

Abstract
Sleep-induced apnea and disordered breathing refers to intermittent, cyclical cessations or reductions of airflow, with or without obstructions of the upper airway (OSA). In the presence of an anatomically compromised, collapsible airway, the sleep-induced loss of compensatory tonic input to the upper airway dilator muscle motor neurons leads to collapse of the pharyngeal airway. In turn, the ability of the sleeping subject to compensate for this airway obstruction will determine the degree of cycling of these events. Several of the classic neurotransmitters and a growing list of neuromodulators have now been identified that contribute to neurochemical regulation of pharyngeal motor neuron activity and airway patency. Limited progress has been made in developing pharmacotherapies with acceptable specificity for the treatment of sleep-induced airway obstruction. We review three types of major long-term sequelae to severe OSA that have been assessed in humans through use of continuous positive airway pressure (CPAP) treatment and in animal models via long-term intermittent hypoxemia (IH): 1) cardiovascular. The evidence is strongest to support daytime systemic hypertension as a consequence of severe OSA, with less conclusive effects on pulmonary hypertension, stroke, coronary artery disease, and cardiac arrhythmias. The underlying mechanisms mediating hypertension include enhanced chemoreceptor sensitivity causing excessive daytime sympathetic vasoconstrictor activity, combined with overproduction of superoxide ion and inflammatory effects on resistance vessels. 2) Insulin sensitivity and homeostasis of glucose regulation are negatively impacted by both intermittent hypoxemia and sleep disruption, but whether these influences of OSA are sufficient, independent of obesity, to contribute significantly to the “metabolic syndrome” remains unsettled. 3) Neurocognitive effects include daytime sleepiness and impaired memory and concentration. These effects reflect hypoxic-induced “neural injury.” We discuss future research into understanding the pathophysiology of sleep apnea as a basis for uncovering newer forms of treatment of both the ventilatory disorder and its multiple sequelae.
I. Introduction
Sleep-disordered breathing refers to momentary, often cyclical, cessations in breathing rhythm (apneas) or momentary or sustained reductions in the breath amplitude (hypopneas), sufficient to cause significant arterial hypoxemia and hypercapnia. These apneas and hypopneas are specific to the sleeping state and are accompanied by 1) a compromised, often even completely closed, extrathoracic upper airway (“obstructive” event); 2) a marked reduction or cessation of brain stem respiratory motor output (“central” event); and 3) a combination of central and obstructive events. These ventilatory inadequacies and their accompanying intermittent hypoxemia often lead to transient arousals from sleep and sleep state fragmentation throughout the night and cause overcompensatory responses of the autonomic nervous system. This phenomenon is now known to occur with varying degrees of severity in literally millions of people throughout the world. In our review we first provide a brief historical perspective of the problem and then detail the pathogenesis and selected long-term consequences of sleep apnea. We have not attempted to cover in depth all of the important research problems in what has become a large field of scientific endeavor. Instead, we refer the interested reader to recent reviews on the epidemiology of sleep-disordered breathing (446, 759), the influence of race and ethnicity (79, 334, 729), sleep apnea in children (543, 670) and the “sudden infant death” syndrome (304, 674), and the neurocognitive effects of sleep apnea (43, 204).
II. History of Sleep Apnea
We have only really begun to study and to understand sleep apnea over the past 40 years, even though there were strong hints of the widespread existence of this problem as early as the 19th Century. Observations of periodic breathing in sleep were first reported in the mid 1850s, and in the 1870s British physicians reported on several cases of obstructed apneas as “fruitless contractions of the inspiratory and expiratory muscles against glottic obstruction with accompanying cyanosis during sleep” (346). During the later half of the 19th century, several cases of obese persons with extreme daytime sleepiness were described (346) and labeled the “Pickwickian syndrome” after Charles Dickens Fat Boy Joe as described in the Pickwick Papers in 1837 (132). Periodic breathing was reported by British physician Hunter and by Irish physicians Cheyne and Stokes in heart failure patients in the early to mid 19th Century (101, 346, 649) and in otherwise healthy subjects sleeping in the hypoxia of high altitudes by the British Physiologists John Scott Haldane, C. G. Douglas, and Mabel Fitzgerald at the turn of the 20th Century (137).
It was not until the mid 1950s that a link between obesity and the control of breathing was fully appreciated as the Pickwickian syndrome was rediscovered. Daytime CO2 retention was observed in obese subjects with daytime sleepiness and without significant lung disease (55). Remarkably, any association with sleep disorders was not considered. In fact, the daytime sleepiness in these patients was ascribed to “CO2 poisoning” accompanying their respiratory failure. Indeed, respiratory physiologists and neurophysiologists studying the control of breathing in these times never considered the extrathoracic upper airway as an important factor in this control system, and we knew little about its neuromuscular regulation. Furthermore, even descriptions of sleep effects on ventilation and ventilatory stability in health were not reported until the comprehensive studies of Bulow in the early 1960s (78). Finally, by the mid 1960s, Gastaut et al. (188) recognized obstructive sleep apnea in obese subjects as intermittent airway obstruction with frequent arousals, thereby providing the first comprehensive links between obesity, sleep-induced airway obstruction, sleep fragmentation, and daytime sleepiness. Following these key observations, research proceeded slowly with case reports of obstructive sleep apnea and the occasional use of chronic tracheostomy for treatment in the early 1970s (213, 375).
The findings of the mid to late 1970s through early 1980s provided a huge impetus to physiological research in this field of sleep and breathing, as highlighted by a series of reports of 1) sleep effects on brain stem respiratory neuronal activity in the unanesthetized, chronically instrumented cat (473, 474); 2) a neuromuscular reflex mechanism maintaining extrathoracic airway patency in the rabbit (74); 3) sleep effects on reflex control of breathing in the dog (514) and identification of a sensitive CO2-induced apneic threshold in sleeping humans (621); 4) description of anatomical and neurophysiological determinants of upper airway occlusion in the sleeping human, which provided a unifying “balance of forces” concept of obstructive sleep apnea (OSA) pathogenesis (549); and 5) the landmark introduction of continuous nasal pressure (CPAP) application as the noninvasive treatment for obstructive sleep apnea (654). Early in the 1990s, simulation of OSA in rodents using cyclic hypoxia was shown to cause a gradual development of daytime hypertension, thereby initiating research into the long-term cardiovascular consequences of sleep apnea (171). At this same time OSA patients were shown to maintain their upper airway patency in wakefulness via a compensatory, augmented EMG activity of their airway dilator muscles (405), which extended an earlier report of more frequently occurring genioglossus EMG activity during wakefulness [and non-rapid eye movement (NREM) sleep] in OSA patients (657). Shortly thereafter, the first population study conducted using in-lab studies of sleep and breathing showed a significant prevalence of sleep apnea or sleep-disordered breathing in a middle-aged, nonclinical population (the Wisconsin Sleep Cohort), and these findings signaled a potentially significant and largely undiagnosed effect of sleep-disordered breathing on public health (758).
From the mid 1990s to the present, we have seen an explosion of basic, clinical, and population research directed toward the prevalence, causes, consequences, and treatment of this long-standing, although only recently appreciated, problem. Sleep apnea has attracted a myriad of researchers from diverse disciplines and clinical subspecialties. At the same time, sleep apnea as a serious, undefined clinical problem has also given birth to many commercial ventures for its diagnosis and treatment, including the building of literally hundreds of sleep medicine clinics throughout the western world with the majority of their business concerned with the diagnosis and treatment of sleep apnea. Finally, given the relatively high prevalence of this sleep-specific problem with potential carryover to daytime pathology, sleep apnea has provided great impetus to the growth of sleep medicine as a clinical and research specialty.
III. Pathogenesis of Sleep Apnea
A. Wakefulness Influences on Ventilatory Control
Remarkably, sleep apnea patients experience little or no problems with their breathing or airway patency while awake. In fact, the great majority of people with sleep apnea possess ventilatory control systems that are capable of precise regulation of their alveolar ventilation and arterial blood gases with extremely small variations from the norm throughout the waking hours. In addition, these healthy control systems, while awake, possess sufficiently sensitive feedback and feedforward controls to ensure precise coordination of chest wall and upper airway “respiratory” muscle recruitment so as to provide maximum airway diameter, low airway resistance and optimum lung volumes and respiratory muscle lengths, regardless of the ventilatory requirement.
To underscore the importance of the “waking stimuli” to breathe and to upper airway patency and to ventilatory control, consider the following qualitative influences of sleep on the control of breathing.
Electrical activity from medullary inspiratory neurons, EMG activity of diaphragm and abductor muscles of the upper airway in healthy humans and/or in cats, show reductions in amplitude upon the transition from awake to NREM sleep, usually accompanied by a mild to moderate hypoventilation (+2 to 8 mmHg PaCO2) and two- to fivefold increases in upper airway resistance (128, 241, 369, 376). Sleep induces consistently greater proportional reductions in the EMG activity in the upper airway versus chest wall pump muscles (471).
A fast and highly variable breathing frequency is a hallmark of rapid eye movement (REM) sleep in mammals, even though postural muscles, including accessory respiratory muscles of the chest wall, are essentially atonic (513). So, an excitatory drive to breathe is common in REM coincident with increased diaphragmatic EMG activity and increased activity in many medullary respiratory neurons above those levels observed in NREM sleep or quiet wakefulness (467, 468, 470).
In cases of congenital central hypoventilation syndrome, the ventilatory response to imposed hypercapnia and to hypoxemia is absent; however, eupneic breathing rhythm is maintained while awake but lost completely in deep NREM or slow wave sleep (15, 160).
PaCO2 can be lowered substantially (using mechanical ventilation) during wakefulness with little or no disruption of breathing pattern; however, in NREM sleep, very small transient reductions in PaCO2 (even only to the waking level) result in significant apnea (239, 404, 621).
Partial ablation of the rat's pre-Bötzinger complex, a major site of respiratory rhythm generation in the medulla, is without effect on breathing pattern or chemoresponsiveness in wakefulness but is accompanied by apnea and ataxic breathing patterns in NREM and REM sleep (401). Furthermore, focal acidification of the retrotrapezoid nucleus, a major site of medullary chemoreception, produces a significant ventilatory response in wakefulness, with no response in sleep (353).
Added resistive or elastic loads to the airway prompt immediate and highly variable increases in the drive to breathe in the waking state, which prevent hypoventilation; however, in sleep, these mechanical loads are not accompanied by an immediate compensatory increase in the drive to breathe, and hypoventilation ensues until chemoreceptor stimuli increase (240, 261, 728).
Mechanical or chemical stimulation of the larynx or intrapulmonary airways causes cough in wakefulness but not in NREM or REM sleep (655), implying that acts requiring complex coordination of glottal, intercostal, diaphragm, abdominal, and tracheobronchial muscles require participation of supramedullary structures that are activated and synchronized while awake but not in NREM sleep.
Studies in chronically instrumented cats and rats have provided findings which demonstrate that the wakefulness stimuli to breathe include tonic excitatory inputs from the reticular formation, brain stem aminergic systems, and hypothalamic orexin-containing neurons (469). In NREM sleep, decrements occur in these excitatory inputs, and in REM sleep, there are both tonic excitatory inputs and phasic inhibitory inputs that account for irregularities in breathing pattern as well as the loss of excitation which contributes to hypotonia of the muscles of the upper airway (163, 259, 467–469, 472, 553, 641). Details concerning sleep effects on the neurochemical control of breathing and airway patency are provided in section iv.
In most healthy human subjects of any age, sleeping at low altitudes, the loss of these wakefulness influences on neurochemical control of breathing and airway patency is of minor physiological consequence. Mild CO2 retention and respiratory acidosis and reductions in alveolar Po2 are not accompanied by significant arterial O2 desaturation or a compromised systemic O2 transport (because of the high affinity of Hb for O2). Furthermore, while loss of upper airway muscle tonic activity results in a doubling or even sometimes a quadrupling of upper airway resistance and intrathoracic pressure swings in sleep in many healthy humans, there are usually only small, inconsequential effects on pulmonary gas exchange, sleep state continuity, autonomic regulation, or ventricular function. However, for many otherwise healthy subjects in whom airway patency is already anatomically compromised in wakefulness and/or whose ventilatory control systems are inappropriately driven by chemical stimuli, simply loss of wakefulness inputs to the control of the upper airway and chest wall muscle motor neurons produces serious, short- and long-term consequences to homeostasis and to health. We shall now discuss the mechanisms contributing to the complex pathogenesis of sleep-disordered breathing.
B. Defining Sleep-Disordered Breathing Events and a Roadmap for Pathogenesis
Sleep-disordered breathing leading to repeated bouts of ventilatory overshoots and undershoots and accompanying swings in arterial blood gases and intrathoracic pressure takes on many forms. Commonly, sleep-disordered breathing is divided into so-called “central” events, denoting an absence or marked reduction in central respiratory motor output to respiratory pump muscles, or “obstructive” events, which are comprised of respiratory efforts against a closed upper airway. However, as we discuss below, most cyclical sleep-disordered breathing events are driven by anomalies in both anatomical and neurochemical control of upper airway and/or chest wall respiratory musculature. Four patterns of sleep-disordered breathing are illustrated in Figure 1, including the waxing and waning of ventilatory responses in the severe heart failure patient (see Fig. 1A), the “cluster” periodic breathing of healthy sea-level natives during sleep in the hypoxia of high altitude (Fig. 1B), and the obstructive and “central” apneas coexisting in an OSA patient during sleep (Fig. 1C). These examples represent the extremes of a broad continuum of sleep-disordered breathing that also includes airway narrowing, rather than complete airway obstruction and periods of transient hypoventilation or “hypopnea” rather than complete apnea.
Fig. 1.
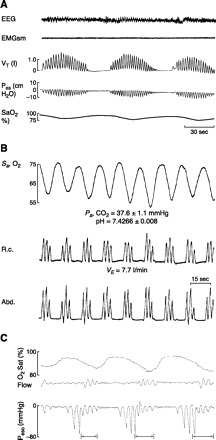
A: periodic (Cheyne-Stokes) breathing in chronic heart failure in non-REM sleep. Note the gradual crescendo and decrescendo of tidal volume (Vt) and esophageal pressure (Pes), the intermittent hypoxemia (SaO2), and the subtle changes in EEG amplitude attending the termination of each periodic breathing cycle. Periodic cycles of apnea plus hyperpnea are fairly uniform and are each 50–60 s in duration. [From Tkacova et al. (681).] B: periodic “cluster-type” breathing in non-REM sleep in a healthy sea-level native during the initial night at 4,300 m altitude. Tidal volume is estimated from expansion of the ribcage (RC) and abdomen (Abd) using inductance plethysmography. Note the abrupt increase in Vt (to 1.5–2.5 times control, steady-state values) at the end of each apneic period. Each periodic cycle is 20–25 s in length. Also note the mild levels of arterial hypocapnia and alkaline pH determined from blood sampling over several periodic cycles. [From Berssenbrugge et al. (53).] C: cyclical “mixed,” i.e., central followed by obstructed apneas, causing intermittent hypoxemia during non-REM sleep. The cessation of airflow denotes the onset of apnea. The absence of cyclical changes in esophageal pressure over the initial 8–10 s of the apnea demonstrates that this initial phase of the apnea is due to the absence of “central” respiratory motor output and inspiratory muscle contractions. Over the latter half of the apnea, flow is still absent but progressive and cyclical increments occur in the negativity of esophageal pressure, indicating increasing inspiratory efforts against a closed airway in response to rising asphyxic chemoreceptor stimuli. The arrows shown at the termination of each apneic period indicate periods of transient cortical arousal.
Population-based cross-sectional and longitudinal studies have specified the dominant risk factors for OSA in the general (nonclinical) population to include excess body weight as the dominant contributor, followed by male gender, and to significant but lesser extents, cranial facial structures and aging (129, 488, 501, 562, 757–759).
We will concentrate on the more common affliction of OSA. In short, the process is initiated because the wakeful state provides compensatory neuronal activation of dilator muscles in an anatomically compromised collapsible pharynx; accordingly, when this activation is lost at sleep onset, the airway narrows and/or collapses. However, the tendency to result (or not to result) in repeated cyclical apneas is the end product of multiple compensatory processes that vary markedly among and within individuals. Concepts have continued to evolve as we learn more about the neurophysiological mechanisms governing control of respiratory rhythm and its coupling with upper airway control and states of consciousness and applying these principles to human patients during sleep.
We discuss OSA pathogenesis in three steps, as outlined in Figure 2. First we detail the varied structural and functional determinants of an anatomical predisposition for airway closure, an absolutely essential component for OSA. The second essential component is sleep. This section emphasizes the effects of the sleeping state on mechanisms underlying both obstructive and central apnea and ventilatory instability. Finally, we attempt to integrate anatomical deficits with mechanisms underlying central neurochemical control of breathing stability and compensatory neuromuscular control of upper airway caliber, to explain the cyclical, repetitive nature of OSA.
Fig. 2.
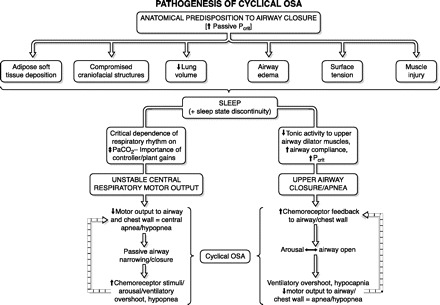
Road map for the discussion of pathogenesis of cyclical obstructive sleep apnea.
C. Anatomical Determinants of Upper Airway Caliber in OSA
1. Unique anatomy of the human airway
The upper airway is a complex structure required to perform deglutation, vocalization, and respiration. In the human, this structure must also perform tightly controlled and complex motor behaviors required for speech. Upper airway obstruction in sleep is most prevalent in the human in part because the hyoid bone, a key anchoring site for pharyngeal dilator muscles, is not rigidly attached to skeletal structures. In other mammals, the hyoid bone is attached to the styloid processes of the skull (425, 756). Thus the human pharynx has no rigid support except at its extreme upper and lower ends where it is anchored to bone (upper) and cartilage (larynx); therefore, pharyngeal cross-sectional area will vary with lumen pressure (271). Humans depend critically on the coordinated actions and interactions of over 20 skeletal muscles that dilate and stent open the oropharynx (see sect. ivC).
Beyond the hyoid arch, Lieberman et al. (361, 362) and Davidson (117) also point to the anatomical changes in the adult human upper airway during the evolutionary development of speech as a potential major contributor to OSA. Specifically, the gradual decent of the larynx to a position greatly inferior to the oropharynx separated the soft palate from the epiglottis. The creation of this “supralaryngeal vocal tract,” like the tube of a clarinet, “filters” the sound produced by the larynx and in turn speech is produced via the changing position of the pharynx, tongue, and lips. The downside is a relatively shortened, compacted face and greatly narrowed oropharynx in which the tongue encroaches significantly on the available space.
2. Sites of airway collapse
Studies using nasal pharyngoscopy, computer tomography and magnetic resonance imaging, or pharyngeal pressure monitoring have shown that one or more sites within the oral pharayngeal region are usually where closure occurs in most subjects with OSA, and this region is also smaller in OSA patients versus controls even during wakefulness (see Fig. 3A) (253, 426, 597, 599). Although the retropalatal region of the oropharynx is the most common site of collapse (see Fig. 3B), airway narrowing is a dynamic process, varying markedly among and within subjects and often includes the retroglossal and hypopharyngeal areas (255, 430, 444). For example, Watanabee et al. (710) have shown that airway closure in obese OSA subjects occurred primarily at the velopharynx, whereas in nonobese OSA patients with a recessed mandible, the closure occurred at both the velo- and oropharynx.
Fig. 3.
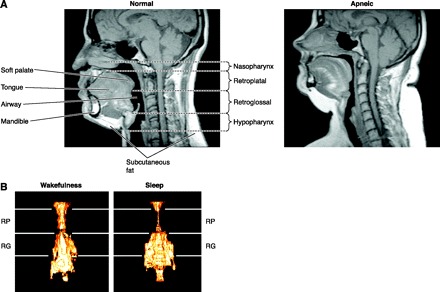
A: midsagittal magnetic resonance image (MRI) in a normal subject (left) and in a patient with severe OSA (right). Highlighted are the four upper airway regions (nasopharynx, retropalatal region, retroglossal region, hypopharynx) and upper airway soft tissue (soft palate, tongue, fat) and craniofacial structures (mandible). Fat deposits are shown in white on the MRI. Note that in the apneic patient: a) the upper airway is smaller, in both the retropalatal and retroglossal region; b) the soft palate is longer and tongue size is larger; and c) the quantity of subcutaneous fat is greater. [From Schwab et al. (597).] B: state dependence of upper airway size in a normal subject as assessed via three-dimensional reconstructions of MRI images. Images represent averages taken over several respiratory cycles during eupneic breathing in sleep and wakefulness. Airway volume during NREM sleep is smaller in the retropalatal (RP) region, not in the retroglossal (RG) region. Such images show the marked effect of sleep, per se, on the loss of upper airway muscle dilator tone and also show that the upper airway does not narrow as a homogeneous tube during sleep. [From Trudo et al. (687).]
3. Soft tissue and bony structure abnormalities
The recent use of quantitative imaging techniques has allowed advances that reveal important differences in both craniofacial and upper airway soft tissue structures in the OSA patient. The reduced size of cranial bony structures in the OSA patient include a reduced mandibular body length, inferior positioned hyoid bone, and retro position of the maxilla, all of which compromise the pharyngeal airspace (21, 556, 557). Airway length, from the top of the hard palate to the base of the epiglottis, is also increased in OSA patients, perhaps reflecting the increased proportion of collapsible airway exposed to collapsing pressures (385, 479). As expected, these craniofacial dimensions are primarily inherited, as the relatives of OSA patients demonstrated retroposed and short mandibles and inferiorly placed hyoid bones, longer soft palates, wider uvulas, and higher narrower hard palates than matched controls (214, 396).
Enlargement of soft tissue structures both within and surrounding the airway contributes significantly to pharyngeal airway narrowing in most cases of OSA. An enlarged soft palate and tongue would encroach on airway diameter in the anterior-posterior plane (107, 254), while the thickened pharyngeal walls would encroach in the lateral plane. Volumetric time overlapped magnetic resonance imaging (MRI) or computer tomography (CT) images strongly implicate the thickness of the lateral pharyngeal walls as a major site of airway compromise, as the airway is narrowed primarily in the lateral dimension in the majority of OSA patients (505, 597). Furthermore, treatment with CPAP, weight loss, or mandibular advancement all show increases in the lateral pharyngeal dimensions (561, 595, 597).
There are many potential causes of lateral wall thickening in OSA patients. First, as shown in both humans and rodent models, obesity is a major contributor to airway compression through increased area and volume of pharyngeal fat deposits (69, 253, 606, 610). This excess fat deposition has also been observed under the mandible and within the tongue, soft palate, or uvula (645). Obesity also gives rise to excess fat-free muscular tissue, thereby increasing the size of many upper airway structures (65, 253, 600, 606) and compressing the lateral airway walls. In children with OSA, tonsillar and adenoid hypertrophy form the major anatomical contributors to airway narrowing (388).
4. Obesity and lung volume
Obesity also contributes indirectly to upper airway narrowing, especially in the hypotonic airway present during sleep, because lung volumes are markedly reduced by a combination of increased abdominal fat mass and the recumbent posture. In turn, the reduced lung volume reduces the “tug” on the trachea induced by the traction exerted via mediastinal structures by negative intrathoracic pressures and by the diaphragm descent, thereby further increasing the thickness of the lateral pharyngeal walls and narrowing the airway.
The highly sensitive “traction” effect of changes in lung volume on upper airway patency and airway resistance was clearly demonstrated in anesthetized animals by Van de Graaff who surgically disconnected the mechanical linkage between chest wall and upper airway by severing all cervical structures anterior and anterolateral to the spine (690). While intact, upper airway resistance (Rua) fell during inspiration, whereas following removal of the mediastinal-tracheal linkages Rua was increased and no longer underwent respiratory modulation. Lung volume effects on Rua are also mediated in part via pharyngeal dilator muscle activity; however, these lung volume effects persist even following denervation of airway muscles in the dog (690) or during muscle paralysis in the human (663). Also with humans sleeping in an iron-lung respirator, variations in box pressure surrounding the chest and abdomen produced small changes in end-expiratory lung volume (EELV), which in turn produced highly sensitive effects on upper airway patency and resistance (45, 235) and on airway closing pressures (see sect. iiiD), even in the paralyzed patient (663). The greater the upper airway collapsibility and airway resistance in these sleeping subjects, the more the airway resistance was reduced as EELV was increased (605, 607, 644). Furthermore, based on endoscopic imaging studies, lung volume effects on airway collapsibility were shown to be more pronounced at the level of the velopharynx versus oropharynx and in obese versus nonobese subjects (663). This sensitive, purely mechanical effect of lung volume changes on passive control of upper airway patency and resistance has not been fully appreciated to date; indeed, this effect may explain at least some of the effect of obesity, sleep itself, and even CPAP treatment on upper airway caliber and collapsibility (605). Finally, the reduced EELV in obese subjects, especially in the recumbent posture, together with increased tissue O2 consumption rates, means that lung O2 stores are more quickly depleted during an apnea resulting in more severe arterial O2 desaturation for any given apneic length (165).
5. Airway edema and surface tension
Accumulation of even relatively small amounts (∼100–200 ml) of edematous fluid enlarges upper airway soft tissue structures in OSA patients and snorers, especially in the soft palate which may be tugged caudally and constricted during apneas (596). Local vascular engorgement may also enlarge soft tissues in the upper airway (709). Furthermore, cephalad displacement of fluid from the lower extremities to the upper airway upon assuming recumbency has been recently documented and associated with sleep-disordered breathing (105, 544, 653).
Surface tension of the liquid lining the mucosa affects collapsibility of the upper airway in the same way as it has been well documented in the lung's airways. A higher surface tension in the upper airway wall of OSA patients has been reported using a method that quantifies surface tension as the force required to separate two surfaces bridged by a droplet of the liquid under study (305, 306). Furthermore, in limited studies, surfactant therapy in OSA patients was shown to significantly reduce airway collapsibility (602) and improve apnea hypopnea index (AHI) by 20–30% (283, 305, 428).
6. Obesity, leptin, and inflammation
Central, or visceral, obesity is associated with the greatest risk for OSA (611). This suggests that factors other than pure mechanical load may contribute to the pathogenesis of respiratory disturbances during sleep. The concept is now emerging that visceral fat depots, which represent a rich source of humoral mediators and inflammatory cytokines, can impact on neural pathways associated with respiratory control (601). Perhaps the most well-studied adipocyte-derived factor affecting respiratory control is leptin, which was initially determined to have a primary role of binding to receptors in the hypothalamus to reduce satiety and increase metabolism (178). Leptin can also act as a respiratory stimulant, and impairment of the leptin signaling pathway, as occurs in leptin-resistant or leptin-deficient states of obesity, causes respiratory depression in mice (453) and is associated with obesity hypoventilation syndrome in humans (515). Even though obesity and OSA are associated with elevated circulating levels of leptin, if centers in the brain impacting on respiratory control act in a similar leptin-resistant manner to hypothalamic regions controlling appetite and metabolism, then impaired leptin signaling in the CNS may contribute to respiratory depression as predicted in murine studies.
In addition to respiratory control, animal studies show leptin is also critical in lung development and affects the distribution of muscle fiber types in the diaphragm (667). However, as yet there is no direct evidence that impaired leptin signaling can impact on the control of respiratory muscles of the upper airway, although it may play a role in nocturnal hypoventilation, particularly in REM sleep where respiration is markedly depressed in leptin-deficient mice (453). Visceral adipose tissue releases many other humoral factors including classical proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin (IL)-6 that are elevated in OSA and can be reduced with CPAP therapy (409, 749). These and other proinflammatory cytokines may impact on sleep (613) or potentially contribute to the local inflammatory response reported in the upper airway tissues of OSA patients (64), but there is little evidence of an effect on respiratory control. Overall, the concept that adipocyte-derived circulating factors can impact on respiratory control of the upper airway or act on the upper airway directly to contribute to the pathogenesis of OSA is intriguing, but currently lacking clear supporting data.
Finally, in our emphasis on the importance of obesity both independently and in its interactions with gender, age, lung volume, and cranial facial dimensions, we need to acknowledge the accumulation of evidence from genome-wide linkage studies of OSA phenotypes (481, 482) pointing to a strong heritability of both AHI (∼33%) and body mass index (BMI) (>50%), with about one-half of the genetic determinants of AHI being obesity related and one-half being independent of obesity (489, 760). A heritable predisposition to this disorder is also suggested by its higher prevalence in males versus females and in African ancestors and East Asians compared with Western populations (269, 543, 758). The wide array of candidate genes that might link genetic mechanisms of obesity with sleep apnea are under investigation and have been recently reviewed (489, 760).
D. Mechanical Determinants of Upper Airway Patency
Mechanical determinants of airway caliber of the human pharynx in sleep are similar to those regulating caliber of any collapsible tube (598). Other well-known biological examples in respiratory physiology include intrathoracic airway collapse upon forced exhalation (531), collapse of pulmonary capillaries in the lung apex (718), and collapse of alae nase at high inspiratory flow rates (70). A Starling resistor model developed by Schwartz and colleagues (603, 628) consists of a collapsible tube with a sealed box interposed between two rigid segments (195) (see Fig. 4). The critical closing pressure (Pcrit) of the passive airway is defined as the pressure inside the airway (Pin) at which the airway collapses. The pressure gradient during airflow through the system is defined by Pupstream − Pcrit and remains independent of Pdownstream. Therefore, with increasing Pcrit, as the differential between Pupstream and Pcrit decreases, inspiratory airflow limitation will eventually develop, and when the Pus falls below Pcrit, complete airway occlusion occurs. Effective therapy for sleep apnea requires that the Pus to Pcrit pressure differential be widened, and this can be accomplished by either 1) an increase in Pus with appropriate amounts of CPAP applied at the airway opening, or 2) by decreasing Pcrit via either reducing the collapsing pressures on the airway (e.g., weight loss or alteration of cranial-facial anatomy or increasing lung volume) or by augmenting “active” neuromuscular control of airway tone (see sect. iiiE) (195, 492).
Fig. 4.
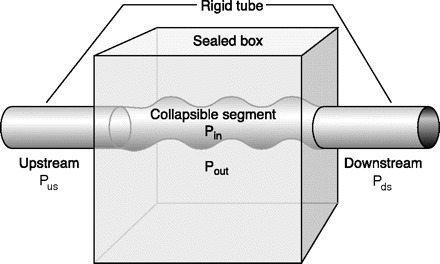
Starling resistor model of obstructive sleep apnea. In the Starling resistor model, the collapsible segment of the tube is bound by an upstream and downstream segment with corresponding upstream pressure (Pus), downstream pressure (Pds), and upstream resistance and downstream resistance. Airway occlusion occurs when the surrounding tissue pressure (Pout), (comprised of pharyngeal muscles and pharyngeal and submucosal fat, mucosal edema, etc.; see sect. iiiC), becomes greater than the intraluminal pressure (Pin), resulting in a transmural pressure of zero. In this model of the upper airway, Pus is atmospheric at the airway opening, and Pds is the tracheal pressure. The critical closing pressure of the collapsible airway (Pcrit) is represented by Pin. When the Pcrit is significantly lower than Pus and Pds, flow through the tube occurs. When Pds falls during inspiration below Pcrit, inspiratory airflow limitation occurs and is independent of further decreases in Pds. Under this condition, the pharynx is in a state of partial collapse, and maximal inspiratory airflow varies linearly as a function of the difference between Pus and Pcrit. Finally, when Pus falls below Pcrit, the upper airway is completely occluded. [Adapted from Gold and Schwartz (195).]
Airway Pcrit due solely to mechanical properties of the airway and its surrounding tissue, termed “passive Pcrit,” has been assessed during sleep and under conditions of complete muscle atonicity in paralyzed, anesthetized patients through the use of pressure control systems connected to nasal masks that are capable of manipulating airway pressure in a stepwise fashion across a wide range (±20 cmH2O) (195, 271, 272, 492). In general, these measures in sleeping or paralyzed humans have shown that passive Pcrit is in the range of <−10 cmH2O in normal subjects with low airway resistance and minimal CO2 retention during NREM sleep, from −10 to −5 cmH2O in snorers, −5 to 0 cmH2O in those with sufficiently high airway resistance to induce airflow limitation and transient hypopneas, and >0 cmH2O in patients with apneas with complete airway obstruction.
Passive Pcrit is significantly associated with the two greatest risk factors for OSA, namely, obesity (see sect. iiiC) and gender. Recent studies using substantial numbers of males and females matched for BMI and age have documented a substantially greater 2–3 cmH2O elevation in passive Pcrit (during NREM sleep) in men versus premenopausal women (285, 307). This gender difference in passive airway collapsibility may be related to the longer pharyngeal airway length and in the mass of soft tissue contained in the soft palate and tongue in males (385, 722). In turn, this difference in pharyngeal soft tissue deposition may be secondary to the tendency toward fat deposition in the upper body and trunk in males versus lower body and extremities in females (407).
E. Neuromuscular Control of Upper Airway Dynamics in Sleep
Clearly then the effects of airway anatomy on airway collapsing pressure in a hypotonic airway are a critical determinant of obstructive apnea. However, several lines of evidence also support neuromuscular factors as significant determinants of airway collapsibility in sleep. First, tonic and phasic EMG activity of pharyngeal airway dilator muscles (genioglossis and tensor palatine) are progressively reduced from wakefulness to NREM to REM sleep and further inhibited coincident with the “phasic” eye movement events in REM. This powerful effect of state has been adequately documented in tracheostomized animal models (374) and recently has been demonstrated in OSA patients in whom the potentially confounding, compensatory responses to sleep-induced changes in upper airway resistance, negative pressure, PaCO2, and respiratory motor output were controlled through the use of either CPAP (148) or positive pressure controlled mechanical ventilation (369). These state effects on the neuromuscular control of the upper airway likely explain, along with reductions in lung volume (see sect. iiiC), why Pcrit is never positive in the waking state, even in OSA patients. Furthermore, genioglossus EMG activity is abnormally high in awake OSA patients (405, 657), and its sleep-induced decrement may be viewed as playing a “permissive” role in explaining (or unmasking) closure of an already anatomically compromised upper airway (see sect. ivD for neurochemical mechanisms underlying these state effects).
Second, neuromuscular factors also play a significant role in the dynamic breath-to-breath and intrabreath regulation of upper airway caliber, through changes in proprioceptive and chemoreceptor feedback. During inspiration, the passive pharynx narrows as intraluminal pressure is progressively reduced because of energy lost in overcoming frictional airway resistance and increases in flow velocity secondary to the Bernoulli effect operating in a reduced lumen size (598). This collapsing effect of a reduced luminal pressure is opposed during inspiration by a reduction in dynamic compliance, i.e., collapsibility, of the airway achieved via reflex activation of pharyngeal dilator muscles. A manifestation of this dilator muscle recruitment is reflected in the markedly higher active Pcrit obtained in OSA patients when they breathe through a tracheotomy versus nasal breathing, pointing to the significant activation of upper airway muscles during inspiration through the intact upper airway (384, 591). In turn, the reflex activation occurs in response to negative pressure airway mechanoreceptors located principally in the larynx and to a lesser extent in the superficial layers of the pharyngeal wall, with their afferent projections located in the superior laryngeal nerve, and also in glossopharyngeal and trigeminal nerves (250, 260, 323, 394, 395, 579, 591, 691). Large changes in negative pressure in the isolated upper airway trigger a dual protective reflex, which restores airway patency by both activating airway dilators (to reduce airway compliance) while inhibiting diaphragm EMG activity (which minimizes intraluminal negative pressure) (224, 394). Vagally mediated feedback influences on laryngeal, tongue, and hyoid muscle via pulmonary stretch receptors also protects against airway collapse as the rate of lung inflation is slowed in the face of increased airway resistance, thereby reflexly activating upper airway motor neurons (323). Finally, chemoreceptor influences also have substantial effects on upper airway muscle recruitment, and in the case of CO2, upper airway motor neurons relative to phrenic motor neurons have been shown to have a substantially higher threshold for inhibition (via hypocapnia) and activation (via hypercapnia) (466, 711).
Dynamic imaging of upper airway caliber as well as breath by breath analysis of airway mechanics during sleep shows that narrowing/closure may occur at end-expiration or during inspiration (25, 426, 574, 575, 658), each of these airway occurrences suggesting quite different mechanisms precipitating collapse. End-expiratory occlusion occurs without the need for generating an inspiratory effort or negative intraluminal pressure and may reflect that at end expiration the airway is no longer held open by phasic inspiratory activation of upper airway dilator muscles or by positive intraluminal pressure (25, 426, 595, 596). On the other hand, closure during inspiration points to an imbalance between the generation of upper airway muscle dilating forces versus an excessive intraluminal negative pressure generated by inspiratory chest wall muscles (549, 598, 658). Circumstances that might favor expiratory over inspiratory phase airway closure have not been thoroughly investigated, although limited data in the anesthetized obese mouse (69) and British bulldog (696) suggest that with anatomically compromised airways, expiratory phase narrowing and inspiratory phase (active) dilation are common in obesity, and the opposite effects occur in the airways of normal, nonobese control animals. Most evidence appears to support a passive closure of the upper airway during expiration as the dominant occurrence in OSA.
In summary, the evidence to date supports important roles for both anatomical and neural control of dilator muscles to the regulation of upper airway caliber in the sleeping human. The relative contributions of these factors will vary widely among and within individuals with, for example, patterns of fat deposition on the one hand and neurochemical sensitivity for dilator muscle recruitment on the other. Important indirect influences on airway caliber may also occur through instabilities in respiratory motor output, as we now discuss below.
F. Instability of Central Respiratory Motor Output and Breathing Pattern in Sleep
1. Unmasking a sensitive apneic threshold
Central apneas and instabilities in humans occur primarily in NREM sleep because of the critical dependence of the ventilatory control system on chemical stimuli, principally PaCO2, in this state. Thus when normal subjects are mechanically ventilated in NREM sleep, transiently induced increases in tidal volume and small reductions in PaCO2 only to waking levels (−3 to 5 mmHg) are sufficient to induce central apnea (239, 404, 621, 677) (see Fig. 5). A similar sensitive hypocapnic-induced apneic threshold can be demonstrated in sleeping, tracheostomized OSA patients (262) or dogs (106) by causing brief airway occlusions which in turn cause chemoreceptor (and often arousal)-driven transient ventilatory overshoots upon termination of the occlusion with subsequent central apneas (upon resumption of sleep). So apnea occurs whether the transient hyperventilation and hypocapnia are produced via “active” or “passive” means. Normally, in wakefulness, a transient increase in central respiratory motor output and hyperventilation are not followed by apneas, because a centrally mediated short-term potentiation of central respiratory motor output lingers following cessation of the ventilatory stimulus and ventilation returns gradually to its control eupneic levels (22, 152). However, in sleep, this stabilizing influence is apparently overridden by a transient reduction in the CO2 drive to breathe, i.e., a sensitive apneic threshold is unmasked. Further evidence for a pivotal role for hypocapnia is the consistent evidence that the prevention of central apnea and periodic breathing during sleep in heart failure (734) or in hypoxic environments (53) is readily achieved by the addition of even very small amounts of inspired Pco2, i.e., just sufficient to prevent the occurrence of transient hypocapnia, regardless of the magnitude of any ventilatory overshoot.
Fig. 5.
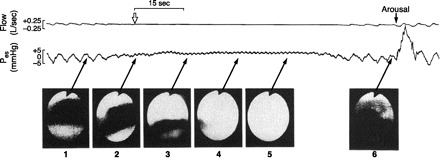
Effects of spontaneous central apnea on upper airway patency during NREM sleep. Fiber optic nasopharyngoscopy was used to determine airway dimensions at the level of the velo- or oropharynx. The initiation of central apnea is identified by the open inverted arrow, with the cessation of both airflow and oscillation of esophageal pressure (Pes). Complete airway occlusion occurred ∼10 s following the onset of central apnea and before an inspiratory effort occurred, as noted by the constant Pes. Central apnea continued and the airway remained closed for 35 s, showing partial return of airflow with resumption of inspiratory effort and then complete airway patency on arousal from sleep with an accompanying ventilatory overshoot. [From Badr et al. (25).]
Of course to reach the apneic threshold we need a source of transient ventilatory overshoots. Transient arousals from sleep provide a common source of this transient extra-drive to breathe, and the accompanying reductions in upper airway resistance permit a greater hyperventilatory manifestation of this increased drive (252, 743, 753). Arousals are especially effective in causing ventilatory overshoots when combined with increasing chemoreceptor drives from the preceding ventilatory undershoot (106, 262). Furthermore, the PaCO2 required to terminate apnea is estimated to be 1–4 mmHg higher than the threshold PaCO2 needed to initiate the apnea, reflecting a so-called control system “inertia” which delays the resumption of breathing rhythm (347, 581). The resultant apnea prolongation presents an enhanced chemoreceptor stimulus to ventilatory overshoot at apnea termination. While the hypocapnic-induced apnea threshold is highly sensitive and reproducible in NREM sleep, there is no apparent threshold in phasic REM sleep even with marked reductions in PetCO2 produced by either mechanical ventilation or by ventilatory overshoots in response to experimental airway occlusion (731). The central apneas and the periodic breathing accompanying heart failure or high-altitude hypoxia are also rarely present in REM sleep (53, 221). Perhaps, analogous to the wakeful state, the erratic, sporadic increases in central inspiratory neural drive during REM (467) override hypocapnic inhibition.
Although hypocapnia is required during the ventilatory overshoot to cause subsequent apnea, lung stretch (732) and/or increases in systemic blood pressure and baroreceptor stimulation (582, 726) accompanying the ventilatory overshoot may also contribute to the ventilatory depression following the overshoot. Several reflex effects of airway negative pressure-sensitive mechanoreceptors on ventilatory control have also been demonstrated in the sleeping canine with an isolated upper airway. These include 1) the inhibitory effects of flow through the upper airway, or lung inflation on the rate of rise of diaphragmatic EMG activity; and 2) the apnea caused by either negative pressure pulses or low-pressure high-frequency pressure oscillations (akin to human snoring) applied to the airway during early expiration (147, 224, 520).
2. Sites of chemoreception causing apnea and instability
At what chemoreceptor site is transient hypocapnia acting in causing central apnea and periodic breathing? Carotid body denervation studies in the sleeping animal showed that neither apnea nor periodic breathing could be elicited even when substantial levels of transient hypocapnia were produced via mechanical ventilation (437). Similarly, using an isolated perfused carotid body preparation in the neurally intact sleeping canine (624, 626), central chemoreceptor hypocapnia (alone) was shown to produce little prolongation of expiratory time (TE) despite marked reductions in PaCO2 (−8 to 15 mmHg), whereas transient hypocapnia of only 3–5 mmHg produced apnea and periodic breathing when both chemoreceptors were able to sense the hypocapnia.
These data are strongly supportive of peripheral over central chemoreceptor in eliciting hypocapnic-induced apneas. On the other hand, we also know that perfusion of the isolated carotid chemoreceptor alone with severely hypocapnic (or hyperoxic) blood will reduce tidal volume (VT) but not cause apnea (627). Furthermore, specific increases in brain extracellular fluid (ECF) [H+] cause substantial increases in ventilation (161, 441, 626, 666). Accordingly, these apparent contradictions point to a new perspective put forward primarily by Guyenet (215) which emphasizes the potential importance of interdependence between peripheral and central chemoreceptors in ventilatory control.
Neuroanatomical evidence in the rodent model shows that Phox 2b gene expression delineates an unbroken chain of neurons in a circuit which includes carotid bodies and their afferent projections (116), chemoreceptor projections of the nucleus tractus solitarius (NTS) to the ventral lateral medulla (VLM) (666), and to CO2 sensitive chemoreceptor neurons in the retrotrapezoid nucleus (RTN) (650). Functionally, an important interdependence between the various chemo- and mechanoreceptor afferents in the respiratory control system has also been demonstrated in reduced preparations by 1) the marked effect of systemic hypoxia increasing the activity of central CO2-sensitive neurons in the RTN, an effect which was subsequently eliminated via carotid body denervation (650, 666); 2) the powerful hypoadditive effects of varying levels of Pco2 in the isolated perfused medulla on the ventilatory response to specific carotid body stimulation in the decerebrate vagotomized rat (121); and 3) the effect of vagal inhibition via lung stretch on carotid chemoreceptor (30) and medullary chemoreceptor responsiveness (215).
Further advances in our understanding of the chemical control of breathing requires that we no longer view the peripheral and central chemosensors as “stand alone” receptors, responding only to changes in the ionic composition of their immediate environment. Rather, we need to determine whether these proposed interdependencies are additive, hypoadditive, or hyperadditive in their influences on the final respiratory motor output (to both upper airway and chest wall) under conditions of transient and steady-state changes in systemic chemical stimuli and during wakefulness and sleep in neurally intact, fully responsive animal models.
3. Variations in susceptibility to central apnea and ventilatory instability
The occurrence of central apneas with repeated cyclic periods of over- and underventilation during sleep varies markedly depending on the gains(s) of the respiratory control system and the stability of the sleeping state. The tendency toward instability depends on the respiratory control system's “loop gain,” an engineering term which defines the “gain” of the negative-feedback loop which regulates ventilation in response to a ventilatory disturbance (302). For example (see Fig. 6), if the magnitude of the increase in ventilation is greater than or equal to the magnitude of the preceding apnea or hypopnea, i.e., a high loop gain, then the system is highly unstable and will fluctuate between under- and overventilation (715).
Fig. 6.
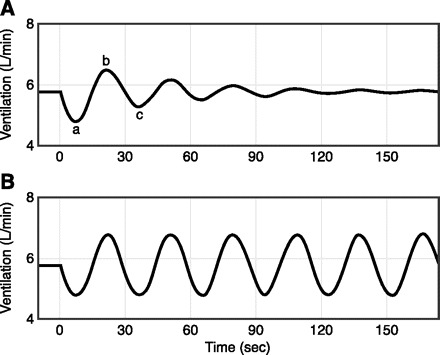
Loop gain (LG) depicts the ratio of ventilatory response to disturbance ratio. A: example of a LG of 0.72. The ventilatory control system is disturbed with a transient reduction in ventilation (a). This produces a response (b) in the opposite direction that is 72% as large as the disturbance. The next response (c) will also be 72% as large as b, etc. Thus a LG of 0.72 produces transient fluctuations in ventilation, but ventilation eventually returns to baseline. B: a LG ≥1 will produce a response that is equal or greater in magnitude to the disturbance. Ventilation, therefore, oscillates without returning to baseline. The system in B is highly unstable. The closer LG is to zero, the smaller the fluctuations in ventilation, and thus the more stable the system (Fig. 7 illustrates how the magnitude of ventilatory overshoots and undershoots, i.e., stability, are determined by two key components of loop gain, namely, controller and plant gains). [From Wellman et al. (715).]
Two types of control system gain, controller gain and plant gain, are major determinants of loop gain and therefore ventilatory stability (99, 300, 302). We illustrate the effects of changing each of these gains on CO2 responsiveness above and below eupnea in Figure 7, which in turn will determine the tendency of ventilatory drive to overshoot in response to a rising chemoreceptor stimulus or to be overly depressed in response to the ensuing hypocapnia (127). For example, Figure 7A illustrates the effect of changing the background drive to breathe which will displace the eupneic PaCO2 along the isometabolic line defining the hyperbolic relationship of PaCO2 to alveolar ventilation. This hyperventilation, per se, protects against apnea and ventilatory instability by requiring a larger additional transient hyperventilation and hypocapnia to reach the apneic threshold (i.e., decreased plant gain), whereas a reduced drive and hypoventilation make one highly susceptible to apnea, requiring only very small further transient ventilatory overshoots (increased plant gain) (436). The other means of changing the magnitude of the CO2 reserve below eupnea is to change the slope of the change in ventilation above and below eupnea, respectively, in response to induced hyper- or hypocapnia (see Fig. 7B, bottom). For example, an increased CO2 response slope above and below eupnea (increased controller gain) has been observed in hypoxic humans and dogs and in chronic heart failure patients who experience periodic breathing in sleep (436, 737, 739, 742). This increased slope of response to changes in PaCO2 results in a reduction of the CO2 reserve and an increased susceptibility to apnea and periodicity despite the background hyperventilation, reduced eupneic PaCO2 and plant gain (127). Thus the magnitude of loop gain and of the CO2 reserve and therefore the propensity for ventilatory instability in sleep is determined by the net effects of any changes or abnormalities in controller gain vs. plant gain.
Fig. 7.
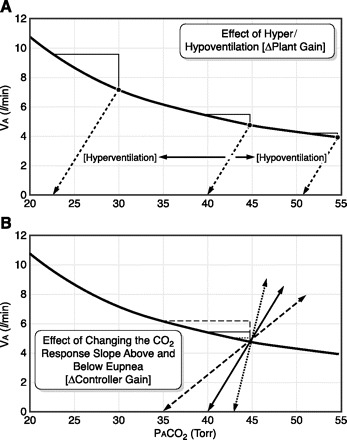
Diagrammatic representation of the relationship between alveolar ventilation (VA) and alveolar Pco2 (PaCO2) at a fixed resting CO2 production (of 250 ml/min); PaCO2 = V̇co2/ V̇A × K. The schematic figure shows how changing plant gain (A, top) or controller gain (B, bottom) will influence the “CO2 reserve” or ΔPaCO2 between eupnea and apnea. A: changing the background drive to breathe without changing the slope of the ΔVA vs. ΔPaCO2 relationship above or below eupnea. For example, background hyperventilation raises VA and lowers PaCO2 along the isometabolic hyperbola. This means that a greater transient increase in VA and reduction in PaCO2 is required to reach the apneic threshold than it would be under control, normocapnic conditions. The reverse is true for conditions which reduce the background drive to breathe and cause hypoventilation. B: at any given level of background PaCO2, changing the slope (or responsiveness) of the ΔVA-ΔPaCO2 relationship below eupnea would alter the CO2 reserve or the amount of reduction in PaCO2 required to cause apnea. Changing the slope of the ventilatory response to CO2 above eupnea would alter the susceptibility for transient ventilatory overshoots. See text for a discussion of conditions which change controller and plant gain and therefore the susceptibility to transient ventilatory overshoots to apnea and ventilatory instability in sleep. [Adapted from Dempsey (127).]
4. Cerebral blood flow and ventilatory instability
Cerebral vascular responsiveness to CO2 is an important protector of brain extracellular fluid Pco2 and [H+] by means of regulating cerebral blood flow (CBF) and the arterial to brain Pco2 difference (587–589). For example, any reduction in cerebral vascular responsiveness and CBF to hypo- or hypercapnia will mean a greater change in brain (and central chemoreceptor) Pco2 (and [H+]) for any given change in arterial Pco2, thereby increasing the slope of the ΔVE/ΔPaCO2 response and increasing controller gain above and below eupnea. The highly sensitive effects of changes in CBF on the control of eupneic ventilation, the ventilatory responsiveness to CO2, and the apneic threshold and CO2 reserve have been demonstrated experimentally during sleep in animal models using mechanical occlusion of carotid inflow (92, 93, 483), and in humans (735, 739) through the use of the cyclooxygenase inhibitor indomethacin to selectively depress cerebral vascular reactivity to CO2. Reductions in baseline CBF and in the cerebrovascular responsiveness to CO2 do occur with aging (50, 266), in severe OSA patients (133, 518), and with congestive heart failure (see sect. iiiG1). In turn, it is likely that these changes contribute to increases in controller gain and to the increased prevalence of sleep-induced ventilatory instability observed in these conditions. Limited clinical findings have shown that the treatment of congestive heart failure with the vasodilator captopril (an angiotensin-converting enzyme inhibitor) both increases CBF (537) and reduces apneic episodes (704).
G. Special Cases of Ventilatory Instability in Sleep
1. Chronic heart failure
The gradual waxing and waning of Cheyne-Stokes respiration (CSR) with cycling periods of 50–60 s duration occurs in about one-third of chronic heart failure (CHF) patients (460, 706) (see Fig. 1A). The causes of periodic breathing in CHF are multifactorial (281, 763). A key contributing abnormality is the increased controller gain in these patients, as defined by an increased ventilatory response slope to CO2, both above and below eupnea. The latter results in an absence of hypoventilation upon transition from wakefulness to sleep and a greatly reduced difference between eupneic PaCO2 and the apneic threshold PaCO2 during sleep (i.e., a narrowed CO2 reserve) (742). There are several potential sources of this increased controller gain as has been documented to occur in human patients and in animal models of CHF, including increased carotid chemoreceptor sensitivity (achieved in part via a reduced expression of nitric oxide and/or an increased expression of angiotensin II at the carotid chemoreceptor) (523, 656) and acute stimulation of lung vascular receptors via increases in left atrial and pulmonary vascular pressures (98, 633). In addition, a reduced cerebrovascular response to CO2 in CHF will 1) increase central chemoreceptor CO2 stimulation at any given level of raised arterial Pco2, thereby enhancing the opportunity for ventilatory overshoot following apneas; and/or 2) reduce central chemoreceptor stimulation for any given reduction in arterial Pco2, thereby enhancing the opportunity for apneas (738). A prolonged circulation time coincident with the reduced cardiac output in CHF will also delay corrective action by chemoreceptors. The resultant lengthening of apnea duration and increasing chemoreceptor stimulus levels enhances the opportunity for arousal, ventilatory overshoot and ventilatory periodicity (281).
So, even though chronic levels of hyperventilation and hypocapnia are common in CHF patients with CSR (280, 442), apparently (as with hypoxia) the stabilizing effects of the reduced plant gain attending the low PaCO2 (see Fig. 7, top) are outweighed by the destabilizing effects of an increased controller gain (above and below eupnea) (see Fig. 7, bottom) and ventilatory instability prevails. Paradoxically, when eupneic PaCO2 is driven even lower via a chronic ventilatory stimulus (acetazolamide), central sleep apnea and periodic breathing are significantly reduced in these patients (276). This is likely due to a further reduction in plant gain, with no change in controller gain, and therefore a widening of the protective CO2 reserve (436). Supplemental O2 also helps stabilize breathing in CHF probably by reducing controller gain (275, 279, 580). Predictably, even very small amounts of inhaled CO2 (or increased dead space) will prevent apneas and period breathing (299, 371), simply because the added CO2 will prevent the patients' arterial Pco2 from falling below the threshold for apnea or hypopnea (53). Finally, a new means of successfully preventing central apneas and periodicity in CHF uses a proportional assist servo-ventilator to provide a breath via inspiratory pressure support (when a need is detected) together with a preset back-up respiratory rate to abort impending apneas (673).
Mixed apneas, i.e., central apneas followed by airway obstruction within the same apneic event, are common in CHF for significant portions of sleep time. This is particularly true in those patients who are obese and/or with a history of snoring, suggesting a high likelihood of a high passive airway Pcrit (278, 282, 682). Accordingly, CPAP treatment also reduces apnea and CSR in some but not all CHF patients (277), possibly because 1) airway narrowing and obstruction are prevented, thereby removing one potential cause of blood gas changes, chemoreceptor stimulation, and arousal that will elicit ventilatory overshoots (262); 2) preventing the reflex central apneas caused by airway closure during early expiration (224, 576); and 3) reducing ventricular afterload and pulmonary vascular pressure.
2. Hypoxic-induced periodic breathing
Sojourners to high altitude commonly experience restlessness and nonrefreshing sleep, in part due to the common occurrence of periodic breathing. During NREM or REM sleep hyperventilation begins immediately upon hypoxic exposure and intensifies with time (see Fig. 1B) (53, 54). After ∼10 min of hypoxia in the sleeping human, tidal volume begins to oscillate in a waxing and waning pattern. These oscillations keep increasing in magnitude as hypoxia is maintained and PaCO2 falls further to the level of the apneic threshold. Commonly then an augmented inspiration occurs and the subject begins overt periodic breathing cycles of ∼15–25 s duration, characterized by two or three huge tidal volumes followed by apneas of 5–15 s duration as well as large swings in cerebral blood flow (130). During these periodic cycles arterial SaO2 swings wildly along the steep part of the oxygen dissociation curve.
As with CHF, the principle reason for apnea and periodic breathing in hypoxic sleep is likely to be the increased controller gain, as evidenced by the steep increase in CO2 response slope above and below eupnea and the greatly narrowed CO2 reserve (436, 742). Accordingly, during apnea elicited via very minimal amounts of transient hypocapnia (∼1–2 mmHg less than eupnea), PaCO2 rises commensurate with sharp reductions in PaO2 below an already hypoxemic baseline, and the interaction of these asphyic stimuli greatly enhances carotid chemoreceptor activity and the drive to breathe. Upon rapid restoration of normoxic SaO2 via increased FiO2, periodic breathing continues with prolonged apneic periods until hyperventilation is gradually reduced and PaCO2 returns to normal.
Unlike the gradual waxing and waning of CSR in CHF, periodic breathing in hypoxia occurs in breath “clusters” with tidal volume increasing from zero to three to four times control, almost instantaneously following each apnea. We believe this implies the presence of a transient arousal at apnea termination which would further augment the responsiveness of the respiratory control system and produce the sudden ventilatory overshoot (177, 301). Although cortical EEG arousals have not been consistently observed throughout periodic breathing in hypoxia (53, 301), it is still feasible that arousal only at the level of the brain stem (249) could greatly magnify this chemoreceptor responsiveness. An additional as yet untested influence in this dynamic, physiologically complex condition would be brain hypoxia acting independently as a ventilatory stimulant (112, 154, 625), an effect which may be sensitized via a simultaneously enhanced carotid chemoreceptor input (see sect. iiiF2).
The amount of periodic breathing in sleep is greatly reduced over time in hypoxia (53, 54) and when chronic respiratory stimulants (such as acetazolamide or doxapram) are administered to sleeping sojourners at high altitude (547, 659). Perhaps then, in short-term hypoxia, the stabilizing effect of a reduction in plant gain associated with a reduced PaCO2 is offset by the marked effect of hypoxia on increasing controller gain and therefore loop gain, whereas with acclimatization or superimposed chronic stimuli, the further reductions in PaCO2 and plant gain override the increased controller gain. Additionally, a few weeks of intermittent hypoxic exposure in canines produced no aftereffects on ventilation 1 day following the cessation of exposure, but did decrease the apneic threshold and widen the CO2 reserve in the sleeping animal (294).
3. Opioid-induced periodicity
Periodic, cluster-type, ataxic breathing patterns have been reported with high prevalence during NREM sleep in patients administered chronic doses of opioid medications (703, 705). The severity of apneic events is dependent on opioid dose (703), occurs predominantly in NREM rather than REM sleep (278), and is only very rarely associated with chronic (daytime) CO2 retention (672). So, while acute opioid administration in animals and awake humans is well known to cause hypoventilation and apnea via mechanisms acting at the level of the medullary respiratory rhythm generator (331, 614), with chronic opioid administration sleep appears to be required to unmask the instabilities in ventilatory control (also see sect. iiiF). Perhaps the central depressant effects of opioids might sensitize the apneic threshold and narrow the CO2 reserve below eupnea. As with other cases of background hypoventilation, plant gain would be enhanced and the CO2 reserve narrowed. However, as with hypoxia, there remains no ready explanation for the abrupt transitions from apnea to transient hyperventilation. Like other forms of predominately central sleep apneas, opioid-induced periodicity is treatable via servo-ventilation (278). Major clinical concerns over opioid-induced sleep apnea are twofold: 1) the dramatic increase in the therapeutic use of opioids (such as methodone, oxycodin, and morphine) over the past decade (519) and 2) the high mortality rates reported for these patients, especially the many sudden deaths occurring in the early morning or in bed (278).
In summary, the causes of cyclical periodic breathing containing predominantly central or mixed apneas are multifactorial, and while we can usually point to specific contributing factors such as those affecting plant and/or controller gains, it is not yet possible to predict with certainty exactly how deficiencies in different elements contained within the control system, especially chemoreceptor control, mesh to produce this repetitive series of ventilatory under- and overshoots in the sleeping state. Since breathing in the sleeping state is so very critically dependent on chemoreceptor control, it is imperative that we follow recent leads from evidence in reduced preparations to more fully understand how peripheral and central chemoreceptors influence each other's responsiveness and in turn overall ventilatory responsiveness in the integrated, intact preparation studied across varying states of consciousness.
H. Interaction of Neurochemical Control Mechanisms and Upper Airway Anatomy in OSA
We now attempt to integrate our previous discussions of airway anatomy, neurochemical control of upper airway dilators, chest wall pump muscles, and ventilatory and sleep state stability into a more cohesive understanding of the pathogenesis of cyclical, repeated airway obstructions. First, there is little doubt that compromised upper airway anatomy plus the loss of wakefulness input increases passive Pcrit, thereby rendering the airway highly susceptible to closure at sleep onset. However, the question remains as to why obstructive sleep apneas recur in a cyclical pattern of ventilatory undershoots and overshoots in OSA patients. Several observations have implicated deficits in neurochemical control and stability of central respiratory motor output and upper airway neuromuscular recruitment as key contributors to cyclical OSA. For example, fluctuations in chemical stimuli, respiratory drive, and ventilation are associated with reciprocal oscillations in EMG and airway resistance, with the airway narrowing the most when drive is at its nadir (8, 23–25, 256, 464, 708). Accordingly, very early in the course of a hypocapnic-induced central apnea, bronchoscopic imaging studies in sleeping humans show airway narrowing and often complete closure in the absence of changes in sleep state or in inspiratory effort (25, 324) (see Fig. 5).
Many patients with severe OSA show a significantly higher than normal loop gain for ventilatory control, as determined by the propensity for periodic breathing observed during ventilatory assist (714, 752) or a greater ventilatory response to single breaths of CO2 (257). Loop gain correlates with OSA severity best in those patients with a passive Pcrit that is near atmospheric pressure, i.e., not too negative or too positive (714) (see also scenario 2 below).
A significant portion of subjects with positive passive Pcrit (i.e., highly collapsible airways) have a relatively low AHI, and many of these patients with high mechanical loads on their airway maintain airway patency for significant periods of time throughout sleep without experiencing repeated, cyclical obstructions (714, 752, 755).
Normal subjects decrease their Pcrit during sustained reductions in airway pressure (i.e., below Pcrit levels determined under passive conditions), whereas many OSA patients do not, implying that the threshold for upper airway muscle activation in response to increased chemostimulation is much higher in the OSA patient (492, 493).
Many OSA patients undergoing either tracheostomy (465) or CPAP treatment (193, 676) continue to show periodic ventilatory cycling for variable periods of time.
Although often difficult to detect via routine, noninvasive polysomnography, “mixed”, i.e., central followed by obstructed apnea within the same event (see Fig. 1C), are common, and the same subject will often experience central and predominantly obstructed apneas within the same night (682).
Animal and human studies show a linear chemoreceptor-driven recruitment of diaphragm EMG as opposed to a highly alinear, threshold-like response of upper airway muscle EMG to increasing chemoreceptor stimuli (227, 251, 370, 517).
In summary, these observations point to strong links between neurocontrol mechanisms and airway obstruction, but the determinants of these links are multifactorial. Accumulating evidence obtained in sleeping patients with highly variable magnitudes of airway mechanical loads points to two scenarios that illustrate the potential importance of compensatory neuromuscular control mechanisms on the one hand and central control instability on the other in determining cyclical OSA, when either or both occur in persons with upper airways that are anatomically susceptible to narrowing and closure. For clarity, we present these examples separately, but clearly the underlying causative mechanisms are common to both scenarios of cycling OSA.
1. Scenario 1: compensatory responses to airway obstruction determine susceptibility to OSA cycling
This scenario is illustrated in Figure 8 from Younes (753) in which CPAP pressure in a sleeping OSA patient is suddenly reduced to less than Pcrit (passive), thereby creating an obstructive apnea. This experimentally imposed obstruction sets the stage to consider the influence of compensatory mechanisms in coping with the resolution of an airway obstruction in response to rising chemoreceptor sensory input. As previously discussed and also recently reviewed (754), these mechanisms include 1) ability to effectively recruit upper airway dilators during the apnea and prior to arousal; 2) effectiveness in converting the neural drive into dilator muscle shortening and airway reopening; 3) arousal threshold, which will vary with a) sleep state, b) the magnitude of sensory input from chemoreceptors, and c) the patient's sensitivity to a given chemosensitive input to the higher CNS; 4) the magnitude of the ventilatory overshoot once airway patency is reestablished, as determined by the rate of rise of chemoreceptor stimulus magnitude and response sensitivity (i.e., controller gain). Transient arousals are major determinants of controller gain and therefore the magnitude of the ventilatory overshoot at end-apnea); and 5) the subsequent ventilatory undershoot will determine the magnitude of the reduction in respiratory motor output to both airway and pump muscles in response to the inhibitory influences of the hypocapnia resulting from the preceding ventilatory overshoot. These factors will vary in magnitude and relative importance among subjects and even within the same subject throughout the night. It is proposed that the nature of the interaction among these factors within and immediately following each obstructive event will determine whether initial obstructive events are followed by stable breathing, slow evolving hypopneas with occasional arousals, or repetitive obstructions (754).
Fig. 8.
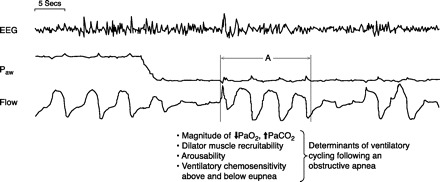
Determinants of how imposed airway obstruction may lead to cyclical obstruction in OSA patients. The figure starts with the OSA patient on sufficient CPAP to ensure a patent airway, optimum airflow and ventilation, blood gases, and stable EEG in NREM sleep. The CPAP level is then quickly removed causing an obstructive apnea with subsequent O2 desaturation and CO2 accumulation. At the termination of the airway obstruction, a transient arousal occurs (A), accompanied by a transient ventilatory overshoot, with subsequent return to sleep and another airway obstruction, thereby beginning the cascade of cyclical ventilatory over- and undershoots and obstructions (see text for explanation of factors which determine the resolution of an airway obstruction and therefore the cyclical nature of OSA). [From Younes (753).]
2. Scenario 2: control system instability determines OSA cycling
A second scenario coupling neuromechanical control mechanisms to cyclical OSA is shown in Figure 9 from Warner et al. (708), which illustrates how oscillations in respiratory motor output (as imposed experimentally in this study by brief hypoxic exposures) may lead to cyclical airway obstruction. A subject is shown in whom airway resistance was already substantially elevated during sleep, but the airway was still patent, prior to imposition of the hypoxia. During the hypoxic-induced ventilatory oscillation period, airway caliber narrowed and then closed at the nadir of central respiratory motor output. This snoring subject likely had a passive pharyngeal closing pressure very close to atmospheric, which is characteristic of groups of subjects who show high, positive correlations between loop gain and AHI (714). In other subjects whose sleep-induced increase in airway resistance was barely measurable (i.e., highly negative passive Pcrit), imposing oscillations in respiratory motor output had no or very little effect on airway resistance, and in patients with highly positive passive Pcrit, cyclical airway obstruction occurred during sleep without the need to superimpose further ventilatory instability.
Fig. 9.
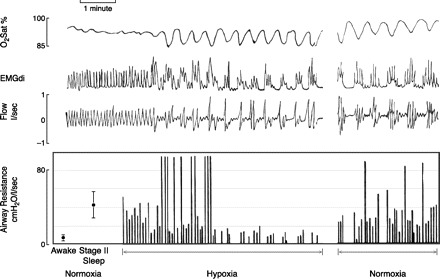
Determinants of how imposed oscillations in respiratory motor output and ventilation (via hypoxia) might lead to cyclical airway obstruction in a subject with an upper airway anatomy susceptible to closure during sleep. Mean values are shown for upper airway resistance during wakefulness and NREM sleep at the left. Breath by breath peak upper airway resistance (Rua) is then shown before, during, and after hypoxic exposure. A subject with a fivefold increase in Rua from awake to sleep but with stable breathing and mild CO2 retention is shown. During early hypoxic exposure, oscillations in respiratory muscle output (EMGdi) but without central apnea occurred, leading to periodic airway obstruction coincident with the nadir of EMGdi. However, with continued hypoxia and fully developed periodic breathing with apneas and profound O2 desaturation and CO2 accumulation, Rua remained very low (even approximating waking levels) during breaths with high levels of respiratory motor output following each central apnea. Then, upon return to normoxia, cyclical airway obstructions returned again at the nadir of EMGdi. [From Warner et al. (708).]
A similar pattern of cyclical obstruction related to an oscillating respiratory drive is prevalent in anatomically susceptible elderly subjects (258, 477). However, in the elderly an unstable sleep state, especially in light stages I and II, is prevalent (477) and appears to be the primary driver to ventilatory periodicity. In these subjects. changing chemoreceptor stimuli would likely also eventually contribute to the periodicity in airway caliber, but only in a secondary role.
3. Summary: treatment implications
All of the causes of these couplings of neural control mechanisms to the severity of cyclical OSA have not yet been determined, although there have been many attempts especially recently to study increasing numbers of OSA patients during sleep (summarized in Refs. 714, 715, 754). The propensity for cyclical OSA will vary within a subject even within the same night as, for example, 1) the majority of all OSA patients will have significant periods during the night of stable breathing without arousals or ventilatory overshoots or undershoots, indicating that they have compensated quite effectively for increases in mechanical loads on the airway; or 2) unstable ventilatory control may result in complete airway obstruction (apnea) or only partial airway narrowing (hypopnea), depending on the magnitude of the oscillating chemical drive, the chemosensitivity to inhibition and excitation of both upper airway and chest wall pump muscles in response to oscillations in chemoreceptor stimuli and the patient's inherent passive Pcrit.
So several interrelated mechanisms clearly contribute to a patient's success or failure in smoothly exiting an obstructive apnea to resume stable breathing or to their experiencing cycling obstructive behavior; or to their propensity to experience oscillations in central respiratory motor output which result in repeated airway obstructions. Accordingly, at this point in our understanding, it seems clear that 1) cycling OSA requires anatomical predisposition to airway closure because no amount of central respiratory motor output instability will result in closure or substantial airway narrowing in normal subjects with highly negative passive Pcrit; and 2) an airway susceptible to closure does not guarantee cycling OSA, because compensatory neuromechanical control mechanisms are an equally important determinant of this repetitive OSA cycling.
Recognizing these complex anatomical-functional relationships in general and recognizing how Pcrit, dilator muscle recruitment, ventilatory response gains, and arousability vary among individual patients may provide clues to OSA treatment by means other than CPAP or by other physical means of manipulating airway caliber, per se. Certainly CPAP clearly prevents airway obstruction in OSA patients; however, it is not tolerated well or at all by some, compliance is low in many others, a residual periodic breathing may ensue in many CPAP users and in some CHF patients CPAP may be contra-indicated (66, 118, 277, 317). To date, attempts to treat OSA with such single entities as increased FiCO2 or FiO2, pharmacological respiratory stimuli, or sedatives (to diminish arousability) have met with very limited success (234, 257, 754). Future attempts in this regard should focus on targeting specific treatments to patients with specific deficits in control system stability or compensatory capabilities in the recruitment of upper airway muscle dilators. Some limited findings provide a basis for future studies, especially in OSA patients with only moderately elevated passive Pcrit (708, 714). For example, use of supplemental FiO2, which is known to reduce CO2 sensitivity above and below eupnea and to increase the CO2 reserve below eupnea (740), was shown to reduce AHI substantially in some OSA patients with high loop gain (715). Furthermore, increasing respiratory motor output via increased FiCO2 stabilizes breathing and greatly reduces upper airway resistance in sleeping subjects with partially obstructed airways (23, 24). Alternatively, pharmacologically induced stimulation of respiratory motor output via such agents as carbonic anhydrase inhibitors (276) or 5-hydroxytryptamine (5-HT)1A receptor agonists (724, 745) reduce plant gain and ventilatory instability. Perhaps application of these agents to patients with only moderately elevated passive Pcrit, combined with high loop gain, may well reduce cycling airway obstructions (708, 714).
IV. Neurochemical Control of Upper Airway Patency
A. Overview
Collectively the very characteristics rendering the pathophysiology of obstructive sleep apnea support the concept that this disorder should be readily amenable to pharmacotherapeutics. First and foremost, obstructive sleep apnea is a remarkably focal process. The site of primary collapse typically lies within a small segment of the oropharynx, where collapse is a consequence of sleep state-dependent reductions in tone in specific upper airway dilator muscles. Second, individuals with obstructive sleep apnea have the capacity in wakefulness to stent open the upper airway through proper activation of the upper airway dilators; it is only in sleep, when neurochemical drive to upper airway motoneurons is altered, that insufficient muscle activity ensues, resulting in pharyngeal collapse. Thus readily reversible changes in neural drive to the upper airway dilator muscles underlie the pathogenesis of obstructive sleep apnea (also see sect. iiiE). Changes in neural drive imply alterations in the amounts or types of neurotransmitters delivered to upper airway motor nuclei. Together, these insights into the pathophysiology of obstructive sleep apnea highlight the importance of sleep state-dependent neurochemical changes reducing the activity of upper airway dilator motoneurons. Over the past two decades tremendous progress has been made in identifying the relevant muscles and their motoneurons and in elucidating the neurochemical control of the upper airway.
B. Upper Airway Dilator Motoneuronal Groups
Multiple muscle groups are involved in the maintenance of upper airway patency in persons anatomically predisposed to obstructive sleep-disordered breathing. Because the oropharynx is so highly collapsible from multiple directions, most individuals with a predisposition to sleep-related collapse of the upper airway rely on opposing muscle groups to work in unison to prevent upper airway collapse, as summarized in Figure 10. Thus it is important to understand the neurochemical control of multiple dilator muscle groups and how the muscles work together. Unfortunately, to date, a tremendous emphasis has been placed on understanding neurochemical control of the genioglossus, or tongue muscle, and its major nerve, the hypoglossal. The genioglossus is the largest and most powerful upper airway dilator. However, genioglossus muscle activation alone may be insufficient to reduce pharyngeal collapsibility (386, 461, 692). Therefore, many of the neurochemical studies of hypoglossal neurochemical control presented below will need to be repeated for the other important dilator groups with different vectors. In summary, then, in individuals with collapsible upper airways, important dilating forces are contributed by several of these groups of motoneurons: motor trigeminal (V), facial (VII), glossopharyngeal (IX) motor vagus (X), and hypoglossal (XII). Muscles regulating lung volume and neck and jaw position will also contribute to upper airway patency (236), and thus cervical ventral horn motoneurons may also influence upper airway patency.
Fig. 10.
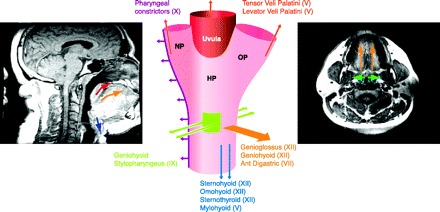
The upper airway in obstructive sleep apnea: a reliance on upper airway dilator muscles for patency. Magnetic resonance images sagittal (left) and coronal (right) of a subject with obstructive sleep apnea. The airway is narrowed but remains patent in wakefulness, in large part because of key dilator muscles, labeled in the center diagram with cranial nerve innervations in parentheses. Arrows indicate overall force vector and are shown on both the diagram and images. Upward directed arrows (red) signify force vectors for levator palatini and tensor veli palatini muscles in raising the soft palate (uvula) and lateral walls. Because the pharynx is collapsible at all tangents, multiple muscle groups must act in concert to prevent collapse of the pharynx.
C. Multiplicity of Function for Upper Airway Motoneurons
One of the greatest challenges in designing therapies to activate upper airway motoneurons is that these muscles also serve critical functions for speech, mastication, propulsion of food into the esophagus, and airway clearance. Thus any attempt to grossly activate one or more upper airway muscles tonically is likely to substantially interfere with these other vital pharyngeal tasks. Central pattern generators are shared for swallowing, sneezing, and breathing (399). Additionally, pharyngeal muscles must also coordinate with ventilatory pump muscle activation. Specifically, the upper airway should be stented open prior to the development of negative intraluminal pressures within the pharynx from activation of the diaphragm and other pump muscles. The coordination of all of these complex processes ultimately merges at pharyngeal motoneurons. Thus an understanding of the neurochemical mechanisms by which sleep state alters resting pharyngeal muscle activity and timing along with an understanding of the neurochemical control of the other pharyngeal functions should be integrated to best develop effective treatments for obstructive sleep apnea.
D. Neurotransmitters and Neuromodulators Influencing Upper Airway Motoneurons
Several classic neurotransmitters and a growing list of neuromodulators (neurochemicals that modulate the activity of a neurotransmitter) show direct (within the motor nucleus) effects on pharyngeal motoneuronal activity. The classic neurotransmitters with direct activity in upper airway motor nuclei are glutamate, glycine, and γ-aminobutyric acid (GABA). The list of neuromodulators continues to grow for both pre- and postsynaptic effects on upper airway motoneurons and now includes acetylcholine (both nicotinic and muscarinic effects), adenosine, ATP, nitric oxide, norepinephrine, orexin, serotonin, substance P, thyrotropin releasing hormone, and vasopressin. Presently, it is estimated that only a fraction of the G protein-coupled receptors in the CNS have been identified, and thus a complete list of promising targets for pharmacotherapeutics eludes us (see Fig. 11 for an overview of key excitatory and inhibitory receptor subtypes in upper airway dilator motor nuclei).
Fig. 11.
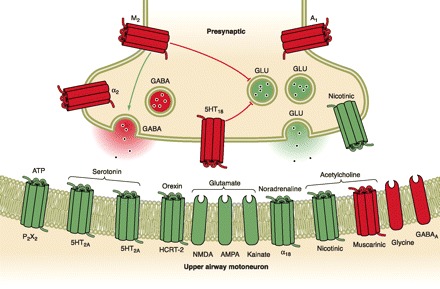
Neurochemical control of the upper airway motoneurons. Presynaptic and postsynaptic excitatory and inhibitory neurochemicals influence the activity of upper airway motoneurons. Numerous excitatory (green) and inhibitory (red) receptor subtypes have been identified using molecular, protein, and physiological studies. Precise roles in motor functions (e.g., respiratory, speech, swallowing, etc.) have not been delineated for any of the receptors. While reductions in noradrenergic tone may contribute to reduced dilator muscle activity in non-REM sleep, the source of reduced motor tone in REM sleep has not been elucidated. Many of the identified receptor subtypes could be targeted pharmacologically, yet none of these is specific to pharyngeal dilator muscles and thus significant side effects including wakefulness are anticipated upon activation of the excitatory targets. M2, muscarinic; α, adrenergic; 5HT, serotonin; P2X2, purinergic; GLY, glycinergic; HCRT, hypocretinergic/orexinergic; GLU, glutamatergic; A, adenosinergic; GABA, γ-aminobutyric acid.
1. Neurotransmitters: glutamate, glycine, and GABA
The precise timing necessary for coordination of rapid complex behaviors, such as swallowing, speaking, or stenting the upper airway prior to inspiration and its resultant negative intraluminal pressures, utilizes rapid onset/offset with an excitatory neurotransmitter, glutamate, and inhibitory neurotransmitters, GABA and glycine. Receptors for the primary excitatory neurotransmitter are α-amino-3-hydroxy-5-methyl-4-isoxasole propionic acid (AMPA), kainite (KA), and N-methyl-d-aspartate (NMDA). Specific receptor subtypes for each of the glutamate receptor groups have been identified for brain stem motoneurons (392, 504). Yet, the relative roles for each receptor subtype in excitation of motoneurons and premotoneurons is just beginning to be explored. Several of these receptor subtypes can be rapidly desensitized by intense activation and/or redox alterations (365). Clearly, this is an important area that deserves further study and one that may contribute to progression of apneic events across the night. In addition to fast action of glutamate at ionotropic receptors, glutamate serves as an excitatory neuromodulator by way of an array of metabotropic receptors.
GABA and glycine are the primary inhibitory neurotransmitters acting at motoneurons, and functional GABAA and glycine receptors are evident on most upper airway dilator motoneurons (480). Glycine plays an essential role in REM sleep postural atonia (631, 733), but the roles of GABA and glycine in atonia of hypoglossal motoneurons are controversial (746). Morrison and co-workers (431, 432) tested the relative contribution of GABA and glycine to genioglossal muscle suppression in an adult rat model with spontaneous sleep and a chronic dialysis probe in the hypoglossal nucleus. Antagonists to both inhibitory neurotransmitters increased baseline nerve activity but did not show a preferential increase for REM sleep, suggesting that neither neurotransmitter contributes significantly to genioglossus muscle atonia in spontaneous REM sleep (368, 431, 432). Of clinical interest, there is a case study of strychnine (a glycine antagonist) effects on genioglossus and tensor veli palatine muscle activity in an individual with obstructive sleep apnea. In this case, strychnine markedly increased tensor veli palatine activity and also increased genioglossus activity relative to arterial oxygen tension (548). For an hour of sleep (at maximum drug effect), apneas were completely abolished and ventilatory efforts were remarkably regular. Thus glycine may not have significant effects on sleep-related respiratory suppression in animals without collapsible airways, but in humans with obstructive sleep apnea, a role of glycinergic tone in sleep-related suppression of muscle tone may be a pharmacological tool for sleep apnea and deserves further study. It is conceivable that glycinergic receptors on upper airway motoneurons could be altered with gene therapy into pharyngeal muscles without disturbing other pharyngeal functions.
2. Neuromodulators
A) SEROTONIN EFFECTS AT UPPER AIRWAY MOTONEURONS.
Serotonin is a powerful modulator of motoneuronal activity (398, 721) and is, perhaps, the best studied for its role in upper airway motoneuronal activity. Activity of the neurons that deliver this neuromodulator throughout the brain have highest firing rates in wakefulness; rates are reduced in NREM sleep, and these neurons are relatively quiescent in REM sleep (243, 696). Leszek Kubin hypothesized that reductions in upper airway dilator muscle activity might be the result of sleep state-dependent decline in serotonin delivery to dilator motoneurons (320). In the decerebrate, vagotomized cat, there is endogenous serotonergic tone in hypoglossal motoneurons (321). Richard Horner confirmed significant intrinsic serotonergic activity in the hypoglossal nucleus in the anesthetized, vagotomized rat (636) yet found little intrinsic serotonergic excitation in the intact, awake rat (637). The Horner group also showed that vagotomy increases serotonergic tone in the hypoglossal nucleus and genioglossus activity; in contrast, in an adult rat with intact vagi, there is little serotonergic tone. (637). Thus animals with unobstructed breathing in sleep may have little to no endogenous 5-HT contributing to resting upper airway motoneuronal tone. Nonetheless, a consistent finding across research labs is the ability of exogenous 5-HT to augment upper airway nerve or muscle activity.
Serotonergic neuromodulation of upper airway motoneurons in individuals with obstructive sleep apnea, however, may differ from the role serotonin plays in animals without obstructive sleep-disordered breathing. Specifically, upper airway muscle activity may be recruited in wakefulness to stent open the upper airway in individuals with obstructive sleep apnea (405). An established animal model with spontaneously occurring sleep state-dependent collapse of the upper airway is the English bulldog (238). In NREM sleep, the dog has hypopneas (partial obstructions, with oxyhemoglobin desaturations and arousals); in REM sleep the dog has significant apneas with more pronounced reductions in arterial oxyhemoglobin saturation (238). Veasey et al. (696) examined the importance of serotonin in the maintenance of the upper airway in the English bulldog, by examining rapid sequencing computerized tomography of the upper airway before and after systemic administration of two serotonin antagonists, methysergide and ritanserin (696). Administration of the serotonin antagonists resulted in temporary collapse of the upper airway (measured with cinematic computerized tomography), associated with reduced upper airway muscle activity and oxyhemoglobin desaturations, without electrographic or behavioral evidence of sleep. Thus, in an animal model of obstructive sleep-disordered breathing, serotonin plays an important role in maintenance of pharyngeal patency. It is important to acknowledge that because the drugs were administered systemically, it is difficult to discern the site of action for the serotonin antagonists. Nonetheless, administration of broad-spectrum serotonergic agonists largely prevents airway collapse in the English bulldog in NREM sleep, supporting the concept that serotonergic neuromodulation could be used to treat obstructive sleep apnea. In REM sleep, serotonergic agents reduced sleep-disordered breathing events in the bulldogs, but are less effective, particularly in preventing events in phasic REM sleep (695).
B) CLINICAL TRIALS OF SEROTONERGIC AGENTS IN OBSTRUCTIVE SLEEP APNEA.
Both serotonergic and serotonin receptor antagonist drugs have been tested for effectiveness in reducing obstructive sleep-disordered breathing, including l-tryptophan, fluoxetine, paroxetine, trazodone, and mirtazapine. In summary, none of the drugs tested has proven universally effective (52, 222, 313, 573, 590). In addition to the excitatory hypoglossal motoneuronal postsynaptic effects of 5-HT2AR and possibly 5-HT2CR, there is an important inhibitory effect observed with 5-HT1BR receptor activation (459). This effect is blockade of glutamate release (62, 618, 619). The effectiveness of 5-HT1B antagonists in obstructive sleep apnea has not been tested, despite the above studies supporting 5-HT1BR as a potential pharmacological target.
C) VENTILATORY EFFECTS OF SEROTONERGIC AGENTS BEYOND UPPER AIRWAY MOTONEURONS.
In considering serotonergic therapies for obstructive sleep apnea, it is important to consider where else activation of various 5-HT receptor subtypes might impact on ventilatory drive to upper airway muscles and overall ventilation (for review, see Ref. 48). 5-HT also has direct effects on medullary premotoneurons. Activation of the 5-HT2 receptor subtypes with iontophoretic application of α-methyl-5-HT depolarizes medullary expiratory and postinspiratory neurons, resulting in a reduction of the persistent and postsynaptically activated potassium currents (333). Systemic administration of a partially selective 5-HT1A agonist, 8-OH DPAT, increases respiratory drive, while ventilation can be suppressed by 5-HT1A antagonist drugs (671). This effect was recently shown to be centrally mediated (662). Like 5-HT2A, 5-HT1A activation promotes wakefulness. Thus 5-HT1A agonists may be helpful with anesthesia and analgesia suppression of ventilation but would likely disrupt or reduce sleep if administered for sleep apnea. The pre-BÖtzinger region contains critical respiratory pattern generator neurons in mammals, and at this site, 5-HT can influence ventilatory drive. Within this respiratory rhythm generator, 5-HT1AR is the predominant 5-HTR. 5-HT1AR agonists applied directly to these respiratory rhythm neurons suppress apneusis and cause a pattern of markedly prolonged inspiratory efforts, and this has been translated into clinical medicine (332, 724). This breathing pattern may occur in barbiturate overdoses, and 5-HT1AR agonists, including buspirone, a partial 5-HT1AR agonist, can reverse the drug-induced apneusis. This partial agonist does not, however, reverse narcotic-induced respiratory suppression in humans (458). Pharmacological blockade of the 5-HT4R within the pre-Botzinger region also directly affects ventilation (554). A highly selective 5-HT4R antagonist, CB113808, induces central apneas when injected into the pre-BÖtzinger area, while a selective agonist, BIMU8, increases respiratory drive and importantly can reverse narcotic-induced apnea without reversing the analgesic effect of narcotics (554). Thus 5-HT4R agonists may have utility in managing opioid-induced ventilatory suppression, but could also have potential for sedative- and sleep-induced ventilatory suppression. The third 5-HTR subtype with activity in the pre-BÖtzinger region that influences abnormal breathing is the 5-HT2AR that is critical for postischemic gasping and recovery of suppressed ventilatory drive. It is imperative as we explore potential pharmacological agents that we take into consideration all possible sites of action and expected effects. For example, 5-HT2AR activation can worsen asthma, hypertension, psychoses, and thromboembolic disease. Thus, while 5-HT2AR activation can increase upper airway dilator tone, 5-HT2AR agonists are not likely to be safe and well-tolerated (352).
5-HT1A effects on ventilation are present in several additional sites within the central nervous system. In the hypothalamus, there are 5-HT1A effects on the ventilatory response to hypoxia. Here, activation of 5-HT1AR inhibits the increased ventilation response to hypoxia. This effect is also observed with active 5-HT7 compounds (186). Activation of the 5-HTR in the nucleus tractus solitarius in the rat increases ventilatory frequency (539). In addition to 5-HT1A activation of ventral respiratory neurons, described above, there is also an excitatory effect of 5-HT1AR activation in the dorsal motor vagal nucleus (76). Specifically, injection of the 5-HT1A agonist 8-OH DPAT into the dorsal motor nucleus increases ventilation without altering upper airway motoneuronal activity.
Serotonergic agents have significant ventilatory effects within the peripheral nervous system. 5-HT applied to the nodose ganglion suppresses respiration and induces apneas (751). 5-HT3 is also responsible for suppression of ventilatory drive at the nodose ganglion (751). Administration of a 5-HT3 antagonist, ondansetron, to normal rats reduced the number of central sleep apneic events (82), and this effect is clearly a peripheral effect, consistent with activation of the nodose ganglion (327). Oral administration of this same 5-HT3 antagonist to the English bulldog reduced the number of sleep-disordered breathing events in REM sleep, without affecting NREM sleep events (693). Therefore, 5-HT3 antagonists may be effective in reducing REM sleep events in some individuals with OSA with sleepiness (OSAS). A recent clinical trial in individuals with OSAS did not observe an overall effect of ondansetron (single dose) on OSAS events (651).
D) SUMMARY.
Serotonin delivered to the upper airway motor nuclei in all animal models tested can increase hypoglossal nerve/genioglossus muscle activity across sleep states. Whether serotonin can augment the activity of all of the motoneurons necessary to stent open the upper airway requires further investigation. Although it is disappointing that the excitatory 5-HT receptor subtypes on upper airway motoneurons are not suitable targets for systemically administered therapies, there may be several ways to locally enhance 5-HT within motoneurons to a level sufficient to maintain critical muscle activity in sleep. In all likelihood, there will be distinct subsets of patients with obstructive sleep apnea who will respond differentially to serotonergic agents, and these groups need careful phenotyping in future drug studies testing effectiveness in sleep apnea.
E) NOREPINEPHRINE.
Norepinephrine has several similarities with 5-HT as a neuromodulator of motoneuronal function and also has several important distinctions. Like 5-HT, norepinephrine has an overall excitatory effect on hypoglossal nerve function when delivered into the hypoglossal nucleus (5, 89, 182, 486), and like 5-HT, noradrenergic neurons show reduced firing rates in NREM sleep and are relatively quiescent in REM sleep (18). Hypoglossal motoneurons are innervated by the A7 noradrenergic group, which shows a similar state dependency in firing (20). In contrast to minimal endogenous 5-HT effects in the hypoglossal nucleus in spontaneously sleeping animals, endogenous norepinephrine contributes to genioglossus tone in wakefulness and in NREM sleep (89). Microinjection of a noradrenergic antagonist into the hypoglossal nucleus reduces genioglossus muscle activity by 25–50%, supporting intrinsic noradrenergic tone in the hypoglossal motor nucleus in both wake and NREM sleep. In contrast, in REM sleep, the noradrenergic antagonist has no effect, suggesting that in REM sleep, there is little to no noradrenergic tone in the hypoglossal nucleus. Moreover, norepinephrine contributes to several important upper airway reflexes (138), while 5-HT does not appear to contribute measurably. Specifically, systemic administration of prazosin, an adrenoreceptor α1B antagonist, prevents the masseter muscle activation to multiple stimuli (termed the jaw closure reflex), while a broad-spectrum serotonin antagonist does not alter this reflex response (642). β-Adrenergic tone contributes to motoneuronal responses to either chemoreflexes or trigeminal diving and superior laryngeal nerve reflexes (209). Whether noradrenergic tone in upper airway dilator motor nuclei contributes to the negative pressure airway reflex should now be examined, and this examination should include, not only hypoglossal, but motor trigeminal and facial nerve responses. The only adrenergic receptor subtype mRNA expressed in the majority of hypoglossal motoneurons was for the α1B receptor (701). The functional significance of the α1B receptor has been established as a powerful excitatory effect present under basal conditions for both hypoglossal and trigeminal motoneurons (89, 162, 164, 642). Thus α1B agonists may have a role in select patients with mild NREM sleep predominant sleep apnea. In light of the excitatory effect at the α1B receptor, there is a concern that prazosin could worsen sleep apnea.
Few clinical trials have examined the effects of noradrenergic agents on obstructive sleep apnea. Clonidine, an α2 agonist antihypertensive, suppresses the activity of noradrenergic neurons, yet the drug also suppresses REM sleep. In light of the REM sleep suppressant effect, clonidine was examined as a potential treatment for sleep apnea in one placebo-controlled trial (n = 8) where two placebo polysomnographies were compared with two clonidine polysomnographies for each subject (273). Overall, 0.2 mg clonidine at bedtime suppressed REM sleep by 40%, had no effect on NREM sleep apnea hypopnea events, but by suppressing REM sleep reduced the total number of REM sleep events. Whether clonidine has efficacy in patients with predominantly REM sleep apnea deserves further study. Two noradrenergic reuptake inhibitors have been tested for effects on sleep apneic events, protriptyline and a newer agent atomoxetine (37, 222). In the earlier study, protriptyline at 10 mg/day was administered for 4 wk and then examined for its effects on apnea frequency. The apnea index dropped for the group from 57 ± 9 to 33 ± 8, P < 0.05. The improvement was only present in NREM sleep, but REM sleep was markedly suppressed (222). In the more recent trial, atomoxetine was administered as 40–80 mg for 4 wk prior to polysomnography in 12 subjects with mild sleep apnea (5–15 apneas-hypopneas/h). Although subjective sleepiness improved, there was no improvement in the apnea/hypopnea index (37).
F) ADENOSINE AND ATP.
Obstructive sleep apneic events (through hypoxia) are expected to alter motoneuronal and extracellular levels of purines: reducing ATP and increasing adenosine. Adenosine modulates motoneuronal activity, where the overall effect of adenosine injected into the hypoglossal nucleus suppresses hypoglossal motoneuronal activity (516). This hyperpolarization effect is mediated through the A1 adenosine receptor and is thought to reduce glutamatergic signaling, as evidenced by A1 agonist-induced reduction in the amplitude of excitatory postsynaptic potentials (46). Like adenosine, ATP modulates glutamatergic neurotransmission (356). The pharmacology of ATP neuromodulation involves >18 ion-gated and G protein-coupled receptor subtypes, and only a few of these have been examined for upper airway dilator motoneurons. Funk et al. (181) examined the neuromodulatory effects of ATP on hypoglossal motoneuronal activity in both medullary slices and in anesthetized adult rats, observing an excitatory effect in both models. This excitatory effect was mediated at least in part by the P2X2 ATP receptor subtype on hypoglossal motoneurons. An excitatory modulation has also been observed for phrenic motoneurons; however, this effect is more complex and is followed by a secondary inhibition of phrenic motoneurons, suggesting the involvement of other purinergic receptors (406). Clearly, the pharmacology of the secondary inhibitory effects should be further developed.
G) ACETYLCHOLINE.
Acetylcholine (ACh) injected directly into the hypoglossal nucleus markedly suppresses hypoglossal nerve activity in medullary slice preparations (88) and genioglossus muscle activity in anesthetized adult rats (367). While the overall effect is inhibitory, activation of select pontomedullary cholinergic neurons can activate or suppress hypoglossal motoneurons. A very thorough description of these opposing effects was recently described (319). Intrinsic ACh inhibitory activity is supported by the observation that administration of an acetylcholinesterase inhibitor into the hypoglossal nucleus suppresses activity (367). Pontine and forebrain cholinergic neurons are least active in NREM sleep, most active in REM sleep, and have intermediate activity in wakefulness (698). The sleep state dependency of medullary cholinergic neurons innervating brain stem motoneurons (563, 684) has not been described. ACh targets two distinct groups of receptors: muscarinic and nicotinic receptors. The inhibitory effect of ACh on hypoglossal motoneurons is muscarinic and can be largely blocked with a muscarinic antagonist, atropine (367). The muscarinic inhibitory effect is likely presynaptic suppression of glutamate release (47). This muscarinic suppression of hypoglossal activity may be one important mechanism by which morphine suppresses genioglossus muscle activity, as dialysis of morphine into the hypoglossal nucleus increases ACh release, and this local increase results in suppression of hypoglossal nerve activity (622). However, there is also an excitatory effect of ACh through activation of nicotinic receptors that is masked by the overwhelming muscarinic effect (367). The nicotinic receptor subtypes include the α4β2 and α7 receptor subtypes (536). Of clinical significance, there is a rapid desensitization of nicotinic receptors in the hypoglossal nucleus within minutes (88, 536).
The first clinical trial of an atropine-like substance involved testing the effectiveness of belladonna on sleep-disordered breathing in infants 4–46 wk of age (289). In this report, apneas were prevented fully in 10 of 15 infants. To date, this has never been reexamined. There are several studies testing the effectiveness of nicotine on obstructive events in adults. In the two controlled trials, transdermal nicotine disrupted sleep without improving the frequency of obstructive breathing events (120, 200, 765). Two studies suggest that anticholinesterase medications can improve obstructive sleep apnea. Physostigmine reduced the AHI by 20% and improved oxygenation (234a). Donepezil, a second anticholinesterase inhibitor, was recently shown to substantially reduce obstructive apneas and oxyhemoglobin desaturation time in individuals with Alzheimer's disease (420). The effect was most pronounced in individuals with severe sleep apnea. Whether this cholinergic therapeutic potential is replicated and whether this will be effective in all individuals with obstructive sleep apnea should now be examined.
H) HYPOCRETIN (OREXIN-A).
Under specific circumstances, orexin plays a critical role in motor control, as evidenced by cataplexy and increased sleep paralysis in patients with narcolepsy (642). Orexinergic neurons project to both trigeminal and hypoglossal motoneuronal soma and dendrites (180), and these neurons, like the monoaminergic groups, have reduced c-fos activation in NREM and REM sleep (157); however, like ACh, levels of orexin in motor nuclei are greatest in wake and REM sleep (308). Peever et al. (494) tested the effects of hypocretin-1 and -2 administered into the motor trigeminal nucleus and hypoglossal nucleus on masseter and genioglossus muscle activity, respectively. Both peptides increased masseter electromyographic activity by a similar magnitude, but only hypocretin-1 increased genioglossus activity. The effect of hypocretin-1 was markedly longer lasting. Blocking glutamate transmission attenuates the excitatory effect of hypocretin in motor nuclei (494). Thus reductions in orexin in upper airway motor nuclei in NREM sleep could contribute to the suppression of upper airway dilator activity in NREM sleep, but this is not expected to contribute to REM sleep atonia in these muscles.
I) LESSER-STUDIED NEUROPEPTIDES.
In addition to the above neurotransmitters and neuromodulators, many other neuropeptides modulate brain stem motoneuron activity. Several of these neuromodulators have significant excitatory effects. Vasopressin binds to the V1A receptor on facial and hypoglossal motoneurons and depolarizes the membrane (552). This neuropeptide may play a greater role in newborn and young animals, as the receptor density declines with age (685). Substance P binding to the NK-1 receptor is evident in the hypoglossal nucleus, where binding sites decline with intermittent hypoxia (328). Substance P (NK-1) agonist applied to neonatal hypoglossal motoneurons in slice increases the phasic amplitude (747). Gatti et al. (189) have shown that substance P terminals appose the protrusor motoneurons within the hypoglossal nucleus. Oxytocin binding sites are present on hypoglossal motoneurons (373), but the physiological significance of these sites has not been established. Histamine is another excitatory neuromodulator with increased neurotransmission in waking. Thus there remain many targets for potential pharmacotherapeutics for obstructive sleep apnea.
E. Clinical Pharmacotherapeutic Trials for Sleep Apnea
Presently, universally effective drug therapy for sleep apnea deludes us. Overall, drugs with serotonin influences have been emphasized in clinical trials. Both serotonergic and serotonin receptor antagonist drugs have been tested for effectiveness in reducing obstructive sleep-disordered breathing, including l-tryptophan, fluoxetine, paroxetine, trazodone, and mirtazapine. In summary, none of the drugs tested has proven universally effective (52, 222, 313, 573, 590). In general, the sample sizes have been too small to prove or disprove the effects of the drug. In addition, there may be subsets of individuals who will respond to serotonergics, particularly patients with REM sleep predominant events, where a serotonergic antagonist may reduce REM sleep time and phasic activity. In light of hypoglossal excitation with 5-HT1B antagonists, effectiveness of 5-HT1B antagonism should be explored. Three drugs with noradrenergic effects have been tested: protriptyline, amoxetine, and clonidine. The overall findings parallel findings with serotonin drugs: no major effect other than REM sleep suppression (37, 222, 273). Individuals with mild NREM sleep apnea may provide a more promising subset of individuals to study. Muscarinic antagonists have yet to be explored. Similarly, genetic therapies that alter glycine receptor function for pharyngeal motoneuron may be promising. The advent of more accurate in-home polysomnographic devices allowing repeated tests in each subject will advance pharmacotherapeutics for obstructive sleep apnea by allowing adequately powered, multidose, repeated trials. As mentioned previously, because the obstruction is present only in sleep, these apneic events should be readily amenable to drug therapies, provided we identify receptors that will not also have prohibitive side effects.
F. Future Directions for the Neurobiology of Upper Airway Control and for the Development of Pharmacotherapies for Obstructive Sleep Apnea
As stated in the beginning of this section, there are likely orphan G protein-coupled receptors that to date have not been identified for brain stem motoneurons and that may not have the widespread brain activation issues present for established excitatory neurochemicals. Identification of these receptors is a tremendously labor intensive endeavor. A more prudent approach would be to complete the testing of promising serotonergic directions. In particular, adults with mild NREM sleep OSAS may benefit from serotonergic therapies that reduce sleep fragmentation (such as 5-HT2C antagonist therapies), and individuals with REM sleep OSAS and without significant oxyhemoglobin desaturations may benefit from a serotonergic therapy that suppresses REM sleep time. The receptor subtypes that seem promising and worthy of further pursuit include 5-HT1B antagonism and/or 5-HT1A, 5-HT4, or 5-HT7 agonists. The latter subtypes may also act on central respiratory neurons and increase drive to pump muscles. It is possible that combination therapies will be helpful, using drugs that increase serotonergic or other excitatory effects at motoneurons with drugs that suppress the vagal inhibitory effects of ventilation. An animal model of upper airway collapse in sleep is needed to expedite identification of safe and effective serotonergic pharmacotherapeutics. Having dissected the rat hyoid arch and its pharyngeal musculature, we believe that such a model can be developed. Present in the rat, but not human, is a series of connected lateral hyoid arch bones that provide tremendous lateral support for the hypopharynx. We predict that removal of the lateral hyoid bones in obese rats, rabbits, or guinea pigs would increase collapsibility of the upper airway and provide animal models with which to test neuromodulator pathways. With successful therapies substantiated in multiple species, large-scale clinical trials with adequate power and sufficient dose range across well-characterized subsets of individuals with OSAS will be able to determine precisely who may benefit from serotonergic and other pharmacotherapies.
V. Cardiovascular Sequelae of Sleep Apnea
A. Introduction
Sleep-disordered breathing (SDB) is recognized as a risk factor for the development of hypertension and other cardiovascular diseases. Because of the associations between SDB and obesity and advancing age, the public health burden of SDB-related cardiovascular disease is expected to rise in the coming years. Fortunately, numerous treatment studies suggest that SDB can be added to the list of “modifiable” risk factors. Although treatment of SDB would seem to be the simplest way to reduce cardiovascular risk, many patients refuse or underutilize the cumbersome state-of-the art treatments and many remain undiagnosed. In the following section, we discuss current concepts of the mechanisms by which SDB contributes to cardiovascular morbidity and mortality. Progress in this area of investigation will ensure a rational approach to prevention and treatment of the cardiovascular sequelae of SDB.
B. Acute Effects of Sleep Apnea on the Cardiovascular System
1. Central hemodynamic effects
Episodes of OSA produce arterial oxygen desaturation, hypercapnia, intrathoracic pressure oscillations, and in most cases, sleep disruption (Fig. 12). The highly negative intrathoracic pressures generated during obstructed inspiratory efforts produce transient decreases in left ventricular stroke volume (612, 683). Inspiratory strains also produce small, transient reductions in systemic arterial pressure. Cardiac output falls during obstructive apnea, secondary to decreased stroke volume and also to reductions in heart rate, which can be marked in some individuals (96, 187, 592, 635).
Fig. 12.
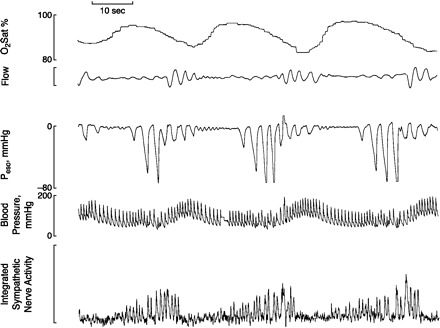
Mixed (central and obstructive) sleep apneas produce marked sympathoexcitation and transient blood pressure elevations in a patient with sleep apnea syndrome. Peso, esophageal pressure; Sat, saturation. [From Skatrud et al. (620).]
Upon resumption of breathing, apnea-induced constraints on stroke volume and heart rate are abruptly removed, allowing release of the augmented cardiac output into a peripheral vascular bed that has been constricted by an increase in sympathetic vasomotor outflow. As a result, the immediate postapnea period is characterized by a marked, transient increase in systemic arterial pressure. This pressor response is caused by sympathetic nervous system activation, because it can be abolished with ganglionic blockade (296, 455, 592). Studies using supplemental oxygen have shown that stimulation of the carotid chemoreceptor by asphyxia (combined hypoxia and hypercapnia) is the most important cause of apnea-induced sympathoexcitation and blood pressure elevation (349, 424). In contrast, the mechanical influence of negative intrathoracic pressure plays little or no role in causing the sympathetically mediated pressor response to apnea (424, 720).
2. Peripheral circulation
Apnea-induced vasoconstriction has been observed in the forearm (13, 264) and the finger (454) of patients with OSA. These findings are surprising because acute exposure to both hypoxia and hypercapnia causes vasodilation in most vascular beds (1, 540), and they suggest that repeated asphyxic exposures, over time, produce alterations in basic mechanisms of neural and local control of vascular resistance. In support of this notion, time-dependent impairments in hypoxic vasodilation have been observed in healthy humans (192) and rats (390) after exposure to hypoxia.
3. Cerebral circulation
The cerebral circulation is exquisitely sensitive to changes in PaO2 and PaCO2; therefore, episodes of OSA have profound effects on flow in this vascular bed. Cerebral blood flow increases progressively during apneas, followed by abrupt decreases in the postapnea hyperventilation period (31, 217, 309). This oscillatory pattern in cerebral blood flow is determined mainly by fluctuations in PaCO2, with a smaller contribution from apnea-induced increases in arterial pressure (532). These cerebral blood flow oscillations, through their influence on washout of CO2 from central chemoreceptors, may produce breathing instability during sleep (738) (see sect. iiiF).
4. Pulmonary circulation
During episodes of OSA, oscillations in PaO2 produce a cyclical pattern of vasoconstrictions and relaxations in the pulmonary circulation that cause marked fluctuations in pulmonary artery pressure (447, 593). These fluctuations are caused by the local vascular effects of alveolar hypoxia and hypoxemia, because they can be abolished by supplemental oxygen (592).
5. Cardiovascular effects of arousal
Most episodes of OSA are accompanied by arousal from sleep. Sleep disruption per se increases sympathetic nerve activity and blood pressure (422), and arousal appears to augment the pressor effects of asphyxia during SDB (423). In experimental animals, “respiratory” arousals caused by upper airway occlusion produce a pressor response accompanied by vasoconstriction in the hindlimb, whereas “nonrespiratory” arousals caused by acoustic or tactile stimulation elicit a smaller pressor response and hindlimb vasodilation (341). Thus it is likely that alterations in sleep state contribute synergistically to sympathetic vasoconstriction and acute blood pressure elevation caused by apnea. In humans, auditory arousals cause transient perturbations in cerebral blood flow that are dependent on the prearousal sleep state (32).
C. Associations Between Sleep Apnea and Cardiovascular Disease
Case control and epidemiological studies indicate that chronic exposure to OSA plays a pathogenetic role in cardiovascular disease. In the following paragraphs we review the evidence linking OSA and specific disease entities. Putative mechanisms will be discussed in a subsequent section.
1. Hypertension
Case-control (168, 725) and cross-sectional (57, 446) studies reveal an association between OSA and hypertension; however, longitudinal data from the Wisconsin Sleep Cohort provide the most compelling evidence for a causal relationship (502). This study demonstrated a dose-response relationship between OSA severity and incident hypertension, with elevated odds ratios for subjects with AHI of 5–15 and those with AHI >15. In contrast to these findings, a larger prospective study of hypertension incidence did not find a dose-response relationship between AHI and incident hypertension that was independent of obesity (452). In both population-based studies, the percentage of subjects in the highest AHI category was small (7 and 4%, respectively), and the severity of SDB was not quantified using indexes of nocturnal hypoxemia, which may predict hypertension risk more accurately than AHI does. Obesity is likely to be an important covariate in correlations based on hypoxemia because SDB events of a given duration would cause greater desaturations in overweight versus normal weight individuals secondary to high metabolic rates and low lung volumes (see sect. iiiC).
The initial experimental support for a causal link between OSA and hypertension came from animal models in which exposure to intermittent hypoxia or intermittent airway occlusion equivalent to severe SDB produced blood pressure elevation that was evident, not only during the exposure period, but also when the animals were normoxic and unperturbed (73, 171). This rise in blood pressure, which was dependent on activation of the sympathetic nervous system by carotid chemoreceptors, was caused mainly by intermittent hypoxia. The addition of intermittent hypercapnia did not augment the hypertensive effect (348), and experimental paradigms that employed repetitive arousals from sleep without hypoxia did not result in blood pressure elevation (33, 73).
More recently, investigators have used chronic intermittent hypoxia (CIH) paradigms that model “mild” (244) and “moderate” (9, 292, 315, 390, 686) levels of SDB. Elevations in blood pressure were observed following most (9, 292, 390, 686) but not all (315) of the studies that employed moderate-level exposures (15–20 events/h). “Mild” CIH (10 events/h) caused a more modest increase in blood pressure in male rats and ovariectomized female rats and only a slight increase in intact females (244).
Reductions in daytime blood pressure following treatment of OSA with nasal CPAP provide further strong evidence for a causal relationship. Some investigators have observed statistically significant reductions in 24-h mean blood pressures following CPAP (Fig. 13) (41, 158, 237, 393, 451, 503), whereas others have not (81, 560). A potential reason for this inconsistency and for the modest blood pressure-lowering effects of CPAP in some of these papers is that many of the subjects were normotensive; CPAP would be expected to have minimal impact in such individuals. In contrast, relatively large CPAP effects on 24-h blood pressure have been demonstrated in hypertensive patients (237), particularly those with resistant hypertension (393).
Fig. 13.
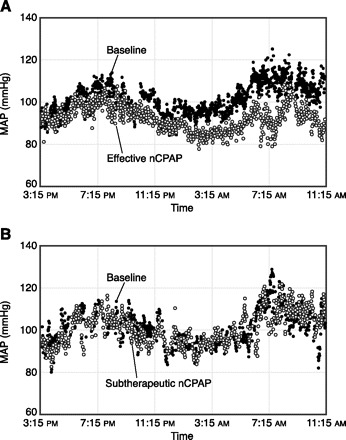
Mean arterial pressures in patients with OSA before and after effective CPAP (A) and subtherapeutic CPAP (B). [From Becker et al. (41).]
The fact that 24-h mean blood pressure does not decrease with CPAP in all patients may be due to inconsistent compliance with treatment, differences in treatment duration or length of exposure to OSA, and the possibility that OSA-related vascular remodeling is irreversible, or it may reflect the multifactorial nature of hypertension. It has recently been suggested that CPAP treatment reduces 24-h mean blood pressures primarily in patients with excessive daytime sleepiness (35, 311, 560). In a multiple regression analysis, baseline blood pressure, improvement in Epworth sleepiness score, BMI, decrease in heart rate, and decrease in time spent at <90% saturation (but not AHI) emerged as important predictors of CPAP-induced decrease in 24-h blood pressure (558).
The effects of CPAP treatment on nighttime blood pressure are unequivocal. Episodes of SDB, even mild apneas and hypopneas, result in substantial acute blood pressure elevations (423); therefore, it is not surprising that elimination of these events lowers average nocturnal values. Numerous investigators have reported CPAP-induced decreases in both systolic and diastolic pressure during sleep and restoration of the nocturnal “dipping” pattern (393). Restoration of this normal circadian blood pressure pattern is an important benefit of CPAP because daytime cardiovascular events and complications are more prevalent in individuals who lack sleep-related falls in blood pressure (134, 697). Moreover, elimination of OSA-related surges in left ventricular afterload may improve cardiac remodeling (140). Thus the benefits of CPAP therapy on cardiovascular risk may extend well beyond demonstrable effects on 24-h mean blood pressure.
The blood pressure-lowering effects of alternative therapies for OSA have recently begun to be tested. In one study, CPAP significantly lowered 24-h blood pressure, whereas supplemental oxygen did not (451). Treatment with dental appliances caused modest yet statistically significant reductions in 24-h mean blood pressure (201), whereas in another study there was no change in blood pressure with any treatment (CPAP, oral device, or placebo pill) (36). It is important to note that in both studies, most of the patients were normotensive or were receiving antihypertensive medications.
The effect of OSA on blood pressure in children and adolescents has received relatively little study. In a clinic-based study, Amin et al. (12) found that 24-h mean blood pressures did not differ according to OSA status; however, children with OSA had smaller nocturnal declines and greater blood pressure variability during sleep and wakefulness. Leung et al. (350) observed that OSA and hypertension were correlated only in obese children. Several recent population-based studies demonstrated that children with SDB (AHI >5) have higher blood pressures versus control subjects, even after adjustment for obesity (56, 155, 354). Alleviation of OSA in children may prevent hypertension; however, at this time no treatment data are available.
In summary, a causal link between severe OSA and hypertension has been firmly established via animal models (73, 171, 390) and CPAP treatment studies that have shown, in hypertensive patients with severe OSA, that blood pressure decreases when SDB is eliminated (41, 503). Whether mild SDB also raises blood pressure is less clear. Multiple epidemiological studies have reported a dose-response relationship between OSA severity, as indicated by AHI, and presence of hypertension (57, 141, 446, 502). In contrast, a larger, more recent prospective study failed to find a statistically significant dose-response relationship that was independent of obesity (452). This discrepancy may be due, at least in part, to demographic differences among these cohorts (e.g., older subjects in the recent study; Ref. 452). A more important reason for inconsistency in these findings regarding the dose-response relationship between SDB and hypertension may be that all but one (446) of these previous studies used AHI as a measure of SDB severity. AHI and other measures of event frequency are inadequate indicators of the amount of intermittent hypoxia, which is likely to be the major cause of cardiovascular dysfunction and disease in the setting of SDB. Furthermore, it may not be possible to firmly establish a dose-response relationship between OSA and hypertension via population-based studies because of the measurement error inherent in such studies (e.g., low reproducibility of single overnight sleep studies and/or measurements of blood pressure, indirect measures of breathing). Confirmation of the dose-response relationship will require prospective studies of the hypertensive effects of mild, moderate, and severe SDB in experimental models and clinical intervention studies in patients across all levels of AHI.
It is commonly believed that more than half of all patients with severe OSA are hypertensive (168, 508, 616, 725). Why do some individuals with OSA develop hypertension while others do not? We believe that the answer lies in the multifactorial nature of this complex disease; OSA is but one of several known risk factors for hypertension. Some individuals may be at increased risk for OSA-related hypertension on the basis of their genetic make-up and/or presence of other prohypertensive characteristics, whereas others may have relative protection from the adverse effects of OSA. An important challenge for future researchers is to identify the patients most at risk for hypertension and other cardiovascular sequelae.
2. Left ventricular dysfunction
Individuals with SDB are more likely to have CHF than those without SDB (608). Conversely, the presence of SDB in patients with CHF is much higher than in the population at large (282). Although central sleep apnea is common in CHF, a significant fraction of patients also experiences obstructive apneas and hypopneas (282, 617) (see sect. iiiG).
Independent associations between OSA and more subtle impairments in ventricular function have been difficult to document. In patients with severe OSA without coexisting heart disease, left ventricular ejection fraction was only modestly reduced relative to control subjects (53 vs. 61%) (7). Ejection fractions below 50% were noted in only 8% of patients with moderate to severe OSA (mean AHI, 47 events/h) (326). Diastolic dysfunction was observed in approximately one-third of patients with severe OSA; however, no comparison group was studied (179). A recent study of obese patients with severe OSA and BMI-matched control subjects revealed depressed diastolic function and increased left atrial volume in OSA patients, whereas left ventricular mass was comparable in the two groups (475). In contrast, several other studies have shown that OSA is associated with left ventricular hypertrophy, even in the absence of systemic hypertension (140, 232, 450). In a population-based study, an association was observed between SDB (AHI >15) and left ventricular hypertrophy using Cornell voltage criteria (87), but not classic voltage criteria (245).
Interestingly, other investigators have not observed decreases in left ventricular systolic function, diastolic function, or increased mass in OSA patients (219, 448). The reasons for this discrepancy are not obvious; however, in one study, SDB was not completely absent in the control subjects (they were nonapneic snorers) (219). In the other study, the expected associations between BMI and hypertension and left ventricular mass were seen, but no independent effect of OSA could be discerned (448).
Nevertheless, studies in experimental animals support the notion that OSA can negatively affect ventricular function. In dogs, several weeks' exposure to severe, repetitive airway obstructions during sleep increased blood pressure, caused left ventricular hypertrophy, and reduced ejection fraction (485). Increased left ventricular mass has been observed in rats exposed to CIH (169).
Limited additional evidence for a causal relationship between OSA and ventricular dysfunction comes from observations of improved heart function following CPAP. In patients with systolic dysfunction and moderate to severe OSA, CPAP treatment improved ejection fraction, functional status, and quality of life (293, 387). However, because neither trial included a placebo treatment arm, these results must be interpreted with caution (478).
Strong, consistent evidence for OSA as a cause of ventricular dysfunction is not available; however, the large increases in ventricular afterload produced by acute episodes of OSA are likely to negatively impact individuals with existing ventricular dysfunction. OSA is often present in patients with severe ventricular dysfunction; however, in many of these individuals CHF is the cause, not the consequence, of OSA.
3. Stroke
An association between OSA and stroke has been observed in numerous cross-sectional studies (17, 39, 143, 198, 413, 487, 608, 716); however, in most cases it was not possible to determine whether OSA preceded the onset of stroke and thus could be involved in pathogenesis. Arzt et al. (17) recently performed a longitudinal analysis of the association between SDB and stroke risk. They found that moderate to severe SDB (AHI ≥20) was associated with increased risk of incident stroke, whereas no increased risk was observed in subjects with mild SDB. The increased risk of stroke associated with SDB appeared to be partially independent of hypertension and confounded by obesity.
Yaggi et al. (744) reported the incidence of stroke (including transient ischemic attack) or death from any cause in individuals with previously diagnosed OSA. They found a significant association between SDB, defined as AHI ≥5, and stroke or death from any cause that persisted after statistical adjustment for confounding factors. The authors reported a dose-response relationship between SDB severity and risk; however, because the combined outcome measure of stroke or death from any cause was used, it is not possible to ascertain the specific effects of OSA on risk of stroke. Munoz et al. (433) conducted a population-based study of OSA and stroke risk in the elderly. They found that incident stroke was nearly three times as likely in subjects with AHI ≥30 versus those with AHI <30. The risk of stroke and transient ischemic attack was assessed prospectively in patients with verified coronary artery disease (417). In this group, OSA (AHI >10) was independently associated with incidence of these cerebrovascular end points. A large clinic-based study assessed the effect of OSA on risk of all-cause mortality in patients with preexisting stroke (568). The odds ratio for death from any cause was higher in patients with OSA (mean AHI ∼35) than in those without OSA (AHI <15). Interestingly, central sleep apnea was not associated with increased risk.
Viewed collectively, these studies demonstrate an independent association between moderate to severe (AHI >30 events/h), but not mild, OSA and risk of stroke. Treatment of OSA appears to decrease the risk of stroke, because the incidence of fatal and nonfatal cardiovascular events is similar in CPAP-treated individuals and control subjects (391). Interestingly, the association between OSA and stroke risk is partially independent of hypertension (17, 744), an established major risk factor for stroke, which suggests that additional pathogenetic mechanisms are operant (see below).
4. Coronary artery disease
Case-control studies have reported a high prevalence of SDB in individuals with clinically verified coronary artery disease (417–419, 495). In these studies, the odds ratios for OSA as an independent predictor of coronary disease approximated those of known risk factors such as diabetes mellitus, hypertension, and obesity. In contrast, large population-based studies have not detected a strong independent association between SDB and coronary disease (245, 608). One possible explanation for divergent findings in clinic- versus population-based studies is differences in the severity of coronary artery disease. All subjects in the former studies had clinically manifest coronary artery disease, whereas in the population studies, subclinical coronary disease may not have been recognized.
Coronary artery calcification, an indicator of subclinical atherosclerosis, was quantified by electron-beam computed tomography in patients referred to a sleep clinic, none of whom had signs or symptoms of coronary artery disease (638). The authors reported a dose-response relationship between OSA severity and coronary artery calcification; however, the odds ratio for calcification was statistically distinct from the reference group only in individuals with severe OSA.
Regardless of whether OSA plays a causal role in the development of coronary atherosclerosis, acute episodes of OSA may influence morbidity and mortality in patients with coexisting coronary disease. In many such individuals, apneas are accompanied by nocturnal angina pectoris and electrocardiographic evidence of myocardial ischemia (2, 176, 220), which can be ameliorated with nasal CPAP (176, 220, 496). Apnea-induced hemodynamic perturbations may have deleterious effects on unstable coronary lesions, leading to plaque rupture and thrombogenesis.
5. Cardiac arrhythmias
Apneas produce multiple arrythmogenic factors: alterations in cardiac sympathetic and parasympathetic activities, myocardial hypoxemia (585), and intrathoracic pressure fluctuations that deform and alter the size of the cardiac chambers (108). In the initial report on this topic, arrhythmias (most commonly bradyarrhythmias, premature ventricular contractions, and atrial fibrillation/flutter) were detected by 24-h ambulatory monitoring in nearly 50% of patients with severe OSA (212). Interestingly, premature ventricular contractions were more common during wakefulness than sleep. Sleep-related bradyarrhythmias and atrial fibrillation/flutter were abolished after treatment, whereas the premature ventricular contractions persisted. In a large, population-based study, the prevalence of complex ventricular ectopy and atrial fibrillation was greater in subjects with severe SDB (≥30 events/h) versus those without SDB (402). In another recent report, patients referred to a cardiology clinic for atrial fibrillation were twice as likely to have OSA, as assessed by the Berlin questionnaire, than patients referred for management of other cardiovascular disease (184). An increased rate of recurrence of atrial fibrillation after cardioversion has been observed in patients with untreated versus treated OSA (291).
Significant associations were observed between presence of bradyarrhythmias and severity of SDB (462). All observed nocturnal bradyarrhythmias (e.g., sinus pause, AV block) occurred during apneas, whereas only about one-third of tachyarrhythmias were temporally related to SDB events (462). In several case series, treatment of OSA reduced the occurrence of nocturnal cardiac rhythm disturbances (40, 207, 223, 310, 680).
Taken together, these findings indicate that patients with moderate to severe OSA are at increased risk for arrhythmias, especially bradyarrhythmias, during sleep. While the clinical significance of OSA-related arrhythmias is not clear, an obvious concern is their potential contribution to sudden cardiac death. Gami et al. (183) reported that OSA causes a shift in the diurnal pattern of sudden cardiac death occurrence from the early morning waking hours (6:00 a.m. to noon) to the sleeping hours (midnight to 6:00 a.m.) (183).
6. Pulmonary hypertension
Episodes of OSA cause marked, acute elevations in pulmonary artery pressure (see above). Over time, structure and function of the pulmonary circulation are altered, resulting in fixed elevations in pressure. CIH causes sustained increases in pulmonary artery pressure in mice (159). Increased prevalence of daytime pulmonary hypertension has been observed in patients with OSA, with estimates ranging from <20 to >70% (26, 90, 174, 318, 325, 330, 570, 572, 577, 713). This wide range of estimates is due in part to methodological differences (i.e., right heart catheterization versus Doppler echocardiography). The independent contribution of OSA to diurnal pulmonary hypertension is impossible to judge from some of these studies, because subjects with coexisting lung and heart disease were not excluded. In studies where such subjects were excluded, the prevalence of OSA-related pulmonary hypertension has been estimated at ∼20% (6, 26, 90, 577). The incidence of pulmonary hypertension in patients with OSA is unknown, because population-based longitudinal data have not been reported.
If every apnea of sufficient duration causes pulmonary vasoconstriction, why isn't fixed daytime pulmonary hypertension a universal finding in patients with OSA? Interindividual differences in hypoxic sensitivity of the pulmonary vasculature (435) probably play a role, as do the frequency and severity of nocturnal desaturations and presence of comorbid conditions. In OSA patients without clinically significant heart and lung disease, pulmonary artery pressure was positively correlated with age, BMI, percentage of total sleep time with SaO2 ≤90% (but not AHI), and daytime PaCO2 (6, 26), and negatively correlated with spirometric measures of pulmonary function (26) and daytime PaO2 (6, 26). These findings suggest that even subtle changes in pulmonary function, in the absence of frank lung disease, can contribute to the development of pulmonary hypertension in patients with OSA. It is important to note, however, that pulmonary hypertension could also be a cause of abnormal arterial blood gases during wakefulness because of its adverse effect on ventilation-perfusion distribution. In addition, left ventricular hypertrophy (232) and diastolic dysfunction (179), which are prevalent in patients with OSA, could cause postcapillary pulmonary arterial hypertension in susceptible individuals.
Even though daytime pulmonary artery pressures may not reach the hypertensive range in all patients with OSA, they are higher than in matched control subjects (6, 16). These elevations are mild relative to those observed in primary pulmonary hypertension, and their clinical significance is unknown. In some patients with OSA, pulmonary artery pressures are within normal limits at rest but increase to hypertensive levels during exercise (242, 521, 679), suggesting that capillary recruitment during exercise, a major mechanism for keeping pulmonary artery pressure low in the face of increasing cardiac output, is limited. A possible explanation for this limitation is a reduction in the total number of vessels in the pulmonary vascular bed (rarefaction), similar to that responsible for capillary pulmonary hypertension in chronic obstructive pulmonary disease (247, 566). OSA-related pulmonary hypertension may be responsible, at least in part, for exertional dyspnea and exercise intolerance in affected individuals (363).
Fixed increases in pulmonary artery pressure caused by OSA may have clinical significance. Increased right ventricular mass and decreased ejection fraction have been observed in OSA patients, particularly those with diurnal arterial blood gas abnormalities (67, 174, 577). The long-term consequences of OSA-related pulmonary hypertension are not known; however, in patients with chronic pulmonary disease, pulmonary hypertension is associated with increased morbidity and mortality (381, 382, 566, 604). Following CPAP treatment, decreases in daytime pulmonary artery pressures have been observed, regardless of whether the diagnosis of pulmonary hypertension was present (6, 16, 571).
7. Summary
Numerous epidemiological and case-control studies have demonstrated statistically significant associations between SDB and cardiovascular disease. In these studies the odds ratios were highest for individuals with moderate to severe SDB; however, dose-response relationships were rarely apparent. Hypopneas that produced >4% desaturation were independently associated with coronary artery disease, whereas those with <4% desaturation were not (533). Viewed collectively, these studies suggest that a threshold level of SDB (∼25–30 events/h of sleep) and significant oxygen desaturation are required to produce cardiovascular disease.
Is OSA-related cardiovascular risk modifiable? Recent nonrandomized long-term treatment studies suggest that it is. Fatal and nonfatal cardiovascular events were less frequent in treated versus untreated patients with severe OSA (391). Other investigators reported a treatment-related enhancement of event-free survival, even in people with mild to moderate OSA; however, in this study, follow-up durations were different in treated and untreated patients, compliance with other therapies (i.e., medications) was not documented, and a substantial fraction of subjects was lost to follow-up (77). Therefore, further study is required before definite conclusions can be reached regarding the effects of treatment on cardiovascular risk in mild to moderate OSA.
D. Pathophysiological Links Between Sleep-Disordered Breathing and Cardiovascular Disease
OSA alters many aspects of cardiovascular structure and function via multiple pathways; however, when the existing evidence is viewed collectively, several common themes emerge. In subsequent paragraphs we highlight the roles played by neurohumoral activation, oxidative stress, and inflammation in OSA-induced cardiovascular morbidity and mortality.
1. Alterations in neurohumoral control of the circulation
Sympathetic nervous system activity is heightened in OSA patients during sleep and during wakefulness (83, 233, 439). Reductions in muscle sympathetic nerve activity (MSNA) have been documented following CPAP treatment (231, 265, 438, 707), indicating a causal relationship between OSA and sympathetic activation.
In healthy humans, brief exposures to continuous or intermittent asphyxia during wakefulness cause increases in MSNA that persist after reestablishment of normoxic, normocapnic conditions (Fig. 14) (113, 421, 736). Long-lasting sympathoexcitation caused by asphyxia is critically dependent on hypoxia, rather than generic chemoreflex stimulation, because it does not occur following hypercapnia alone (741). The mechanisms that maintain high levels of MSNA after withdrawal of chemical stimuli are not known; however, available evidence suggests that this long-lasting sympathoexcitation has both reflex and central nervous system origins.
Fig. 14.
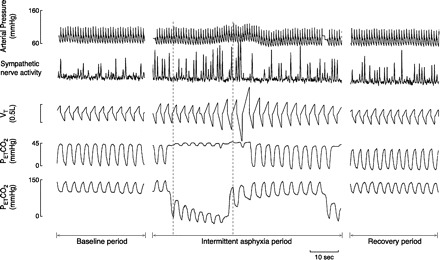
In healthy humans, brief (20-min) exposure to intermittent asphyxia causes sympathetic activation that persists after normalization of blood gases, not only during the interasphyxia phases, but also in the room air recovery period. [From Xie et al. (736).]
In patients with OSA, plasma levels of angiotensin II (ANG II) and aldosterone are elevated, and CPAP treatment causes decreases in plasma renin and ANG II that correlate with reductions in blood pressure (414). The blood pressure elevation produced by CIH in rats can be prevented by blockade of ANG II type I receptors (AT1R), by suppression of the renin-angiotensin-aldosterone system with high-salt diet, and by renal nerve denervation (166, 173). The latter finding suggests an important role for circulating ANG II; however, production of ANG II in the vascular wall and in other tissues may also contribute importantly to the blood pressure raising effect of CIH.
2. Augmentation of carotid chemoreflex function
In rats, CIH increases diurnal blood pressure, but only when the sympathetic nervous system and the carotid chemoreceptors are intact (170, 171). CIH exposure also increases basal sympathetic outflow and results in enhanced sympathetic activation during subsequent acute hypoxic exposures (131, 206, 389, 529, 615). Moreover, CIH enhances carotid body sensory activity, as evidenced by increased rates of carotid sinus nerve discharge during normoxia and upon reexposure to hypoxia (498, 551, 615). These findings, which indicate sensitization of the carotid chemoreceptor, are consistent with human data showing that 1) supplemental oxygen reduces sympathetic outflow in OSA patients but not control subjects (439), and 2) hypoxia-induced sympathetic activation is enhanced in OSA patients versus controls (440).
Reactive oxygen species (ROS) generated during hypoxia-reoxygenation cycles play a role in the carotid chemoreflex sensitization caused by CIH. Pretreatment with a superoxide anion scavenger prevented CIH-induced sensory long-term facilitation of the carotid body (498) and long-term facilitation of respiratory motor output (499). The mitochondrion appears to be a source of ROS in this model (322, 528); however, oxidases in the plasma membrane and cytoplasm (e.g., NADPH oxidase and xanthine oxidase) may also contribute importantly.
Several lines of evidence suggest that ANG II-induced activation of NADPH oxidase and subsequent ROS production may contribute importantly to the chemoreflex sensitization produced by CIH. Infusion of ANG II into the isolated carotid body increases carotid sinus nerve activity (336), and exposure to hypoxia upregulates AT1R in the carotid body (335). Systemic infusion of ANG II increases carotid sinus nerve discharge and augments the amount of sympathoexcitation elicited by acute exposure to hypoxia (360). Both of these effects can be abolished by concomitant infusion of an AT1R antagonist, a superoxide dismutase mimetic, or an inhibitor of NADPH oxidase. Taken together, these findings indicate that ANG II sensitizes the carotid chemoreflex via AT1R activation and NADPH oxidase-derived oxidative stress (359, 360). Exposure of rats to CIH augments chemoreflex control of sympathetic outflow and results in increased AT1R expression and increased superoxide production in carotid body, all of which can be prevented by in vivo treatment with losartan, an AT1R antagonist (389). A potential stimulus for upregulation of carotid body ANG II/AT1R by CIH is an altered expression of oxygen-sensitive potassium channels (288).
Endothelin-1 (ET-1), which is present in glomus cells of the carotid body, produces chemoexcitation by binding to ET-1A receptors (94, 527). In cats, 4 days of CIH produced a 10-fold increase in the expression of ET-1 receptors in the carotid body, increased basal carotid sinus nerve discharge, and an augmentation in the response to acute hypoxia (551). This potentiation, which could be abolished by the endothelin receptor antagonist bosentan, was larger in perfused versus superfused isolated carotid bodies, which suggests that the excitatory effect of ET-1 is mediated, at least in part, by its vasoconstrictor effect.
Intermittent (but not continuous) application of 5-HT elicits sensory long-term facilitation of the isolated carotid body of rats and mice (500). Although 5-HT is a potent vasoconstrictor, this adaptation was not dependent on vascular responses because it occurred in the superfused carotid body. This sensory long-term facilitation could be prevented by concomitant application of ketanserin (5-HT2 receptor antagonist), apocynin (an inhibitor of NADPH oxidase), and N-acetylcysteine (an antioxidant).
In contrast to ANG II, ET-1, and 5-HT, nitric oxide (NO) is an inhibitor of carotid body chemosensitivity (656). In rabbits, intravenous administration of NG-nitro-l-arginine (l-NNA), a NO synthase inhibitor, increased the basal rate of carotid sinus nerve discharge and enhanced the response to hypoxia (656). Conversely, administration of l-arginine, the substrate for NO synthase, decreased baseline discharge and attenuated hypoxic chemosensitivity (656). The carotid body contains two isoforms of NO synthase: eNOS expressed in blood vessels and nNOS expressed in ganglion cells (526). In rats, exposure to CIH causes downregulation of nNOS mRNA (712) and protein (389) in carotid body; thus removal of the inhibitory influence of NO may be responsible, at least in part, for CIH-induced increases in carotid chemoreflex sensitivity.
3. Decrements in baroreflex function
Depressed baroreflex control of heart rate has been documented in patients with OSA (28, 84, 449), and in one study, impaired baroreflex control of MSNA was also observed (84). However, patients and control subjects were not always matched on the basis of hypertension, which can per se alter the set-point and sensitivity of sinoaortic baroreceptors (91). In matched OSA patients and control subjects, phenylephrine-induced increases in blood pressure elicited comparable heart rate and MSNA responses (439). In the same study, nitroprusside-induced decreases in blood pressure revealed OSA-related impairment of the sympathetic, but not the heart rate, component of the baroreflex response (439). When hypertensive and normotensive OSA patients were compared, decreased baroreflex control of heart rate was observed in hypertensive but not normotensive patients (28, 769). Taken together, these findings suggest that depressed baroreflex function in OSA is attributable, at least in part, to coexisting hypertension.
Baroreflex sensitivity has been studied in experimental models of OSA. In healthy humans, 30-min exposure to voluntary, repetitive, end-expiratory apneas failed to alter the gain of baroreflex control of heart rate and MSNA, even though both operating points were shifted toward higher heart rates and higher levels of sympathetic activity (415). A dog model of intermittent airway obstructions during sleep also caused baroreflex resetting, but failed to alter baroreflex sensitivity (72). In rats exposed to CIH, a decrease in baroreflex sensitivity was observed after 2 wk; however, arterial pressure became elevated after only 5 days, which suggests that decreased baroreflex sensitivity was a consequence, not the cause, of the blood pressure elevation (329).
It is not clear whether decreased baroreflex control of heart rate in OSA patients represents a neural adaptation or whether it is secondary to decreased arterial compliance in the carotid sinuses and aortic arch. Several investigators have observed OSA-related increases in arterial stiffness (28, 140, 311, 509, 669, 688). In OSA patients who were treated with CPAP, improvements in baroreflex sensitivity occurred in parallel with decrements in arterial stiffness (449).
It is unlikely that alterations in baroreflex function, per se, are responsible for elevations in diurnal blood pressure in OSA; however, they may result in impaired ability to buffer the pressor responses generated by episodes of apnea. This notion is supported by the observation that the surges in MSNA caused by obstructive apneas are, at least in part, resistant to baroreceptor inhibition (378).
4. Increases in central sympathetic outflow
In addition to effects on chemoreflex function, chronic exposure to intermittent hypoxia may augment central sympathetic outflow. Again, ANG II is a likely contributor to this process. Sympathetic premotor neurons in the brain stem, which are important modulators of postganglionic sympathetic discharge, receive excitatory inputs from higher centers such as the paraventricular nucleus (PVN) of the hypothalamus and the circumventricular organs (115, 185, 541).
In its role as a regulator of central sympathetic outflow, ANG II has important inhibitory interactions with NO (366, 770). CIH exposure decreases nNOS expression in the PVN and increases AT1R expression in the circumventricular organs (712). Cortical regions that modulate central sympathetic outflow may also be involved in CIH-induced sympathoexcitation. Increased Fos-like immunoreactivity was observed in medial prefrontal and insular cortex following 30 days of CIH (615).
5. Alterations in local vascular regulation
Several lines of evidence indicate that endothelial function is impaired in patients with OSA. Reductions in endothelium-dependent vasodilation in the forearm have been demonstrated by invasive and noninvasive means (110, 295, 312, 445). In both cases, indicators of nocturnal hypoxemia (e.g., minimum arterial oxygen saturation, amount of time with saturation <90%) better predicted the degree of endothelial dysfunction than did the frequency of apneas and hypopneas. A causal relationship between OSA and endothelial dysfunction was demonstrated by a study in which flow-mediated dilation in the forearm was improved by CPAP treatment (270). This beneficial effect was lost when CPAP was temporarily withheld. l-NMMA, a NO synthase inhibitor, caused greater reduction in forearm blood flow after versus before CPAP treatment, which suggests that elimination of OSA augmented resting NO availability (340).
Pulmonary artery responses to ACh, sodium nitroprusside (SNP), and l-NMMA were assessed in OSA patients before and after CPAP treatment (339). The decrease in pulmonary artery blood flow produced by l-NMMA was more pronounced after treatment, suggesting that NO levels in the pulmonary circulation were depressed in the untreated condition. In addition, small increases in ACh-induced vasodilation were observed after treatment (339).
Several other observations suggest that OSA reduces the bioavailability of NO. Decreased plasma levels of NO derivatives and normalization of these levels following CPAP treatment have been observed in patients with OSA (11, 267). Scavenging of NO by ROS is a potential explanation for the decrease in its bioavailability. In patients with OSA, increased production of superoxide by neutrophils (594), increased biomarkers of lipid peroxidation (345), and increased levels of 8-isoprostanes (11, 86) have been observed.
Data from animal models are consistent with these findings of decreased NO bioavailability. In rats exposed to CIH, acetylcholine-induced dilation in cremaster arterioles is diminished and the constrictor response to acute NO synthase inhibition is smaller than in control rats (664). CIH also attenuates ACh-induced vasodilation in the cerebral and skeletal muscle circulations, whereas it has no effect on nitroprusside-induced vasodilation (512).
Interestingly, CIH in rats also attenuates norepinephrine-induced vasoconstriction (511). In vivo treatment with a superoxide dismutase mimetic prevented this attenuation, which suggests that the impairment was caused by excess superoxide ion and oxidative/nitrosative stress (511). Other investigators have observed that exposure to intermittent asphyxia enhances ET-1-induced vasoconstriction in the mesenteric circulation (10). In lungs isolated from rats exposed to CIH, vasoconstriction produced by a thromboxane mimetic that was augmented (630). In the forearms of patients with OSA, Hedner and colleagues observed impaired norepinephrine-induced vasoconstriction (210) and enhanced ANG II-induced vasoconstriction (314). Thus it appears that the effects of CIH exposure on vasoconstrictor responsiveness are specific to the vascular bed and vasoactive substance under study.
There is some evidence that ET-1 contributes to CIH-induced vascular dysfunction. ET-1 receptor blockade lowers blood pressure in rats previously exposed to CIH (292), and CIH increases plasma levels of ET-1 (686). Elevated plasma levels of ET-1 are not consistently seen in OSA patients (194, 208, 286, 507, 567, 764); nevertheless, high local concentrations of ET-1 could adversely affect vascular function, as has been shown in the carotid body (551), in the absence of elevated plasma levels.
How does CIH exposure cause oxidative/nitrosative stress in the vascular wall? Recent evidence points to two enzymes, NADPH oxidase and xanthine oxidase, as potential sources of ROS that contribute to CIH-induced vascular dysfunction. Although the mechanism is unclear, exposure to CIH has been shown to enhance NADPH-stimulated superoxide production in rat mesenteric arteries (686). It is possible that CIH-induced activation of the renin-angiotensin system contributes to vascular oxidative stress via the known stimulatory effects of ANG II on NADPH oxidase- (342) and xanthine oxidase-dependent superoxide production (337). Superoxide generated in this manner would be expected to react with NO to form peroxynitrite, thereby reducing NO availability. Peroxynitrite, in turn, can oxidize tetrahydrobiopterin (BH4), a critical cofactor for NO synthase, causing the enzyme to produce superoxide instead of NO (so-called “uncoupling”) (297). In patients with OSA, flow-mediated dilation in the forearm was enhanced by allopurinol treatment (151), which suggests that xanthine oxidase-derived superoxide plays an important role in OSA-induced endothelial dysfunction.
Reactive oxygen species may also contribute to OSA-induced cardiovascular morbidity via their role as initiators of the inflammatory response (343). Increased levels of C-reactive protein and various proinflammatory cytokines have been reported in OSA patients versus control subjects (564, 609, 699, 749). In a rat model, exposure to intermittent airway obstruction elicited a systemic inflammatory response (434).
The mechanisms by which inflammation contributes to OSA-induced vascular dysfunction are not known; however, recent evidence points to the involvement of the T-lymphocyte. This cell is known to play an important role in ANG II-induced hypertension and endothelial dysfunction via NADPH oxidase-induced superoxide production (216). Furthermore, ET-1 is a potent activator of T-cells (122).
Inflammation may be an important link between increased sympathetic nervous system activity and vascular dysfunction in OSA. Chronically elevated sympathetic activity evokes an inflammatory cascade in several organs and vascular beds (761), and T-cell-rich tissues like the liver and spleen are densely innervated with sympathetic nerves. Moreover, hypoxia-induced increases in renal sympathetic nerve activity activate the renin-angiotensin-aldosterone system. Mineralocorticoid receptor stimulation induces not only inflammation, but also oxidative stress, leading to endothelial dysfunction and vascular remodeling (75). It is therefore tempting to speculate that inflammation is an important link between the neurohumoral and vascular manifestations of OSA in the pathogenesis of hypertension and atherosclerosis.
What is the impact of OSA-induced endothelial dysfunction on vascular regulation? In patients with OSA, attenuated hypoxic vasodilation in the forearm (546a, 550) and the cerebral circulation (175, 546a) have been observed, as have attenuated cerebrovascular responses to hypercapnia (135, 546a). In a large population-based study, decrements in cerebrovascular CO2 reactivity were associated with measures of nocturnal oxygen saturation, but not AHI, suggesting that the degree of hypoxemia is a more important contributor to these functional impairments than frequency of events (545). These alterations in vascular regulation may negatively impact tissue perfusion during acute episodes of apnea. Moreover, the resulting enhanced pressor responses to apneas may contribute to loss of the normal sleep-related decline in blood pressure (i.e., “nondipping”). The functional consequences of OSA-induced endothelial dysfunction are not well understood; however, OSA-induced impairments in vascular function may compromise blood flow regulation during other stressors, such as exercise.
6. Alterations in arterial wall structure and biomechanics
Increased carotid intima-media thickness (412, 669) and increased arterial stiffness (28, 140, 311, 509, 669, 688) have been observed in individuals with OSA. Blood levels of NO and ET-1, endothelium-derived regulators of vascular stiffness with opposing actions, are decreased and increased, respectively (267, 507). In rats, 14-day exposure to CIH increases vascular wall stiffness in skeletal muscle resistance arteries (511).
Mitogenic factors known to participate in remodeling (e.g., vascular endothelial growth factor, basic fibroblast growth factor, platelet-derived growth factor) are upregulated during hypoxia (85, 142) and during inflammation (75). Inflammation triggers secretion of enzymes that disrupt the balance between matrix metalloproteinases and their inhibitors (274). ANG II (197, 767) and aldosterone (75) are well-established promoters of vascular inflammation and remodeling; therefore, CIH-induced activation of the renin-angiotensin-aldosterone system may contribute importantly to CIH-induced alterations in vascular structure and biomechanics.
Chronic sympathetic activation may contribute to vascular remodeling via inflammation (761) and/or release of catecholamines that induce vascular wall growth (58). It has recently become evident that adventitial fibroblasts contribute to hypoxia-induced vascular remodeling (648). The release of ATP from adrenergic nerve terminals during hypoxia-induced sympathetic stimulation causes proliferation and migration of adventitial fibroblasts into the intima and media of pulmonary arteries (190) and may also be a stimulus for remodeling in the systemic circulation. In addition, surges in sympathetic outflow during episodes of apnea produce cyclical increases in arterial pressure and blood flow. The cyclical stretch caused by these surges may trigger adaptations in endothelial cells, vascular smooth muscle, and extracellular matrix aimed at normalizing wall stress (689, 727).
Remodeling of the pulmonary circulation has been investigated in animal models of OSA. In mice, CIH raises pulmonary artery pressure (80, 159), produces right ventricular hypertrophy (80, 159), and causes muscularization of pulmonary arteries similar to that caused by continuous hypoxia (159). In rats, CIH-induced right ventricular hypertrophy has been documented in many previous investigations (167, 169, 171, 316, 400, 630). In addition to its effect on NO bioavailability in pulmonary resistance vessels (see above), there is preliminary evidence to suggest that CIH causes pulmonary hypertension via inflammatory pathways (63, 196, 647).
7. Development of atherosclerotic lesions
Independent associations exist between OSA and major risk factors for atherosclerosis, including hypertension (502) as well as insulin resistance (268) and hypercholesterolemia (104) (see sect. vi). Moreover, early signs of atherosclerosis are evident in OSA patients, even in the absence of traditional risk factors, and CPAP treatment results in reversal of these changes (139). In an animal model, CIH has been shown to cause atherosclerotic lesions in mice fed a high-cholesterol diet (583). In this study, marked progression of dyslipidemia and increased serum markers of lipid peroxidation were also observed.
OSA-induced oxidative stress and inflammation, which were discussed in relation to impaired vascular function and structure (above), also are likely contributors to the development of atherosclerotic lesions. High levels of ROS in endothelial, vascular smooth muscle, and adventitial cells are known to initiate atherogenic processes (225). ROS-activated proinflammatory transcription factors, such as activator protein-1 and nuclear factor-κB, stimulate the production of inflammatory cytokines that cause proliferation of vascular smooth muscle cells in the intimal layer (75) and adhesion of leukocytes to the endothelium (3).
Systemic inflammation is a well-established feature of OSA (144, 345, 412, 564, 609, 699, 749). Increases in some OSA-related markers of inflammation, especially tumor necrosis factor-α (TNF-α), are thought to be caused by sleep disruption because they are correlated with daytime sleepiness (564, 699, 700). Nevertheless, the primary proatherogenic feature of OSA appears to be intermittent hypoxia. In a large group of OSA patients without known cardiovascular disease, nocturnal oxygen saturation levels were predictive of carotid artery thickening and plaque occurrence, independently of hypertension (27). In another study, desaturation index (not AHI) was the strongest predictor of plasma levels TNF-α and interleukin-8 in patients with OSA (565).
In addition to its role as an initiator of atherogenesis, OSA may contribute to progression of established lesions. Multiple sources of evidence suggest that OSA promotes thrombosis, because enhanced platelet activation and aggregation (60, 149, 411, 559, 578, 702), enhanced erythrocyte adhesiveness and aggregation (497), increased fibrinogen levels (103, 145, 497, 646, 717), and diminished fibrinolytic activity (538) have all been observed in patients with OSA. Increased endothelial cell apoptosis in patients with OSA may also contribute to coagulation abnormalities (150). OSA-induced inflammatory chemokines and cytokines are potential contributors to plaque rupture via their effects on extracellular matrix (75). The hemodynamic perturbations caused by repetitive apneas may destabilize vulnerable plaques and may enhance oscillatory shear stress and superoxide production (225) at areas of the vascular tree predisposed to atherosclerosis.
8. Cerebrovascular disease
An association exists between OSA and atrial fibrillation (184, 212, 291); therefore, thromboembolism may be an important cause of stroke in OSA patients. Moreover, Beelke et al. (44) recently demonstrated that apnea-induced changes in intrathoracic pressure cause interatrial shunting in patients with patent foramen ovale. In the setting of hypercoagulability, this anomaly could give rise to embolization. Recent case reports implicate OSA as a possible cause of cryptogenic stroke (476).
Individual episodes of OSA cause marked fluctuations in cerebral blood flow (31, 217, 309) (see above). Diminished cerebrovascular reactivity to hypoxia and hypercapnia has been observed in patients with OSA (133, 175, 545). Similar impairments are seen in patients with ischemic cerebrovascular disease (111, 383); however, whether these alterations in vascular regulation contribute to the pathogenesis of stroke in the setting of OSA is unknown.
9. Alterations in cardiac function and structure
Episodes of OSA activate hypoxia-related neurohumoral pathways, increase transmural pressures and afterloads, and lead to chronically elevated blood pressure, any of which could contribute to the development or worsening of ventricular dysfunction. In addition, fixed increases in large artery stiffness (139, 510) may lead to cardiac remodeling. Increased left ventricular mass and diastolic dysfunction have been observed in patients with moderate to severe OSA (140, 179, 232, 475), but not in those with less frequent apneas and hypopneas (448). In some of these studies, left atrial size was increased as well, which could explain the association between OSA and atrial fibrillation (184, 484).
What is the trigger for OSA-related ventricular dysfunction? Negative intrathoracic pressures resulting from OSA episodes, via their effects on left and right ventricular afterloads, may contribute to ventricular remodeling. Nevertheless, animal models indicate that chronic exposure to intermittent hypoxia per se, in the absence of airway obstruction, is also sufficient to impair ventricular function (95, 97, 230). In these models, ventricular dysfunction and remodeling were accompanied by markers of oxidative stress (95, 230). On the cellular level, both hypertrophy and apoptosis of cardiac myocytes have been observed following CIH in rats (97).
10. Genetic aspects of OSA-related cardiovascular disease
The fact that cardiovascular disease is not a universal finding in patients with OSA suggests a role for genetic predisposition. On the basis of what is known about the mechanisms of cardiovascular disease in OSA, several candidate genes have been investigated.
Several investigators have studied the role of angiotensin converting enzyme (ACE) gene polymorphisms (61, 490, 766). In aggregate, these studies suggest complex, potentially important interactions between ACE gene insertion/deletion polymorphisms and SDB as mechanisms for OSA-related hypertension. The ACE D allele is associated with hypertension in subjects with mild-moderate OSA, whereas in patients with severe OSA, the ACE D allele may have a protective influence (61, 490).
An association between OSA and a leptin receptor gene polymorphism has recently been reported (524). In this study, total cholesterol, high-density lipoprotein (HDL), low-density lipoprotein (LDL), and triglyceride levels were higher in OSA patients with the Arg/Arg genotype. OSA is also associated with TNF-α and β2-adrenergic receptor polymorphisms (38, 555).
Polymorphism in the haptoglobin gene, which is associated with cardiovascular risk in diabetes (351), was recently recognized as a risk factor for OSA-related cardiovascular disease (344). Putative mechanisms for this association include impairments in the immunomodulatory and antioxidant properties of haptoglobin (344). Another potentially fruitful target of investigation is the extracellular superoxide dismutase gene. A common variant of this gene is associated with increased risk of coronary disease in non-SDB populations (287); however, the relationship between this gene variant and cardiovascular disease in OSA has not, to our knowledge, been explored. Clearly, further studies are required to establish the genetic variations and the gene-environment interactions responsible for increasing the risk of cardiovascular disease in patients with SDB.
E. Summary: Cardiovascular Sequelae of Sleep Apnea
A schematic representation of the current concept of interdependent mechanisms by which OSA in humans and CIH in animal models led to cardiovascular dysfunction and disease is shown in Figure 15. In the carotid body and in brain regions influencing sympathetic outflow, intermittent hypoxia causes upregulation of ANG II/NADPH oxidase and downregulation of NOS. The resultant excess of superoxide ion results in chronically elevated sympathetic outflow. Sympathetic overactivity, in turn, produces trophic and pro-atherosclerotic effects on resistance vessels via oxidative stress and inflammation. Increased sympathetic outflow to the kidney stimulates renin release and leads to elevated circulating levels of ANG II and aldosterone, two hormones with oxidative stress and inflammatory effects of their own. Sympathetic activation of liver and spleen, T-cell-rich tissues, may play a role in the inflammatory response. In OSA, episodic hypercapnia and arousal from sleep play secondary roles by potentiating the hypoxia-induced increase in sympathetic outflow (248, 422, 423).
Fig. 15.
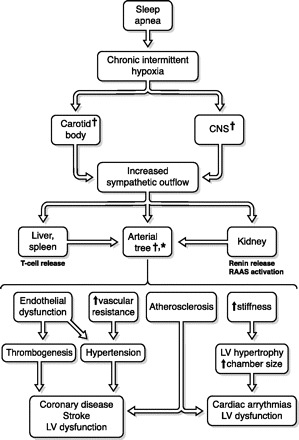
Putative mechanisms by which OSA activates the sympathetic nervous system, initiating a cascade of events that results in cardiovascular disease. CNS, central nervous system; RAAS, renin-angiotensin-aldosterone system. *Inflammation; †oxidant stress.
In the years since identification of the syndrome, much has been learned about the cardiovascular sequelae of OSA; nevertheless, many questions remain unanswered. Do these sequelae have functional, as well as disease-related, consequences (e.g., do they impair regulation of the systemic or pulmonary circulation during exercise)? Can OSA patients with increased risk for development of cardiovascular disease be identified on the basis of genotype or phenotype, thereby enabling rational decisions about who to treat? Can the cardiovascular consequences of OSA be prevented or ameliorated by pharmacological interventions (e.g., antioxidant or anti-inflammatory agents)?
VI. Obstructive Sleep Apnea and Insulin Resistance
A. Introduction
Insulin resistance is a central part of the metabolic syndrome, a condition that is reaching epidemic proportions in Western Society and now emerging in developing countries (298, 530). The metabolic syndrome has many features in common with OSA including obesity, hyperlipidemia, hypertension, and insulin resistance. OSA is so interwoven in the fabric of the metabolic syndrome, or Syndrome X, that the combination of OSA and metabolic syndrome has been labeled “Syndrome Z” (723). Consequently, the causal nature of the relationship between OSA and various components of the metabolic syndrome, in particular insulin resistance, has been difficult to untangle.
As detailed above, there is now a considerable body of evidence indicating that OSA can independently contribute to the development of sustained daytime hypertension. In contrast, the concept that OSA potentially impairs insulin sensitivity (i.e., causes insulin resistance), is a much more recent development. Earliest reports that OSA may impair glucose homeostasis, independent of obesity, began to surface in the early 1990s, and the field has slowly gained momentum since. However, the lack of preclinical data to guide and support the clinical studies, in addition to the technical difficulties associated with assessing metabolic end points, has hampered progress in the field of OSA and insulin resistance.
The measurement of insulin sensitivity, or surrogates of insulin sensitivity, require invasive measurements that are often difficult and technically challenging. Even the simplest marker of insulin sensitivity, the homeostasis model assessment (HOMA) index, requires measurement of blood glucose and plasma insulin. Other metabolic assessments utilized in OSA patients include the standard clinical assessment of oral glucose tolerance test (OGTT) and the predominantly research-focused, frequently sampled intravenous glucose tolerance test (IVGTT). The latter test provides several parameters of insulin and glucose homeostasis, including a modeled estimate of insulin sensitivity (51). Due to technical demands, very few studies have utilized the gold standard measurement of insulin sensitivity: the hyperinsulinemic euglycemic clamp (125). Thus the challenges of measuring insulin sensitivity and other metabolic parameters involved in glucose homeostasis have acted to impede research in this relatively new field.
B. Prevalence and Incidence of Insulin Resistance Type 2 Diabetes in OSA
The first studies examining the prevalence of glucose dysregulation and OSA appeared in 1993. Since this time, a group of studies, generally involving large epidemiological cohorts, have used questionnaire-based surrogates of OSA, such as snoring or witnessed apnea, and assessed the prevalence or incidence of diabetes based on elevated glucose levels, medication use, or self-reported diabetes. The largest of these studies was a prospective cohort from the Nurses' Health Study (4) that followed 69,852 women without diagnosed diabetes over a 10-yr period. During the course of the 10-yr follow-up period, 1,957 women were diagnosed with the development of type 2 diabetes. The presence of snoring was associated with a 2.25 increase in relative risk of developing diabetes compared with nonsnorers, and the association remained significant after adjustment for covariates including BMI, activity, smoking, and family history of diabetes. These results indicated that in a large population-based study snoring is independently associated with an elevated risk of developing type 2 diabetes (4).
Since snoring is only a surrogate marker of OSA, other epidemiological studies have used polysomnography to objectively classify the presence and severity of OSA. In general, these studies have involved smaller cohorts recruited from clinic populations. Two of the larger studies with clinic-based sample sizes of 250–300 subjects both demonstrated a positive association, independent of obesity, between the severity of OSA and indexes of insulin resistance determined by fasting insulin and glucose (268, 652). Similar findings were reported by Punjabi et al. (535) from a community-based sample of overweight or obese, but otherwise asymptomatic, males. In this study by Punjabi et al. (535), subjects with mild or moderate to severe OSA had significantly increased odds ratios for elevated fasting and 2-h glucose levels from the OGTT, after adjustment for both BMI and percent body fat. A large community-based pediatric study of 907 children also found a significant association between an AHI of >5 and elevated circulating insulin and HOMA index of insulin resistance (542). By far the largest epidemiological study to date that directly assessed OSA by polysomnography and measured glucose and insulin levels under fasting conditions and after an OGTT was based on a subset of 2,656 subjects from the ongoing Sleep-Heart-Health study (534). In subjects with an AHI of 15 events/h or greater, there were small, but statistically significant, elevated odds ratios of 1.46 for fasting glucose and 1.44 for 2-h glucose levels in the OGTT, after adjustment for age, BMI, waist girth, race, sex, and smoking. In addition, the HOMA index of insulin sensitivity was also significantly elevated in subjects exhibiting an AHI of 15 events/h or greater. In combination, the prevalence data from multiple epidemiological studies support an independent association between OSA and impaired glucose homeostasis.
Despite several positive prevalence studies of OSA and indexes of insulin resistance, there is so far only one prospective study examining the association between OSA, determined by polysomnography, and the development of type 2 diabetes (546). Comparable to previous cross-sectional studies, they showed a positive association between clinically significant OSA and a diagnosis of type 2 diabetes in 1,387 participants in the Wisconsin Sleep Cohort after adjustment for age, sex, and waist girth. However, in a follow-up study of 978 subjects, the odds ratio for developing type 2 diabetes within a 4-yr period for those with an AHI of >15 events/h did not reach statistical significance after adjustment for waist girth. Although many factors may account for the potential disparity between the cross-sectional and longitudinal findings of this study, the results suggest that epidemiologically there is a lack of strong support for a causal relationship between OSA and the development of insulin resistance and type 2 diabetes. An alternative approach using prospective studies in clinical populations to examine the existence of causality is to determine the impact of therapeutic treatment of OSA on glucose and insulin regulation.
C. Effect of Treatment of OSA on Insulin Sensitivity
Nasal continuous positive airway pressure (nCPAP) is the predominant therapeutic treatment for OSA and is highly effective at eliminating periods of airway obstruction during sleep. The relationship between the use of nCPAP and indices of insulin resistance has been a focus of several recent studies in OSA patients. The studies have been predominantly of small sample size with subjects recruited from sleep or diabetes clinics; the length of treatment as variable as 1 day to 6 mo; the nCPAP intervention uncontrolled with limited adherence data; and few studies utilized the hyperinsulinemic euglycemic clamp as the major outcome variable. In general, the studies were largely negative with variable lengths of nCPAP treatment having no effect on fasting insulin levels (119), glucose tolerance (104), or insulin sensitivity (629). One exception was the 4-mo nCPAP trial by Brooks et al. (71), which did show a 32% increase in insulin sensitivity using the hyperinsulinemic euglycemic clamp in subjects with effective nCPAP treatment. Thus this initial small and disparate group of treatment studies did not appear to support the relatively larger body of prevalence studies, suggesting an independent effect of OSA on disrupting glucose homeostasis.
However, a more recent and larger study by Harsch et al. in 2004 (226) demonstrated a positive effect of nCPAP on insulin sensitivity. In an uncontrolled longitudinal study in 40 OSA patients, insulin sensitivity improved 18% after just 2 days of treatment and by 31% after 3 mo of treatment. Interestingly, a post hoc analysis showed that subjects with a BMI of under 30 kg/m2 showed much greater improvements in insulin sensitivity within 2 days of treatment compared with subjects with a BMI at or above 30 kg/m2. In a follow-up study in nine of the subjects who were adherent to nCPAP, the improvement in insulin sensitivity was still evident on average 2.9 yr later (586). Thus the concept has emerged that the detrimental effects of OSA on insulin sensitivity are potentially more apparent in the absence of comorbidities associated with obesity. Interestingly, in a prepubertal pediatric population, the reverse scenario was recently reported in response to a therapeutic intervention. Circulating insulin and the insulin-glucose ratio were significantly decreased 6–12 mo following adenotonsillectomy in obese subjects, but not nonobese subjects (202). The interaction between the effects of therapy and comorbidities on insulin resistance in OSA will require careful consideration in future studies.
A randomized, placebo-controlled nCPAP study by West et al. (719) has recently challenged the positive findings of Harsch et al. (226). Three months of nCPAP therapy in patients with known type 2 diabetes and newly diagnosed OSA showed an improvement in measures of sleepiness, but there was no change in HbA1c, HOMA index, or insulin sensitivity as measured by the clamp procedure in either the therapeutic or placebo nCPAP groups. A difference between these two conflicting studies is that West et al. (719) used more obese subjects with preexisting type 2 diabetes. However, a similar 6-wk randomized, placebo-controlled nCPAP study in nondiabetic patients reported no improvements in metabolic outcomes with therapy, although there was a trend (P = 0.08) for HOMA to improve by ∼15% (109). Both nCPAP studies reported average compliance rates between 3–4 h per night, which may be critical given the report by Dorkova et al. (136) demonstrating significant improvements in HOMA-assessed insulin resistance in nCPAP compliant (average 5.07 h/night) but not noncompliant (average 3.49 h/night) subjects after 8 wk. It remains to be seen whether an appropriately powered, placebo-controlled, nCPAP study in OSA patients without overt diabetes and long-term follow-up can show significant benefits of therapy on insulin resistance. In summary, the clinical evidence for a causal pathway between OSA and insulin resistance remains equivocal. Although prevalence data for an association between OSA and insulin resistance exists, incidence studies and interventional therapeutic studies have not consistently supported a role for OSA inducing insulin resistance.
D. Experimental Evidence That OSA Can Lead to Insulin Resistance
Two primary physiological disturbances that characterize OSA, acute periods of hypoxic stress and disruption of sleep, can potentially impair glucose homeostasis. Several epidemiological studies including the Sleep-Heart-Health Study (534) have reported that fasting basal glucose levels, the HOMA index, or blood glucose levels during the OGTT are elevated as a function of the degree of nighttime hypoxemia in OSA. Exposure to chronic hypoxia, as occurs with ascent to altitude, can, at least initially over the first 48 h, lead to insulin resistance (338). However, by 7 days of exposure to 4,559 m above sea level, insulin sensitivity is correcting back to sea level values. Experimentally, acute insulin resistance, as determined by a euglycemic clamp, reached a maximum 20 min after a continuous 30-min exposure to hypoxia that lowered arterial oxygen saturation to ∼75% in normal individuals (Fig. 16) (463). Thus short-term exposure to sustained hypoxia produces insulin resistance in humans, but the acute, repetitive hypoxic stress that occurs during sleep in OSA may have different metabolic consequences.
Fig. 16.
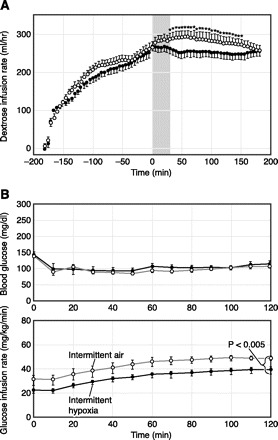
Hyperinsulinemic euglycemic clamps performed in healthy humans and mice during exposure to hypoxia. A: 30 min of sustained hypoxia (vertical gray bar; arterial oxyhemoglobin saturation to 75%) reduced whole body glucose uptake (solid circles) compared with the same healthy human subjects under normoxic conditions (open circles) with plasma glucose clamped at ∼80–100 mg/dl (463). B: 9 h of exposure to intermittent hypoxia (FiO2 reduced to 0.050–0.060 over 30 s and returned to 0.209 in the subsequent 30 s resulting in 60 hypoxic episodes/h) reduced whole body glucose uptake (solid squares) compared with a comparable group of lean healthy C57BL/6J mice under control conditions (open squares) with plasma glucose clamped at ∼100 mg/dl (263).
The potential specificity of the paradigm of hypoxic stress associated with OSA has prompted studies of glucose homeostasis and insulin resistance in rodent models of IH. Interestingly, it appears that glucose homeostasis in mice is a time-dependent phenomena affected by the presence or absence of the IH stimulus (748). Blood glucose levels and insulin resistance as assessed by the hyperinsulinemic euglycemic clamp are elevated during exposure to IH during the light or sleeping phase (Fig. 16) (263). In contrast, during the 12-h dark or active period in which animals are maintained in a nonhypoxic constant room air environment, blood glucose and insulin resistance may actually be decreased below control levels (522), suggesting an overcompensation in insulin sensitivity. Thus the degree of insulin resistance may fluctuate on a diurnal basis dependent on the presence or absence of the IH stress. Diurnal fluctuations in insulin resistance represent a very different pattern of response compared with the development of hypertension in rodent models of IH, where arterial blood pressure remains continuously elevated (329, 664). Thus the mechanisms by which IH chronically elevates blood pressure, but phasically impacts on insulin resistance, suggest differing downstream mechanisms produce the cardiovascular and metabolic disruptions.
Since OSA and obesity frequently coexist, the disruptive effects of IH stress on glucose homeostasis may be potentially exacerbated by adiposity. The comorbid impact of obesity was examined in a rodent model of IH. Genetically obese ob/ob mice exhibited increasing basal insulin levels associated with impaired glucose tolerance over a 3-mo period of exposure to IH compared with weight-matched control ob/ob mice (522). The confounding or interacting effects of obesity have likely contributed to much of the inconsistency in the clinical literature where comorbidity associated with obesity is often present.
Recently, techniques have been developed to assess the impact of experimental IH on glucose homeostasis in healthy humans free of OSA and metabolic dysfunction. The study by Louis et al. (372) showed that a 5-h period of experimental IH in normal sleeping humans (20–30 hypoxic events/h) decreased insulin sensitivity, as assessed by the IVGTT, from 3.8 to 2.6 mU·l−1·min−1. These data, in combination with the rodent studies, suggest that acute exposure to IH can cause insulin resistance in both humans and animals even in the absence of comorbid conditions.
In addition to hypoxic stress, impaired sleep is also a potential candidate for metabolic dysfunction. Reducing sleep time to 4 h/night over a 6-day period decreases the rate of glucose clearance, glucose effectiveness, as well as the acute insulin response to glucose (639). There are also several prospective epidemiological studies, including the large Nurses Health Study mentioned above (19), demonstrating that short sleep duration increases the risk of developing diabetes. Furthermore, sleep deprivation can reduce leptin levels and increase ghrelin levels, potentially acting to stimulate appetite (640, 665). Thus the impact of sleep deprivation on compromising metabolic function may be exacerbated by a secondary effect to increase appetite, which itself may lead to weight gain and further metabolic dysfunction.
Sleep deprivation, or sleep restriction, may not reflect the disturbances in sleep that occur in OSA. Typically, total sleep time is not significantly restricted in OSA, but rather sleep is fragmented by repetitive arousals resulting from the impaired breathing during sleep. The study by Stamatakis et al. (643) attempted to model the sleep fragmentation of OSA by arousing normal healthy humans from sleep over two nights (30–40 times/h) using auditory and mechanical stimuli. This form of experimentally induced sleep fragmentation caused a 20.4% decrease in the insulin sensitivity index as assessed by the IVGTT. There does not appear to be any comparable animal models of sleep fragmentation that have demonstrated metabolic abnormalities. Clearly there is a need for more clinical and translational research to determine whether sleep fragmentation can contribute to the development of insulin resistance.
E. Are There Plausible Mechanisms for OSA to Cause Insulin Resistance?
Determining the mechanisms that cause insulin resistance is a focus for a large number of researchers in the area of type 2 diabetes, although currently few approach the issue from the perspective of OSA. For the purposes of this review, potential mechanistic pathways of insulin resistance are split into two broad categories defined as “classical” and “lipotoxic,” respectively, in Figure 17. These categories and pathways should be considered neither exhaustive nor mutually exclusive, but rather provide a framework for exploring the mechanisms through which OSA can putatively cause insulin resistance. In the absence of obesity, several factors listed under the classical pathways that lead to insulin resistance are relevant to OSA.
Fig. 17.
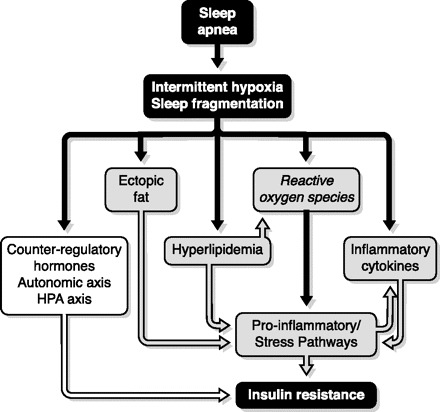
Putative pathways for the physiological disturbances of intermittent hypoxia and sleep fragmentation to cause insulin resistance through activation of “classical” (white) or “lipotoxic” (grey) pathways.
The ability of OSA to activate the sympathetic nervous system is now well characterized. Increased sympathetic nerve activity is implicated as a primary mechanism in the development of sustained hypertension in OSA patients (83, 634) and rodent models of IH (170). Since activating the sympathetic nervous system can also potentially impact on insulin sensitivity (246), it has been proposed that increased sympathetic nerve activity may lead to insulin resistance in OSA patients (226). Although no clinical data exist to support or refute this hypothesis, a study in conscious mice demonstrated that the development of insulin resistance over a 9-h period of IH exposure persisted even after complete pharmacological denervation of both the sympathetic and parasympathetic nervous systems with a ganglionic blocking agent (263). This animal study suggests, at least in the short term, that activation of the sympathetic nervous system is not required for the development of insulin resistance in response to hypoxic stress. However, the possibility remains that hypoxic activation of the sympathetic nervous system, or an increase in circulating catecholamines, contributes to the long-term progression of insulin resistance and metabolic function that may occur over decades in patients exhibiting OSA and obesity.
Increased circulating catecholamines are just one of a group of circulating hormones that act in a counterregulatory fashion to the blood glucose-lowering actions of insulin (246), as well as inhibit insulin release from the pancreas (525). Clinical studies in OSA patients (172, 408) and rodent studies of IH (348) have demonstrated increased levels of circulating catecholamines. However, no study has attempted to link an increase in catecholamines to changes in insulin sensitivity or glucose homeostasis. Growth hormone is another counterregulatory hormone that is affected by sleep and OSA. In contrast to catecholamines, growth hormone levels appear, if anything, to be reduced in the presence of OSA (191, 569), suggesting that this hormone does not play any direct role in the development of insulin resistance. However, growth hormone is the predominant factor controlling release from the liver of insulin-like growth factor I (IGF-I), a peptide with insulin-sensitizing actions. Studies in adult and pediatric patients show that OSA decreases IGF-I and that treatment with nCPAP in adults (211) or surgical adenotonsillectomy in children (34) can increase IGF-I. In contrast to these positive therapeutic effects, a more recent study in adult male OSA patients was unable to detect an independent effect of treatment to increase IGF-I in a parallel, randomized, sham placebo-controlled 1-mo nCPAP trial (403). Furthermore, the relationship between any OSA-induced lowering of IGF-I and the development of insulin resistance has not been directly explored.
Intuitively, the hypoxic stress of OSA would likely activate the hypothalamic-pituitary-adrenal (HPA) axis, elevate cortisol levels, and putatively contribute to insulin resistance. Rodent studies of IH have demonstrated sensitization of the HPA axis (377) as well as increased levels of corticosterone, the predominant glucocorticoid in rodents, that peak during the 12 h of the light or sleeping period when the hypoxic stimulus is present (748). Moreover, these spikes in corticosterone mirrored spikes in glucose, an association that suggests a potential contribution to hypoxic-mediated insulin resistance. However, there are few human studies that have examined cortisol changes in OSA. What few studies have been conducted indicate that OSA does not affect cortisol levels (156, 211, 403). However, one study has shown an increase in cortisol levels in OSA patients relative to weight-matched controls, and the elevated cortisol levels in patients were reduced with nCPAP (68). Interestingly, the study by Speigel et al. (639) in normal healthy young adults demonstrated that sleep restriction to 4 h/night changed the circadian profile of circulating cortisol with elevated levels in the afternoon and early evening. Thus similar to the somatotropic axis, it may be necessary to assess any impact of OSA on the HPA axis hormonal profiles across the entire circadian cycle.
The development of insulin resistance and type 2 diabetes is largely dependent on the presence of obesity. There is now a growing body of evidence that the “lipotoxic” effects of obesity play an important role in the pathogenesis of insulin resistance. Supporting this hypothesis are observations that acute hyperlipidemia induces insulin resistance (20, 59) and that decreasing the metabolic availability of lipids in vivo increases insulin sensitivity (457, 661). Proinflammatory/stress pathways have been proposed as an important link between lipotoxicity and the development of insulin resistance (Fig. 17). Cellular responses to inflammatory and stress signals are mediated by a number of ubiquitously expressed signaling cascades, including the NF-κB pathway. Increased activity in proinflammatory/stress pathways has been implicated in impairing insulin action in peripheral tissues (762).
Multiple factors have been proposed to activate proinflammatory/stress pathways in obesity, including generation of reactive oxygen species, release of inflammatory cytokines, hyperlipidemia, and ectoptic deposition of fat. Interestingly, all of these pathways are potentially activated by the hypoxic stress of OSA in patients or experimentally induced IH in rodents. The hypoxic stress of OSA is a unique stimulus that incorporates a rapid period of tissue deoxygenation immediately followed by rapid tissue reoxygenation. These rapid and repetitive swings in deoxygenation/reoxygenation have the potential to generate reactive oxygen species (144, 594) and lead to lipid peroxidation (410) in OSA patients and in multiple organs including liver (357), heart (95), and brain (694) in rodents exposed to IH. In addition to any direct actions of hypoxic stress from OSA, lipotoxicity may also generate reactive oxygen species and ultimately activate proinflammatory/stress pathways that lead to insulin resistance. Now evidence is emerging that OSA produces a proinflammatory state, and specifically that OSA and IH can activate the NF-κB pathway (205, 564, 565).
Adipocytes are a major source of circulating cytokines that both induce and respond to proinflammatory stress pathways. In general, cytokines are secreted into the circulation as a function of the size of an adipocyte, consequently establishing a positive relationship between adiposity and circulating cytokines (29, 623). For example, two important inflammatory cytokines, TNF-α and IL-6, have elevated circulating levels in obesity and are decreased by weight loss (416). Clinical studies suggest that OSA may have an independent role in further increasing circulating levels of TNF-α, IL-6, as well as the general inflammatory marker C-reactive protein, above the levels seen in obese, nonapneic control subjects, as well as in OSA patients treated with nCPAP (409, 699, 749). Apart from leptin (522), an insulin-sensitizing cytokine, there are little data assessing whether hypoxic stress increases circulating levels of inflammatory cytokines in rodent models of IH.
Both clinical studies and animal studies suggest that an independent relationship may exist between OSA and hyperlipidemia. In a sample of nearly 5,000 subjects from the Sleep Heart Health study, there was a positive association between the severity of OSA and increased serum total cholesterol and triglycerides, as well as decreased serum HDL, in men and women aged less than 65 yr (443). Other smaller clinical studies involving nCPAP support a role for OSA increasing HDL and lowering LDL cholesterol (102, 559). Furthermore, hyperlipidemia can result from exposure to IH in rodent models (358). Thus the hypoxic stress of OSA potentially increases the risk of hyperlipidemia.
In addition to increasing circulating lipid levels, there is evidence that hypoxic stress may affect the accumulation of lipids and subsequent inflammation in organs, as well as the overall distribution of body fat. Exposure of mice to IH can lead to lipid accumulation in the liver (358), upregulation of transcription factors controlling lipid biosynthesis in the liver (355), cause lipid peroxidation and activation of the NF-κB pathway (355), and cause steatohepatitis in a mouse model of diet-induced fatty liver (584). Clinical studies also suggest that OSA is a potential risk factor for steatosis, elevated liver enzymes, and steatohepatitis (668). In addition to producing ectopic fat accumulation and subsequent inflammation in the liver, there is indirect evidence that OSA may predispose to weight gain (506) and potentially influence the distribution of visceral versus subcutaneous fat accumulation (104). Although no studies have focused on whether OSA or IH can lead to ectopic fat accumulation in muscle, the predominant source of insulin-mediated glucose uptake, current evidence suggests that lipid accumulation can occur in the liver and is associated with a proinflammatory state.
In summary, the downstream sequelae of OSA impact a vast array of organ systems and cellular processes in ways that could lead to the development of insulin resistance. The question becomes not “are there plausible mechanisms for OSA to cause insulin resistance?” but rather “what is the relative importance of the many mechanistic pathways through which OSA may cause insulin resistance?” Despite the challenges imposed by tackling this question, there remain many other issues that hinder the development of this area of research.
F. Future Challenges
There is an immediate need for large-scale incidence studies of insulin resistance and type 2 diabetes in well-characterized patients with polysomnographic determination of OSA. The more sophisticated the assessment of insulin resistance (e.g., hyperinsulinemic euglycemic clamp or IVGTT), the more significance such studies will carry. Similarly, long-term correctly controlled nCPAP studies are required to assess the relative burden of disease and determine the potential therapeutic benefits associated with treatment, as well as address the fundamental question of whether insulin resistance and type 2 diabetes are reversible by a therapy that is highly effective at eliminating respiratory disturbances during sleep. Moreover, are there subgroups of pediatric and adult patients, such as those with preexisting type 2 diabetes or increased visceral fat accumulation, who are more resistant to the therapeutic metabolic benefits of nCPAP? The effectiveness of nCPAP for improving insulin sensitivity should be compared quantitatively with the insulin-sensitizing effects of pharmacological therapies and behavioral therapies of weight loss and physical activity. Study designs need to become more comprehensive with respect to multiple sampling across the day and night, as well as controlling potentially confounding factors such as food intake and physical activity. However, despite all these challenges, the issue that restricts progress the most is the overriding and pervasive influence of obesity in both metabolic disorders and OSA. Solving the algebraic equation “Z − X” and extracting the Z component from Syndrome X is proving extremely difficult. Whereas animal models can provide unique insights and control for the effects of obesity, ultimately discoveries need to be translated back into the clinical arena. The data gathered to date suggest the potential for disturbances in sleep and breathing that occur in OSA to possess some degree of independent action, but any action likely represents the tip of a metabolic iceberg buoyed by obesity and insulin resistance.
VII. Neural Injury in Obstructive Sleep Apnea
A. Introduction
The majority of adults with untreated OSA present to the clinician with one or more neurobehavioral impairments, including sleepiness, fatigue, depressed mood, impaired memory, and/or poor concentration. Less commonly, individuals presenting with OSA note motor and/or sensory impairments. It has been difficult to discern in clinical trials whether OSA directly contributes to any one of the associated neurological complaints. This is, in part, because many individuals with OSA have comorbidities that are associated with neural injury, including diabetes, hypertension, and cerebrovascular disease, as described in the previous sections. Moreover, the onset of sleep apnea is insidious, and many individuals present to the physician for evaluation of sleep apnea only years after symptoms were first noted. Nonetheless, the concept that OSA contributes to neural injury is strongly supported by the observation that effective treatment of OSA with continuous airway pressure frequently improves many of the neurological signs and symptoms. Severe neurobehavioral impairments, however, typically do not fully reverse. This raises an important clinical question: does OSA result in irreversible neurobehavioral sequelae?
B. Insight From Neuroimaging Studies
Neuroradiography can provide important insight into structural and functional differences associated with disease in a noninvasive manner. In the case of obstructive sleep apnea, where CPAP affords effective treatment of the disease, neuroimaging before and after treatment may also provide insight into direct effects (reversible effects) of the disease on the CNS. Before reviewing the research findings of neuroimaging in sleep apnea, it is important to consider the difficulties and limitations of such studies. One of the challenges in interpreting findings in neuroradiological studies concerning the effects of sleep apnea on brain structure or function is that comorbid conditions (e.g., cardiovascular disease, diabetes) also negatively impact on brain function. For example, there is no general consensus whether subjects should be matched for all other diseases so that the only difference across groups is a high or low apnea index, or whether studies should simply compare all subjects, regardless of comorbidities, with high versus low apnea hypopnea indexes. As you will see below, the two strategies can lead to very different interpretations of the impact of sleep apnea on brain function. A second challenge is an inherent challenge with associative and cross-sectional studies: whether the neural injury preceded sleep apnea or vice versa. A critical obstacle is that there may well be interindividual differences with susceptibility to neural injury such that not all subjects with sleep apnea will have significant neural injury. Presently for sleep apnea we cannot define these vulnerable subsets. Until the groups of individuals at risk are defined, very large sample sizes will be required to detect overall differences; unfortunately, to date, most imaging studies involve sample sizes smaller than 30. With these limitations in mind, there have been several important, insightful studies.
Three groups of researchers have examined grey matter loss in sleep apnea. Macey and co-workers (379, 380) used MRI to examine grey matter in 21 individuals with sleep apnea and 21 controls (all male, ranging from 28–70 yr of age). They found significant reductions in grey matter in several brain regions, including the hippocampus and cingulate cortex. More importantly, grey matter loss correlated positively with apnea severity. Morrell et al. (429) also found grey matter loss in the hippocampus in OSA. In contrast, O'Donoghue et al. (456) examined 27 males with severe OSA and 24 male controls and did not find differences in grey matter. In this study, subjects were matched for diabetes, hypertension, and other cardiovascular diseases. It is possible, as shown below in animal models, that mechanisms involved in cardiovascular and diabetic injury in sleep apnea are the same as brain injury. Thus O'Donoghue et al. (456) may have selected out the individuals who are most predisposed to neural injury. An alternative explanation for the negative results is that the O'Donoghue study recruited younger adults and thus may have missed effects that require a longer course of sleep apnea or an older age for measurable injury.
Positron emission tomography (PET) of the brain can provide insight into regional brain metabolism and may also provide insight into abnormalities in specific neurochemical transmission. While the latter has yet to be taken advantage of in sleep apnea studies, there is a recent report highlighting the value of PET scanning in sleep apnea (14). In this study, subjects with unexplained residual sleepiness despite effective therapy for sleep apnea with CPAP were examined for fluorodeoxyglucose uptake in the prefrontal cortex in parallel with polysomnography and a vigilance test. Four of the seven subjects had impaired glucose utilization in the frontal and/or temporal cortex; one additional subject had reduced glucose utilization in the parietal cortex. The remaining two subjects had no obvious PET scan abnormalities. Of clinical significance, this study supports the concept that some cognitive impairments, including sleepiness, are not fully reversible in all patients with sleep apnea.
Vascular injury and disturbances in blood flow may contribute to neural loss. In a recent study by Minoguchi et al. (411), brain MRI was used to compare the percentage of silent brain infarctions in subjects with and without OSA. Remarkably, 25% of individuals with severe sleep apnea had infarctions. In contrast, only 7% of obese matched nonapneics had infarcts evident. In support of a direct relationship, the group measured two serum markers for cerebrovascular disease, sCD40L and sP-selectin. Both of these markers were high in sleep apnea and declined upon effective treatment with nasal CPAP. Recently, single photon emission tomography (SPECT) was implemented to measure regional blood flow in sleep apnea (284). In this study 27 subjects with severe OSA (AHI, 30–104/h) were compared with 27 controls, age and sex matched. As with lesions, blood flow appears regionally modified in awake subjects with OSA, with lowest levels in the parahippocampal gyri, pericentral gyri and cuneus. Whether this impacts cognitive function during wakefulness or is secondary to neural injury is not clear.
MRI with spectroscopy may be used to examine metabolic disturbances, where disturbances include neuronal loss, gliosis, and other causes of altered metabolism. A reduced N-acetylaspartate(NAA)-to-choline ratio was found for the cerebral white matter, where the AHI correlated negatively with the NAA-to-choline ratio (290).
These studies prompt the question: Is this loss of brain tissue associated with impaired cognitive performance? Several recent studies have begun to address this issue. Thomas et al. (675) examined cognitive function in parallel with functional MRI in individuals with severe OSA. Ages of the enrolled subjects ranged from 21 to 50 yr, with a male predominance. Subjects with OSA had less activation of the prefrontal cortex while performing a working memory task. A similar decrement was observed in hypoxic and nonhypoxic subjects with sleep apnea, suggesting that hypoxia does not influence the decrement in prefrontal cortical activation during learning. In contrast, hypoxic subjects showed far less activation in the parietal cortex. This suggests that within the brain there are regional differences for hypoxia-sensitive neural tissue and arousal or sleep disruption-sensitive neuronal tissue. One of the more alarming findings from recent studies is that young children with sleep apnea may also have neuronal loss and cognitive impairments. Halbower et al. (218) examined 19 children with sleep apnea and 12 controls, matched for age, gender, ethnicity, and socioeconomic class. Children with severe sleep apnea had significant decrements in their IQ (15 points) and significant decrements in verbal working memory and verbal fluency. These cognitive impairments paralleled reductions in the NAA-to-choline ratio in the hippocampus and frontal cortex by spectroscopy. These findings have serious clinical implications and are worthy of confirmation and then determination of reversibility. At the very least, these findings should prompt careful screening for sleep apnea in all obese children and an aggressive campaign to educate parents on the risks of childhood obesity and to increase efforts to reduce childhood obesity. If these findings prove irreversible, the sleep community should play an important role in campaigning these issues.
C. Evidence of Neural Injury From Animal Models
While there are numerous physiological perturbances in OSA that might disturb neuronal homeostasis and function, David Gozal was one of the first researchers to explore whether the frequent hypoxia/reoxygenation patterns in sleep apnea result in lasting neural injury and impaired neural function. To test this, Gozal et al. (203) exposed young adult rats to either 2 wk of fluctuating ambient oxygen patterns modeling oxygenation in severe sleep apnea, constant hypoxia of the same duration, or room air oxygen tension and then examined the effects of varied oxygenation on learning and neuronal health (203). Gozal et al. (203) identified learning impairments and increased apoptosis within the CA1 region of the hippocampus only in rats exposed to intermittent hypoxia. Similarly, spatial memory impairment was observed several months after intermittent hypoxia exposure to newborn rat pups (123, 124). Thus memory impairments may occur as a result of hypoxia/reoxygenation patterns modeling OSA, and these may persist well beyond the hypoxia/reoxygenation exposure. Future clinical trials should now focus on spatial memory function in individuals with OSA and oxyhemoglobin desaturations. At the same time, animal studies should identify the mechanisms by which frequent hypoxia/reoxygenation events result in hippocampal injury and dysfunction.
D. Daytime Sleepiness
One of the most common neurobehavioral impairments in OSA is sleepiness. In fact, two-thirds of adults with OSA complain of significant sleepiness and/or fatigue (100). When treated for OSA, patients typically report less somnolence (153, 491). Despite marked improvements in subjective sleepiness, randomized controlled trials show small improvements in objective sleepiness (<1 min increase in mean sleep latency overall) despite effective therapy for OSA (491). This objective measure is the average latency to fall asleep during four or five nap opportunities distributed across the morning and afternoon. Thus falling asleep 1 min later across four or five nap opportunities is not a clinically significant improvement in wake function. Although apneic events result in many physiological disturbances as reviewed in previous sections, the oxygen desaturation indexes most strongly predict sleepiness, relative to other polysomnographic parameters, including sleep time, AHI, or arousal index (42, 114, 153, 303, 678). While clinical studies show a strong association with hypoxia/reoxygenation and sleepiness, as discussed above for memory function, whether hypoxia/reoxygenation can induce irreversible sleepiness must be explored in animal models, without the confounds of obesity, diabetes, and other comorbidities.
E. Effects of Hypoxia/Reoxygenation Exposures on Wake Function
Sleepiness has been assessed in mice 2 wk after exposure to 8 wk of IH and resulted in marked reductions in the average sleep latency across the day and reductions in total wake time for 24 h (694). The magnitude of these effects was similar to changes observed in humans with sleep apnea. Most importantly, these wake impairments in mice were not reversible, even after a 6 mo recovery period in normal oxygen conditions (768). The Veasey laboratory (768) has recently identified the groups of neurons implicated in wakefulness (wake-active neurons) injured by intermittent hypoxia. Forty percent of the noradrenergic neurons in the locus coeruleus and dopaminergic wake-active neurons in the periaqueductal grey were lost in this model, and most remaining wake-active neurons in these nuclei showed impaired wake responses (768). In contrast, orexinergic, histaminergic, serotonergic, and cholinergic wake neurons appeared unperturbed. This differential susceptibility was used to determine the mechanisms by which the wake-active neurons are injured by hypoxia/reoxygenation events. The susceptible catecholaminergic neurons showed increased oxidative injury in response to long-term intermittent hypoxia, relative to resistant neurons. Furthermore, a unique feature of the susceptible wake-active neurons is that they contain NADPH oxidase. Inhibition of this enzyme throughout exposure to hypoxia/reoxygenation or transgenic disruption of the enzyme's activity largely prevents the hypoxia/reoxygenation wake impairments, supporting the importance of this enzyme in the injury to these neurons (768) (see Fig. 18). Whether a similar catecholaminergic wake neural injury is found in humans with OSA should now be advanced through post mortem neuroanatomical studies of wake active neurons in individuals with and without obstructive sleep apnea.
Fig. 18.
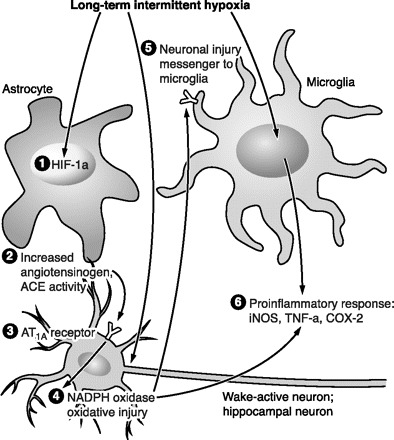
Proposed model of NADPH oxidase injury from hypoxia reoxygenation. Hypoxia/reoxygenation events increase the production of angiotensin II peripherally or in astrocytes, resulting in activation of angiotensin 1A receptors on catecholaminergic neurons. AT receptor activation upregulates NADPH oxidase activity, resulting in oxidative injury. Sleep apnea and intermittent hypoxia are associated with marked inflammation in the brain including inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX2), and tumor necrosis factor-α (TNF-α). Whether this proinflammatory response occurs in neurons or adjacent microglial cells should now be advanced.
F. Upper Airway Dilator Motoneurons May Also Be Injured in OSA
Clinical studies have identified neural dysfunction and injury in adults with obstructive sleep apnea. Sensory nerve action potential amplitudes are reduced in individuals with OSA, and treatment of sleep apnea partially reverses this defect, supporting the concept that sleep apnea contributes to this neural dysfunction (146). Electromyographic studies of the palatopharyngeus muscle in individuals with sleep apnea show long polyphasic potentials and reduced amplitude at maximum voluntary effort (660). Consistent with functional impairment, histological studies have identified demyelination of motoneurons in resected palatal tissue in OSA (64, 364, 730). The severity of peripheral nerve dysfunction correlates with oxyhemoglobin desaturations in sleep apnea (397). Whether OSA, independent of obesity, diabetes, and other comorbidities, injures peripheral neurons requires study in animal models. Long-term exposure to hypoxia/reoxygenation impairs hypoglossal whole nerve responsiveness to both glutamatergic and serotonergic excitation of hypoglossal motoneurons (694).
There is increasing evidence that the endoplasmic reticulum (ER) plays a central role in both adaptive responses to and injury from ischemia-reperfusion challenges (49, 126, 228, 229). The unfolded protein response (UPR) in the ER represents an adaptive response to minimize accumulation of misfolded proteins that would be toxic to the cell. This is accomplished by reducing overall protein translation, increasing the production of chaperones, upregulating clearance of improperly folded proteins, and increasing antioxidant capacity (750). Several components of this protective response are mediated by phosphorylation of eIF-2α. However, when this stress is insurmountable, the ER may take on the role of executioner, activating proapoptotic proteins, including CHOP/GADD153 and caspase-7 (199, 632).
We predicted that IH might activate the UPR and that this response might be injurious in select motoneurons. IH for 8 wk induces significant ER stress in select upper airway motoneurons, including the facial and hypoglossal motoneurons and sparing motor trigeminal (768a). This differential susceptibility was then used to identify the molecular basis of IH susceptibility. Here motoneurons with higher ER stress at baseline develop uncompensated ER stress with exposure to long-term intermittent hypoxia, and many of these motoneurons succumb to apoptosis (768a; Fig. 19).
Fig. 19.
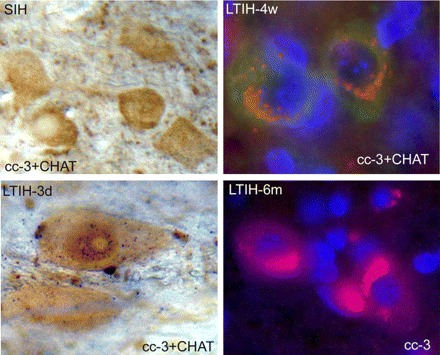
Activation of caspase-3 in motoneurons following exposure to intermittent hypoxia. Top left: DAB stained (brown) hypoglossal motoneurons with minimal cleaved caspase-3 (black). In contrast, even after 3 days of IH exposure, caspase is evident in the nucleus (bottom left). Cleaved caspase-3 is persistently elevated across intermittent hypoxia at 4 wk (top right) and 6 mo (bottom right), but little enters the nucleus, suggesting that the observed loss of neurons occurs largely through nonapoptotic means.
In summary, intermittent hypoxia, modeling oxygenation patterns of moderate-severe sleep apnea, injures select populations of neurons, including hippocampal, catecholaminergic wake-active, and hypoglossal and facial upper airway motoneurons. It is time to translate these findings to human studies, examining brain tissue from individuals with and without OSA who have died suddenly without a specific neurological diagnosis. As an initial exploration, a focus on the above groups is justified. In light of the mechanisms uncovered in wake and motor neurons, activation of these pathways should also be examined in humans to begin to identify promising therapeutic avenues for the prevention and possibly partial reversal of these injuries.
VIII. Future Directions
Substantial advances in several areas of physiology have been made over the past two to three decades, driven by the need to understand the causes, consequences, and treatment of sleep apnea. Major advances in this regard include the neurochemical regulation of upper airway motor neurons and airway caliber; the causes of long-lasting alterations in cardiovascular, biochemical, and neuronal structure and function elicited via intermittent hypoxemia; and the vital importance of the wakeful state on the one hand and variations in sleep state on the other on the regulation of respiratory stability. We close our review of the pathophysiology of sleep apnea by suggesting a few outstanding, fundamental questions for further research.
An experimental approach is needed to provide a more rigorous evaluation of the true prevalence of clinically significant sleep-disordered breathing, than the widely disparate estimates currently provided via epidemiological, correlational approaches. Interventional treatment trials need to be conducted, preferably at the earliest detectable stages of sleep apnea. Also, prospective trials designed to test the effects of mild to moderate levels of intermittent hypoxia on cardiovascular outcomes and daytime neurocognitive functions need to be applied, using the methods currently available in both animals and humans (see sect. v).
How can we best prevent sleep state effects specifically on upper airway collapsibility via pharmacological targets? Do we now have sufficient evidence in support of targets with cholinesterase inhibitors? Do we need to further identify key receptor subtypes?
Obesity complicates defining a causal relationship between OSA and insulin resistance. How can we best design large-scale, controlled studies to determine CPAP treatment effects on insulin resistance and type II diabetes in carefully phenotyped subjects?
Can we identify, on the basis of genotype or phenotype, OSA patients most at risk for development of cardiovascular disease? Can pharmaceutical interventions be used, alone or in combination with traditional therapies, to prevent or redress the cardiovascular consequences of OSA?
Alternative treatments aimed at minimizing respiratory control system loop gain and breathing instabilities need to be tested specifically in those OSA patients with upper airways that are only moderately susceptible to collapse.
Grants
Financial support for the research contained in this manuscript was provided by the National Heart, Lung, and Blood Institute, including HL-15469 and HL-50531 (to J. A. Dempsey), HL-80492 and HL-79555 (to S. C. Veasey), HL-74072 (to B. J. Morgan), and HL-63767 (to C. P. O'Donnell).
ACKNOWLEDGMENTS
J. A. Dempsey and B. J. Morgan are indebted to their colleague the late Dr. James B. Skatrud for his 30 years of contributions to and leadership of the University of Wisconsin-Madison respiratory research team. Jennifer Montoya and Anthony Jacques are acknowledged for invaluable administrative assistance in preparing the manuscript and Dr. Robert C. Molthen, Dr. Alan Schwartz, and Dr. Paul Peppard for important scientific input.
Present addresses: S. C. Veasey, Center for Sleep and Respiratory Neurobiology and Dept. of Medicine, School of Medicine, Univ. of Pennsylvania, Philadelphia, PA; B. J. Morgan, The John Rankin Laboratory of Pulmonary Medicine, Depts. of Population Health Sciences and of Orthopedics and Rehabilitation, School of Medicine and Public Health, Univ. of Wisconsin, Madison, WI; C. P. O'Donnell, Dept. of Medicine, Div of Pulmonary, Alergy and Critical Care Medicine, Univ. of Pittsburgh School of Medicine, Pittsburgh, PA.
Address for reprint requests and other correspondence: J. A. Dempsey, Univ. of Wisconsin-Madison, 4245 MSC, 1300 University Ave., Madison, WI 53706 (e-mail: jdempsey@wisc.edu).
REFERENCES
- 1. Aalkjaer C, Poston L. Effects of pH on vascular tension: which are the important mechanisms? J Vasc Res 33: 347–359, 1996 [DOI] [PubMed] [Google Scholar]
- 2. Abinader EG, Peled N, Sharif D, Lavie P. ST-segment depression during obstructive sleep apnea. Am J Cardiol 73: 727, 1994 [DOI] [PubMed] [Google Scholar]
- 3. Adams MR, Kinlay S, Blake GJ, Orford JL, Ganz P, Selwyn AP. Atherogenic lipids and endothelial dysfunction: mechanisms in the genesis of ischemic syndromes. Annu Rev Med 51: 149–167, 2000 [DOI] [PubMed] [Google Scholar]
- 4. Al Delaimy WK, Manson JE, Willett WC, Stampfer MJ, Hu FB. Snoring as a risk factor for type II diabetes mellitus: a prospective study. Am J Epidemiol 155: 387–393, 2002 [DOI] [PubMed] [Google Scholar]
- 5. Al-Zubaidy ZA, Erickson RL, Greer JJ. Serotonergic and noradrenergic effects on respiratory neural discharge in the medullary slice preparation of neonatal rats. Pflügers Arch 431: 942–949, 1996 [DOI] [PubMed] [Google Scholar]
- 6. Alchanatis M, Tourkohoriti G, Kakouros S, Kosmas E, Podaras S, Jordanoglou JB. Daytime pulmonary hypertension in patients with obstructive sleep apnea: the effect of continuous positive airway pressure on pulmonary hemodynamics. Respiration 68: 566–572, 2001 [DOI] [PubMed] [Google Scholar]
- 7. Alchanatis M, Tourkohoriti G, Kosmas EN, Panoutsopoulos G, Kakouros S, Papadima K, Gaga M, Jordanoglou JB. Evidence for left ventricular dysfunction in patients with obstructive sleep apnoea syndrome. Eur Respir J 20: 1239–1245, 2002 [DOI] [PubMed] [Google Scholar]
- 8. Alex CG, Onal E, Lopata M. Upper airway occlusion during sleep in patients with Cheyne-Stokes respiration. Am Rev Respir Dis 133: 42–45, 1986 [DOI] [PubMed] [Google Scholar]
- 9. Allahdadi KJ, Cherng TW, Pai H, Silva AQ, Walker BR, Nelin LD, Kanagy NL. Endothelin type A receptor antagonist normalizes blood pressure in rats exposed to eucapnic intermittent hypoxia. Am J Physiol Heart Circ Physiol 295: H434–H440, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Allahdadi KJ, Walker BR, Kanagy NL. Augmented endothelin vasoconstriction in intermittent hypoxia-induced hypertension. Hypertension 45: 705–709, 2005 [DOI] [PubMed] [Google Scholar]
- 11. Alonso-Fernandez A, Garcia-Rio F, Arias MA, Hernanz A, de la Pena M, Pierola J, Barcelo A, Lopez-Collazo E, Agusti A. Effects of CPAP on oxidative stress and nitrate efficiency in sleep apnoea: a randomised trial. Thorax 64: 581–586, 2009 [DOI] [PubMed] [Google Scholar]
- 12. Amin RS, Carroll JL, Jeffries JL, Grone C, Bean JA, Chini B, Bokulic R, Daniels SR. Twenty-four-hour ambulatory blood pressure in children with sleep-disordered breathing. Am J Respir Crit Care Med 169: 950–956, 2004 [DOI] [PubMed] [Google Scholar]
- 13. Anand A, Remsburg-Sailor S, Launois SH, Weiss JW. Peripheral vascular resistance increases after termination of obstructive apneas. J Appl Physiol 91: 2359–2365, 2001 [DOI] [PubMed] [Google Scholar]
- 14. Antczak J, Popp R, Hajak G, Zulley J, Marienhagen J, Geisler P. Positron emission tomography findings in obstructive sleep apnea patients with residual sleepiness treated with continuous positive airway pressure. J Physiol Pharmacol 58 Suppl 5: 25–35, 2007 [PubMed] [Google Scholar]
- 15. Antic NA, Malow BA, Lange N, McEvoy RD, Olson AL, Turkington P, Windisch W, Samuels M, Stevens CA, Berry-Kravis EM, Weese-Mayer DE. PHOX2B mutation-confirmed congenital central hypoventilation syndrome: presentation in adulthood. Am J Respir Crit Care Med 174: 923–927, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Arias MA, Garcia-Rio F, Alonso-Fernandez A, Martinez I, Villamor J. Pulmonary hypertension in obstructive sleep apnoea: effects of continuous positive airway pressure: a randomized, controlled cross-over study. Eur Heart J 27: 1106–1113, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Arzt M, Young T, Finn L, Skatrud JB, Bradley TD. Association of sleep-disordered breathing and the occurrence of stroke. Am J Respir Crit Care Med 172: 1447–1451, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci 1: 876–886, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ayas NT, White DP, Al Delaimy WK, Manson JE, Stampfer MJ, Speizer FE, Patel S, Hu FB. A prospective study of self-reported sleep duration and incident diabetes in women. Diabetes Care 26: 380–384, 2003 [DOI] [PubMed] [Google Scholar]
- 20. Bachmann OP, Dahl DB, Brechtel K, Machann J, Haap M, Maier T, Loviscach M, Stumvoll M, Claussen CA, Schick F, Haring HU, Jacob S. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes 50: 2579–2584, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Bacon WH, Turlot JC, Krieger J, Stierle JL. Cephalometric evaluation of pharyngeal obstructive factors in patients with sleep apneas syndrome. Angle Orthod 60: 115–122, 1990 [DOI] [PubMed] [Google Scholar]
- 22. Badr MS, Skatrud JB, Dempsey JA. Determinants of poststimulus potentiation in humans during NREM sleep. J Appl Physiol 73: 1958–1971, 1992 [DOI] [PubMed] [Google Scholar]
- 23. Badr MS, Skatrud JB, Dempsey JA. Effect of chemoreceptor stimulation and inhibition on total pulmonary resistance in humans during NREM sleep. J Appl Physiol 76: 1682–1692, 1994 [DOI] [PubMed] [Google Scholar]
- 24. Badr MS, Skatrud JB, Simon PM, Dempsey JA. Effect of hypercapnia on total pulmonary resistance during wakefulness and during NREM sleep. Am Rev Respir Dis 144: 406–414, 1991 [DOI] [PubMed] [Google Scholar]
- 25. Badr MS, Toiber F, Skatrud JB, Dempsey J. Pharyngeal narrowing/occlusion during central sleep apnea. J Appl Physiol 78: 1806–1815, 1995 [DOI] [PubMed] [Google Scholar]
- 26. Bady E, Achkar A, Pascal S, Orvoen-Frija E, Laaban JP. Pulmonary arterial hypertension in patients with sleep apnoea syndrome. Thorax 55: 934–939, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baguet JP, Hammer L, Levy P, Pierre H, Launois S, Mallion JM, Pepin JL. The severity of oxygen desaturation is predictive of carotid wall thickening and plaque occurrence. Chest 128: 3407–3412, 2005 [DOI] [PubMed] [Google Scholar]
- 28. Baguet JP, Levy P, Barone-Rochette G, Tamisier R, Pierre H, Peeters M, Mallion JM, Pepin JL. Masked hypertension in obstructive sleep apnea syndrome. J Hypertens 26: 885–892, 2008 [DOI] [PubMed] [Google Scholar]
- 29. Bahceci M, Gokalp D, Bahceci S, Tuzcu A, Atmaca S, Arikan S. The correlation between adiposity and adiponectin, tumor necrosis factor alpha, interleukin-6 and high sensitivity C-reactive protein levels. Is adipocyte size associated with inflammation in adults? J Endocrinol Invest 30: 210–214, 2007 [DOI] [PubMed] [Google Scholar]
- 30. Bajic J, Zuperku EJ, Tonkovic-Capin M, Hopp FA. Interaction between chemoreceptor and stretch receptor inputs at medullary respiratory neurons. Am J Physiol Regul Integr Comp Physiol 266: R1951–R1961, 1994 [DOI] [PubMed] [Google Scholar]
- 31. Balfors EM, Franklin KA. Impairment of cerebral perfusion during obstructive sleep apneas. Am J Respir Crit Care Med 150: 1587–1591, 1994 [DOI] [PubMed] [Google Scholar]
- 32. Bangash MF, Xie A, Skatrud JB, Reichmuth KJ, Barczi SR, Morgan BJ. Cerebrovascular response to arousal from NREM and REM sleep. Sleep 31: 321–327, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bao G, Metreveli N, Fletcher EC. Acute and chronic blood pressure response to recurrent acoustic arousal in rats. Am J Hypertens 12: 504–510, 1999 [DOI] [PubMed] [Google Scholar]
- 34. Bar A, Tarasiuk A, Segev Y, Phillip M, Tal A. The effect of adenotonsillectomy on serum insulin-like growth factor-I and growth in children with obstructive sleep apnea syndrome. J Pediatr 135: 76–80, 1999 [DOI] [PubMed] [Google Scholar]
- 35. Barbe F, Mayoralas LR, Duran J, Masa JF, Maimo A, Montserrat JM, Monasterio C, Bosch M, Ladaria A, Rubio M, Rubio R, Medinas M, Hernandez L, Vidal S, Douglas NJ, Agusti AG. Treatment with continuous positive airway pressure is not effective in patients with sleep apnea but no daytime sleepiness. A randomized, controlled trial. Ann Intern Med 134: 1015–1023, 2001 [DOI] [PubMed] [Google Scholar]
- 36. Barnes M, McEvoy RD, Banks S, Tarquinio N, Murray CG, Vowles N, Pierce RJ. Efficacy of positive airway pressure and oral appliance in mild to moderate obstructive sleep apnea. Am J Respir Crit Care Med 170: 656–664, 2004 [DOI] [PubMed] [Google Scholar]
- 37. Bart Sangal R, Sangal JM, Thorp K. Atomoxetine improves sleepiness and global severity of illness but not the respiratory disturbance index in mild to moderate obstructive sleep apnea with sleepiness. Sleep Med 9: 506–510, 2008 [DOI] [PubMed] [Google Scholar]
- 38. Bartels NK, Borgel J, Wieczorek S, Buchner N, Hanefeld C, Bulut D, Mugge A, Rump LC, Sanner BM, Epplen JT. Risk factors and myocardial infarction in patients with obstructive sleep apnea: impact of beta2-adrenergic receptor polymorphisms. BMC Med 5: 1, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bassetti C, Aldrich MS. Sleep apnea in acute cerebrovascular diseases: final report on 128 patients. Sleep 22: 217–223, 1999 [DOI] [PubMed] [Google Scholar]
- 40. Becker H, Brandenburg U, Peter JH, von Wichert P. Reversal of sinus arrest and atrioventricular conduction block in patients with sleep apnea during nasal continuous positive airway pressure. Am J Respir Crit Care Med 151: 215–218, 1995 [DOI] [PubMed] [Google Scholar]
- 41. Becker HF, Jerrentrup A, Ploch T, Grote L, Penzel T, Sullivan CE, Peter JH. Effect of nasal continuous positive airway pressure treatment on blood pressure in patients with obstructive sleep apnea. Circulation 107: 68–73, 2003 [DOI] [PubMed] [Google Scholar]
- 42. Bedard MA, Montplaisir J, Malo J, Richer F, Rouleau I. Persistent neuropsychological deficits and vigilance impairment in sleep apnea syndrome after treatment with continuous positive airways pressure (CPAP). J Clin Exp Neuropsychol 15: 330–341, 1993 [DOI] [PubMed] [Google Scholar]
- 43. Beebe DW, Gozal D. Obstructive sleep apnea and the prefrontal cortex: towards a comprehensive model linking nocturnal upper airway obstruction to daytime cognitive and behavioral deficits. J Sleep Res 11: 1–16, 2002 [DOI] [PubMed] [Google Scholar]
- 44. Beelke M, Angeli S, Del SM, De CF, Canovaro P, Nobili L, Ferrillo F. Obstructive sleep apnea can be provocative for right-to-left shunting through a patent foramen ovale. Sleep 25: 856–862, 2002 [PubMed] [Google Scholar]
- 45. Begle RL, Badr S, Skatrud JB, Dempsey JA. Effect of lung inflation on pulmonary resistance during NREM sleep. Am Rev Respir Dis 141: 854–860, 1990 [DOI] [PubMed] [Google Scholar]
- 46. Bellingham MC, Berger AJ. Adenosine suppresses excitatory glutamatergic inputs to rat hypoglossal motoneurons in vitro. Neurosci Lett 177: 143–146, 1994 [DOI] [PubMed] [Google Scholar]
- 47. Bellingham MC, Berger AJ. Presynaptic depression of excitatory synaptic inputs to rat hypoglossal motoneurons by muscarinic M2 receptors. J Neurophysiol 76: 3758–3770, 1996 [DOI] [PubMed] [Google Scholar]
- 48. Benarroch EE. Brainstem respiratory control: substrates of respiratory failure of multiple system atrophy. Mov Disord 22: 155–161, 2007 [DOI] [PubMed] [Google Scholar]
- 49. Benavides A, Pastor D, Santos P, Tranque P, Calvo S. CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia 52: 261–275, 2005 [DOI] [PubMed] [Google Scholar]
- 50. Bentourkia M, Bol A, Ivanoiu A, Labar D, Sibomana M, Coppens A, Michel C, Cosnard G, De Volder AG. Comparison of regional cerebral blood flow and glucose metabolism in the normal brain: effect of aging. J Neurol Sci 181: 19–28, 2000 [DOI] [PubMed] [Google Scholar]
- 51. Bergman RN, Prager R, Volund A, Olefsky JM. Equivalence of the insulin sensitivity index in man derived by the minimal model method and the euglycemic glucose clamp. J Clin Invest 79: 790–800, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Berry RB, Yamaura EM, Gill K, Reist C. Acute effects of paroxetine on genioglossus activity in obstructive sleep apnea. Sleep 22: 1087–1092, 1999 [DOI] [PubMed] [Google Scholar]
- 53. Berssenbrugge A, Dempsey J, Iber C, Skatrud J, Wilson P. Mechanisms of hypoxia-induced periodic breathing during sleep in humans. J Physiol 343: 507–526, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berssenbrugge AD, Dempsey JA, Skatrud JB. Effects of sleep state on ventilatory acclimatization to hypoxia in humans. J Appl Physiol 57: 1089–1096, 1984 [DOI] [PubMed] [Google Scholar]
- 55. Bickelmann AG, Burwell CS, Robin ED, Whaley RD. Extreme obesity associated with alveolar hypoventilation: a Pickwickian syndrome. Am J Med 21: 811–818, 1956 [DOI] [PubMed] [Google Scholar]
- 56. Bixler EO, Vgontzas AN, Lin HM, Liao D, Calhoun S, Fedok F, Vlasic V, Graff G. Blood pressure associated with sleep-disordered breathing in a population sample of children. Hypertension 52: 841–846, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bixler EO, Vgontzas AN, Lin HM, Ten HT, Leiby BE, Vela-Bueno A, Kales A. Association of hypertension and sleep-disordered breathing. Arch Intern Med 160: 2289–2295, 2000 [DOI] [PubMed] [Google Scholar]
- 58. Bleeke T, Zhang H, Madamanchi N, Patterson C, Faber JE. Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ Res 94: 37–45, 2004 [DOI] [PubMed] [Google Scholar]
- 59. Boden G, Lebed B, Schatz M, Homko C, Lemieux S. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes 50: 1612–1617, 2001 [DOI] [PubMed] [Google Scholar]
- 60. Bokinsky G, Miller M, Ault K, Husband P, Mitchell J. Spontaneous platelet activation and aggregation during obstructive sleep apnea and its response to therapy with nasal continuous positive airway pressure. A preliminary investigation. Chest 108: 625–630, 1995 [DOI] [PubMed] [Google Scholar]
- 61. Bostrom KB, Hedner J, Melander O, Grote L, Gullberg B, Rastam L, Groop L, Lindblad U. Interaction between the angiotensin-converting enzyme gene insertion/deletion polymorphism and obstructive sleep apnoea as a mechanism for hypertension. J Hypertens 25: 779–783, 2007 [DOI] [PubMed] [Google Scholar]
- 62. Bouryi VA, Lewis DI. The modulation by 5-HT of glutamatergic inputs from the raphe pallidus to rat hypoglossal motoneurones, in vitro. J Physiol 553: 1019–1031, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 169: 764–769, 2004 [DOI] [PubMed] [Google Scholar]
- 64. Boyd JH, Petrof BJ, Hamid Q, Fraser R, Kimoff RJ. Upper airway muscle inflammation and denervation changes in obstructive sleep apnea. Am J Respir Crit Care Med 170: 541–546, 2004 [DOI] [PubMed] [Google Scholar]
- 65. Bradley TD, Brown IG, Grossman RF, Zamel N, Martinez D, Phillipson EA, Hoffstein V. Pharyngeal size in snorers, nonsnorers, and patients with obstructive sleep apnea. N Engl J Med 315: 1327–1331, 1986 [DOI] [PubMed] [Google Scholar]
- 66. Bradley TD, Logan AG, Kimoff RJ, Series F, Morrison D, Ferguson K, Belenkie I, Pfeifer M, Fleetham J, Hanly P, Smilovitch M, Tomlinson G, Floras JS. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med 353: 2025–2033, 2005 [DOI] [PubMed] [Google Scholar]
- 67. Bradley TD, Rutherford R, Grossman RF, Lue F, Zamel N, Moldofsky H, Phillipson EA. Role of daytime hypoxemia in the pathogenesis of right heart failure in the obstructive sleep apnea syndrome. Am Rev Respir Dis 131: 835–839, 1985 [DOI] [PubMed] [Google Scholar]
- 68. Bratel T, Wennlund A, Carlstrom K. Pituitary reactivity, androgens and catecholamines in obstructive sleep apnoea. Effects of continuous positive airway pressure treatment (CPAP). Respir Med 93: 1–7, 1999 [DOI] [PubMed] [Google Scholar]
- 69. Brennick MJ, Pack AI, Ko K, Kim E, Pickup S, Maislin G, Schwab RJ. Altered upper airway and soft tissue structures in the New Zealand Obese mouse. Am J Respir Crit Care Med 179: 158–169, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bridger GP, Proctor DF. Maximum nasal inspiratory flow and nasal resistance. Ann Otol Rhinol Laryngol 79: 481–488, 1970 [DOI] [PubMed] [Google Scholar]
- 71. Brooks B, Cistulli PA, Borkman M, Ross G, McGhee S, Grunstein RR, Sullivan CE, Yue DK. Obstructive sleep apnea in obese noninsulin-dependent diabetic patients: effect of continuous positive airway pressure treatment on insulin responsiveness. J Clin Endocrinol Metab 79: 1681–1685, 1994 [DOI] [PubMed] [Google Scholar]
- 72. Brooks D, Horner RL, Floras JS, Kozar LF, Render-Teixeira CL, Phillipson EA. Baroreflex control of heart rate in a canine model of obstructive sleep apnea. Am J Respir Crit Care Med 159: 1293–1297, 1999 [DOI] [PubMed] [Google Scholar]
- 73. Brooks D, Horner RL, Kozar LF, Render-Teixeira CL, Phillipson EA. Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J Clin Invest 99: 106–109, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Brouillette RT, Thach BT. A neuromuscular mechanism maintaining extrathoracic airway patency. J Appl Physiol 46: 772–779, 1979 [DOI] [PubMed] [Google Scholar]
- 75. Brown NJ. Aldosterone and vascular inflammation. Hypertension 51: 161–167, 2008 [DOI] [PubMed] [Google Scholar]
- 76. Browning KN, Travagli RA. Characterization of the in vitro effects of 5-hydroxytryptamine (5-HT) on identified neurones of the rat dorsal motor nucleus of the vagus (DMV). Br J Pharmacol 128: 1307–1315, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Buchner NJ, Sanner BM, Borgel J, Rump LC. Continuous positive airway pressure treatment of mild to moderate obstructive sleep apnea reduces cardiovascular risk. Am J Respir Crit Care Med 176: 1274–1280, 2007 [DOI] [PubMed] [Google Scholar]
- 78. Bulow K. Respiration and wakefulness in man. Acta Physiol Scand Suppl 209: 1–110, 1963 [PubMed] [Google Scholar]
- 79. Buxbaum SG, Elston RC, Tishler PV, Redline S. Genetics of the apnea hypopnea index in Caucasians and African Americans. I. Segregation analysis. Genet Epidemiol 22: 243–253, 2002 [DOI] [PubMed] [Google Scholar]
- 80. Campen MJ, Shimoda LA, O'Donnell CP. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J Appl Physiol 99: 2028–2035, 2005 [DOI] [PubMed] [Google Scholar]
- 81. Campos-Rodriguez F, Grilo-Reina A, Perez-Ronchel J, Merino-Sanchez M, Gonzalez-Benitez MA, Beltran-Robles M, Almeida-Gonzalez C. Effect of continuous positive airway pressure on ambulatory BP in patients with sleep apnea and hypertension: a placebo-controlled trial. Chest 129: 1459–1467, 2006 [DOI] [PubMed] [Google Scholar]
- 82. Carley DW, Radulovacki M. Role of peripheral serotonin in the regulation of central sleep apneas in rats. Chest 115: 1397–1401, 1999 [DOI] [PubMed] [Google Scholar]
- 83. Carlson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest 103: 1763–1768, 1993 [DOI] [PubMed] [Google Scholar]
- 84. Carlson JT, Hedner JA, Sellgren J, Elam M, Wallin BG. Depressed baroreflex sensitivity in patients with obstructive sleep apnea. Am J Respir Crit Care Med 154: 1490–1496, 1996 [DOI] [PubMed] [Google Scholar]
- 85. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 407: 249–257, 2000 [DOI] [PubMed] [Google Scholar]
- 86. Carpagnano GE, Kharitonov SA, Resta O, Foschino-Barbaro MP, Gramiccioni E, Barnes PJ. 8-Isoprostane, a marker of oxidative stress, is increased in exhaled breath condensate of patients with obstructive sleep apnea after night and is reduced by continuous positive airway pressure therapy. Chest 124: 1386–1392, 2003 [DOI] [PubMed] [Google Scholar]
- 87. Casale PN, Devereux RB, Alonso DR, Campo E, Kligfield P. Improved sex-specific criteria of left ventricular hypertrophy for clinical and computer interpretation of electrocardiograms: validation with autopsy findings. Circulation 75: 565–572, 1987 [DOI] [PubMed] [Google Scholar]
- 88. Chamberlin NL, Bocchiaro CM, Greene RW, Feldman JL. Nicotinic excitation of rat hypoglossal motoneurons. Neuroscience 115: 861–870, 2002 [DOI] [PubMed] [Google Scholar]
- 89. Chan E, Steenland HW, Liu H, Horner RL. Endogenous excitatory drive modulating respiratory muscle activity across sleep-wake states. Am J Respir Crit Care Med 174: 1264–1273, 2006 [DOI] [PubMed] [Google Scholar]
- 90. Chaouat A, Weitzenblum E, Krieger J, Oswald M, Kessler R. Pulmonary hemodynamics in the obstructive sleep apnea syndrome. Results in 220 consecutive patients. Chest 109: 380–386, 1996 [DOI] [PubMed] [Google Scholar]
- 91. Chapleau MW. Arterial baroreflexes. In: Hypertension Primer, edited by Izzo JL, Jr., Sica DA, Black HR. Dallas, TX: American Heart Association, 2008 [Google Scholar]
- 92. Chapman RW, Santiago TV, Edelman NH. Effects of graded reduction of brain blood flow on chemical control of breathing. J Appl Physiol 47: 1289–1294, 1979 [DOI] [PubMed] [Google Scholar]
- 93. Chapman RW, Santiago TV, Edelman NH. Effects of graded reduction of brain blood flow on ventilation in unanesthetized goats. J Appl Physiol 47: 104–111, 1979 [DOI] [PubMed] [Google Scholar]
- 94. Chen J, He L, Dinger B, Fidone S. Cellular mechanisms involved in rabbit carotid body excitation elicited by endothelin peptides. Respir Physiol 121: 13–23, 2000 [DOI] [PubMed] [Google Scholar]
- 95. Chen L, Einbinder E, Zhang Q, Hasday J, Balke CW, Scharf SM. Oxidative stress and left ventricular function with chronic intermittent hypoxia in rats. Am J Respir Crit Care Med 172: 915–920, 2005 [DOI] [PubMed] [Google Scholar]
- 96. Chen L, Scharf SM. Systemic and myocardial hemodynamics during periodic obstructive apneas in sedated pigs. J Appl Physiol 84: 1289–1298, 1998 [DOI] [PubMed] [Google Scholar]
- 97. Chen L, Zhang J, Gan TX, Chen-Izu Y, Hasday JD, Karmazyn M, Balke CW, Scharf SM. Left ventricular dysfunction and associated cellular injury in rats exposed to chronic intermittent hypoxia. J Appl Physiol 104: 218–223, 2008 [DOI] [PubMed] [Google Scholar]
- 98. Chenuel BJ, Smith CA, Skatrud JB, Henderson KS, Dempsey JA. Increased propensity for apnea in response to acute elevations in left atrial pressure during sleep in the dog. J Appl Physiol 101: 76–83, 2006 [DOI] [PubMed] [Google Scholar]
- 99. Cherniack NS, Longobardo GS. Mathematical models of periodic breathing and their usefulness in understanding cardiovascular and respiratory disorders. Exp Physiol 91: 295–305, 2006 [DOI] [PubMed] [Google Scholar]
- 100. Chervin RD, Archbold KH, Dillon JE, Panahi P, Pituch KJ, Dahl RE, Guilleminault C. Inattention, hyperactivity, and symptoms of sleep-disordered breathing. Pediatrics 109: 449–456, 2002 [DOI] [PubMed] [Google Scholar]
- 101. Cheyne J. A case of apoplexy in which the fleshy part of the heart was connected to fat. Dublin Hospital Report 2: 216–223, 1818 [Google Scholar]
- 102. Chin K, Nakamura T, Shimizu K, Mishima M, Nakamura T, Miyasaka M, Ohi M. Effects of nasal continuous positive airway pressure on soluble cell adhesion molecules in patients with obstructive sleep apnea syndrome. Am J Med 109: 562–567, 2000 [DOI] [PubMed] [Google Scholar]
- 103. Chin K, Ohi M, Kita H, Noguchi T, Otsuka N, Tsuboi T, Mishima M, Kuno K. Effects of NCPAP therapy on fibrinogen levels in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 153: 1972–1976, 1996 [DOI] [PubMed] [Google Scholar]
- 104. Chin K, Shimizu K, Nakamura T, Narai N, Masuzaki H, Ogawa Y, Mishima M, Nakao K, Ohi M. Changes in intra-abdominal visceral fat and serum leptin levels in patients with obstructive sleep apnea syndrome following nasal continuous positive airway pressure therapy. Circulation 100: 706–712, 1999 [DOI] [PubMed] [Google Scholar]
- 105. Chiu KL, Ryan CM, Shiota S, Ruttanaumpawan P, Arzt M, Haight JS, Chan CT, Floras JS, Bradley TD. Fluid shift by lower body positive pressure increases pharyngeal resistance in healthy subjects. Am J Respir Crit Care Med 174: 1378–1383, 2006 [DOI] [PubMed] [Google Scholar]
- 106. Chow CM, Xi L, Smith CA, Saupe KW, Dempsey JA. A volume-dependent apneic threshold during NREM sleep in the dog. J Appl Physiol 76: 2315–2325, 1994 [DOI] [PubMed] [Google Scholar]
- 107. Ciscar MA, Juan G, Martinez V, Ramon M, Lloret T, Minguez J, Armengot M, Marin J, Basterra J. Magnetic resonance imaging of the pharynx in OSA patients and healthy subjects. Eur Respir J 17: 79–86, 2001 [DOI] [PubMed] [Google Scholar]
- 108. Condos WR, Jr, Latham RD, Hoadley SD, Pasipoularides A. Hemodynamics of the Mueller maneuver in man: right and left heart micromanometry and Doppler echocardiography. Circulation 76: 1020–1028, 1987 [DOI] [PubMed] [Google Scholar]
- 109. Coughlin SR, Mawdsley L, Mugarza JA, Wilding JP, Calverley PM. Cardiovascular and metabolic effects of CPAP in obese males with OSA. Eur Respir J 29: 720–727, 2007 [DOI] [PubMed] [Google Scholar]
- 110. Cross MD, Mills NL, Al-Abri M, Riha R, Vennelle M, Mackay TW, Newby DE, Douglas NJ. Continuous positive airway pressure improves vascular function in obstructive sleep apnoea/hypopnoea syndrome: a randomised controlled trial. Thorax 63: 578–583, 2008 [DOI] [PubMed] [Google Scholar]
- 111. Cupini LM, Diomedi M, Placidi F, Silvestrini M, Giacomini P. Cerebrovascular reactivity and subcortical infarctions. Arch Neurol 58: 577–581, 2001 [DOI] [PubMed] [Google Scholar]
- 112. Curran AK, Rodman JR, Eastwood PR, Henderson KS, Dempsey JA, Smith CA. Ventilatory responses to specific CNS hypoxia in sleeping dogs. J Appl Physiol 88: 1840–1852, 2000 [DOI] [PubMed] [Google Scholar]
- 113. Cutler MJ, Swift NM, Keller DM, Wasmund WL, Smith ML. Hypoxia-mediated prolonged elevation of sympathetic nerve activity after periods of intermittent hypoxic apnea. J Appl Physiol 96: 754–761, 2004 [DOI] [PubMed] [Google Scholar]
- 114. Dahlof P, Norlin-Bagge E, Hedner J, Ejnell H, Hetta J, Hallstrom T. Improvement in neuropsychological performance following surgical treatment for obstructive sleep apnea syndrome. Acta Otolaryngol 122: 86–91, 2002 [DOI] [PubMed] [Google Scholar]
- 115. Dampney RA, Horiuchi J, Killinger S, Sheriff MJ, Tan PS, McDowall LM. Long-term regulation of arterial blood pressure by hypothalamic nuclei: some critical questions. Clin Exp Pharmacol Physiol 32: 419–425, 2005 [DOI] [PubMed] [Google Scholar]
- 116. Dauger S, Pattyn A, Lofaso F, Gaultier C, Goridis C, Gallego J, Brunet JF. Phox2b controls the development of peripheral chemoreceptors and afferent visceral pathways. Development 130: 6635–6642, 2003 [DOI] [PubMed] [Google Scholar]
- 117. Davidson TM. The Great Leap Forward: the anatomic basis for the acquisition of speech and obstructive sleep apnea. Sleep Med 4: 185–194, 2003 [DOI] [PubMed] [Google Scholar]
- 118. Davies RJ, Harrington KJ, Ormerod OJ, Stradling JR. Nasal continuous positive airway pressure in chronic heart failure with sleep-disordered breathing. Am Rev Respir Dis 147: 630–634, 1993 [DOI] [PubMed] [Google Scholar]
- 119. Davies RJ, Turner R, Crosby J, Stradling JR. Plasma insulin and lipid levels in untreated obstructive sleep apnoea and snoring: their comparison with matched controls and response to treatment. J Sleep Res 3: 180–185, 1994 [DOI] [PubMed] [Google Scholar]
- 120. Davila DG, Hurt RD, Offord KP, Harris CD, Shepard JW., Jr Acute effects of transdermal nicotine on sleep architecture, snoring, and sleep-disordered breathing in nonsmokers. Am J Respir Crit Care Med 150: 469–474, 1994 [DOI] [PubMed] [Google Scholar]
- 121. Day TA, Wilson RJ. A negative interaction between brainstem and peripheral respiratory chemoreceptors modulates peripheral chemoreflex magnitude. J Physiol 587: 883–896, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. De Frutos S, Duling L, Alo D, Berry T, Jackson-Weaver O, Walker M, Kanagy N, Gonzalez BL. NFATc3 is required for intermittent hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol 294: H2382–H2390, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Decker MJ, Hue GE, Caudle WM, Miller GW, Keating GL, Rye DB. Episodic neonatal hypoxia evokes executive dysfunction and regionally specific alterations in markers of dopamine signaling. Neuroscience 117: 417–425, 2003 [DOI] [PubMed] [Google Scholar]
- 124. Decker MJ, Rye DB. Neonatal intermittent hypoxia impairs dopamine signaling and executive functioning. Sleep Breath 6: 205–210, 2002 [DOI] [PubMed] [Google Scholar]
- 125. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab Gastrointest Physiol 237: E214–E223, 1979 [DOI] [PubMed] [Google Scholar]
- 126. DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem 91: 1–8, 2004 [DOI] [PubMed] [Google Scholar]
- 127. Dempsey JA. Crossing the apnoeic threshold: causes and consequences. Exp Physiol 90: 13–24, 2005 [DOI] [PubMed] [Google Scholar]
- 128. Dempsey JA, Skatrud JB. A sleep-induced apneic threshold and its consequences. Am Rev Respir Dis 133: 1163–1170, 1986 [DOI] [PubMed] [Google Scholar]
- 129. Dempsey JA, Skatrud JB, Jacques AJ, Ewanowski SJ, Woodson BT, Hanson PR, Goodman B. Anatomic determinants of sleep-disordered breathing across the spectrum of clinical and nonclinical male subjects. Chest 122: 840–851, 2002 [DOI] [PubMed] [Google Scholar]
- 130. Dempsey JA, Smith CA, Przybylowski T, Chenuel B, Xie A, Nakayama H, Skatrud JB. The ventilatory responsiveness to CO2 below eupnoea as a determinant of ventilatory stability in sleep. J Physiol 560: 1–11, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dick TE, Hsieh YH, Wang N, Prabhakar N. Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp Physiol 92: 87–97, 2007 [DOI] [PubMed] [Google Scholar]
- 132. Dickens CJH. The Posthumous Papers of the Pickwick Club. London: Chapman and Hall, 1837 [Google Scholar]
- 133. Diomedi M, Placidi F, Cupini LM, Bernardi G, Silvestrini M. Cerebral hemodynamic changes in sleep apnea syndrome and effect of continuous positive airway pressure treatment. Neurology 51: 1051–1056, 1998 [DOI] [PubMed] [Google Scholar]
- 134. Dolan E, Stanton A, Thijs L, Hinedi K, Atkins N, McClory S, Den HE, McCormack P, Staessen JA, O'Brien E. Superiority of ambulatory over clinic blood pressure measurement in predicting mortality: the Dublin outcome study. Hypertension 46: 156–161, 2005 [DOI] [PubMed] [Google Scholar]
- 135. Dopp J, Reichmuth K, Puleo D, Hayes D, Skatrud J, Morgan B. Vascular response to hypercapnia in patients with obstructive sleep apnea. Am J Respir Crit Care Med 173: A517, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dorkova Z, Petrasova D, Molcanyiova A, Popovnakova M, Tkacova R. Effects of CPAP on cardiovascular risk profile in patients with severe obstructive sleep apnea and metabolic syndrome. Chest 134: 686–692, 2008 [DOI] [PubMed] [Google Scholar]
- 137. Douglas CG, Haldane JS, Henderson Y, Schneider EC. Physiological observations made on pikes peak CO, with special reference to adaptaion to low barometric pressure. Phil Trans R Soc Lond Ser B 203: 185–318, 1913 [Google Scholar]
- 138. Douse MA, White DP. Serotonergic effects on hypoglossal neural activity and reflex responses. Brain Res 726: 213–222, 1996 [PubMed] [Google Scholar]
- 139. Drager LF, Bortolotto LA, Figueiredo AC, Krieger EM, Lorenzi GF. Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med 176: 706–712, 2007 [DOI] [PubMed] [Google Scholar]
- 140. Drager LF, Bortolotto LA, Figueiredo AC, Silva BC, Krieger EM, Lorenzi-Filho G. Obstructive sleep apnea, hypertension, and their interaction on arterial stiffness and heart remodeling. Chest 131: 1379–1386, 2007 [DOI] [PubMed] [Google Scholar]
- 141. Duran J, Esnaola S, Rubio R, Iztueta A. Obstructive sleep apnea-hypopnea and related clinical features in a population-based sample of subjects aged 30 to 70 yr. Am J Respir Crit Care Med 163: 685–689, 2001 [DOI] [PubMed] [Google Scholar]
- 142. Duyndam MC, Hulscher TM, Fontijn D, Pinedo HM, Boven E. Induction of vascular endothelial growth factor expression and hypoxia-inducible factor 1alpha protein by the oxidative stressor arsenite. J Biol Chem 276: 48066–48076, 2001 [DOI] [PubMed] [Google Scholar]
- 143. Dyken ME, Somers VK, Yamada T, Ren ZY, Zimmerman MB. Investigating the relationship between stroke and obstructive sleep apnea. Stroke 27: 401–407, 1996 [DOI] [PubMed] [Google Scholar]
- 144. Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med 165: 934–939, 2002 [DOI] [PubMed] [Google Scholar]
- 145. Dziewas R, Ritter M, Kruger L, Berger S, Langer C, Kraus J, Dittrich R, Schabitz WR, Ringelstein EB, Young P. C-reactive protein and fibrinogen in acute stroke patients with and without sleep apnea. Cerebrovasc Dis 24: 412–417, 2007 [DOI] [PubMed] [Google Scholar]
- 146. Dziewas R, Schilling M, Engel P, Boentert M, Hor H, Okegwo A, Ludemann P, Ringelstein EB, Young P. Treatment for obstructive sleep apnoea: effect on peripheral nerve function. J Neurol Neurosurg Psychiatry 78: 295–297, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Eastwood PR, Satoh M, Curran AK, Zayas MT, Smith CA, Dempsey JA. Inhibition of inspiratory motor output by high-frequency low-pressure oscillations in the upper airway of sleeping dogs. J Physiol 517: 259–271, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Eckert DJ, Malhotra A, Lo YL, White DP, Jordan AS. The influence of obstructive sleep apnea and gender on genioglossus activity during rapid eye movement sleep. Chest 135: 957–964, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Eisensehr I, Ehrenberg BL, Noachtar S, Korbett K, Byrne A, McAuley A, Palabrica T. Platelet activation, epinephrine, and blood pressure in obstructive sleep apnea syndrome. Neurology 51: 188–195, 1998 [DOI] [PubMed] [Google Scholar]
- 150. El Solh AA, Akinnusi ME, Baddoura FH, Mankowski CR. Endothelial cell apoptosis in obstructive sleep apnea: a link to endothelial dysfunction. Am J Respir Crit Care Med 175: 1186–1191, 2007 [DOI] [PubMed] [Google Scholar]
- 151. El Solh AA, Saliba R, Bosinski T, Grant BJ, Berbary E, Miller N. Allopurinol improves endothelial function in sleep apnoea: a randomised controlled study. Eur Respir J 27: 997–1002, 2006 [DOI] [PubMed] [Google Scholar]
- 152. Eldridge FL. Central neural respiratory stimulatory effect of active respiration. J Appl Physiol 37: 723–735, 1974 [DOI] [PubMed] [Google Scholar]
- 153. Engleman HM, Douglas NJ. Sleep. 4: Sleepiness, cognitive function, and quality of life in obstructive sleep apnoea/hypopnoea syndrome. Thorax 59: 618–622, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Engwall MJ, Smith CA, Dempsey JA, Bisgard GE. Ventilatory afterdischarge and central respiratory drive interactions in the awake goat. J Appl Physiol 76: 416–423, 1994 [DOI] [PubMed] [Google Scholar]
- 155. Enright PL, Goodwin JL, Sherrill DL, Quan JR, Quan SF. Blood pressure elevation associated with sleep-related breathing disorder in a community sample of white and Hispanic children: the Tucson Children's Assessment of Sleep Apnea study. Arch Pediatr Adolesc Med 157: 901–904, 2003 [DOI] [PubMed] [Google Scholar]
- 156. Entzian P, Linnemann K, Schlaak M, Zabel P. Obstructive sleep apnea syndrome and circadian rhythms of hormones and cytokines. Am J Respir Crit Care Med 153: 1080–1086, 1996 [DOI] [PubMed] [Google Scholar]
- 157. Estabrooke IV, McCarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, Saper CB, Scammell TE. Fos expression in orexin neurons varies with behavioral state. J Neurosci 21: 1656–1662, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Faccenda JF, Mackay TW, Boon NA, Douglas NJ. Randomized placebo-controlled trial of continuous positive airway pressure on blood pressure in the sleep apnea-hypopnea syndrome. Am J Respir Crit Care Med 163: 344–348, 2001 [DOI] [PubMed] [Google Scholar]
- 159. Fagan KA. Selected contribution: pulmonary hypertension in mice following intermittent hypoxia. J Appl Physiol 90: 2502–2507, 2001 [DOI] [PubMed] [Google Scholar]
- 160. Farmer WC, Glenn WW, Gee JB. Alveolar hypoventilation syndrome. Studies of ventilatory control in patients selected for diaphragm pacing. Am J Med 64: 39–49, 1978 [DOI] [PubMed] [Google Scholar]
- 161. Fencl V, Miller TB, Pappenheimer JR. Studies on the respiratory response to disturbances of acid-base balance, with deductions concerning the ionic composition of cerebral interstitial fluid. Am J Physiol 210: 459–472, 1966 [DOI] [PubMed] [Google Scholar]
- 162. Fenik VB, Davies RO, Kubin L. Noradrenergic, serotonergic and GABAergic antagonists injected together into the XII nucleus abolish the REM sleep-like depression of hypoglossal motoneuronal activity. J Sleep Res 14: 419–429, 2005 [DOI] [PubMed] [Google Scholar]
- 163. Fenik VB, Davies RO, Kubin L. REM sleep-like atonia of hypoglossal (XII) motoneurons is caused by loss of noradrenergic and serotonergic inputs. Am J Respir Crit Care Med 172: 1322–1330, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Fenik VB, Davies RO, Kubin L. REM sleep-like atonia of hypoglossal (XII) motoneurons is caused by loss of noradrenergic and serotonergic inputs. Am J Respir Crit Care Med 172: 1322–1330, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Findley LJ, Ries AL, Tisi GM, Wagner PD. Hypoxemia during apnea in normal subjects: mechanisms and impact of lung volume. J Appl Physiol 55: 1777–1783, 1983 [DOI] [PubMed] [Google Scholar]
- 166. Fletcher EC, Bao G, Li R. Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension 34: 309–314, 1999 [DOI] [PubMed] [Google Scholar]
- 167. Fletcher EC, Bao G, Miller CC., III Effect of recurrent episodic hypocapnic, eucapnic, and hypercapnic hypoxia on systemic blood pressure. J Appl Physiol 78: 1516–1521, 1995 [DOI] [PubMed] [Google Scholar]
- 168. Fletcher EC, DeBehnke RD, Lovoi MS, Gorin AB. Undiagnosed sleep apnea in patients with essential hypertension. Ann Intern Med 103: 190–195, 1985 [DOI] [PubMed] [Google Scholar]
- 169. Fletcher EC, Lesske J, Behm R, Miller CC, III, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol 72: 1978–1984, 1992 [DOI] [PubMed] [Google Scholar]
- 170. Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension 20: 612–619, 1992 [DOI] [PubMed] [Google Scholar]
- 171. Fletcher EC, Lesske J, Qian W, Miller CC, III, Unger T. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension 19: 555–561, 1992 [DOI] [PubMed] [Google Scholar]
- 172. Fletcher EC, Miller J, Schaaf JW, Fletcher JG. Urinary catecholamines before and after tracheostomy in patients with obstructive sleep apnea and hypertension. Sleep 10: 35–44, 1987 [DOI] [PubMed] [Google Scholar]
- 173. Fletcher EC, Orolinova N, Bader M. Blood pressure response to chronic episodic hypoxia: the renin-angiotensin system. J Appl Physiol 92: 627–633, 2002 [DOI] [PubMed] [Google Scholar]
- 174. Fletcher EC, Schaaf JW, Miller J, Fletcher JG. Long-term cardiopulmonary sequelae in patients with sleep apnea and chronic lung disease. Am Rev Respir Dis 135: 525–533, 1987 [DOI] [PubMed] [Google Scholar]
- 175. Foster GE, Hanly PJ, Ostrowski M, Poulin MJ. Effects of continuous positive airway pressure on cerebral vascular response to hypoxia in patients with obstructive sleep apnea. Am J Respir Crit Care Med 175: 720–725, 2007 [DOI] [PubMed] [Google Scholar]
- 176. Franklin KA, Nilsson JB, Sahlin C, Naslund U. Sleep apnoea and nocturnal angina. Lancet 345: 1085–1087, 1995 [DOI] [PubMed] [Google Scholar]
- 177. Franklin KA, Sandstrom E, Johansson G, Balfors EM. Hemodynamics, cerebral circulation, and oxygen saturation in Cheyne-Stokes respiration. J Appl Physiol 83: 1184–1191, 1997 [DOI] [PubMed] [Google Scholar]
- 178. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature 395: 763–770, 1998 [DOI] [PubMed] [Google Scholar]
- 179. Fung JW, Li TS, Choy DK, Yip GW, Ko FW, Sanderson JE, Hui DS. Severe obstructive sleep apnea is associated with left ventricular diastolic dysfunction. Chest 121: 422–429, 2002 [DOI] [PubMed] [Google Scholar]
- 180. Fung SJ, Yamuy J, Sampogna S, Morales FR, Chase MH. Hypocretin (orexin) input to trigeminal and hypoglossal motoneurons in the cat: a double-labeling immunohistochemical study. Brain Res 903: 257–262, 2001 [DOI] [PubMed] [Google Scholar]
- 181. Funk GD, Kanjhan R, Walsh C, Lipski J, Comer AM, Parkis MA, Housley GD. P2 receptor excitation of rodent hypoglossal motoneuron activity in vitro and in vivo: a molecular physiological analysis. J Neurosci 17: 6325–6337, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Funk GD, Smith JC, Feldman JL. Development of thyrotropin-releasing hormone and norepinephrine potentiation of inspiratory-related hypoglossal motoneuron discharge in neonatal and juvenile mice in vitro. J Neurophysiol 72: 2538–2541, 1994 [DOI] [PubMed] [Google Scholar]
- 183. Gami AS, Howard DE, Olson EJ, Somers VK. Day-night pattern of sudden death in obstructive sleep apnea. N Engl J Med 352: 1206–1214, 2005 [DOI] [PubMed] [Google Scholar]
- 184. Gami AS, Pressman G, Caples SM, Kanagala R, Gard JJ, Davison DE, Malouf JF, Ammash NM, Friedman PA, Somers VK. Association of atrial fibrillation and obstructive sleep apnea. Circulation 110: 364–367, 2004 [DOI] [PubMed] [Google Scholar]
- 185. Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Sympathoexcitation by central ANG II: roles for AT1 receptor upregulation and NAD(P)H oxidase in RVLM. Am J Physiol Heart Circ Physiol 288: H2271–H2279, 2005 [DOI] [PubMed] [Google Scholar]
- 186. Gargaglioni LH, Bicego KC, Nucci TB, Branco LG. Serotoninergic receptors in the anteroventral preoptic region modulate the hypoxic ventilatory response. Respir Physiol Neurobiol 153: 1–13, 2006 [DOI] [PubMed] [Google Scholar]
- 187. Garpestad E, Katayama H, Parker JA, Ringler J, Lilly J, Yasuda T, Moore RH, Strauss HW, Weiss JW. Stroke volume and cardiac output decrease at termination of obstructive apneas. J Appl Physiol 73: 1743–1748, 1992 [DOI] [PubMed] [Google Scholar]
- 188. Gastaut H, Tassinari CA, Duron B. Polygraphic study of the episodic diurnal and nocturnal (hypnic and respiratory) manifestations of the Pickwick syndrome. Brain Res 1: 167–186, 1966 [DOI] [PubMed] [Google Scholar]
- 189. Gatti PJ, Llewellyn-Smith IJ, Sun QJ, Chalmers J, Pilowsky P. Substance P-immunoreactive boutons closely appose inspiratory protruder hypoglossal motoneurons in the cat. Brain Res 834: 155–159, 1999 [DOI] [PubMed] [Google Scholar]
- 190. Gerasimovskaya EV, Ahmad S, White CW, Jones PL, Carpenter TC, Stenmark KR. Extracellular ATP is an autocrine/paracrine regulator of hypoxia-induced adventitial fibroblast growth. Signaling through extracellular signal-regulated kinase-1/2 and the Egr-1 transcription factor. J Biol Chem 277: 44638–44650, 2002 [DOI] [PubMed] [Google Scholar]
- 191. Gianotti L, Pivetti S, Lanfranco F, Tassone F, Navone F, Vittori E, Rossetto R, Gauna C, Destefanis S, Grottoli S, De Giorgi R, Gai V, Ghigo E, Maccario M. Concomitant impairment of growth hormone secretion and peripheral sensitivity in obese patients with obstructive sleep apnea syndrome. J Clin Endocrinol Metab 87: 5052–5057, 2002 [DOI] [PubMed] [Google Scholar]
- 192. Gilmartin G, Tamisier R, Anand A, Cunnington D, Weiss JW. Evidence of impaired hypoxic vasodilation after intermediate-duration hypoxic exposure in humans. Am J Physiol Heart Circ Physiol 291: H2173–H2180, 2006 [DOI] [PubMed] [Google Scholar]
- 193. Gilmartin GS, Daly RW, Thomas RJ. Recognition and management of complex sleep-disordered breathing. Curr Opin Pulm Med 11: 485–493, 2005 [DOI] [PubMed] [Google Scholar]
- 194. Gjorup PH, Sadauskiene L, Wessels J, Nyvad O, Strunge B, Pedersen EB. Abnormally increased endothelin-1 in plasma during the night in obstructive sleep apnea: relation to blood pressure and severity of disease. Am J Hypertens 20: 44–52, 2007 [DOI] [PubMed] [Google Scholar]
- 195. Gold AR, Schwartz AR. The pharyngeal critical pressure. The whys and hows of using nasal continuous positive airway pressure diagnostically. Chest 110: 1077–1088, 1996 [DOI] [PubMed] [Google Scholar]
- 196. Golembeski SM, Fagan KA. Oxidative stress, inflammation and gene expression in intermittent hypoxic pulmonary hypertension. Am J Respir Crit Care Med 171: A881, 2005 [Google Scholar]
- 197. Gonzalez NC, Allen J, Schmidt EJ, Casillan AJ, Orth T, Wood JG. Role of the renin-angiotensin system in the systemic microvascular inflammation of alveolar hypoxia. Am J Physiol Heart Circ Physiol 292: H2285–H2294, 2007 [DOI] [PubMed] [Google Scholar]
- 198. Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 27: 252–259, 1996 [DOI] [PubMed] [Google Scholar]
- 199. Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal 8: 1391–1418, 2006 [DOI] [PubMed] [Google Scholar]
- 200. Gothe B, Strohl KP, Levin S, Cherniack NS. Nicotine: a different approach to treatment of obstructive sleep apnea. Chest 87: 11–17, 1985 [DOI] [PubMed] [Google Scholar]
- 201. Gotsopoulos H, Kelly JJ, Cistulli PA. Oral appliance therapy reduces blood pressure in obstructive sleep apnea: a randomized, controlled trial. Sleep 27: 934–941, 2004 [DOI] [PubMed] [Google Scholar]
- 202. Gozal D, Capdevila OS, Kheirandish-Gozal L. Metabolic alterations and systemic inflammation in obstructive sleep apnea among nonobese and obese prepubertal children. Am J Respir Crit Care Med 177: 1142–1149, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci 21: 2442–2450, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Gozal D, Kheirandish-Gozal L. Neurocognitive and behavioral morbidity in children with sleep disorders. Curr Opin Pulm Med 13: 505–509, 2007 [DOI] [PubMed] [Google Scholar]
- 205. Greenberg H, Ye X, Wilson D, Htoo AK, Hendersen T, Liu SF. Chronic intermittent hypoxia activates nuclear factor-kappaB in cardiovascular tissues in vivo. Biochem Biophys Res Commun 343: 591–596, 2006 [DOI] [PubMed] [Google Scholar]
- 206. Greenberg HE, Sica A, Batson D, Scharf SM. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol 86: 298–305, 1999 [DOI] [PubMed] [Google Scholar]
- 207. Grimm W, Koehler U, Fus E, Hoffmann J, Menz V, Funck R, Peter JH, Maisch B. Outcome of patients with sleep apnea-associated severe bradyarrhythmias after continuous positive airway pressure therapy. Am J Cardiol 86: 688–692, 2000 [DOI] [PubMed] [Google Scholar]
- 208. Grimpen F, Kanne P, Schulz E, Hagenah G, Hasenfuss G, Andreas S. Endothelin-1 plasma levels are not elevated in patients with obstructive sleep apnoea. Eur Respir J 15: 320–325, 2000 [DOI] [PubMed] [Google Scholar]
- 209. Grogaard J, Sundell H. Effect of beta-adrenergic agonists on apnea reflexes in newborn lambs. Pediatr Res 17: 213–219, 1983 [DOI] [PubMed] [Google Scholar]
- 210. Grote L, Kraiczi H, Hedner J. Reduced alpha- and beta(2)-adrenergic vascular response in patients with obstructive sleep apnea. Am J Respir Crit Care Med 162: 1480–1487, 2000 [DOI] [PubMed] [Google Scholar]
- 211. Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 68: 352–358, 1989 [DOI] [PubMed] [Google Scholar]
- 212. Guilleminault C, Connolly SJ, Winkle RA. Cardiac arrhythmia and conduction disturbances during sleep in 400 patients with sleep apnea syndrome. Am J Cardiol 52: 490–494, 1983 [DOI] [PubMed] [Google Scholar]
- 213. Guilleminault C, Eldridge FL, Dement WC. Insomnia with sleep apnea: a new syndrome. Science 181: 856–858, 1973 [DOI] [PubMed] [Google Scholar]
- 214. Guilleminault C, Partinen M, Hollman K, Powell N, Stoohs R. Familial aggregates in obstructive sleep apnea syndrome. Chest 107: 1545–1551, 1995 [DOI] [PubMed] [Google Scholar]
- 215. Guyenet PG. The 2008 Carl Ludwig Lecture: retrotrapezoid nucleus, CO2 homeostasis, and breathing automaticity. J Appl Physiol 105: 404–416, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Hajak G, Klingelhofer J, Schulz-Varszegi M, Sander D, Ruther E. Sleep apnea syndrome and cerebral hemodynamics. Chest 110: 670–679, 1996 [DOI] [PubMed] [Google Scholar]
- 218. Halbower AC, Degaonkar M, Barker PB, Earley CJ, Marcus CL, Smith PL, Prahme MC, Mahone EM. Childhood obstructive sleep apnea associates with neuropsychological deficits and neuronal brain injury. PLoS Med 3: e301, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Hanly P, Sasson Z, Zuberi N, Alderson M. Ventricular function in snorers and patients with obstructive sleep apnea. Chest 102: 100–105, 1992 [DOI] [PubMed] [Google Scholar]
- 220. Hanly P, Sasson Z, Zuberi N, Lunn K. ST-segment depression during sleep in obstructive sleep apnea. Am J Cardiol 71: 1341–1345, 1993 [DOI] [PubMed] [Google Scholar]
- 221. Hanly PJ, Millar TW, Steljes DG, Baert R, Frais MA, Kryger MH. Respiration and abnormal sleep in patients with congestive heart failure. Chest 96: 480–488, 1989 [DOI] [PubMed] [Google Scholar]
- 222. Hanzel DA, Proia NG, Hudgel DW. Response of obstructive sleep apnea to fluoxetine and protriptyline. Chest 100: 416–421, 1991 [DOI] [PubMed] [Google Scholar]
- 223. Harbison J, O'Reilly P, McNicholas WT. Cardiac rhythm disturbances in the obstructive sleep apnea syndrome: effects of nasal continuous positive airway pressure therapy. Chest 118: 591–595, 2000 [DOI] [PubMed] [Google Scholar]
- 224. Harms CA, Zeng YJ, Smith CA, Vidruk EH, Dempsey JA. Negative pressure-induced deformation of the upper airway causes central apnea in awake and sleeping dogs. J Appl Physiol 80: 1528–1539, 1996 [DOI] [PubMed] [Google Scholar]
- 225. Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol 91: 7A–11A, 2003 [DOI] [PubMed] [Google Scholar]
- 226. Harsch IA, Schahin SP, Radespiel-Troger M, Weintz O, Jahreiss H, Fuchs FS, Wiest GH, Hahn EG, Lohmann T, Konturek PC, Ficker JH. Continuous positive airway pressure treatment rapidly improves insulin sensitivity in patients with obstructive sleep apnea syndrome. Am J Respir Crit Care Med 169: 156–162, 2004 [DOI] [PubMed] [Google Scholar]
- 227. Haxhiu MA, van Lunteren E, Mitra J, Cherniack NS. Comparison of the response of diaphragm and upper airway dilating muscle activity in sleeping cats. Respir Physiol 70: 183–193, 1987 [DOI] [PubMed] [Google Scholar]
- 228. Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Oxidative injury to the endoplasmic reticulum in mouse brains after transient focal ischemia. Neurobiol Dis 15: 229–239, 2004 [DOI] [PubMed] [Google Scholar]
- 229. Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J Cereb Blood Flow Metab 25: 41–53, 2005 [DOI] [PubMed] [Google Scholar]
- 230. Hayashi T, Yamashita C, Matsumoto C, Kwak CJ, Fujii K, Hirata T, Miyamura M, Mori T, Ukimura A, Okada Y, Matsumura Y, Kitaura Y. Role of gp91phox-containing NADPH oxidase in left ventricular remodeling induced by intermittent hypoxic stress. Am J Physiol Heart Circ Physiol 294: H2197–H2203, 2008 [DOI] [PubMed] [Google Scholar]
- 231. Hedner J, Darpo B, Ejnell H, Carlson J, Caidahl K. Reduction in sympathetic activity after long-term CPAP treatment in sleep apnoea: cardiovascular implications. Eur Respir J 8: 222–229, 1995 [DOI] [PubMed] [Google Scholar]
- 232. Hedner J, Ejnell H, Caidahl K. Left ventricular hypertrophy independent of hypertension in patients with obstructive sleep apnoea. J Hypertens 8: 941–946, 1990 [DOI] [PubMed] [Google Scholar]
- 233. Hedner J, Ejnell H, Sellgren J, Hedner T, Wallin G. Is high and fluctuating muscle nerve sympathetic activity in the sleep apnoea syndrome of pathogenetic importance for the development of hypertension? J Hypertens 6 Suppl: S529–S531, 1988 [DOI] [PubMed] [Google Scholar]
- 234. Hedner J, Grunstein R, Eriksson B, Ejnell H. A double-blind, randomized trial of sabeluzole, a putative glutamate antagonist, in obstructive sleep apnea. Sleep 19: 287–289, 1996 [DOI] [PubMed] [Google Scholar]
- 234a. Hedner J, Kraiczi H, Peker Y, Murphy P. Reduction of sleep-disordered breathing after physostigmine. Am J Respir Crit Care Med 168: 1246–1251, 2003 [DOI] [PubMed] [Google Scholar]
- 235. Heinzer RC, Stanchina ML, Malhotra A, Fogel RB, Patel SR, Jordan AS, Schory K, White DP. Lung volume and continuous positive airway pressure requirements in obstructive sleep apnea. Am J Respir Crit Care Med 172: 114–117, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Heinzer RC, Stanchina ML, Malhotra A, Jordan AS, Patel SR, Lo YL, Wellman A, Schory K, Dover L, White DP. Effect of increased lung volume on sleep disordered breathing in patients with sleep apnoea. Thorax 61: 435–439, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Heitmann J, Ehlenz K, Penzel T, Becker HF, Grote L, Voigt KH, Peter JH, Vogelmeier C. Sympathetic activity is reduced by nCPAP in hypertensive obstructive sleep apnoea patients. Eur Respir J 23: 255–262, 2004 [DOI] [PubMed] [Google Scholar]
- 238. Hendricks JC, Kline LR, Kovalski RJ, O'Brien JA, Morrison AR, Pack AI. The English bulldog: a natural model of sleep-disordered breathing. J Appl Physiol 63: 1344–1350, 1987 [DOI] [PubMed] [Google Scholar]
- 239. Henke KG, Arias A, Skatrud JB, Dempsey JA. Inhibition of inspiratory muscle activity during sleep. Chemical and nonchemical influences. Am Rev Respir Dis 138: 8–15, 1988 [DOI] [PubMed] [Google Scholar]
- 240. Henke KG, Badr MS, Skatrud JB, Dempsey JA. Load compensation and respiratory muscle function during sleep. J Appl Physiol 72: 1221–1234, 1992 [DOI] [PubMed] [Google Scholar]
- 241. Henke KG, Dempsey JA, Kowitz JM, Skatrud JB. Effects of sleep-induced increases in upper airway resistance on ventilation. J Appl Physiol 69: 617–624, 1990 [DOI] [PubMed] [Google Scholar]
- 242. Hetzel M, Kochs M, Marx N, Woehrle H, Mobarak I, Hombach V, Hetzel J. Pulmonary hemodynamics in obstructive sleep apnea: frequency and causes of pulmonary hypertension. Lung 181: 157–166, 2003 [DOI] [PubMed] [Google Scholar]
- 243. Heym J, Steinfels GF, Jacobs BL. Activity of serotonin-containing neurons in the nucleus raphe pallidus of freely moving cats. Brain Res 251: 259–276, 1982 [DOI] [PubMed] [Google Scholar]
- 244. Hinojosa-Laborde C, Mifflin SW. Sex differences in blood pressure response to intermittent hypoxia in rats. Hypertension 46: 1016–1021, 2005 [DOI] [PubMed] [Google Scholar]
- 245. Hla KM, Young T, Finn LA, Peppard PE, Kinsey TJ, Ende D. Electrocardiographically indicated cardiovascular disease in sleep-disordered breathing. Sleep Breath 12: 251–258, 2008 [DOI] [PubMed] [Google Scholar]
- 246. Hoffman RP, Sinkey CA, Dopp JM, Phillips BG. Systemic and local adrenergic regulation of muscle glucose utilization during hypoglycemia in healthy subjects. Diabetes 51: 734–742, 2002 [DOI] [PubMed] [Google Scholar]
- 247. Hopkins N, McLoughlin P. The structural basis of pulmonary hypertension in chronic lung disease: remodelling, rarefaction or angiogenesis? J Anat 201: 335–348, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Hornbein TF, Griffo ZJ, Roos A. Quantitation of chemoreceptor activity: interrelation of hypoxia and hypercapnia. J Neurophysiol 24: 561–568, 1961 [DOI] [PubMed] [Google Scholar]
- 249. Horner RL. Arousal mechanisms and autonomic consequences. In: Sleep Apnea: Pathogenesis, Diagnosis and Treatment, edited by Pack AI. New York: Dekker, 2002 [Google Scholar]
- 250. Horner RL, Innes JA, Holden HB, Guz A. Afferent pathway(s) for pharyngeal dilator reflex to negative pressure in man: a study using upper airway anaesthesia. J Physiol 436: 31–44, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Horner RL, Liu X, Gill H, Nolan P, Liu H, Sood S. Effects of sleep-wake state on the genioglossus vs. diaphragm muscle response to CO2 in rats. J Appl Physiol 92: 878–887, 2002 [DOI] [PubMed] [Google Scholar]
- 252. Horner RL, Sanford LD, Pack AI, Morrison AR. Activation of a distinct arousal state immediately after spontaneous awakening from sleep. Brain Res 778: 127–134, 1997 [DOI] [PubMed] [Google Scholar]
- 253. Horner RL, Shea SA, McIvor J, Guz A. Pharyngeal size and shape during wakefulness and sleep in patients with obstructive sleep apnoea. Q J Med 72: 719–735, 1989 [PubMed] [Google Scholar]
- 254. Huang Y, White DP, Malhotra A. The impact of anatomic manipulations on pharyngeal collapse: results from a computational model of the normal human upper airway. Chest 128: 1324–1330, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Hudgel DW. Variable site of airway narrowing among obstructive sleep apnea patients. J Appl Physiol 61: 1403–1409, 1986 [DOI] [PubMed] [Google Scholar]
- 256. Hudgel DW, Chapman KR, Faulks C, Hendricks C. Changes in inspiratory muscle electrical activity and upper airway resistance during periodic breathing induced by hypoxia during sleep. Am Rev Respir Dis 135: 899–906, 1987 [DOI] [PubMed] [Google Scholar]
- 257. Hudgel DW, Gordon EA, Thanakitcharu S, Bruce EN. Instability of ventilatory control in patients with obstructive sleep apnea. Am J Respir Crit Care Med 158: 1142–1149, 1998 [DOI] [PubMed] [Google Scholar]
- 258. Hudgel DW, Hamilton HB. Respiratory muscle activity during sleep-induced periodic breathing in the elderly. J Appl Physiol 77: 2285–2290, 1994 [DOI] [PubMed] [Google Scholar]
- 259. Hugelin A, Cohen MI. The reticular activating system and respiratory regulation in the cat. Ann NY Acad Sci 109: 586–603, 1963 [DOI] [PubMed] [Google Scholar]
- 260. Hwang JC, St John WM, Bartlett D., Jr Respiratory-related hypoglossal nerve activity: influence of anesthetics. J Appl Physiol 55: 785–792, 1983 [DOI] [PubMed] [Google Scholar]
- 261. Iber C, Berssenbrugge A, Skatrud JB, Dempsey JA. Ventilatory adaptations to resistive loading during wakefulness and non-REM sleep. J Appl Physiol 52: 607–614, 1982 [DOI] [PubMed] [Google Scholar]
- 262. Iber C, Davies SF, Chapman RC, Mahowald MM. A possible mechanism for mixed apnea in obstructive sleep apnea. Chest 89: 800–805, 1986 [DOI] [PubMed] [Google Scholar]
- 263. Iiyori N, Alonso LC, Li J, Sanders MH, Garcia-Ocana A, O'Doherty RM, Polotsky VY, O'Donnell CP. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med 175: 851–857, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Imadojemu VA, Gleeson K, Gray KS, Sinoway LI, Leuenberger UA. Obstructive apnea during sleep is associated with peripheral vasoconstriction. Am J Respir Crit Care Med 165: 61–66, 2002 [DOI] [PubMed] [Google Scholar]
- 265. Imadojemu VA, Mawji Z, Kunselman A, Gray KS, Hogeman CS, Leuenberger UA. Sympathetic chemoreflex responses in obstructive sleep apnea and effects of continuous positive airway pressure therapy. Chest 131: 1406–1413, 2007 [DOI] [PubMed] [Google Scholar]
- 266. Inoue K, Ito H, Goto R, Nakagawa M, Kinomura S, Sato T, Sato K, Fukuda H. Apparent CBF decrease with normal aging due to partial volume effects: MR-based partial volume correction on CBF SPECT. Ann Nucl Med 19: 283–290, 2005 [DOI] [PubMed] [Google Scholar]
- 267. Ip MS, Lam B, Chan LY, Zheng L, Tsang KW, Fung PC, Lam WK. Circulating nitric oxide is suppressed in obstructive sleep apnea and is reversed by nasal continuous positive airway pressure. Am J Respir Crit Care Med 162: 2166–2171, 2000 [DOI] [PubMed] [Google Scholar]
- 268. Ip MS, Lam B, Ng MM, Lam WK, Tsang KW, Lam KS. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med 165: 670–676, 2002 [DOI] [PubMed] [Google Scholar]
- 269. Ip MS, Lam B, Tang LC, Lauder IJ, Ip TY, Lam WK. A community study of sleep-disordered breathing in middle-aged Chinese women in Hong Kong: prevalence and gender differences. Chest 125: 127–134, 2004 [DOI] [PubMed] [Google Scholar]
- 270. Ip MS, Tse HF, Lam B, Tsang KW, Lam WK. Endothelial function in obstructive sleep apnea and response to treatment. Am J Respir Crit Care Med 169: 348–353, 2004 [DOI] [PubMed] [Google Scholar]
- 271. Isono S, Feroah TR, Hajduk EA, Brant R, Whitelaw WA, Remmers JE. Interaction of cross-sectional area, driving pressure, and airflow of passive velopharynx. J Appl Physiol 83: 851–859, 1997 [DOI] [PubMed] [Google Scholar]
- 272. Isono S, Morrison DL, Launois SH, Feroah TR, Whitelaw WA, Remmers JE. Static mechanics of the velopharynx of patients with obstructive sleep apnea. J Appl Physiol 75: 148–154, 1993 [DOI] [PubMed] [Google Scholar]
- 273. Issa FG. Effect of clonidine in obstructive sleep apnea. Am Rev Respir Dis 145: 435–439, 1992 [DOI] [PubMed] [Google Scholar]
- 274. Jacob MP, Badier-Commander C, Fontaine V, Benazzoug Y, Feldman L, Michel JB. Extracellular matrix remodeling in the vascular wall. Pathol Biol 49: 326–332, 2001 [DOI] [PubMed] [Google Scholar]
- 275. Javaheri S. Pembrey's dream: the time has come for a long-term trial of nocturnal supplemental nasal oxygen to treat central sleep apnea in congestive heart failure. Chest 123: 322–325, 2003 [DOI] [PubMed] [Google Scholar]
- 276. Javaheri S. Acetazolamide improves central sleep apnea in heart failure: a double-blind, prospective study. Am J Respir Crit Care Med 173: 234–237, 2006 [DOI] [PubMed] [Google Scholar]
- 277. Javaheri S. CPAP should not be used for central sleep apnea in congestive heart failure patients. J Clin Sleep Med 2: 399–402, 2006 [PubMed] [Google Scholar]
- 278. Javaheri S. Adaptive pressure support servo-ventilation: a novel treatment for sleep apnea associated with use of opioids. Int J Cardiol. In press [Google Scholar]
- 279. Javaheri S, Ahmed M, Parker TJ, Brown CR. Effects of nasal O2 on sleep-related disordered breathing in ambulatory patients with stable heart failure. Sleep 22: 1101–1106, 1999 [DOI] [PubMed] [Google Scholar]
- 280. Javaheri S, Corbett WS. Association of low PaCO2 with central sleep apnea and ventricular arrhythmias in ambulatory patients with stable heart failure. Ann Intern Med 128: 204–207, 1998 [DOI] [PubMed] [Google Scholar]
- 281. Javaheri S, Dempsey JA. Mechanisms of sleep apnea and periodic breathing in systemic heart failure. In: Sleep Medicine Clinics, edited by Javaheri S. Philadelphia, PA: Saunders, 2007 [Google Scholar]
- 282. Javaheri S, Parker TJ, Liming JD, Corbett WS, Nishiyama H, Wexler L, Roselle GA. Sleep apnea in 81 ambulatory male patients with stable heart failure. Types and their prevalences, consequences, and presentations. Circulation 97: 2154–2159, 1998 [DOI] [PubMed] [Google Scholar]
- 283. Jokic R, Klimaszewski A, Mink J, Fitzpatrick MF. Surface tension forces in sleep apnea: the role of a soft tissue lubricant: a randomized double-blind, placebo-controlled trial. Am J Respir Crit Care Med 157: 1522–1525, 1998 [DOI] [PubMed] [Google Scholar]
- 284. Joo EY, Tae WS, Han SJ, Cho JW, Hong SB. Reduced cerebral blood flow during wakefulness in obstructive sleep apnea-hypopnea syndrome. Sleep 30: 1515–1520, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Jordan AS, Wellman A, Edwards JK, Schory K, Dover L, MacDonald M, Patel SR, Fogel RB, Malhotra A, White DP. Respiratory control stability and upper airway collapsibility in men and women with obstructive sleep apnea. J Appl Physiol 99: 2020–2027, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Jordan W, Reinbacher A, Cohrs S, Grunewald RW, Mayer G, Ruther E, Rodenbeck A. Obstructive sleep apnea: plasma endothelin-1 precursor but not endothelin-1 levels are elevated and decline with nasal continuous positive airway pressure. Peptides 26: 1654–1660, 2005 [DOI] [PubMed] [Google Scholar]
- 287. Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, Jensen G, Nordestgaard BG. Genetically reduced antioxidative protection and increased ischemic heart disease risk: The Copenhagen City Heart Study. Circulation 109: 59–65, 2004 [DOI] [PubMed] [Google Scholar]
- 288. Kaab S, Miguel-Velado E, Lopez-Lopez JR, Perez-Garcia MT. Down regulation of Kv34 channels by chronic hypoxia increases acute oxygen sensitivity in rabbit carotid body. J Physiol 566: 395–408, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Kahn A, Rebuffat E, Sottiaux M, Muller MF, Bochner A, Grosswasser J. Prevention of airway obstructions during sleep in infants with breath-holding spells by means of oral belladonna: a prospective double-blind crossover evaluation. Sleep 14: 432–438, 1991 [DOI] [PubMed] [Google Scholar]
- 290. Kamba M, Inoue Y, Higami S, Suto Y, Ogawa T, Chen W. Cerebral metabolic impairment in patients with obstructive sleep apnoea: an independent association of obstructive sleep apnoea with white matter change. J Neurol Neurosurg Psychiatry 71: 334–339, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Kanagala R, Murali NS, Friedman PA, Ammash NM, Gersh BJ, Ballman KV, Shamsuzzaman AS, Somers VK. Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation 107: 2589–2594, 2003 [DOI] [PubMed] [Google Scholar]
- 292. Kanagy NL, Walker BR, Nelin LD. Role of endothelin in intermittent hypoxia-induced hypertension. Hypertension 37: 511–515, 2001 [DOI] [PubMed] [Google Scholar]
- 293. Kaneko Y, Floras JS, Usui K, Plante J, Tkacova R, Kubo T, Ando S, Bradley TD. Cardiovascular effects of continuous positive airway pressure in patients with heart failure and obstructive sleep apnea. N Engl J Med 348: 1233–1241, 2003 [DOI] [PubMed] [Google Scholar]
- 294. Katayama K, Smith CA, Henderson KS, Dempsey JA. Chronic intermittent hypoxia increases the CO2 reserve in sleeping dogs. J Appl Physiol 103: 1942–1949, 2007 [DOI] [PubMed] [Google Scholar]
- 295. Kato M, Roberts-Thomson P, Phillips BG, Haynes WG, Winnicki M, Accurso V, Somers VK. Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation 102: 2607–2610, 2000 [DOI] [PubMed] [Google Scholar]
- 296. Katragadda S, Xie A, Puleo D, Skatrud JB, Morgan BJ. Neural mechanism of the pressor response to obstructive and nonobstructive apnea. J Appl Physiol 83: 2048–2054, 1997 [DOI] [PubMed] [Google Scholar]
- 297. Katusic ZS. Vascular endothelial dysfunction: does tetrahydrobiopterin play a role? Am J Physiol Heart Circ Physiol 281: H981–H986, 2001 [DOI] [PubMed] [Google Scholar]
- 298. Kelishadi R. Childhood overweight, obesity, and the metabolic syndrome in developing countries. Epidemiol Rev 29: 62–76, 2007 [DOI] [PubMed] [Google Scholar]
- 299. Khayat RN, Xie A, Patel AK, Kaminski A, Skatrud JB. Cardiorespiratory effects of added dead space in patients with heart failure and central sleep apnea. Chest 123: 1551–1560, 2003 [DOI] [PubMed] [Google Scholar]
- 300. Khoo MC. Determinants of ventilatory instability and variability. Respir Physiol 122: 167–182, 2000 [DOI] [PubMed] [Google Scholar]
- 301. Khoo MC, Anholm JD, Ko SW, Downey R, III, Powles AC, Sutton JR, Houston CS. Dynamics of periodic breathing and arousal during sleep at extreme altitude. Respir Physiol 103: 33–43, 1996 [DOI] [PubMed] [Google Scholar]
- 302. Khoo MC, Kronauer RE, Strohl KP, Slutsky AS. Factors inducing periodic breathing in humans: a general model. J Appl Physiol 53: 644–659, 1982 [DOI] [PubMed] [Google Scholar]
- 303. Kingshott RN, Vennelle M, Hoy CJ, Engleman HM, Deary IJ, Douglas NJ. Predictors of improvements in daytime function outcomes with CPAP therapy. Am J Respir Crit Care Med 161: 866–871, 2000 [DOI] [PubMed] [Google Scholar]
- 304. Kinney HC, Filiano JJ, White WF. Medullary serotonergic network deficiency in the sudden infant death syndrome: review of a 15-year study of a single dataset. J Neuropathol Exp Neurol 60: 228–247, 2001 [DOI] [PubMed] [Google Scholar]
- 305. Kirkness JP, Madronio M, Stavrinou R, Wheatley JR, Amis TC. Relationship between surface tension of upper airway lining liquid and upper airway collapsibility during sleep in obstructive sleep apnea hypopnea syndrome. J Appl Physiol 95: 1761–1766, 2003 [DOI] [PubMed] [Google Scholar]
- 306. Kirkness JP, Madronio M, Stavrinou R, Wheatley JR, Amis TC. Surface tension of upper airway mucosal lining liquid in obstructive sleep apnea/hypopnea syndrome. Sleep 28: 457–463, 2005 [DOI] [PubMed] [Google Scholar]
- 307. Kirkness JP, Schwartz AR, Schneider H, Punjabi NM, Maly JJ, Laffan AM, McGinley BM, Magnuson T, Schweitzer M, Smith PL, Patil SP. Contribution of male sex, age, and obesity to mechanical instability of the upper airway during sleep. J Appl Physiol 104: 1618–1624, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Kiyashchenko LI, Mileykovskiy BY, Maidment N, Lam HA, Wu MF, John J, Peever J, Siegel JM. Release of hypocretin (orexin) during waking and sleep states. J Neurosci 22: 5282–5286, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Klingelhofer J, Hajak G, Sander D, Schulz-Varszegi M, Ruther E, Conrad B. Assessment of intracranial hemodynamics in sleep apnea syndrome. Stroke 23: 1427–1433, 1992 [DOI] [PubMed] [Google Scholar]
- 310. Koehler U, Fus E, Grimm W, Pankow W, Schafer H, Stammnitz A, Peter JH. Heart block in patients with obstructive sleep apnoea: pathogenetic factors and effects of treatment. Eur Respir J 11: 434–439, 1998 [DOI] [PubMed] [Google Scholar]
- 311. Kohler M, Craig S, Nicoll D, Leeson P, Davies RJ, Stradling JR. Endothelial function and arterial stiffness in minimally symptomatic obstructive sleep apnea. Am J Respir Crit Care Med 178: 984–988, 2008 [DOI] [PubMed] [Google Scholar]
- 312. Kraiczi H, Caidahl K, Samuelsson A, Peker Y, Hedner J. Impairment of vascular endothelial function and left ventricular filling: association with the severity of apnea-induced hypoxemia during sleep. Chest 119: 1085–1091, 2001 [DOI] [PubMed] [Google Scholar]
- 313. Kraiczi H, Hedner J, Dahlof P, Ejnell H, Carlson J. Effect of serotonin uptake inhibition on breathing during sleep and daytime symptoms in obstructive sleep apnea. Sleep 22: 61–67, 1999 [PubMed] [Google Scholar]
- 314. Kraiczi H, Hedner J, Peker Y, Carlson J. Increased vasoconstrictor sensitivity in obstructive sleep apnea. J Appl Physiol 89: 493–498, 2000 [DOI] [PubMed] [Google Scholar]
- 315. Kraiczi H, Magga J, Sun XY, Ruskoaho H, Zhao X, Hedner J. Hypoxic pressor response, cardiac size, and natriuretic peptides are modified by long-term intermittent hypoxia. J Appl Physiol 87: 2025–2031, 1999 [DOI] [PubMed] [Google Scholar]
- 316. Kraiczi H, Magga J, Sun XY, Ruskoaho H, Zhao X, Hedner J. Hypoxic pressor response, cardiac size, and natriuretic peptides are modified by long-term intermittent hypoxia. J Appl Physiol 87: 2025–2031, 1999 [DOI] [PubMed] [Google Scholar]
- 317. Kribbs NB, Pack AI, Kline LR, Smith PL, Schwartz AR, Schubert NM, Redline S, Henry JN, Getsy JE, Dinges DF. Objective measurement of patterns of nasal CPAP use by patients with obstructive sleep apnea. Am Rev Respir Dis 147: 887–895, 1993 [DOI] [PubMed] [Google Scholar]
- 318. Krieger J, Sforza E, Apprill M, Lampert E, Weitzenblum E, Ratomaharo J. Pulmonary hypertension, hypoxemia, and hypercapnia in obstructive sleep apnea patients. Chest 96: 729–737, 1989 [DOI] [PubMed] [Google Scholar]
- 319. Kubin L, Fenik V. Pontine cholinergic mechanisms and their impact on respiratory regulation. Respir Physiol Neurobiol 143: 235–249, 2004 [DOI] [PubMed] [Google Scholar]
- 320. Kubin L, Kimura H, Tojima H, Pack AI, Davies RO. Behavior of VRG neurons during the atonia of REM sleep induced by pontine carbachol in decerebrate cats. Brain Res 592: 91–100, 1992 [DOI] [PubMed] [Google Scholar]
- 321. Kubin L, Reignier C, Tojima H, Taguchi O, Pack AI, Davies RO. Changes in serotonin level in the hypoglossal nucleus region during carbachol-induced atonia. Brain Res 645: 291–302, 1994 [DOI] [PubMed] [Google Scholar]
- 322. Kumar GK, Rai V, Sharma SD, Ramakrishnan DP, Peng YJ, Souvannakitti D, Prabhakar NR. Chronic intermittent hypoxia induces hypoxia-evoked catecholamine efflux in adult rat adrenal medulla via oxidative stress. J Physiol 575: 229–239, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Kuna ST. Inhibition of inspiratory upper airway motoneuron activity by phasic volume feedback. J Appl Physiol 60: 1373–1379, 1986 [DOI] [PubMed] [Google Scholar]
- 324. Kuna ST, McCarthy MP, Smickley JS. Laryngeal response to passively induced hypocapnia during NREM sleep in normal adult humans. J Appl Physiol 75: 1088–1096, 1993 [DOI] [PubMed] [Google Scholar]
- 325. Laaban JP, Cassuto D, Orvoen-Frija E, Iliou MC, Mundler O, Leger D, Oppert JM. Cardiorespiratory consequences of sleep apnoea syndrome in patients with massive obesity. Eur Respir J 11: 20–27, 1998 [DOI] [PubMed] [Google Scholar]
- 326. Laaban JP, Pascal-Sebaoun S, Bloch E, Orvoen-Frija E, Oppert JM, Huchon G. Left ventricular systolic dysfunction in patients with obstructive sleep apnea syndrome. Chest 122: 1133–1138, 2002 [DOI] [PubMed] [Google Scholar]
- 327. Lacolley P, Owen JR, Sandock K, Lewis TH, Bates JN, Robertson TP, Lewis SJ. 5-HT activates vagal afferent cell bodies in vivo: role of 5-HT2 and 5-HT3 receptors. Neuroscience 143: 273–287, 2006 [DOI] [PubMed] [Google Scholar]
- 328. Laferriere A, Moss IR. Respiratory responses to intermittent hypoxia in unsedated piglets: relation to substance P binding in brainstem. Respir Physiol Neurobiol 143: 21–35, 2004 [DOI] [PubMed] [Google Scholar]
- 329. Lai CJ, Yang CC, Hsu YY, Lin YN, Kuo TB. Enhanced sympathetic outflow and decreased baroreflex sensitivity are associated with intermittent hypoxia-induced systemic hypertension in conscious rats. J Appl Physiol 100: 1974–1982, 2006 [DOI] [PubMed] [Google Scholar]
- 330. Laks L, Lehrhaft B, Grunstein RR, Sullivan CE. Pulmonary hypertension in obstructive sleep apnoea. Eur Respir J 8: 537–541, 1995 [PubMed] [Google Scholar]
- 331. Lalley PM. Opiate slowing of feline respiratory rhythm and effects on putative medullary phase-regulating neurons. Am J Physiol Regul Integr Comp Physiol 290: R1387–R1396, 2006 [DOI] [PubMed] [Google Scholar]
- 332. Lalley PM, Bischoff AM, Richter DW. Serotonin 1A-receptor activation suppresses respiratory apneusis in the cat. Neurosci Lett 172: 59–62, 1994 [DOI] [PubMed] [Google Scholar]
- 333. Lalley PM, Bischoff AM, Schwarzacher SW, Richter DW. 5-HT2 receptor-controlled modulation of medullary respiratory neurones in the cat. J Physiol 487: 653–661, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Lam B, Ip MS, Tench E, Ryan CF. Craniofacial profile in Asian and white subjects with obstructive sleep apnoea. Thorax 60: 504–510, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Lam SY, Fung ML, Leung PS. Regulation of the angiotensin-converting enzyme activity by a time-course hypoxia in the carotid body. J Appl Physiol 96: 809–813, 2004 [DOI] [PubMed] [Google Scholar]
- 336. Lam SY, Leung PS. A locally generated angiotensin system in rat carotid body. Regul Pept 107: 97–103, 2002 [DOI] [PubMed] [Google Scholar]
- 337. Landmesser U, Spiekermann S, Preuss C, Sorrentino S, Fischer D, Manes C, Mueller M, Drexler H. Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler Thromb Vasc Biol 27: 943–948, 2007 [DOI] [PubMed] [Google Scholar]
- 338. Larsen JJ, Hansen JM, Olsen NV, Galbo H, Dela F. The effect of altitude hypoxia on glucose homeostasis in men. J Physiol 504: 241–249, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Lattimore JD, Wilcox I, Adams MR, Kilian JG, Celermajer DS. Treatment of obstructive sleep apnoea leads to enhanced pulmonary vascular nitric oxide release. Int J Cardiol 126: 229–233, 2008 [DOI] [PubMed] [Google Scholar]
- 340. Lattimore JL, Wilcox I, Skilton M, Langenfeld M, Celermajer DS. Treatment of obstructive sleep apnoea leads to improved microvascular endothelial function in the systemic circulation. Thorax 61: 491–495, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Launois SH, Abraham JH, Weiss JW, Kirby DA. Patterned cardiovascular responses to sleep and nonrespiratory arousals in a porcine model. J Appl Physiol 85: 1285–1291, 1998 [DOI] [PubMed] [Google Scholar]
- 342. Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation 95: 588–593, 1997 [DOI] [PubMed] [Google Scholar]
- 343. Lavie L, Lavie P. Molecular mechanisms of cardiovascular disease in OSAHS: the oxidative stress link. Eur Respir J 33: 1467–1484, 2009 [DOI] [PubMed] [Google Scholar]
- 344. Lavie L, Lotan R, Hochberg I, Herer P, Lavie P, Levy AP. Haptoglobin polymorphism is a risk factor for cardiovascular disease in patients with obstructive sleep apnea syndrome. Sleep 26: 592–595, 2003 [DOI] [PubMed] [Google Scholar]
- 345. Lavie L, Vishnevsky A, Lavie P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep 27: 123–128, 2004 [PubMed] [Google Scholar]
- 346. Lavie P. Restless Nights: Understanding Snoring and Sleep Apnea. New Haven, CT: Yale Univ. Press, 2003 [Google Scholar]
- 347. Leevers AM, Simon PM, Dempsey JA. Apnea after normocapnic mechanical ventilation during NREM sleep. J Appl Physiol 77: 2079–2085, 1994 [DOI] [PubMed] [Google Scholar]
- 348. Lesske J, Fletcher EC, Bao G, Unger T. Hypertension caused by chronic intermittent hypoxia–influence of chemoreceptors and sympathetic nervous system. J Hypertens 15: 1593–1603, 1997 [DOI] [PubMed] [Google Scholar]
- 349. Leuenberger U, Jacob E, Sweer L, Waravdekar N, Zwillich C, Sinoway L. Surges of muscle sympathetic nerve activity during obstructive apnea are linked to hypoxemia. J Appl Physiol 79: 581–588, 1995 [DOI] [PubMed] [Google Scholar]
- 350. Leung LC, Ng DK, Lau MW, Chan CH, Kwok KL, Chow PY, Cheung JM. Twenty-four-hour ambulatory BP in snoring children with obstructive sleep apnea syndrome. Chest 130: 1009–1017, 2006 [DOI] [PubMed] [Google Scholar]
- 351. Levy AP, Hochberg I, Jablonski K, Resnick HE, Lee ET, Best L, Howard BV. Haptoglobin phenotype is an independent risk factor for cardiovascular disease in individuals with diabetes: The Strong Heart Study. J Am Coll Cardiol 40: 1984–1990, 2002 [DOI] [PubMed] [Google Scholar]
- 352. Leysen JE. 5-HT2 receptors. Curr Drug Targets CNS Neurol Disord 3: 11–26, 2004 [DOI] [PubMed] [Google Scholar]
- 353. Li A, Randall M, Nattie EE. CO2 microdialysis in retrotrapezoid nucleus of the rat increases breathing in wakefulness but not in sleep. J Appl Physiol 87: 910–919, 1999 [DOI] [PubMed] [Google Scholar]
- 354. Li AM, Au CT, Sung RY, Ho C, Ng PC, Fok TF, Wing YK. Ambulatory blood pressure in children with obstructive sleep apnoea: a community based study. Thorax 63: 803–809, 2008 [DOI] [PubMed] [Google Scholar]
- 355. Li J, Nanayakkara A, Jun J, Savransky V, Polotsky VY. The effect of deficiency in SREBP cleavage-activating protein (SCAP) on lipid metabolism during intermittent hypoxia. Physiol Genomics 31: 273–280, 2007 [DOI] [PubMed] [Google Scholar]
- 356. Li J, Perl ER. ATP modulation of synaptic transmission in the spinal substantia gelatinosa. J Neurosci 15: 3357–3365, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Li J, Savransky V, Nanayakkara A, Smith PL, O'Donnell CP, Polotsky VY. Hyperlipidemia and lipid peroxidation are dependent on the severity of chronic intermittent hypoxia. J Appl Physiol 102: 557–563, 2007 [DOI] [PubMed] [Google Scholar]
- 358. Li J, Thorne LN, Punjabi NM, Sun CK, Schwartz AR, Smith PL, Marino RL, Rodriguez A, Hubbard WC, O'Donnell CP, Polotsky VY. Intermittent hypoxia induces hyperlipidemia in lean mice. Circ Res 97: 698–706, 2005 [DOI] [PubMed] [Google Scholar]
- 359. Li YL, Gao L, Zucker IH, Schultz HD. NADPH oxidase-derived superoxide anion mediates angiotensin II-enhanced carotid body chemoreceptor sensitivity in heart failure rabbits. Cardiovasc Res 75: 546–554, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Li YL, Xia XH, Zheng H, Gao L, Li YF, Liu D, Patel KP, Wang W, Schultz HD. Angiotensin II enhances carotid body chemoreflex control of sympathetic outflow in chronic heart failure rabbits. Cardiovasc Res 71: 129–138, 2006 [DOI] [PubMed] [Google Scholar]
- 361. Lieberman DE, McCarthy RC. The ontogeny of cranial base angulation in humans and chimpanzees and its implications for reconstructing pharyngeal dimensions. J Hum Evol 36: 487–517, 1999 [DOI] [PubMed] [Google Scholar]
- 362. Lieberman DE, McCarthy RC, Hiiemae KM, Palmer JB. Ontogeny of postnatal hyoid and larynx descent in humans. Arch Oral Biol 46: 117–128, 2001 [DOI] [PubMed] [Google Scholar]
- 363. Lin CC, Lin CK, Wu KM, Chou CS. Effect of treatment by nasal CPAP on cardiopulmonary exercise test in obstructive sleep apnea syndrome. Lung 182: 199–212, 2004 [DOI] [PubMed] [Google Scholar]
- 364. Lindman R, Stal PS. Abnormal palatopharyngeal muscle morphology in sleep-disordered breathing. J Neurol Sci 195: 11–23, 2002 [DOI] [PubMed] [Google Scholar]
- 365. Lipton SA. Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation. Curr Drug Targets 8: 621–632, 2007 [DOI] [PubMed] [Google Scholar]
- 366. Liu JL, Murakami H, Zucker IH. Angiotensin II-nitric oxide interaction on sympathetic outflow in conscious rabbits. Circ Res 82: 496–502, 1998 [DOI] [PubMed] [Google Scholar]
- 367. Liu X, Sood S, Liu H, Horner RL. Opposing muscarinic and nicotinic modulation of hypoglossal motor output to genioglossus muscle in rats in vivo. J Physiol 565: 965–980, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Liu X, Sood S, Liu H, Nolan P, Morrison JL, Horner RL. Suppression of genioglossus muscle tone and activity during reflex hypercapnic stimulation by GABA(A) mechanisms at the hypoglossal motor nucleus in vivo. Neuroscience 116: 249–259, 2003 [DOI] [PubMed] [Google Scholar]
- 369. Lo YL, Jordan AS, Malhotra A, Wellman A, Heinzer RA, Eikermann M, Schory K, Dover L, White DP. Influence of wakefulness on pharyngeal airway muscle activity. Thorax 62: 799–805, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Lo YL, Jordan AS, Malhotra A, Wellman A, Heinzer RC, Schory K, Dover L, Fogel RB, White DP. Genioglossal muscle response to CO2 stimulation during NREM sleep. Sleep 29: 470–477, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Lorenzi-Filho G, Rankin F, Bies I, Douglas Bradley T. Effects of inhaled carbon dioxide and oxygen on Cheyne-Stokes respiration in patients with heart failure. Am J Respir Crit Care Med 159: 1490–1498, 1999 [DOI] [PubMed] [Google Scholar]
- 372. Louis M, Pujabi NM. Effects of acute intermittent hypoxia on glucose metabolism in awake healthy volunteers. J Appl Physiol 106: 1538–1544, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Loup F, Tribollet E, Dubois-Dauphin M, Pizzolato G, Dreifuss JJ. Localization of oxytocin binding sites in the human brainstem and upper spinal cord: an autoradiographic study. Brain Res 500: 223–230, 1989 [DOI] [PubMed] [Google Scholar]
- 374. Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature 441: 589–594, 2006 [DOI] [PubMed] [Google Scholar]
- 375. Lugaresi E, Coccagna G, Mantovani M, Brignani F. Effect of tracheotomy in hypersomnia with periodic respiration. Electroencephalogr Clin Neurophysiol 30: 373–374, 1971 [PubMed] [Google Scholar]
- 376. Lydic R, Orem J. Respiratory neurons of the pneumotaxic center during sleep and wakefulness. Neurosci Lett 15: 187–192, 1979 [DOI] [PubMed] [Google Scholar]
- 377. Ma S, Mifflin SW, Cunningham JT, Morilak DA. Chronic intermittent hypoxia sensitizes acute hypothalamic-pituitary-adrenal stress reactivity and Fos induction in the rat locus coeruleus in response to subsequent immobilization stress. Neuroscience 154: 1639–1647, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Macefield VG, Elam M. Prolonged surges of baroreflex-resistant muscle sympathetic drive during periodic breathing. Clin Auton Res 12: 165–169, 2002 [DOI] [PubMed] [Google Scholar]
- 379. Macey PM, Henderson LA, Macey KE, Alger JR, Frysinger RC, Woo MA, Harper RK, Yan-Go FL, Harper RM. Brain morphology associated with obstructive sleep apnea. Am J Respir Crit Care Med 166: 1382–1387, 2002 [DOI] [PubMed] [Google Scholar]
- 380. Macey PM, Kumar R, Woo MA, Valladares EM, Yan-Go FL, Harper RM. Brain structural changes in obstructive sleep apnea. Sleep 31: 967–977, 2008 [PMC free article] [PubMed] [Google Scholar]
- 381. MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part one. Am J Respir Crit Care Med 150: 833–852, 1994 [DOI] [PubMed] [Google Scholar]
- 382. MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part two. Am J Respir Crit Care Med 150: 1158–1168, 1994 [DOI] [PubMed] [Google Scholar]
- 383. Maeda H, Matsumoto M, Handa N, Hougaku H, Ogawa S, Itoh T, Tsukamoto Y, Kamada T. Reactivity of cerebral blood flow to carbon dioxide in various types of ischemic cerebrovascular disease: evaluation by the transcranial Doppler method. Stroke 24: 670–675, 1993 [DOI] [PubMed] [Google Scholar]
- 384. Malhotra A, Fogel RB, Edwards JK, Shea SA, White DP. Local mechanisms drive genioglossus activation in obstructive sleep apnea. Am J Respir Crit Care Med 161: 1746–1749, 2000 [DOI] [PubMed] [Google Scholar]
- 385. Malhotra A, Huang Y, Fogel RB, Pillar G, Edwards JK, Kikinis R, Loring SH, White DP. The male predisposition to pharyngeal collapse: importance of airway length. Am J Respir Crit Care Med 166: 1388–1395, 2002 [DOI] [PubMed] [Google Scholar]
- 386. Mann EA, Burnett T, Cornell S, Ludlow CL. The effect of neuromuscular stimulation of the genioglossus on the hypopharyngeal airway. Laryngoscope 112: 351–356, 2002 [DOI] [PubMed] [Google Scholar]
- 387. Mansfield DR, Gollogly NC, Kaye DM, Richardson M, Bergin P, Naughton MT. Controlled trial of continuous positive airway pressure in obstructive sleep apnea and heart failure. [see comment]. Am J Respir Crit Care Med 169: 361–366, 2004 [DOI] [PubMed] [Google Scholar]
- 388. Marcus CL. Sleep-disordered breathing in children. Am J Respir Crit Care Med 164: 16–30, 2001 [DOI] [PubMed] [Google Scholar]
- 389. Marcus NJ, Li YL, Bird CE, Olson EB, Smith SS, Sorenson KI, Schultz HD, Morgan BJ. Chronic intermittent hypoxia alters chemoreflex control of lumbar sympathetic nerve activity and carotid body protein expression. FASEB J 23: 1008.1, 2009 [Google Scholar]
- 390. Marcus NJ, Olson EB, Jr, Bird CE, Philippi NR, Morgan BJ. Time-dependent adaptation in the hemodynamic response to hypoxia. Respir Physiol Neurobiol 165: 90–96, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 365: 1046–1053, 2005 [DOI] [PubMed] [Google Scholar]
- 392. Martin MR, Lodge D, Headley PM, Biscoe TJ. Pharmacological studies of facial motoneurones in the rat. Eur J Pharmacol 42: 291–298, 1977 [DOI] [PubMed] [Google Scholar]
- 393. Martinez-Garcia MA, Gomez-Aldaravi R, Soler-Cataluna JJ, Martinez TG, Bernacer-Alpera B, Roman-Sanchez P. Positive effect of CPAP treatment on the control of difficult-to-treat hypertension. Eur Respir J 29: 951–957, 2007 [DOI] [PubMed] [Google Scholar]
- 394. Mathew OP. Upper airway negative-pressure effects on respiratory activity of upper airway muscles. J Appl Physiol 56: 500–505, 1984 [DOI] [PubMed] [Google Scholar]
- 395. Mathew OP, Abu-Osba YK, Thach BT. Influence of upper airway pressure changes on genioglossus muscle respiratory activity. J Appl Physiol 52: 438–444, 1982 [DOI] [PubMed] [Google Scholar]
- 396. Mathur R, Douglas NJ. Family studies in patients with the sleep apnea-hypopnea syndrome. Ann Intern Med 122: 174–178, 1995 [DOI] [PubMed] [Google Scholar]
- 397. Mayer P, Dematteis M, Pepin JL, Wuyam B, Veale D, Vila A, Levy P. Peripheral neuropathy in sleep apnea. A tissue marker of the severity of nocturnal desaturation. Am J Respir Crit Care Med 159: 213–219, 1999 [DOI] [PubMed] [Google Scholar]
- 398. McCall RB, Aghajanian GK. Pharmacological characterization of serotonin receptors in the facial motor nucleus: a microiontophoretic study. Eur J Pharmacol 65: 175–183, 1980 [DOI] [PubMed] [Google Scholar]
- 399. McFarland DH, Lund JP. Modification of mastication and respiration during swallowing in the adult human. J Neurophysiol 74: 1509–1517, 1995 [DOI] [PubMed] [Google Scholar]
- 400. McGuire M, Bradford A. Chronic intermittent hypoxia increases haematocrit and causes right ventricular hypertrophy in the rat. Respir Physiol 117: 53–58, 1999 [DOI] [PubMed] [Google Scholar]
- 401. McKay LC, Feldman JL. Unilateral ablation of pre-Botzinger complex disrupts breathing during sleep but not wakefulness. Am J Respir Crit Care Med 178: 89–95, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Mehra R, Benjamin EJ, Shahar E, Gottlieb DJ, Nawabit R, Kirchner HL, Sahadevan J, Redline S. Association of nocturnal arrhythmias with sleep-disordered breathing: The Sleep Heart Health Study. Am J Respir Crit Care Med 173: 910–916, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Meston N, Davies RJ, Mullins R, Jenkinson C, Wass JA, Stradling JR. Endocrine effects of nasal continuous positive airway pressure in male patients with obstructive sleep apnoea. J Intern Med 254: 447–454, 2003 [DOI] [PubMed] [Google Scholar]
- 404. Meza S, Mendez M, Ostrowski M, Younes M. Susceptibility to periodic breathing with assisted ventilation during sleep in normal subjects. J Appl Physiol 85: 1929–1940, 1998 [DOI] [PubMed] [Google Scholar]
- 405. Mezzanotte WS, Tangel DJ, White DP. Waking genioglossal electromyogram in sleep apnea patients versus normal controls (a neuromuscular compensatory mechanism). J Clin Invest 89: 1571–1579, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Miles GB, Parkis MA, Lipski J, Funk GD. Modulation of phrenic motoneuron excitability by ATP: consequences for respiratory-related output in vitro. J Appl Physiol 92: 1899–1910, 2002 [DOI] [PubMed] [Google Scholar]
- 407. Millman RP, Carlisle CC, McGarvey ST, Eveloff SE, Levinson PD. Body fat distribution and sleep apnea severity in women. Chest 107: 362–366, 1995 [DOI] [PubMed] [Google Scholar]
- 408. Minemura H, Akashiba T, Yamamoto H, Akahoshi T, Kosaka N, Horie T. Acute effects of nasal continuous positive airway pressure on 24-hour blood pressure and catecholamines in patients with obstructive sleep apnea. Intern Med 37: 1009–1013, 1998 [DOI] [PubMed] [Google Scholar]
- 409. Minoguchi K, Tazaki T, Yokoe T, Minoguchi H, Watanabe Y, Yamamoto M, Adachi M. Elevated production of tumor necrosis factor-alpha by monocytes in patients with obstructive sleep apnea syndrome. Chest 126: 1473–1479, 2004 [DOI] [PubMed] [Google Scholar]
- 410. Minoguchi K, Yokoe T, Tanaka A, Ohta S, Hirano T, Yoshino G, O'Donnell CP, Adachi M. Association between lipid peroxidation and inflammation in obstructive sleep apnoea. Eur Respir J 28: 378–385, 2006 [DOI] [PubMed] [Google Scholar]
- 411. Minoguchi K, Yokoe T, Tazaki T, Minoguchi H, Oda N, Tanaka A, Yamamoto M, Ohta S, O'Donnell CP, Adachi M. Silent brain infarction and platelet activation in obstructive sleep apnea. Am J Respir Crit Care Med 175: 612–617, 2007 [DOI] [PubMed] [Google Scholar]
- 412. Minoguchi K, Yokoe T, Tazaki T, Minoguchi H, Tanaka A, Oda N, Okada S, Ohta S, Naito H, Adachi M. Increased carotid intima-media thickness and serum inflammatory markers in obstructive sleep apnea. Am J Respir Crit Care Med 172: 625–630, 2005 [DOI] [PubMed] [Google Scholar]
- 413. Mohsenin V, Valor R. Sleep apnea in patients with hemispheric stroke. Arch Phys Med Rehabil 76: 71–76, 1995 [DOI] [PubMed] [Google Scholar]
- 414. Moller DS, Lind P, Strunge B, Pedersen EB. Abnormal vasoactive hormones and 24-hour blood pressure in obstructive sleep apnea. Am J Hypertens 16: 274–280, 2003 [DOI] [PubMed] [Google Scholar]
- 415. Monahan KD, Leuenberger UA, Ray CA. Effect of repetitive hypoxic apnoeas on baroreflex function in humans. J Physiol 574: 605–613, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Monzillo LU, Hamdy O, Horton ES, Ledbury S, Mullooly C, Jarema C, Porter S, Ovalle K, Moussa A, Mantzoros CS. Effect of lifestyle modification on adipokine levels in obese subjects with insulin resistance. Obesity Res 11: 1048–1054, 2003 [DOI] [PubMed] [Google Scholar]
- 417. Mooe T, Franklin KA, Holmstrom K, Rabben T, Wiklund U. Sleep-disordered breathing and coronary artery disease: long-term prognosis. Am J Respir Crit Care Med 164: 1910–1913, 2001 [DOI] [PubMed] [Google Scholar]
- 418. Mooe T, Rabben T, Wiklund U, Franklin KA, Eriksson P. Sleep-disordered breathing in men with coronary artery disease. Chest 109: 659–663, 1996 [DOI] [PubMed] [Google Scholar]
- 419. Mooe T, Rabben T, Wiklund U, Franklin KA, Eriksson P. Sleep-disordered breathing in women: occurrence and association with coronary artery disease. Am J Med 101: 251–256, 1996 [DOI] [PubMed] [Google Scholar]
- 420. Moraes W, Poyares D, Sukys-Claudino L, Guilleminault C, Tufik S. Donepezil improves obstructive sleep apnea in Alzheimer disease: a double-blind, placebo-controlled study. Chest 133: 677–683, 2008 [DOI] [PubMed] [Google Scholar]
- 421. Morgan BJ, Crabtree DC, Palta M, Skatrud JB. Combined hypoxia and hypercapnia evokes long-lasting sympathetic activation in humans. J Appl Physiol 79: 205–213, 1995 [DOI] [PubMed] [Google Scholar]
- 422. Morgan BJ, Crabtree DC, Puleo DS, Badr MS, Toiber F, Skatrud JB. Neurocirculatory consequences of abrupt change in sleep state in humans. J Appl Physiol 80: 1627–1636, 1996 [DOI] [PubMed] [Google Scholar]
- 423. Morgan BJ, Dempsey JA, Pegelow DF, Jacques A, Finn L, Palta M, Skatrud JB, Young TB. Blood pressure perturbations caused by subclinical sleep-disordered breathing. Sleep 21: 737–746, 1998 [DOI] [PubMed] [Google Scholar]
- 424. Morgan BJ, Denahan T, Ebert TJ. Neurocirculatory consequences of negative intrathoracic pressure vs. asphyxia during voluntary apnea. J Appl Physiol 74: 2969–2975, 1993 [DOI] [PubMed] [Google Scholar]
- 425. Morgan TD, Remmers JE. Phylogeny and animal models: An Uninhibited Survey. In: Obstructive Sleep Apnea, edited by Kushida CA. New York: Informa Healthcare USA, 2007 [Google Scholar]
- 426. Morrell MJ, Arabi Y, Zahn B, Badr MS. Progressive retropalatal narrowing preceding obstructive apnea. Am J Respir Crit Care Med 158: 1974–1981, 1998 [DOI] [PubMed] [Google Scholar]
- 428. Morrell MJ, Arabi Y, Zahn BR, Meyer KC, Skatrud JB, Badr MS. Effect of surfactant on pharyngeal mechanics in sleeping humans: implications for sleep apnoea. Eur Respir J 20: 451–457, 2002 [DOI] [PubMed] [Google Scholar]
- 429. Morrell MJ, McRobbie DW, Quest RA, Cummin AR, Ghiassi R, Corfield DR. Changes in brain morphology associated with obstructive sleep apnea. Sleep Med 4: 451–454, 2003 [DOI] [PubMed] [Google Scholar]
- 430. Morrison DL, Launois SH, Isono S, Feroah TR, Whitelaw WA, Remmers JE. Pharyngeal narrowing and closing pressures in patients with obstructive sleep apnea. Am Rev Respir Dis 148: 606–611, 1993 [DOI] [PubMed] [Google Scholar]
- 431. Morrison JL, Sood S, Liu H, Park E, Liu X, Nolan P, Horner RL. Role of inhibitory amino acids in control of hypoglossal motor outflow to genioglossus muscle in naturally sleeping rats. J Physiol 552: 975–991, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432. Morrison JL, Sood S, Liu H, Park E, Nolan P, Horner RL. GABAA receptor antagonism at the hypoglossal motor nucleus increases genioglossus muscle activity in NREM but not REM sleep. J Physiol 548: 569–583, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Munoz R, Duran-Cantolla J, Martinez-Vila E, Gallego J, Rubio R, Aizpuru F, De La TG. Severe sleep apnea and risk of ischemic stroke in the elderly. Stroke 37: 2317–2321, 2006 [DOI] [PubMed] [Google Scholar]
- 434. Nacher M, Serrano-Mollar A, Farre R, Panes J, Segui J, Montserrat JM. Recurrent obstructive apneas trigger early systemic inflammation in a rat model of sleep apnea. Respir Physiol Neurobiol 155: 93–96, 2007 [DOI] [PubMed] [Google Scholar]
- 435. Naeije R, Melot C, Mols P, Hallemans R. Effects of vasodilators on hypoxic pulmonary vasoconstriction in normal man. Chest 82: 404–410, 1982 [DOI] [PubMed] [Google Scholar]
- 436. Nakayama H, Smith CA, Rodman JR, Skatrud JB, Dempsey JA. Effect of ventilatory drive on carbon dioxide sensitivity below eupnea during sleep. Am J Respir Crit Care Med 165: 1251–1260, 2002 [DOI] [PubMed] [Google Scholar]
- 437. Nakayama H, Smith CA, Rodman JR, Skatrud JB, Dempsey JA. Carotid body denervation eliminates apnea in response to transient hypocapnia. J Appl Physiol 94: 155–164, 2003 [DOI] [PubMed] [Google Scholar]
- 438. Narkiewicz K, Kato M, Phillips BG, Pesek CA, Davison DE, Somers VK. Nocturnal continuous positive airway pressure decreases daytime sympathetic traffic in obstructive sleep apnea. Circulation 100: 2332–2335, 1999 [DOI] [PubMed] [Google Scholar]
- 439. Narkiewicz K, van de Borne PJ, Montano N, Dyken ME, Phillips BG, Somers VK. Contribution of tonic chemoreflex activation to sympathetic activity and blood pressure in patients with obstructive sleep apnea. Circulation 97: 943–945, 1998 [DOI] [PubMed] [Google Scholar]
- 440. Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation 99: 1183–1189, 1999 [DOI] [PubMed] [Google Scholar]
- 441. Nattie E. Multiple sites for central chemoreception: their roles in response sensitivity and in sleep and wakefulness. Respir Physiol 122: 223–235, 2000 [DOI] [PubMed] [Google Scholar]
- 442. Naughton M, Benard D, Tam A, Rutherford R, Bradley TD. Role of hyperventilation in the pathogenesis of central sleep apneas in patients with congestive heart failure. Am Rev Respir Dis 148: 330–338, 1993 [DOI] [PubMed] [Google Scholar]
- 443. Newman AB, Nieto FJ, Guidry U, Lind BK, Redline S, Pickering TG, Quan SF. Relation of sleep-disordered breathing to cardiovascular disease risk factors: the Sleep Heart Health Study. Am J Epidemiol 154: 50–59, 2001 [DOI] [PubMed] [Google Scholar]
- 444. Nguyen ATD, Yim S, Malhotra Pathogenesis A. In: Obstructive Sleep Apnea, edited by Kushida CA. New York: Informa Healthcare USA, 2007 [Google Scholar]
- 445. Nieto FJ, Herrington DM, Redline S, Benjamin EJ, Robbins JA. Sleep apnea and markers of vascular endothelial function in a large community sample of older adults. Am J Respir Crit Care Med 169: 354–360, 2004 [DOI] [PubMed] [Google Scholar]
- 446. Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 283: 1829–1836, 2000 [DOI] [PubMed] [Google Scholar]
- 447. Niijima M, Kimura H, Edo H, Shinozaki T, Kang J, Masuyama S, Tatsumi K, Kuriyama T. Manifestation of pulmonary hypertension during REM sleep in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 159: 1766–1772, 1999 [DOI] [PubMed] [Google Scholar]
- 448. Niroumand M, Kuperstein R, Sasson Z, Hanly PJ. Impact of obstructive sleep apnea on left ventricular mass and diastolic function. Am J Respir Crit Care Med 163: 1632–1636, 2001 [DOI] [PubMed] [Google Scholar]
- 449. Noda A, Nakata S, Koike Y, Miyata S, Kitaichi K, Nishizawa T, Nagata K, Yasuma F, Murohara T, Yokota M. Continuous positive airway pressure improves daytime baroreflex sensitivity and nitric oxide production in patients with moderate to severe obstructive sleep apnea syndrome. Hypertens Res 30: 669–676, 2007 [DOI] [PubMed] [Google Scholar]
- 450. Noda A, Okada T, Yasuma F, Nakashima N, Yokota M. Cardiac hypertrophy in obstructive sleep apnea syndrome. Chest 107: 1538–1544, 1995 [DOI] [PubMed] [Google Scholar]
- 451. Norman D, Loredo JS, Nelesen RA, Ancoli-Israel S, Mills PJ, Ziegler MG, Dimsdale JE. Effects of continuous positive airway pressure versus supplemental oxygen on 24-hour ambulatory blood pressure. Hypertension 47: 840–845, 2006 [DOI] [PubMed] [Google Scholar]
- 452. O'Connor GT, Caffo B, Newman AB, Quan SF, Rapoport DM, Redline S, Resnick HE, Samet J, Shahar E. Prospective study of sleep-disordered breathing and hypertension: the Sleep Heart Health Study. Am J Respir Crit Care Med 179: 1159–1164, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453. O'Donnell C, Schaub CD, Haines AS, Berkowitz DE, Tankersley CG, Schwartz AR, Smith PL. Leptin prevents respiratory depression in obesity. Am J Respir Crit Care Med 159: 1477–1484, 1999 [DOI] [PubMed] [Google Scholar]
- 454. O'Donnell CP, Allan L, Atkinson P, Schwartz AR. The effect of upper airway obstruction and arousal on peripheral arterial tonometry in obstructive sleep apnea. Am J Respir Crit Care Med 166: 965–971, 2002 [DOI] [PubMed] [Google Scholar]
- 455. O'Donnell CP, Ayuse T, King ED, Schwartz AR, Smith PL, Robotham JL. Airway obstruction during sleep increases blood pressure without arousal. J Appl Physiol 80: 773–781, 1996 [DOI] [PubMed] [Google Scholar]
- 456. O'Donoghue FJ, Briellmann RS, Rochford PD, Abbott DF, Pell GS, Chan CH, Tarquinio N, Jackson GD, Pierce RJ. Cerebral structural changes in severe obstructive sleep apnea. Am J Respir Crit Care Med 171: 1185–1190, 2005 [DOI] [PubMed] [Google Scholar]
- 457. Oakes ND, Bell KS, Furler SM, Camilleri S, Saha AK, Ruderman NB, Chisholm DJ, Kraegen EW. Diet-induced muscle insulin resistance in rats is ameliorated by acute dietary lipid withdrawal or a single bout of exercise: parallel relationship between insulin stimulation of glucose uptake and suppression of long-chain fatty acyl-CoA. Diabetes 46: 2022–2028, 1997 [DOI] [PubMed] [Google Scholar]
- 458. Oertel BG, Schneider A, Rohrbacher M, Schmidt H, Tegeder I, Geisslinger G, Lotsch J. The partial 5-hydroxytryptamine1A receptor agonist buspirone does not antagonize morphine-induced respiratory depression in humans. Clin Pharmacol Ther 81: 59–68, 2007 [DOI] [PubMed] [Google Scholar]
- 459. Okabe S, Kubin L. Role of 5HT1 receptors in the control of hypoglossal motoneurons in vivo. Sleep 19: S150–S153, 1996 [DOI] [PubMed] [Google Scholar]
- 460. Oldenburg O, Lamp B, Faber L, Teschler H, Horstkotte D, Topfer V. Sleep-disordered breathing in patients with symptomatic heart failure: a contemporary study of prevalence in and characteristics of 700 patients. Eur J Heart Fail 9: 251–257, 2007 [DOI] [PubMed] [Google Scholar]
- 461. Oliven A, Tov N, Geitini L, Steinfeld U, Oliven R, Schwartz AR, Odeh M. Effect of genioglossus contraction on pharyngeal lumen and airflow in sleep apnoea patients. Eur Respir J 30: 748–758, 2007 [DOI] [PubMed] [Google Scholar]
- 462. Olmetti F, La Rovere MT, Robbi E, Taurino AE, Fanfulla F. Nocturnal cardiac arrhythmia in patients with obstructive sleep apnea. Sleep Med 9: 471–472, 2008 [DOI] [PubMed] [Google Scholar]
- 463. Oltmanns KM, Gehring H, Rudolf S, Schultes B, Rook S, Schweiger U, Born J, Fehm HL, Peters A. Hypoxia causes glucose intolerance in humans. Am J Respir Crit Care Med 169: 1231–1237, 2004 [DOI] [PubMed] [Google Scholar]
- 464. Onal E, Burrows DL, Hart RH, Lopata M. Induction of periodic breathing during sleep causes upper airway obstruction in humans. J Appl Physiol 61: 1438–1443, 1986 [DOI] [PubMed] [Google Scholar]
- 465. Onal E, Lopata M, O'Connor T. Pathogenesis of apneas in hypersomnia-sleep apnea syndrome. Am Rev Respir Dis 125: 167–174, 1982 [DOI] [PubMed] [Google Scholar]
- 466. Onal E, Lopata M, O'Connor TD. Diaphragmatic and genioglossal electromyogram responses to isocapnic hypoxia in humans. Am Rev Respir Dis 124: 215–217, 1981 [DOI] [PubMed] [Google Scholar]
- 467. Orem J. Central respiratory activity in rapid eye movement sleep: augmenting and late inspiratory cells. Sleep 17: 665–673, 1994 [DOI] [PubMed] [Google Scholar]
- 468. Orem J. Excitatory drive to the respiratory system in REM sleep. Sleep 19: S154–S156, 1996 [DOI] [PubMed] [Google Scholar]
- 469. Orem J, Kubin L. Respiratory physiology: central neural control. In: Principles and Practice of Sleep Medicine, edited by Kryger MH, Roth J, Dement WC. Philadelphia, PA: Saunders, 2005 [Google Scholar]
- 470. Orem J, Lovering AT, Dunin-Barkowski W, Vidruk EH. Endogenous excitatory drive to the respiratory system in rapid eye movement sleep in cats. J Physiol 527: 365–376, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Orem J, Lovering AT, Dunin-Barkowski W, Vidruk EH. Tonic activity in the respiratory system in wakefulness, NREM and REM sleep. Sleep 25: 488–496, 2002 [PubMed] [Google Scholar]
- 472. Orem J, Lydic R. Upper airway function during sleep and wakefulness: experimental studies on normal and anesthetized cats. Sleep 25: 49–68, 2002 [DOI] [PubMed] [Google Scholar]
- 473. Orem J, Montplaisir J, Dement WC. Changes in the activity of respiratory neurons during sleep. Brain Res 82: 309–315, 1974 [DOI] [PubMed] [Google Scholar]
- 474. Orem J, Osorio I, Brooks E, Dick T. Activity of respiratory neurons during NREM sleep. J Neurophysiol 54: 1144–1156, 1985 [DOI] [PubMed] [Google Scholar]
- 475. Otto ME, Belohlavek M, Romero-Corral A, Gami AS, Gilman G, Svatikova A, Amin RS, Lopez-Jimenez F, Khandheria BK, Somers VK. Comparison of cardiac structural and functional changes in obese otherwise healthy adults with versus without obstructive sleep apnea. Am J Cardiol 99: 1298–1302, 2007 [DOI] [PubMed] [Google Scholar]
- 476. Ozdemir O, Beletsky V, Hachinski V, Spence JD. Cerebrovascular events on awakening, patent foramen ovale and obstructive sleep apnea syndrome. J Neurol Sci 268: 193–194, 2008 [DOI] [PubMed] [Google Scholar]
- 477. Pack AI, Cola MF, Goldszmidt A, Ogilvie MD, Gottschalk A. Correlation between oscillations in ventilation and frequency content of the electroencephalogram. J Appl Physiol 72: 985–992, 1992 [DOI] [PubMed] [Google Scholar]
- 478. Packer M. The placebo effect in heart failure. Am Heart J 120: 1579–1582, 1990 [DOI] [PubMed] [Google Scholar]
- 479. Pae EK, Lowe AA, Fleetham JA. A role of pharyngeal length in obstructive sleep apnea patients. Am J Orthod Dentofacial Orthop 111: 12–17, 1997 [DOI] [PubMed] [Google Scholar]
- 480. Pagnotta SE, Lape R, Quitadamo C, Nistri A. Pre- and postsynaptic modulation of glycinergic and GABAergic transmission by muscarinic receptors on rat hypoglossal motoneurons in vitro. Neuroscience 130: 783–795, 2005 [DOI] [PubMed] [Google Scholar]
- 481. Palmer LJ, Buxbaum SG, Larkin E, Patel SR, Elston RC, Tishler PV, Redline S. A whole-genome scan for obstructive sleep apnea and obesity. Am J Hum Genet 72: 340–350, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Palmer LJ, Buxbaum SG, Larkin EK, Patel SR, Elston RC, Tishler PV, Redline S. Whole genome scan for obstructive sleep apnea and obesity in African-American families. Am J Respir Crit Care Med 169: 1314–1321, 2004 [DOI] [PubMed] [Google Scholar]
- 483. Parisi RA, Neubauer JA, Frank MM, Santiago TV, Edelman NH. Linkage between brain blood flow and respiratory drive during rapid-eye-movement sleep. J Appl Physiol 64: 1457–1465, 1988 [DOI] [PubMed] [Google Scholar]
- 484. Parkash R, Green MS, Kerr CR, Connolly SJ, Klein GJ, Sheldon R, Talajic M, Dorian P, Humphries KH. The association of left atrial size and occurrence of atrial fibrillation: a prospective cohort study from the Canadian Registry of Atrial Fibrillation. Am Heart J 148: 649–654, 2004 [DOI] [PubMed] [Google Scholar]
- 485. Parker JD, Brooks D, Kozar LF, Render-Teixeira CL, Horner RL, Douglas BT, Phillipson EA. Acute and chronic effects of airway obstruction on canine left ventricular performance. Am J Respir Crit Care Medicine 160: 1888–1896, 1999 [DOI] [PubMed] [Google Scholar]
- 486. Parkis MA, Bayliss DA, Berger AJ. Actions of norepinephrine on rat hypoglossal motoneurons. J Neurophysiol 74: 1911–1919, 1995 [DOI] [PubMed] [Google Scholar]
- 487. Parra O, Arboix A, Bechich S, Garcia-Eroles L, Montserrat JM, Lopez JA, Ballester E, Guerra JM, Sopena JJ. Time course of sleep-related breathing disorders in first-ever stroke or transient ischemic attack. Am J Respir Crit Care Med 161: 375–380, 2000 [DOI] [PubMed] [Google Scholar]
- 488. Partinen M, Guilleminault C, Quera-Salva MA, Jamieson A. Obstructive sleep apnea and cephalometric roentgenograms. The role of anatomic upper airway abnormalities in the definition of abnormal breathing during sleep. Chest 93: 1199–1205, 1988 [DOI] [PubMed] [Google Scholar]
- 489. Patel SR. Shared genetic risk factors for obstructive sleep apnea and obesity. J Appl Physiol 99: 1600–1606, 2005 [DOI] [PubMed] [Google Scholar]
- 490. Patel SR, Larkin EK, Mignot E, Lin L, Redline S. The association of angiotensin converting enzyme (ACE) polymorphisms with sleep apnea and hypertension. Sleep 30: 531–533, 2007 [DOI] [PubMed] [Google Scholar]
- 491. Patel SR, White DP, Malhotra A, Stanchina ML, Ayas NT. Continuous positive airway pressure therapy for treating sleepiness in a diverse population with obstructive sleep apnea: results of a meta-analysis. Arch Intern Med 163: 565–571, 2003 [DOI] [PubMed] [Google Scholar]
- 492. Patil SP, Schneider H, Marx JJ, Gladmon E, Schwartz AR, Smith PL. Neuromechanical control of upper airway patency during sleep. J Appl Physiol 102: 547–556, 2007 [DOI] [PubMed] [Google Scholar]
- 493. Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 132: 325–337, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Peever JH, Lai YY, Siegel JM. Excitatory effects of hypocretin-1 (orexin-A) in the trigeminal motor nucleus are reversed by NMDA antagonism. J Neurophysiol 89: 2591–2600, 2003 [DOI] [PubMed] [Google Scholar]
- 495. Peker Y, Kraiczi H, Hedner J, Loth S, Johansson A, Bende M. An independent association between obstructive sleep apnoea and coronary artery disease. Eur Respir J 14: 179–184, 1999 [DOI] [PubMed] [Google Scholar]
- 496. Peled N, Abinader EG, Pillar G, Sharif D, Lavie P. Nocturnal ischemic events in patients with obstructive sleep apnea syndrome and ischemic heart disease: effects of continuous positive air pressure treatment. J Am Coll Cardiol 34: 1744–1749, 1999 [DOI] [PubMed] [Google Scholar]
- 497. Peled N, Kassirer M, Kramer MR, Rogowski O, Shlomi D, Fox B, Berliner AS, Shitrit D. Increased erythrocyte adhesiveness and aggregation in obstructive sleep apnea syndrome. Thromb Res 121: 631–636, 2008 [DOI] [PubMed] [Google Scholar]
- 498. Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci USA 100: 10073–10078, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499. Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J Appl Physiol 94: 2342–2349, 2003 [DOI] [PubMed] [Google Scholar]
- 500. Peng YJ, Yuan G, Jacono FJ, Kumar GK, Prabhakar NR. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. J Physiol 576: 289–295, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501. Peppard PE, Young T, Palta M, Dempsey J, Skatrud J. Longitudinal study of moderate weight change and sleep-disordered breathing. JAMA 284: 3015–3021, 2000 [DOI] [PubMed] [Google Scholar]
- 502. Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 342: 1378–1384, 2000 [DOI] [PubMed] [Google Scholar]
- 503. Pepperell JC, Ramdassingh-Dow S, Crosthwaite N, Mullins R, Jenkinson C, Stradling JR, Davies RJ. Ambulatory blood pressure after therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised parallel trial. Lancet 359: 204–210, 2002 [DOI] [PubMed] [Google Scholar]
- 504. Petralia RS, Wang YX, Wenthold RJ. Histological and ultrastructural localization of the kainate receptor subunits, KA2 and GluR6/7, in the rat nervous system using selective antipeptide antibodies. J Comp Neurol 349: 85–110, 1994 [DOI] [PubMed] [Google Scholar]
- 505. Pevernagie DA, Stanson AW, Sheedy PF, Daniels BK, Shepard JW., Jr Effects of body position on the upper airway of patients with obstructive sleep apnea. Am J Respir Crit Care Med 152: 179–185, 1995 [DOI] [PubMed] [Google Scholar]
- 506. Phillips BG, Hisel TM, Kato M, Pesek CA, Dyken ME, Narkiewicz K, Somers VK. Recent weight gain in patients with newly diagnosed obstructive sleep apnea. J Hypertens 17: 1297–1300, 1999 [DOI] [PubMed] [Google Scholar]
- 507. Phillips BG, Narkiewicz K, Pesek CA, Haynes WG, Dyken ME, Somers VK. Effects of obstructive sleep apnea on endothelin-1 and blood pressure. J Hypertens 17: 61–66, 1999 [DOI] [PubMed] [Google Scholar]
- 508. Phillips BG, Somers VK. Hypertension and obstructive sleep apnea. Curr Hypertens Rep 5: 380–385, 2003 [DOI] [PubMed] [Google Scholar]
- 509. Phillips C, Hedner J, Berend N, Grunstein R. Diurnal and obstructive sleep apnea influences on arterial stiffness and central blood pressure in men. Sleep 28: 604–609, 2005 [DOI] [PubMed] [Google Scholar]
- 510. Phillips CL, Yee B, Yang Q, Villaneuva AT, Hedner J, Berend N, Grunstein R. Effects of continuous positive airway pressure treatment and withdrawal in patients with obstructive sleep apnea on arterial stiffness and central BP. Chest 134: 94–100, 2008 [DOI] [PubMed] [Google Scholar]
- 511. Phillips SA, Olson EB, Lombard JH, Morgan BJ. Chronic intermittent hypoxia alters NE reactivity and mechanics of skeletal muscle resistance arteries. J Appl Physiol 100: 1117–1123, 2006 [DOI] [PubMed] [Google Scholar]
- 512. Phillips SA, Olson EB, Morgan BJ, Lombard JH. Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol 286: H388–H393, 2004 [DOI] [PubMed] [Google Scholar]
- 513. Phillipson EA, Bowes G. Control of breathing during sleep. In: Handbook of Physiology. The Respiratory System. Control of Breathing. Bethesda, MD: Am. Physiol. Soc., 1986, sect. 3, vol. II, pt. 2, chapt. 19, p. 649–690 [Google Scholar]
- 514. Phillipson EA, Murphy E, Kozar LF. Regulation of respiration in sleeping dogs. J Appl Physiol 40: 688–693, 1976 [DOI] [PubMed] [Google Scholar]
- 515. Phipps PR, Starritt E, Caterson I, Grunstein RR. Association of serum leptin with hypoventilation in human obesity. Thorax 57: 75–76, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Pierrefiche O, Bischoff AM, Richter DW, Spyer KM. Hypoxic response of hypoglossal motoneurones in the in vivo cat. J Physiol 505: 785–795, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517. Pillar G, Malhotra A, Fogel RB, Beauregard J, Slamowitz DI, Shea SA, White DP. Upper airway muscle responsiveness to rising PCO(2) during NREM sleep. J Appl Physiol 89: 1275–1282, 2000 [DOI] [PubMed] [Google Scholar]
- 518. Placidi F, Diomedi M, Cupini LM, Bernardi G, Silvestrini M. Impairment of daytime cerebrovascular reactivity in patients with obstructive sleep apnoea syndrome. J Sleep Res 7: 288–292, 1998 [DOI] [PubMed] [Google Scholar]
- 519. Pletcher MJ, Kertesz SG, Kohn MA, Gonzales R. Trends in opioid prescribing by race/ethnicity for patients seeking care in US emergency departments. JAMA 299: 70–78, 2008 [DOI] [PubMed] [Google Scholar]
- 520. Plowman L, Lauff DC, Berthon-Jones M, Sullivan CE. Waking and genioglossus muscle responses to upper airway pressure oscillation in sleeping dogs. J Appl Physiol 68: 2564–2573, 1990 [DOI] [PubMed] [Google Scholar]
- 521. Podszus T, Bauer W, Mayer J, Penzel T, Peter JH, von Wichert P. Sleep apnea and pulmonary hypertension. Klin Wochenschr 64: 131–134, 1986 [DOI] [PubMed] [Google Scholar]
- 522. Polotsky VY, Li J, Punjabi NM, Rubin AE, Smith PL, Schwartz AR, O'Donnell CP. Intermittent hypoxia increases insulin resistance in genetically obese mice. J Physiol 552: 253–264, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523. Ponikowski P, Chua TP, Anker SD, Francis DP, Doehner W, Banasiak W, Poole-Wilson PA, Piepoli MF, Coats AJ. Peripheral chemoreceptor hypersensitivity: an ominous sign in patients with chronic heart failure. Circulation 104: 544–549, 2001 [DOI] [PubMed] [Google Scholar]
- 524. Popko K, Gorska E, Wasik M, Stoklosa A, Plywaczewski R, Winiarska M, Gorecka D, Sliwinski P, Demkow U. Frequency of distribution of leptin receptor gene polymorphism in obstructive sleep apnea patients. J Physiol Pharmacol 58 Suppl 5: 551–561, 2007 [PubMed] [Google Scholar]
- 525. Porte D, Jr, Williams RH. Inhibition of insulin release by norepinephrine in man. Science 152: 1248–1250, 1966 [DOI] [PubMed] [Google Scholar]
- 526. Prabhakar NR. NO and CO as second messengers in oxygen sensing in the carotid body. Respir Physiol 115: 161–168, 1999 [DOI] [PubMed] [Google Scholar]
- 527. Prabhakar NR, Jacono FJ. Cellular and molecular mechanisms associated with carotid body adaptations to chronic hypoxia. High Alt Med Biol 6: 112–120, 2005 [DOI] [PubMed] [Google Scholar]
- 528. Prabhakar NR, Kumar GK. Oxidative stress in the systemic and cellular responses to intermittent hypoxia. Biol Chem 385: 217–221, 2004 [DOI] [PubMed] [Google Scholar]
- 529. Prabhakar NR, Peng YJ, Jacono FJ, Kumar GK, Dick TE. Cardiovascular alterations by chronic intermittent hypoxia: importance of carotid body chemoreflexes. Clin Exp Pharmacol Physiol 32: 447–449, 2005 [DOI] [PubMed] [Google Scholar]
- 530. Prentice AM. The emerging epidemic of obesity in developing countries. Int J Epidemiol 35: 93–99, 2006 [DOI] [PubMed] [Google Scholar]
- 531. Pride NB, Permutt S, Riley RL, Bromberger-Barnea B. Determinants of maximal expiratory flow from the lungs. J Appl Physiol 23: 646–662, 1967 [DOI] [PubMed] [Google Scholar]
- 532. Przybylowski T, Bangash MF, Reichmuth K, Morgan BJ, Skatrud JB, Dempsey JA. Mechanisms of the cerebrovascular response to apnoea in humans. J Physiol 548: 323–332, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533. Punjabi NM, Newman AB, Young TB, Resnick HE, Sanders MH. Sleep-disordered breathing and cardiovascular disease: an outcome-based definition of hypopneas. Am J Respir Crit Care Med 177: 1150–1155, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534. Punjabi NM, Shahar E, Redline S, Gottlieb DJ, Givelber R, Resnick HE. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol 160: 521–530, 2004 [DOI] [PubMed] [Google Scholar]
- 535. Punjabi NM, Sorkin JD, Katzel LI, Goldberg AP, Schwartz AR, Smith PL. Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. Am J Respir Crit Care Med 165: 677–682, 2002 [DOI] [PubMed] [Google Scholar]
- 536. Quitadamo C, Fabbretti E, Lamanauskas N, Nistri A. Activation and desensitization of neuronal nicotinic receptors modulate glutamatergic transmission on neonatal rat hypoglossal motoneurons. Eur J Neurosci 22: 2723–2734, 2005 [DOI] [PubMed] [Google Scholar]
- 537. Rajagopalan B, Raine AE, Cooper R, Ledingham JG. Changes in cerebral blood flow in patients with severe congestive cardiac failure before and after captopril treatment. Am J Med 76: 86–90, 1984 [DOI] [PubMed] [Google Scholar]
- 538. Rangemark C, Hedner JA, Carlson JT, Gleerup G, Winther K. Platelet function and fibrinolytic activity in hypertensive and normotensive sleep apnea patients. Sleep 18: 188–194, 1995 [DOI] [PubMed] [Google Scholar]
- 539. Raul L. Serotonin2 receptors in the nucleus tractus solitarius: characterization and role in the baroreceptor reflex arc. Cell Mol Neurobiol 23: 709–726, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540. Ray CJ, Abbas MR, Coney AM, Marshall JM. Interactions of adenosine, prostaglandins and nitric oxide in hypoxia-induced vasodilatation: in vivo and in vitro studies. J Physiol 544: 195–209, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541. Reddy MK, Patel KP, Schultz HD. Differential role of the paraventricular nucleus of the hypothalamus in modulating the sympathoexcitatory component of peripheral and central chemoreflexes. Am J Physiol Regul Integr Comp Physiol 289: R789–R797, 2005 [DOI] [PubMed] [Google Scholar]
- 542. Redline S, Storfer-Isser A, Rosen CL, Johnson NL, Kirchner HL, Emancipator J, Kibler AM. Association between metabolic syndrome and sleep-disordered breathing in adolescents. Am J Respir Crit Care Med 176: 401–408, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543. Redline S, Tishler PV, Schluchter M, Aylor J, Clark K, Graham G. Risk factors for sleep-disordered breathing in children. Associations with obesity, race, and respiratory problems. Am J Respir Crit Care Med 159: 1527–1532, 1999 [DOI] [PubMed] [Google Scholar]
- 544. Redolfi S, Yumino D, Ruttanaumpawan P, Yau B, Su MC, Lam J, Bradley TD. Relationship between overnight rostral fluid shift and obstructive sleep apnea in nonobese men. Am J Respir Crit Care Med 179: 241–246, 2009 [DOI] [PubMed] [Google Scholar]
- 545. Reichmuth K, Austin D, Peppard P, Nieto J, Young T, Barczi S, Skatrud J, Morgan B. Sleep disordered breathing and cerebral vasoreactivity to CO2. Sleep 30 Suppl: A157, 2007 [Google Scholar]
- 546. Reichmuth KJ, Austin D, Skatrud JB, Young T. Association of sleep apnea and type II diabetes: a population-based study. Am J Respir Crit Care Med 172: 1590–1595, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546a. Reichmuth KJ, Dopp JM, Barczi SR, Skatrud JB, Wojdyla P, Hayes JD, Morgan BJ. Impaired vascular regulation in patients with obstructive sleep apnea: effects of CPAP treatment. Am J Respir Crit Care Med. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Reite M, Jackson D, Cahoon RL, Weil JV. Sleep physiology at high altitude. Electroencephalogr Clin Neurophysiol 38: 463–471, 1975 [DOI] [PubMed] [Google Scholar]
- 548. Remmers JE, Anch AM, deGroot WJ, Baker JP, Jr, Sauerland EK. Oropharyngeal muscle tone in obstructive sleep apnea before and after strychnine. Sleep 3: 447–453, 1980 [DOI] [PubMed] [Google Scholar]
- 549. Remmers JE, deGroot WJ, Sauerland EK, Anch AM. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol 44: 931–938, 1978 [DOI] [PubMed] [Google Scholar]
- 550. Remsburg S, Launois SH, Weiss JW. Patients with obstructive sleep apnea have an abnormal peripheral vascular response to hypoxia. J Appl Physiol 87: 1148–1153, 1999 [DOI] [PubMed] [Google Scholar]
- 551. Rey S, Del RR, Iturriaga R. Contribution of endothelin-1 to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Brain Res 1086: 152–159, 2006 [DOI] [PubMed] [Google Scholar]
- 552. Reymond-Marron I, Tribollet E, Raggenbass M. The vasopressin-induced excitation of hypoglossal and facial motoneurons in young rats is mediated by V1a but not V1b receptors, is independent of intracellular calcium signalling. Eur J Neurosci 24: 1565–1574, 2006 [DOI] [PubMed] [Google Scholar]
- 553. Richard CA, Harper RM. Respiratory-related activity in hypoglossal neurons across sleep-waking states in cats. Brain Res 542: 167–170, 1991 [DOI] [PubMed] [Google Scholar]
- 554. Richter DW, Manzke T, Wilken B, Ponimaskin E. Serotonin receptors: guardians of stable breathing. Trends Mol Med 9: 542–548, 2003 [DOI] [PubMed] [Google Scholar]
- 555. Riha RL, Brander P, Vennelle M, McArdle N, Kerr SM, Anderson NH, Douglas NJ. Tumour necrosis factor-alpha (−308) gene polymorphism in obstructive sleep apnoea-hypopnoea syndrome. Eur Respir J 26: 673–678, 2005 [DOI] [PubMed] [Google Scholar]
- 556. Riley R, Guilleminault C, Herran J, Powell N. Cephalometric analyses and flow-volume loops in obstructive sleep apnea patients. Sleep 6: 303–311, 1983 [DOI] [PubMed] [Google Scholar]
- 557. Rivlin J, Hoffstein V, Kalbfleisch J, McNicholas W, Zamel N, Bryan AC. Upper airway morphology in patients with idiopathic obstructive sleep apnea. Am Rev Respir Dis 129: 355–360, 1984 [DOI] [PubMed] [Google Scholar]
- 558. Robinson GV, Langford BA, Smith DM, Stradling JR. Predictors of blood pressure fall with continuous positive airway pressure (CPAP) treatment of obstructive sleep apnoea (OSA). Thorax 63: 855–859, 2008 [DOI] [PubMed] [Google Scholar]
- 559. Robinson GV, Pepperell JC, Segal HC, Davies RJ, Stradling JR. Circulating cardiovascular risk factors in obstructive sleep apnoea: data from randomised controlled trials. Thorax 59: 777–782, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560. Robinson GV, Smith DM, Langford BA, Davies RJ, Stradling JR. Continuous positive airway pressure does not reduce blood pressure in nonsleepy hypertensive OSA patients. Eur Respir J 27: 1229–1235, 2006 [DOI] [PubMed] [Google Scholar]
- 561. Rodenstein DO, Dooms G, Thomas Y, Liistro G, Stanescu DC, Culee C, Aubert-Tulkens G. Pharyngeal shape and dimensions in healthy subjects, snorers, and patients with obstructive sleep apnoea. Thorax 45: 722–727, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562. Rosen CL, Larkin EK, Kirchner HL, Emancipator JL, Bivins SF, Surovec SA, Martin RJ, Redline S. Prevalence and risk factors for sleep-disordered breathing in 8- to 11-year-old children: association with race and prematurity. J Pediatr 142: 383–389, 2003 [DOI] [PubMed] [Google Scholar]
- 563. Rukhadze I, Kubin L. Mesopontine cholinergic projections to the hypoglossal motor nucleus. Neurosci Lett 413: 121–125, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 112: 2660–2667, 2005 [DOI] [PubMed] [Google Scholar]
- 565. Ryan S, Taylor CT, McNicholas WT. Predictors of elevated nuclear factor-kappaB-dependent genes in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 174: 824–830, 2006 [DOI] [PubMed] [Google Scholar]
- 566. Ryland D, Reid L. The pulmonary circulation in cystic fibrosis. Thorax 30: 285–292, 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Saarelainen S, Seppala E, Laasonen K, Hasan J. Circulating endothelin-1 in obstructive sleep apnea. Endothelium 5: 115–118, 1997 [DOI] [PubMed] [Google Scholar]
- 568. Sahlin C, Sandberg O, Gustafson Y, Bucht G, Carlberg B, Stenlund H, Franklin KA. Obstructive sleep apnea is a risk factor for death in patients with stroke: a 10-year follow-up. Arch Intern Med 168: 297–301, 2008 [DOI] [PubMed] [Google Scholar]
- 569. Saini J, Krieger J, Brandenberger G, Wittersheim G, Simon C, Follenius M. Continuous positive airway pressure treatment. Effects on growth hormone, insulin and glucose profiles in obstructive sleep apnea patients. Horm Metab Res 25: 375–381, 1993 [DOI] [PubMed] [Google Scholar]
- 570. Sajkov D, Cowie RJ, Thornton AT, Espinoza HA, McEvoy RD. Pulmonary hypertension and hypoxemia in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 149: 416–422, 1994 [DOI] [PubMed] [Google Scholar]
- 571. Sajkov D, Wang T, Saunders NA, Bune AJ, McEvoy RD. Continuous positive airway pressure treatment improves pulmonary hemodynamics in patients with obstructive sleep apnea. Am J Respir Crit Care Med 165: 152–158, 2002 [DOI] [PubMed] [Google Scholar]
- 572. Sajkov D, Wang T, Saunders NA, Bune AJ, Neill AM, Douglas MR. Daytime pulmonary hemodynamics in patients with obstructive sleep apnea without lung disease. Am J Respir Crit Care Med 159: 1518–1526, 1999 [DOI] [PubMed] [Google Scholar]
- 573. Salazar-Grueso EF, Rosenberg RS, Roos RP. Sleep apnea in olivopontocerebellar degeneration: treatment with trazodone. Ann Neurol 23: 399–401, 1988 [DOI] [PubMed] [Google Scholar]
- 574. Sanders MH, Kern N. Obstructive sleep apnea treated by independently adjusted inspiratory and expiratory positive airway pressures via nasal mask. Physiologic and clinical implications. Chest 98: 317–324, 1990 [DOI] [PubMed] [Google Scholar]
- 575. Sanders MH, Moore SE. Inspiratory and expiratory partitioning of airway resistance during sleep in patients with sleep apnea. Am Rev Respir Dis 127: 554–558, 1983 [DOI] [PubMed] [Google Scholar]
- 576. Sanders MH, Rogers RM, Pennock BE. Prolonged expiratory phase in sleep apnea. A unifying hypothesis. Am Rev Respir Dis 131: 401–408, 1985 [DOI] [PubMed] [Google Scholar]
- 577. Sanner BM, Doberauer C, Konermann M, Sturm A, Zidek W. Pulmonary hypertension in patients with obstructive sleep apnea syndrome. Arch Intern Med 157: 2483–2487, 1997 [PubMed] [Google Scholar]
- 578. Sanner BM, Konermann M, Tepel M, Groetz J, Mummenhoff C, Zidek W. Platelet function in patients with obstructive sleep apnoea syndrome. Eur Respir J 16: 648–652, 2000 [DOI] [PubMed] [Google Scholar]
- 579. Sant'Ambrogio G, Mathew OP, Fisher JT, Sant'Ambrogio FB. Laryngeal receptors responding to transmural pressure, airflow and local muscle activity. Respir Physiol 54: 317–330, 1983 [DOI] [PubMed] [Google Scholar]
- 580. Sasayama S, Izumi T, Seino Y, Ueshima K, Asanoi H. Effects of nocturnal oxygen therapy on outcome measures in patients with chronic heart failure and Cheyne-Stokes respiration. Circ J 70: 1–7, 2006 [DOI] [PubMed] [Google Scholar]
- 581. Satoh M, Eastwood PR, Smith CA, Dempsey JA. Nonchemical elimination of inspiratory motor output via mechanical ventilation in sleep. Am J Respir Crit Care Med 163: 1356–1364, 2001 [DOI] [PubMed] [Google Scholar]
- 582. Saupe KW, Smith CA, Henderson KS, Dempsey JA. Respiratory and cardiovascular responses to increased and decreased carotid sinus pressure in sleeping dogs. J Appl Physiol 78: 1688–1698, 1995 [DOI] [PubMed] [Google Scholar]
- 583. Savransky V, Nanayakkara A, Li J, Bevans S, Smith PL, Rodriguez A, Polotsky VY. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med 175: 1290–1297, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584. Savransky V, Nanayakkara A, Vivero A, Li J, Bevans S, Smith PL, Torbenson MS, Polotsky VY. Chronic intermittent hypoxia predisposes to liver injury. Hepatology 45: 1007–1013, 2007 [DOI] [PubMed] [Google Scholar]
- 585. Schafer H, Koehler U, Ploch T, Peter JH. Sleep-related myocardial ischemia and sleep structure in patients with obstructive sleep apnea and coronary heart disease. Chest 111: 387–393, 1997 [DOI] [PubMed] [Google Scholar]
- 586. Schahin SP, Nechanitzky T, Dittel C, Fuchs FS, Hahn EG, Konturek PC, Ficker JH, Harsch IA. Long-term improvement of insulin sensitivity during CPAP therapy in the obstructive sleep apnoea syndrome. Med Sci Monit 14: CR117–CR121, 2008 [PubMed] [Google Scholar]
- 587. Schmidt CF. The influence of cerebral blood-flow on respiration. I. The respiratory responses to changes in cerebral blood-flow. Am J Physiol 84: 202–222, 1928 [Google Scholar]
- 588. Schmidt CF. The influence of cerebral blood-flow on respiration. II. The gaseous metabolism of the brain. Am J Physiol 84: 223–241, 1928 [Google Scholar]
- 589. Schmidt CF. The influence of cerebral blood-flow on respiration. III. The interplay of factors concerned in the regulation of respiration. Am J Physiol 84: 242–259, 1928 [Google Scholar]
- 590. Schmidt HS. l-Tryptophan in the treatment of impaired respiration in sleep. Bull Eur Physiopathol Respir 19: 625–629, 1983 [PubMed] [Google Scholar]
- 591. Schneider H, Boudewyns A, Smith PL, O'Donnell CP, Canisius S, Stammnitz A, Allan L, Schwartz AR. Modulation of upper airway collapsibility during sleep: influence of respiratory phase and flow regimen. J Appl Physiol 93: 1365–1376, 2002 [DOI] [PubMed] [Google Scholar]
- 592. Schneider H, Schaub CD, Chen CA, Andreoni KA, Schwartz AR, Smith PL, Robotham JL, O'Donnell CP. Neural and local effects of hypoxia on cardiovascular responses to obstructive apnea. J Appl Physiol 88: 1093–1102, 2000 [DOI] [PubMed] [Google Scholar]
- 593. Schroeder JS, Motta J, Guilleminault C. Hemodynamic studies in sleep apnea. In: Sleep Apnea Syndromes, edited by Guilleminault C, Dement WC. New York: Liss, 1978 [Google Scholar]
- 594. Schulz R, Mahmoudi S, Hattar K, Sibelius U, Olschewski H, Mayer K, Seeger W, Grimminger F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med 162: 566–570, 2000 [DOI] [PubMed] [Google Scholar]
- 595. Schwab RJ, Gefter WB, Hoffman EA, Gupta KB, Pack AI. Dynamic upper airway imaging during awake respiration in normal subjects and patients with sleep disordered breathing. Am Rev Respir Dis 148: 1385–1400, 1993 [DOI] [PubMed] [Google Scholar]
- 596. Schwab RJ, Gefter WB, Pack AI, Hoffman EA. Dynamic imaging of the upper airway during respiration in normal subjects. J Appl Physiol 74: 1504–1514, 1993 [DOI] [PubMed] [Google Scholar]
- 597. Schwab RJ, Gupta KB, Gefter WB, Metzger LJ, Hoffman EA, Pack AI. Upper airway and soft tissue anatomy in normal subjects and patients with sleep-disordered breathing. Significance of the lateral pharyngeal walls. Am J Respir Crit Care Med 152: 1673–1689, 1995 [DOI] [PubMed] [Google Scholar]
- 598. Schwab RJ, Kuna ST, Remmers JE. Anatomy and physiology of upper airway obstruction. In: Principles and Practice of Sleep Medicine, edited by Kryger MH, Roth J, Dement WC. Philadelphia, PA: Saunders, 2005 [Google Scholar]
- 599. Schwab RJ, Pasirstein M, Pierson R, Mackley A, Hachadoorian R, Arens R, Maislin G, Pack AI. Identification of upper airway anatomic risk factors for obstructive sleep apnea with volumetric magnetic resonance imaging. Am J Respir Crit Care Med 168: 522–530, 2003 [DOI] [PubMed] [Google Scholar]
- 600. Schwartz AR, Gold AR, Schubert N, Stryzak A, Wise RA, Permutt S, Smith PL. Effect of weight loss on upper airway collapsibility in obstructive sleep apnea. Am Rev Respir Dis 144: 494–498, 1991 [DOI] [PubMed] [Google Scholar]
- 601. Schwartz AR, Patil SP, Laffan AM, Polotsky V, Schneider H, Smith PL. Obesity and obstructive sleep apnea: pathogenic mechanisms and therapeutic approaches. Proc Am Thorac Soc 5: 185–192, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602. Schwartz AR, Schneider H, Smith PL. Upper airway surface tension: is it a significant cause of airflow obstruction during sleep? J Appl Physiol 95: 1759–1760, 2003 [DOI] [PubMed] [Google Scholar]
- 603. Schwartz AR, Smith PL, Wise RA, Gold AR, Permutt S. Induction of upper airway occlusion in sleeping individuals with subatmospheric nasal pressure. J Appl Physiol 64: 535–542, 1988 [DOI] [PubMed] [Google Scholar]
- 604. Semmens M, Reid L. Pulmonary arterial muscularity and right ventricular hypertrophy in chronic bronchitis and emphysema. Br J Dis Chest 68: 253–263, 1974 [DOI] [PubMed] [Google Scholar]
- 605. Series F, Cormier Y, Desmeules M. Influence of passive changes of lung volume on upper airways. J Appl Physiol 68: 2159–2164, 1990 [DOI] [PubMed] [Google Scholar]
- 606. Series F, Cote C, Simoneau JA, Gelinas Y, St Pierre S, Leclerc J, Ferland R, Marc I. Physiologic, metabolic, and muscle fiber type characteristics of musculus uvulae in sleep apnea hypopnea syndrome and in snorers. J Clin Invest 95: 20–25, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Series F, Marc I. Influence of lung volume dependence of upper airway resistance during continuous negative airway pressure. J Appl Physiol 77: 840–844, 1994 [DOI] [PubMed] [Google Scholar]
- 608. Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, Javier NF, O'connor GT, Boland LL, Schwartz JE, Samet JM. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med 163: 19–25, 2001 [DOI] [PubMed] [Google Scholar]
- 609. Shamsuzzaman AS, Winnicki M, Lanfranchi P, Wolk R, Kara T, Accurso V, Somers VK. Elevated C-reactive protein in patients with obstructive sleep apnea. Circulation 105: 2462–2464, 2002 [DOI] [PubMed] [Google Scholar]
- 610. Shelton KE, Woodson H, Gay S, Suratt PM. Pharyngeal fat in obstructive sleep apnea. Am Rev Respir Dis 148: 462–466, 1993 [DOI] [PubMed] [Google Scholar]
- 611. Shinohara E, Kihara S, Yamashita S, Yamane M, Nishida M, Arai T, Kotani K, Nakamura T, Takemura K, Matsuzawa Y. Visceral fat accumulation as an important risk factor for obstructive sleep apnoea syndrome in obese subjects. J Intern Med 241: 11–18, 1997 [DOI] [PubMed] [Google Scholar]
- 612. Shiomi T, Guilleminault C, Stoohs R, Schnittger I. Leftward shift of the interventricular septum and pulsus paradoxus in obstructive sleep apnea syndrome. Chest 100: 894–902, 1991 [DOI] [PubMed] [Google Scholar]
- 613. Shoham S, Davenne D, Cady AB, Dinarello CA, Krueger JM. Recombinant tumor necrosis factor and interleukin 1 enhance slow-wave sleep. Am J Physiol Regul Integr Comp Physiol 253: R142–R149, 1987 [DOI] [PubMed] [Google Scholar]
- 614. Shook JE, Watkins WD, Camporesi EM. Differential roles of opioid receptors in respiration, respiratory disease, and opiate-induced respiratory depression. Am Rev Respir Dis 142: 895–909, 1990 [DOI] [PubMed] [Google Scholar]
- 615. Sica AL, Greenberg HE, Ruggiero DA, Scharf SM. Chronic-intermittent hypoxia: a model of sympathetic activation in the rat. Respir Physiol 121: 173–184, 2000 [DOI] [PubMed] [Google Scholar]
- 616. Silverberg DS, Oksenberg A, Radwan H, Iaina A. Is obstructive sleep apnea a common cause of essential hypertension? Isr J Med Sci 31: 527–535, 1995 [PubMed] [Google Scholar]
- 617. Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD. Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. Am J Respir Crit Care Med 160: 1101–1106, 1999 [DOI] [PubMed] [Google Scholar]
- 618. Singer JH, Bellingham MC, Berger AJ. Presynaptic inhibition of glutamatergic synaptic transmission to rat motoneurons by serotonin. J Neurophysiol 76: 799–807, 1996 [DOI] [PubMed] [Google Scholar]
- 619. Singer JH, Berger AJ. Presynaptic inhibition by serotonin: a possible mechanism for switching motor output of the hypoglossal nucleus. Sleep 19: S146–S149, 1996 [DOI] [PubMed] [Google Scholar]
- 620. Skatrud JB, Badr MS, Morgan BJ. Control of breathing during sleep and sleep disordered breathing. In: Control of Breathing in Health and Disease, edited by Altose M, Kawakami Y. New York: Dekker, 1999 [Google Scholar]
- 621. Skatrud JB, Dempsey JA. Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. J Appl Physiol 55: 813–822, 1983 [DOI] [PubMed] [Google Scholar]
- 622. Skulsky EM, Osman NI, Baghdoyan HA, Lydic R. Microdialysis delivery of morphine to the hypoglossal nucleus of Wistar rat increases hypoglossal acetylcholine release. Sleep 30: 566–573, 2007 [DOI] [PubMed] [Google Scholar]
- 623. Skurk T, Alberti-Huber C, Herder C, Hauner H. Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab 92: 1023–1033, 2007 [DOI] [PubMed] [Google Scholar]
- 624. Smith CA, Chenuel BJ, Henderson KS, Dempsey JA. The apneic threshold during non-REM sleep in dogs: sensitivity of carotid body vs. central chemoreceptors. J Appl Physiol 103: 578–586, 2007 [DOI] [PubMed] [Google Scholar]
- 625. Smith CA, Engwall MJ, Dempsey JA, Bisgard GE. Effects of specific carotid body and brain hypoxia on respiratory muscle control in the awake goat. J Physiol 460: 623–640, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 626. Smith CA, Rodman JR, Chenuel BJ, Henderson KS, Dempsey JA. Response time and sensitivity of the ventilatory response to CO2 in unanesthetized intact dogs: central vs. peripheral chemoreceptors. J Appl Physiol 100: 13–19, 2006 [DOI] [PubMed] [Google Scholar]
- 627. Smith CA, Saupe KW, Henderson KS, Dempsey JA. Ventilatory effects of specific carotid body hypocapnia in dogs during wakefulness and sleep. J Appl Physiol 79: 689–699, 1995 [DOI] [PubMed] [Google Scholar]
- 628. Smith PL, Wise RA, Gold AR, Schwartz AR, Permutt S. Upper airway pressure-flow relationships in obstructive sleep apnea. J Appl Physiol 64: 789–795, 1988 [DOI] [PubMed] [Google Scholar]
- 629. Smurra M, Philip P, Taillard J, Guilleminault C, Bioulac B, Gin H. CPAP treatment does not affect glucose-insulin metabolism in sleep apneic patients. Sleep Med 2: 207–213, 2001 [DOI] [PubMed] [Google Scholar]
- 630. Snow JB, Kitzis V, Norton CE, Torres SN, Johnson KD, Kanagy NL, Walker BR, Resta TC. Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. J Appl Physiol 104: 110–118, 2008 [DOI] [PubMed] [Google Scholar]
- 631. Soja PJ, Lopez-Rodriguez F, Morales FR, Chase MH. Effects of excitatory amino acid antagonists on the phasic depolarizing events that occur in lumbar motoneurons during REM periods of active sleep. J Neurosci 15: 4068–4076, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632. Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci 27: 901–908, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633. Solin P, Bergin P, Richardson M, Kaye DM, Walters EH, Naughton MT. Influence of pulmonary capillary wedge pressure on central apnea in heart failure. Circulation 99: 1574–1579, 1999 [DOI] [PubMed] [Google Scholar]
- 634. Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 96: 1897–1904, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635. Somers VK, Dyken ME, Mark AL, Abboud FM. Parasympathetic hyperresponsiveness and bradyarrhythmias during apnoea in hypertension. Clin Auton Res 2: 171–176, 1992 [DOI] [PubMed] [Google Scholar]
- 636. Sood S, Liu X, Liu H, Nolan P, Horner RL. 5-HT at hypoglossal motor nucleus and respiratory control of genioglossus muscle in anesthetized rats. Respir Physiol Neurobiol 138: 205–221, 2003 [DOI] [PubMed] [Google Scholar]
- 637. Sood S, Morrison JL, Liu H, Horner RL. Role of endogenous serotonin in modulating genioglossus muscle activity in awake and sleeping rats. Am J Respir Crit Care Med 172: 1338–1347, 2005 [DOI] [PubMed] [Google Scholar]
- 638. Sorajja D, Gami AS, Somers VK, Behrenbeck TR, Garcia-Touchard A, Lopez-Jimenez F. Independent association between obstructive sleep apnea and subclinical coronary artery disease. Chest 133: 927–933, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet 354: 1435–1439, 1999 [DOI] [PubMed] [Google Scholar]
- 640. Spiegel K, Tasali E, Penev P, Van Cauter E. Brief communication: sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, increased hunger and appetite. Ann Intern Med 141: 846–850, 2004 [DOI] [PubMed] [Google Scholar]
- 641. St John WM. Influence of reticular mechanisms upon hypoglossal, trigeminal and phrenic activities. Respir Physiol 66: 27–40, 1986 [DOI] [PubMed] [Google Scholar]
- 642. Stafford IL, Jacobs BL. Noradrenergic modulation of the masseteric reflex in behaving cats. I. Pharmacological studies. J Neurosci 10: 91–98, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643. Stamatakis K, Punjabi NM. Effects of sleep fragmentation on glucose metabolism in normal subjects. Chest July 19, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644. Stanchina ML, Malhotra A, Fogel RB, Trinder J, Edwards JK, Schory K, White DP. The influence of lung volume on pharyngeal mechanics, collapsibility, and genioglossus muscle activation during sleep. Sleep 26: 851–856, 2003 [DOI] [PubMed] [Google Scholar]
- 645. Stauffer JL, Buick MK, Bixler EO, Sharkey FE, Abt AB, Manders EK, Kales A, Cadieux RJ, Barry JD, Zwillich CW. Morphology of the uvula in obstructive sleep apnea. Am Rev Respir Dis 140: 724–728, 1989 [DOI] [PubMed] [Google Scholar]
- 646. Steiner S, Jax T, Evers S, Hennersdorf M, Schwalen A, Strauer BE. Altered blood rheology in obstructive sleep apnea as a mediator of cardiovascular risk. Cardiology 104: 92–96, 2005 [DOI] [PubMed] [Google Scholar]
- 647. Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006 [DOI] [PubMed] [Google Scholar]
- 648. Stenmark KR, Gerasimovskaya E, Nemenoff RA, Das M. Hypoxic activation of adventitial fibroblasts: role in vascular remodeling. Chest 122: 326S–334S, 2002 [DOI] [PubMed] [Google Scholar]
- 649. Stokes W. The Diseases of the Heart and Aorta. Dublin: 1854 [Google Scholar]
- 650. Stornetta RL, Moreira TS, Takakura AC, Kang BJ, Chang DA, West GH, Brunet JF, Mulkey DK, Bayliss DA, Guyenet PG. Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. J Neurosci 26: 10305–10314, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651. Stradling J, Smith D, Radulovacki M, Carley D. Effect of ondansetron on moderate obstructive sleep apnoea, a single night, and placebo-controlled trial. J Sleep Res 12: 169–170, 2003 [DOI] [PubMed] [Google Scholar]
- 652. Strohl KP, Novak RD, Singer W, Cahan C, Boehm KD, Denko CW, Hoffstein VS. Insulin levels, blood pressure and sleep apnea. Sleep 17: 614–618, 1994 [DOI] [PubMed] [Google Scholar]
- 653. Su MC, Chiu KL, Ruttanaumpawan P, Shiota S, Yumino D, Redolfi S, Haight JS, Bradley TD. Lower body positive pressure increases upper airway collapsibility in healthy subjects. Respir Physiol Neurobiol 161: 306–312, 2008 [DOI] [PubMed] [Google Scholar]
- 654. Sullivan CE, Issa FG, Berthon-Jones M, Eves L. Reversal of obstructive sleep apnoea by continuous positive airway pressure applied through the nares. Lancet 1: 862–865, 1981 [DOI] [PubMed] [Google Scholar]
- 655. Sullivan CE, Murphy E, Kozar LF, Phillipson EA. Waking and ventilatory responses to laryngeal stimulation in sleeping dogs. J Appl Physiol 45: 681–689, 1978 [DOI] [PubMed] [Google Scholar]
- 656. Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced activity of carotid body chemoreceptors in rabbits with heart failure: role of nitric oxide. J Appl Physiol 86: 1273–1282, 1999 [DOI] [PubMed] [Google Scholar]
- 657. Suratt PM, McTier RF, Wilhoit SC. Upper airway muscle activation is augmented in patients with obstructive sleep apnea compared with that in normal subjects. Am Rev Respir Dis 137: 889–894, 1988 [DOI] [PubMed] [Google Scholar]
- 658. Suratt PM, Wilhoit SC, Cooper K. Induction of airway collapse with subatmospheric pressure in awake patients with sleep apnea. J Appl Physiol 57: 140–146, 1984 [DOI] [PubMed] [Google Scholar]
- 659. Sutton JR, Houston CS, Mansell AL, McFadden MD, Hackett PM, Rigg JR, Powles AC. Effect of acetazolamide on hypoxemia during sleep at high altitude. N Engl J Med 301: 1329–1331, 1979 [DOI] [PubMed] [Google Scholar]
- 660. Svanborg E. Impact of obstructive apnea syndrome on upper airway respiratory muscles. Respir Physiol Neurobiol 147: 263–272, 2005 [DOI] [PubMed] [Google Scholar]
- 661. Swinburn BA, Metcalf PA, Ley SJ. Long-term (5-year) effects of a reduced-fat diet intervention in individuals with glucose intolerance. Diabetes Care 24: 619–624, 2001 [DOI] [PubMed] [Google Scholar]
- 662. Szereda-Przestaszewska M, Kaczynska K. Peripheral 5-HT1A receptors are not essential for increased ventilation evoked by systemic 8-OH-DPAT challenge in anaesthetized rats. Exp Physiol 92: 953–961, 2007 [DOI] [PubMed] [Google Scholar]
- 663. Tagaito Y, Isono S, Remmers JE, Tanaka A, Nishino T. Lung volume and collapsibility of the passive pharynx in patients with sleep-disordered breathing. J Appl Physiol 103: 1379–1385, 2007 [DOI] [PubMed] [Google Scholar]
- 664. Tahawi Z, Orolinova N, Joshua IG, Bader M, Fletcher EC. Altered vascular reactivity in arterioles of chronic intermittent hypoxic rats. J Appl Physiol 90: 2007–2013, 2001 [DOI] [PubMed] [Google Scholar]
- 665. Taheri S, Lin L, Austin D, Young T, Mignot E. Short sleep duration is associated with reduced leptin, elevated ghrelin, increased body mass index (BMI). Sleep 27: 146–147, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666. Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL, Guyenet PG. Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2-sensitive neurons in rats. J Physiol 572: 503–523, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667. Tankersley CG, O'Donnell C, Daood MJ, Watchko JF, Mitzner W, Schwartz A, Smith P. Leptin attenuates respiratory complications associated with the obese phenotype. J Appl Physiol 85: 2261–2269, 1998 [DOI] [PubMed] [Google Scholar]
- 668. Tanne F, Gagnadoux F, Chazouilleres O, Fleury B, Wendum D, Lasnier E, Lebeau B, Poupon R, Serfaty L. Chronic liver injury during obstructive sleep apnea. Hepatology 41: 1290–1296, 2005 [DOI] [PubMed] [Google Scholar]
- 669. Tanriverdi H, Evrengul H, Kara CO, Kuru O, Tanriverdi S, Ozkurt S, Kaftan A, Kilic M. Aortic stiffness, flow-mediated dilatation and carotid intima-media thickness in obstructive sleep apnea: non-invasive indicators of atherosclerosis. Respiration 73: 741–750, 2006 [DOI] [PubMed] [Google Scholar]
- 670. Tauman R, Gozal D. Obesity and obstructive sleep apnea in children. Paediatr Respir Rev 7: 247–259, 2006 [DOI] [PubMed] [Google Scholar]
- 671. Taylor NC, Li A, Nattie EE. Medullary serotonergic neurones modulate the ventilatory response to hypercapnia, but not hypoxia in conscious rats. J Physiol 566: 543–557, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672. Teichtahl H, Wang D, Cunnington D, Kronborg I, Goodman C, Prodromidis A, Drummer O. Cardiorespiratory function in stable methadone maintenance treatment (MMT) patients. Addict Biol 9: 247–253, 2004 [DOI] [PubMed] [Google Scholar]
- 673. Teschler H, Dohring J, Wang YM, Berthon-Jones M. Adaptive pressure support servo-ventilation: a novel treatment for Cheyne-Stokes respiration in heart failure. Am J Respir Crit Care Med 164: 614–619, 2001 [DOI] [PubMed] [Google Scholar]
- 674. Thach BT. Some aspects of clinical relevance in the maturation of respiratory control in infants. J Appl Physiol 104: 1828–1834, 2008 [DOI] [PubMed] [Google Scholar]
- 675. Thomas RJ, Rosen BR, Stern CE, Weiss JW, Kwong KK. Functional imaging of working memory in obstructive sleep-disordered breathing. J Appl Physiol 98: 2226–2234, 2005 [DOI] [PubMed] [Google Scholar]
- 676. Thomas RJ, Terzano MG, Parrino L, Weiss JW. Obstructive sleep-disordered breathing with a dominant cyclic alternating pattern: a recognizable polysonographic variant with practical clinical implications. Sleep 27: 229–234, 2004 [DOI] [PubMed] [Google Scholar]
- 677. Thomson S, Morrell MJ, Cordingley JJ, Semple SJ. Ventilation is unstable during drowsiness before sleep onset. J Appl Physiol 99: 2036–2044, 2005 [DOI] [PubMed] [Google Scholar]
- 678. Tiihonen M, Partinen M. Polysomnography and maintenance of wakefulness test as predictors of CPAP effectiveness in obstructive sleep apnea. Electroencephalogr Clin Neurophysiol 107: 383–386, 1998 [DOI] [PubMed] [Google Scholar]
- 679. Tilkian AG, Guilleminault C, Schroeder JS, Lehrman KL, Simmons FB, Dement WC. Hemodynamics in sleep-induced apnea. Studies during wakefulness and sleep. Ann Intern Med 85: 714–719, 1976 [DOI] [PubMed] [Google Scholar]
- 680. Tilkian AG, Guilleminault C, Schroeder JS, Lehrman KL, Simmons FB, Dement WC. Sleep-induced apnea syndrome. Prevalence of cardiac arrhythmias and their reversal after tracheostomy. Am J Med 63: 348–358, 1977 [DOI] [PubMed] [Google Scholar]
- 681. Tkacova R, Hall MJ, Liu PP, Fitzgerald FS, Bradley TD. Left ventricular volume in patients with heart failure and Cheyne-Stokes respiration during sleep. Am J Respir Crit Care Med 156: 1549–1555, 1997 [DOI] [PubMed] [Google Scholar]
- 682. Tkacova R, Niroumand M, Lorenzi-Filho G, Bradley TD. Overnight shift from obstructive to central apneas in patients with heart failure: role of Pco2 and circulatory delay. Circulation 103: 238–243, 2001 [DOI] [PubMed] [Google Scholar]
- 683. Tolle FA, Judy WV, Yu PL, Markand ON. Reduced stroke volume related to pleural pressure in obstructive sleep apnea. J Appl Physiol 55: 1718–1724, 1983 [DOI] [PubMed] [Google Scholar]
- 684. Travers JB, Yoo JE, Chandran R, Herman K, Travers SP. Neurotransmitter phenotypes of intermediate zone reticular formation projections to the motor trigeminal and hypoglossal nuclei in the rat. J Comp Neurol 488: 28–47, 2005 [DOI] [PubMed] [Google Scholar]
- 685. Tribollet E, Goumaz M, Raggenbass M, Dreifuss JJ. Appearance and transient expression of vasopressin and oxytocin receptors in the rat brain. J Recept Res 11: 333–346, 1991 [DOI] [PubMed] [Google Scholar]
- 686. Troncoso Brindeiro CM, da Silva AQ, Allahdadi KJ, Youngblood V, Kanagy NL. Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. Am J Physiol Heart Circ Physiol 293: H2971–H2976, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687. Trudo FJ, Gefter WB, Welch KC, Gupta KB, Maislin G, Schwab RJ. State-related changes in upper airway caliber and surrounding soft-tissue structures in normal subjects. Am J Respir Crit Care Med 158: 1259–1270, 1998 [DOI] [PubMed] [Google Scholar]
- 688. Tsioufis C, Thomopoulos K, Dimitriadis K, Amfilochiou A, Tousoulis D, Alchanatis M, Stefanadis C, Kallikazaros I. The incremental effect of obstructive sleep apnoea syndrome on arterial stiffness in newly diagnosed essential hypertensive subjects. J Hypertens 25: 141–146, 2007 [DOI] [PubMed] [Google Scholar]
- 689. Tulis DA, Prewitt RL. Medial and endothelial platelet-derived growth factor A chain expression is regulated by in vivo exposure to elevated flow. J Vasc Res 35: 413–420, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690. Van de Graaff WB. Thoracic influence on upper airway patency. J Appl Physiol 65: 2124–2131, 1988 [DOI] [PubMed] [Google Scholar]
- 691. Van LE, Strohl KP, Parker DM, Bruce EN, Van de Graaff WB, Cherniack NS. Phasic volume-related feedback on upper airway muscle activity. J Appl Physiol 56: 730–736, 1984 [DOI] [PubMed] [Google Scholar]
- 692. Veasey SC. Molecular and physiologic basis of obstructive sleep apnea. Clin Chest Med 24: 179–193, 2003 [DOI] [PubMed] [Google Scholar]
- 693. Veasey SC, Chachkes J, Fenik P, Hendricks JC. The effects of ondansetron on sleep-disordered breathing in the English bulldog. Sleep 24: 155–160, 2001 [DOI] [PubMed] [Google Scholar]
- 694. Veasey SC, Davis CW, Fenik P, Zhan G, Hsu YJ, Pratico D, Gow A. Long-term intermittent hypoxia in mice: protracted hypersomnolence with oxidative injury to sleep-wake brain regions. Sleep 27: 194–201, 2004 [DOI] [PubMed] [Google Scholar]
- 695. Veasey SC, Fenik P, Panckeri K, Pack AI, Hendricks JC. The effects of trazodone with l-tryptophan on sleep-disordered breathing in the English bulldog. Am J Respir Crit Care Med 160: 1659–1667, 1999 [DOI] [PubMed] [Google Scholar]
- 696. Veasey SC, Panckeri KA, Hoffman EA, Pack AI, Hendricks JC. The effects of serotonin antagonists in an animal model of sleep-disordered breathing. Am J Respir Crit Care Med 153: 776–786, 1996 [DOI] [PubMed] [Google Scholar]
- 697. Verdecchia P. Prognostic value of ambulatory blood pressure: current evidence and clinical implications. Hypertension 35: 844–851, 2000 [DOI] [PubMed] [Google Scholar]
- 698. Vertes RP. Brain stem gigantocellular neurons: patterns of activity during behavior and sleep in the freely moving rat. J Neurophysiol 42: 214–228, 1979 [DOI] [PubMed] [Google Scholar]
- 699. Vgontzas AN, Papanicolaou DA, Bixler EO, Kales A, Tyson K, Chrousos GP. Elevation of plasma cytokines in disorders of excessive daytime sleepiness: role of sleep disturbance and obesity. J Clin Endocrinol Metab 82: 1313–1316, 1997 [DOI] [PubMed] [Google Scholar]
- 700. Vgontzas AN, Zoumakis E, Lin HM, Bixler EO, Trakada G, Chrousos GP. Marked decrease in sleepiness in patients with sleep apnea by etanercept, a tumor necrosis factor-alpha antagonist. J Clin Endocrinol Metab 89: 4409–4413, 2004 [DOI] [PubMed] [Google Scholar]
- 701. Volgin DV, Mackiewicz M, Kubin L. Alpha(1B) receptors are the main postsynaptic mediators of adrenergic excitation in brainstem motoneurons, a single-cell RT-PCR study. J Chem Neuroanat 22: 157–166, 2001 [DOI] [PubMed] [Google Scholar]
- 702. Von KR, Loredo JS, Ancoli-Israel S, Dimsdale JE. Association between sleep apnea severity and blood coagulability: treatment effects of nasal continuous positive airway pressure. Sleep Breath 10: 139–146, 2006 [DOI] [PubMed] [Google Scholar]
- 703. Walker JM, Farney RJ, Rhondeau SM, Boyle KM, Valentine K, Cloward TV, Shilling KC. Chronic opioid use is a risk factor for the development of central sleep apnea and ataxic breathing. J Clin Sleep Med 3: 455–461, 2007 [PMC free article] [PubMed] [Google Scholar]
- 704. Walsh JT, Andrews R, Starling R, Cowley AJ, Johnston ID, Kinnear WJ. Effects of captopril and oxygen on sleep apnoea in patients with mild to moderate congestive cardiac failure. Br Heart J 73: 237–241, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 705. Wang D, Teichtahl H, Drummer O, Goodman C, Cherry G, Cunnington D, Kronborg I. Central sleep apnea in stable methadone maintenance treatment patients. Chest 128: 1348–1356, 2005 [DOI] [PubMed] [Google Scholar]
- 706. Wang H, Parker JD, Newton GE, Floras JS, Mak S, Chiu KL, Ruttanaumpawan P, Tomlinson G, Bradley TD. Influence of obstructive sleep apnea on mortality in patients with heart failure. J Am Coll Cardiol 49: 1625–1631, 2007 [DOI] [PubMed] [Google Scholar]
- 707. Waradekar NV, Sinoway LI, Zwillich CW, Leuenberger UA. Influence of treatment on muscle sympathetic nerve activity in sleep apnea. Am J Respir Crit Care Med 153: 1333–1338, 1996 [DOI] [PubMed] [Google Scholar]
- 708. Warner G, Skatrud JB, Dempsey JA. Effect of hypoxia-induced periodic breathing on upper airway obstruction during sleep. J Appl Physiol 62: 2201–2211, 1987 [DOI] [PubMed] [Google Scholar]
- 709. Wasicko MJ, Leiter JC, Erlichman JS, Strobel RJ, Bartlett D., Jr Nasal and pharyngeal resistance after topical mucosal vasoconstriction in normal humans. Am Rev Respir Dis 144: 1048–1052, 1991 [DOI] [PubMed] [Google Scholar]
- 710. Watanabe T, Isono S, Tanaka A, Tanzawa H, Nishino T. Contribution of body habitus and craniofacial characteristics to segmental closing pressures of the passive pharynx in patients with sleep-disordered breathing. Am J Respir Crit Care Med 165: 260–265, 2002 [DOI] [PubMed] [Google Scholar]
- 711. Weiner D, Mitra J, Salamone J, Cherniack NS. Effect of chemical stimuli on nerves supplying upper airway muscles. J Appl Physiol 52: 530–536, 1982 [DOI] [PubMed] [Google Scholar]
- 712. Weiss JW, Liu MD, Huang J. Sleep apnoea and hypertension: physiological basis for a causal relationship of obstructive sleep apnoea to hypertension. Exp Physiol 92: 21–26, 2007 [DOI] [PubMed] [Google Scholar]
- 713. Weitzenblum E, Krieger J, Apprill M, Vallee E, Ehrhart M, Ratomaharo J, Oswald M, Kurtz D. Daytime pulmonary hypertension in patients with obstructive sleep apnea syndrome. Am Rev Respir Dis 138: 345–349, 1988 [DOI] [PubMed] [Google Scholar]
- 714. Wellman A, Jordan AS, Malhotra A, Fogel RB, Katz ES, Schory K, Edwards JK, White DP. Ventilatory control and airway anatomy in obstructive sleep apnea. Am J Respir Crit Care Med 170: 1225–1232, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 715. Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respir Physiol Neurobiol 162: 144–151, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716. Wessendorf TE, Teschler H, Wang YM, Konietzko N, Thilmann AF. Sleep-disordered breathing among patients with first-ever stroke. J Neurol 247: 41–47, 2000 [DOI] [PubMed] [Google Scholar]
- 717. Wessendorf TE, Thilmann AF, Wang YM, Schreiber A, Konietzko N, Teschler H. Fibrinogen levels and obstructive sleep apnea in ischemic stroke. Am J Respir Crit Care Med 162: 2039–2042, 2000 [DOI] [PubMed] [Google Scholar]
- 718. West JB, Dollery CT, Naimar KA. Distribution of blood flow in isolated lung: relation to vascular and alveolar pressures. J Appl Physiol 19: 713–724, 1964 [DOI] [PubMed] [Google Scholar]
- 719. West SD, Nicoll DJ, Wallace TM, Matthews DR, Stradling JR. The effect of CPAP on insulin resistance and HbA1c in men with obstructive sleep apnoea and type 2 diabetes. Thorax 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720. White SG, Fletcher EC, Miller CC., III Acute systemic blood pressure elevation in obstructive and nonobstructive breath hold in primates. J Appl Physiol 79: 324–330, 1995 [DOI] [PubMed] [Google Scholar]
- 721. White SR. Serotonin and co-localized peptides: effects on spinal motoneuron excitability. Peptides 6 Suppl 2: 123–127, 1985 [DOI] [PubMed] [Google Scholar]
- 722. Whittle AT, Marshall I, Mortimore IL, Wraith PK, Sellar RJ, Douglas NJ. Neck soft tissue and fat distribution: comparison between normal men and women by magnetic resonance imaging. Thorax 54: 323–328, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723. Wilcox I, McNamara SG, Collins FL, Grunstein RR, Sullivan CE. “Syndrome Z”: the interaction of sleep apnoea, vascular risk factors and heart disease. Thorax 53 Suppl 3: S25–S28, 1998 [PMC free article] [PubMed] [Google Scholar]
- 724. Wilken B, Lalley P, Bischoff AM, Christen HJ, Behnke J, Hanefeld F, Richter DW. Treatment of apneustic respiratory disturbance with a serotonin-receptor agonist. J Pediatr 130: 89–94, 1997 [DOI] [PubMed] [Google Scholar]
- 725. Williams AJ, Houston D, Finberg S, Lam C, Kinney JL, Santiago S. Sleep apnea syndrome and essential hypertension. Am J Cardiol 55: 1019–1022, 1985 [DOI] [PubMed] [Google Scholar]
- 726. Wilson CR, Manchanda S, Crabtree D, Skatrud JB, Dempsey JA. An induced blood pressure rise does not alter upper airway resistance in sleeping humans. J Appl Physiol 84: 269–276, 1998 [DOI] [PubMed] [Google Scholar]
- 727. Wilson E, Mai Q, Sudhir K, Weiss RH, Ives HE. Mechanical strain induces growth of vascular smooth muscle cells via autocrine action of PDGF. J Cell Biol 123: 741–747, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728. Wilson PA, Skatrud JB, Dempsey JA. Effects of slow wave sleep on ventilatory compensation to inspiratory elastic loading. Respir Physiol 55: 103–120, 1984 [DOI] [PubMed] [Google Scholar]
- 729. Wong ML, Sandham A, Ang PK, Wong DC, Tan WC, Huggare J. Craniofacial morphology, head posture, and nasal respiratory resistance in obstructive sleep apnoea: an inter-ethnic comparison. Eur J Orthod 27: 91–97, 2005 [DOI] [PubMed] [Google Scholar]
- 730. Woodson BT, Garancis JC, Toohill RJ. Histopathologic changes in snoring and obstructive sleep apnea syndrome. Laryngoscope 101: 1318–1322, 1991 [DOI] [PubMed] [Google Scholar]
- 731. Xi L, Chow CM, Smith CA, Dempsey JA. Effects of REM sleep on the ventilatory response to airway occlusion in the dog. Sleep 17: 674–687, 1994 [DOI] [PubMed] [Google Scholar]
- 732. Xi L, Smith CA, Saupe KW, Henderson KS, Dempsey JA. Effects of rapid-eye-movement sleep on the apneic threshold in dogs. J Appl Physiol 75: 1129–1139, 1993 [DOI] [PubMed] [Google Scholar]
- 733. Xi MC, Morales FR, Chase MH. The motor inhibitory system operating during active sleep is tonically suppressed by GABAergic mechanisms during other states. J Neurophysiol 86: 1908–1915, 2001 [DOI] [PubMed] [Google Scholar]
- 734. Xie A, Rankin F, Rutherford R, Bradley TD. Effects of inhaled CO2 and added dead space on idiopathic central sleep apnea. J Appl Physiol 82: 918–926, 1997 [DOI] [PubMed] [Google Scholar]
- 735. Xie A, Skatrud JB, Barczi SR, Reichmuth K, Morgan BJ, Mont S, Dempsey JA. Influence of cerebral blood flow on breathing stability. J Appl Physiol 106: 850–856, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 736. Xie A, Skatrud JB, Crabtree DC, Puleo DS, Goodman BM, Morgan BJ. Neurocirculatory consequences of intermittent asphyxia in humans. J Appl Physiol 89: 1333–1339, 2000 [DOI] [PubMed] [Google Scholar]
- 737. Xie A, Skatrud JB, Dempsey JA. Effect of hypoxia on the hypopnoeic and apnoeic threshold for CO2 in sleeping humans. J Physiol 535: 269–278, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 738. Xie A, Skatrud JB, Khayat R, Dempsey JA, Morgan B, Russell D. Cerebrovascular response to carbon dioxide in patients with congestive heart failure. Am J Respir Crit Care Med 172: 371–378, 2005 [DOI] [PubMed] [Google Scholar]
- 739. Xie A, Skatrud JB, Morgan B, Chenuel B, Khayat R, Reichmuth K, Lin J, Dempsey JA. Influence of cerebrovascular function on the hypercapnic ventilatory response in healthy humans. J Physiol 577: 319–329, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 740. Xie A, Skatrud JB, Puleo DS, Dempsey JA. Influence of arterial O2 on the susceptibility to posthyperventilation apnea during sleep. J Appl Physiol 100: 171–177, 2006 [DOI] [PubMed] [Google Scholar]
- 741. Xie A, Skatrud JB, Puleo DS, Morgan BJ. Exposure to hypoxia produces long-lasting sympathetic activation in humans. J Appl Physiol 91: 1555–1562, 2001 [DOI] [PubMed] [Google Scholar]
- 742. Xie A, Skatrud JB, Puleo DS, Rahko PS, Dempsey JA. Apnea-hypopnea threshold for CO2 in patients with congestive heart failure. Am J Respir Crit Care Med 165: 1245–1250, 2002 [DOI] [PubMed] [Google Scholar]
- 743. Xie A, Wong B, Phillipson EA, Slutsky AS, Bradley TD. Interaction of hyperventilation and arousal in the pathogenesis of idiopathic central sleep apnea. Am J Respir Crit Care Med 150: 489–495, 1994 [DOI] [PubMed] [Google Scholar]
- 744. Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 353: 2034–2041, 2005 [DOI] [PubMed] [Google Scholar]
- 745. Yamauchi M, Dostal J, Kimura H, Strohl KP. Effects of buspirone on posthypoxic ventilatory behavior in the C57BL/6J and A/J mouse strains. J Appl Physiol 105: 518–526, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746. Yamuy J, Fung SJ, Xi M, Morales FR, Chase MH. Hypoglossal motoneurons are postsynaptically inhibited during carbachol-induced rapid eye movement sleep. Neuroscience 94: 11–15, 1999 [DOI] [PubMed] [Google Scholar]
- 747. Yasuda K, Robinson DM, Selvaratnam SR, Walsh CW, McMorland AJ, Funk GD. Modulation of hypoglossal motoneuron excitability by NK1 receptor activation in neonatal mice in vitro. J Physiol 534: 447–464, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 748. Yokoe T, Alonso LC, Romano LC, Rosa TC, O'Doherty RM, Garcia-Ocana A, Minoguchi K, O'Donnell CP. Intermittent hypoxia reverses the diurnal glucose rhythm and causes pancreatic beta-cell replication in mice. J Physiol 586: 899–911, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749. Yokoe T, Minoguchi K, Matsuo H, Oda N, Minoguchi H, Yoshino G, Hirano T, Adachi M. Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation 107: 1129–1134, 2003 [DOI] [PubMed] [Google Scholar]
- 750. Yoshida H. ER stress and diseases. FEBS J 274: 630–658, 2007 [DOI] [PubMed] [Google Scholar]
- 751. Yoshioka M, Goda Y, Togashi H, Matsumoto M, Saito H. Pharmacological characterization of 5-hydroxytryptamine-induced apnea in the rat. J Pharmacol Exp Ther 260: 917–924, 1992 [PubMed] [Google Scholar]
- 752. Younes M. Contributions of upper airway mechanics and control mechanisms to severity of obstructive apnea. Am J Respir Crit Care Med 168: 645–658, 2003 [DOI] [PubMed] [Google Scholar]
- 753. Younes M. Role of arousals in the pathogenesis of obstructive sleep apnea. Am J Respir Crit Care Med 169: 623–633, 2004 [DOI] [PubMed] [Google Scholar]
- 754. Younes M. Pathogenesis of obstructive sleep disorders. J Appl Physiol 105: 1389–1405, 2008 [DOI] [PubMed] [Google Scholar]
- 755. Younes M, Ostrowski M, Atkar R, Laprairie J, Siemens A, Hanly P. Mechanisms of breathing instability in patients with obstructive sleep apnea. J Appl Physiol 103: 1929–1941, 2007 [DOI] [PubMed] [Google Scholar]
- 756. Young JW, McDonald JP. An investigation into the relationship between the severity of obstructive sleep apnoea/hypopnoea syndrome and the vertical position of the hyoid bone. Surgeon 2: 145–151, 2004 [DOI] [PubMed] [Google Scholar]
- 757. Young T, Finn L, Austin D, Peterson A. Menopausal status and sleep-disordered breathing in the Wisconsin Sleep Cohort Study. Am J Respir Crit Care Med 167: 1181–1185, 2003 [DOI] [PubMed] [Google Scholar]
- 758. Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 328: 1230–1235, 1993 [DOI] [PubMed] [Google Scholar]
- 759. Young T, Peppard PE, Taheri S. Excess weight and sleep-disordered breathing. J Appl Physiol 99: 1592–1599, 2005 [DOI] [PubMed] [Google Scholar]
- 760. Young T, Shahar E, Nieto FJ, Redline S, Newman AB, Gottlieb DJ, Walsleben JA, Finn L, Enright P, Samet JM. Predictors of sleep-disordered breathing in community-dwelling adults: the Sleep Heart Health Study. Arch Intern Med 162: 893–900, 2002 [DOI] [PubMed] [Google Scholar]
- 761. Yu HJ, Lin BR, Lee HS, Shun CT, Yang CC, Lai TY, Chien CT, Hsu SM. Sympathetic vesicovascular reflex induced by acute urinary retention evokes proinflammatory and proapoptotic injury in rat liver. Am J Physiol Renal Physiol 288: F1005–F1014, 2005 [DOI] [PubMed] [Google Scholar]
- 762. Yuan MS, Konstantopoulos N, Lee JS, Hansen L, Li ZW, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of IKK beta. Science 293: 1673–1677, 2001 [DOI] [PubMed] [Google Scholar]
- 763. Yumino D, Bradley TD. Central sleep apnea and cheyne-stokes respiration. Proc Am Thorac Soc 5: 226–236, 2008 [DOI] [PubMed] [Google Scholar]
- 764. Zamarron-Sanz C, Ricoy-Galbaldon J, Gude-Sampedro F, Riveiro-Riveiro A. Plasma levels of vascular endothelial markers in obstructive sleep apnea. Arch Med Res 37: 552–555, 2006 [DOI] [PubMed] [Google Scholar]
- 765. Zevin S, Swed E, Cahan C. Clinical effects of locally delivered nicotine in obstructive sleep apnea syndrome. Am J Ther 10: 170–175, 2003 [DOI] [PubMed] [Google Scholar]
- 766. Zhang J, Zhao B, Gesongluobu Sun Y, Wu Y, Pei W, Ye J, Hui R, Liu L. Angiotensin-converting enzyme gene insertion/deletion (I/D) polymorphism in hypertensive patients with different degrees of obstructive sleep apnea. Hypertens Res 23: 407–411, 2000 [DOI] [PubMed] [Google Scholar]
- 767. Zhao Q, Ishibashi M, Hiasa K, Tan C, Takeshita A, Egashira K. Essential role of vascular endothelial growth factor in angiotensin II-induced vascular inflammation and remodeling. Hypertension 44: 264–270, 2004 [DOI] [PubMed] [Google Scholar]
- 768. Zhu Y, Fenik P, Zhan G, Mazza E, Kelz M, Aston-Jones G, Veasey SC. Selective loss of catecholaminergic wake active neurons in a murine sleep apnea model. J Neurosci 27: 10060–10071, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 768a. Zhu Y, Fenik P, Zhan G, Sanfillipo-Cohn B, Naidoo N, Veasey SC. Eif-2a protects brain stem motoneurons in a murine model of sleep apnea. J Neurosci 28: 2168–2178, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 769. Ziegler MG, Nelesen RA, Mills PJ, Ancoli-Israel S, Clausen JL, Watkins L, Dimsdale JE. The effect of hypoxia on baroreflexes and pressor sensitivity in sleep apnea and hypertension. Sleep 18: 859–865, 1995 [PubMed] [Google Scholar]
- 770. Zucker IH. Brain angiotensin II: new insights into its role in sympathetic regulation. Circ Res 90: 503–505, 2002 [DOI] [PubMed] [Google Scholar]