

Abstract
The azanucleotides azacitidine and decitabine have been shown to induce hematologic response and prolong survival in higher-risk myelodysplastic syndromes. They are inhibitors of DNA methyltransferase-1 and induce DNA-hypomethylation. Induction of apoptosis is also clinically relevant, in particular during the first treatment cycles, when cytopenia is a frequent side-effect. Since the hypomethylating effect is reversible, and the malignant clone has been shown to persist in most responding patients, several cycles are necessary to achieve and maintain responses, while treatment interruption is associated with rapid relapse. Methylation studies have shown global and gene-specific hypermethylation in myelodysplastic syndromes, but there seems to be little relation between the degree of demethylation following hypomethylating treatment and hematologic response. The presence of concurrent genomic hypermethylation and hypomethylation may impair the predictive power of current detection techniques. This scenario has been complicated by the identification of epigenetic enzyme mutations, including TET2, IDH1/2, DNMT3A and EZH2, which are important for response to hypomethylating treatment. Changes in azanucleotide metabolism genes may also play a role. In the future, methylation analysis concentrating not only on promoters, but also on gene bodies and intergenic regions, may identify key genes in patients with the highest probability of response to azanucleotides and allow a patient-tailored approach.
Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of myeloid disorders, characterized by deeply variable clinical behavior due to disease-specific characteristics, such as aberrant karyotype, depth of peripheral blood cytopenias and of bone marrow infiltration, but also recurrent mutations, and DNA hypermethylation. Furthermore, patients’ characteristics such as age, performance status and comorbidities have been shown to significantly impact prognosis.1–5 The natural history of higher risk MDS has been recently modified by the use of hypomethylating (HMT) drugs, including azacitidine (AZA, Vidaza®, Celgene) and decitabine (DAC, Dacogen®, Janssen). Azacitidine used at the standard dose of 75 mg/m2/7 days/month has been shown to prolong survival and delay leukemic transformation compared to supportive treatment in higher risk MDS.6 Decitabine has been shown to induce responses and prolong progression-free survival when compared to best supportive care.7,8
Despite 20–30% probability of complete and partial remission, achievement of hematologic improvement in 20–50% of patients and perhaps also stable disease, lead to improved survival. Also poor prognostic patient groups, including therapy-related myeloid neoplasms,9 respond to HMT, but median duration of response is 12–18 months and disease relapse appears almost inevitable.6–9
Treatment initiation is then a commitment for patients and physicians, since the subcutaneous or intravenous administration of the drugs requires patient admission to outpatient clinics 5–7 days per month for several years. AZA and DAC have been shown to actively demethylate DNA, but to date none of the methylation markers indicated as predictor of response has been validated in large prospective studies. In this review, we will try to discuss possible causes of this failure.
DNA methylation
Methylation of cytosines is due to the enzymes DNA-methyl transferase (DNMT3A and 3B), which add CH3 methyl-groups mainly in the context of CpG residues to newly synthesized DNA molecules. The enzyme DNMT1 is a maintenance methyl-transferase, assigned to methylation of the replicated DNA. The drugs AZA and DAC are known to induce gene expression through DNA hypomethylation, following DNMT1 sequestration. This action is reversible since the drug does not influence de novo DNMT synthesis. This has been shown in vitro and in vivo and is seemingly the rationale for the necessity of repeated cycles to maximize the number of responses.10–12
The reversibility of methylation also explains the necessity for a maintenance treatment as long as response persists, since treatment interruption is associated to rapid relapse.10–12 The molecular basis for disease persistence during HMT and eventual relapse have been recently postulated by Itzykson et al. who used next generation sequencing to study the prevalence of several gene mutations in single-cell-derived colonies from chronic myelomonocytic leukemia patients.13 This study showed early clonal dominance and expansion of the more mutated clones during the course of the disease, which were not influenced by the type of treatment. Loss of response after stopping treatment is further favored by the fact that leukemic stem cells included in the Lin-CD34+ compartment seem to be spared from the activity of the drugs, probably also due to their non-proliferating status. In 15 MDS and AML patients achieving complete remission following AZA and valproic acid treatment, Craddock et al. showed that leukemic stem cells were substantially reduced, but were never eradicated, and expansion of this population took place before morphological relapse.14 Leukemic stem cells have been shown to over-express multidrug resistance (MDR) transporters, including P-glycoprotein (P-gp).15 Hypomethylating drugs have been shown to down-regulate P-gp expression in myeloid cell lines, with decitabine demethylating the repressor binding site of the MDR-1 gene.16–17
Higher-risk MDS are characterized by a high degree of global methylation compared to normal CD34+ cells and de novo acute myeloid leukemia (AML).18,19 In MDS, different methods have been used to study global methylation. Some of them, including liquid chromatography-mass spectroscopy/mass spectroscopy (LC-MS/MS), capture of methylated DNA by methyl-binding protein (MBD2), and the LINE-1 and the LUMA assays, evaluate genomic methylation without distinguishing regulatory from structural regions. After the seminal studies in which specific gene promoters were evaluated for methylation by MSP, global methylation at promoter regions has been mainly studied by methyl-CpG island enrichment using immune-precipitation (MeDIP) by an anti-5-methylcytidine antibody, coupled to a microarray platform, and more recently by next generation bisulfite sequencing approaches.19–21
Normal cells are characterized by an open chromatin structure at promoter regions, which are transcriptionally active, and by hypermethylation and histone deacetylation at repetitive regions, with the goal of maintaining chromosome structure and genomic integrity. Conversely, hypermethylation is a general feature of cancer and of aging.22 Tumor cells are frequently hypermethylated at promoter regions, leading to downregulation of expression tumor suppressor genes and hypomethylated at repetitive DNA regions, leading to chromosomal instability and to malignant transformation. Given this, ‘global’ methylation, assessed by current techniques, may depict the sum of hyper- and hypomethylation at different regions. Actually, there were no differences in global methylation in MDS patients achieving complete and partial remission versus those resistant to hypomethylating treatment.23,24 Similarly, in acute myeloid leukemia (AML), the reduction in marrow blasts did not correlate with the decrease in global methylation levels, suggesting that hypomethylation was related to the activity of decitabine rather than cytoreduction leading to a decrease in leukemia burden (Figure 1).20 The lack of correlation between hypermethylation and HMT activity is confirmed by data on chronic myelomonocytic leukemia (CMML) patients. Comparing the DNA methylation profiles of CMML at diagnosis, we have shown that patients responding to decitabine presented both gains and losses of methylation compared to non-responders.25 Only 10% of differentially methylated regions were located at promoter regions, underscoring the importance of using platforms with extended, extra-promoter, genomic coverage.25 This is consistent with the recent knowledge that long non-coding DNA regions and the corresponding RNAs may have a critical role in gene expression regulation at the histone level.26 Analysis of the methylation status of these long non-coding regions may in the future help to predict response to HMT.
Figure 1.
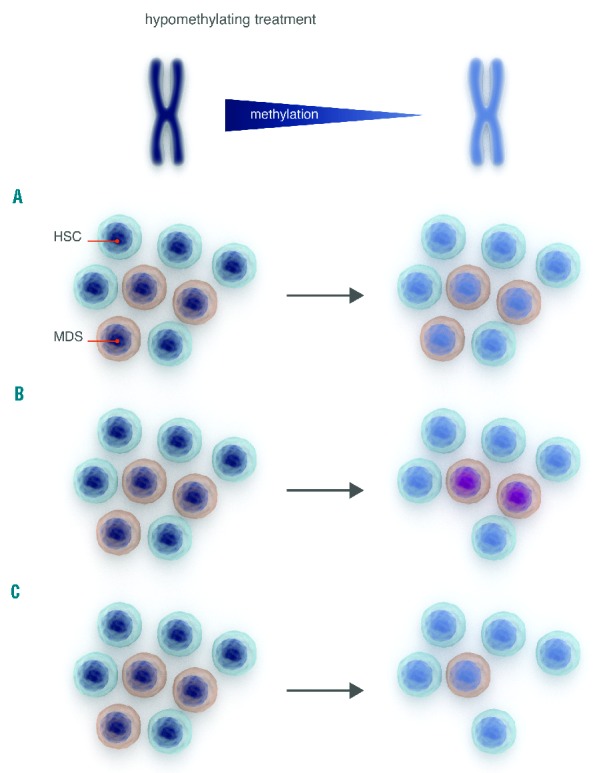
Limits of global methylation studies during hypomethylating treatment. Following HMT there is a decrease in global methylation. At least three scenarios can be hypothesized. (A) The number of MDS-progenitor cells does not change, changes in methylation reflect the sum of hyper- and hypomethylation at different regions. Critical genes in MDS cells may maintain their methylation status and there is no clinical response. Nuclei in dark blue indicate methylated DNA, light blue indicates demethylation. (B) MDS progenitor cells are demethylated, but critical genes maintain their methylation status (illustrated by violet nuclei), lineage commitment and apoptosis are induced, leading to hematologic improvement, but the abnormal clone persists and there is no cytogenetic response. (C) There is global demethylation, associated to reduction of MDS-progenitor cells and the patient achieves complete remission (ideal situation).
‘Gene-specific’ hypermethylation has been shown to be a negative prognostic factor for the natural history of MDS, also in the context of epigenetic treatment.23,24,27–30
Several putative tumor suppressor genes have been shown to be hypermethylated in MDS, including cell cycle, signal transduction, lineage commitment, apoptosis, immune response and cytoskeletal remodeling genes, which may be then induced by AZA or DAC treatment.29 In addition to ‘absolute’ methylation, also methylation density seems to play a significant role. We have shown that high (over 50%) methylation levels of the apoptosis regulator BCL2L10 are associated to reduced survival in patients with MDS treated with azacitidine.30 These data may indicate that critical methylation levels may counteract demethylation by azanucleotides and reduce treatment efficacy. The role of the BCL2L10 gene as response predictor has been confirmed by Cluzeau et al., although results are in part conceptually conflicting.31 These authors actually showed that “low” BCL2L10 expression quantified by flow cytometry is predictive of azacitidine response.31 The reasons for this discrepancy are not clear but may rely on mechanisms regulating BCL2L10 expression, in addition to methylation.
A methylation panel of ten promoter regions studied by bisulfite-pyrosequencing, including E- and N-cadherin, estrogen receptor, NOR1, nucleoplasmin 2, oligodendrocyte OLIG2, p15INK4B, progesterone receptor A and B, PDZ and LIM domain 4 (RIL) has been indicated as negative prognostic factor for MDS.32 This was a very large study including over 300 patients from three independent protocols, and defined a methylation score based on all genes for each patient. Although there was no significant association between methylation at baseline and clinical response to decitabine or azacitidine, reduced methylation over time correlated with clinical response. The methylation score was an independent prognosticator for survival and AML progression in the multivariable analysis, including also age and IPSS.32 Another proposed marker for HMT response is the enzyme phosphoinositide-phospholipase Cbeta1(PLCbeta1), which is known to regulate proliferation and motility.33 PLCbeta1 mRNA has been shown to increase in responder patients, along with PLCbeta1 promoter demethylation. This correlated to and anticipated the clinical outcome, suggesting that PI-PLCbeta1 gene reactivation may be used as predictor of azacitidine response.
Unlike minimal residual disease markers in other hematologic malignancies, it is not clear whether these ‘dynamic’ markers analyzed before and after HMT reflect hypomethylation of the MDS clone or if they are an expression of normal bone marrow recovery at the time of disease remission (Figure 1).
Epigenetic regulation of gene expression relies also on microRNAs (miR), which are known to cooperate with known genetic lesions. In particular, the microRNA miR-22 has been shown to be over-expressed and to play a role in myelodysplastic syndromes.34 Mouse models conditionally expressing miR-22 in the hematopoietic compartment, show reduced global 5-hydroxymethylcytosine (5-hmC) levels through TET2 downregulation, increased hematopoietic stem cell self-renewal and defective differentiation, mimicking an MDS-like disorder.34 There are no data concerning whether hypomethylating treatment can induce changes in miR expression. However, overexpression of the microRNA miR-29b has been associated to DNA hypomethylation in AML cell lines, due to reduced DNMT3a and 3b, and, indirectly, DNMT1 expression.35 Blum et al. studied elderly, previously untreated, AML patients treated with intravenous (IV) low-dose decitabine at 20 mg/m2 for ten days, which induced 64% overall responses, including 47% CR. Clinical response was associated to higher miR-29b pre-treatment levels, which probably lead to increased sensitivity of the leukemic cells to HMT in the presence of reduced DNMT1 expression.36
Mutations of epigenetic regulators
Recently, mutations of epigenetic modifiers, including DNMT3A, TET methylcytosine dioxygenase 2 (TET2), isocitrate dehydrogenase 1 (IDH1) and 2, additional sex combs-like 1 (ASXL1), enhancer of zeste homolog 2 (EZH2), have been shown to play a significant role in the pathogenesis of myeloid disorders (reviewed by Shih et al.37).
DNMT3A and B are de novo methyltransferase (Figure 2). DNMT3A mutations at exons 18, 19, 21, 22 and 24–26 have been identified in approximately 20% of AML and 8–10% MDS patients. They are predicted to be associated with DNA hypomethylation at certain CpG sites and have been shown to confer unfavorable prognosis in AML treated with conventional chemotherapy.38 On the other hand, AML carriers of DNMT3A mutations treated with decitabine had significantly higher complete remission rates, when compared to wild-type DNMT3A.39 This indicates that inactivation may potentiate the hypomethylating activity of the drug in AML.
Figure 2.
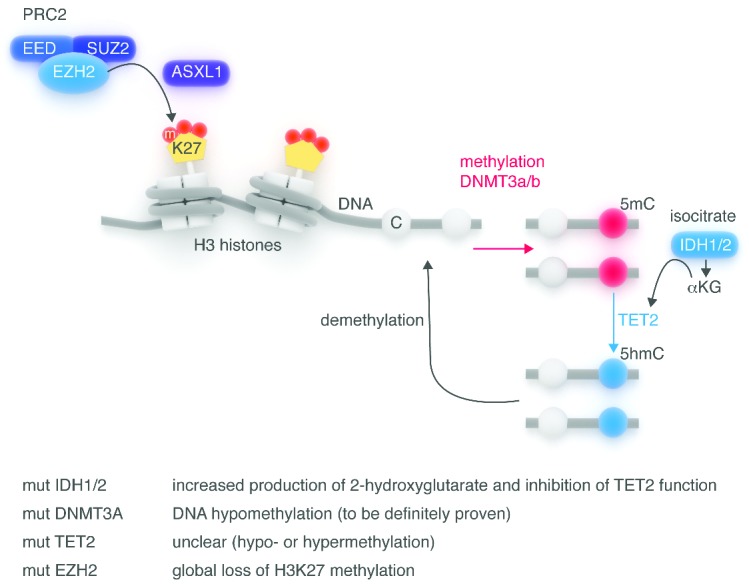
General overview of the epigenetic enzyme network. Epigenetic regulation of gene expression involves several enzymatic systems acting on DNA and on histones.37 De novo methylation of DNA is due to DNMT3A and 3B, which catalyze the formation of 5-methyl-cytosine (5mC) in the context of CpG islands, blocking access to transcription factors. The TET2 family of enzymes converts 5mC in 5-hydroxymethyl-cytosine (5hmC) in the presence of a-ketoglutarate (αKG), which is produced by hydroxylation of isocytrate due to IDH1/2 enzymes. This results in DNA hypomethylation. Histone modifications, in particular tri-methylation of Histone H3 lysine 27 by EZH2, a key component of the polycomb repressive complex 2 (PRC2), takes active part in the network. ASXL1 has been shown to be important for recruitment and/or stabilization of the PRC2 complex at key locations in the genome. DNMT3A, TET2 and EZH2 mutations induce loss of function of the enzymes, while IDH1/2 are gain-of-function mutations.
TET2 is the first discovered active demethylating enzyme (Figure 2). TET2 loss-of-function mutations are recurrent in MDS, myeloproliferative neoplasms (MPN), CMML, and AML.40–42 TET2 mutations have been shown to impair the catalytic activity of TET2 and diminish hydroxylation of 5-methylcytosine (5mC), probably leading to accumulation of 5mC at various genomic locations.41 In this line, Figueroa et al. reported a correlation between TET2 mutations and DNA hypermethylation at HpaII sites in de novo AML.42 In contrast, Ko et al. found that TET2 mutations and low 5hmC levels were associated with DNA hypomethylation at most differentially methylated CpG sites in myeloid malignancies.41 This discrepancy may reflect differences in the genomic CpG sites examined, or in the techniques used for 5mC quantification. Despite this, TET2 mutations have often been associated to HMT response in MDS, AML and CMML.30,43,44 These data have been confirmed in a very recent report on 92 MDS patients treated with HMT, where TET2 and/or DNMT3A mutations were independent predictors of better response and prolonged progression-free survival.45
ASXL1 and EZH2 are polycomb group protein (PcG) complexes and influence chromatin configuration by ‘directing’ methylation at specific chromatin marks, inducing activation or repression. Decitabine treatment has been shown to significantly reduce EZH2 expression in vivo.46 These data show that epigenetic modifications induced by HMT are complex and influence expression of key enzymes, whose mutations in turn may impact HMT activity.
Apoptosis induction
Direct effects of azacitidine and decitabine, such as apoptosis, may not be of secondary importance in terms of drug efficacy, especially in AML (Figure 3). Several groups, including ours, have shown significant induction of apoptosis in vitro early after treatment start, before the hypomethylating effect becomes evident.24,47 The rate of cell death is dose-dependent, as also shown by the inhibition of erythroid and granulocytic-monocytic colony-formation capacity of bone-marrow CD34+ progenitor cells (E Fabiani et al., unpublished observations, 2010). Activation of the extrinsic apoptotic pathway seems to be important, as shown by the induction of TNF-related apoptosis-inducing ligand (TRAIL).48 Upregulation of TRAIL following DAC treatment was associated with DNA demethylation, increased DNaseI sensitivity, and histone acetylation of a non-CpG island, CpG-rich, region located 2kb upstream to the transcription start site.
Figure 3.

Candidate pathways for azanucleotide activity. Following uptake into the cell by nucleoside-specific transport, AZA and DAC enter the cell and after phosphorylation and reduction, are incorporated into DNA. Excess azanucleotides are rapidly deaminated to uracil by cytidine deaminase and excreted by the kidneys. DNMT1 is then trapped by 5-Aza-deoxinucleotides, resulting in DNA hypomethylation following replication. Mutations of epigenetic enzymes alter chromatin configuration (i.e. EZH2), methylation (DNMT1, 3A and 3B) and the production of 5hmC (IDH1/2, TET2). Azacitidine and decitabine treatment reactivate expression of most oncogenic pathways, including among others lineage commitment, cell adhesion and cycle, signal transduction, immune responses and apoptosis. Upregulation of cancer antigens has also been shown, together with induction of T-cell responses. Changes in any of these stages may influence patients’ response to hypomethylating treatment.
Immunoregulatory effects are also induced, in particular T-cell responses. Azacitidine treatment has been shown to induce a cytotoxic CD8+ T-cell response to several tumor antigens, and to induce at the same time expansion of regulatory T cells (Treg).49 On the other hand, it has been recently reported that Treg numbers decrease following continued azacitidine treatment, despite a brief increase during the first month.50 Indeed Treg expansion at MDS diagnosis was associated with a lack of response to azacitidine in 68 patients with higher-risk MDS. To date, the final effect of azacitidine in the immunomodulatory setting remains unclear.
HMT induces significant cytopenia, especially during the first 2–3 cycles, probably due to the abovementioned apoptotic effect.6,51 This may be due to toxicity of HMT on the neoplastic clone and on residual hematopoietic cells, which have been shown to carry similar epigenetic and genetic alterations.52 Later on, despite the rarity of cytogenetic responses, the incidence of cytopenia tends to decrease, pointing also to a differentiating role of HMT on MDS progenitor cells.
Azanucleoside metabolism
AZA and DAC are pro-drugs; following uptake into the cell by nucleoside-specific transport mechanisms, they are phosphorylated to monophosphate derivative by uridine-cytidine kinase (UCK) and deoxycytidine-kinase (DCK), respectively, and then to diphosphate and triphosphate derivatives by pyrimidine monophosphate and diphosphate kinases. Decitabine exerts its action through direct incorporation into DNA, while only 10–20% of azacitidine is incorporated into DNA after reduction due to the ribonucleotide-reductase (RR) enzyme, which is a ‘limiting’ reaction. Most of the AZA is indeed incorporated into RNA, causing inhibition of RNA and protein synthesis.53 Excess azanucleotides are rapidly deaminated to uracil by cytidine deaminase (CDA), and after opening of the ring structure, are excreted by the kidneys (Figure 3). Many of the enzymes involved in azanucleotide metabolism display single nucleotide polymorphisms (SNP). The first ATP-dependent phosphorylation, catalyzed by the enzyme UCK for AZA and DCK for DAC is a limiting step for drug activation. Accordingly, we demonstrated that UCK1 expression was significantly decreased in azacitidine-resistant patients, independent of UCK1 promoter methylation. In addition, lower UCK1 levels were associated to shorter overall survival.54
Concerning drug catabolism, the CDA SNP A79C has been shown to reduce plasma CDA activity, especially in females, corresponding to higher decitabine plasma levels.55 In this study, survival was significantly worse in male versus female patients with MDS treated with 5-azacytidine/decitabine. We have shown that CDA SNP, including A92G, C451T and -897C>A, do not play a role as predictors of toxicity, response or survival in patients treated with azacitidine combined to valproic acid.56
Another enzyme essential for the methyl cycle and folate cell metabolism is methylenetetrahydrofolate reductase (MTHFR), which acts as a co-substrate of methionine synthetase for the production of S-adenosyl-methionine, a methyl group donor for CpG island and global methylation. We found that the neither MTHFR 677TT or 1298CC SNP, which have been shown to reduce DNA methylation, play a role as marker of response to azacitidine.57
Conclusions
Azanucleotides impact expression of genes belonging to most oncogenic pathways, including, among others, lineage commitment, cell cycle and adhesion, signal transduction, immune responses and apoptosis. A very recent paper by Di Ruscio et al. identifies a new class of RNA capable of controlling genomic methylation and influence transcription of specific genes by interacting with DNMT1 in a locus-specific way.58 Interaction of azanucleotides with this new mechanism of epigenetic regulation of gene expression may further impact drug efficacy, but may also lead the way to the identification of new vehicles for targeted therapy. Furthermore, since present methylation analysis is hampered by the heterogeneity of bone marrow samples, including neoplastic and normal bone marrow progenitor cells, new methods, able to probe DNA-methylation at the single cell level,59 may better define MDS-specific methylation markers predictive of azanucleotide response. In the near future, the identification of combinations of HMT to other drugs, targeting other epigenetic mechanisms, such as histone-deacetylase inhibitors, immunomodulatory agents or new compounds targeting frequently mutated genes in MDS, may improve azacitidine response and hopefully achieve a cure of the disease.
Acknowledgments
Authors would like to acknowledge Prof. Sante Tura for inspiring discussions.
Footnotes
Funding
This work was supported by grants from Associazione Italiana Ricerca sul Cancro (A.I.R.C.) and FIRB (RBAP11TF7Z).
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Della Porta MG, Malcovati L, Strupp C, Ambaglio I, Kuendgen A, Zipperer E, et al. Risk stratification based on both disease status and extra-hematologic comorbidities in patients with myelodysplastic syndrome. Haematologica. 2011;96(3):441–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malcovati L, Della Porta MG, Strupp C, Ambaglio I, Kuendgen A, Nachtkamp K, et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based Prognostic Scoring System (WPSS). Haematologica. 2011;96(10):1433–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kantarjian H, O’Brien S, Ravandi F, Cortes J, Shan J, Bennett JM, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113(6):1351–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voso MT, Fenu S, Latagliata R, Buccisano F, Piciocchi A, Aloe-Spiriti MA, et al. The revised IPSS predicts survival and leukemic evolution of myelodysplastic syndromes significantly better than IPSS and WPSS: validation by the GROM Italian regional database. J Clin Oncol. 2013;31(21):2671–7 [DOI] [PubMed] [Google Scholar]
- 6.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lübbert M, Suciu S, Baila L, Rüter BH, Platzbecker U, Giagounidis A, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol. 2011;29(15):1987–96 [DOI] [PubMed] [Google Scholar]
- 8.Steensma DP, Baer MR, Slack JL, Buckstein R, Godley LA, Garcia-Manero G, et al. Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: the alternative dosing for outpatient treatment (ADOPT) trial. J Clin Oncol. 2009;27(23):3842–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fianchi L, Criscuolo M, Lunghi M, Gaidano G, Breccia M, Levis A, et al. Outcome of therapy-related myeloid neoplasms treated with azacitidine. J Hematol Oncol. 2012;5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stresemann C, Bokelmann I, Mahlknecht U, Lyko F. Azacytidine causes complex DNA methylation responses in myeloid leukemia. Mol Cancer Ther. 2008;7(9):2998–3005 [DOI] [PubMed] [Google Scholar]
- 11.Voso MT, Breccia M, Lunghi M, Poloni A, Niscola P, Finelli C, et al. Rapid loss of response after withdrawal of treatment with azacitidine: a case series in patients with higher-risk myelodysplastic syndromes or chronic myelomonocytic leukemia. Eur J Haematol 2013;90(4):345–8 [DOI] [PubMed] [Google Scholar]
- 12.Silverman LR, Fenaux P, Mufti GJ, Santini V, Hellström-Lindberg E, Gattermann N, et al. Continued azacitidine therapy beyond time of first response improves quality of response in patients with higher-risk myelodysplastic syndromes. Cancer 2011;117(12):2697–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, et al. Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121(12):2186–98 [DOI] [PubMed] [Google Scholar]
- 14.Craddock C, Quek L, Goardon N, Freeman S, Siddique S, Raghavan M, et al. Azacitidine fails to eradicate leukemic stem/progenitor cell populations in patients with acute myeloid leukemia and myelodysplasia. Leukemia. 2013;27(5):1028–36 [DOI] [PubMed] [Google Scholar]
- 15.de Figueiredo-Pontes LL, Pintão MC, Oliveira LC, Dalmazzo LF, Jácomo RH, Garcia AB, et al. Determination of P-glycoprotein, MDR-related protein 1, breast cancer resistance protein, and lung-resistance protein expression in leukemic stem cells of acute myeloid leukemia. Cytometry B Clin Cytom. 2008;74(3):163–8 [DOI] [PubMed] [Google Scholar]
- 16.Onda K, Suzuki R, Tanaka S, Oga H, Oka K, Hirano T. Decitabine, a DNA methyltransferase inhibitor, reduces P-glycoprotein mRNA and protein expressions and increases drug sensitivity in drug-resistant MOLT4 and Jurkat cell lines. Anticancer Res. 2012;32(10):4439–44 [PubMed] [Google Scholar]
- 17.Efferth T, Futscher BW, Osieka R. 5-Azacytidine modulates the response of sensitive and multidrug-resistant K562 leukemic cells to cytostatic drugs. Blood Cells Mol Dis. 2001;27(3):637–48 [DOI] [PubMed] [Google Scholar]
- 18.Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E, et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood. 2009;114(16):3448–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe C, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009;113(6):1315–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan P, Frankhouser D, Murphy M, Tam HH, Rodriguez B, Curfman J, et al. Genome-wide methylation profiling in decitabine-treated patients with acute myeloid leukemia. Blood. 2012;120(12):2466–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borgel J, Guibert S, Weber M. Methylated DNA immunoprecipitation (MeDIP) from low amounts of cells. Methods Mol Biol. 2012;925:149–58 [DOI] [PubMed] [Google Scholar]
- 22.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20(4):440–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blum W, Klisovic RB, Hackanson B, Liu Z, Liu S, Devine H, et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J Clin Oncol. 2007;25(25):3884–91 [DOI] [PubMed] [Google Scholar]
- 24.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110(7):2302–8 [DOI] [PubMed] [Google Scholar]
- 25.Figueroa M, Sotzen J, Abdel-Wahab O, Levis A, Masala E, Santini V. A DNA methylation signature at diagnosis distinguishes CMML patients who respond to decitabine Leukemia Res. 2013;37(S1):S77 [Google Scholar]
- 26.Spitale RC, Tsai MC, Chang HY. RNA templating the epigenome: long noncoding RNAs as molecular scaffolds. Epigenetics. 2011;6(5):539–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quesnel B, Guillerm G, Vereecque R, Wattel E, Preudhomme C, Bauters F, et al. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood. 1998;91(8):2985–90 [PubMed] [Google Scholar]
- 28.Khan H, Vale C, Bhagat T, Verma A. Role of DNA methylation in the pathogenesis and treatment of myelodysplastic syndromes. Semin Hematol. 2013;50(1):16–37 [DOI] [PubMed] [Google Scholar]
- 29.Tsai HC, Li H, Van Neste L, Cai Y, Robert C, Rassool FV, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21(3):430–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voso MT, Fabiani E, Piciocchi A, Matteucci C, Brandimarte L, Finelli C, et al. Role of BCL2L10 methylation and TET2 mutations in higher risk myelodysplastic syndromes treated with 5-azacytidine. Leukemia. 2011;25(12):1910–3 [DOI] [PubMed] [Google Scholar]
- 31.Cluzeau T, Robert G, Mounier N, Karsenti JM, Dufies M, Puissant A, et al. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget. 2012;3(4):490–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X, et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol. 2010;28(4):605–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Follo MY, Finelli C, Mongiorgi S, Clissa C, Bosi C, Testoni N, et al. Reduction of phosphoinositide-phospholipase C beta1 methylation predicts the responsiveness to azacitidine in high-risk MDS. Proc Natl Acad Sci USA. 2009;106(39):16811–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM, et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell. 2013;13(1):87–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113(25):6411–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blum W, Garzon R, Klisovic RB, Schwind S, Walker A, Geyer S, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci USA. 2010;107(16):7473–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599–612 [DOI] [PubMed] [Google Scholar]
- 38.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metzeler KH, Walker A, Geyer S, Garzon R, Klisovic RB, Bloomfield CD, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26(5):1106–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838–42 [DOI] [PubMed] [Google Scholar]
- 41.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, et al. Groupe Francophone des Myelodysplasies (GFM). Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25(7):1147–52 [DOI] [PubMed] [Google Scholar]
- 44.Braun T, Itzykson R, Renneville A, de Renzis B, Dreyfus F, Laribi K, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood. 2011;118(14):3824–31 [DOI] [PubMed] [Google Scholar]
- 45.Traina F, Visconte V, Elson P, Tabarroki A, Jankowska AM, Hasrouni E, et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia. 2014;28(1):78–87 [DOI] [PubMed] [Google Scholar]
- 46.Xu F, Li X, Wu L, Zhang Q, Yang R, Yang Y, et al. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: relation to adverse epigenetic alteration and poor prognostic scoring. Ann Hematol. 2011;90(6):643–53 [DOI] [PubMed] [Google Scholar]
- 47.Fabiani E, Leone G, Giachelia M, D’alo’ F, Greco M, Criscuolo M, et al. Analysis of genome-wide methylation and gene expression induced by 5-aza-2′-deoxycytidine identifies BCL2L10 as a frequent methylation target in acute myeloid leukemia. Leuk Lymphoma. 2010;51(12):2275–84 [DOI] [PubMed] [Google Scholar]
- 48.Soncini M, Santoro F, Gutierrez A, Frigè G, Romanenghi M, Botrugno OA, et al. The DNA demethylating agent decitabine activates the TRAIL pathway and induces apoptosis in acute myeloid leukemia. Biochim Biophys Acta. 2013;1832(1):114–20 [DOI] [PubMed] [Google Scholar]
- 49.Goodyear OC, Dennis M, Jilani NY, Loke J, Siddique S, Ryan G, et al. Azacitidine augments expansion of regulatory T cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (AML). Blood. 2012;119(14):3361–9 [DOI] [PubMed] [Google Scholar]
- 50.Costantini B, Kordasti SY, Kulasekararaj AG, Jiang J, Seidl T, Abellan PP, et al. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica. 2013;98(8):1196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santini V, Fenaux P, Mufti GJ, Hellström-Lindberg E, Silverman LR, List A, et al. Management and supportive care measures for adverse events in patients with myelodysplastic syndromes treated with azacitidine. Eur J Haematol. 2010;85(2):130–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Will B, Zhou L, Vogler TO, Ben-Neriah S, Schinke C, Tamari R, et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood. 2012;120(10):2076–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123(1):8–13 [DOI] [PubMed] [Google Scholar]
- 54.Valencia A, Martino A, Sanna A, Buchi F, Masala E, Canzian F, et al. Expression of nucleoside metabolizing enzymes in myelodysplastic syndromes and modulation of response to azacitidine. Leukemia. 2013; 10.1038/leu.2013.330 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahfouz RZ, Jankowska A, Ebrahem Q, Gu X, Visconte V, Tabarroki A, et al. Increased CDA expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clin Cancer Res. 2013;19(4):938–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voso MT, Santini V, Finelli C, Musto P, Pogliani E, Angelucci E, et al. Valproic acid at therapeutic plasma levels may increase 5-azacytidine efficacy in higher risk myelodysplastic syndromes. Clin Cancer Res. 2009;15(15):5002–7 [DOI] [PubMed] [Google Scholar]
- 57.Criscuolo M, Chiusolo P, Giachelia M, Fianchi L, Fabiani E, Giammarco S, et al. MTHFR polymorphisms in patients with myelodysplastic syndromes and therapy-related myeloid neoplasms. Leuk Lymphoma. In press [DOI] [PubMed] [Google Scholar]
- 58.Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. 2013;503(7476):371–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lorthongpanich C, Cheow LF, Balu S, Quake SR, Knowles BB, Burkholder WF, et al. Single-cell DNA-methylation analysis reveals epigenetic chimerism in preimplantation embryos. Science. 2013;341(6150):1110–2 [DOI] [PubMed] [Google Scholar]