

Abstract
Alcohol abuse is associated with the development of fatty liver disease and also with significant osteopenia in both genders. In this study, we examined ethanol-induced pathology in response to diets with differing fat/carbohydrate ratios. Male Sprague-Dawley rats were fed intragastrically with isocaloric liquid diets. Dietary fat content was either 5% (high carbohydrate, HC) or 45% (high fat, HF), with or without ethanol (12–13 g/kg/day). After 14, 28, or 65 days, livers were harvested and analyzed. In addition, bone morphology was analyzed after 65 days. HC rats gained more weight and had larger fat pads than HF rats with or without ethanol. Steatosis developed in HC + ethanol (HC+EtOH) compared to HF + ethanol (HF+EtOH) rats, accompanied by increased fatty acid (FA) synthesis and increased nuclear carbohydrate response element binding protein (ChREBP) (p < 0.05), but in the absence of effects on hepatic silent mating type information regulation 2 homolog (SIRT-1) or nuclear sterol regulatory binding element protein (SREBP-1c). Ethanol reduced serum leptin (p < 0.05) but not adiponectin. Over time, HC rats developed fatty liver independent of ethanol. FA degradation was significantly elevated by ethanol in both HC and HF groups (p < 0.05). HF+EtOH rats had increased oxidative stress from 28 days, increased necrosis compared to HF controls and higher expression of cytochromes P450, CYP2E1, and CYP4A1 compared to HC+EtOH rats (p < 0.05). In contrast, HC+EtOH rats had no significant increase in oxidative stress until day 65 with no observed increase in necrosis. Unlike liver pathology, no dietary differences were observed on ethanol-induced osteopenia in HC compared to HF groups. These data demonstrate that interactions between diet composition and alcohol are complex, dependent on the length of exposure, and are an important influence in development of fatty liver injury. Importantly, it appears that diet composition does not affect alcohol-associated skeletal toxicity.
Keywords: alcohol, ethanol, diet, carbohydrate, polyunsaturated fat, liver, fatty liver disease, steatosis, fatty acid synthesis, bone
Introduction
Chronic alcohol consumption is well known to result in a variety of lipid-associated pathologies in different tissues, including dysfunction of white adipose tissue leading to loss of fat and altered adipokine secretion, development of fatty liver disease which can progress to cirrhosis, and osteopenia accompanied by increased bone marrow adiposity which is a risk factor for development of osteoporosis (Addolorato, Capristo, Greco, Stefanini, & Gasbarrini, 1997; Alvisa-Negrín et al., 2009; Reynard et al., 2002; Shankar et al., 2008; Sun et al., 2012). It is possible that these pathologies are all linked. Disorders in adipose fat storage function can result in excess fatty acid influx into the liver and reduced production of the adipokine adiponectin, both of which will result in steatosis (Cusi, 2010; You & Rogers, 2009). Maintenance of adipose tissue triglyceride buffering capacity after alcohol treatment in the presence of the peroxisome- proliferator activated receptor (PPAR)γ agonist rosiglitazone has been shown to improve lipid homeostasis in the adipose-tissue liver axis (Sun et al., 2012). Similarly, loss of adipose tissue in anorexia has been shown to result in increased bone marrow adiposity (Ecklund et al., 2010).
However, alcohol-associated morbidity and mortality is a low penetrance disease and there is a significant degree of inter-individual difference in susceptibility to ethanol toxicity (Potts & Verma, 2012; Tsuchiya et al., 2012). Diet is an important factor contributing to the diversity of toxic responses to ethanol (Baumgardner, Shankar, Korourian, Badger, & Ronis, 2007; Korourian et al., 1999; Ronis, Hakkak, et al., 2004; Ronis, Korourian, Zipperman, Hakkak, & Badger, 2004). We have previously demonstrated that diets rich in carbohydrates appear to protect against alcoholic liver damage (ALD) (Korourian et al., 1999). However, it is unclear if such diets can also protect other alcohol target tissues such as the skeleton. The first stage in development of ALD is fatty liver (steatosis), and this appears necessary, although not sufficient, for further necroinflammatory and fibrotic injury (Korourian et al., 1999; Tsuchiya et al., 2012). However, despite intensive study, the molecular mechanisms underlying ethanol-induced steatosis remain controversial. Moreover, obesity, excess caloric intake, and dietary imbalance in favor of fats or simple carbohydrates can result in steatosis by themselves (Baumgardner, Shankar, Hennings, Badger, & Ronis, 2008; Marecki, Ronis, & Badger, 2011; Mouzaki & Allard, 2012; Ronis et al., 2012) and the pathology of non-alcoholic fatty liver disease (NAFLD) shares many features with ALD (Baumgardner et al., 2008; Nagata, Suzuki, & Sakaguchi, 2007; Ronis et al., 2012).
The rate-limiting steps in de novo fatty acid (FA) synthesis are the expression and activity of the enzymes fatty acid synthase (FASN) and acyl CoA carboxylase (ACC-1) (Dentin et al., 2004). Regulation of FA synthesis is complex and incompletely understood. Regulation has been suggested to involve the interaction of a large number of transcription factors responsive to endocrine factors including insulin, adipokines, thyroid hormone, and sex steroids, as well as nutrients including glucose, fatty acid, and cholesterol metabolites (Dentin et al., 2004; Filhoulaud, Guilmeau, Dentin, Girard, & Postic, 2013; Poupeau & Postic, 2011). However, the major transcriptional regulators appear to be sterol regulatory binding protein (SREBP)-1c and carbohydrate response element binding protein (ChREBP) (Dentin et al., 2004). FA degradation can also be catalyzed by several pathways. This process is coordinately regulated though control of mitochondrial fatty acid transport via carnitine palmitoyl transferase (CPT-1) and enzymes involved in β- and ω-oxidation in the mitochondria and peroxisomes, respectively, such as acyl CoA oxidase (ACO) via the transcription factor PPARα (Contreras, Torres, & Tovar, 2013). It has been suggested that ethanol increases FA synthesis and suppresses FA degradation as a result of impaired secretion of adiponectin from adipose tissue and subsequent downstream inhibition of hepatic expression of the histone deacetylase silent mating type information regulation 2 homolog (SIRT-1) (Chen, Sebastian, & Nagy, 2007; Chen, Sebastian, Tang, et al., 2009; Tang et al., 2012; You, Considine, Leone, Kelly, & Crabb, 2005; You, Liang, Ajmo, & Ness, 2008; You & Rogers, 2009). Down-regulation of SIRT-1 has also been suggested to increase expression and activation of SREBP-1c and to suppress signaling through PPARα (Dominy, Lee, Gerhart-Hines, & Puigserver, 2010; You & Rogers, 2009). However, ethanol-induced steatosis has also been reported to occur in the presence of suppressed SREBP-1c signaling, reduced lipogenesis, and increased FA degradation (Baumgardner et al., 2007; He, Simmen, Ronis, & Badger, 2004; Ronis et al., 2011).
In addition to FA synthesis and degradation, other pathways can also influence hepatic triglyceride content. These include FA transport via membrane receptors CD36 and the fatty acid transport proteins (FATPs); hydrolysis of fat droplets catalyzed by adiponutrin (PNPLA3), and the synthesis and export of very low density lipoprotein (VLDL) which is regulated in part through expression of the microsomal triglyceride transport protein (MTP). Each of these pathways has been implicated in development of fatty liver (Baumgardner et al., 2008; He et al., 2010; Ronis et al., 2011; Sugimoto et al., 2002). Also, increased expression of FATP2, impaired expression of MTP, and reduced VLDL secretion have all been suggested as possible mechanisms underlying ethanol-induced steatosis (Kharbanda, Todero, Ward, Cannella, & Tuma, 2009; Ronis et al., 2011; Sugimoto et al., 2002; Sun et al., 2012; Tan et al., 2012). However, it remains unclear whether the responses of these pathways to ethanol are also dependent on dietary fat or carbohydrate content.
The current study was designed to examine the molecular mechanisms underlying the development of hepatic steatosis and the progression of liver pathology in response to feeding isocaloric diets with differing fat/carbohydrate ratios in the presence or absence of ethanol, and to determine if alcohol-induced osteopenia is also influenced by the dietary fat/carbohydrate ratio.
Methods
Animals and experimental protocol
Groups of n = 8–11 male Sprague-Dawley rats age 9 weeks (300 g) were cannulated intragastrically and allowed to recover to presurgical weight for 14 days prior to being isocalorically fed control liquid diets high in carbohydrate (HC) (16% protein [hydrolyzed whey], 79% carbohydrate [dextrose/maltodextran], and 5% fat [corn oil]) or high in fat (HF) (16% protein, 39% carbohydrate, 45% fat) at a level of 187 kcal/kg3/4/day for 14, 28, and 65 days via total enteral nutrition (TEN) at a rate of 3 mL/h, 23 h/day (Badger, Ronis, Lumpkin, Valentine, et al., 1993). Additional HC and HF groups were fed diets with ethanol isocalorically substituted for carbohydrate calories to produce diets with a final caloric composition of 16% protein, 45% carbohydrate, 5% fat, and 34% ethanol (HC+EtOH), or 16% protein, 5% carbohydrate, 45% fat, and 34% ethanol (HF+EtOH), respectively. At sacrifice, after 14, 28, and 65 days of TEN, trunk blood and liver were collected from fed animals immediately after being removed from TEN feeding. Serum was prepared for endocrine and biochemical measurements and stored frozen at −20 °C until use. In addition, at the 65-day time point, abdominal fat, tibias, and femurs were also collected. All tissues were flash-frozen in liquid nitrogen and stored at −80 °C until use.
Biochemical and endocrine analysis of serum
Blood ethanol concentration (BEC) at sacrifice was measured by Analox (Ronis et al., 2011). Non-esterified free fatty acids (NEFA) were measured using the NEFA C kit from Waco Chemicals (Richmond, VA). Serum triglycerides were measured using Triglyceride Reagent (IR141; Synermed, Westfield, IN). Serum glucose levels were measured using Glucose Reagent (IR071-072; Synermed, Westfield, IN). Serum insulin and leptin levels were measured using ELISA kits from Linco Research (St. Charles, MO) according to manufacturer’s protocols. Serum adiponectin levels were measured using an ELISA kit from B-Bridge International (Sunnyvale, CA), according to manufacturer’s protocols.
Hepatic steatosis and progression of ALD
Hepatic steatosis was measured by image analysis of Oil Red O staining and biochemically using Triglyceride Reagent (Synermed). Liver lipid peroxidation (thiobarbituric acid reactive substances, TBARS) was assessed as a measure of oxidative stress as described by Ohkawa, Ohishi, & Yagi (1979). Serum alanine amino transferase activities (ALTs) were measured to assess liver necrosis using the Infinity™ ALT liquid stable reagent (Thermo Electron Corp., Waltham, MA) according to manufacturer’s protocols.
Analysis of gene expression
mRNAs of genes involved in FA transport (CD36 and FATP2), FA synthesis (FASN and ACC1), and FA degradation (CPT1 and ACO) (in triglyceride droplet hydrolysis [adiponeutrin] and in VLDL export [MTP]) were quantified by real time RT-PCR as previously described (Ronis et al., 2011, 2012).
Chromatin immunoprecipitation analysis (ChIP)
Recruitment of SREBP-1c to the FASN promoter was assessed by ChIP. The ChIP-IT enzymic kit (Active Motif) was used with minor modifications for in vivo samples as described previously (Shankar et al., 2010). Three pools of liver samples (each pool representing 2–3 separate animals) were used for the analysis. Immunoprecipitation was performed using 5 μg of anti-SREBP-1c (Novus Biologicals, Littleton, CO). The SREBP-1c target binding region on the FASN promoter was amplified by PCR.
Western immunoblot analysis of protein expression
Antibodies against SIRT-1deactylase (sc. 15404), GCN1 acetylase (sc. 20698), and the transcription factor carbohydrate response element binding protein (ChREBP) (sc. 21189) were obtained from Santa Cruz Biotechnology. Protein expression of SIRT-1, GCN-1, SREBP-1c, and ChREBP in nuclear extracts and of FASN in whole liver lysates was measured by Western immunoblot and expression of apoproteins for the cytochrome P450 enzymes CYP2E1 and CYP4A1 were measured in microsomal fractions by Western immunoblot as described previously (Borengasser et al., 2011; Ronis, Hakkak, et al., 2004; Ronis, Korourian, et al., 2004; Ronis et al., 2011, 2012). Amido black staining of total protein on transfer membranes was used to control for loading.
Analysis of bone strength and architecture
The effects of fat/carbohydrate ratio and ethanol on bone mechanical strength was assessed in whole tibia by three-point bending using an MTS 858 Bionix test system load frame (MTS, Eden Prairie, MN) as described previously (Brown et al., 2002). The loading point was displaced at 0.1 mm/s until failure, and load displacement data were recorded at 100 Hz. Test curves were analyzed using TestWorks software (MTS) to determine peak load and stiffness. The microarchitecture of trabecular bone was assessed by μCT at the distal femur using ex vivo fixed femurs as described previously (Bouxsein et al., 2010; Sebastian, Suva, & Friedman, 2008; Suva et al., 2008). All μCT analyses were consistent with current guidelines for the assessment of bone microstructure in rodents using micro-computed tomography (Bouxsein et al., 2010). Formalin-fixed femora were imaged using a MicroCT 40 (Scanco Medical AG, Bassersdorf, Switzerland) using a 16-μm isotropic voxel size in all dimensions. Semi-automated contouring was used to select a region of interest (ROI) extending 3.2 mm proximal to the distal femoral metaphysis, but excluding the cortex and subcortical bone, composed of 250 adjacent slices. Three-dimensional reconstructions were created by stacking the regions of interest from each two-dimensional slice and then applying a gray-scale threshold and Gaussian noise filter as described (Suva et al., 2008), using a consistent and pre-determined threshold (300) with all data acquired at 55 kVp, 145 mA, and 275-ms integration time. Fractional bone volume (bone volume/tissue volume; BV/TV) and architectural properties of trabecular bone, trabecular thickness (Tb.Th.), trabecular separation (Tb.Sp.), trabecular number (Tb.N.), and connectivity density (Conn. D., mm 3), were calculated using previously published methods (Suva et al., 2008). Similarly, for cortical bone assessment, μCT slices were segmented into bone and marrow regions by applying a visually chosen, fixed threshold for all samples after smoothing the image with a three-dimensional Gaussian low-pass filter (σ = 0.8, support = 1.0) to remove noise, and a fixed threshold. Cortical geometry was assessed in a 1.5-mm-long region centered at the femoral midshaft. The outer contour of the bone was found automatically using the built-in Scanco contouring tool. Total area was calculated by counting all voxels within the contour, bone area was calculated by counting all voxels that were segmented as bone, and marrow area was calculated as total area - bone area. This calculation was performed on all 40 slices using the average for the final calculation. The outer and inner perimeter of the cortical midshaft was determined by a three-dimensional triangulation of the bone surface (BS) of the 40 slices, and cortical thickness and other cortical parameters were determined as described (Suva et al., 2008). Parameters assessed included total cross sectional area (Tt.CSA), cortical area (Ct.CSA), cortical thickness (Ct.Th.), medullary area (Me.Ar.), periosteal perimeter (Ps. Pm.), and endocortical perimeter (Ec.Pm).
Statistical analysis
Data for continuous outcomes are presented as mean ± standard errors of the mean (SE). Two-factor analysis of variance (ANOVA) was used to compare mean effects of diet (HF vs. HC), EtOH (control vs. EtOH), and their interaction on each outcome at each time point. Post hoc comparisons were performed using an all-pairs Student Neuman-Keuls comparison test and was considered significant at p < 0.05. In addition, one-way ANOVA was utilized to compare the effects of time of TEN diet infusion on each outcome within each diet-EtOH combination. Whenever the Bonferroni test-corrected significant probability of a time-dependent effect was < 0.05, Tukey-Kramer post hoc testing was utilized to determine differences between means at different time points. Statistical analyses were performed using the Stata statistical package version 12.0 (Stata Corporation, College Station, TX) and Sigma Stat software package 11.0 (Systat Software, Inc., San Jose, CA).
Results
Growth, body composition, and serum biochemistry
Isocaloric feeding of HC and HF diets resulted in a significant difference in growth of male rats fed via TEN for 65 days. Weight gain in the HF group was smaller than that of the HC group: 4.2 g/day vs. 5.4 g/day (p < 0.05) (Table 1). However, although there was a decrease in absolute fat pad and liver weights in the HF compared to the HC group (p < 0.05), there was no effect of fat/carbohydrate ratio on the relative weight of the abdominal fat pads or of the liver (Table 1). Ethanol treatment resulted in similar blood ethanol concentrations in both the HC+EtOH and HF+EtOH groups (175 ± 75 and 200 ± 60 mg/dL, respectively). Consistent with previous reports that ethanol suppresses growth hormone secretion (Badger, Ronis, & Lumpkin, 1993; Badger, Ronis, Lumpkin, Valentine, et al., 1993), ethanol-fed groups had suppressed weight gain in both diet groups (p < 0.05), but growth suppression was greater in the HF+EtOH group compared to the HC+EtOH group: 1.60 vs. 2.75 g/day (p < 0.05). This latter effect appeared to be due to reductions in adiposity since there was a corresponding reduction in the absolute and relative abdominal fat pad weights in the HF+EtOH group compared to the HC+EtOH group (p < 0.05). Although absolute liver weight was decreased (p < 0.05), ethanol treatment also significantly increased relative liver weight in both diet groups (p < 0.05) (Table 1).
Table 1.
Effects of Fat/Carbohydrate Ratio and Ethanol on Growth, Organ Weights and Leptin in the Male Rat
| HC | HF | |||
|---|---|---|---|---|
| (−) EtOH | (+) EtOH | (−) EtOH | (+) EtOH | |
| Weight Gain (g) | 352 ± 15.11
Diet Effect |
179 ± 11.8*
p<0.001 |
278 ± 13.1#
EtOH Effect |
104 ± 10.7*,#
p<0.001 |
| Abdominal Fat Weight (g) | 20.5 ± 1.54 Diet Effect |
12.8 ± 1.01*
p<0.001 |
15.3 ± 0.61#
EtOH Effect |
7.4 ± 0.95*,#
p<0.001 |
| Abdominal Fat Weight (%) | 3.30 ± 0.18 Diet Effect |
2.86 ± 0.16 p<0.001 |
2.93 ± 0.16 EtOH Effect |
1.98 ± 0.17*,#
p<0.001 |
| Liver Weight (g) | 19.9 ± 0.66 Diet Effect |
18.1 ± 0.72*
p<0.001 |
17.5 ± 0.63#
EtOH Effect |
14.1 ± 0..67*,#
p<0.001 |
| Liver Weight (%) | 3.24 ± 0.08 Diet Effect |
4.09 ± 0.11*
p=0.039 |
3.15 ± 0.06 EtOH Effect |
3.85 ± 0..06*,#
p<0.001 |
| Leptin (ng/ml) | 63.7 ± 6.26 Diet Effect |
26.6 ± 2.09*
p=0.003 |
40.5 ± 4.71#
EtOH Effect |
14.7 ± 4.66*
p<0.001 |
S.E.M., HC–high carbohydrate, HF–high fat,
Diet effect within group p<0.05,
EtOH effect within group p<0.05
The HC diet resulted in higher levels of serum insulin and lower levels of plasma glucose than the HF diet after 14 days (p < 0.05). Calculated Homeostasis Model Assessment (HOMA) (Levy, Matthews, & Hermans, 1998) values indicated significantly greater insulin resistance after consumption of the HC diet than the HF diet at this time point (p < 0.05). However, this difference disappeared after longer periods of TEN. Ethanol reduced serum insulin and HOMA in the HF+EtOH group compared to the HC+EtOH group at 14 days, but this difference disappeared after longer periods of feeding. After 65 days of TEN, ethanol treatment significantly reduced serum insulin and HOMA (p < 0.05) independent of diet, but serum glucose values were significantly elevated in the HF+EtOH group relative to the HC+EtOH group (Table 2). Mean serum non-esterified free fatty acids (NEFA) values increased over time in all groups but this only reached statistical significance in the HC and HF+EtOH groups. The consumption of the HF diet resulted in overall higher serum levels of NEFA relative to the HC diet (p < 0.05) (Table 2). Ethanol treatment had no effects on serum NEFA values in either diet group except at the day-28 time point (Table 2).
Table 2.
Effects of Fat/Carbohydrate Ratio and Ethanol on Serum Biochemistry in the Male Rat
| Day 14 | Day 28 | Day 65 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HC | HF | HC | HF | HC | HF | |||||||
| (−) EtOH | (+) EtOH | (−) EtOH | (+) EtOH | (−) EtOH | (+) EtOH | (−) EtOH | (+) EtOH | (−) EtOH | (+) EtOH | (−) EtOH | (+) EtOH | |
| Insulin (ng/ml) | 5.06 ± 0.511
Diet Effect |
4.54 ± 0.69 p<0.001 |
2.57 ± 0.46# EtOH Effect |
1.65 ± 0.17#,a p=0.185 |
6.70 ± 0.72 Diet Effect |
6.87 ± 2.97 p=0.426 |
5.28 ± 1.62 EtOH Effect |
5.66 ± 0.93b
p=0.867 |
7.04 ± 1.30 Diet Effect |
2.90 ± 0.47* p=0.351 |
5.86 ± 1.25 EtOH Effect |
2.10 ± 0.32*,a p<0.001 |
| Glucose (mg/dL) | 152 ± 12.0 Diet Effect |
169 ± 9.4 p=0.025 |
181 ± 9.5a,b EtOH Effect |
189 ± 8.9 p=0.228 |
182 ± 9.1 Diet Effect |
205 ± 18.0 p=0.008 |
216 ± 14.2b
EtOH Effect |
261 ± 20.4#
p=0.041 |
138 ± 10.4 Diet Effect |
144 ± 19.7 p=0.004 |
149 ± 15.0a
EtOH Effect |
233 ± 18.6*,#
p=0.009 |
| HOMA | 46.4 ± 5.87 Diet Effect |
37.3 ± 4.61 p<0.001 |
28.4 ± 4.41# EtOH Effect |
21.3 ± 2.63#,a p=0.084 |
73.1 ± 7.01 Diet Effect |
44.9 ± 9.98 p=0.149 |
68.7 ± 15.9 EtOH Effect |
89.9 ± 19.6b
p=0.803 |
47.7 ± 7.50 Diet Effect |
25.3 ± 6.01 p=0.466 |
59.1 ± 15.1 EtOH Effect |
30.4 ± 5.40a
p=0.030 |
| NEFA (mmol/L) | 0.47 ± 0.05a
Diet Effect |
0.52 ± 0.04 p=0.004 |
0.62 ± 0.05# EtOH Effect |
0.66 ± 0.05#,a p=0.301 |
0.58 ± 0.04a Diet Effect |
0.99 ± 0.21 p=0.067 |
0.92 ± 0.10 EtOH Effect |
1.23 ± 0.19a,b p=0.027 |
1.23 ± 0.30b Diet Effect |
0.94 ± 0.12 p=0.024 |
2.22 ± 0.56 EtOH Effect |
1.78 ± 0.28b
p=0.357 |
S.E.M., HC–high carbohydrate, HF–high fat, EtOH – ethanol.
Diet effect within group, p<0.05,
EtOH effect within group p<0.05,
Superscript letter -time course effect, a<b; p<0.05
Development of steatosis and progression of liver injury
The appearance of fat droplets in the liver was assessed by Oil Red O staining of frozen sections (Fig. 1). Quantification of staining by digital imaging and biochemical analysis of triglyceride accumulation is also shown (Figs. 2A and 2B). After 14 days of diet, little fat accumulation was present in either HC or HF control groups. Ethanol treatment significantly increased Oil Red O staining only in the HC+EtOH group (p < 0.05). Similar results were obtained when hepatic triglyceride content was assessed biochemically (Fig. 2A). A similar pattern was observed after 28 days. In contrast, after 65 days, significant steatosis developed in the HC controls themselves relative to the shorter periods of TEN feeding (p < 0.05). Although average triglyceride values remained significantly greater in the HC+EtOH group than the HC control group after 65 days of feeding (p < 0.05), the difference in fat droplet accumulation between HC and HC+EtOH groups disappeared (Figs. 1 and 2B). The HF+EtOH group had lower levels of steatosis than the HC+EtOH group at all time points (p < 0.05) with no significant increase in mean levels of triglycerides and lipid droplets relative to the HF control groups at any time point (p < 0.1) (Figs. 2A and 2B). Despite the lower overall level of steatosis in the HF groups compared to the HC groups and lower ethanol-associated fat accumulation in the HF+EtOH compared to the HC+EtOH group, the HF+EtOH group developed significant oxidative stress earlier than the HC+EtOH group as measured by TBARS (Ohkawa et al., 1979) (p < 0.05) (Fig. 2C). In addition, only the HF+EtOH group had evidence of necrosis relative to its appropriate dietary control, indicated by significant increases in serum ALT values after 65 days of TEN feeding (p < 0.05) (Fig. 2D). Serum ALT values doubled in both the HC and HC+ETOH groups after 65 days of TEN feeding relative to values after 14 days or 28 days of feeding (p < 0.05) (Fig. 2D).
Figure 1.
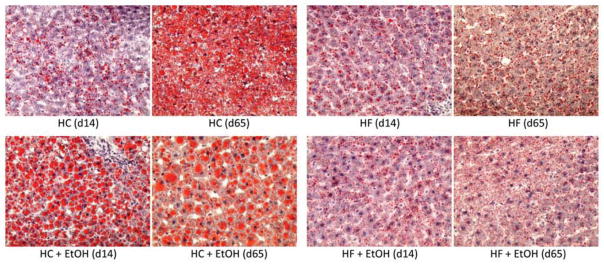
Effects of dietary fat/carbohydrate ratio and ethanol on development of hepatic steatosis. Representative pictures of Oil Red O stained livers (×10) after 14 or 65 days of TEN feeding. HC – high carbohydrate, HF – high fat, EtOH – ethanol.
Figure 2.

Effects of dietary fat/carbohydrate ratio and ethanol on A) liver triglycerides, B) lipid droplet accumulation based on area of Oil Red O staining, C) liver lipid peroxidation (TBARs – thiobarbituric acid reactive products), and D) necrosis indicator, serum alanine aminotransferase (ALT) activity. HC – high carbohydrate, HF – high fat, EtOH – ethanol. Data are mean ± SEM. Statistical significance for the effects of length of TEN feeding on each parameter was determined by one-way ANOVA followed by Tukey-Kramer post hoc analysis. Means with different letters are significantly different, a < b, p < 0.05. Statistical significance with regard to the effects of diet and ethanol within each time point was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Means with # are significantly different by diet within the respective control or ethanol group; means with * are significantly different after EtOH treatment within each diet group, p < 0.05.
Serum adipokines and hepatic acetylation/deacetylation pathways
Since effects on adipokine secretion and hepatic acetylation pathways have both been implicated in the development of alcoholic steatosis (Choudhury et al., 2011; Tan et al., 2012; You et al., 2007; You & Rogers, 2009), we next examined the effects of fat/carbohydrate ratio and ethanol treatment on plasma leptin and adiponectin levels and on hepatic expression of deacetylase SIRT-1 and acetylase GCN5. Isocalorically increasing the dietary polyunsaturated fat/carbohydrate ratio significantly reduced both plasma leptin and adiponectin concentrations after 65 days of TEN feeding in the HF compared to the HC group (p < 0.05) (Table 1, Fig. 3A). In addition, ethanol consumption significantly reduced serum leptin levels relative to the appropriate control group in both diets (p < 0.05) (Table 1). However, no significant ethanol effect was observed on serum adiponectin levels after either 14 days or 65 days of TEN feeding as part of either the HC or HF diet. Despite lower serum adiponectin after high fat feeding, hepatic expression of SIRT-1 protein was increased in the HF compared to the HC groups after both 14 and 65 days of TEN feeding (p < 0.05) (Figs. 3B and 3D). There was no significant effect of dietary fat/carbohydrate ratio on expression of hepatic GCN5 protein after either 14 days or 65 days of TEN feeding (Figs. 3B and 3D). SIRT-1 protein expression was increased after both 14 days and 65 days of TEN feeding in the HF+EtOH groups compared to HF controls (p < 0.05). In contrast, ethanol had no effect on hepatic SIRT-1 protein expression when fed as part of the HC diet (Figs. 3B and 3D). Effects of ethanol on GCN5 expression were both diet- and time-dependent. At 14 days of TEN feeding, ethanol significantly increased GCN5 expression only in the HF group (p < 0.05). After 65 days of TEN feeding, ethanol significantly increased GCN5 expression only in the HC group (p < 0.05) (Figs. 3C and 3D).
Figure 3.

Effects of dietary fat/carbohydrate ratio and ethanol on serum adiponectin and hepatic acylation/deacetylation pathways. A) serum adiponectin, B) immunoquantitation of SIRT-1 protein expression, C) immunoquantitation of GCN5 protein expression, D) Western immunoblots from day 14 and day 65 groups. Each lane represents a different animal, n = 3/group. AB – amido black stain loading control, HC – high carbohydrate, HF – high fat, EtOH – ethanol. Data are mean ± SEM. Statistical significance with regard to the effects of diet and ethanol at each time point was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Means with # are significantly different by diet within the respective control or EtOH group; means with * are significantly different after ethanol treatment within each diet group, p < 0.05.
Effects on fatty acid synthesis and degradation pathways
Since changes in both FA synthesis and degradation pathways have been suggested to underlie the development of alcoholic steatosis, we examined expression of FASN mRNA and protein as well as mRNAs encoding ACC-1, CPT-1, and ACO in the liver (Figs. 4 and 5). Isocalorically increasing dietary polyunsaturated fats from 5% to 45% resulted in significantly lower expression of both FASN and ACC-1 mRNA and FASN protein at all time points in the HF compared to HC groups (p < 0.05). Moreover, the HC diet by itself significantly increased the expression of both enzymes over the 65 days of TEN, compared to the early 14-day and 28-day time points. Similarly, the HF diet also significantly increased FASN protein expression on day 65 relative to the HF group on day 14 of TEN (p < 0.05). Ethanol consumption rapidly increased FASN mRNA and protein expression and ACC-1 mRNA in the HC+EtOH group compared to HC controls after only 14 days, and increased FA synthesis continued to be observed in the HC+EtOH group compared to the HC group after 28 days (p < 0.05). However, over the longer 65-day period, the HC diet by itself increased expression of these rate-limiting enzymes in FA synthesis to the same degree as in the HC+EtOH group. Ethanol consumption also significantly increased FASN protein expression at 14 days and ACC-1 mRNA at 28 days in the HF+EtOH groups relative to HF controls. However, the ethanol-induced increases in these enzymes in the context of HF feeding were modest and inconsistent across all measures and time points.
Figure 4.

Effects of dietary fat/carbohydrate ratio and ethanol on expression of mRNAs encoding enzymes in hepatic FA synthesis and degradation pathways. A) fatty acid synthase (Fasn), B) acyl CoA carboxylase (ACC-1), C) carnitine palmitoyl transferase (Cpt-1), D) acyl CoA oxidase (Aco). HC – high carbohydrate, HF – high fat, EtOH – ethanol. Data are mean ± SEM. Statistical significance for the effects of length of TEN feeding on each parameter was determined by one-way ANOVA followed by Tukey-Kramer post hoc analysis. Means with different letters are significantly different, a < b, p < 0.05. Statistical significance with regard to the effects of diet and ethanol within each time point was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Means with # are significantly different by diet within the respective control or ethanol group; means with * are significantly different after ethanol treatment within each diet group, p < 0.05.
Figure 5.
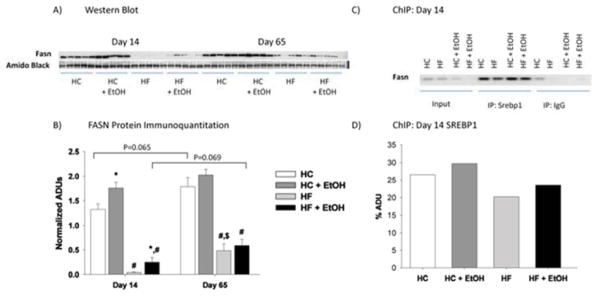
Effects of dietary fat/carbohydrate ratio and ethanol on hepatic fatty acid synthase (FASN) protein expression A) as determined by Western immunoblot and B) by binding of SREBP-1c to its response element on the FASN promoter as determined by ChIP. HC – high carbohydrate, HF – high fat, EtOH – ethanol. Immunoquantitation data are mean ± SEM. Statistical significance with regard to the effects of diet and EtOH at each time point was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Means with # are significantly different by diet within the respective control or ethanol group; means with * are significantly different after ethanol treatment within each diet group, p < 0.05.
Since signaling via SREBP-1c and CHREBP are the main drivers of FA synthesis, the effects of fat/carbohydrate ratio and ethanol on nuclear localization of these transcription factors and on the binding of SREBP-1c to its response element on the FASN promoter were next measured by ChIP analysis (Figs. 5 and 6). Nuclear ChREBP was higher and SREBP-1c was lower in the HF compared to the HC group after 14 days of TEN feeding (p < 0.05) (Fig. 6). However, after 65 days of feeding, ChREB was elevated in the HC group relative to earlier feeding periods (p < 0.05), and differences in nuclear ChREBP between HC and HF groups was no longer significant. In contrast, reduced expression of nuclear SREBP-1c protein in HF compared to HC groups persisted and increased with length of diet (p < 0.05) (Fig. 6). Decreased nuclear SREBP-1c protein coincided with decreased binding of SREBP-1c to the FASN promoter in ChIP analysis (Fig. 5). Ethanol consumption resulted in increased nuclear localization of ChREBP protein in both the HC+EtOH and HF+ETOH groups compared to HC and HF controls after 14 days of TEN (p < 0.05), but this effect was lost by day 65. In contrast, no significant effect of ethanol treatment was observed on nuclear SREBP-1c protein expression or SREBP-1c binding to the FASN promoter after 14 days feeding of either diet. After 65 days of TEN diets, ethanol reduced nuclear expression of SREBP-1c in the HC+EtOH compared to the HC group (p < 0.05) (Fig. 6).
Figure 6.
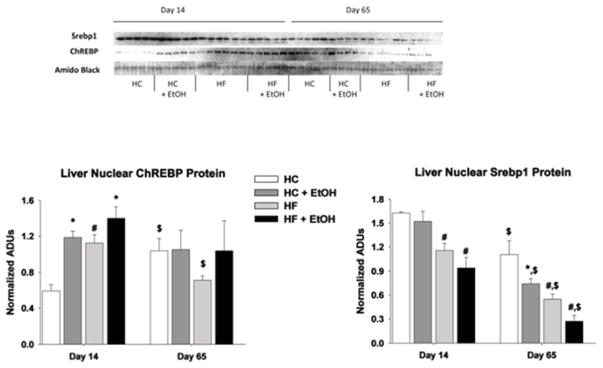
Effects of dietary fat/carbohydrate ratio and ethanol on hepatic expression of transcription factor proteins SREBP-1c and ChREBP in nuclear extracts. HC – high carbohydrate, HF – high fat, EtOH – ethanol. Immunoquantitation data are mean ± SEM. Statistical significance for the effects of length of TEN feeding on each parameter was determined by one-way ANOVA followed by Student Newman-Keuls post hoc analysis. Means with $ are significantly different from the day-14 time point, p < 0.05. Statistical significance with regard to the effects of diet and ethanol at each time point was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Means with # are significantly different by diet within the respective control or ethanol group; means with * are significantly different after ethanol treatment within each diet group, p < 0.05.
CPT-1 mRNA and ACO mRNA are involved in FA degradation. CPT-1 mRNA encodes carnitine palmitoyl transferase, the rate-limiting transport protein for FA into the mitochondria for β-oxidation. ACO mRNA encodes acyl CoA oxidase, an enzyme important in peroxisomal oxidation of FAs. CPT-1 mRNA and ACO mRNA were both elevated after 65 days of TEN feeding of either control diet relative to earlier time points (p < 0.05) (Fig. 4). In addition, CPT-1 mRNA was elevated by ethanol in the CHO+EtOH groups relative to CHO controls at 14 days and 65 days of feeding, and was elevated to a greater degree in the HF+EtOH group compared to HF or HC+EtOH groups at both time points. ACO mRNA was also increased by EtOH in the HF but not HC group at 14 days (p < 0.05), but was elevated by ethanol treatment in both diets after 65 days of TEN (p < 0.05) (Fig. 4).
Effects on other pathways regulating hepatic triglyceride accumulation
We next examined the effects of changing fat/carbohydrate ratio on expression of mRNAs encoding proteins important in pathways regulating FA transport, triglyceride hydrolysis and VLDL secretion. These included CD36 and FATP2 (transport), adiponutrin (hydrolysis) and MTP (VLDL secretion) (Fig. 7). The expression of CD36 was elevated over time of TEN for both HC and HF diets with or without ethanol (p < 0.05). In addition, CD36 mRNA was significantly increased in the HF+EtOH group compared to its HF control at the 14-day time point (p < 0.05). Likewise, ethanol significantly increased the expression of FATP2 mRNA in the HF group at both day 14 and day 65 of TEN (p < 0.05). In contrast, adiponutrin mRNA was low in all HF groups compared to HC groups (p < 0.05), and its expression was not consistently affected by ethanol treatment at any time point. MTP mRNA expression was not affected across time or by diet and was not consistently affected by ethanol treatment.
Figure 7.

Effects of dietary fat/carbohydrate ratio and ethanol on expression of mRNAs encoding enzymes in hepatic FA transport, triglyceride hydrolysis and VLDL secretion pathways. A) CD36, B) fatty acid transport protein 2 (Fatp-2), C) adiponutrin, D) microsomal triglyceride transport protein (Mttp). HC – high carbohydrate, HF – high fat, EtOH – ethanol. Data are mean ± SEM. Statistical significance for the effects of length of TEN feeding on each parameter was determined by one-way ANOVA followed by Tukey-Kramer post hoc analysis. Means with different letters are significantly different, a < b < c, p < 0.05. Statistical significance with regard to the effects of diet and ethanol within each time point was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Means with # are significantly different by diet within the respective control or ethanol group; means with * are significantly different after ethanol treatment within each diet group, p < 0.05.
Effects on hepatic cytochrome P450 expression
Cytochrome P450 CYP2E1, the major ethanol-inducible form, and CYP4A1, the major form involved in ω-hydroxylation of FAs, have both been shown to be “leaky” redox systems which generate reactive oxygen species (Albano et al., 1991; Dai, Rashba-Step, & Cederbaum, 1993; Leclercq et al., 2000). Both enzymes have been reported to be involved in the generation of oxidative stress after alcohol consumption and to be involved in progression of liver pathology from simple steatosis to steatohepatitis (Leung & Nieto, 2013; Maher, 2001; Ronis, Lindros, & Ingelman-Sundberg, 1996). We next measured the expression of CYP2E1 and CYP4A1 apoproteins in microsomes prepared from the livers of rats after 65 days of TEN. The expression of both enzymes was significantly higher in animals fed HF compared to HC diets (p < 0.05) (Fig. 8). In addition, ethanol induced the expression of both enzymes in the HC+EtOH compared to the HC group (p < 0.05), and expression of both enzymes was higher in the HF+EtOH group compared to the HC+EtOH group (p < 0.05).
Figure 8.
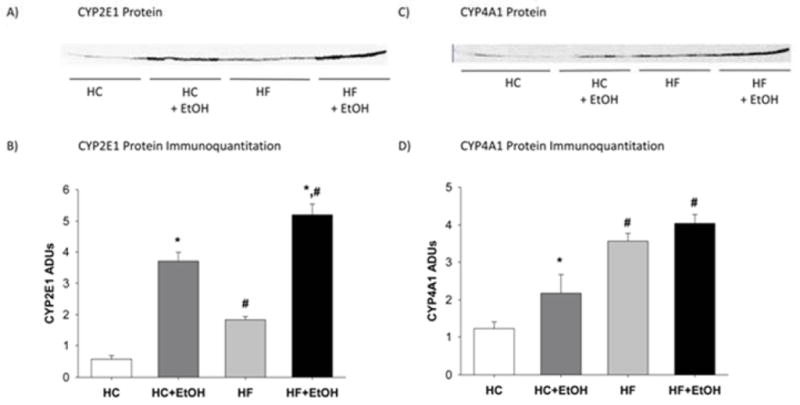
Effects of dietary fat/carbohydrate ratio and ethanol on hepatic microsomal cytochrome P450 protein expression after 65 days of TEN feeding as determined by Western immunoblot: A) Western blot of CYP2E1, B) immunoquantification of CYP2E1, C) Western blot of CYP4A1, and D) immunoquantification of CYP4A1. HC – high carbohydrate, HF – high fat, EtOH – ethanol. Immunoquantitation data are mean ± SEM. Statistical significance with regard to the effects of diet and ethanol was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Means with # are significantly different by diet within the respective control or ethanol group; means with * are significantly different after EtOH treatment within each diet group, p < 0.05.
Effects on bone architecture and strength
The effects of dietary fat/carbohydrate ratio and ethanol on bone architecture and strength were measured across all treatment groups after 65 days of TEN feeding (Table 3). No significant effects of diet alone were observed on these parameters. In contrast, ethanol treatment significantly reduced trabecular bone volume and connectivity density as well as cortical thickness, cortical surface area, and periosteal perimeter of the femur (p < 0.05). These diet-independent changes were associated with reduced bone strength in the tibia (p = 0.06).
Table 3.
Effects of Fat/Carbohydrate Ratio and Ethanol on Bone Quality in the Male Rat
| Parameter | HC | HC+EtOH | HF | HF+EtOH | Two-Way ANOVA (P value) | ||
|---|---|---|---|---|---|---|---|
| Diet | EtOH | Interaction | |||||
| Tibia1 | |||||||
| Load to Failure | 150 ± 16 | 134 ± 18 | 162 ± 14 | 108 ± 21 | 0.69 | 0.06 | 0.29 |
| Stiffness | 432 ± 32 | 399 ± 36 | 441 ± 29 | 350 ± 43 | 0.58 | 0.09 | 0.42 |
| Femur2 | |||||||
| BV/TV | 0.13 ± 0.02 | 0.10 ± 0.01* | 0.13 ± 0.01 | 0.10 ± 0.01* | 0.95 | 0.01 | 0.62 |
| Tb. No. | 1.96 ± 0.14 | 1.71 ± 0.17 | 1.80 ± 0.14 | 1.62 ± 0.15 | 0.42 | 0.16 | 0.81 |
| Tb. Sp. | 0.53 ± 0.05 | 0.67 ± 0.06 | 0.63 ± 0.05 | 0.67 ± 0.06 | 0.33 | 0.10 | 0.37 |
| Tb. Th. | 0.10 ± 0.004 | 0.10 ± 0.005 | 0.09 ± 0.004 | 0.09 ± 0.004 | 0.07 | 0.74 | 0.78 |
| DA | 1.47 ± 0.04 | 1.50 ± 0.04 | 1.51 ± 0.03 | 1.45 ± 0.04 | 0.84 | 0.65 | 0.28 |
| Conn. D. | 28.8 ± 3.0 | 18.8 ± 3.4* | 30.0 ± 2.8 | 21.2 ± 3.2* | 0.55 | 0.005 | 0.84 |
| SMI | 2.42 ± 0.1 | 2.53 ± 0.1 | 2.47 ± 0.1 | 2.53 ± 0.1 | 0.79 | 0.42 | 0.81 |
| Tot. CSA | 11.8 ± 0.47 | 10.5 ± 0.54 | 11.3 ± 0.44 | 10.9 ± 0.50 | 0.97 | 0.10 | 0.40 |
| Me. Ar. | 4.06 ± 0.23 | 4.10 ± 0.26 | 4.01 ± 0.21 | 4.20 ± 0.24 | 0.92 | 0.65 | 0.78 |
| Ct. CSA | 7.34 ± 0.31 | 6.08 ± 0.36* | 6.95 ± 0.30 | 6.35 ± 0.34* | 0.87 | 0.009 | 0.33 |
| Ct. Th. | 0.72 ± 0.02 | 0.64 ± 0.02* | 0.69 ± 0.02 | 0.63 ± 0.22* | 0.42 | 0.002 | 0.74 |
| Av. Diam. | 1.78 ± 0.03 | 1.75 ± 0.03 | 1.78 ± 0.03 | 1.78 ± 0.03 | 0.85 | 0.63 | 0.71 |
| Me. Diam. | 1.08 ± 0.03 | 1.13 ± 0.03 | 1.07 ± 0.03 | 1.10 ± 0.03 | 0.57 | 0.21 | 0.66 |
| Ec. Pm. | 7.38 ± 0.26 | 7.11 ± 0.31 | 7.34 ± 0.25 | 7.45 ± 0.28 | 0.58 | 0.77 | 0.49 |
| Ps. Pm. | 12.9 ± 0.35 | 11.7 ± 0.40* | 12.5 ± 0.33 | 12.0 ± 0.37* | 0.90 | 0.03 | 0.35 |
Data are mean ± SEM. Statistical significance was determined by two-way ANOVA. HC – high carbohydrate; HF – high fat; Ethanol –EtOH. EtOH means with * are significantly different from diet controls. 1Whole tibia mechanical strength testing as described in Materials and Methods. Load to Failure (N), Stiffness (N/mm3). 2Bone morphology of the femur determined by μCT analysis as described in Material and Methods. BV/TV: Bone volume/tissue volume; Tb. No.: trabecular number (1/mm); Tb Spp.: trabecular spacing (μM); Tb.Th: trabecular thickness (μM); DA: degree of anisotropy; Conn. D.: connectivity density (mm3); SMI: structure model index; Tot. CSA: total cross sectional area (cm2); Me.Ar.: medullary area (cm2); Ct. CSA: cortical cross sectional area (cm2); Ct. Th.: cortical thickness (mm); Av. Diam: average diameter (cm); Me. Diam.: medullary diameter (cm); Ec. Pm.: endocortical perimeter (cm); Ps.Pm.: periosteal perimeter (cm).
Discussion
Despite intensive investigation, the molecular mechanisms underlying development of fatty liver following alcohol abuse remain the subject of considerable dispute (Baumgardner et al., 2007; Ronis et al., 2011; You & Rogers, 2009). Ethanol has previously been reported to cause white adipose tissue dysfunction and to reduce adiposity without decreasing lean body mass in both rodents and humans, at least in part, as a result of induction of CYP2E1, generation of oxidative stress, and inhibition of PPARγ signaling (Addolorato et al., 1997; Chen et al., 2011; Chen et al., 2009; Crabb, Zeng, Liangpunsakul, Jones, & Considine, 2011; Liangpunsakul, Crabb, & Qi, 2010; Mercer et al., 2012; Sun et al., 2012; Tan et al., 2012). In addition, several investigators have published data suggesting a central role for reduced secretion of the adipokine adiponectin and inhibition of downstream pathways in the liver involving the SIRT-1-AMP kinase axis in the development of alcoholic steatosis (Yin et al., 2012; You, Considine, Leone, Kelly, & Crabb, 2005; You, Liang, et al., 2008, You & Rogers, 2009). If this pathway is indeed central, then it would be predicted that reduced adiponectin, decreased hepatic SIRT-1, increased hepatic nuclear SREBP-1c, increased de novo FA synthesis, and increased FA degradation would be a universal feature of animal models of alcoholic liver disease. However, a variety of other studies including the current one have failed to observe that ethanol decreases serum adiponectin despite decreased adiposity, and have shown increases in steatosis, despite no change or suppressed nuclear SREBP-1c protein and despite increased PPARα-dependent FA degradation (Baumgardner et al., 2007; He et al., 2004; Ronis et al., 2011). It is possible that these differences reflect differences in species, strain, and animal models of alcohol administration. It is also possible that differences in published results reflect complex interactions between alcohol and other dietary components in regulation of the pathways regulating endocrine communication between the adipose tissue and liver, and hepatic pathways controlling lipid homeostasis that are dependent on the duration of alcohol exposure. Dietary fat composition has previously been suggested to alter interactions between ethanol and the SIRT-1 pathway (You, Cao, Liang, Ajmo, & Ness, 2008; You, Liang, et al., 2008), and dietary fat type and levels of dietary carbohydrate have also previously been suggested to influence progression of ALD from simple steatosis to steatohepatitis (Korourian et al., 1999; Ronis, Hakkak, et al., 2004).
After short periods of TEN diet infusion up to 28 days, hepatic triglyceride accumulation was minor and fat droplets were small and rare in rats fed both the HC and HF control diets despite the large difference in fat/carbohydrate ratio. This was not surprising since it is well established that diets containing polyunsaturated fats result in little hepatic lipid accumulation, even though levels as low as the 5% fat in the HC diet have suppressed lipogenesis and FA transport relative to diets containing saturated or monounsaturated fats (Clarke, Turini, Jump, Abraham, & Reedy, 1998; Iritani & Fukuda, 1995; Jeffcoat, Roberts, & James, 1979; Jump, Thelen, & Mater, 1999; Ronis et al., 2012). The molecular mechanisms underlying this effect are complex and have been suggested to be related to signaling mediated via dietary linoleic acid, resulting in relatively low levels of nuclear ChREBP and SREBP-1c (Dentin et al., 2004; Jump et al., 1999). The HF diet had significantly reduced levels of FA synthesis and increased FA degradation compared to the HC diet as indicated by expression of FASN, ACC, CPT-1, and ACO mRNAs. Decreased FA synthesis occurred despite higher nuclear ChREBP expression in the HF than the HC diet at day 14. However, it was accompanied by greater insulin resistance and significantly reduced nuclear SREBP-1c and SREBP-1c binding to the FASN promoter in the HF diet group at this time point. Increased FA degradation in the HF group could be related to increased PPARα signaling through polyunsaturated fat metabolites. Consistent with this, we also observed increases in expression of another PPARα target, CYP4A1, after longer periods of TEN feeding in the HF group relative to the HC group. Increased FA transport via FATP2 and decreased triglyceride hydrolysis via adiponeutrin in the HF group compared to the HC group appear to compensate for the effects of HF on FA turnover, and levels of fat accumulation in the HC and HF control groups at day 14 and day 28 were comparable.
Although ethanol significantly increased overall hepatic triglyceride accumulation and fat droplets in both diet groups even after short term 14-day and 28-day TEN feeding, steatosis was dramatically greater in the HC+EtOH groups than the HF+EtOH groups and was associated with increases in FA synthesis in the HC+EtOH group relative to the other groups. This effect of ethanol was not associated with any change in serum adiponectin, hepatic expression of either SIRT-1 or GCN5 deacetylase or acetylase proteins, or changes in nuclear accumulation of SREBP-1c protein relative to HC controls. These data are not consistent with the hypothesis that suppressed adiponection-SIRT-1-AMP kinase signaling underlies alcoholic steatosis when consumed as part of a high carbohydrate diet. In contrast, we report that ethanol treatment increased hepatic accumulation of ChREBP which could drive increased transcription of FASN and ACC-1. These data are consistent with a recent report from Liangpunsakul et al. (2013) demonstrating increased nuclear accumulation of ChREBP in ethanol-treated hepatoma cells in vitro and in ethanol-fed mice.
Interestingly, over longer periods of 65 days of TEN diet infusion, triglycerides accumulated and steatosis developed in the rats fed the HC control diet to the same degree as in the HC+EtOH group. This also appeared to be related to increases in FA synthesis correlating with nuclear accumulation of ChREBP, and suggests that ethanol and simple carbohydrates activate the same lipogenic pathways but with a different time course. Additional increases in FA transport via CD36 with time also appear to contribute to fat accumulation in the HC group. In the HF groups, the more modest accumulation of hepatic triglycerides after chronic ethanol treatment was associated with little or no increase in FA synthesis pathways. A smaller increase in nuclear ChREBP was counteracted by decreased nuclear SREBP-1c and increased expression of mRNAs encoding genes involved in FA degradation. Consistent with our previous studies (Ronis et al., 2011) in the context of HF feeding, ethanol appeared to increase fat accumulation, in part as a result of increased FA transport through increasing FATP2 expression.
Despite the much lower level of steatosis, progression of necrotic alcohol liver injury beyond simple steatosis as measured by increases in ALT over the appropriate dietary controls was only evident in the HF+EtOH group and was accompanied by earlier appearance of oxidative stress as measured by TBARS and by increased expression of CYP2E1 and CYP4A1 relative to the HC+EtOH group. This is consistent with previous data from our laboratory suggesting protection against EtOH hepatotoxicity by high carbohydrate diets (Korourian et al., 1999; Ronis, Hakkak, et al., 2004) and may reflect availability of energy from glucose for use in ongoing cellular repair processes in response to alcoholic injury. Interestingly, ALTs increased to the same degree in both HC and HC+EtOH groups after 65 days of TEN feeding relative to earlier time points and appeared to be associated with the high levels of steatosis found in both these groups. These data support the important role of dietary polyunsaturated fats and lipid peroxidation in the progression of liver injury after consumption of diets high in ethanol and fat, but suggest that when consumed in the context of high carbohydrate diets, the mechanisms underlying development of alcoholic and non-alcoholic steatohepatitis may be indistinguishable.
In contrast to the dramatic differences in development of alcoholic steatosis and liver injury, dietary manipulation had no effect on alcohol-induced osteopenia. As has been reported, ethanol consumption reduced both trabecular and cortical bone (Alvisa-Negrin et al., 2009; Chen et al., 2011; Mercer et al., 2012). Despite the fact that increased bone marrow adiposity has been linked to reduced adipose tissue mass in anorexia (Ecklund et al., 2010), and that ethanol inhibition of bone formation has been shown to result in increased bone marrow fat as the result of trans-differentiation of marrow mesenchymal stem cells into adipocytes (Chen et al., 2010; Shankar et al., 2008), the different effects of fat/carbohydrate ratio on ethanol-associated loss of abdominal fat were not reflected in differences in ethanol-induced skeletal toxicity. These data suggest that the molecular mechanisms underlying alcohol effects in liver, adipose tissue, and bone tissues are strikingly distinct.
In summary, our data demonstrate that the development of steatosis and progression of ALD differs depending on the relative levels of dietary carbohydrate and polyunsaturated fat even when ethanol exposure is identical. In addition, the data are consistent with the idea that in the context of diets high in carbohydrate, stimulation of FA synthesis by ethanol is rapid and underlies the development of steatosis as a result of the activation of hepatic ChREBP signaling. Importantly, feeding diets unbalanced by the presence of excess simple carbohydrates is not benign, but over time also results in the development of fatty liver disease even in the absence of alcohol via the same pathway. In the context of high levels of polyunsaturated fats, ALD progressed despite relatively little accumulation of hepatic triglycerides. This points to the importance of lipid peroxidation in development of hepatic necroinflammatory injury (Nanji, 2004). Finally our data suggest that mechanisms underlying alcohol pathology are tissue specific. This is the focus of ongoing investigations.
Acknowledgments
Supported in part by NIH AA08645 (TMB), NIH AA018282 (MJR), Carl L. Nelson Chair in Orthopaedic Creativity (LJS), the Marion B. Lyon Revocable Trust, and USDA Agricultural Service CRIS 6251-51999-007-04S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addolorato G, Capristo E, Greco AV, Stefanini GF, Gasbarrini G. Energy expenditure, substrate oxidation, and body composition in subjects with chronic alcoholism: new findings from metabolic assessment. Alcoholism: Clinical and Experimental Research. 1997;21:962–967. [PubMed] [Google Scholar]
- Albano E, Tomasi A, Persson JO, Terelius Y, Goria-Gatti L, Ingelman-Sundberg M, et al. Role of ethanol-inducible cytochrome P450 (P450IIE1) in catalysing the free radical activation of aliphatic alcohols. Biochemical Pharmacology. 1991;41:1895–1902. doi: 10.1016/0006-2952(91)90129-s. [DOI] [PubMed] [Google Scholar]
- Alvisa-Negrín J, González-Reimers E, Santolaria-Fernández F, García-Valdecasas-Campelo E, Valls MR, Pelazas-González R, et al. Osteopenia in alcoholics: effect of alcohol abstinence. Alcohol and Alcoholism. 2009;44:468–475. doi: 10.1093/alcalc/agp038. [DOI] [PubMed] [Google Scholar]
- Badger TM, Ronis MJ, Lumpkin CK, Valentine CR, Shahare M, Irby D, et al. Effects of chronic ethanol on growth hormone secretion and hepatic cytochrome P450 isozymes of the rat. The Journal of Pharmacology and Experimental Therapeutics. 1993;264:438–447. [PubMed] [Google Scholar]
- Badger TM, Ronis MJJ, Lumpkin CK. The Effects of Alcohol on Growth Hormone and Growth Hormone-Regulated Systems. In: Zakhari S, editor. NIAAA Research Monograph 23, Alcohol and the Endocrine System. NIH; 1993. pp. 339–361. [Google Scholar]
- Baumgardner JN, Shankar K, Korourian S, Badger TM, Ronis MJ. Undernutrition enhances alcohol-induced hepatocyte proliferation in the liver of rats fed via total enteral nutrition. American Journal of Physiology, Gastrointestinal and Liver Physiology. 2007;293:G355–G364. doi: 10.1152/ajpgi.00038.2007. [DOI] [PubMed] [Google Scholar]
- Baumgardner JN, Shankar K, Hennings L, Badger TM, Ronis MJ. A new model for nonalcoholic steatohepatitis utilizing total enteral nutrition to overfeed a high-polyunsaturated fat diet. American Journal of Physiology, Gastrointestinal and Liver Physiology. 2008;294:G27–G38. doi: 10.1152/ajpgi.00296.2007. [DOI] [PubMed] [Google Scholar]
- Borengasser SJ, Lau F, Kang P, Blackburn ML, Ronis MJ, Badger TM, et al. Maternal obesity during gestation impairs fatty acid oxidation and mitochondrial SIRT3 expression in rat offspring at weaning. PLos One. 2011;6:e24068. doi: 10.1371/journal.pone.0024068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using microcomputed tomography. Journal of Bone and Mineral Research. 2010;25:1468–1486. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- Brown EC, Perrien DS, Fletcher TW, Irby DJ, Aronson J, Gao GG, et al. Skeletal toxicity associated with chronic ethanol exposure in a rat model using total enteral nutrition. The Journal of Pharmacology and Experimental Therapeutics. 2002;301:1132–1138. doi: 10.1124/jpet.301.3.1132. [DOI] [PubMed] [Google Scholar]
- Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Badger TM, Ronis MJ. A role for ethanol-induced oxidative stress in controlling lineage commitment of mesenchymal stromal cells through inhibition of Wnt/beta-catenin signaling. Journal of Bone and Mineral Research. 2010;25:1117–1127. doi: 10.1002/jbmr.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Lumpkin CK, Badger TM, et al. Inhibition of NADPH oxidases prevents chronic ethanol-induced bone loss in female rats. The Journal of Pharmacology and Experimental Therapeutics. 2011;336:734–742. doi: 10.1124/jpet.110.175091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Sebastian BM, Nagy LE. Chronic ethanol feeding to rats decreases adiponectin secretion by subcutaneous adipocytes. American Journal of Physiology, Endocrinology, and Metabolism. 2007;292:E621–E628. doi: 10.1152/ajpendo.00387.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Sebastian BM, Tang H, McMullen MM, Axhemi A, Jacobsen DW, et al. Taurine supplementation prevents ethanol-induced decrease in serum adiponectin and reduces hepatic steatosis in rats. Hepatology. 2009;49:1554–1562. doi: 10.1002/hep.22811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury M, Pandey RS, Clemens DL, Davis JW, Lim RW, Shukla SD. Knock down of GCN5 histone acetyltransferase by siRNA decreases ethanol-induced histone acetylation and affects differential expression of genes in human hepatoma cells. Alcohol. 2011;45:311–324. doi: 10.1016/j.alcohol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SD, Turini M, Jump DB, Abraham S, Reedy M. Polyunsaturated fatty acid inhibition of fatty acid synthase transcription is independent of PPAR activation. Zeitschrift für Ernährungswissenschaft. 1998;37:S14–S20. [PubMed] [Google Scholar]
- Contreras AV, Torres N, Tovar AR. PPAR-α as a key nutritional and environmental sensor for metabolic adaptation. Advances in Nutrition. 2013;4:439–452. doi: 10.3945/an.113.003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabb DW, Zeng Y, Liangpunsakul S, Jones R, Considine R. Ethanol impairs differentiation of human adipocyte stromal cells in culture. Alcoholism: Clinical and Experimental Research. 2011;35:1584–1592. doi: 10.1111/j.1530-0277.2011.01504.x. [DOI] [PubMed] [Google Scholar]
- Cusi K. The role of adipose tissue and lipotoxicity in the pathogenesis of type 2 diabetes. Current Diabetes Research. 2010;10:306–315. doi: 10.1007/s11892-010-0122-6. [DOI] [PubMed] [Google Scholar]
- Dai Y, Rashba-Step J, Cederbaum AI. Stable expression of human cytochrome P4502E1 in HepG2 cells: characterization of catalytic activities and production of reactive oxygen intermediates. Biochemistry. 1993;32:6928–6937. doi: 10.1021/bi00078a017. [DOI] [PubMed] [Google Scholar]
- Dentin R, Pégorier JP, Benhamed F, Foufelle F, Ferré P, Fauveau V, et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. The Journal of Biological Chemistry. 2004;279:20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- Dominy JE, Jr, Lee Y, Gerhart-Hines Z, Puigserver P. Nutrient-dependent regulation of PGC-1alpha’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochimica et Biophysica Acta. 2010;1804:1676–1683. doi: 10.1016/j.bbapap.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecklund K, Vajapeyam S, Feldman HA, Buzney CD, Mulkern RV, Kleinman PK, et al. Bone marrow changes in adolescent girls with anorexia nervosa. Journal of Bone and Mineral Research. 2010;25:298–304. doi: 10.1359/jbmr.090805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filhoulaud G, Guilmeau S, Dentin R, Girard J, Postic C. Novel insights into ChREBP regulation and function. Trends in Endocrinology and Metabolism. 2013;24:257–268. doi: 10.1016/j.tem.2013.01.003. [DOI] [PubMed] [Google Scholar]
- He L, Simmen FA, Ronis MJ, Badger TM. Post-transcriptional regulation of sterol regulatory element-binding protein-1 by ethanol induces class I alcohol dehydrogenase in rat liver. The Journal of Biological Chemistry. 2004;279:28113–28121. doi: 10.1074/jbc.M400906200. [DOI] [PubMed] [Google Scholar]
- He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. The Journal of Biological Chemistry. 2010;285:6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iritani N, Fukuda H. Polyunsaturated fatty acid-mediated suppression of insulin-dependent gene expression of lipogenic enzymes in rat liver. Journal of Nutritional Science and Vitaminology. 1995;41:207–216. doi: 10.3177/jnsv.41.207. [DOI] [PubMed] [Google Scholar]
- Jeffcoat R, Roberts PA, James AT. The control of lipogenesis by dietary linoleic acid and its influence on the deposition of fat. European Journal of Biochemistry. 1979;101:447–453. doi: 10.1111/j.1432-1033.1979.tb19738.x. [DOI] [PubMed] [Google Scholar]
- Jump DB, Thelen A, Mater M. Dietary polyunsaturated fatty acids and hepatic gene expression. Lipids. 1999;34:S209–S212. doi: 10.1007/BF02562292. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK, Todero SL, Ward BW, Cannella JJ, 3rd, Tuma DJ. Betaine administration corrects ethanol-induced defective VLDL secretion. Molecular and Cellular Biochemistry. 2009;327:75–78. doi: 10.1007/s11010-009-0044-2. [DOI] [PubMed] [Google Scholar]
- Korourian S, Hakkak R, Ronis MJJ, Shelnut S, Waldren JA, Badger TM. The Effects of Enteral Nutrition and Chronic Alcohol on Hepatic Necrosis. Toxological Sciences. 1999;47:110–118. [Google Scholar]
- Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. The Journal of Clinical Investigation. 2000;105:1067–1075. doi: 10.1172/JCI8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. Journal of Hepatology. 2013;58:395–398. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- Levy JC, Matthews DR, Hermans MP. Correct homeostasis assessment (HOMA) evaluation uses the computer program. Diabetes Care. 1998;21:2191–2192. doi: 10.2337/diacare.21.12.2191. [DOI] [PubMed] [Google Scholar]
- Liangpunsakul S, Crabb DW, Qi R. Relationship among alcohol intake, body fat, and physical activity: a population-based study. Annals of Epidemiology. 2010;20:670–675. doi: 10.1016/j.annepidem.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liangpunsakul S, Ross RA, Crabb DW. Activation of carbohydrate response element-binding protein by ethanol. Journal of Investigative Medicine. 2013;61:270–277. doi: 10.231/JIM.0b013e31827c2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher J. The CYP2E1 knockout delivers another punch: first ASH, now NASH. Alcoholic steatohepatitis. Nonalcoholic steatohepatitis. Hepatology. 2001;33:311–312. doi: 10.1053/jhep.2001.0330311. [DOI] [PubMed] [Google Scholar]
- Marecki JC, Ronis MJ, Shankar K, Badger TM. Hyperinsulinemia and ectopic fat deposition can develop in the face of hyperadiponectinemia in young obese rats. The Journal of Nutritional Biochemistry. 2011;22:142–152. doi: 10.1016/j.jnutbio.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Mercer KE, Wynne RA, Lazarenko OP, Lumpkin CK, Hogue WR, Suva LJ, et al. Vitamin D supplementation protects against bone loss associated with chronic alcohol administration in female mice. The Journal of Pharmacology and Experimental Therapeutics. 2012;343:401–412. doi: 10.1124/jpet.112.197038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouzaki M, Allard JP. The role of nutrients in the development, progression, and treatment of nonalcoholic fatty liver disease. Journal of Clinical Gastroenterology. 2012;46:457–467. doi: 10.1097/MCG.0b013e31824cf51e. [DOI] [PubMed] [Google Scholar]
- Nagata K, Suzuki H, Sakaguchi S. Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. The Journal of Toxicological Sciences. 2007;32:453–468. doi: 10.2131/jts.32.453. [DOI] [PubMed] [Google Scholar]
- Nanji AA. Role of dietary fatty acids in the pathogenesis of experimental alcoholic liver disease. Alcohol. 2004;34:21–25. doi: 10.1016/j.alcohol.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Ohkawa H, Ohishi N, Yagi K. Reaction of linoleic acid hydroperoxide with thiobarbituric acid. Journal of Lipid Research. 1978;19:1053–1057. [PubMed] [Google Scholar]
- Potts JR, Verma S. Alcoholic hepatitis: diagnosis and management in 2012. Expert Review of Gastroenterology & Hepatology. 2012;6:695–710. doi: 10.1586/egh.12.57. [DOI] [PubMed] [Google Scholar]
- Poupeau A, Postic C. Cross-regulation of hepatic glucose metabolism via ChREBP and nuclear receptors. Biochimica et Biophysica Acta. 2011;1812:995–1006. doi: 10.1016/j.bbadis.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Reynard B, Balian A, Falik D, Capron F, Bedossa P, Chaput JC, et al. Risk factors of fibrosis in alcohol-induced liver. Hepatology. 2002;35:635–638. doi: 10.1053/jhep.2002.31782. [DOI] [PubMed] [Google Scholar]
- Ronis MJ, Baumgardner JN, Marecki JC, Hennings L, Wu X, Shankar K, et al. Dietary fat source alters hepatic gene expression profile and determines the type of liver pathology in rats overfed via total enteral nutrition. Physiological Genomics. 2012;44:1073–1089. doi: 10.1152/physiolgenomics.00069.2012. [DOI] [PubMed] [Google Scholar]
- Ronis MJ, Hakkak R, Korourian S, Albano E, Yoon S, Ingelman-Sundberg M, et al. Alcoholic liver disease in rats fed ethanol as part of oral or intragastric low-carbohydrate liquid diets. Experimental Biology and Medicine. 2004;229:351–360. doi: 10.1177/153537020422900410. [DOI] [PubMed] [Google Scholar]
- Ronis MJ, Hennings L, Stewart B, Basnakian AG, Apostolov EO, Albano E, et al. Effects of long-term ethanol administration in a rat total enteral nutrition model of alcoholic liver disease. American Journal of Physiology, Gastrointestinal and Liver Physiology. 2011;300:G109–G119. doi: 10.1152/ajpgi.00145.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronis MJ, Korourian S, Zipperman M, Hakkak R, Badger TM. Dietary saturated fat reduces alcoholic hepatotoxicity in rats by altering fatty acid metabolism and membrane composition. The Journal of Nutrition. 2004;134:904–912. doi: 10.1093/jn/134.4.904. [DOI] [PubMed] [Google Scholar]
- Ronis MJJ, Lindros KO, Ingelman-Sundberg M. The CYP2E Subfamily. In: Ioannides C, editor. Cytochromes P450: Metabolic and Toxicological Aspects. Boca Raton, FL: CRC Press; 1996. pp. 211–239. [Google Scholar]
- Sebastian EM, Suva LJ, Friedman PA. Differential effects of intermittent PTH(1-34) and PTH(7-34) on bone microarchitecture and aortic calcification in experimental renal failure. Bone. 2008;43:1022–1030. doi: 10.1016/j.bone.2008.07.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar K, Hidestrand M, Liu X, Chen JR, Haley R, Perrien DS, et al. Chronic ethanol consumption inhibits postlactational anabolic bone rebuilding in female rats. Journal of Bone and Mineral Research. 2008;23:338–349. doi: 10.1359/jbmr.071023. [DOI] [PubMed] [Google Scholar]
- Shankar K, Kang P, Harrell A, Zhong Y, Marecki JC, Ronis MJ, et al. Maternal overweight programs insulin and adiponectin signaling in the offspring. Endocrinology. 2010;151:2577–2589. doi: 10.1210/en.2010-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto T, Yamashita S, Ishigami M, Sakai N, Hirano K, Tahara M, et al. Decreased microsomal triglyceride transfer protein activity contributes to initiation of alcoholic liver steatosis in rats. Journal of Hepatology. 2002;36:157–162. doi: 10.1016/s0168-8278(01)00263-x. [DOI] [PubMed] [Google Scholar]
- Sun X, Tang Y, Tan X, Li Q, Zhong W, Sun X, et al. Activation of peroxisome proliferator-activated receptor-γ by rosiglitazone improves lipid homeostasis at the adipose tissue-liver axis in ethanol-fed mice. American Journal of Physiology, Gastrointestinal and Liver Physiology. 2012;302:G548–G557. doi: 10.1152/ajpgi.00342.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva LJ, Hartman E, Dilley JD, Russell S, Akel NS, Skinner RA, et al. Platelet dysfunction and a high bone mass phenotype in a murine model of platelet-type von Willebrand disease. The American Journal of Pathology. 2008;172:430–439. doi: 10.2353/ajpath.2008.070417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Sun X, Li Q, Zhao Y, Zhong W, Sun X, et al. Leptin deficiency contributes to the pathogenesis of alcoholic fatty liver disease in mice. The American Journal of Pathology. 2012;181:1279–1286. doi: 10.1016/j.ajpath.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Sebastian BM, Axhemi A, Chen X, Hillian AD, Jacobsen DW, et al. Ethanol-induced oxidative stress via the CYP2E1 pathway disrupts adiponectin secretion from adipocytes. Alcoholism: Clinical and Experimental Research. 2012;36:214–222. doi: 10.1111/j.1530-0277.2011.01607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya M, Ji C, Kosyk O, Shymonyak S, Melnyk S, Kono H, et al. Interstrain differences in live injury and one-carbon metabolism in alcohol-fed mice. Hepatology. 2012;56:130–139. doi: 10.1002/hep.25641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Hu M, Zhang R, Shen Z, Flatow L, You M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. The Journal of Biological Chemistry. 2012;287:9817–9826. doi: 10.1074/jbc.M111.333534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Cao Q, Liang X, Ajmo JM, Ness GC. Mammalian sirtuin 1 is involved in the protective action of dietary saturated fat against alcoholic fatty liver in mice. The Journal of Nutrition. 2008;138:497–501. doi: 10.1093/jn/138.3.497. [DOI] [PubMed] [Google Scholar]
- You M, Considine RV, Leone TC, Kelly DP, Crabb DW. Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology. 2005;42:568–577. doi: 10.1002/hep.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Liang X, Ajmo JM, Ness GC. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. American Journal of Physiology, Gastrointestinal and Liver Physiology. 2008;294:G892–G898. doi: 10.1152/ajpgi.00575.2007. [DOI] [PubMed] [Google Scholar]
- You M, Rogers CQ. Adiponectin: a key adipokine in alcoholic fatty liver. Experimental Biology and Medicine. 2009;234:850–859. doi: 10.3181/0902-MR-61. [DOI] [PubMed] [Google Scholar]