regions (Fig. 3e), with ~ 8% of mCpHs having no neighboring CpGs within 500 bp flanking sequences, raising the possibility of a mCpG-independent role for mCpHs in these low CpG-density regions.
Relationship between mCpH, protein-DNA interaction and gene expression
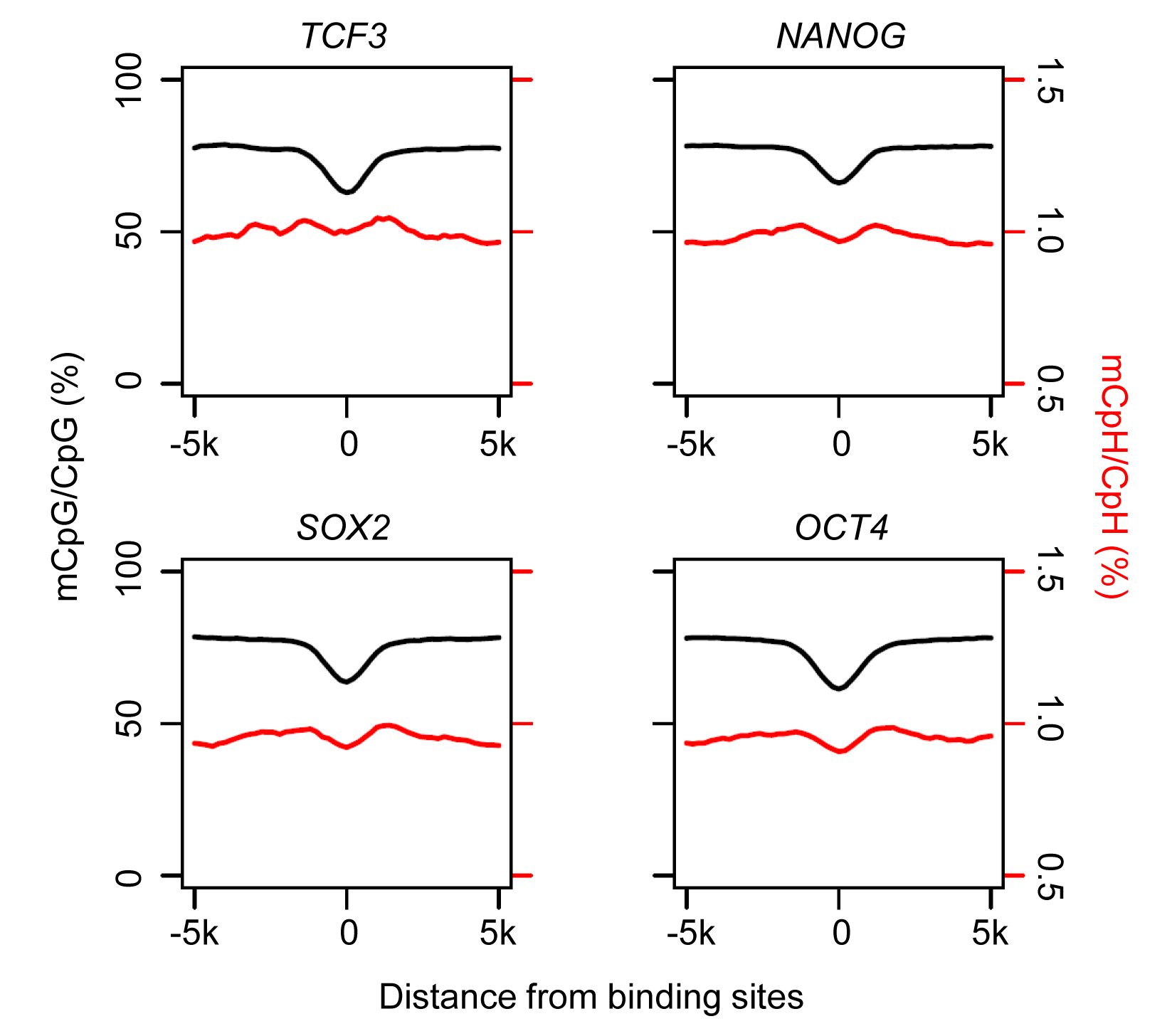
To begin to understand the potential function of CpH methylation in neurons, we first examined its relationship with neuronal protein-DNA interactions. Both CpGs and CpHs were hypomethylated around neuronal transcription factor binding sites previously identified in neurons36 (Fig. 4a and Supplementary Table 2), suggesting that protein-DNA interaction exhibited similar effects on CpGs and CpHs in preventing de novo DNA methylation and/or causing active DNA demethylation30. In contrast, hypomethylation of neuronal DNA at ESC-specific transcription factor binding sites37 was much less pronounced in neurons (Supplementary Fig. 6). Similar to neuronal transcription factor binding sites, both CpGs and CpHs were hypomethylated around transcription start sites (TSSs; Fig. 4b).
Figure 4. Relationship between CpH methylation and protein-DNA interaction or gene expression in vivo.

(a) Averaged methylation levels at previously profiled protein-DNA interaction sites. Data references for protein-DNA interactions are listed in Supplementary Table 2. CBP: CREB-binding protein; CREB: cAMP responsive element-binding protein; NPAS4: neuronal PAS domain protein 4; POL2: RNA polymerase II; SRF: serum response element-binding transcription factor; CTCF: CCCTC-binding factor. (b) Averaged methylation levels across all annotated genes, stratified by their mRNA levels in the adult mouse dentate gyrus. Both CpG and CpH methylation were anti-correlated with expression levels of associated genes. TSS: transcription start site; PAS: poly(A) site; RPKM: reads per kilobase of exon model per million mapped reads. Please see anti-correlation between CpG-far CH methylation in regulatory regions and gene expression in Supplementary Fig. 7.
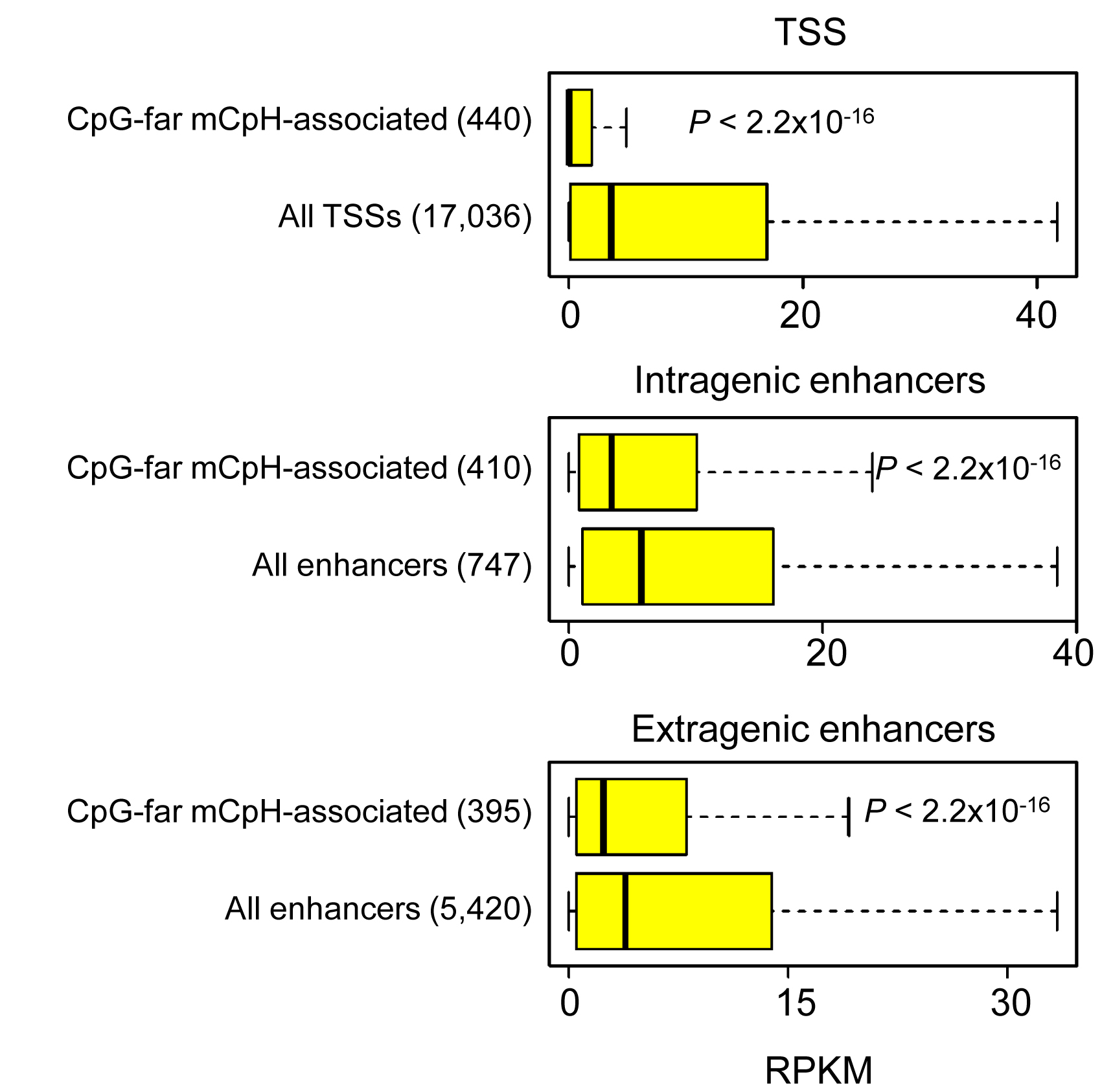
To further understand the potential role of CpH methylation in transcriptional regulation, we profiled gene expression in the adult mouse dentate gyrus by mRNA-Seq (~ 92 million reads from three biological replicates). In contrast to ESCs23 and consistent with previous results from a mixed population of neural cells28, 29, both neuronal CpG and CpH methylation anti-correlated with associated gene expression levels throughout the 5′ upstream, gene body and 3′ downstream regions in dentate granule neurons (Fig. 4b). To minimize the contribution from nearby mCpGs, we focused on the ~ 8% or 70,364 mCpHs that did not have any neighboring CpGs within 500 bp flanking sequences (Fig. 3e). 395, 410 and 440 of these CpG-far mCpHs were located within 2 kb of extragenic enhancers, intragenic enhancers36 and TSSs, respectively. Importantly, these CpG-far mCpH-containing regulatory regions were also associated with significantly lower nearest-gene expression levels compared to corresponding background gene sets (Supplementary Fig. 7), further suggesting a potential function of CpH methylation in transcriptional repression.
Repression of reporter gene expression by mCpH
To directly assess the intrinsic capacity of CpH methylation in transcriptional repression, we modified a well-established quantitative reporter assay using in vitro methylated plasmids31, 32, 38, 39. GFP-expressing plasmids were methylated at specific locations using a panel of bacterial DNMTs before co-transfection with unmethylated RFP-expression plasmids into HEK293 cells (Supplementary Fig. 8a) or cultured mouse hippocampal neurons (Fig. 5a). Importantly, with the same mC density, CpH methylation by M.MspI (mCCGG) caused a similar degree of repression compared to CpG methylation by M.HpaII (CmCGG; Fig. 5b and Supplementary Fig. 8b). In addition, GpC methylation by M.CviPI, most of which resided in the CpH context, caused similar repression as CpG methylation by M.SssI (Fig. 5b and Supplementary Fig. 8b). The strong repression caused by GpC methylation was not simply due to the overlapping CpGs (GmCGs), because the repression caused by M.SssI-catalyzed CpG methylation could be further strengthened by either M.MspI or M.CviPI methylation (Supplementary Fig. 8b). None of these methylation patterns showed detectable effects on transfection efficiencies (Fig. 5b and Supplementary Fig. 8b). Bisulfite sequencing of plasmid DNA recovered after transfection showed high efficiencies of all types of in vitro methylation and no de novo CpG methylation in CpH-methylated plasmids (Supplementary Fig. 8c), indicating that CpH methylation did not repress GFP expression indirectly by inducing CpG methylation. Although in vitro methylation did not fully recapitulate endogenous CpH methylation patterns, these results suggested that mCpGs and mCpHs both have the intrinsic capacity to repress transcription in mammalian cells, including in neurons.
Figure 5. Repression of reporter gene expression by CpH methylation in neurons.

(a) A schematic diagram of experimental design of the in vitro methylated plasmid reporter assay. (b) Representative phase-contrast and immunofluorescent images of cultured hippocampal neurons co-transfected with an unmethylated RFP plasmid and GFP plasmids with different methylation patterns (left). Scale bar: 50 μm. Also shown is quantification of percentages of GFP+RFP+ neurons among all RFP+ neurons (right). Values represent mean ± s.e.m. (n = 3;P values are indicated for each condition; one-way ANOVA and Tukey’s test).
Recognition of mCpH by MeCP2 in vitro and in vivo
MeCP2 has been well established to recognize mCpGs and regulate gene expression in the nervous system13, 14. Across the genome both CpGs and CpHs were hypermethylated near MeCP2-bound regions previously determined by chromatin-immunoprecipitation (ChIP) in neurons40 (Fig. 6a). We directly tested the binding capacity of recombinant MeCP2 protein to synthetically methylated oligos by electrophoretic mobility shift assay (EMSA). As expected, MeCP2 exhibited clear binding to mCpG-containing oligos (Fig. 6b). Notably, MeCP2 also bound to CpH-methylated oligos with a lower affinity in the absence of any mCpGs. Furthermore, the presence of both mCpGs and mCpHs greatly enhanced MeCP2 binding (Fig. 6b). In contrast, another MBD protein MBD2b exhibited higher selectivity towards mCpGs and bound minimally to CpH-methylated oligos in this assay, although the co-existence of mCpGs and mCpHs also enhanced MBD2b binding (Supplementary Fig. 9).
Figure 6. Recognition of CpH methylation by MeCP2 in vitro and in vivo.

(a) Neuronal CpG (top) and CpH (bottom) methylation levels were averaged around MeCP2-bound regions previously determined using whole-mouse brains40. (b) EMSA analysis using four oligo probes methylated at specific positions indicated in bold (top). Quantification of MeCP2-bound oligos is also shown (bottom). Values represent mean ± s.e.m. (n = 3). See Supplementary Fig. 9 for results of MBD2b. (c) MeCP2 ChIP-bisulfite-sequencing analysis of unmethylated, CpG-methylated/CpH-unmethylated, and CpH-methylated regions in the adult mouse hippocampus. The levels of CpH methylation in the latter regions were significantly higher in ChIP samples than in the input DNA from adult mouse hippocampus (P values are indicated; Fisher’s exact test).
To determine physiologically whether endogenous MeCP2 recognizes mCpHs in vivo, we examined the methylation patterns of endogenous MeCP2-bound DNA by bisulfite sequencing of the MeCP2-ChIP DNA from adult mouse hippocampus. Consistent with previous studies48, MeCP2-ChIP selectively enriched CpG-methylated alleles at a given locus (Fig. 6c). Importantly, CpH-methylated regions that were depleted of CpGs were also highly enriched in the MeCP2-bound chromatin (Fig. 6c), supporting the in vivo interaction between MeCP2 and mCpHs in neurons.
mCpH establishment and maintenance in vivo
We next examined the developmental timing of CpH methylation in neurons. Consistent with a recent study of the cortex29, time-course analyses revealed that CpH methylation at the selected loci was established during postnatal development of the hippocampus and then present throughout life, whereas CpG methylation was established during early development (Fig. 7a). Maturing mouse hippocampal neurons in vitro also showed a gradual increase in CpH methylation over time, but not in CpG methylation (Fig. 7b). Furthermore, CpH methylation levels in the adult human brain DNA were much higher than those in fetal brain samples, and adult and fetal human heart DNA (Fig. 7c). Together, these results indicated that, distinct from most CpG methylation25, CpH methylation was established during postnatal neuronal maturation in both mouse and human brains.
Figure 7. Establishment of CpH methylation during neuronal maturation.

(a) Progression of CpH (left) and CpG (right) methylation levels in the mouse brain at eight developmental time points. Values represent means (n = 3). (b) CpG and CpH methylation in mouse hippocampal neuronal cultures. Methylation levels were measured after 7 or 14 days in vitro (DIV). Values represent mean + s.e.m. (n = 3; P values are indicated; Student’s t test). (c) CpG and CpH methylation in fetal and adult human tissues.
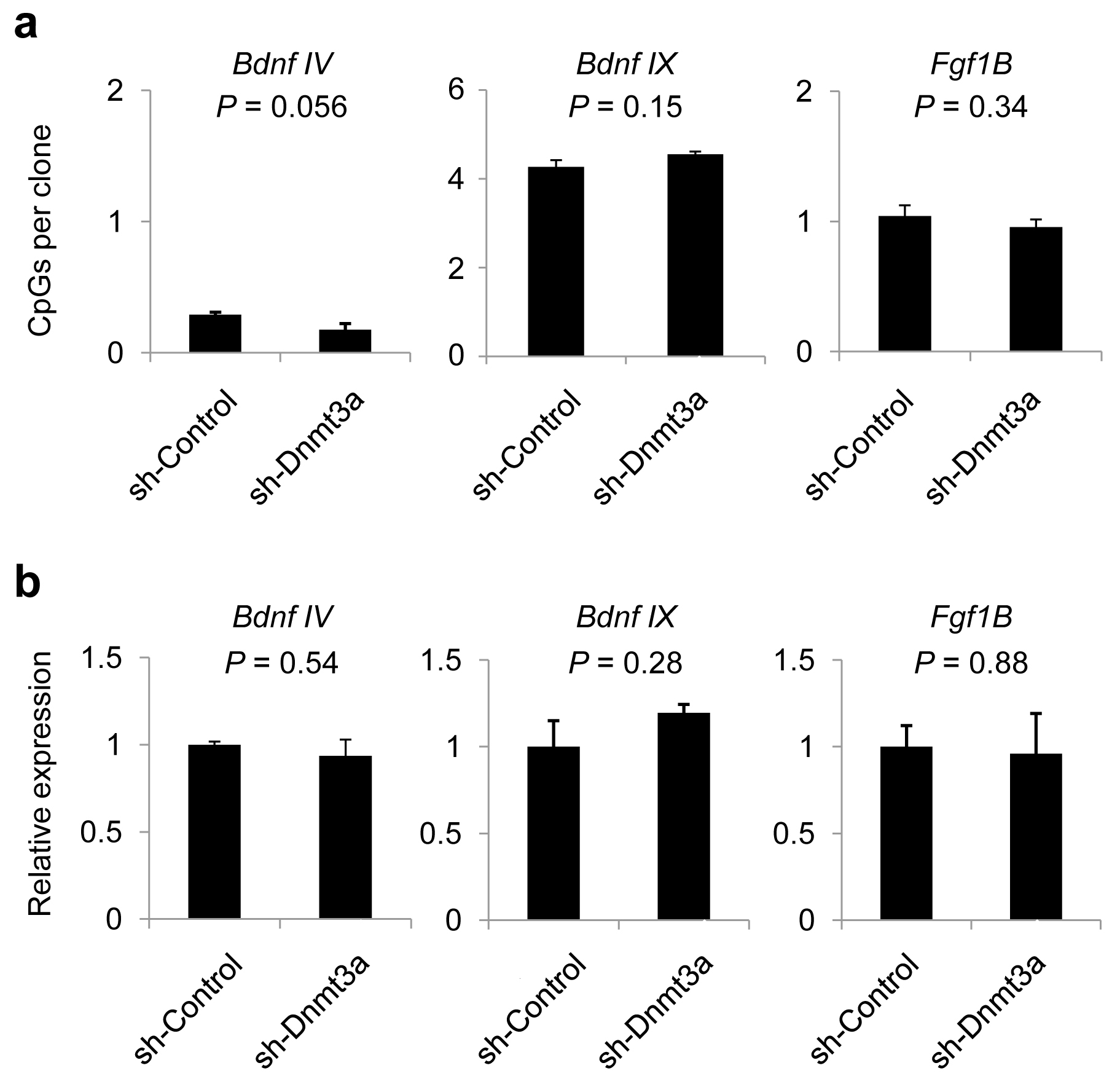
Finally, we examined the regulation of CpH methylation in post-mitotic neurons in vivo. Conditional neuronal triple knockout of Dnmt1/3a/3b (CamKIIα-Cre) led to a preferential loss of CpH methylation in the postnatal hippocampus at the same set of loci examined, whereas CpG methylation was only modestly reduced as previously observed41 (Fig. 8a). Among the three DNMTs, DNMT1 and DNMT3A are mostly prominently expressed in post-mitotic neurons41. To determine their individual roles in maintaining neuronal CpH methylation in vivo, we injected adeno-associated viruses (AAVs) expressing short-hairpin RNAs (shRNAs) that specifically targeted DNMT1 or DNMT3A into the dentate gyrus of adult wild-type mice (Supplementary Fig. 10). DNMT3A knock-down led to a significant reduction in CpH methylation, but not CpG methylation at these loci, whereas DNMT1 knock-down showed little effect (Fig. 8b). Importantly, the preferential loss of CpH methylation caused by DNMT3A knock-down was accompanied by the significant de-repression of the mRNA expression of these CpH-methylated genes (Fig. 8c), whereas no methylation or mRNA expression changes were observed in a unmethylated gene (Bdnf IV) or CpG-methylated/CpH-unmethylated genes (Bdnf IX and Fgf1B; Supplementary Fig. 11). Furthermore, DNMT3A-ChIP analysis showed that CpH-methylated regions were bound by DNMT3A in dentate neurons in vivo (Supplementary Fig. 12 and Supplementary Table 1e). These results strongly supported a physiological role of CpH methylation in repression of gene expression in neurons in vivo. To examine whether differences in de novo DNMT expression could explain the brain-specific accumulation of CpH methylation, we measured the relative expression levels of DNMTs in a variety of mouse tissues. Surprisingly, we found a similar expression level of DNMT3A, and much lower expression levels of DNMT3A2 and DNMT3B in the brain, compared to other tissues without CpH methylation (Supplementary Fig. 13). Collectively, these results suggested that continuous DNMT3A expression is required but not sufficient to accumulate neuronal CpH methylation in vivo.
Figure 8. Neuronal CpH methylation is actively maintained by DNMT3A and regulates endogenous gene expression in vivo.

(a) CpH methylation levels were significantly decreased in postnatal neuron-specific DNMT1/3a/3b triple knockout (cTKO; CamKIIa-Cre). Samples from adult hippocampus were examined (P values are indicated; Fisher’s exact test). (b) Requirement of DNMT3A for maintenance of CpH methylation in the adult dentate gyrus. AAVs expressing different shRNAs were stereotaxically injected into the adult mouse dentate gyrus. DNA methylation was measured one week later from micro-dissected dentate gyri. Values represent mean ± s.e.m. (n = 3; P values are indicated; Student’s t-test). (c) mRNA expression of CpH-methylated genes following DNMT3A knock-down. Values represent mean ± s.e.m. (n = 3; P values are indicated; Student’s t-test). See also Supplementary Fig. 11 for results on unmethylated and CpG-methylated/CpH-unmethylated genes.
DISCUSSION
Our systematic analysis of the in vivo neuronal methylome from adult mouse dentate gyrus at single-base-resolution and subsequent mechanistic studies generated novel insights into the genomic features, conservation, potential function, reader and regulator of non-CpG methylation in the adult mammalian brain. Given that CpGs only represent 4% of the metazoan genome, CpH methylation greatly expands the proportion of the neuronal genome that is subject to regulation by cytosine methylation and represents a new mechanism of neuronal transcription regulation. Our study provided a number of novel findings that have significant implications for the future understanding of this epigenetic modification in the nervous system.
Different from previous profiling studies28-30, we obtained single-base resolution DNA methylomes from the in vivo adult dentate gyrus preparation with the vast majority of cells consisting of a single neuronal subtype, therefore providing a unique resource for the field. In adult dentate neurons, CpH methylation accounted for ~ 25% of all mCs, which was similar to recent findings in the mouse cortex28 and human pluripotent stem cells in vitro19-24. Our study revealed some clear distinctions between mCpHs in the brain and those in pluripotent stem cells (Supplementary Fig. 14). For example, the preference of mCHGs over mCHHs and the 8, 21, 29 bp spacing pattern of mCHGs in proliferating pluripotent stem cells23 are absent in post-mitotic neurons. Instead, their highly correlated genomic distributions, identical 8 bp spacing (reminiscent of the DNMT3A:DNMT3L complex42) and requirement for DNMT3A for active maintenance suggested that mCHG and mCHH are similarly regulated in neurons. Though the bisulfite sequencing approach does not differentiate between mC and hydroxymethylcytosine (hmC), the vast majority of steady-state mCpHs are unlikely to be hydroxymethylated. Recent single-base resolution hmC studies have shown a lack of significant hmCpHs in pluripotent stem cells and in the brain29, 43, 44. In addition, mCpHs and hmCs exhibit opposite correlations with gene expression43-45.
Our study provided the first evidence for the potential function of CpH methylation in the nervous system. In a plasmid reporter assay, mCpHs were sufficient to repress gene transcription in cultured neurons, surprisingly, as effectively as mCpGs. Although in vitro methylation was complete and symmetric while endogenous CpH methylation was mostly intermediate and asymmetric, our results suggested an intrinsic capacity of mCpHs to repress transcription independently of CpG contexts. The de-repression of CpH-methylated genes upon DNMT3A knockdown, which preferentially decreased CpH methylation, but not CpG methylation, provided physiological evidence for a critical role of CpH methylation in repressing gene expression in vivo. We further showed a genome-wide anti-correlation between CpH methylation levels and the associated gene expression, including mCpHs in regulatory genomic regions without any nearby CpGs.
Recent studies have shown dynamic changes of neuronal DNA methylation in development, maturation, plasticity, learning memory and brain disorders7, 8. Notably, both CpGs and CpHs are substrates for methylation46, 47 and demethylation32. Our result suggested that mCpHs are a more dynamic population in the neuronal DNA methylome in vivo. When de novo DNMT activities were decreased, CpH methylation was preferentially lost before CpG methylation showed any changes. The apparently higher turnover of mCpHs than mCpGs could be due to either or both of the two following possibilities. First, demethylation of mCpHs could be more efficient than that of mCpGs. Our previous in vitro study using fully hydroxymethylated DNA indicated that hmCpHs were more efficiently demethylated than hmCpGs by ~4 fold in mammalian cells32. Second, re-methylation of CpG could be more efficient than that of CpH. Because demethylation is mediated by a base-excision repair mechanism in a strand-specific manner32, demethylation of mCpG would first generate a hemi-methylated CpG, which could be readily re-methylated by DNMT1. In contrast, mCpH, once demethylated, can only be re-methylated de novo by DNMT3A. The apparent heterogeneous methylation patterns of mCpHs in neurons (Fig. 3a) may be a result of their dynamic turnover, with only a fraction of all alleles captured in the methylated status at the steady state. It will be interesting for future studies to address how DNMT3A and DNA demethylases are specifically targeted to mCpH loci and the biological significance of the dynamic turnover of mCpHs in the neuronal gene expression program. It also remains to be investigated to what extent CpH methylation mediates the unique and essential role of DNMT3A in post-natal development and functions of the nervous system41, 48. Notably, DNMT3A has been shown to regulate emotional behavior and spine plasticity in the nucleus accumbens49.
Our study pinpointed the developmental timing of CpH methylation during neuronal maturation and identified MeCP2 as the first mCpH binding protein in vitro and in vivo. Despite the widely recognized importance of MeCP2 as a DNA methylation reader for mCpGs50, and more recently for hmCpGs45, little is known about how it actually reads DNA methylation on a fine scale. Our results from the in vitro binding assay and in vivo ChIP-bisulfite sequencing assays directly shed light onto this fundamental question. Intriguingly, the timing of postnatal onset of Rett syndrome coincides with the emergence of CpH methylation, but not CpG methylation in neurons. Our finding of a strong influence of mCpH influence on MeCP2 binding in neurons in vivo indicates a new direction for understanding the biology of MeCP2 and mechanisms underlying MeCP2-related mental disorders.
Cytosine methylation plays critical roles in regulating gene expression under both physiological and pathological conditions. While CpG methylation occurs more frequently on average across the genome, large fractions of mammalian genomes are devoid of CpGs. Our observation of preferential CpH methylation in these CpG-depleted regions suggested that CpH methylation might at least partly compensate for the lack of CpGs and increase the local mC density in neurons without adding constitutively methylated new CpG dinucleotides in the genome. Such a compensatory effect was observed not only in the transcriptional regulatory effects of mCpGs and mCpHs, but also in MeCP2 binding, which was greatly enhanced when mCpG and mCpH were adjacent to each other. In summary, CpH methylation greatly expands the proportion of the neuronal genome that is under regulation by cytosine methylation and represents a new layer of epigenetic modulation of the neuronal genome. The novel characteristics of CpH methylation point to new directions for future understanding this epigenetic modification in neuronal identity, development, plasticity, and psychiatric and neurological disorders.
ONLINE METHODS
Sample preparation
Mice (E12 to two years old, male, C57BL/6 background, housed in a vivarium with a 12/12 light/dark cycle, with no more than five mice per cage) were used for analysis in accordance with protocols approved by the Institutional Animal Care and Use Committee of Johns Hopkins University School of Medicine. For post-natal day 21 (P21) to two year old animals, micro-dissection of dentate granule cell layers was rapidly performed bilaterally from hippocampus. This preparation was highly enriched for mature neurons as shown by immunohistochemistry to contain ~ 90% NeuN+ neurons, most of which were Prox1+ dentate granule cells31. Previous studies at selective loci showed quantitatively very similar CpG methylation status with FACS purified NeuN+ mature neurons33. The current study also showed quantitatively similar levels of CpG and CpH methylation at multiple loci examined between micro-dissected samples and FACS purified NeuN+ neuronal samples (Fig. 1c). For embryonic day 12 and P1 studies, whole brain and spleen were used. For early postnatal tissues (P3-7) and for adult CaMKIIα-Cre::Dnmt1f/f3af/f3bf/f mice41, whole hippocampi were used. For AAV-mediated knockdown of endogenous Dnmt1 or Dnmt3a expression, engineered AAV co-expressing GFP under the Ubi promoter and shRNA against mouse Dnmt1 or Dnmt3a or control shRNA under the U6 promoter were stereotaxcially injected into the dentate gyrus of adult wild-type mice as previously described32. The following shRNA sequences were used: GTTCAGATGTGCGGCGAGT (control); GCTGACACTAAGCTGTTTGTA (Dnmt1); CATCCACTGTGAATGATAA (Dnmt3a). We have previously shown widespread expression across the whole dentate gyrus via AAV-mediated infection32. Dentate gyri were micro-dissected one week later for quantitative PCR analysis of endogenous gene expression and for DNA methylation analysis. Adult and fetal human brain, spleen and heart genomic DNA samples were from BioChain. Hippocampal neuron cultures were prepared from mouse E18 hippocampus as previously described51. These cultures were highly enriched with neurons (> 99%) and contained very few other cell types.
Genomic DNA was extracted using DNeasy (Qiagen). Total RNA was extracted using RNeasy (Qiagen) with DNaseI digestion.
Bisulfite-Seq
Three μg genomic DNA spiked-in with 0.5% unmethylated lambda DNA (Promega) from each sample was fragmented to ~ 300 bp DNA fragments using Covaris Acoustic System. The ends of fragmented DNA were repaired with T4 DNA polymerase, Klenow DNA polymerase, and T4 PNK were used to convert the overhangs into phosphorylated blunt ends. Klenow Fragment (3’ to 5’ exo minus) was used to add an ‘A’ base to the 3’ end of the blunt phosphorylated DNA fragments. Following ligation of adapters (TruSeq index, 12 indexes), bisulfite conversion was performed using an EZ DNA Methylation-Direct kit (Zymo). PCR was then used to enrich bisulfite-converted DNA fragments to obtain the DNA library suitable for HiSeq2000 sequencing. Paired-end sequencing reads (100 bp) were aligned to the in silico bisulfite converted mouse reference genome mm9 using Bismark52 in a strand-specific manner not allowing any mismatches or multiple alignments. In-house Perl scripts were used to summarize methylation levels of individual cytosines.
Identification of mC loci
A conservative binomial model was used to establish the threshold for mC calling. Specifically, at each locus, we randomly generated the C (methylated) and T (unmethylated) read numbers with the actual total read number at this locus, using a binomial distribution of P = 0.006 (the non-conversion rate determined by spiked-in unmethylated lambda DNA results). For each different threshold, we calculated the false discovery rate (FDR) using the ratio between the number of mC loci called from the “null” data and that of the actual data. We chose a threshold of P < 10-5, under which the FDR for all three classes of mCs are less than 3%. We further validated this model by applying it to the dataset from Lister et al. (2009)20, which confirmed previously published results.
Sanger bisulfite sequencing and ChIP-bisulfite sequencing
Strand-specific primers were designed for the bisulfite-converted genome sequence, and synthesized by IDTDNA (Supplementary Table 1b, c). Sanger bisulfite sequencing was carried out as previously described31-33. Briefly, 2 μg genomic DNA for each sample was bisulfite-treated and column-purified (Zymo). Fifty ng bisulfite-converted DNA was used in each PCR (Invitrogen). Amplicons were gel-purified and TA-cloned into pCR2.1 vectors (Invitrogen). Bacterial colonies were sent for Sanger sequencing using M13R primer (Genewiz). Typically, 10-20 clones were examined for each sample. Notably, the primers designed for Sanger bisulfite sequencing only avoided CpGs and would therefore preferentially amplify those alleles with no or low levels of CpH methylation in the priming regions.
For DNMT3A-ChIP, freshly microdissected dentate gyri were cut into fine pieces and incubated in 1% methanol-free formaldehyde (Pierce) in DPBS for 20 minutes with continuous rocking. 10x glycine (Cell Signaling) was added to the solution and incubated for 5 minutes. After washed with ice-cold DPBS, tissue pieces were homogenized using Douning homogenizer (Kimble Chase) on ice. After centrifugation, the pellet was resuspended in cell lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% Igepal) and rotated at 4°C for 10 minutes. After centrifugation, the pellet was resuspended with shearing buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris pH 8.0) and sonicated by Bioruptor plus (Diagenode) for 21 cycles (30s on, 90s off) at the high intensity setting. After centrifugation, NaCl (150 mM) and Triton X-100 (1%) were added to the supernatant and incubated with protein G Dynabeads (Life Technologies) for 30 minutes. After removing the beads, anti-Dnmt3a (sc-20703, Santa Cruz) or rabbit IgG (2729, Cell Signaling) antibodies were added and incubated at 4°C overnight. Next, protein G beads were added to the samples and incubated at 4°C for two hours. The beads were washed once with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.0, 150 mM NaCl), once with high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.0, 500 mM NaCl), once with LiCl wash buffer (1% Sodium Deoxycholate, 1% Igepal, 10 mM Tris pH 8.0, 1 mM EDTA, 250 mM LiCl), and twice with 1x TE buffer. Freshly made elution buffer (1% SDS, 0.1M NaHCO3) was added to the beads and chromatin was eluted at 65°C thermomixer for 1 hour. After removing beads, cross-linking was reversed at 65°C overnight. Finally, Protease K (NEB) was added to the de-crosslinked chromatin solution and incubated additional 2 hour at 55°C. The eluted DNA fragments were purified using PCR purification kit (Qiagen), following by qPCR using region specific primers (Supplementary Table 1e).
For MeCP2 ChIP-bisulfite sequencing analyses, hippocampi were microdissected from knock-in mice in which a FLAG tag was fused to endogenous MeCP2 protein in frame (H.L. and Q.C., in preparation). ChIP was performed as described above using FLAG antibodies (Sigma). Input and ChIP DNA were subsequently subject to Sanger bisulfite sequencing as described above.
mRNA-Seq
For each sample, poly(A)-containing mRNA was purified from 1 μg DNase-treated total RNA. Following the purification, the mRNA was fragmented into small pieces using divalent cations under elevated temperature. Reverse transcriptase and random primers were used to generate the first-strand cDNA. The second-strand cDNA was synthesized using DNA polymerase I and RNaseH. These cDNA fragments went through an end-repair process using T4 DNA polymerase, T4 PNK and Klenow DNA polymerase, and the addition of a single ‘A’ base using Klenow Fragment (3' to 5' exo minus), then the ligation of the Illumina PE adapters using T4 DNA ligase. An index (up to 12) was inserted into Illumina adapters so that multiple samples can be sequenced in one lane of 8-lane flow cell if necessary. These products were then purified and enriched with PCR to create the final cDNA library for Illumina HiSeq2000 sequencing. Pair-end reads (97 bp) of cDNA sequences were aligned to mouse reference genome mm9 by TopHat53 with reference gene annotations. The relative abundances of each transcript were estimated by Cufflinks54 using Ensembl gene annotation (Build NCBIM37).
Methylated reporter assay
Two μg cFUGW plasmids32 were used in each in vitro methylation reaction with 4 units of methyltransferase (NEB) in their respective buffer conditions (37°C, > 8 hours). After purification by phenol extraction and ethanol precipitation, 0.4 μg mock-treated or methylated plasmids were transfected in HEK293 cells using 1.5 μl X-tremeGENE HP (Roche). After 48 hours, cells were suspended and GFP+ cells were quantified by a FACSCalibur flow cytometer (BD Biosciences) as previously described32. For experiments using primary neuronal cultures, 2 μg of in vitro methylated GFP plasmids were transfected by electroporation with 2 μg of unmethylated RFP control plasmid. After 7 days, RFP+ and GFP+RFP+ neurons were quantified.
Electrophoretic mobility shift assay
Each binding reaction was carried out as previously described55. A mixture of 100 fmol of biotinylated dsDNA probe and different amounts of recombinant MBD domain of human MeCP2 (Diagenode) were incubated in 20 μl of binding buffer (10 mM Tris-Cl at pH 7.5 with 50 mM KCl, 1 mM DTT) at room temperature for 30 minutes. Reaction products were loaded onto 6% Novex TBE polyacrylamide gels (Invitrogen) and ran at 10 mA for ~30 minutes until the bromophenol blue dye migrated halfway to the bottom of the gel. Nucleic acids were transferred to nylon membranes (Amersham) and detected with the LightShift EMSA Kit (Pierce) according to the manufacturer’s protocol.
Statistical Analysis
We performed two-sided Student’s t-tests for comparing two sample groups, and one-way ANOVA for comparing more than two sample groups unless indicated otherwise. Data distribution was assumed to be normal but this was not formally tested. For comparing numbers of methylated/unmethylated cytosines from two samples, two-sided Fisher’s exact tests were performed (Figs. 6c and 8a). For comparing large numbers of binary outcomes, χ2 tests were performed (Fig. 2d).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank S. Baylin, D. Ginty and members of Song and Ming laboratories for comments and suggestions, Y. Cai and L. Liu for technical support. This work was supported by NIH (NS047344, ES021957, MH087874) and Simons Foundation to H.S., by NIH (HD069184, NS048271), Maryland Stem Cell Research Fund (MSCRF), and Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to G-l. M., and Lieber Institute fund to Y.G.; Y.S. and C.Z were supported by MSCRF postdoctoral fellowships; J.S was supported by a Samsung Scholarship.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Footnotes
Accession codes
NCBI Gene Expression Omnibus GSE52331
Supplemental Information
Supplemental Information includes online methods, two tables and fourteen figures.
REFERENCES
- 1.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–74. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng J, Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int Rev Neurobiol. 2009;89:67–84. doi: 10.1016/S0074-7742(09)89004-1. [DOI] [PubMed] [Google Scholar]
- 3.Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci. 2010;13:1299–1440. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang TY, Meaney MJ. Epigenetics and the environmental regulation of the genome and its function. Annu Rev Psychol. 2010;61:439–66. doi: 10.1146/annurev.psych.60.110707.163625. C1-3. [DOI] [PubMed] [Google Scholar]
- 5.Ma DK, et al. Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat Neurosci. 2010;13:1338–1344. doi: 10.1038/nn.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson ED, Monteggia LM. Epigenetics in the mature mammalian brain: Effects on behavior and synaptic transmission. Neurobiol Learn Mem. 2011;96:53–60. doi: 10.1016/j.nlm.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. 2011;91:603–49. doi: 10.1152/physrev.00012.2010. [DOI] [PubMed] [Google Scholar]
- 8.Telese F, Gamliel A, Skowronska-Krawczyk D, Garcia-Bassets I, Rosenfeld MG. "Seq-ing" insights into the epigenetics of neuronal gene regulation. Neuron. 2013;77:606–23. doi: 10.1016/j.neuron.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 10.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–20. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607–20. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–37. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–52. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- 15.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–32. [PubMed] [Google Scholar]
- 16.Jones PA, Liang G. Rethinking how DNA methylation patterns are maintained. Nat Rev Genet. 2009;10:805–11. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yokochi T, Robertson KD. Preferential methylation of unmethylated DNA by Mammalian de novo DNA methyltransferase Dnmt3a. J Biol Chem. 2002;277:11735–45. doi: 10.1074/jbc.M106590200. [DOI] [PubMed] [Google Scholar]
- 18.Yoder JA, Soman NS, Verdine GL, Bestor TH. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J Mol Biol. 1997;270:385–95. doi: 10.1006/jmbi.1997.1125. [DOI] [PubMed] [Google Scholar]
- 19.Ramsahoye BH, et al. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237–42. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laurent L, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–31. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lister R, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziller MJ, et al. Genomic Distribution and Inter-Sample Variation of Non-CpG Methylation across Human Cell Types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stadler MB, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–5. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 25.Smith ZD, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–44. doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomizawa S, et al. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development. 2011;138:811–20. doi: 10.1242/dev.061416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ichiyanagi T, Ichiyanagi K, Miyake M, Sasaki H. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie W, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–31. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lister R, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varley KE, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23:555–67. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma DK, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–7. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo JU, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–51. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng Y, et al. A unique family of Mrr-like modification-dependent restriction endonucleases. Nucleic Acids Res. 2010;38:5527–34. doi: 10.1093/nar/gkq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chodavarapu RK, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–92. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–7. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marson A, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–33. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barreto G, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–5. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 39.Hu XV, et al. Identification of RING finger protein 4 (RNF4) as a modulator of DNA demethylation through a functional genomics screen. Proc Natl Acad Sci U S A. 2010;107:15087–92. doi: 10.1073/pnas.1009025107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skene PJ, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–68. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng J, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–30. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248–51. doi: 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–80. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Booth MJ, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–7. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 45.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–30. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suetake I, Miyazaki J, Murakami C, Takeshima H, Tajima S. Distinct enzymatic properties of recombinant mouse DNA methyltransferases Dnmt3a and Dnmt3b. J Biochem. 2003;133:737–44. doi: 10.1093/jb/mvg095. [DOI] [PubMed] [Google Scholar]
- 47.Gowher H, Jeltsch A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J Mol Biol. 2001;309:1201–8. doi: 10.1006/jmbi.2001.4710. [DOI] [PubMed] [Google Scholar]
- 48.Wu H, et al. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329:444–8. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LaPlant Q, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–43. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lewis JD, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–14. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 51.Song H, Stevens CF, Gage FH. Astroglia induce neurogenesis from adult neural stem cells. Nature. 2002;417:39–44. doi: 10.1038/417039a. [DOI] [PubMed] [Google Scholar]
- 52.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–2. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu S, et al. DNA methylation presents distinct binding sites for human transcription factors. Elife. 2013;2:e00726. doi: 10.7554/eLife.00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.