

Abstract
Antifolates inhibit de novo folate biosynthesis, whereas ethionamide targets the mycolate synthetic pathway in Mycobacterium tuberculosis. These antibiotics are effective against M. tuberculosis but their use has been hampered by concerns over toxicity and low therapeutic indexes. With the increasing spread of drug-resistant forms, interest in using old drugs for tuberculosis treatment has been renewed. Specific inhibitors targeting resistance mechanisms could sensitize M. tuberculosis to these available, clinically approved drugs. This review discusses recently developed strategies to boost the antituberculous activity of ethionamide and antifolates. These approaches might help broaden the currently limited chemotherapeutic options of not only drug-resistant but also drug-susceptible tuberculosis, which still remains one of the most common infectious diseases in the developing world.
Keywords: antifolates, ethionamide, mycobacterium, potentiation, targeting resistance, tuberculosis
The currently available options for chemotherapeutic treatment of tuberculosis (TB) are severely restricted [1]. The five available first-line TB drugs are over 60 years old, and treatment regimens usually consist of daily doses of four out of the five drugs for 6–9 months. Poor patient adherence leads to the repeated exposure of the bacterium to subinhibitory drug concentrations that promote the acquisition of resistance mutations [1,2]. As a result, multiple drug-resistant (MDR) and extensively drug resistant (XDR) TB strains are now emerging [101]. Treatment of drug-resistant TB requires even longer regimens using second-line drugs that are expensive, difficult to administer and typically lead to severe toxic side-effects in patients [1,3]. Some XDR strains are virtually impossible to treat with the current TB drugs [4–6].
Despite the profound intrinsic resistance of Mycobacterium tuberculosis, available ‘non-TB’ drugs that are active against this bacillus may not be as rare as commonly assumed. Many antibiotics, which are routinely used in other infections, have never undergone trials for TB treatment for varying reasons: cytotoxicity, low therapeutic indexes and ignorance due to lack of motivation. Indeed, many of them were considered promising against M. tuberculosis but later abandoned and never explored again after more effective drugs, such as isoniazid and rifampicin, were introduced [7,8]. Other drugs have never even been examined for activity against TB [8,9]. Experiences with resistance in other bacteria also discouraged trials for some drugs [8,9]. Considering the urgent need for alternative TB treatment strategies and the fact that new drug development is lengthy and costly [10], such old, abandoned drugs need to be re-examined, repurposed or reused in more effective ways [7,11]. These drugs would at least be useful in cases of drug-resistant TB to which first-line drugs have become inactive. Furthermore, concerns over toxicity and low therapeutic indexes might be addressed through chemical modifications or potentiation approaches [12–15]. Drug potentiation through inactivation of resistance mechanisms has been used for antibiotics in the β-lactam family. β-lactams are now commonly prescribed for the treatment of many non-mycobacterial infections in combination with inhibitors of β-lactamases that are key determinants of β-lactam resistance. This approach has extended the life of β-lactams for more than 30 years and will continue to for many more years to come [15,16]. With similar strategies applied to other drugs, potentiators, which are inhibitors of resistance mechanisms, might not only help to prevent loss of drug efficacy due to emerging resistant strains but also to make available for the first time a large pool of well-characterized, US FDA-approved antibiotics. This approach, therefore, presents an attractive solution that could provide quick relief to the current epidemic of drug-resistant TB [15,17,18].
Ethionamide
Ethionamide (ETH or 2-ethylthioisonicotinamide) is a thioamide analog of the first-line tuberculosis drug isoniazid (INH), and like INH, ETH is a prodrug that must be activated within the M. tuberculosis cytosol to exert anti-TB activity. The gene responsible for this activation step is ethA, which encodes an NADPH-specific FAD-containing monooxygenase that oxidizes ETH [19–23]. The oxidized form of ETH can then form adducts with NAD+, which bind and inhibit InhA (Figure 1) [19,22,24–26]. The targeted enzyme, InhA, which is also targeted by INH, is an NADH-dependent enoyl-acyl carrier protein reductase of the fatty acid biosynthesis II system required for mycolic acid synthesis [19,22,24–27]. Inhibition of InhA typically leads to cell wall defects that rapidly kill M. tuberculosis [19,22,24–27]. Interestingly, ETH and INH cross-resistance occurs in only 13% of the cases, indicating that different sites are affected within InhA and that inhA mutations are not the main ETH resistance mechanism [28,29]. Similarly, most INH resistance mutations in clinical isolates of M. tuberculosis have been mapped to other chromosomal loci (katG, ndh and aphC), while mutations in inhA only account for 15–43% of mutations [30,31]. Although ETH is quite effective in killing both drug-susceptible and drug-resistant strains of M. tuberculosis, its current use in the clinic remains restricted. The biggest concern lies in its high toxicity, caused by toxic S-oxides that are produced through the oxidation catalyzed by host flavin-containing monooxygenase, even at the lowest doses required to kill M. tuberculosis [12–14,32]. Common side effects include hepatitis and gastrointestinal discomfort, which make ETH difficult to use and often lead to poor patient adherence, thereby granting opportunity for acquired resistance [33]. Therefore, ETH is currently used only as a second-line drug to treat TB cases caused by MDR and XDR M. tuberculosis strains.
Figure 1. Potentiation of ethionamide by targeting EthR.
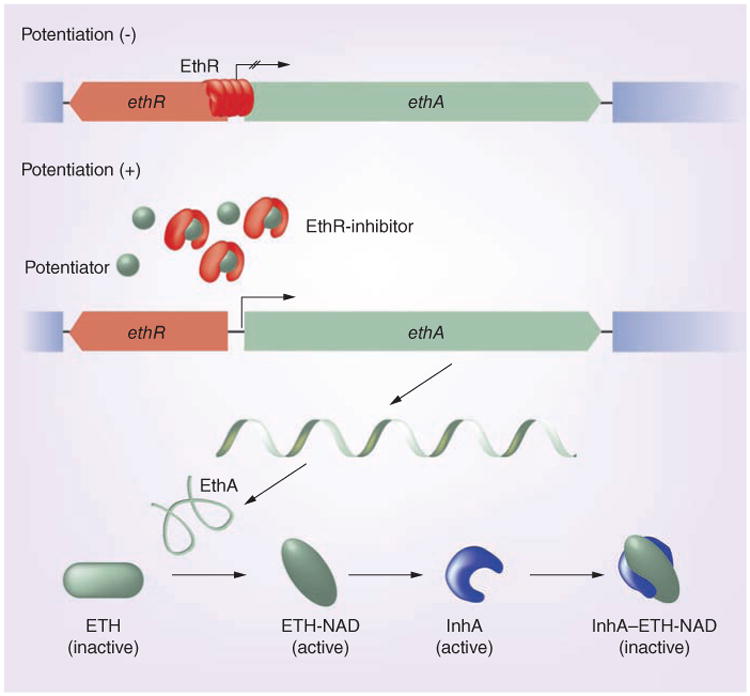
The binding of inhibitors releases EthR from its interaction with the ethA promoter. This derepresses the flavoprotein EthA, which is responsible for oxidizing and thus converting ETH to its active form, ETH-NAD. The activated drug then binds to InhA and inhibits its activity in mycolate biosynthesis. EthR inhibitors could thereby function as ETH potentiators.
ETH: Ethionamide.
Adapted with permission from [15].
Ethionamide resistance mechanisms in M. tuberculosis
Clinically acquired mutations conferring ETH resistance are commonly mapped in one or more of four chromosomal loci: ethA, ethR, inhA, or its promoter region [19,20,22,28,34]. Specifically, mutations within the ethA open reading frame reduce the catalytic activity of the encoded enzyme, leading to lowered activation of the prodrug ETH, hence reducing InhA inhibition [19,20]. Similarly, ethR encodes a repressor of ethA transcription, and mutations in ethR typically produce mutant EthR proteins with higher binding affinity to the ethA promoter, resulting in reduced production of EthA and lowered ETH activation [19,28]. Besides the ETH activation, mutations in inhA produce mutant enzymes with lowered binding activity to activated ETH, whereas mutations in the inhA promoter lead to InhA overexpression, thus deluging ETH-NAD with massive amounts of its target (Figure 1) [19,22,34]. Studies have also attributed ETH resistance to some other genes: ndh, dfrA and mshA [19]. ndh, a gene encoding an NADH dehydrogenase, might be overexpressed in resistant strains, resulting in higher levels of NAD+ that outcompetes ETH-NAD in the binding to InhA [35]. This mechanism was shown to increase ETH resistance in Mycobacterium bovis BCG and Mycobacterium smegmatis, but it has never been observed in M. tuberculosis [19]. mshA, a gene encoding a glycosyltransferase in mycothiol synthesis, has been suggested to be involved in the EthA-mediated activation of ETH [19,25,36]. Mutations in mshA conferring ETH resistance were observed in M. tuberculosis grown in suboptimal ETH levels in vitro, but no clinical strains with mshA mutations have been isolated [19].
The limiting step of ETH antimycobacterial action is its activation by EthA. If activation within the M. tuberculosis cytosol is limited, higher amounts of ETH would be required to produce a bactericidal level of ETH-NAD, which results in higher cytotoxicity to the host cell. By contrast, given the fact that EthA-mediated activation of ETH is absolutely required for antimycobacterial activity, pharmaceutical stimulation of EthA expression could be used to enhance ETH activity. In fact, it has been shown that in trans overexpression of EthA led to higher susceptibility to ETH and deficient mycolic acid synthesis in M. smegmatis [22,37]. However, attempts at overexpression of EthA in M. tuberculosis have so far failed [20,22]. The most attractive step for enhanced activation of ETH is the inhibition of ethA transcription by EthR (Figure 1). EthR controls expression of ethA by binding to the ethA promoter located within the intergenic region between ethR and ethA, thus preventing its transcription [28,29]. Indeed, in trans overexpression of ethR leads to reduced levels of intracellular EthA and reduced ETH susceptibility, whereas deletion of ethR leads to increased ETH susceptibility [22,28,38,39]. In vitro studies showed that multimers of EthR, a member of the TetR/CamR transcription-repressor family, assemble cooperatively on a 55 base pair operator, OethR, located within the ethA promoter to block access of RNA polymerase to the ethA promoter [21,24,37,39,40]. Like all TetR/CamR repressors, EthR forms a homodimer with two functional domains, each composed of nine α-helices (Figure 2) [21,37,38]. Helices 1–3 form the classical helix-turn-helix motif of DNA-binding proteins and interact with OethR, while helices 4–9 at the C-terminus of EthR form a hydrophobic tunnel (Figure 2) [21,37,41]. This hydrophobic tunnel comprises the ligand-binding site of the otherwise hydrophilic protein, thereby controlling EthR conformation and its DNA-binding capacity [21,37, 41]. Dimerization occurs through interaction of helices from both monomers in a 4-helix bundle. Earlier studies showed that EthR forms crystals with hexadecyl octanoate (HexOc), a hydrophobic molecule that occupies the hydrophobic tunnel of EthR through hydrogen bonds as well as hydrophobic interactions (Figure 2) [21,37,41]. The binding tunnel is buried within the monomer cores and contains numerous aromatic residues, resulting in a hydrophobic milieu [37,38]. Binding of EthR to HexOc, which results in a conformational alteration that increases the distance of the DNA-binding domains of the monomers by 18 Å, results in decreased binding ability of EthR to OEthR [21,37,41]. Recent mutagenesis studies showed that residues phenylalanine 110 (located in helix 5) and alanine 95 (located near helices 4 and 5) of M. tuberculosis EthR contribute to the ligand-binding domain and the point mutations altered the ligand-binding capacity of EthR [19]. More importantly, glycine 106 (located in helix 5) functions as a molecular switch to shift between conformations of EthR upon ligand binding [42]. The mutation of glycine 106 to tryptophan mimics the ligand binding that releases EthR from OethR and thus derepresses ethA transcription (Figure 1) [42]. Ligands such as HexOc could therefore prove to be powerful potentiators of ETH by inhibiting EthR repression of ethA transcription. Effective EthR inhibitors would help to resensitize ETH resistant M. tuberculosis strains, or lower the intrinsic resistance level of susceptible strains. Following this, the enhanced therapeutic index would allow for the lowering of effective dosages that would minimize toxic effects to the host. Furthermore, reduced toxicity would also improve patient adherence, thus preventing further acquisition of resistance mutations.
Figure 2. Crystal structure of a hexadecyl octanoate-bound EthR homodimer.
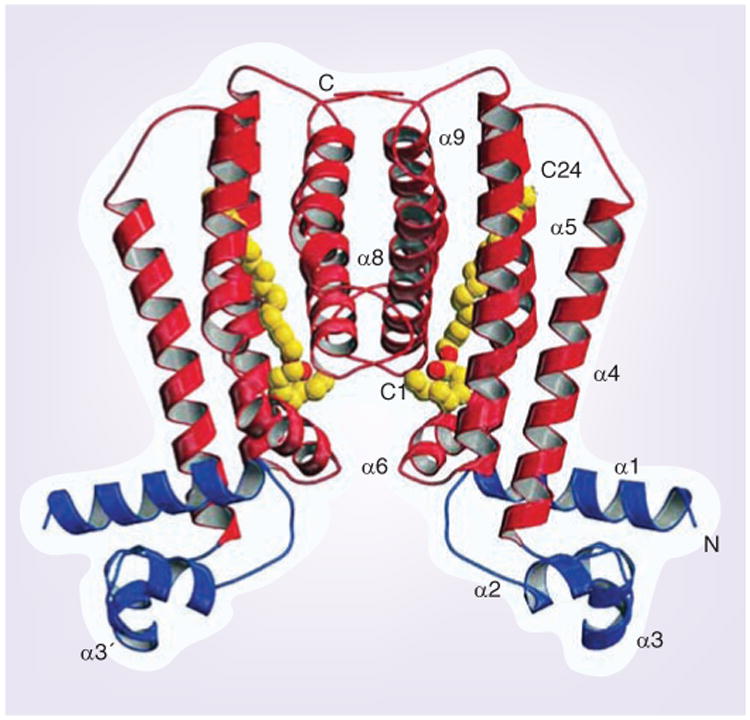
Each EthR monomer consists of nine helices (α1–α9). The first three helices at the N-terminus (blue) form a three-helix bundle DNA-binding domain in which α2–α3 form a helix–turn–helix motif that is stabilized by α1. The six C-terminal helices (α4–α9, red) are involved in dimerization. They form a hydrophobic tunnel that binds ligands. The DNA recognition helices (α3 and α3′) are separated by 52 Å. The hexadecyl octanoate ligand is shown as yellow and red spheres.
Reproduced with permission from [21].
Development of ethionamide potentiators
To design such clinically applicable ETH potentiators, it is crucial to understand optimal conditions for ligand interactions within the EthR binding pocket, as well as chemical compositions required for maximal bioavailability. For example, the hydro-phobic ester ligand HexOc co-crystalized with EthR might be too hydrophobic for its delivery to the intracellular M. tuberculosis. Nevertheless, HexOc serves well as a lead in the search for more effective inhibitors. Since the structure of the EthR-HexOc complex was solved, several groups have tried to derive hydrophobic esters to identify more effective EthR inhibitors. Initial studies using more-hydrophilic ketones as EthR ligands identified benzylacetone, which exhibits pronounced inhibition of M. smegmatis growth at 5 μg ml-1 ETH, threefold lower than the normal ETH concentration required to kill the bacterium [21,27]. However, benzylacetone was still far from ideal, considering that the chemical would have to travel through the membranes of the macrophage and the phagosome as well as the impermeable mycobacterial cell wall before it reaches its target EthR [40,43].
An elegant mammalian cell-based reporter system was introduced by the Fussenegger group to screen for compounds that not only derepress ethA transcription but are also able to reach their target effectively [40]. Conveniently, the system can also monitor cell toxicity at the same time [40]. In this reporter system, the ethA promoter was fused to a reporter gene and introduced into the mammalian genome. An efficient ligand of EthR, upon binding, would release EthR from OethR, thus resulting in expression of the measurable reporter protein. Screening a library of synthesized hydrophilic esters using this reporter system, a licensed food additive, 2-phenylethyl-butyrate, was found to be an efficient EthR inhibitor [40]. 2-phenylethyl-butyrate could clearly sensitize M. tuberculosis to ETH in vitro, although the level of sensitivity increase remains unknown [40]. To demonstrate bioavailability of 2-phenylethyl-butyrate at whole body level, human embryonic kidney cells carrying the reporter system were implanted into mice. The chemical was fed to the mice and was able to reach EthR in the human embryonic kidney cells to activate the reporter system [40]. More recently, it was demonstrated that 2-phenylethyl-butyrate effectively increases susceptibility of not only susceptible strains but also ETH-resistant M. tuberculosis strains with mutations leading to overexpression of inhA [44].
In an independent study, a pharmacore model was devised to screen for EthR ligands. Knowledge of the ligand-binding tunnel of EthR, together with its residues required for hydrogen bond formation and hydrophobic interactions, was used to design a model for a ligand consisting of a 4.6-Å linker between two hydrophilic ends that should bind inside the hydrophobic cavity and form hydrogen bonds with asparagine residues 176 and 179 [37]. Screening of a drug-like chemical library identified 131 compounds fitting the designed model [37]. After surface plasmon resonance analysis and cocrystallization, compound BDM14500 emerged as a lead that reduces binding of EthR to the ethA promoter by more than 50% [37]. BDM14500 significantly increases ETH susceptibility of M. tuberculosis in vitro, indicating potential for optimization to produce more efficient inhibitors [37]. Optimization of this 1,2,4-oxadiazole lead compound produced two thiophen-2-yl-1,2,4-oxadiazole compounds with increased stability and binding affinity, BDM31343 and BDM31381 [33,37]. Plasmon surface resonance analysis and cocrystallization with EthR confirmed efficient binding [33,37]. Kinetic studies showed that BDM31343 and BDM31381 inhibit EthR binding with nanomolar and micromolar IC50 values, respectively [33,37]. More importantly, both compounds boosted ETH activity against M. tuberculosis, achieving a 10- and 20-fold reduction of ETH minimum inhibitory concentrations (MICs) [33]. BDM31381 treatment increased the level of ethA mRNA in M. tuberculosis cultures by 35-fold, suggesting highly efficient binding between the compound and EthR, which may be due to a novel hydrogen bond formed with the asparagine 179 residue of EthR [33,40]. Combinations of ETH and either of these compounds were used to treat mice infected with M. tuberculosis. Compared with mice treated with ETH alone, the BDM31381-ETH combination exhibited moderate decreases in bacterial loads in the lungs after 3 weeks of treatment, whereas the BDM31343-ETH combination tripled the effect of ETH, indicating effective potentiating activity [33,40]. To further optimize EthR inhibition, a series of modifications of the heterocycles of this compound were instilled and tested for EthR binding and ETH potentiation in vitro and in vivo. This process finally yielded compound BDM41906, which showed improved stability in mice [45]. This compound potentiated ETH activity tenfold against intracellular M. tuberculosis [45]. While ETH potentiation in mice will still have to be determined, stability in mice livers indicated that this compound may be a promising, efficient potentiator of ETH [45].
Antifolates
Cells need folate as a vitamin. This is because folate species are essential cofactors in a wide variety of one-carbon transfer reactions that are involved in important cellular processes, such as the synthesis of purines, thymidine, pantothenate and amino acids (Figure 3A) [15,46–48]. Folate deficiency results in suspension of key metabolic pathways; DNA and protein synthesis, methylations, homocysteine homeostatic control and others, leading to an arrest of cell division and eventually cell death. Folate molecules are characterized by a two-ring pteridine group to which para-aminobenzoic acid (pABA) and one or more glutamate residues are fused sequentially during tetrahydrofolate (H4PteGlu) biosynthesis (Figure 3). Cellular tetrahydrofolate species differ by one-carbon groups attached at positions N5 and/or N10 (Figure 3B).
Figure 3. Folate metabolism and antagonism in mycobacteria.

(A) Simplified interconversions of folate derivatives in de novo folate synthesis and one-carbon metabolic network. DHPS and DHFR are inhibited by current antifolates. (B) Chemical structure of monoglutamylated tetrahydrofolate and its derivatives carrying one-carbon groups at various levels of oxidation attached to N10 and/or N5.
DHFR: Dihydrofolate reductase; DHFS: Dihydrofolate synthase; DHPS: Dihydropteroate synthase; ETH: Ethionamide; INH: Isoniazid; MS: Methionine synthase; MTHFS: 5,10-methenyltetrahydrofolate synthase; pABA: Para-aminobenzoic acid; SHMT: Serine hydroxymethyltransferase; TIM: Trimethoprim; TS: Thymidylate synthase.
Adapted with permission from [15].
Unlike most bacteria and plants, which are able to synthesize folate de novo, mammals lack enzymes for complete de novo folate biosynthesis, and thus have to rely on folate from their diet. Therefore, de novo folate synthesis presents an attractive target for development of antibacterial drugs [49]. Antifolates were the frst chemotherapeutic agents effectively used to treat bacterial infections. They were used extensively between the 1930s and 1960s, after which new antibiotics with lower toxicity and better efficiency replaced them in standard treatment regimens [50,51]. Nevertheless, antifolate combinations such as co-trimoxazole (sulfamethoxazole plus trimethoprim) are still commonly used to treat many bacterial infections, including urinary tract infection and those caused by Shigella, Pneumococcus and Staphylococcus [8,51–53]. While reactions involved in folate metabolism are well known, currently only two steps of the de novo synthesis are targeted by antibacterial antifolates. Dihydrofolate reductase (DHFR) is inhibited by trimethoprim, while dihydropteroate synthase (DHPS) is targeted by sulfonamides and sulfones that outcompete pABA in the condensation with the pteridine moiety (Figure 3A) [49,54,55].
Antifolates as antimycobacterial drugs
Antifolates were regarded as promising anti-TB drugs during the 1930s. Indeed, early experiments using animal models proved that sulfonamides are effective in curing TB [8]. However, the 1943 discovery of streptomycin, which is less toxic and more effective against TB, brought an end to these efforts [8]. These drugs were then abandoned and no longer considered as useful against M. tuberculosis [8]. However, with the current worldwide epidemic of MDR and XDR TB, the search for alternative treatment strategies has re-awakened interest in this group of forgotten compounds [15]. In fact, the sulfone drug dapsone has been used to treat leprosy for many decades, and interestingly, some of the current frontline TB drugs may target enzymes of folate metabolism. Despite its toxicity causing gastrointestinal ailments, para-aminosalicylic acid (PAS) has been commonly used to treat TB [56,102]. Its mechanism of action has remained unknown, but recent work suggested that PAS resistance is mediated through the thymidylate synthase ThyA [57] that catalyzes two chemical conversions; deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), and 5,10-methylene-tetrahydrofolate to dihydrofolate [58]. A lack of dTMP has been known to cause the so-called ‘thymineless death’ [59], whereas the ability to convert 5,10-methylene-tetrahydrofolate back to dihydrofolate might contribute to cellular folate homeostasis (Figure 3A) [57]. This study indicated that PAS inhibits unknown components in folate metabolism. This would further prove that folate metabolism is a valid target for drug development for bacterial infections including TB. It is important to note that M. tuberculosis simultaneously expresses another thymidilate synthase, ThyX, at lower levels (Figure 3A) [60,61]. However, ThyX is both structurally and mechanistically distinct from ThyA [62,63]. Whereas thyX is essential, deletion of thyA confers PAS resistance [60,64]. Besides the thymidylate synthase enzymatic activity, ThyX probably catalyzes other cellular functions that are essential in M. tuberculosis [60,63]. Whether ThyX is involved in the PAS resistance of M. tuberculosis remains to be established.
Interestingly, recent reports suggested that INH, similar to ETH, may not only target the InhA enzyme of mycolate synthesis [65], but also DHFR, thus inhibiting folate biosynthesis in the same step affected by trimethoprim [66,67]. Proteomic studies showed that expression of DHFR in M. tuberculosis is induced by INH exposure [66,67]. Moreover, INH was shown to be able to inhibit DHFR in vitro [66,67]. When the M. tuberculosis DHFR-encoding gene was in trans overexpressed in M. smegmatis, INH resistance was increased by twofold in one study [66]. However, a contrary paper showed that overexpression of M. tuberculosis DHFR in M. tuberculosis or M. smegmatis did not alter INH resistance, while trimethoprim resistance was clearly increased in M. smegmatis [68]. Furthermore, the latter paper showed that no interaction between activated INH and DHFR could be observed in an Escherichia coli cell-based system [68], thus suggesting that DHFR may not be a target for INH in M. tuberculosis. Further work will need to clarify this controversial issue.
Many recent studies provided convincing evidence that sulfonamides could be used for TB treatment [69–72]. Clinical isolates of M. tuberculosis were shown to be commonly susceptible to co-trimoxazole (sulfamethoxazole plus trimethoprim) [69], or even sulfamethoxazole alone [71] at clinically achievable levels. These works clearly confirmed the initial studies done with Promin, a sulfa drug used in the 1930s [8]. In addition, clinical isolates, including MDR strains, collected over 12 years in Taiwan all exhibited sensitivity to sulfamethoxazole without acquisition of resistance [70]. Interestingly, a case report of an 81-year-old patient described treatment with co-trimoxazole without the knowledge of his TB condition [69]; over 2.5 weeks of treatment, his condition improved steadily. Although the treatment was switched to a standard TB regimen and co-trimoxazole was discontinued after TB was found, the symptomatic improvement that resulted from the 2.5 weeks of co-trimoxazole treatment indicates a curing effect of antifolates. This was supported by the fact that the M. tuberculosis strain isolated from the patient was found susceptible to co-trimoxazole in vitro [69].
New sulfonamide potentiators are needed for M. tuberculosis
The antifolate combination of co-trimoxazole is based on the idea of potentiation. Sulfamethoxazole, which targets DHPS in de novo folate biosynthesis, is synergized by trimethoprim that inhibits DHFR required for the reduction of dihydrofolate (Figure 3A) [49,51,54]. Trimethoprim is the only sulfonamide potentiator currently available. In addition, potentiation of sulfonamides by trimethoprim does not work for some bacteria, including M. tuberculosis for unknown reasons. Recent studies have revisited the synergy of trimethoprim–sulfonamides in M. tuberculosis. While Forgacs et al. showed that co-trimoxazole is effective against M. tuberculosis, two other studies indicated that the anti-mycobacterial activity of co-trimoxazole is solely attributable to sulfamethoxazole [69–71].
The M. tuberculosis genome clearly contains a copy of dfrA (rv2763c or folA) [73,74]. It has also been shown in numerous studies that the gene product is a functional DHFR, which can be inhibited by trimethoprim in vitro [66,68,73,75,76]. Therefore, it remains unknown why trimethoprim does not work against M. tuberculosis [71]. MICs of trimethoprim required to inhibit M. tuberculosis exceed 128 μg ml-1, at least 100-fold higher than the MICs of M. smegmatis [77,78]. It is possible that M. tuberculosis is not entirely dependent on DHFR for the reduction of dihydrofolate to tetrahydrofolate. Similar to bacteria lacking genes encoding DHFRs [79], other as-yet-unidentified trimethoprim-resistant reductases may help catalyze the dihydrofolate reduction activity. It is important to note that dfrA was implicated to be essential, and expression of DHFR in M. tuberculosis was detected by mass spectrometric analysis [80,81]. Another possibility is that expression of DHFR in M. tuberculosis vastly exceeds the amount required for its function in folate biosynthesis. This high cellular concentration of DHFR, together with other intrinsic resistance mechanisms, such as cell wall exclusion, efflux pumps or modifying enzymes, neutralize the activity of trimethoprim. In M. smegmatis, up to 97% of the wild-type DHFR expression levels could be depleted with no effect on growth, but the depletion led to trimethoprim hypersusceptibility [82]. Metabolic profiling of DHFR-depleted cells showed a pattern similar to that obtained after exposure of nondepleted M. smegmatis to subinhibitory concentrations of trimethoprim [82]. In vitro kinetic studies also suggested that M. tuberculosis DHFR might have a lower affinity to trimethoprim compared with the homologous enzymes in other bacteria. The IC50 of M. tuberculosis DHFR inhibited by trimethoprim is approximately 100-fold higher than E. coli DHFR, and its ki is 20-fold higher than that of the M. smegmatis enzyme [74,78,83,84].
The lack of effective sulfonamide potentiators poses a potential risk for rapid emergence of resistant strains. In fact, it has been shown in other bacteria commonly treated with sulfonamides that a single point mutation at conserved residues corresponding to serine 53 or proline 55 of M. tuberculosis DHPS confers complete sulfonamide resistance [85,86]. Resistant strains of Mycobacterium leprae with these mutations have been isolated, indicating the possibility that M. tuberculosis may rapidly acquire them as well [85,86]. Since the WHO has issued recommendations of using co-trimoxazole for prophylactic treatment of AIDS patients [87], who are often coinfected with M. tuberculosis [88], sulfonamide resistance in M. tuberculosis may arise even before they can be widely used for TB treatment [89]. Clearly, it is critical to identify alternative sulfonamide potentiators that would allow us to not only prolong the lifespan of these valuable drugs against M. tuberculosis, but also to lower treatment doses, thus reducing side effects.
As the structure of M. tuberculosis DHFR, with and without various inhibitors, has been obtained and compared with human DHFR, a wealth of information for targeted design of DHFR inhibitors is available [66,73]. Indeed, several efforts to develop inhibitors of M. tuberculosis DHFR have been reported [74,75,76,78,90,91]. Derivatives of pyrimidine-2,4-diamines designed from crystal structure information displayed improved inhibition of M. tuberculosis DHFR expressed in Saccharomyces cerevisiae compared with trimethoprim, but showed low activity towards the human DHFR [76]. Several lipophilic deazapteridine compounds also showed improved in vitro MICs (10- to 100-fold lower) compared with trimethoprim, while displaying low affinity for mammalian DHFR [78,91]. While such studies are encouraging, many of them were done outside the M. tuberculosis cell, therefore excluding M. tuberculosis specific resistance mechanisms as well as the issue of excessive DHFR expression.
Future directions in sulfonamide potentiator development
Given the possibility that DHFR might not be the major reductase responsible for the reduction of dihydrofolate to tetrahydrofolate, future studies should address the identity of the unknown reductases responsible for this essential reaction, followed by the development of inhibitors and testing their sulfonamides-potentiating activity against M. tuberculosis. A recent study of whole-genome antifolate-resistance determinants in mycobacteria may help to identify these alternate DHFRs [92]. Besides targeting DHFR, this work could help to identify nontraditional targets for sulfonamide potentiation in M. tuberculosis [92]. Fifty antifolate-resistance determinants have been identified thus far, and many of these determinants proved to be responsible for the high intrinsic sulfonamide-resistance in mycobacteria including M. tuberculosis [Unpublished Data]. A recent paper reported the role of 5,10-methenyltetrahydrofolate synthase (MTHFS) as an enzyme required for the utilization of storage form of folate, called 5-formyltetrahydrofolate [92]. It was proposed that bacteria including M. tuberculosis use this storage form to survive during the limited new synthesis caused by classical antifolates. Lack of MTHFS results in 64-fold increased susceptibility to sulfonamides–trimethoprim combinations [92]. Inhibitors of MTHFS could, therefore, block the conversion of 5-formyltetrahydrofolate to metabolically active folate species, thereby rapidly exhausting cellular folate species required for metabolically essential reactions.
Finally, as the current frontline TB drugs INH, ETH and PAS were shown as potential inhibitors of the folate biosynthetic pathway [66–68], they should be tested in combinations with sulfonamides to determine potentiation effects. These combinations, in which effective doses and side effects of sulfonamides are reduced, may quickly find their way into clinical trials because they are all already-approved drugs.
Conclusion
With the current crisis of the drug-resistant TB epidemic, we need to focus not only on the development of new drugs, but also on the systematic re-evaluation and repurposing of available drugs that have been thus far disregarded for clinical trials and possible optimization. Among these forgotten drugs, ETH and classical antifolates are emerging as promising candidates for pharmaceutical potentiation. They have been shown to be active against M. tuberculosis, and strategies are already in place for further development of effective potentiators. While much remains to be done in terms of compound optimization and clinical trials, many compounds have been identified as promising leads. Studies searching for potential methods to reapply these drugs to the clinic should be encouraged and supported. Potentiators of ETH and antifolates would help to boost the efficacy of these drugs, thus allowing for lower doses and reduction of side effects. This would provide a safer and more effective implementation of these drugs in TB treatment.
Expert commentary
Many old drugs such as ETH and antifolates have been vastly underused or even abandoned, as there is a lack of effective methods for enhancing efficacy. We must reverse our old way of thinking and attempt to make use of the potential held by these compounds. By developing inhibitors to known resistance mechanisms employed by M. tuberculosis, it is possible to circumvent the bacterial defense mechanisms to both increase efficacy of existing drugs and lower cytotoxicity. While EthR inhibition proved to be a valid strategy for ETH potentiation, chemical optimization to increase both target binding affinity and bioavailability must be the foremost focus in this area. Concerning antifolates, research must clarify which targets are most promising for potentiation. DHFR might not be an effective target given differential outcomes observed with trimethoprim activity in vitro and in whole-cell assays. Alternatively, efforts could be focused on the development of inhibitors to other steps in both de novo folate biosynthesis and one-carbon metabolic reactions. Development of novel cocktails composed of sulfonamides and folate-targeting TB drugs such as INH, ETH and PAS might hold exciting future discoveries.
Five-year view
Much work has been done laying important groundwork that establishes ETH and antifolates as potential old drugs to be recharged against M. tuberculosis, but equally much remains to be done to implement the potentiation strategies for these drugs into TB therapies. The next 5 years will probably see EthR inhibitors moving into animal experiments and clinical trials. Reliability of DHFR as a valid target for development of sulfonamide potentiators should be investigated. At the same time, alternative targets should be identified and their potential investigated. This area of research could bring important breakthroughs to the implementation of sulfonamides into the treatment of both drug-resistant and drug-susceptible TB. Novel regimens in which PAS, ETH or INH are used to synergize sulfonamides might be explored to shorten the current TB treatment courses. Finally, further testing of candidates from the antifolate-resistance determinant screen and design of inhibitors against good candidates will be focused on in the next 5 years.
Key issues.
Potentiation uses knowledge of resistance mechanisms to develop inhibitors to resistance mechanisms in order to (re)sensitize bacteria to available drugs.
Due to the severely limited arsenal of drugs effective against multiple drug-resistant and extensively drug resistant tuberculosis (TB), old drugs need to be repurposed for the treatment of these infections.
Ethionamide (ETH) is effective against TB; but due to its high toxicity, it is only used as a second-line drug. Potentiation could lower required ETH concentrations and decrease toxic side effects.
EthR represses activation of the prodrug ETH, and inhibitors to EthR potentiate ETH by increasing levels of active drug through increased expression of EthA.
EthR inhibitors show ETH potentiation in vitro, but only a few have been tested with varying success in mice. Further testing in animal models and patients is required.
Classical antifolates were considered effective against Mycobacterium tuberculosis in animal models, and are used to treat mycobacterial infections such as leprosy, but were disregarded due to the emergence of more effective, less toxic anti-TB drugs.
Sulfonamides have been shown to be useful against multiple strains of drug resistant and extensively drug resistant TB, but the only available potentiator, trimethoprim, does not have a synergistic effect against M. tuberculosis.
M. tuberculosis dihydrofolate reductase may not be a good target for development of sulfonamide potentiators.
Other antifolate resistance determinants could provide effective targets for sulfonamide potentiators.
Acknowledgments
The authors thank Kien Nguyen and Henry Boom for critical reading and comments on the manuscript.
Work in the Nguyen laboratory is supported by the US NIH (R01AI087903) and a STERIS Infectious Diseases Research Support Grant.
Footnotes
Financial & competing interests disclosure: The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
•• of considerable interest
- 1.Nguyen L, Thompson CJ. Foundations of antibiotic resistance in bacterial physiology: the mycobacterial paradigm. Trends Microbiol. 2006;14(7):304–312. doi: 10.1016/j.tim.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen L, Pieters J. Mycobacterial subversion of chemotherapeutic reagents and host defense tactics: challenges in tuberculosis drug development. Annu Rev Pharmacol Toxicol. 2009;49:427–453. doi: 10.1146/annurev-pharmtox-061008-103123. [DOI] [PubMed] [Google Scholar]
- 3.Dye C. Tuberculosis 2000–2010: control, but not elimination. Int J Tuberculosis Lung Dis. 2000;4(12 Suppl. 2):S146–S152. [PubMed] [Google Scholar]
- 4.Gandhi NR, Moll A, Sturm AW, et al. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. 2006;368(9547):1575–1580. doi: 10.1016/S0140-6736(06)69573-1. [DOI] [PubMed] [Google Scholar]
- 5.Jassal M, Bishai WR. Extensively drug-resistant tuberculosis. Lancet Infect Dis. 2009;9(1):19–30. doi: 10.1016/S1473-3099(08)70260-3. [DOI] [PubMed] [Google Scholar]
- 6.LoBue P. Extensively drug-resistant tuberculosis. Curr Opin Infect Dis. 2009;22(2):167–173. doi: 10.1097/qco.0b013e3283229fab. [DOI] [PubMed] [Google Scholar]
- 7.Andronis C, Sharma A, Virvilis V, Deftereos S, Persidis A. Literature mining, ontologies and information visualization for drug repurposing. Brief Bioinformatics. 2011;12(4):357–368. doi: 10.1093/bib/bbr005. [DOI] [PubMed] [Google Scholar]
- 8.Barr J. A short history of dapsone, or an alternative model of drug development. J Hist Med Allied Sci. 2011;66(4):425–467. doi: 10.1093/jhmas/jrq068. [DOI] [PubMed] [Google Scholar]
- 9.Global Alliance for TB Drug Development. Handbook of anti-tuberculosis agents. Tuberculosis. 2008;88(2):85–170. doi: 10.1016/S1472-9792(08)70002-7. [DOI] [PubMed] [Google Scholar]
- 10.Bolten BM, DeGregorio T. From the analyst's couch. Trends in development cycles. Nat Rev Drug Discov. 2002;1(5):335–336. doi: 10.1038/nrd805. [DOI] [PubMed] [Google Scholar]
- 11.Deftereos SN, Andronis C, Friedla EJ, Persidis A, Persidis A. Drug repurposing and adverse event prediction using high-throughput literature analysis. Wiley Interdiscip Rev Syst Biol Med. 2011;3(3):323–334. doi: 10.1002/wsbm.147. [DOI] [PubMed] [Google Scholar]
- 12.Burns M. Management of narrow therapeutic index drugs. J Thromb Thrombolysis. 1999;7(2):137–143. doi: 10.1023/a:1008829403320. [DOI] [PubMed] [Google Scholar]
- 13.Sood A, Panchagnula R. Design of controlled release delivery systems using a modified pharmacokinetic approach: a case study for drugs having a short elimination half-life and a narrow therapeutic index. Int J Pharm. 2003;261(1–2):27–41. doi: 10.1016/s0378-5173(03)00267-9. [DOI] [PubMed] [Google Scholar]
- 14.Zimmerman TJ, Kooner KS, Kandarakis AS, Ziegler LP. Improving the therapeutic index of topically applied ocular drugs. Arch Ophthalmol. 1984;102(4):551–553. doi: 10.1001/archopht.1984.01040030429017. [DOI] [PubMed] [Google Scholar]
- 15.Wolff KA, Sherman MB, Nguyen L. Potentiation of available antibiotics by targeting resistance – an emerging trend in tuberculosis drug development. In: Rundfeldt C, editor. Drug Development – A Case Study Based Insight into Modern Strategies. InTech; NY, USA: 2011. [Google Scholar]
- 16.Drawz SM, Bonomo RA. Three decades of beta-lactamase inhibitors. Clin Microbiol Rev. 2010;23(1):160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright GD. Resisting resistance: new chemical strategies for battling superbugs. Chem Biol. 2000;7(6):R127–R132. doi: 10.1016/s1074-5521(00)00126-5. [DOI] [PubMed] [Google Scholar]
- 18.Wright GD, Sutherland AD. New strategies for combating multidrug-resistant bacteria. Trends Mol Med. 2007;13(6):260–267. doi: 10.1016/j.molmed.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Brossier F, Veziris N, Truffot-Pernot C, Jarlier V, Sougakoff W. Molecular investigation of resistance to the antituberculous drug ethionamide in multidrug-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2011;55(1):355–360. doi: 10.1128/AAC.01030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE., 3rd Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2000;97(17):9677–9682. doi: 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21••.Frénois F, Engohang-Ndong J, Locht C, Baulard AR, Villeret V. Structure of EthR in a ligand bound conformation reveals therapeutic perspectives against tuberculosis. Mol Cell. 2004;16(2):301–307. doi: 10.1016/j.molcel.2004.09.020. First crystal structure of EthR with ligand provides insights and leads for EthR inhibitor development. [DOI] [PubMed] [Google Scholar]
- 22.Morlock GP, Metchock B, Sikes D, Crawford JT, Cooksey RC. ethA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2003;47(12):3799–3805. doi: 10.1128/AAC.47.12.3799-3805.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F, Langley R, Gulten G, et al. Mechanism of thioamide drug action against tuberculosis and leprosy. J Exp Med. 2007;204(1):73–78. doi: 10.1084/jem.20062100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vannelli TA, Dykman A, Ortiz de Montellano PR. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J Biol Chem. 2002;277(15):12824–12829. doi: 10.1074/jbc.M110751200. [DOI] [PubMed] [Google Scholar]
- 25.Vilchèze C, Av-Gay Y, Attarian R, et al. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol Microbiol. 2008;69(5):1316–1329. doi: 10.1111/j.1365-2958.2008.06365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y. The magic bullets and tuberculosis drug targets. Annu Rev Pharmacol Toxicol. 2005;45:529–564. doi: 10.1146/annurev.pharmtox.45.120403.100120. [DOI] [PubMed] [Google Scholar]
- 27.Fraaije MW, Kamerbeek NM, Heidekamp AJ, Fortin R, Janssen DB. The prodrug activator EtaA from Mycobacterium tuberculosis is a Baeyer-Villiger monooxygenase. J Biol Chem. 2004;279(5):3354–3360. doi: 10.1074/jbc.M307770200. [DOI] [PubMed] [Google Scholar]
- 28••.Baulard AR, Betts JC, Engohang-Ndong J, et al. Activation of the pro-drug ethionamide is regulated in mycobacteria. J Biol Chem. 2000;275(36):28326–28331. doi: 10.1074/jbc.M003744200. First report showing EthR regulation of EthA expression and ethionamide activation. [DOI] [PubMed] [Google Scholar]
- 29.Schaaf HS, Victor TC, Venter A, et al. Ethionamide cross- and co-resistance in children with isoniazid-resistant tuberculosis. Int J Tuberc Lung Dis. 2009;13(11):1355–1359. [PubMed] [Google Scholar]
- 30.Zhang Y, Vilcheze C, Jacobs WR., Jr . Mechanisms of drug resistance in Mycobacterium tuberculosis. In: Cole ST, Eisenach K, Mcmurray D, Jacobs WR Jr, editors. Tuberculosis and the Tubercle Bacillus. ASM Press; DC, USA: 2005. pp. 115–140. [Google Scholar]
- 31.Barry CE, 3rd, Slayden RA, Mdluli K. Mechanisms of isoniazid resistance in Mycobacterium tuberculosis. Drug Resist Updat. 1998;1(2):128–134. doi: 10.1016/s1368-7646(98)80028-9. [DOI] [PubMed] [Google Scholar]
- 32.Francois AA, Nishida CR, de Montellano PR, Phillips IR, Shephard EA. Human flavin-containing monooxygenase 2.1 catalyzes oxygenation of the antitubercular drugs thiacetazone and ethionamide. Drug Metab Dispos. 2009;37(1):178–186. doi: 10.1124/dmd.108.024158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flipo M, Desroses M, Lecat-Guillet N, et al. Ethionamide boosters: synthesis, biological activity, and structure–activity relationships of a series of 1,2,4-oxadiazole EthR inhibitors. J Med Chem. 2011;54(8):2994–3010. doi: 10.1021/jm200076a. [DOI] [PubMed] [Google Scholar]
- 34.Banerjee A, Dubnau E, Quemard A, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263(5144):227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 35.Vilchèze C, Weisbrod TR, Chen B, et al. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob Agents Chemother. 2005;49(2):708–720. doi: 10.1128/AAC.49.2.708-720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu X, Vilchèze C, Av-Gay Y, Gómez-Velasco A, Jacobs WR., Jr Precise null deletion mutations of the mycothiol synthesis genes reveal their role in isoniazid and ethionamide resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother. 2011;55(7):3133–3139. doi: 10.1128/AAC.00020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Willand N, Dirié B, Carette X, et al. Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat Med. 2009;15(5):537–544. doi: 10.1038/nm.1950. Development of pharmacore prediction model for synthetic EthR inhibitor screening. [DOI] [PubMed] [Google Scholar]
- 38.Dover LG, Corsino PE, Daniels IR, et al. Crystal structure of the TetR/CamR family repressor Mycobacterium tuberculosis EthR implicated in ethionamide resistance. J Mol Biol. 2004;340(5):1095–1105. doi: 10.1016/j.jmb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Engohang-Ndong J, Baillat D, Aumercier M, et al. EthR, a repressor of the TetR/CamR family implicated in ethionamide resistance in mycobacteria, octamerizes cooperatively on its operator. Mol Microbiol. 2004;51(1):175–188. doi: 10.1046/j.1365-2958.2003.03809.x. [DOI] [PubMed] [Google Scholar]
- 40••.Weber W, Schoenmakers R, Keller B, et al. A synthetic mammalian gene circuit reveals antituberculosis compounds. Proc Natl Acad Sci USA. 2008;105(29):9994–9998. doi: 10.1073/pnas.0800663105. Development of mammalian cell-based reporter assay for EthR inhibitor screening. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frénois F, Baulard AR, Villeret V. Insights into mechanisms of induction and ligands recognition in the transcriptional repressor EthR from Mycobacterium tuberculosis. Tuberculosis (Edinb) 2006;86(2):110–114. doi: 10.1016/j.tube.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Carette X, Blondiaux N, Willery E, et al. Structural activation of the transcriptional repressor EthR from Mycobacterium tuberculosis by single amino acid change mimicking natural and synthetic ligands. Nucleic Acids Res. 2012;40(7):3018–3030. doi: 10.1093/nar/gkr1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen L, Pieters J. The Trojan horse: survival tactics of pathogenic mycobacteria in macrophages. Trends Cell Biol. 2005;15(5):269–276. doi: 10.1016/j.tcb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 44.Grau T, Selchow P, Tigges M, et al. Phenylethyl butyrate enhances the potency of second-line drugs against clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56(2):1142–1145. doi: 10.1128/AAC.05649-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flipo M, Desroses M, Lecat-Guillet N, et al. Ethionamide boosters. 2. Combining bioisosteric replacement and structure-based drug design to solve pharmacokinetic issues in a series of potent 1,2,4-oxadiazole EthR inhibitors. J Med Chem. 2012;55(1):68–83. doi: 10.1021/jm200825u. [DOI] [PubMed] [Google Scholar]
- 46.Blakeley RL. The Biochemistry of Folic Acid and Related Pteridines. John Wiley and Sons, Inc.; NY, USA: 1969. [Google Scholar]
- 47.Green J, Nichols B, Matthews R. Folate biosynthesis, reduction, and polyglutamylation. In: Neidhardt F, Curtiss Iii R, Ingraham J, et al., editors. Escherichia coli and Salmonella typhimurium - Cellular and Molecular Biology. ASM Press; DC, USA: 1996. pp. 665–673. [Google Scholar]
- 48.Selhub J. Folate, vitamin B12 and vitamin B6 and one carbon metabolism. J Nutr Health Aging. 2002;6(1):39–42. [PubMed] [Google Scholar]
- 49.Bermingham A, Derrick JP. The folic acid biosynthesis pathway in bacteria: evaluation of potential for antibacterial drug discovery. Bioessays. 2002;24(7):637–648. doi: 10.1002/bies.10114. [DOI] [PubMed] [Google Scholar]
- 50.Bertino JR. Folate Antagonists as Chemotherapeutic Agents. New York Academy of Sciences; NY, USA: 1971. New York Academy of Sciences. Section of Biological and Medical Sciences. [Google Scholar]
- 51.Libecco JA, Powell KR. Trimethoprim/sulfamethoxazole: clinical update. Pediatr Rev. 2004;25(11):375–380. [PubMed] [Google Scholar]
- 52.Doull JA. Sulfone therapy of leprosy Background, early history and present status. Int J Lepr. 1963;31:143–160. [PubMed] [Google Scholar]
- 53.Proctor RA. Role of folate antagonists in the treatment of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46(4):584–593. doi: 10.1086/525536. [DOI] [PubMed] [Google Scholar]
- 54.Gangjee A, Jain HD, Kurup S. Recent advances in classical and non-classical antifolates as antitumor and antiopportunistic infection agents: part I. Anticancer Agents Med Chem. 2007;7(5):524–542. doi: 10.2174/187152007781668724. [DOI] [PubMed] [Google Scholar]
- 55.Gangjee A, Jain HD, Kurup S. Recent advances in classical and non-classical antifolates as antitumor and antiopportunistic infection agents: part II. Anticancer Agents Med Chem. 2008;8(2):205–231. doi: 10.2174/187152008783497064. [DOI] [PubMed] [Google Scholar]
- 56.Caminero JA, Sotgiu G, Zumla A, Migliori GB. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect Dis. 2010;10(9):621–629. doi: 10.1016/S1473-3099(10)70139-0. [DOI] [PubMed] [Google Scholar]
- 57.Rengarajan J, Sassetti CM, Naroditskaya V, Sloutsky A, Bloom BR, Rubin EJ. The folate pathway is a target for resistance to the drug para-aminosalicylic acid (PAS) in mycobacteria. Mol Microbiol. 2004;53(1):275–282. doi: 10.1111/j.1365-2958.2004.04120.x. [DOI] [PubMed] [Google Scholar]
- 58.Hardy LW, Finer-Moore JS, Montfort WR, Jones MO, Santi DV, Stroud RM. Atomic structure of thymidylate synthase: target for rational drug design. Science. 1987;235(4787):448–455. doi: 10.1126/science.3099389. [DOI] [PubMed] [Google Scholar]
- 59.Kwon YK, Higgins MB, Rabinowitz JD. Antifolate-induced depletion of intracellular glycine and purines inhibits thymineless death in E. coli. ACS Chem Biol. 2010;5(8):787–795. doi: 10.1021/cb100096f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fivian-Hughes AS, Houghton J, Davis EO. Mycobacterium tuberculosis thymidylate synthase gene thyX is essential and potentially bifunctional, while thyA deletion confers resistance to p-aminosalicylic acid. Microbiology (Reading, Engl) 2012;158(Pt 2):308–318. doi: 10.1099/mic.0.053983-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sampathkumar P, Turley S, Ulmer JE, Rhie HG, Sibley CH, Hol WG. Structure of the Mycobacterium tuberculosis flavin dependent thymidylate synthase (MtbThyX) at 2.0A resolution. J Mol Biol. 2005;352(5):1091–1104. doi: 10.1016/j.jmb.2005.07.071. [DOI] [PubMed] [Google Scholar]
- 62.Koehn EM, Fleischmann T, Conrad JA, et al. An unusual mechanism of thymidylate biosynthesis in organisms containing the thyX gene. Nature. 2009;458(7240):919–923. doi: 10.1038/nature07973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hunter JH, Gujjar R, Pang CK, Rathod PK. Kinetics and ligand-binding preferences of Mycobacterium tuberculosis thymidylate synthases, ThyA and ThyX. PLoS ONE. 2008;3(5):e2237. doi: 10.1371/journal.pone.0002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mathys V, Wintjens R, Lefevre P, et al. Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2009;53(5):2100–2109. doi: 10.1128/AAC.01197-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subba Rao G, Vijayakrishnan R, Kumar M. Structure-based design of a novel class of potent inhibitors of InhA, the enoyl acyl carrier protein reductase from Mycobacterium tuberculosis: a computer modelling approach. Chem Biol Drug Des. 2008;72(5):444–449. doi: 10.1111/j.1747-0285.2008.00722.x. [DOI] [PubMed] [Google Scholar]
- 66••.Argyrou A, Vetting MW, Aladegbami B, Blanchard JS. Mycobacterium tuberculosis dihydrofolate reductase is a target for isoniazid. Nat Struct Mol Biol. 2006;13(5):408–413. doi: 10.1038/nsmb1089. First report indicating that the first-line tuberculosis drug isoniazid may target folate biosynthesis. [DOI] [PubMed] [Google Scholar]
- 67.Argyrou A, Jin L, Siconilfi-Baez L, Angeletti RH, Blanchard JS. Proteome-wide profiling of isoniazid targets in Mycobacterium tuberculosis. Biochemistry. 2006;45(47):13,947–13953. doi: 10.1021/bi061874m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang F, Jain P, Gulten G, et al. Mycobacterium tuberculosis dihydrofolate reductase is not a target relevant to the antitubercular activity of isoniazid. Antimicrob Agents Chemother. 2010;54(9):3776–3782. doi: 10.1128/AAC.00453-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69••.Forgacs P, Wengenack NL, Hall L, Zimmerman SK, Silverman ML, Roberts GD. Tuberculosis and trimethoprim-sulfamethoxazole. Antimicrob Agents Chemother. 2009;53(11):4789–4793. doi: 10.1128/AAC.01658-08. First report indicating usefulness of classical antifolates for treatment of multiple drug-resistant and extensively drug-resistant TB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang TS, Kunin CM, Yan BS, Chen YS, Lee SS, Syu W., Jr Susceptibility of Mycobacterium tuberculosis to sulfamethoxazole, trimethoprim and their combination over a 12 year period in Taiwan. J Antimicrob Chemother. 2012;67(3):633–637. doi: 10.1093/jac/dkr501. [DOI] [PubMed] [Google Scholar]
- 71.Ong W, Sievers A, Leslie DE. Mycobacterium tuberculosis and sulfamethoxazole susceptibility. Antimicrob Agents Chemother. 2010;54(6):2748. doi: 10.1128/AAC.00029-10. author reply 2748; author reply 2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Young LS. Reconsidering some approved antimicrobial agents for tuberculosis. Antimicrob Agents Chemother. 2009;53(11):4577–4579. doi: 10.1128/AAC.00887-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li R, Sirawaraporn R, Chitnumsub P, et al. Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J Mol Biol. 2000;295(2):307–323. doi: 10.1006/jmbi.1999.3328. [DOI] [PubMed] [Google Scholar]
- 74.Sirawaraporn W, Sirawaraporn R, Chanpongsri A, Jacobs WR, Jr, Santi DV. Purification and characterization of dihydrofolate reductase from wild-type and trimethoprim-resistant Mycobacterium smegmatis. Exp Parasitol. 1991;72(2):184–190. doi: 10.1016/0014-4894(91)90136-k. [DOI] [PubMed] [Google Scholar]
- 75.Kumar M, Vijayakrishnan R, Subba Rao G. In silico structure-based design of a novel class of potent and selective small peptide inhibitor of Mycobacterium tuberculosis dihydrofolate reductase, a potential target for anti-TB drug discovery. Mol Divers. 2010;14(3):595–604. doi: 10.1007/s11030-009-9172-6. [DOI] [PubMed] [Google Scholar]
- 76.El-Hamamsy MH, Smith AW, Thompson AS, Threadgill MD. Structure-based design, synthesis and preliminary evaluation of selective inhibitors of dihydrofolate reductase from Mycobacterium tuberculosis. Bioorg Med Chem. 2007;15(13):4552–4576. doi: 10.1016/j.bmc.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 77.Malik R, Wigney DI, Dawson D, Martin P, Hunt GB, Love DN. Infection of the subcutis and skin of cats with rapidly growing mycobacteria: a review of microbiological and clinical findings. J Feline Med Surg. 2000;2(1):35–48. doi: 10.1053/jfms.2000.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suling WJ, Reynolds RC, Barrow EW, Wilson LN, Piper JR, Barrow WW. Susceptibilities of Mycobacterium tuberculosis and Mycobacterium avium complex to lipophilic deazapteridine derivatives, inhibitors of dihydrofolate reductase. J Antimicrob Chemother. 1998;42(6):811–815. doi: 10.1093/jac/42.6.811. [DOI] [PubMed] [Google Scholar]
- 79.Myllykallio H, Leduc D, Filee J, Liebl U. Life without dihydrofolate reductase FolA. Trends Microbiol. 2003;11(5):220–223. doi: 10.1016/s0966-842x(03)00101-x. [DOI] [PubMed] [Google Scholar]
- 80.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7(9):e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Souza GA, Leversen NA, Målen H, Wiker HG. Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J Proteomics. 2011;75(2):502–510. doi: 10.1016/j.jprot.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 82••.Wei JR, Krishnamoorthy V, Murphy K, et al. Depletion of antibiotic targets has widely varying effects on growth. Proc Natl Acad Sci USA. 2011;108(10):4176–4181. doi: 10.1073/pnas.1018301108. Excessive expression makes dihydrofolate reductase a less attractive target for development of sulfonamide potentiators against Mycobacterium tuberculosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nelson RG, Rosowsky A. Dicyclic and tricyclic diaminopyrimidine derivatives as potent inhibitors of Cryptosporidium parvum dihydrofolate reductase: structure–activity and structure–selectivity correlations. Antimicrob Agents Chemother. 2001;45(12):3293–3303. doi: 10.1128/AAC.45.12.3293-3303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.White EL, Ross LJ, Cunningham A, Escuyer V. Cloning, expression, and characterization of Mycobacterium tuberculosis dihydrofolate reductase. FEMS Microbiol Lett. 2004;232(1):101–105. doi: 10.1016/S0378-1097(04)00038-2. [DOI] [PubMed] [Google Scholar]
- 85.Baca AM, Sirawaraporn R, Turley S, Sirawaraporn W, Hol WG. Crystal structure of Mycobacterium tuberculosis 7,8-dihydropteroate synthase in complex with pterin monophosphate: new insight into the enzymatic mechanism and sulfa-drug action. J Mol Biol. 2000;302(5):1193–1212. doi: 10.1006/jmbi.2000.4094. [DOI] [PubMed] [Google Scholar]
- 86.Kai M, Matsuoka M, Nakata N, et al. Diaminodiphenylsulfone resistance of Mycobacterium leprae due to mutations in the dihydropteroate synthase gene. FEMS Microbiol Lett. 1999;177(2):231–235. doi: 10.1111/j.1574-6968.1999.tb13737.x. [DOI] [PubMed] [Google Scholar]
- 87.Date AA, Vitoria M, Granich R, Banda M, Fox MY, Gilks C. Implementation of co-trimoxazole prophylaxis and isoniazid preventive therapy for people living with HIV. Bull World Health Organ. 2010;88(4):253–259. doi: 10.2471/BLT.09.066522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.WHO. Tuberculosis Fact Sheet 2011. 2011 Feb 26; 2010. [Google Scholar]
- 89.Vinetz JM. Intermittent preventive treatment for malaria in sub-Saharan African: a halfway technology or a critical intervention? Am J Trop Med Hyg. 2010;82(5):755–756. doi: 10.4269/ajtmh.2010.825editorial. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reynolds RC, Campbell SR, Fairchild RG, et al. Novel boron-containing, nonclassical antifolates: synthesis and preliminary biological and structural evaluation. J Med Chem. 2007;50(14):3283–3289. doi: 10.1021/jm0701977. [DOI] [PubMed] [Google Scholar]
- 91.Suling WJ, Maddry JA. Antimycobacterial activity of 1-deaza-7,8-dihydropteridine derivatives against Mycobacterium tuberculosis and Mycobacterium avium complex in vitro. J Antimicrob Chemother. 2001;47(4):451–454. doi: 10.1093/jac/47.4.451. [DOI] [PubMed] [Google Scholar]
- 92.Ogwang S, Nguyen HT, Sherman M, et al. Bacterial conversion of folinic acid is required for antifolate resistance. J Biol Chem. 2011;286(17):15377–15390. doi: 10.1074/jbc.M111.231076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 101.WHO. Frequently asked questions – XDR-TB. www.who.int/tb/challenges/xdr/faqs/en/index.html.
- 102.WHO. Guidelines for the programmatic management of drug-resistant tuberculosis. http://whqlibdoc.who.int/publications/2008/9789241547581_eng.pdf.