

Abstract
Platelet release by megakaryocytes is regulated by a concert of environmental and autocrine factors. We previously showed that constitutively released adenosine diphosphate by human megakaryocytes leads to platelet production. Here we show that adenosine diphosphate elicits, in human megakaryocytes, an increase in cytosolic calcium concentration, followed by a plateau, which is lowered in the absence of extracellular calcium, suggesting the involvement of Store-Operated Calcium Entry. Indeed, we demonstrate that megakaryocytes express the major candidates to mediate Store-Operated Calcium Entry, stromal interaction molecule 1, Orai1 and canonical transient receptor potential 1, which are activated upon either pharmacological or physiological depletion of the intracellular calcium pool. This mechanism is inhibited by phospholipase C or inositol-3-phosphate receptor inhibitors and by a specific calcium entry blocker. Studies on megakaryocyte behavior, on extracellular matrix proteins that support proplatelet extension, show that calcium mobilization from intracellular stores activates signaling cascades that trigger megakaryocyte adhesion and proplatelet formation, and promotes extracellular calcium entry which is primarily involved in the regulation of the contractile force responsible for megakaryocyte motility. These findings provide the first evidence that both calcium mobilization from intracellular stores and extracellular calcium entry specifically regulate human megakaryocyte functions.
Introduction
Megakaryopoiesis is the process by which bone marrow megakaryocytes (Mks) are derived from pluripotent hematopoietic stem cells to produce platelets. During differentiation, Mks migrate from the osteoblastic to the vascular niche in response to Stromal-derived factor-1α (SDF-1α).1 Once in the proximity of the vasculature, Mks convert their cytoplasm into branching filaments called proplatelets, which protrude into the sinusoid lumen where platelets are released.2 A growing body of evidence indicates that the characteristics of the extracellular matrix (ECM) structure and composition surrounding Mks play an important role in the regulation of platelet production.1,3,4 Moreover, early during megakaryopoiesis, Mks develop platelet-specific granules, which release their contents supporting Mk development.5,6 Among these, we recently demonstrated that Mks constitutively release adenosine diphosphate (ADP) which promotes proplatelet formation by interacting with its receptor P2Y13.7 Most importantly, we described that patients with Delta-Storage Pool Deficiency (δ-SPD), a congenital bleeding diathesis characterized by deficiency of dense granules and their constituents (including ADP) in Mks and platelets, display in vivo a significantly higher prevalence of thrombocytopenia than those observed in other disorders of primary hemostasis.7 However, the exact mechanisms by which matrix components and soluble factors coordinate to regulate platelet release are still unknown. The increase in cytosolic calcium concentration ([Ca2+]i) has been described to be required for interaction with the extracellular environment in different human cell types.8,9 Interestingly, extracellular nucleotides and purinergic receptors are critically involved in mediating this interaction with changes in [Ca2+]i.10,11 Mks express all P2Y G-protein-coupled receptors (GPCRs) for purine and pyrimidine nucleotides7 and all G-proteins coupled to Ca2+ signaling activation.12 Among the known P2Y receptors that bind ADP, only P2Y1 and P2Y13 have been recognized to mediate an increase in [Ca2+]i via the phosholipase C (PLC) pathway.13–17 Specifically, PLC activation leads to the generation of the second messengers inositol 1,4,5-trisphosphate (IP3) which diffuses within the cytosol to bind to and activate the IP3 receptors (IP3Rs) in the endoplasmic reticulum (ER), the most abundant intracellular Ca2+ store.18 This results in cytoplasmic Ca2+ elevation that can be separated into two distinct phases. In the first phase, Ca2+ is released from intracellular stores via the IP3Rs. In the second phase, the decrease in ER Ca2+ content causes the activation of plasma membrane Ca2+ channels resulting in the influx of extracellular Ca2+ inside the cells, a mechanism that has been termed Store-Operated Ca2+ Entry (SOCE).19 The prime candidates to mediate SOCE in hematopoietic cells are STIM and Orai proteins.19 More specifically, STIM1 (stromal interaction molecule 1) is the transmembrane ER Ca2+ sensor that translocates in close proximity to the plasma membrane upon store depletion and activates Orai1 (also known as calcium-release-activated calcium-modulator, CRACM), the pore-forming subunit of store-operated Ca2+ channels.19 The role of the additional STIM1 and Orai1 paralogs, i.e. STIM2 and Orai2–3, is far from being fully understood in naïve cell systems, albeit they may recapitulate SOCE when ectopically expressed.19 Moreover, several evidences demonstrated that members of the canonical transient receptor potential (TRPC) family of cation channels may represent additional candidates for SOCE.20 Agonist-induced elevation of intracellular Ca2+ levels is essential for platelet activation. In this regard, STIM1 and Orai1 proteins have been shown to be key players in human platelet SOCE during aggregation,21,22 whereas TRPC involvement has been questioned.23,24 Importantly, it has been demonstrated that SOCE plays a major role in mediating adhesion and motility onto ECM components of different cell types, including hematopoietic stem cells.9,25 Thus we hypothesized that purinergic signaling and SOCE may be responsible for Mk interaction with the ECM environment and consequent regulation of platelet production. Our results demonstrate that ADP induces both intracellular Ca2+ mobilization and extracellular Ca2+ inflow in human Mks, which in turn support the cytoskeletal reorganization responsible for cellular adhesion and migration and final proplatelet formation on ECM components that promote such a dynamic process, such as fibrinogen and fibronectin. These findings provide the first evidence that Ca2+ signaling is a fundamental regulator of human Mk functions.
Methods
Human cord blood was collected from the local blood bank following normal pregnancies and deliveries with informed consent of the parents, in accordance with the ethical committee of the IRCCS Policlinico San Matteo Foundation, Pavia, Italy, and the principles of the Declaration of Helsinki. CD34+ cells from cord blood samples were separated by immunomagnetic bead selection (Miltenyi Biotec, Bologna, Italy) and differentiated, as previously described.4
At the end of cell culture, Mks were harvested and plated onto glass cover-slips previously coated with different ECM components in order to evaluate cell adhesion, migration and proplatelet formation. All images were acquired by Olympus BX51 microscope (Olympus, Deutschland GmbH, Hamburg, Germany). In some experiments, before being seeded, cells were pre-incubated with the following substances, at the indicated final concentrations: apyrase 1 U/mL, ADP 25 μM, 2-APB 20 μM, U-73122 10 μM, BTP-2 20 μM.
We employed Ca2+ imaging to investigate the expression and functionality of SOCE on the same extracellular matrix components. Specifically, Mks were loaded with 4 μM fura-2 acetoxymethyl ester (AM) or 5 mM FLUO-3 AM and observed using an upright epifluorescence Axiolab microscope (Carl Zeiss) equipped with a Zeiss X63 Achroplan objective or a laser-scanning confocal microscope (Nikon, Eclipse TE300).
For all the experiments, values are expressed as mean±standard deviation (SD). Student’s t-test was performed for paired observations. ANOVA, followed by the post hoc Bonferroni t-test, was performed for grouped observations. P<0.01 or P<0.05 were considered statistically significant. All experiments were independently replicated at least 3 times, unless otherwise specified.
Complete details of methods are provided in the Online Supplementary Appendix.
Results
ADP regulates proplatelet formation in adhesion on extracellular matrix components
During differentiation, human Mks release ADP that support proplatelet formation.7 In order to demonstrate whether ADP plays a role also in regulating Mk interaction with extracellular matrix components, Mks were differentiated in vitro starting from human cord blood derived CD34+ cells. At Day 13 of culture, the percentage of mature CD41+ Mks was 93±5% and cells were able to extend long and branched proplatelet structure ending with platelet-sized tips (Online Supplementary Figure S1A-C). Fully differentiated Mks were plated on two different bone marrow ECM components, fibrinogen and fibronectin, known to support Mk maturation and proplatelet formation.3,4 A time course analysis revealed that on fibrinogen, Mks exhibit actin stress fibers, focal contacts and convoluted microtubules throughout the cytoplasm after 3 h of incubation, while after 8 h they started to extend proplatelet-like pseudopods that became more branched when incubation was prolonged to 16 h (Figure 1A and Online Supplementary Figure S1D-F). The addition of the ADP scavenger apyrase 1 U/mL7 markedly inhibited cytoskeletal reorganization and Mk adhesion (Figure 1B–D and Online Supplementary Figure S2). Conversely, treatment with ADP 25 μM augmented the number of adherent Mks exhibiting stress fibers and microtubule assembly (Figure 1B and C), with a consequent significant increase of adherent proplatelet forming Mks after 16 h of incubation (Figure 1D). Similar results were obtained on fibronectin (Online Supplementary Figure S3). Conversely, no significant differences were observed in the same conditions in adhesion on type I collagen, which has been described to inhibit platelet release,3 or polylysine, a neutral control substrate (data not shown). Thus, these results suggest that ADP promotes proplatelet formation in adhesion on extracellular matrix components that support this process.
Figure 1.
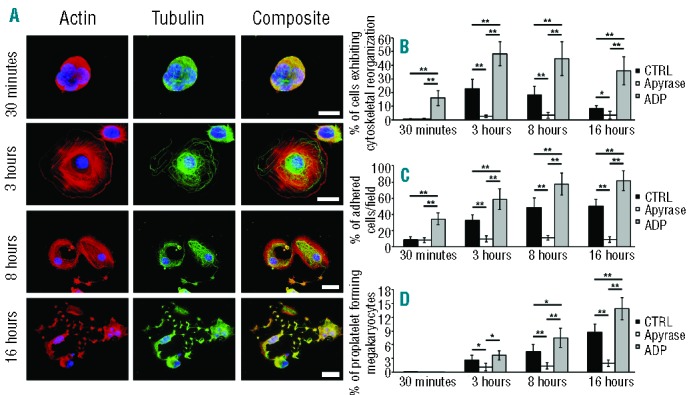
Analysis of Mk-matrix interaction during proplatelet formation. Cord blood derived-Mks at Day 13 of culture were plated on fibrinogen-coated cover-slips, at 37° in a 5% CO2. (A) After different time-points (30 minutes, 3-8-16 hours) adherent cells were fixed and stained for immunofluorescence analysis with TRITC-phalloidin (red) and antibody against α-tubulin (green). Nuclei were counterstained with Hoechst 33258 in blue. Images were acquired by an Olympus BX51, magnification 60× and 100×, scale bar=20 μm. In some experiments Mks were seeded in the presence or absence of the ADP scavenger apyrase (1 U/mL) or ADP (25 μM) and analyzed as described for control. (B) Cytoskeletal reorganization, (C) adhesion and (D) proplatelet formation (B) were analyzed with respect to non-treated controls (CTRL) (mean±SD, n=5 independent experiments; *P<0.05, **P<0.01).
ADP induces the activation of Ca2+ signaling in human megakaryocytes
Human Mks were exposed to ADP (25 μM) in physiological salt solution (PSS) containing 1.5 mM Ca2+ and fluorescence measurements of [Ca2+]i were carried out in Mks loaded with the Ca2+ sensitive fluorochrome Fura-2. We showed that ADP elicited a large increase in [Ca2+]i with a clear plateau following the initial Ca2+ peak (Figure 2A). Pre-incubation with MRS 2179 (10 μM), a highly specific inhibitor of P2Y1, or MRS 2211 (10 μM), the specific P2Y13 inhibitor, significantly affected both the initial peak, and the extent of the plateau (Figure 2A and E). However, the strongest inhibition was obtained by the latter, confirming a primary role for P2Y13 in mediating Mk response to ADP.7 Subsequently, we focused on the study of Ca2+ signaling in order to understand its role in regulating cell response to agonist stimulation. Thus, Mks were pre-incubated with BAPTA-AM (20 μM), a membrane-permeable intracellular Ca2+ chelator. Here ADP stimulation did not evoke any detectable Ca2+ signal (Figure 2B and E). Interestingly, when ADP was applied in the absence of extracellular Ca2+ (0Ca2+), the initial increase in [Ca2+]i remained unchanged while the plateau phase totally disappeared (Figure 2C and E). Thus, intracellular Ca2+ release plays a major role in eliciting the initial Ca2+ peak induced by ADP, while extracellular Ca2+influx determines a sustained Ca2+ signaling, the hallmark of SOCE activation.26 In accordance with this evidence, removing extracellular Ca2+ during the plateau phase, in the presence of ADP, caused a rapid drop of [Ca2+]i to baseline values (Figure 2D).
Figure 2.
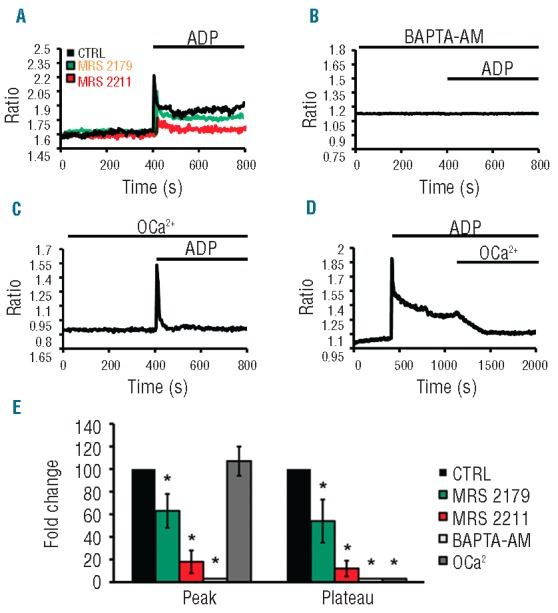
ADP elicited Ca2+ mobilization in human Mks. (A) ADP (25 μM) evoked an initial rise in [Ca2+]i (peak) followed by a signal of lesser magnitude (plateau) in human Mks (CTRL). Pre-incubation with the P2Y1 inhibitor, MRS 2179 (10 μM) and the P2Y13 inhibitor, MRS 2211 (10 μM) inhibited ADP-induced Ca2+ signaling. (B) Pre-treatment with BAPTA-AM caused the complete lack of Ca2+ response to ADP. (C) When ADP was applied in the absence of extracellular Ca2+, the intracellular Ca2+ displayed a fast kinetic with no plateau phase. (D) Removal of extracellular Ca2+ (0Ca2+) caused the rapid recovery of [Ca2+]i to resting levels. (E) Statistical evaluation of the effect of the different treatment on the amplitudes of the two phases of Ca2+ signaling (n=5 independent experiments; *P<0.01).
mRNA and protein expression of the putative mediators of SOCE in human megakaryocytes
We investigated, in human Mks, the expression of the molecules that have been proposed to mediate SOCE. Specifically, we analyzed the expression of mRNA encoding for the recently cloned Orai (Orai1, Orai2 and Orai3) and STIM (STIM1 and STIM2) genes and for all the known TRPC expressed in humans (TRPC1, TRPC3, TRPC4, TRPC5, TRPC6, TRPC7). Orai1, Orai2, Orai3, STIM1, STIM2, TRPC1 and TRPC6 transcripts were detectable in human Mks (Figure 3A and B), whereas we did not detect any significant amount of mRNAs for TRPC3/4/5/7. Expression of STIM1, Orai1, and TRPC1, the best characterized effectors of SOCE, was also confirmed in human CD61+ Mks by Western blot (Figure 3C).
Figure 3.
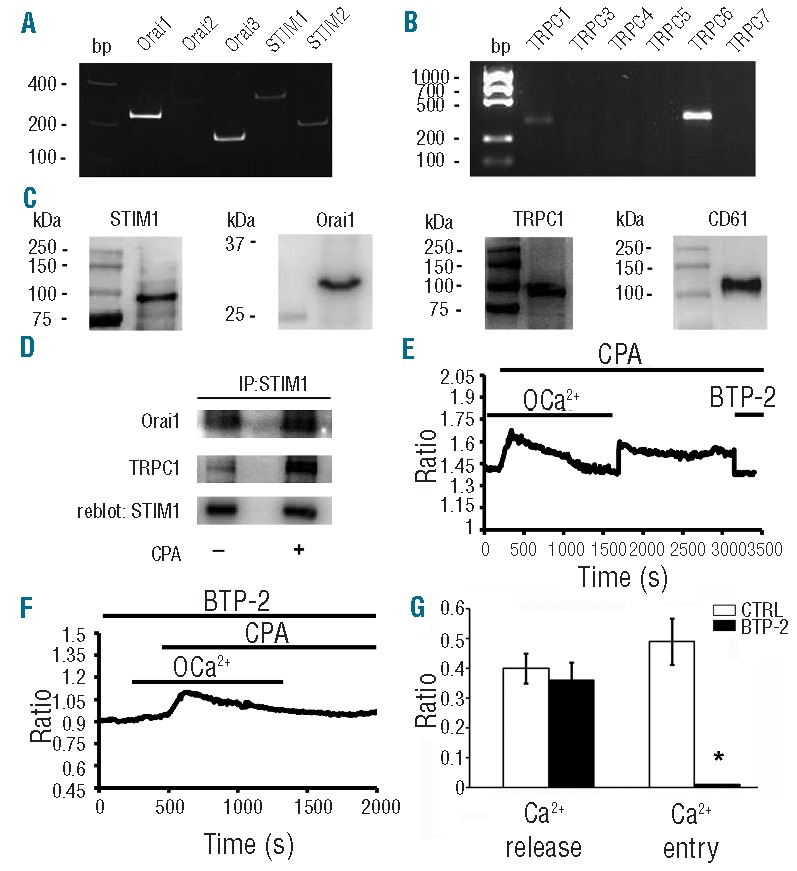
Expression and function of molecular mediators of SOCE in human Mks. (A–B) Gel electrophoresis of the PCR products for Orai, STIM and TRPC expression. PCR products were of expected size: Orai1, 257bp; Orai2, 334bp; Orai3, 159bp; Stim1, 347bp; Stim2 186bp TRPC1, 307bp, TRPC6, 341bp. No signal was observed for TRPC3/5/6/7. Representative of 3 independent experiments. (C) Protein expression of STIM1, Orai1 and TRPC1 in human CD61+ Mks at Day 13 of culture. (D) Mature Mks were treated (+) or not (−) with CPA (10 μM) and lysed. Lysates were immunoprecipitated (IP) with an anti-STIM1 antibody and subjected to Western blotting. Membranes were stained with antibodies against Orai1 and TRPC1 and re-blot with antibody against STIM1 to ensure equal immunoprecipitation of the protein. Representative of 3 independent experiments. (E) Intracellular Ca2+ pools were depleted by exposing the cells to CPA (10 μM) in 0Ca2+ solution. Re-addition of extracellular Ca2+ led to an increase in [Ca2+]i which was indicative of SOCE. Subsequently, acute application of BTP-2 (50 μM) inhibited CPA-elicited Ca2+ inflow, thus confirming the store-dependent nature of Ca2+ entry. (F) Pre-incubation with BTP-2 (20 μM) prevented CPA-induced Ca2+ signal on Ca2+ readdition to extracellular solution. (G) Statistical analysis of the effect exerted by BTP-2 on the peak amplitudes of both Ca2+ release and Ca2+ entry stimulated by CPA (mean±SD, n=5 independent experiments; *P<0.01).
SOCE is functional in human megakaryocytes
In order to demonstrate the presence of a functional SOCE in human Mks, we exposed cells to cyclopiazonic acid (CPA, 10 μM), a widely employed activator of this mechanism.27 Specifically, CPA blocks Sarco-Endoplasmic Reticulum Ca2+ ATPase (SERCA) activity, thereby preventing Ca2+sequestration into the stores and leading to their depletion and SOCE activation. Figure 3D shows that STIM1 co-immunoprecipitates with both Orai1 and TRPC1, with a significant increase in cells subjected to store depletion as compared to non-treated controls, thus suggesting the formation of a molecular complex among the three molecules under these conditions. In order to further confirm this evidence, we next carried out fluorescence measurements of [Ca2+]i. Depleting the stores with CPA (10 μM), without extracellular Ca2+ (0Ca2+), evoked a transient rise in [Ca2+]i because of passive emptying of Ca2+ stores through leakage channels in ER membrane. Thereafter, Ca2+ levels dropped to base-line values as the plasma membrane transporters (i.e. plasma membrane Ca2+-ATPase and Na+-Ca2+exchanger) extruded Ca2+ from the cytosol. When external Ca2+ was restored to 1.5 mM, a second increase in intracellular Ca2+ levels was observed due to activated influx through store-operated channels (Figure 3E). Acute application of BTP-2 (50 μM), a specific inhibitor of SOCE,26 strongly inhibited CPA-dependent Ca2+ inflow (Figure 3E). Accordingly, 30 min pre-incubation with BTP-2 (20 μM) prevented SOCE (Figure 3F), whereas it did not affect the ER Ca2+ content (Figure 3F and G).
SOCE is engaged by human megakaryocytes in response to ADP and regulates cell adhesion and migration
SOCE recruitment by GPCRs is mediated by the PLCβ/IP3 pathway.18 Thus, human Mks were treated with the selective pan PLCβ inhibitor, U-73122 (10 μM),28 and that of IP3Rs, 2-APB (20 μM), at a concentration that did not affect SOCE.29,30 U-73122 has been shown to prevent Ca2+ release by interfering with SERCA-dependent Ca2+ sequestration into ER lumen. However, the acute addition of this drug never caused the slow increase in [Ca2+]i which is typical of SERCA-inhibiting drugs (see, for example, the tracing shown in Figure 4B). Moreover, we have previously utilized 10 μM U-73122 to selectively impair PLC activity in a variety of cell types.26,31 Both molecules prevented ADP-induced [Ca2+]i elevation, compared to control (Figure 4A–C), demonstrating that the IP3-dependent Ca2+ pool shapes initial Mk response to ADP. Importantly, Mk pre-treatment with BTP-2 (20 μM) for 30 min prevented the plateau phase of the Ca2+ signal after ADP stimulation without affecting the transient Ca2+ peak (Figure 4D). SOCE activation following treatment with ADP was also confirmed by the evidence that STIM1 association with Orai1 and TRPC1 was notably enhanced upon stimulation with the agonist (Figure 4E). The aforementioned inhibitors were also tested for their effects on Mk functions. After 16 h, the percentages of Mks displaying cytoskeletal reorganization on fibrinogen was significantly inhibited by 2-APB (20 μM), U-73122 (10 μM) and BTP-2 (20 μM) (Figure 4F) with a decreased number of both adherent and proplatelet forming Mks on ECM components (Figure 4G and H). Interestingly, after treatment with 2-APB and U-73122, adherent proplatelets exhibited altered structure and decreased branching, while, in the presence of BTP-2, the few adherent Mks were still able to extend normal structured and branched proplatelets (data not shown). Therefore, we hypothesized that the observed role of Ca2+ inflow was above all to promote Mk interaction with ECM components, suggesting a possible role for SOCE in triggering in vivo Mk migration at the site of platelet release. Actually, ADP enhanced migration of CD41+ Mks in response to SDF-1α compared to control, while addition of apyrase, 2-APB, U-73122 or BTP-2 significantly reduced it (Figure 4I and Online Supplementary Figure S4A and B). Interestingly, some of the Mks that had passed through the filter in presence of BTP-2 elongated normal branched proplatelet, further supporting the hypothesis that extracellular Ca2+ entry regulates mainly Mk active interaction with the ECM components, while intracellular Ca2+ release is sufficient to activate proplatelet formation (Online Supplementary Figure S4C). The same results were obtained on fibronectin (data not shown).
Figure 4.
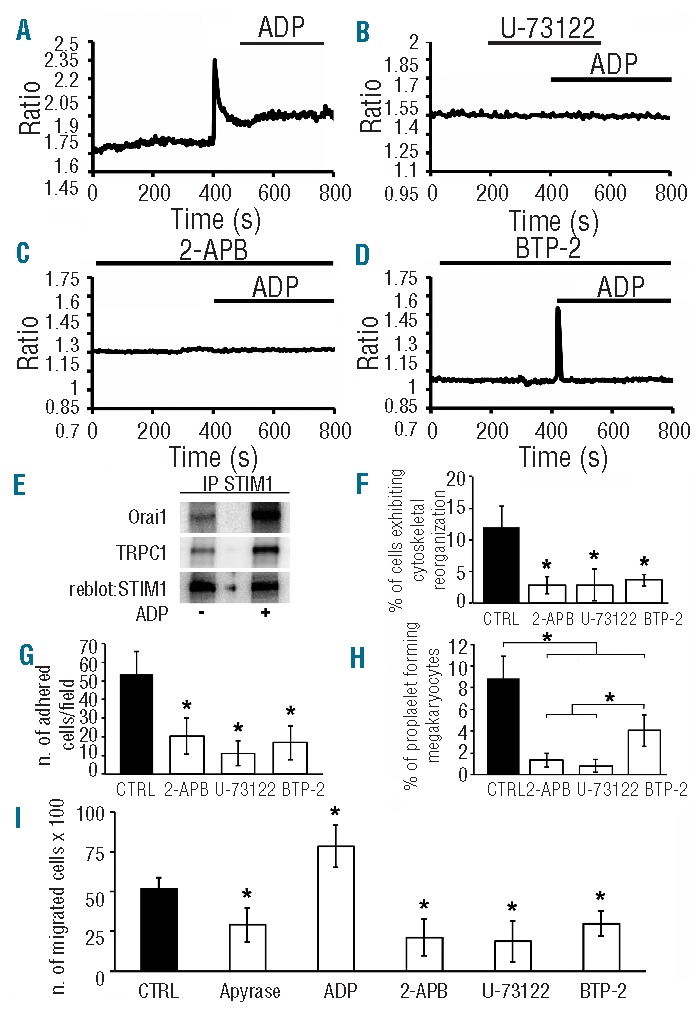
ADP induces SOCE in human Mks. (A) ADP (25 μM) evoked the described [Ca2+]i signaling in control Mks. Sample pre-treatment with U-73122 (B) or 2-APB (C) caused the complete lack of response to ADP. (D) 20-min pre-incubation with BTP-2 inhibited ADP-elicited Ca2+ inflow, but not intracellular Ca2+ mobilization, confirming the store-dependent nature of the response to ADP. (E) Mature Mks were treated (+) or not (−) with ADP (25 μM) and lysed. Lysates were immunoprecipitated (IP) with an anti-STIM1 antibody and subjected to Western blotting. Membranes were stained with antibodies against Orai1 and TRPC1 and re-blot with an antibody against STIM1 to ensure equal immunoprecipitation of the protein. Representative of 3 independent experiments. Mks at Day 13 of culture were pre-treated or not with 2-APB, U-73122 and BTP-2, before being seeded on fibrinogen. The percentage of Mks displaying cytoskeletal reorganization (F), the number of adherent Mks (G) and the percentage of proplatelet formation in adhesion on fibrinogen (H) were analyzed compared to untreated controls (CTRL) (mean±SD, n=5 independent experiments; *P<0.01). (I) Mks at Day 13 of differentiation were treated with the same compounds described above and left to migrate in a Transwell plate, previously coated with fibrinogen, for 16 hours. Mks that had passed in the lower chamber were collected and counted by phase contrast microscopy (n=3 independent experiments; *P<0.01).
ADP-induced Ca2+ mobilization activates megakaryocyte downstream signaling
To assess the role of ADP-induced Ca2+ signals we investigated the activation of molecules relevant for proplatelet formation and/or Mk adhesion and motility (i.e. the serine/threonine-specific kinase Akt, the mitogen-activated protein kinases ERK, the tyrosine kinases FAK, Src Syk and the myosin light chain, MLC).7,32–35 We showed that, upon stimulation with ADP (25 μM), the extent of phosphorylation of all tested molecules increased in adherent Mks on both fibrinogen and fibronectin substrates (Figure 5A). We next focused our attention exclusively on the role of Ca2+ on ADP signaling. We confirmed that ADP drives the activation of these pathways (Figure 5B, lane 2) over the basal level (Figure 5B, lane 1). At the same time, some Mks were incubated in the presence of the intracellular Ca2+ chelator BAPTA-AM (20 μM) to prevent the overall Ca2+ activation, or in the absence of extracellular Ca2+ (0Ca2+), to selectively abrogate Ca2+ influx and thus SOCE activation. Interestingly, intracellular Ca2+buffering prevented ADP-mediated activation of all molecules, with the exception of Syk (Figure 5B, lane 3). Conversely, 0Ca2+ prevented only phosphorylation of MLC (Figure 5B, lane 4).
Figure 5.
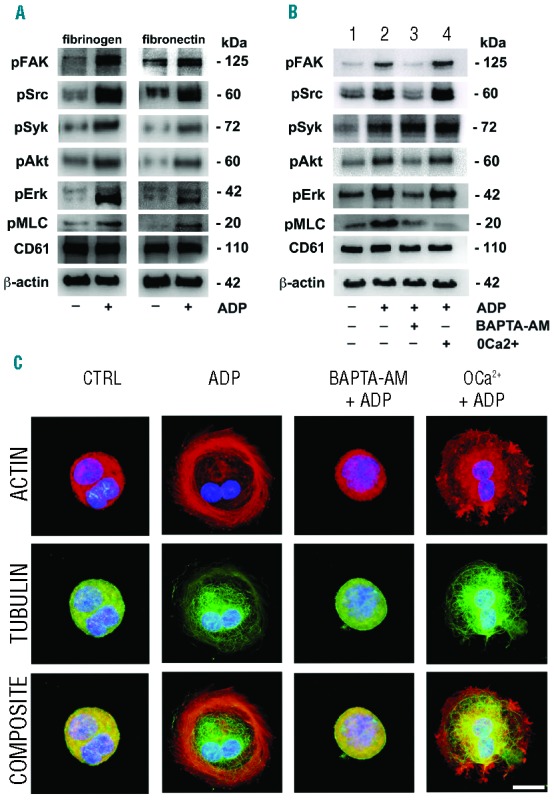
The role of intracellular and extracellular Ca2+ in regulating Mk response to ADP. (A) Human Mks at Day 13 of culture were stimulated (+) or not (−) with ADP and let to adhere on fibrinogen or fibronectin before being lysed. (B) Parallel samples were suspended in the PSS or Ca2+ free solution (0Ca2+) and pre-treated (+) or not (−) with BAPTA-AM, before being stimulated (+) or not (−) with ADP and finally lysed. All samples were subjected to Western blot analysis for evaluation of FAK, Src Syk, Akt, Erk and MLC phosphorylation. Samples were also probed with anti-CD61 and anti–β-actin antibodies to ensure equal loading. (C) Some cells were allowed to adhere in the same conditions on fibrinogen. Mks were fixed and stained for immunofluorescence analysis with TRITC-phalloidin (red) and antibody against α-tubulin (green). Nuclei were counterstained with Hoechst 33258 in blue. Images were acquired by an Olympus BX51, magnification 100×, scale bar=20 μm. Representative of 3 independent experiments.
We examined the cytoskeleton structure under the same conditions upon adhesion on fibrinogen. Treatment with ADP induced actin stress fiber formation and microtubule assembly, whereas pre-treatment with BAPTA-AM abolished these responses (Figure 5C). Removal of extracellular Ca2+ prevented actin stress fiber development, but did not affect microtubule assembly (Figure 5C). Results were perfectly comparable on fibronectin (data not shown). Importantly, Mk behavior was monitored, during stimulation with ADP (25 μM), using a flow chamber system. During perfusion experiments, in the presence of ADP, Mks showed a sustained Ca2+ oscillation with different spatial and temporal organization of Ca2+ in an observation time frame of approximately 5 min (Figure 6A). The same Mks showed remodeling of their cytoplasm (Figure 6B).
Figure 6.
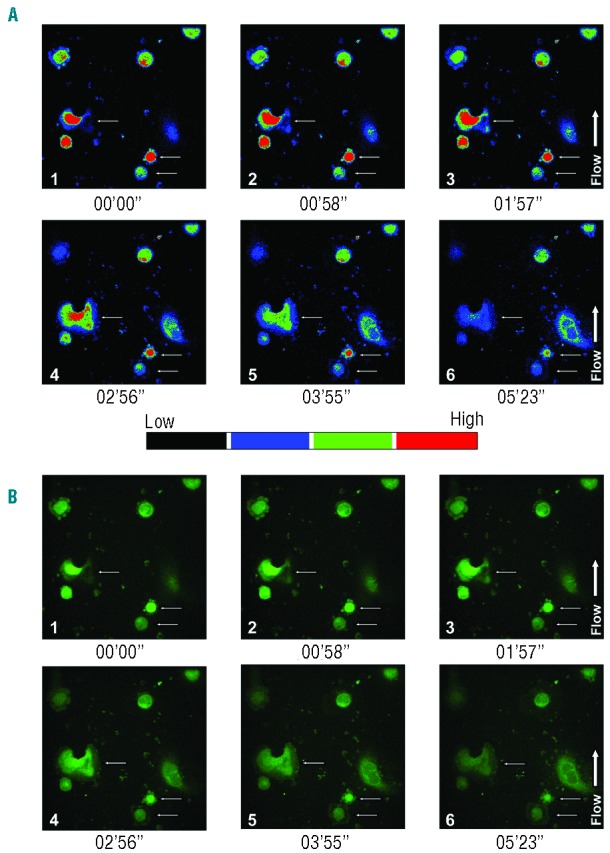
ADP elicited Ca2+ mobilization in human Mks in flow conditions. Mks at Day 13 of culture were plated on fibrinogen-coated coverslips, at 37°C in a 5% CO2 atmosphere and then loaded with 5 μM FLUO 3-AM. Perfusion experiments were started at a shear rate of 50 sec-1 and Mks behavior was followed for 22 min. (A) Images of Mks in pseudo-colors captured in an observation time of 5 min and 23 seconds. Associated single-cell calcium flux recording demonstrate the duration and amplitude of the calcium response. Shear rate and ADP (25 μM) evoked an initial intracellular Ca2+ mobilization and a subsequent spatiotemporal redistribution of Ca2+. (B) The same frames were recorded in green fluorescence to better demonstrate the remodeling of human Mks cytoplasm in those cells undergoing to calcium fluxes.
Inhibition of extracellular Ca2+ inflow affects megakaryocyte interaction with type I collagen
Stimulation or inhibition of ADP signaling in human Mks did not affect interaction with type I collagen, as described above. However, it has been demonstrated that human Mks display a significant increase in [Ca2+]i during interaction with this ECM component.36 Importantly, we demonstrated that only type I collagen is able to inhibit proplatelet formation by sustained phosphorylation of MLC in human Mks.34 Thus we decided to test the role of SOCE on Mk behavior on this ECM component. Interestingly, treatment with BTP-2 (20 μM) significantly inhibited Mk adhesion with a decreased number of cells displaying cytoskeleton reorganization with respect to non-treated controls (Figure 7A–C).
Figure 7.
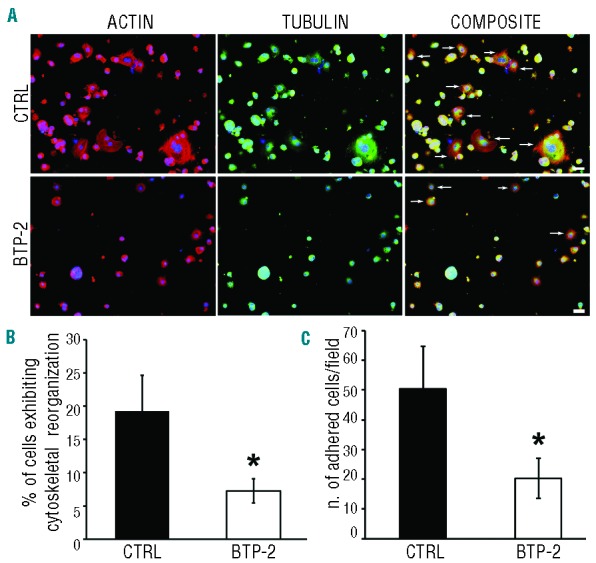
A role for Ca2+ inflow in regulating adhesion on type I collagen. Cord blood derived-Mks at Day 13 of culture were plated on type I collagen-coated cover-slips, at 37°C in a 5% CO2 in presence or absence of BTP-2. (A) After 16 h adherent cells were fixed and stained for immunofluorescence analysis with TRITC-phalloidin (red) and antibody against α-tubulin (green). Nuclei were counterstained with Hoechst 33258 in blue. Arrows inidicate Mks exhibiting cytoskeletal reorganization. Images were acquired by an Olympus BX51, magnification 20×, scale bar=20 μm). The percentages of Mk displaying reorganized cytoskeleton (B) and the numbers of adherent Mks (C) were analyzed compared to untreated controls (CTRL) (mean±SD, n=5 independent experiments; *P<0.01).
Discussion
Mk maturation and proplatelet formation in the bone marrow are consequent to an integrated concert of signals belonging to both extracellular environment and Mks themselves.3,4,34 Importantly, it is known that different autocrine loops are fundamental modulators of Mk development.5,6 With regard to this, we demonstrated that ADP is constitutively released by human Mks in vitro and that it is a crucial molecule in the regulation of platelet production.7 Importantly, we also described that patients with Delta-Storage Pool Deficiency, display in vivo a significantly higher prevalence of thrombocytopenia than those observed in other disorders of primary hemostasis.7 Despite this knowledge, it has not yet been shown whether or not soluble factors that promote platelet release can regulate Mk interaction with ECM components that support such a dynamic process.
In this study, we aimed to unravel the mechanism underlying platelet production within a bone marrow ECM environment. We demonstrated that ADP promotes Ca2+ release from intracellular stores which is responsible for the regulation of proplatelet formation and for the activation of SOCE. This latter promotes Mk adhesion and migration.
First, we showed that Mk interaction over fibrinogen and fibronectin requires the co-ordination of different cellular processes which operate in a cycle that can be divided into four critical steps: 1) early passive adhesion to ECM components; 2) active reorganization of the cytoskeleton and assembly of focal adhesions; 3) dynamic contraction of the cytoplasm with appearance of proplatelet-like pseudopod; 4) final proplatelet branching in adhesion on ECM components. Interestingly, all these events were regulated by constitutively released ADP, whereas its scavenger apyrase significantly inhibited them. It is worthy of note that exogenous administrated ADP markedly enhanced adhesion of proplatelet forming Mks. Thus ADP plays a role in favoring the establishment of the tight interaction with extracellular substrates that support proplatelet formation and helps them in this process.
ADP-dependent platelet activation relies on the increase in cytosolic Ca2+ concentration, achieved by SOCE activation.21 The same mechanism is responsible for promoting interaction with the ECM environment in different human cell types.9,25 In order to investigate the role of Ca2+ signaling in regulating ADP-dependent human Mk functions, we performed real-time measurements of intracellular Ca2+ movements. We demonstrated an increase in [Ca2+]i upon ADP stimulation in static conditions and a direct correlation between Ca2+ signaling and Mk cytoskeleton remodeling under flow conditions.
The application of MRS 2211, a selective P2Y13 antagonist, inhibited the ADP-induced Ca2+ mobilization to a greater extent than MRS 2179, a selective P2Y1 antagonist. These results confirmed our previous data that P2Y13, which is expressed by human Mks but not by human platelets, represents a crucial and specific modulator of Mk function.7 Interestingly, ADP elicited a large increase in [Ca2+]i with a clear plateau following the initial peak. Treatment with BAPTA-AM totally abolished this response, suggesting that first mobilization is sustained by intracellular Ca2+ release. Conversely, exposure to ADP in the absence of extracellular Ca2+ prevents the plateau phase. Thus, in human Mks, ADP-induced depletion of the intracellular stores activates Ca2+ inflow, a feature that suggests SOCE involvement.
We demonstrated that human Mks express both mRNA and protein for the most characterized SOCE mediators, STIM1, Orai1 and TRPC1. Moreover, we showed that Mks express also mRNA for Stim2 and Orai2 and 3, which are involved in SOCE, and for two members of the TRPC family, TRPC1 and 6.19,20 However, whether or not TRPC1 is an additional candidate for SOCE is still a matter of debate.21,23 Conversely, TRPC6 activation is completely independent of store depletion.24 Importantly, we demonstrated that SOCE was functionally activated in response to the emptying of the IP3-sensitive Ca2+ reservoir. Specifically, passive depletion of the ER Ca2+ content with CPA, led to the formation of a ternary molecular complex between STIM1, Orai1 and TRPC1 which is likely to be involved in SOCE.21 For instance, in platelets, Orai1 mediates the store-dependent interaction between STIM1 and TRPC1 which regulates SOCE in response to both pharmacological (i.e. SERCA inhibition) and IP3-dependent depletion of the ER Ca2+ content.37 BTP-2, a selective inhibitor of store-dependent Ca2+ inflow, abrogated CPA-and ADP-induced Ca2+ inflow. Moreover, blocking PLC or the IP3Rs, prevented depletion of the ER Ca2+ pool and abrogated SOCE. Interestingly, PLC and IP3Rs inhibitors prevented both adhesion and proplatelet formation, while BTP-2 significantly decreased Mk-ECM interaction, but exerted a minor impact on Mks that, if adherent, were still able to extend normal branched proplatelets. Consistently, ADP-dependent IP3-dependent Ca2+ mobilization was sufficient to activate all signaling molecules known to be relevant for both Mk adhesion and proplatelet formation, including FAK, Src, Akt and ERK,7,32–35 indicating that intracellular Ca2+ release can promote these events. Conversely, MLC activation was selectively driven by store-dependent Ca2+ entry, as more extensively discussed below.
Recently, Chen et al. described the unique role of STIM1-dependent Ca2+ signaling in controlling cell migration by the regulation of actomyosin contractility.38 A number of studies have demonstrated SOCE involvement in biological and pathological migration of multiple cell types, including CD133+ hematopoietic stem cells9 and blood cells.39 Moreover, the critical role for extracellular nucleotides and purinergic receptors in mediating cell motility is well known.10,11 Dynamic regulation of Mk migration, within the bone marrow environment, is critical for platelet production.1 In fact, patients with MYH9-related disease, caused by mutations of the gene for the heavy chain of the non-muscle myosin, present thrombocytopenia, probably caused by impaired migration and premature proplatelet formation within the osteoblastic niche due to the lack of proper interaction with ECM components.40,41 Here, we showed that the ADP-mediated Ca2+ inflow is required for MLC phosphorylation, actin polymerization and formation of focal complexes in human Mks, but not for microtubule assembly. It has been well described that actomyosin contraction plays a major role in regulating the structure of the focal complexes, which serve as cytoskeletal organizing centers42 as well as surface-sensing entities that coordinate cellular migration.43,44 Thus, we hypothesized that the functional counterpart of the observed role of ADP-induced SOCE could be to support actomyosin cytoskeleton reorganization responsible for Mk migration. Consistently, we showed that absence of ADP or SOCE inhibition strongly impaired Mks motility toward SDF-1α. Therefore, these results show the role of Ca2+ compartmentalization in regulating different Mk function, in greater detail: Ca2+ mobilization from intracellular stores drives both Mk adhesion and proplatelet formation (by activating FAK, Src, ERK and Akt), while Ca2+ inflow following store depletion is primarily responsible for promoting cytoskeletal reorganization and subsequent migration on ECM components (through MLC-mediated remodeling of actomyosin cytoskeleton). Thus, intracellular and extracellular Ca2+ are both needed to ensure Mk functions.
Importantly, rare cases of patients with mutated STIM1 and Orai1 accompanied by thrombocytopenia have been described,45,46 though there are no reports regarding bone marrow phenotype and/or Mk development and function. Stim1–/– or Orai1–/– mice have also been described.47,48 Both these models present normal platelet counts. However, mice, as well as humans, express different STIM, Orai and TRPC isoforms that mediate SOCE activation. Thus, we do not expect that single knockdown could totally affect SOCE either in mice or in humans. Actually, both Stim1–/– and Orai1–/– platelets aggregate normally to ADP even at very low concentrations of agonists and ADP is still able to induce unaltered cellular activation despite the defect in [Ca2+]i signaling. However, the importance of [Ca2+]i in the regulation of platelet release in vivo has been recently suggested by two different groups that described new somatic mutations of calreticulin, a protein that binds Ca2+ in the ER contributing to Ca2+homeostasis, in patients affected by myeloproliferative neoplasms. Interestingly, these mutations impair Ca2+ binding affinity and are expressed in all hematopoietic progenitors, including megakaryocyte-erythroid progenitors.49,50 Given the evidence that all these patients are characterized by an excessive platelet production, it is possible that an impaired Ca2+ homeostasis in bone marrow Mks may be responsible for the altered platelet production.
In summary, we report the first evidence that SOCE activators and Ca2+ mobilization are involved in the regulation of human Mk functions. These data, along with the most recent clinical discoveries on calreticulin mutations, open new perspectives in the study of the signals that in vivo concur in promoting platelet production.
Acknowledgments
The authors would like to thank Marco Cattaneo for critical review of the manuscript, Cesare Perotti for the supply of human cord blood, Valentina Boz and Maria Rita Cozzi for their help during calcium measurements under flow, and Gianluca Viarengo for flow cytometry analysis.
Footnotes
The online version of this article has a Supplementary Appendix.
Funding
This paper was supported by the Cariplo Foundation (grant 2010.0807), Regione Lombardia - Project SAL-45, Almamater Foundation (Pavia) and Regione Lombardia - Progetto di Cooperazione Scientifica e Tecnologica Internazionale, National Institutes of Health (grant EB016041-01) and Italian Ministry of Health (grant RF-2009-1550218). The funding organizations had no role in study design, data collection and analysis, or preparation of the manuscript.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Avecilla ST, Hattori K, Heissig B, Tejada R, Liao F, Shido K, et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med. 2004;10(1):64–71 [DOI] [PubMed] [Google Scholar]
- 2.Italiano JE, Lecine P, Shivdasani R, Hartwig JH. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol. 1999;147(6):1299–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malara A, Gruppi C, Rebuzzini P, Visai L, Perotti C, Moratti R, et al. Megakaryocytematrix interaction within bone marrow: new roles for fibronectin and factor XIII-A. Blood. 2011;117(8):2476–83 [DOI] [PubMed] [Google Scholar]
- 4.Balduini A, Pallotta I, Malara A, Lova P, Pecci A, Viarengo G, et al. Adhesive receptors, extracellular proteins and myosin IIA orchestrate proplatelet formation by human megakaryocytes. J Thromb Haemost. 2008;6(11):1900–7 [DOI] [PubMed] [Google Scholar]
- 5.Casella I, Feccia T, Chelucci C, Samoggia P, Castelli G, Guerriero R, et al. Autocrineparacrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood. 2003;101(4):1316–23 [DOI] [PubMed] [Google Scholar]
- 6.Lambert MP, Rauova L, Bailey M, Sola-Visner MC, Kowalska MA, Poncz M. Platelet factor 4 is a negative autocrine in vivo regulator of megakaryopoiesis: clinical and therapeutic implications. Blood. 2007;110(4):1153–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balduini A, Di Buduo CA, Malara A, Lecchi A, Rebuzzini P, Currao M, et al. Constitutively released adenosine diphosphate regulates proplatelet formation by human megakaryocytes. Haematologica. 2012;97(11):1657–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Neill CA, Galasko CS. Calcium mobilization is required for spreading in human osteoblasts. Calcif Tissue Int. 2000;67(1):53–9 [DOI] [PubMed] [Google Scholar]
- 9.Seidel J, Niggemann B, Punzel M, Fischer J, Zänker KS, Dittmar T. The neurotransmitter GABA is a potent inhibitor of the stromal cell-derived factor-1alpha induced migration of adult CD133+ hematopoietic stem and progenitor cells. Stem Cells Dev. 2007;16(5):827–36 [DOI] [PubMed] [Google Scholar]
- 10.Kaczmarek E, Erb L, Koziak K, Jarzyna R, Wink MR, Guckelberger O, et al. Modulation of endothelial cell migration by extracellular nucleotides: involvement of focal adhesion kinase and phosphatidylinositol 3-kinase-mediated pathways. Thromb Haemost. 2005;93(4):735–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jantaratnotai N, McLarnon JG. Calcium dependence of purinergic subtype P2Y1 receptor modulation of C6 glioma cell migration. Neurosci Lett. 2011;497(2):80–4 [DOI] [PubMed] [Google Scholar]
- 12.den Dekker E, Gorter G, van der Vuurst H, Heemskerk JW, Akkerman JW. Biogenesis of G-protein mediated calcium signaling in human megakaryocytes. Thromb Haemost. 2001;86(4):1106–13 [PubMed] [Google Scholar]
- 13.Schachter JB, Li Q, Boyer JL, Nicholas RA, Harden TK. Second messenger cascade specificity and pharmacological selectivity of the human P2Y1-purinoceptor. Br J Pharmacol. 1996;118(1):167–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazzucato M, Cozzi MR, Pradella P, Ruggeri ZM, De Marco L. Distinct roles of ADP receptors in von Willebrand factormediated platelet signaling and activation under high flow. Blood. 2004;104(10):3221–7 [DOI] [PubMed] [Google Scholar]
- 15.Communi D, Gonzalez NS, Detheux M, Brézillon S, Lannoy V, Parmentier M, et al. Identification of a novel human ADP receptor coupled to Gi. J Biol Chem. 2001;276(44):41479–85 [DOI] [PubMed] [Google Scholar]
- 16.Zhang FL, Luo L, Gustafson E, Palmer K, Qiao X, Fan X, et al. P2Y13: identification and characterization of a novel Galphaicoupled ADP receptor from human and mouse. J Pharmacol Exp Ther. 2002;301(2):705–13 [DOI] [PubMed] [Google Scholar]
- 17.Lyubchenko T, Woodward H, Veo KD, Burns N, Nijmeh H, Liubchenko GA, et al. P2Y1 and P2Y13 purinergic receptors mediate Ca2+ signaling and proliferative responses in pulmonary artery vasa vasorum endothelial cells. Am J Physiol Cell Physiol. 2011;300(2):C266–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–29 [DOI] [PubMed] [Google Scholar]
- 19.Parekh AB. Store-operated CRAC channels: function in health and disease. Nat Rev Drug Discov. 2010;9(5):399–410 [DOI] [PubMed] [Google Scholar]
- 20.Yuan JP, Lee KP, Hong JH, Muallem S. The closing and opening of TRPC channels by Homer1 and STIM1. Acta Physiol (Oxf). 2012;204(2):238–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galán C, Zbidi H, Bartegi A, Salido GM, Rosado JA. STIM1, Orai1 and hTRPC1 are important for thrombin- and ADP-induced aggregation in human platelets. Arch Biochem Biophys. 2009;490(2):137–44 [DOI] [PubMed] [Google Scholar]
- 22.Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. 2009;113(9):2056–63 [DOI] [PubMed] [Google Scholar]
- 23.Varga-Szabo D, Authi KS, Braun A, Bender M, Ambily A, Hassock SR. Store-operated Ca2+ entry in platelets occurs independently of transient receptor potential (TRP) C1. Pflugers Arch. 2008;457(2):377–87 [DOI] [PubMed] [Google Scholar]
- 24.Hassock SR, Zhu MX, Trost C, Flockerzi V, Authi KS. Expression and role of TRPC proteins in human platelets: evidence that TRPC6 forms the store-independent calcium entry channel. Blood. 2002;100(8):2801–11 [DOI] [PubMed] [Google Scholar]
- 25.Masiero L, Lapidos KA, Ambudkar I, Kohn EC. Regulation of the RhoA pathway in human endothelial cell spreading on type IV collagen: role of calcium influx. J Cell Sci. 1999;112(Pt 19):3205–13 [DOI] [PubMed] [Google Scholar]
- 26.Sánchez-Hernández Y, Laforenza U, Bonetti E, Fontana J, Dragoni S, Russo M, et al. Store-operated Ca(2+) entry is expressed in human endothelial progenitor cells. Stem Cells Dev. 2010;19(12):1967–81 [DOI] [PubMed] [Google Scholar]
- 27.Bird GS, DeHaven WI, Smyth JT, Putney JW., Jr.Methods for studying store-operated calcium entry. Methods. 2008;46(3):204–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ, et al. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther. 1990;255(2):756–68 [PubMed] [Google Scholar]
- 29.Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxy-diphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem. 1997;122(3):498–505 [DOI] [PubMed] [Google Scholar]
- 30.DeHaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW., Jr.Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J Biol Chem. 2008;283(28):19265–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dragoni S, Laforenza U, Bonetti E, Lodola F, Bottino C, Berra-Romani R, et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells. 2011;29(11):1898–907 [DOI] [PubMed] [Google Scholar]
- 32.Mazharian A, Watson SP, Séverin S. Critical role for ERK1/2 in bone marrow and fetal liver-derived primary megakaryocyte differentiation, motility, and proplatelet formation. Exp Hematol. 2009;37(10):1238–49 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazharian A, Thomas SG, Dhanjal TS, Buckley CD, Watson SP. Critical role of Src-Syk-PLC{gamma}2 signaling in megakaryocyte migration and thrombopoiesis. Blood. 2010;116(5):793–800 [DOI] [PubMed] [Google Scholar]
- 34.Malara A, Gruppi C, Pallotta I, Spedden E, Tenni R, Raspanti M, et al. Extracellular matrix structure and nano-mechanics determine megakaryocyte function. Blood. 2011;118(16):4449–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Badalucco S, Di Buduo CA, Campanelli R, Pallotta I, Catarsi P, Rosti V, et al. Involvement of TGF 1 in autocrine regulation of proplatelet formation in healthy subjects and patients with primary myelofibrosis. Haematologica. 2013;98(4):514–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mountford JC, Melford SK, Bunce CM, Gibbins J, Watson SP. Collagen or collagen-related peptide cause (Ca2+)i elevation and increased tyrosine phosphorylation in human megakaryocytes. Thromb Haemost. 1999;82(3):1153–9 [PubMed] [Google Scholar]
- 37.Jardin I, Lopez JJ, Salido GM, Rosado JA. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J Biol Chem. 2008;283(37):25296–304 [DOI] [PubMed] [Google Scholar]
- 38.Chen YT, Chen YF, Chiu WT, Wang YK, Chang HC, Shen MR. The ER Ca2+ sensor STIM1 regulates actomyosin contractility of migratory cells. J Cell Sci. 2013;126(Pt 5):1260–7 [DOI] [PubMed] [Google Scholar]
- 39.Schaff UY, Dixit N, Procyk E, Yamayoshi I, Tse T, Simon SI. Orai1 regulates intracellular calcium, arrest, and shape polarization during neutrophil recruitment in shear flow. Blood. 2010;115(3):657–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pecci A, Malara A, Badalucco S, Bozzi V, Torti M, Balduini CL, et al. Megakaryocytes of patients with MYH9-related thrombocytopenia present an altered proplatelet formation. Thromb Haemost. 2009;102(1):90–6 [DOI] [PubMed] [Google Scholar]
- 41.Pecci A, Bozzi V, Panza E, Barozzi S, Gruppi C, Seri M, et al. Mutations responsible for MYH9-related thrombocytopenia impair SDF-1-driven migration of megakaryoblastic cells. Thromb Haemost. 2011;106(4):693–704 [DOI] [PubMed] [Google Scholar]
- 42.Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518 [DOI] [PubMed] [Google Scholar]
- 43.Geiger B, Bershadsky AD. Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol. 2001;13(5):584–92 [DOI] [PubMed] [Google Scholar]
- 44.Zaidel-Bar R, Cohen M, Addadi L, Geiger B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem Soc Trans. 2004;32(Pt3):416–20 [DOI] [PubMed] [Google Scholar]
- 45.McCarl CA, Picard C, Khalil S, Kawasaki T, Röther J, Papolos A, et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol. 2009;124(6):1311–8 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360(19):1971–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M, et al. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. 2008;205(7):1583–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. 2009;113(9):2056–63 [DOI] [PubMed] [Google Scholar]
- 49.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N Engl J Med. 2013;369(25):2379–90 [DOI] [PubMed] [Google Scholar]
- 50.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N Engl J Med. 2013;369(25):2391–405 [DOI] [PMC free article] [PubMed] [Google Scholar]