

Abstract
Neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease trigger neuronal cell death through endogenous suicide pathways. Surprisingly, although the cell death itself may occur relatively late in the course of the degenerative process, the mediators of the underlying cell-death pathways have shown promise as potential therapeutic targets.
Thank Heaven! The crisis — the danger, is past, and the lingering illness, is over at last — and the fever called “Living” is conquered at last.
from For Annie, by Edgar Allan Poe
Programmed cell death (PCD) has a critical role in the development of the nervous system, and both anti-PCD and pro-PCD modulators feature prominently in the establishment of neural architecture. It has been 100 years since the first description of developmental neuronal-cell death1, and more than 50 years since Levi-Montalcini showed that such physiological cell death is inhibited by soluble factors such as nerve growth factor2. Dysregulation of cell-death programmes features in developmental and neoplastic disorders of the nervous system, and there is increasing evidence to suggest that such dysregulation may also occur in neurodegenerative, infectious, traumatic, ischaemic, metabolic and demyelinating disorders.
In 1964, Lockshin and his colleagues introduced the term programmed cell death to describe the apparently predetermined pattern by which specific cells die during insect development3. In 1966, it was shown that this process requires protein synthesis, at least in some cases2, indicating that it is the result of an active cellular suicide process. Then, in 1972, John Kerr and his colleagues coined the term apoptosis to describe a morphologically relatively uniform set of cell deaths seen in many different situations, from development to insult response to cell turnover4.
Although PCD has often been equated with apoptosis, non-apoptotic forms also exist5–9, and neurodegenerative conditions such as Huntington’s disease, amyotrophic lateral sclerosis (ALS) and ischaemia show cell deaths that do not fulfil the criteria for apoptosis7.
Classical developmental studies support the view that at least three different forms of PCD are distinguishable (Table 1): type I, also known as nuclear or apoptotic; type II, also known as autophagic3,5,8; and type III, also known as cytoplasmic5,6,8,10. These occur reproducibly in specific nuclei and with specific frequencies, at particular times of nervous- system development. But these cell-death pathways may also be activated by various insults, such as DNA damage or the accumulation of misfolded proteins.
Table 1.
Characteristics of dying cells
| Characteristics | Established forms of cell death | Atypical forms of cell death | ||||
|---|---|---|---|---|---|---|
| Apoptosis | Autophagic | Paraptosis | Calcium-mediated | AIF/PARP-dependent | Oncosis | |
| Morphology |
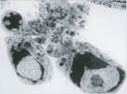 Chromatin condensation, nuclear fragmentation, apoptotic bodies |
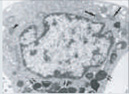 Autophagic vacuoles |
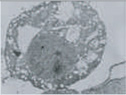 ER swelling, mitochondrial swelling |
 Membrane whorls |
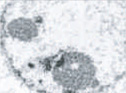 Mild chromatin condensation |
 Cellular swelling |
| Triggers | Include death receptors, trophic-factor withdrawal, DNA damage, viral infections | Serum, amino-acid starvation, protein aggregates | Trophotoxicity | Calcium entry, C. elegans deg mutants | DNA damage, glutamate, nitric oxide | Ischaemia, excitotoxicity |
| Mediators | Caspases, BH1–3, BH3 proteins | JNK1? MKK7? ATG orthologues | ERK2, NUR77 | Calpains, cathepsins | PARP, AIF | JNK |
| Inhibitors | Caspase inhibitors, BH1–4 proteins | JNK inhibitors? | U0126 (MEK), DN NUR77 | Calreticulin, Some calpain inhibitors? | PARP inhibitors | JNK inhibitors |
| Examples | Type I PCD, nuclear PCD | Type II PCD | Type III PCD, cytoplasmic PCD | C. elegans deg mutants | Some excitotoxic PCD | Ischaemic PCD |
Note the difference in morphology present in each form, as well as the differences in biochemical mediators, inducers, and inhibitors. At present, only apoptosis and autophagic PCD are generally accepted as being legitimate forms of PCD; however, ongoing research should reveal which of the additional candidates represent novel pathways of PCD. DN, dominant-negative; DEG, degeneration; U0126, a mitogen-activated protein kinase inhibitor. (Images reproduced, with permission, from refs 58–62.)
Neurodegenerative diseases are associated with a number of insults that may trigger PCD: misfolded proteins, reactive oxygen and nitrogen species, mitochondrial-complex inhibition, calcium entry, excitotoxicity, trophic-factor withdrawal, and death-receptor activation to name a few. In some cases, however, deaths occur that do not fit neatly into any of the three classes of PCD, and these more controversial forms of death are also discussed below.
Temporal studies of neurodegenerative models suggest, however, that PCD may be a relatively late event in the neurodegenerative process, and that death is preceded by early functional alterations (for example, electrophysiological deficits and cellular-stress-pathway activation) and microanatomical deficits (such as neurite retraction and synapse loss; see page 768). Surprisingly, then, various approaches aimed at inhibiting PCD have led to improved outcomes in neurodegenerative models, indicating that these pathways could have an important role in neurodegenerative diseases. Furthermore, recent studies have suggested that death in the nervous system may trigger stem-cell proliferation and survival, and so the work on cell death pathways — the subject of this review — offers many potential points of entry into the therapeutics of neurodegenerative disease states.
Classical apoptosis
Apoptosis (from the Greek, ‘falling away’), also referred to as nuclear or type I PCD, is the best-characterized type of PCD (Box 1). Morphologically, cells typically round up, form blebs, undergo zeiosis (an appearance of boiling due to rapid bleb formation), chromatin condensation, nuclear fragmentation and the budding off of apoptotic bodies. Phosphatidylserine, which is usually located at the plasma membrane and faces inwards on live cells, now faces both inwards and outwards11. These morphological and histochemical changes are largely the result of activation of a set of cell-suicide cysteine proteases, the caspases12,13.
Box 1 | Intrinsic and extrinsic apoptotic pathways.
In the best-characterized example of the extrinsic pathway (panel a) Fas is bound by the trimeric Fas ligand (FasL). This results in the recruitment of FADD though Fas’s death domain (DD) and caspase-8 through FADD’s death effector domain (DED)63. The proximity of the initiator caspase, which results from its recruitment to the trimeric Fas–FADD complex, leads to its activation, and the subsequent activation of effector caspases (such as caspase- 3 and 7) by cleavage. Furthermore, the extrinsic pathway interacts with the intrinsic pathway via caspase-8 cleavage of BID to produce tBID.
Whereas the extrinsic pathway uses caspase-8 (or caspase-10) to initiate cell death, the intrinsic pathway relies on caspase-9 (panel b). The propensity of cells to undergo apoptosis, known as the apostat, is largely determined by the balance between anti-apoptotic and pro-apoptotic members of the Bcl-2 family of proteins. There are three types of proapoptotic Bcl-2-family protein. First, the multidomain proteins, such as BAX and BAK, have Bcl-2 homology domains 1–3 (BH1–3), which can permeabilize mitochondrial outer membranes64. Second, the BH3-only proteins, such as BIM and tBID, activate BAX and BAK and may participate in pore formation. Third, the BH3-only derepressors such as PUMA, NOXA and BAD sequester the anti-apoptotic Bcl-2 and Bcl-XL (and related proteins with BH1–4 domains), freeing up BH1–3 proteins to permeabilize the mitochondria. This causes the release of various pro-apoptotic mitochondrial proteins, such as cytochrome c and SMAC (also known as DIABLO), into the cytosol. Other proteins that are unrelated to Bcl-2 can also influence the actions of the Bcl-2 family members65–67.
After release from the mitochondria, cytochrome c induces conformational change and heptamerization of the cytosolic protein APAF-1. The heptamer binds caspase-9, which results in its activation and the cleavage of effector caspases such as caspase-3 and 7. These active caspases can still be held in check by inhibitor of apoptosis proteins (IAPs) such as XIAP68, which may function as direct inhibitors of caspase activity and as E3 ligases that mediate caspase degradation69. This IAP-mediated block may be released by proteins such as SMAC70,71 and OMI (also known as HTRA2)72, which are also released from the mitochondria.
Fission and fusion of mitochondria73,74 also seem to function in cell death. Fission is mediated by DRP1 and FIS1, and inhibition of these proteins blocks the induction of apoptosis by staurosporine74. Pro-apoptotic proteins may be recruited to the mitochondria by DRP1, and mitochondrial remodelling may enhance the release of cytochrome c and other mitochondrial proteins. This could have special relevance to PCD during neurodegeneration, because mutations in the mitochondrial fusion mediator OPA1 are associated with optic atrophy75,76. Ongoing work should clarify the roles of mitochondrial fission and fusion, and their mediators, in PCD and neurodegeneration.

The biochemical activation of classical apoptosis occurs through two main pathways (Box 1). These are the extrinsic pathway, which originates through the activation of cell-surface death receptors such as Fas, and results in the activation of caspase-8 or -10 (ref. 14), and the intrinsic pathway, which originates from mitochondrial release of cytochrome c and associated activation of caspase-9. A third, less well-characterized pathway — essentially a second intrinsic pathway — originates from the endoplasmic reticulum (ER) and also results in the activation of caspase-9 (refs 15–17). Other organelles, such as the nucleus and Golgi apparatus, have damage sensors that link to apoptotic pathways18.
Various biochemical responders, both physiological and pathological, act on the intrinsic pathway of apoptosis. Whether triggered by DNA damage or by sensors associated with other organelles, the typical outcome is that the balance between pro-apoptotic members of the Bcl-2 family and anti-apoptotic members is shifted toward the pro-apoptotic members (Box 1), which leads (although not invariably) to the cell’s demise. But both the intrinsic and the extrinsic apoptotic pathways ultimately rely on the activation of caspases for death to ensue.
Caspases are cysteine-aspartyl-specific proteases that cleave with remarkable specificity at a small subset of aspartic acid residues. There are two types of apoptotic caspase: initiators and effectors. The initiator caspases cleave inactive forms of effector caspases, thereby activating them; effector caspases (for example, caspase-3 and 7) in turn cleave other protein substrates in the cell, resulting in the apoptotic process.
Both type of caspase are relatively inactive (the effectors more so than the initiators) when first synthesized, but they differ markedly in terms of their activation. Initiator caspases (caspase-8, 9 and 10) exist as monomers and bind to other proteins by means of what is known as the caspase activation and recruitment domain (CARD) in caspase-9 and a death effector domain (DED) in caspase-8 and 10. This protein–protein interaction results in dimerization of the caspases, which leads to their activation. Contrary to earlier models, cleavage of initiator caspases is neither required nor sufficient for activation19. However, the effector caspases exist as dimers in the cell and are activated by cleavage rather than induced proximity. Cleavage produces a tetramer with two large subunits and two small subunits with a substrate specificity that differs from initiator caspases.
The caspase substrates — the number of which is unknown but is probably somewhere between 0.5% and 5% of proteins — contribute to the apoptotic phenotype in various ways, such as by activation of proteolytic cascades, inactivation of repair, DNA cleavage, mitochondrial permeabilization and initiation of the process of phagytosis to clear up the dying cells, apoptotic bodies and debris.
Origami meets apoptosis
Misfolded proteins are constantly being produced, and for some proteins such misfolded species represent a significant fraction of the overall output. Misfolded proteins trigger a protective stress response, known as the unfolded-protein response (UPR; Fig. 1). Although this response may put off a cellular catastrophe for a short time, prolonged ER stress and UPR activation completely overwhelm the cellular protective mechanism, ultimately resulting in the activation of suicide pathways20–23 (Box 2). Morover, misfolded proteins also aggregate as oligomers and higher-order multimers, both of which may interact with critical cellular targets such as chaperones and transcription factors, among others.
Figure 1. Misfolded proteins and endoplasmic-reticulum stress.

a, In addition to its role as an ER chaperone protein, BiP also regulates the activation of the three proximal ER-stress transducers — IRE1, PERK and ATF6. All three transducers contain an amino-terminal luminal domain that interacts with BiP. Under normal conditions, BiP serves as a negative regulator of IRE1, PERK and ATF6 activation. When misfolded proteins accumulate and result in ER stress, BiP binds to these proteins. BiP is thus released from the transducers, which are consequently activated. The activation of all three proximal sensors results in reduction in the amount of new protein translocated into the ER lumen, increased degradation of ER-localized proteins and increased proteinfolding capacity of the ER. b, BiP release from PERK correlates with oligomerization, trans-autophosphorylation and activation of downstream signalling by PERK. PERK-dependent phosphorylation of eIF2α on Ser 51 leads to attenuation of protein translation and thereby reduces the workload of the ER. c, BiP release from ATF6 permits transport of AFT6 to the Golgi compartment for regulated intramembrane proteolysis by site-1 protease (S1P) and site-2 protease (S2P) to generate a 50-kDa cytosolic b-ZIP-containing fragment (ATF6DC). ATF6DC translocates to the nucleus to activate ER-stress inducible target genes that code for proteins involved in protein folding. This BiP-regulated activation provides a direct mechanism to sense the folding capacity of the ER. d, Accumulation of misfolded proteins in the ER causes BiP to release IRE1. IRE1 is a type I transmembrane protein that contains both a serine/threonine kinase domain and an endoribonuclease domain; the latter processes an intron from X-box protein 1 (XBP1) mRNA, rendering it competent for translation to produce the 41-kDa XBP1(S) protein. Unspliced XBP1 mRNA encodes a basic leucine-zipper protein, XBP1(U), that lacks transactivation activity and is more labile than XBP1(S). XBP1(S) binds to the promoters of several genes involved in retrograde transport of misfolded proteins from the ER to the cytosol and in ER-induced protein degradation.
Box 2 | Endoplasmic-reticulum stress and cell death.
ER stress is coupled to specific independent death pathways, as well as demonstrating involvement with the intrinsic and extrinsic apoptotic pathways. Studies from various laboratories have disclosed the roles of several ER-stress-induced cell-death modulators and effectors through the use of biochemical, pharmacological and genetic tools. These ER-stress-induced cell-death modulators include various members of the Bcl-2 family (Bcl-2, Bcl-XL, BAX, BAK, BI-1 and BIK), BAP31 and p53- dependent gene products such as NOXA and PUMA.
BAP31, an ER-membrane protein, binds Bcl-2 (or Bcl-XL) and a caspase-8-containing pro-apoptotic complex77. When BAP31 is cleaved, a proapoptotic p20 fragment is derived, which, among other effects, induces mitochondrial fission, enhancing cytochrome c release78. Conversely, BAR, which is expressed primarily in neurons of the central nervous system, also bridges Bcl-2 and caspase-8 but functions as an antiapoptotic protein79.
CHOP (also known as GADD153), a transcription factor induced during ER stress and activated by p38 mitogen-activated protein kinase, may also function as an ER-stress-induced cell-death modulator. It has been proposed to promote ER-stress-induced cell death by downregulating Bcl-2 expression.
Recent data have also implicated the calcium-binding protein apoptosis-linked gene-2 (ALG-2), and valosin-containing protein (VCP) as mediators of ER-stress-induced PCD. p23 interacts with PUMA and inhibits apoptosis, an effect that is reversed after cleavage. An ALG-2-interacting protein known as ALIX (or AIP-1) links motor-neuron-cell death during development as well as neuronal death in a HD model to the endolysosomal system80.
In addition to the two main pathways that initiate the caspase cascade — namely, the extrinsic and intrinsic pathways — some studies point to a caspase-12-mediated apoptotic pathway that involves the ER and the UPR (at least in murine cells), although this is a controversial area of research. Caspase-12 may be activated by calpain or caspase-7, or by ER-stress-activated IRE1, which may recruit caspase-12 through tumour-necrosis-factor receptor-associated factor 2 protein (TRAF2).
Oligomerized IRE1 also binds TRAF2, signalling downstream kinases (ASK1 and JNK) that, in turn, cause activation of pro-apoptotic proteins and cell death.

These pathways are of particular interest in neurodegenerative disease studies because they are implicated in all the main examples of such diseases24. Disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), ALS and prion-protein diseases all share one common feature: accumulation and aggregation of misfolded proteins20,21 (see pages 774 and 803).
Mediators of cell death induced by misfolded proteins have recently been identified (Box 2). Bcl-2 family proteins also seem to have a key role in the cellular-suicide decision process25, and in the communication between the ER and the mitochondria. BAX/BAK double knockout cells demonstrate no caspase activation following ER stress, indicating that these are required mediators26. BIK may function to activate BAX and BAK in this pathway, whereas BI-1 inhibits BAX activation and translocation to the ER27. Other Bcl-2 family proteins are also involved, such as PUMA and NOXA, as well as p53 (ref. 28).
Rather than true protein misfolding, prion proteins may trigger PCD when they are in an alternative, physiologically relevant conformation29. The prion protein exists in three different topologies: a secreted form, a transmembrane form in which the amino terminus is extracellular (NTM), and a transmembrane form in which the carboxy terminus is extracellular (CTM). The latter form is pro-apoptotic and associated with neurodegeneration in vivo, whereas the secreted form is antiapoptotic. It remains to be seen whether other proteins have similar features.
Autophagic cell death
Desperate times call for desperate measures, and cells have a host of protective stress responses, most of which switch into execution mode during prolonged activation. During starvation, cells may only be able to survive by a process known as autophagy (from the Greek, ‘self eating’). Autophagy occurs in diverse organisms and is subdivided into macroautophagy, microautophagy and chaperone-mediated autophagy. It complements the proteasomal pathway in that long-lived proteins, protein aggregates, and organelles are degraded by this regulated lysosomal pathway of degradation (see page 780). Targets for degradation — for example, damaged mitochondria or aggregates of misfolded proteins — are encircled by a process that is essentially an intracellular form of phagocytosis. The newly membrane-delimited structure — an autophagosome — then fuses with a lysosome, resulting in the degradation of the contents of the autophagosome. The molecular details of this process have been best characterized in yeast, in which a number of ATG (autophagy) genes have been identified, most of which have clear orthologues in higher eukaryotes.
Because the degradation of molecules and organelles by autophagy results in the production of energy and amino acids for protein synthesis, it is a cellular protective pathway that, although constitutively active at a low level, can be markedly upregulated by nutrient starvation. Nutrient withdrawal inactivates TOR (target of rapamycin), activating an ATG complex30. In a second step, vesicle nucleation occurs, and then, in a third step, vesicle expansion occurs, followed finally by the recycling of ATG proteins. The importance of this pathway in vivo has been illustrated by Atg7 conditionally null mice31, which show various cellular abnormalities such as ubiquitin-positive aggregates and apparently damaged mitochondria. Furthermore, mice deficient in autophagy due to knockout of beclin 1 are nonviable32.
Although the roles of the autophagic process in protein and organellar degradation, and in cellular protection during nutrient starvation, are well accepted, the role of autophagy in PCD is more controversial30,32,33. This is in part because the term ‘autophagic cell death’ has been used for two potentially distinct observations: cell death associated with autophagy and cell death requiring autophagy. Most examples of autophagic cell death represent the former. However, increasing evidence suggests that the autophagic process is required for at least some cell deaths. For example, haploinsufficiency of beclin 1 leads to a tumour predisposition phenotype, indicating that autophagy is tumour suppressive32. More direct evidence has been provided by cells whose apoptotic machinery has been inhibited: when mouse embryonic fibroblasts (MEFs) that are null for both Bax and Bak are treated with the apoptosis inducers staurosporine or etoposide they undergo a form of cell death that is associated with autophagosomes, is dependent on Atg5 and beclin 1 and is inhibited by the autophagy/class III phosphatidylinositol-3 kinase inhibitor 3-methyladenine33.
Furthermore, Lenardo’s group found that caspase inhibition by zVAD-fmk (a general caspase inhibitor) in L929 cells results in autophagy-dependent PCD34, which has been proposed to be mediated by the selective degradation of catalase35. On the one hand, this may alert us to the possibility that anti-apoptotic therapies carry the potential risk of inducing non-apoptotic PCD; on the other hand, it may argue that therapeutics directed at multiple cell-death pathways will be required for optimal efficacy in diseases that involve PCD. Because cell death associated with some neurodegenerative diseases (or disease-associated mutants, such as α-synuclein) is associated with an autophagic morphology, these implications are potentially important for the development of effective therapies to prevent or ameliorate neurodegenerative disorders36.
It is clear that in vivo testing to establish whether the autophagic process is required for any form(s) of PCD is needed, and the required genetic and pharmacological tools are increasingly available. Many questions in this area remain unanswered. If autophagy is a cellular-protective programme that — like the UPR — at some point activates PCD, what is the signal that initiates PCD? How important is the role of autophagic PCD in neurodegeneration? Does autophagic PCD occur in vivo in the absence of apoptosis inhibition? Are there ‘executioners’ analogous to caspases in autophagic PCD? Given the common finding of protein aggregates in neurodegenerative diseases, is a defect in autophagy a common underlying problem in these diseases? If so, does this contribute to the triggering of cell death in neurodegenerative diseases?
Alternative cell-death programmes
In comparison with apoptosis, little is known about autophagic PCD, and even less is known about other non-apoptotic forms of PCD. In fact, so far, other forms of PCD have not been generally accepted by the scientific community. However, as has previously been noted37, such forms of cell death have been observed repeatedly, although their genetics and biochemical pathways are poorly understood. Type III PCD5, or cytoplasmic cell death, is a ‘necrosis-like’ form of PCD that includes swelling of the ER and mitochondria, and lacks typical apoptotic features such as apoptotic bodies and nuclear fragmentation. It has recently been noted that the hyperactivation of the tyrosine-kinase receptor insulin-like growth factor I receptor (IGFR) induces a non-apoptotic form of cell death known as paraptosis9. This was shown to be programmatic — in that it required transcription and translation — and was found to be morphologically indistinguishable from type III PCD. Neither Bcl-2 nor caspase inhibitors block this form of PCD, nor are caspases activated, but inhibitors of extracellular signal-regulated kinase 2 (ERK2) — but not ERK1 — were found to inhibit paraptosis38, as was AIP-1 (a PCD-interacting protein also known as ALIX).
The idea that PCD might be induced by hyperactivation of a trophic factor receptor — trophotoxicity — is compatible with earlier observations that some trophic factors may increase neuronal cell death, for example that induced by excitotoxicity39. Such an effect might be protective against neoplasia, in that it may eliminate cells that would otherwise undergo autocrine-loop-stimulated oncogenesis. The resulting programme would necessarily be non-apoptotic, because trophic factors inactivate apoptotic signalling.
Aponecrosis is a term applied to a combination of apoptosis and necrosis40. Many cytotoxins induce PCD at low concentrations but at higher concentrations induce necrotic cell death, presumably because the cell’s homeostatic processes are overwhelmed before the cell-death programmes can be completed. In fact, this is the most common pattern seen with cellular toxins, from hydrogen peroxide and other oxidants to mitochondrial toxins such as antimycin A40. Nicotera, Lipton and their colleagues showed that glutamate-induced neuronal-cell death could proceed through apoptosis or necrosis, depending on mitochondrial membrane potential and cellular energy state41. However, in most cases, the necrotic morphology associated with aponecrosis has not been proved not to be programmed. So, it is still not clear whether aponecrosis represents a combination of apoptosis and a non-apoptotic form of PCD, or whether it represents a combination of apoptosis and non-programmatic cell death.
Forms of cell death have been described that do not fit the criteria for any of the three types of developmental cell death (Table 1). For example, a non-apoptotic, caspase-independent form that does not resemble type II or type III developmental PCD has been described by Driscoll and her colleagues42in C. elegans expressing mutant channel proteins such as MEC-4(d). A uniform, necrosis-like cell death — characterized morphologically by membranous whorls not seen in other types of cell death — is triggered by calcium entry, mediated by specific calpains and cathepsins, and inhibited by calreticulin.
A fifth apparent form of PCD has been described by the Dawsons and their colleagues, who showed that a non-apoptotic form of cell death depends on the activation of poly-(ADP-ribose) polymerase (PARP) and the consequent translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus43. AIF is a flavoprotein described by Kroemer and colleagues44 that is involved with DNA fragmentation, along with endonuclease G and DNA-fragmentation factor. This form of PCD was shown to be activated by agents that induce DNA damage, and shows a morphology and biochemistry that is, as far as we know, distinct from PCD types I–III.
It is likely that, as additional data are gathered from other cell-death paradigms, novel biochemical pathways of PCD will be characterized. For example, there is an extensive literature on the morphological criteria for another potential form of PCD — oncosis — but the biochemical underpinnings of oncosis have not yet been described. Oncosis refers to a specific morphology of cell death — cellular swelling — that is typically induced by ischaemia and is thought to be mediated by the failure of plasma-membrane ionic pumps. One potential mediator of oncosis is a calpain-family protease (possibly a mitochondrial calpain45).
Triggering cell death in neurodegeneration
As noted above, a number of different and potentially interrelated insults may occur as part of the neurodegenerative process. The role of the cell-death machinery in neurodegenerative diseases is controversial, especially because synaptic loss and electrophysiological abnormalities typically precede cell loss in these disease states (see page 768). However, recent studies suggest a critical role for cell-death mediators in neurodegenerative diseases, even before the reduction in neuronal number. For example, in a transgenic mouse model of AD that features senile plaques, synapse and memory loss, but little or no neuronal loss, mutation of the caspase cleavage site at Asp 664 in the amyloid precursor protein (APP) completely suppressed synapse loss, dentate gyral atrophy, astrogliosis and memory loss, even though senile plaque number and amyloid-β concentrations were unaffected46. In an analogous study with an HD-transgenic mouse model, mutation of the caspase-6 site (but not the caspase-3 sites) in polyglutamine-expanded huntingtin prevented both the neurodegeneration and motor abnormalities characteristic of Huntington’s disease47.
More direct evidence for caspase activation in neurodegeneration (Fig. 2) comes from the employment of antibodies directed against neo-epitopes derived by caspase cleavage46,48 and from the inhibition of neurodegeneration by caspase inhibitors49,50. However, some neurodegenerative models and diseases clearly demonstrate non-apoptotic forms of PCD7. Determining which PCD pathways are triggered in each neurodegenerative disease, which pathway accounts for each fraction of cell death, the mechanism(s) by which each pathway is triggered, and the interactions between the various pathways should shed new light on the degenerative process and its potential treatment or prevention. Perhaps even more important will be dissecting the pathways mediating sub apoptotic events such as synaptosis and Wallerian degeneration — likely to be important features in the neurodegenerative process — and understanding how the interplay between these various processes results in the neurodegenerative phenotype.
Figure 2. Caspase cleavage in Alzheimer’s disease.

An antibody specific for the neoepitope generated by cleavage of APP at the caspase-cleavage site, Asp 664, demonstrates reactivity (brown) in the hippocampus of a patient with Alzheimer’s disease.
The ability to initiate the neurodegenerative process, with widely varying insults — from misfolded proteins to reactive oxygen species to caspase recruitment complexes, as well as other mechanisms — and yet produce a relatively small number of syndromes indicates the existence of a death network that can be entered from many different sites but once triggered follows similar interdependent biochemical pathways with little dependence on the point of entry. This idea is compatible with the findings that therapeutics aimed at different pathways (for example, caspase activation, mitochondrial release of cytochrome c, metal binding and reactive-oxygen-species scavenging) all have partly salutary effects. However, it also suggests that a complete halt of the neurodegenerative process may require therapeutics that address all the interacting pathways of the network.
Targeting programmed cell death
Neuronal loss is a relatively late event in neurodegenerative diseases, following neuronal dysfunction, synapse loss and, often, somal atrophy. But targeting PCD has been successful in at least some model systems of neurodegeneration, and it is possible that targeting multiple PCD pathways will be even more effective. Caspase inhibition in vivo retarded the degeneration in transgenic mouse models of both ALS and HD, despite the fact that the morphological description of neuronal cell death in these diseases is not compatible with apoptosis49,50. Bcl-2 expression in a transgenic model of ALS delayed symptom onset and increased lifespan, but did not alter the disease duration51.
Minocycline, a second-generation tetracycline that inhibits mitochondrial cytochrome c release, effected neuroprotection in mouse models of HD, PD and ALS52. Minocycline is orally bioavailable, penetrates the blood–brain barrier and has been proved safe for use in humans. It is currently being evaluated in clinical trials in patients with HD and ALS52.
Rasagiline, which has been approved for the treatment of PD, has also been proposed to target apoptosis, but because it is a potent, selective, irreversible inhibitor of monoamine oxidase type B, its therapeutic effect on PD may have nothing to do with effects on apoptosis.
Trophic factors have multiple effects, including the inhibition of apoptosis, the stimulation of neural precursors and the stimulation of neurite outgrowth. The literature is rife with examples of trophic-factor therapy for various neurological conditions, and although many have been unsuccessful, the delivery of the correct factor(s) to the right target in the correct concentration for a particular disease still holds great promise (although hyperactivation of at least some trophic-factor receptors may induce PCD). A recent study of fibroblast growth factor 2 (FGF2) in a mouse model of HD prolonged survival, improved motor performance and reduced polyglutamine aggregates53. A number of approaches have been taken to deliver nerve growth factor (NGF) to patients with AD, the most recent being by means of genetically engineered fibroblasts in a phase I trial54. In this study, improvements in cognition and positron-emission tomography (used to monitor fluorodeoxyglucose uptake) were documented. Glial-derived neurotrophic factor (GDNF) has shown promise in the treatment of PD55.
HD may represent one of the best possibilities for trophic-factor therapy: presymptomatic diagnosis is readily available, and although a number of different trophic factors have shown promise in animal models (for example, FGF2, NGF and ciliary neurotrophic factor56), brain-derived neurotrophic factor (BDNF) is both reduced in the disease and therapeutic in models. Furthermore, cysteamine has been shown to increase BDNF levels in brain57.
A lifeline
Exciting recent evidence suggests that pathological processes may stimulate neurogenesis in the brain and may redirect the migration of nascent neurons towards the site of pathology. And, under certain circumstances, stimulating neurogenesis can improve the performance and survival of mice with neurodegenerative disease. These and similar results raise the question of whether stem-cell exhaustion or senescence after prolonged stimulation might have a role in the long-term course of neurodegenerative disease. Furthermore, although the factors linking neurodegeneration to neural-stem-cell proliferation and inhibition of PCD are largely unknown, the potential for therapy using these putative factors is likely to be significant. Reconciliation of cell-death pathways with neurodegenerative and regenerative mechanisms should offer an improved understanding of disease and open avenues for therapeutic intervention.
Acknowledgements
We apologize to all colleagues whose papers we were unable to cite owing to space limitations. We thank S. Rabizadeh, A. Kurakin, V. Galvan, D. Madden, L. Egger, J. Fombonne, T.-V. Nguyen, N. Rooke, K. Niazi, A. Swistowski, K. Poksay and S. Chen for critical reading of the manuscript, and M. Susag, L. Sheridan and R. Abulencia for manuscript preparation.
Footnotes
The authors declare no competing financial interests.
References
- 1.Studnicka FK. In: Lehrbuch der vergleichende mikroskopischen Anatomie der Wirbeltiere. Oppel A, editor. Germany: Fischer; 1905. pp. 1–256. [Google Scholar]
- 2.Levi-Montalcini R. The nerve growth factor: its mode of action on sensory and sympathetic nerve cells. Harvey Lect. 1966;60:217–259. [PubMed] [Google Scholar]
- 3.Lockshin RA, Williams CM. Programmed cell death. II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J. Insect Physiol. 1964;10:643–649. [Google Scholar]
- 4.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 6.Cunningham TJ. Naturally occurring neuron death and its regulation by developing neural pathways. Int. Rev. Cytol. 1982;74:163–186. doi: 10.1016/s0074-7696(08)61172-9. [DOI] [PubMed] [Google Scholar]
- 7.Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am. J. Pathol. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- 8.Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–266. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- 9.Sperandio S, de Belle I, Bredesen DE. An alternative, non-apoptotic form of programmed cell death. Proc. Natl Acad. Sci. USA. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oppenheim RW. Naturally occurring cell death during neural development. Trends Neurosci. 1985;17:487–493. [Google Scholar]
- 11.Fadok VA, et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 12.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 13.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 14.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 15.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 16.Rao RV, et al. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J. Biol. Chem. 2002;277:21836–21842. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- 17.Yuan J, Yankner BA. Caspase activity sows the seeds of neuronal death. Nature Cell Biol. 1999;1:E44–E45. doi: 10.1038/10037. [DOI] [PubMed] [Google Scholar]
- 18.Green DR, Kroemer G. Pharmacological manipulation of cell death: clinical applications in sight? J. Clin. Invest. 2005;115:2610–2617. doi: 10.1172/JCI26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopito RR, Ron D. Conformational disease. Nature Cell Biol. 2000;2:E207–E209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- 21.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 22.Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891–894. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 23.Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 24.Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004;16:653–662. doi: 10.1016/j.ceb.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scorrano L, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 26.Ruiz-Vela A, Opferman JT, Cheng EH, Korsmeyer SJ. Proapoptotic BAX and BAK control multiple initiator caspases. EMBO Rep. 2005;6:379–385. doi: 10.1038/sj.embor.7400375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chae HJ, et al. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol. Cell. 2004;15:355–366. doi: 10.1016/j.molcel.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 29.Hegde RS, et al. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- 30.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J. Clin. Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl Acad. Sci. USA. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nature Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 34.Yu L, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 35.Yu L, et al. Autophagic programmed cell death by selective catalase degradation. Proc. Natl Acad. Sci. USA. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez-Santos C, et al. Dopamine induces autophagic cell death and α-synuclein increase in human neuroblastoma SH-SY5Y cells. J. Neurosci. Res. 2003;73:341–350. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- 37.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 38.Sperandio S, et al. Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004;11:1066–1075. doi: 10.1038/sj.cdd.4401465. [DOI] [PubMed] [Google Scholar]
- 39.Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- 40.Formigli L, et al. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J. Cell Physiol. 2000;182:41–49. doi: 10.1002/(SICI)1097-4652(200001)182:1<41::AID-JCP5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 41.Ankarcrona M, et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 42.Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature. 2002;419:939–944. doi: 10.1038/nature01108. [DOI] [PubMed] [Google Scholar]
- 43.Yu SW, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 44.Susin SA, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 45.Liu X, Van Vleet T, Schnellmann RG. The role of calpain in oncotic cell death. Annu. Rev. Pharmacol. Toxicol. 2004;44:349–370. doi: 10.1146/annurev.pharmtox.44.101802.121804. [DOI] [PubMed] [Google Scholar]
- 46.Galvan V, et al. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc. Natl Acad. Sci. USA. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graham RK, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 48.Yang F, et al. Antibody to caspase-cleaved actin detects apoptosis in differentiated neuroblastoma and plaque-associated neurons and microglia in Alzheimer’s disease. Am. J. Pathol. 1998;152:379–389. [PMC free article] [PubMed] [Google Scholar]
- 49.Friedlander RM, Brown RH, Gagliardini V, Wang J, Yuan J. Inhibition of ICE slows ALS in mice. Nature. 1997;388:31. doi: 10.1038/40299. [DOI] [PubMed] [Google Scholar]
- 50.Ona VO, et al. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 51.Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- 52.Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 53.Jin K, et al. FGF-2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington’s disease. Proc. Natl Acad. Sci. USA. 2005;102:18189–18194. doi: 10.1073/pnas.0506375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tuszynski MH, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nature Med. 2005;11:551–555. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- 55.Lang AE, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006;59:459–466. doi: 10.1002/ana.20737. [DOI] [PubMed] [Google Scholar]
- 56.Kordower JH, Isacson O, Emerich DF. Cellular delivery of trophic factors for the treatment of Huntington’s disease: is neuroprotection possible? Exp. Neurol. 1999;159:4–20. doi: 10.1006/exnr.1999.7156. [DOI] [PubMed] [Google Scholar]
- 57.Borrell-Pages M, et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J. Clin. Invest. 2006;116:1410–1424. doi: 10.1172/JCI27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kerr JFR, Harmon BV. In: Apoptosis: The Molecular Basis of Cell Death. Tomei LD, Cope FO, editors. Vol. 321. Plainview, New York: Cold Spring Harbor Laboratory Press; 1991. [Google Scholar]
- 59.Bursch W, et al. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J. Cell Sci. 2000;113:1189–1198. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- 60.Hall IH, Elkins AL, Karthikeyan S, Spielvogel BF. The cytotoxicity of 1-(phenylmethyl)-4,7,10-tris-[(4’methylphenyl) sulfonyl]-1,4,7,10-tetraazacyclododecane in human Tmolt3 T leukemic cells. Anticancer Res. 1997;17:1195–1198. [PubMed] [Google Scholar]
- 61.Susin SA, et al. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ohno M, et al. ‘Apoptotic’ myocytes in infarct area in rabbit hearts may be oncotic myocytes with DNA fragmentation: analysis by immunogold electron microscopy combined with in situ nick end-labeling. Circulation. 1998;98:1422–1430. doi: 10.1161/01.cir.98.14.1422. [DOI] [PubMed] [Google Scholar]
- 63.Muzio M, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 64.Kuwana T, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 65.Guo B, et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- 66.Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P, Green DR. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J. Biol. Chem. 2000;275:7337–7342. doi: 10.1074/jbc.275.10.7337. [DOI] [PubMed] [Google Scholar]
- 67.Lin B, et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell. 2004;116:527–540. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- 68.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 69.Holley CL, Olson MR, Colon-Ramos DA, Kornbluth S. Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nature Cell Biol. 2002;4:439–444. doi: 10.1038/ncb798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 71.Verhagen AM, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 72.Martins LM, et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J. Biol. Chem. 2002;277:439–444. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 73.Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr. Opin. Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 74.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frezza C, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 76.Cipolat S, et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126:163–175. doi: 10.1016/j.cell.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 77.Ng FW, et al. p28 Bap31, a Bcl-2/Bcl-XL- and procaspase-8-associated protein in the endoplasmic reticulum. J. Cell Biol. 1997;139:327–338. doi: 10.1083/jcb.139.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003;160:1115–1127. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roth W, et al. Bifunctional apoptosis inhibitor (BAR) protects neurons from diverse cell death pathways. Cell Death Differ. 2003;10:1178–1187. doi: 10.1038/sj.cdd.4401287. [DOI] [PubMed] [Google Scholar]
- 80.Mahul-Mellier AL, Hemming FJ, Blot B, Fraboulet S, Sadoul R. Alix, making a link between apoptosis-linked gene-2, the endosomal sorting complexes required for transport, and neuronal death in vivo. J. Neurosci. 2006;26:542–549. doi: 10.1523/JNEUROSCI.3069-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]