

Abstract
5β-Reduced steroids are non-planar steroids that have 90° bend in their structure to create an A/B cis-ring junction. This novel property is required for bile-acids to act as emulsifiers, but in addition 5β-reduced steroids have remarkable physiology and may act as potent tocolytic agents, endogenous cardiac glycosides, neurosteroids, and can act as ligands for orphan and membrane bound receptors. In humans there is only a single 5β-reductase gene AKR1D1, which encodes Δ4-3-ketosteroid-5β-reductase (AKR1D1). This enzyme is a member of the aldoketo reductase superfamily, but possesses an altered catalytic tetrad, in which Glu120 replaces the conserved His residue. This predominant liver enzyme generates all 5β-dihydrosteroids in the C19–C27 steroid series. Mutations exist in the AKR1D1 gene, which result in loss of protein stability and are causative in bile-acid deficiency.
Keywords: bile acid biosynthesis, steroid metabolism, enzyme mechanism, genetics
1. Introduction
The C-4/C-5 double bond at the A-ring of Δ4-3-ketosteroids is a characteristic structure of nearly all steroid hormones. This double bond can be saturated by either 5α- or 5β-reduction catalyzed by steroid 5α-reductases (SRD5A1-SRD5A3) or by steroid 5β-reductases (e.g. AKR1D1) [1]. 5α-Reduction generates planar steroids with an A/B trans ring-junction. This reaction produces the most potent androgen 5α-dihydrotestosterone and is essential for the development and regulation of male secondary sex-characteristics and the normal growth of the prostate [2]. By contrast, 5β-reduction introduces a 90° bend at the A/B ring junction forming a cis-configuration. In vertebrates, the most important and well-studied 5β-reduced steroids are bile acids. Bile acids are powerful emulsifying agents that facilitate transportation and absorption of fat and fat-soluble vitamins. Bile acid biosynthesis also serves as a major pathway for cholesterol metabolism [3].
In addition to being an essential step in bile acid biosynthesis, 5β-reduction inactivates steroid hormones and initiates steroid hormone clearance. However, 5β-reduction may also be involved in the pre-receptor regulation of nuclear receptors by controlling ligand availability. For example, 5β-reduced steroids act as ligands for orphan nuclear receptors, the farnesoid X receptor (FXR) and the pregnane X receptor (PXR). 5β-Reduction also generates neuroactive 5β-pregnanes that modulate the activities of the GABAA and NMDA receptors. 5β-Reduced steroids play important roles in other species. In plants, 5β-reduction produces cardiac glycosides as an important defense mechanism against herbivores [4]. Cardiac glycosides such as digoxin obtained from Digitalis lanata are the mainstay therapy for congestive heart failure. In insects, 5β-reduction leads to the production of ecdysone and 20-hydroxyecdysone, which are essential for metamorphsis [5].
Steroid 5β-reductases in vertebrates and plants have been identified and characterized. Vertebrate 5β-reductases belong to the aldo-keto reductase (AKR) superfamily, 1D subfamily [1]. The plant 5β-reductase belongs to the short chain dehydrogenase/reductase family [6]. Even though both enzymes perform the same function and utilize NADPH as cofactor, the two enzyme families have evolved from different ancestors and do not share sequence or structure homology. Insect 5β-reductase has not been identified as yet. In this review, we focus on the function of 5β-steroids in humans and human 5β-reductase AKR1D1. As will be seen below 5β-reduced steroids are not inactive steroid metabolites and have their own unique physiology and pharmacology.
2. Functions of 5β-steroids
Bile acids
Bile acids are the most abundant 5β-reduced steroids. About 500 mg of cholesterol is converted to bile acids in adult human liver each day (~2 g total bile acid pool) [3, 7]. Bile acids solubilize dietary cholesterol, lipids, and fat soluble vitamins (A, D, E, and K) by forming mixed micelles and facilitate absorption of nutrients [8]. Compared to the 5α-reduced allo-bile acids, the 5β-configuration augments the facial amphipathicity of bile acids by providing a larger surface area of the hydrophobic β-face. This is greatly favored for micelle formation and makes the 5β-reduced bile acids superior emulsifiers [9, 10]. The mixed micelles enhance anchorage of pancreatic lipases to the micelle surface, where bile acids also act at activators for lipases to facilitate fat hydrolysis [11–14].
The introduction of the A/B cis-ring configuration into the bile acid structure can be traced back very early in vertebrate evolution, as C27 bile alcohols are found in cartilaginous fish [15]. In human, two primary bile acids are C24 chenodeoxycholic acid and cholic acid (Figure 1), which account for ~70% of total bile acids [16]. The other 30% are composed of secondary bile acids produced mainly by 7α-dehydroxylation of the primary bile acids by intestinal bacteria. Bile acids are synthesized in liver and released into the small intestine in conjugated forms with glycine or taurine [3, 7]. Bile acids are passively or actively reabsorbed in the small intestine and recycled in the liver [8].
Figure 1.

Structures of biologically active 5β-reduced sterols and steroids.
For a long time, bile acids were considered solely as steroidal detergents and emulsifying agents. But now their roles as regulatory/signaling molecules have been recognized. They are ligands for the orphan nuclear receptors FXR and PXR. Through activating FXR, bile acids regulate many genes in the liver and intestine, which modulate the biosynthesis and metabolism of bile acids and lipoproteins and determine the composition of the intestinal flora and fuana [3, 7, 8, 17, 18]. Recently, δ-aminolevulinate synthase, the rate limiting enzyme of porphyrin-heme synthesis, has also been identified as a gene regulated by FXR, indicating a role of bile acids and their precursors in regulating hepatic heme biosynthesis [19]. The secondary bile acid, lithocholic acid activates PXR [20] and the vitamin D receptor [21, 22]. PXR in turn regulates the expression of CYP3A4, which encodes the major drug and xenobiotic catabolizing P450 isozyme in human liver. The secondary bile acid, ursodeoxycholic acid activates the glucocorticoid receptor and exerts immunomodulatory effects [23–25]. Bile acids also activate several signaling pathways including the c-Jun NH2-terminal kinase (JNK) 1/2 pathway (to feedback inhibit bile acid biosynthesis) [8], the protein kinase B (AKT) pathway (to regulate glucose metabolism) [8, 26], FXR, short heterodimer partner (SHP), liver X receptor (LXR), and the sterol regulatory element-binding protein (SREBP)-1c pathway (to regulate lipid metabolism) [8, 27], the extracellular-signal-regulated kinases (ERK) pathway (to prevent apoptosis) [28, 29], and the epidermal growth factor receptor (to modulate intestinal permeability) [30]. A recently identified G protein-coupled bile acid receptor (TGR5) [31] further expands the function of bile acids and their roles in energy metabolism [32], inflammation [33, 34], and gallbladder contractility [35]. Bile acids also affect cardiac function by regulating vascular tone and myocardial contractility, but the underlying mechanism for this remains largely unknown [36].
With their fat emulsification properties and regulatory functions, bile acid analogs and agents that control their levels have potential as therapeutic agents for many diseases [10]. Owning to their structural rigidity and chirality, bile acids/bile acid derivatives also represent chiral auxiliaries and building blocks in supramolecular and materials chemistry [37].
5β-Androstanes
Androgens have been used as a mainstay treatment for anemia until the introduction of recombinant human erythropoietin and are still used today as adjuvant therapy to enhance the effectiveness of recombinant human erythropoietin [38]. The mechanism, by which androgens stimulate erythropoiesis has not been completely elucidated but involves increased synthesis of erythropoetin in the kidney. 5β-Androstanes are more potent stimulators than 5α-androstanes to enhance growth and survival of colony forming unit-erythroid [39]. 5β-Androstanes function similarly to 5β-pregnanes in heme biosynthesis by inducing δ-aminolevulinate synthase expression [39–41]. Since 5β-androstanes are devoid of androgenic effects that are responsible for the major side effects associated with androgen replacement therapy, 5β-androstanes have been recommended as a substitute for testosterone in the treatment of anemia, especially for female and young patients.
Some steroids with the 3α-hydroxy-5β configuration, especially etiocholanolone, are pyrogenic in humans [42]. When injected intramuscularly, etiocholanolone elicits a latent fever through a local inflammatory response by activating leukocytes [43]. The symptom is more prominent in men than in women [44]. Elevated plasma levels of unconjugated etiocholanolone have been associated with periodic disease manifested by recurrent episodes of fever and serious membrane inflammation [45]. This clinical syndrome is denoted etiocholanolone fever. Aberrant steroid metabolism and extrahepatic production of etiocholanolone, where steroid conjugation is limited, were suggested to be responsible for the symptom [45]. However, there were reports questioning the validity of the disease by showing high but similar levels of etiocholanolone were present during both febrile and afebrile periods in patients [46].
5β-Pregnanes
5β-Pregnanes are neurosteroids devoid of progestogenic effects but modulate GABAA receptor and N-methyl-D-aspartate (NMDA) receptor activity. Both GABAA and NMDA receptors are ligand-gated neurotransmitter receptors required for normal brain function and development. The GABAA receptor mediates fast synaptic inhibition and functions as a chloride ion conducting channel that opens upon γ-aminobutyric acid (GABA) binding [47]. Pregnanolone (3α-hydroxy-5β-pregnan-20-one) potentiates the GABA response by promoting the open state of the GABAA receptor, and at higher concentrations the steroid directly activates the GABAA receptor in the absence of GABA [48]. The 3β enantiomers of pregnanolone as well as 5β-pregnane sulfates inhibit potentiation and may also act as direct GABAA receptor antagonists [49]. The NMDA receptor regulates excitatory neurotransmission and functions as a cation channel that requires binding of both glutamate and the co-agonist glycine for full activation [50]. Pregnanolone sulfate inhibits NMDA receptors by binding to active receptors and shifting the active receptors into conformations that cause NMDA desensitization [51]. However, whether de novo synthesis of 5β-reduced pregnanes occurs in the central nervous system remains to be proven.
5β-Pregnanes, especially 5β-dihydroprogsterone, have also been reported as potent tocolytic agents and maybe responsible for the uterine quiescence maintained by progesterone during pregnancy [52, 53]. 5β-Pregnanes inhibit myometrial contractions in vitro [54, 55]. Interestingly, plasma 5β-pregnane concentrations or the 5β-pregnane/progesterone ratio decreases during late pregnancy until post-partum [53, 56, 57] with a concurrent decrease in 5β-reductase expression in the uterus [53]. The mechanism through which the 5β-pregnanes exert the tocolytic effect remains controversial, and possibly involves PXR [58], the GABAA receptor [59], calcium signaling [60, 61], and the oxytocin receptor [62, 63].
5β-Pregnanes are involved in erythropoiesis in birds [40], rodents [64], and primates [65] and have also been reported to stimulate the growth of erythroid progenitor cells in human [41]. 5β-Pregnanes promote iron uptake in human bone marrow culture [66] and experiments on avian liver suggest that the steroids exert stimulatory effects by inducing δ-aminolevulinate synthase, which augments heme synthesis [41].
5β-pregnanes are also agonists for PXR and human constitutive androstane receptor (CAR). PXR and CAR are orphan nuclear receptors most abundantly expressed in liver. The function of PXR solely depends on ligand binding, whereas CAR is active in the absence of ligand but can be further regulated by activators and repressors [67]. Both receptors can be activated by a variety of xenobiotics and exert a xenoprotective function by regulating phase I and II detoxification enzymes and transporters. 5β-Pregnanes are among the most potent agonists for PXR and CAR, indicating their important xenoprotective role and potential to mediate liver xenobiotic metabolism [68, 69].
5β-Dihydrocortisol
5β-Reduced cortisol was once thought inactive until studies revealed the abnormal accumulation of 5β-dihydrocortisol in patients with primary open angle glaucoma [70]. 5β-Reduced glucocorticoid metabolites do not bind to the glucocorticoid receptor [71]. But 5β-dihydrocortisol sensitizes ocular tissue to glucocorticoids by triggering glucocorticoid receptor nuclear translocation [72]. The hypersensitivity to glucocorticoids was proposed to be the basis of ocular hypertension associated with primary open angle glaucoma. By contrast, 3α,5β-tetrahydrocortisol, a metabolite of 5β-dihydrocortisol, reduces intraocular pressure. Mechanisms for both actions have not been elucidated. However, the reduction of intraocular pressure by tetrahydrocortisol may be related to its ability to modify the cytoskeleton through actin organizing proteins, which causes drainage through the trabecular meshwork [73]. 3α,5β-Tetrahydrocortisol also exhibits antagonist properties on the GABAA receptor [74].
Cardiac glycosides
Cardiac glycosides are predominantly produced by plants and have also been identified in toads and insects [4]. Cardiac glycosides bind to the α-subunit of Na+/K+-ATPase and inhibit ion transport. In plants, these compounds are used as a defense mechanism against herbivores. In humans, cardiac glycosides increase myocardial contraction and exhibit natriuretic and vasoconstrictive effects. Exogenous cardiac glycosides are well known for their use in treatment of congestive heart failure. In the past two decades the presence of endogenous cardiac glycosides in human and other mammals has also been reported [75–79]. Though the origin of the endogenous glycosides remains controversial as these substances could also be obtained from diet, radiotracer studies in rats using both [14C]-labeled acetic acid and cholesterol supports that the adrenal gland could at least generate some of the glycosides endogenously through cholesterol biosynthesis [80]. Endogenous cardiac glycosides function as regulatory molecules, which influence Na+/H+ and Na+/Ca2+ exchange and affect various signaling pathways mediated by Na+/K+-ATPase. Increased plasma concentrations of endogenous cardiac glycosides were observed in patients with asymptomatic left ventricular dysfunction [81]. The steroids may have the potential to be developed into a biomarker for predicting heart failure.
Ecdysteroids
Ecdysteroids are nonvertebrate 5β-steroids produced by insects, crustaceans (zooecdysteroids), plants (phytoecdysteroids), and fungi (mycoecdysteroids). There are over 300 ecdysteroids identified to date [82]. Zooecdysteroids, especially ecdysone and 20-hydroxyecdysone, are crucial hormones for insect and crustaceans, which regulate molting and metamorphosis during arthropod development and also affect reproduction, behavior, and stress resistance in adults [83]. In plants and fungi, phytoecdysteroids and mycoecdysteroids function as protective agents against insect predators and soil nematodes. The ecdysteroidogenesis pathway has not been fully elucidated. A Δ4-3-ketosteroid and a Δ4-diketol, have been proposed as intermediates [5]. Ecdysteroids exert many beneficial effects on mammals, including anabolic, hepatoprotective, neuroprotective, and immunoprotective properties [82]. The steroids stimulate protein synthesis, increase muscle mass, influence cholesterol absorption, and exhibit anti-tumor effects. Ecdysteroids are devoid of androgenic, estrogenic, or glucocorticoid effects and are considered safe for human consumption. Ecdysteroids have been developed as food supplements for their anabolic properties but have not been thoroughly evaluated for therapeutic uses. Ecdysteroids are not ligands for mammalian steroid nuclear receptors due to their substantially higher dissociation constants compared to the natural ligands for these receptors. Ecdysteroids are proposed to act through membrane bound receptors that influence signal transduction pathways [82].
3. Human Δ4-3-Ketosteroid 5β-Reductase (AKR1D1)
The metabolism of steroid hormones that contain the Δ4-3-ketosteroid functionality proceeds in the liver by either 5α- or 5β-reduction. Subsequently the 3-ketone group is metabolized to yield four stereoisomeric tetrahydrosteroids, in which the alcohol can assume either a 3α-axial or 3β-equatorial configuration. This route of steroid metabolism was originally proposed by Tomkins for glucocorticoids even though the discrete enzymes involved had not yet been identified [84]. Remarkably, the introduction of the A/B cis-ring junction in 5β-reduced steroids is a reaction that is difficult to perform chemically, since common reductants produce the allylic-3β-alcohol. The enzyme, which catalyzes the 5β-reduction of Δ4-3-ketosteroids, was first purified from rat liver to homogeneity [85] and in 1991 its cDNA was cloned [86]. The rat enzyme not only exhibits activity towards Δ4-steroid hormones like testosterone and progesterone, but also turns over glucocorticoids and bile acid precursors with lower catalytic efficiency. 5β-Reductases were later annotated as belonging to the 1D subfamily of the aldo-keto reductase superfamily. The rat enzyme is denoted as AKR1D2. In 1994, human steroid 5β-reductase AKR1D1 was cloned using a rat cDNA as a probe [87]. Initially it was postulated that AKR1D1 might not be the only human 5β-reductase due to discrepancies in its substrate preference reported by different studies using different assay conditions or transfection conditions [88]. This debate was settled when recombinant AKR1D1 was purified to homogeneity and shown to have activity towards all tested Δ4-3-ketosteroids, including steroid hormones (testosterone, progesterone, glucocorticoids, and mineralcorticoids) and bile acid precursors [89]. In addition, genetic defects in AKR1D1 not only cause bile acid deficiency but also elicits a significant decrease in all the 5β-reduced steroid metabolites in humans, suggesting AKR1D1 is the only enzyme capable of performing 5β-reduction [90].
Gene structure and regulation
The AKR1D1 gene is located on chromosome 7, and is in close proximity to the aldose reductases, AKR1B1 and AKR1B10 [91]. AKR1D1 shares the highest sequence identity (>50%) with hydroxysteroid dehydrogenases, AKR1C1-AKR1C4. But the AKR1C genes are located on chromosome 10 [91]. The AKR1D1 gene consists of nine exons and eight sizable introns that make the AKR1D1 the largest gene in the AKR superfamily [92]. The mRNA of the gene possesses a long 3’-untranslated region containing AT-rich sequences signifying that it may undergo rapid degradation [92]. Transcriptional regulation of AKR1D1 has not been fully investigated. The FXR has been reported to upregulate rat 5β-reductase (AKR1D2) expression [93]. In the presence of the synthetic FXR agonist GW4064, AKR1D2 gene transcription was greatly enhanced in rat primary hepatocytes. The fold induction was similar to that observed with the SHP, a well-known nuclear receptor corepressor induced upon FXR activation. However, expression of SHP inhibits bile acid biosynthesis by repressing CYP7A1 gene transcription as part of the feedback inhibition of bile acid biosynthesis. Thus it is uncertain why FXR enhances 5β-reductase expression. In addition, the promoter region of the AKR1D1 gene contains consensus sequences for cis-regulatory elements, including an osmotic response element, an estrogen response element, a phorbol ester response element, and an anti-oxidant response element [91]. Though there is still no direct evidence to prove that these consensus response elements regulate AKR1D1 expression in human, it has been reported in hamster, rat, and dove that 5β-reductase activity is induced by estrogens [94–96]. Rat 5β-reductase has been shown to be modestly induced by dietary antioxidants [97].
Tissue distribution
AKR1D1 is predominantly expressed in the liver where it is present at least 10-fold higher than in any other tissue [98, 99]. This is in agreement with the major biological function of AKR1D1 in bile acid biosynthesis and steroid hormone clearance, both of which take place in the liver. Expression of AKR1D1 in brain, uterus, and placenta has also been reported and this would be consistent with the function of active 5β-reduced metabolites acting as neurosteroids and tocolytic agents [53, 100]. In addition, AKR1D1 has also been detected in testis, colon, skeletal muscle, prostate, lymph node, breast, thyroid, adipose tissue, and blood cells [92, 101–103]. There is no sex-related differences in AKR1D1 expression in humans [104], but differences in 5β-reductase activity towards glucocorticoids between men and women has been reported [105]. Sexual dimorphism for 5β-reductase expression/activity has been reported in mouse, rat, fish, frog, and bird [1, 106–108].
Alternative splice variants, single-nucleotide polymorphisms, and genetic defects
Six transcripts of AKR1D1 have been reported in the Ensembl database, four of which can be translated into protein variants (Figure 2) [109]. Besides the transcript that encodes the full length protein of 326 residues (AKR1D1-002, ENSP00000242375, CCDS5846), the other three splice variants code for protein sequences that are of 290, 285, and 96 residues in length. None of the splice variants have been biochemically characterized. Based on structural analyses, the 290-residue-long variant AKR1D1-006 (ENSP00000389197, CCDS55169) lacks the last α helix and the C-terminal flexible loop. The variant may still be able to fold properly but since the missing loop is involved in steroid binding, the isoform would likely have low activity due to decreased affinity for steroid substrates. On the other hand, the 285-residue-long variant AKR1D1-001 (ENSP00000402374, CCDS55170) lacks residues 153–193 and the 96-residue-long variant AKR1D1-009 (ENSP00000397042) lacks major parts of the protein, both of which would compromise the structural integrity of the (α/β)8 barrel structure and would render the variants inactive.
Figure 2.

AKR1D1 gene structures and splice variants. The nine exons are represented as filled boxes and numbered. The constitutive exons are shown in black and the alternatively spliced exons are in red.
In addition to the splice variants, there are also numerous single-nucleotide polymorphisms (SNPs) reported for AKR1D1. The majority of the SNPs are located in the 5’- and 3’-untranslated regions and introns. There are 42 non-synonymous mutations in the protein coding region. None of these non-synonymous mutations have a reported minor allelic frequency so their frequency in the population is unknown. There are studies associating several of these polymorphic variants with serious genetic defects, which could lead to neonatal lethal bile acid deficiency as described below. But most of the SNPs have not been functionally characterized.
AKR1D1 associated bile acid deficiency is an autosomal recessive condition first recognized in 1988 [110]. It is now considered one of the most common genetic defects in bile acid biosynthesis. Clinical representation of the defect includes neonatal cholestatic jaundice and fat soluble vitamin malabsorption, which may rapidly progress to cirrhosis, hemochromatosis, and neonatal liver failure [111, 112]. But these manifestations are not necessarily specific to 5β-reductase deficiency. Diagnosis of the defect needs to be confirmed with steroid urine analysis by mass spectrometry. Patients with inborn errors in AKR1D1 exhibit low or undetectable levels of plasma and urine primary bile acids (chenodeoxycholic acid and cholic acid) but significantly elevated concentrations and higher ratios of unsaturated bile acids that retain the Δ4-3-oxo-functionality (>70 %) and 5α-reduced allo-bile acids [113, 114]. These abnormal bile acids are hepatotoxic and less soluble than the primary bile acids, which could be responsible for their lack of canalicular secretion [3]. In addition, the lack of primary bile acids prevents the feed-back inhibition of cholesterol 7α-hydroxylase (CYP7A1) and sterol 12α-hydroxylase (CYP8B1), which will exacerbate the built up of Δ4-3-oxo- and allo-bile-acids (Figure 3).
Figure 3.

Bile acid biosynthesis. AKR1D1 associated bile acid deficiency causes accumulation of Δ4- and allo-bile acids and prevents feedback inhibition dependent upon the primary bile acids, cholic acid and chenodeoxycholic acid. CYP7A1, cholesterol 7α-hydroxylase; CYP8B1 sterol 12α-hydroxylase; HSD3B7, 3β-hydroxysteroid dehydrogenase. Enzymes are italicized as their gene names.
Genetic analyses have revealed three obvious inborn errors in AKR1D1. Two caused nonsense mutations in exon 2 and 3 [115, 116], the other caused a frameshift mutation in exon 5, which produces a non-functional AKR1D1 with a premature stop-codon [117]. In addition, a growing list of missense mutations have also been associated with inherited 5β-reductase deficiency, including Leu106Phe, Pro133Arg, Pro198Leu, Gly223Glu, Asp241Val, Arg261Cys, and Arg266Gln [115–118]. These residues are evolutionary conserved among all AKR1 family members [119] and a review of the AKR1D1 structure shows that none of these residues are involved in catalysis, substrate binding, or cofactor binding [120]. However, when expressed in human embryonic kidney cells, these AKR1D1 mutants showed moderate (Pro133Arg) to severe (Leu106Phe, Pro198Leu, Gly223Glu, Arg261Cys) decreases in expression and enzyme activity, suggesting that the mutations impair protein folding and stability [121]. Thus far the Asp241Val and the Arg266Gln mutants have not been characterized. Of the natural mutations that cause bile acid deficiency, only the Pro133Arg mutant could be expressed in E. coli and obtained in homogeneous form for biochemical characterization [121]. The Pro133Arg mutant exhibited a much higher Kd value for the cofactor NADPH, and transient kinetic studies showed that the mutant was compromised in its ability to carry out the chemical reaction (M. Chen and Y. Jin, unpublished data). The bile acid deficiency caused by the missense AKR1D1 mutants may result from diminishment of both enzyme expression and activity. Clinical observations also suggest that AKR1D1 is a labile enzyme since hepatocyte damage causes a loss in 5β-reductase activity (secondary 5β-reductase deficiency), which could be due to either low expression or rapid degradation of AKR1D1 [122]. Fortunately, bile-acid deficiency can be treated by supplementation with primary bile-acids [117].
AKR1D1 Enzymology
a) Kinetic mechanism
The detailed kinetic mechanism of AKR1D1 has not been fully elucidated. Based on the previous studies on the AKR1C enzymes and the similarity between AKR1C enzymes and AKR1D1, AKR1D1 likely utilizes the same ordered bi-bi reaction mechanism, in which the cofactor is the first to be bound and the last to be released (Figure 4) [119]. For AKR1C enzymes, the release of oxidized cofactor product is characteristically slow, whereas the rates of hydride transfer and release of reduced steroid product vary greatly and are vulnerable to perturbation/mutation in the steroid binding site [123–125]. Depending on the steroid substrate used, the rate-determining step in AKR1C catalyzed ketosteroid reduction can be chemistry, steroid release, or cofactor release [123]. AKR1D1 is anticipated to utilize a similar kinetic mechanism for 5β-reduction. AKR1D1 binds NADPH tightly with a Kd value of 320 nM (100 mM potassium phosphate buffer, pH 7.0, 37 °C, unpublished data). This number is four-fold lower than that of AKR1C2 under the same conditions (unpublished data), indicating AKR1D1 retains the slow cofactor release characteristics as the other AKR1 enzymes. In the stopped-flow under multiple turnover conditions, burst-phase kinetics were observed at saturating substrate concentration for the “fast” substrates (aldosterone, cortisone, and testosterone, kcat > 8 min−1, pH 6) but not for the “slow” substrates 7α-hydroxy-4-cholesten-3-one and cholestenone, kcat < 2 min−1, pH 6) (Y. Jin, unpublished data), indicating the rate limiting step of AKR1D1 is also substrate dependent. For the fast substrates, the overall rate of 5β-reduction is limited by the release of either the steroid or the cofactor product, whereas for the slow substrates the chemical event governs the reaction rate.
Figure 4.
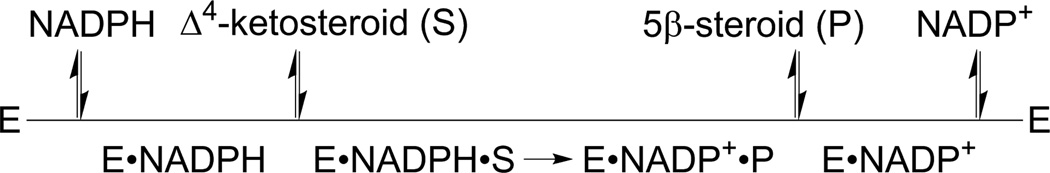
Ordered bi bi kinetic mechanism for AKR1D1. (S) = substrate, and (P) = product.
b) Substrate specificity
AKR1D1 accepts various C18–C27 Δ4-3-ketosteroids, including sex steroids, glucocorticoids, mineralocorticoids, and bile acid precursors as substrates with a pH optimum at 6.0 [89]. The enzyme is promiscuous with respect to the side chain at the C-17 position. In addition, AKR1D1 also reduces Δ1,4-dienes and likely plays a role in the metabolism of synthetic androgens and glucocorticoids. Under steady-state conditions, AKR1D1 exhibits significantly different kcat (2.0–11.7 min−1) and Km (0.3–15.1 µM) values for different steroid substrates. The catalytic efficiency of the most favored substrate Δ4-androstene-3,17-dione is over 30-fold higher than the least favored substrate cortisol [89]. Substrate inhibition was observed for C18, C19, and some of the C21 steroids but not C27 steroids. More extensive substrate screening to include nonsteroidal substrates has not been performed with AKR1D1. But plant progesterone 5β-reductase was shown to use the 5β-reduction machinery to reduce monocyclic enones and acyclic enoate esters [126]. In addition, AKR1C enzymes, the closest structural homologs to AKR1D1, demonstrate that their active site can accommodate small nonsteroidal molecules such as S-tetralol and 1-acenaphthenol. Thus, it is reasonable to assume that the double bond reductase activity of AKR1D1 may not be limited to steroid substrates.
c) Inhibition
Several inhibitors developed for AKR1C enzymes and 5β-reductases have been tested with AKR1D1. AKR1D1 is not inhibited by non-steroidal anti-inflammatory drugs (NSAIDs) such as indomethacin, mefenamic acid, and the related 4-benzoylbenzoic acid [89]. Recently NSAID analogs have been pursued as AKR1C3 inhibitors, where AKR1C3 inhibitors are desirable for treatment of castrate resistant prostate cancer [127]. The lack of cross inhibition of AKR1D1 by AKR1C inhibitors is desired. The 5α-reductase inhibitor finasteride acts as a competitive inhibitor for AKR1D1 [128]. Even though the inhibition potency is much lower for AKR1D1 than for the 5α-reductases, it may be involved in some side effects elicited by finasteride. Primary bile acid chenodexoycholic acid and secondary bile acid ursodeoxycholic acid both act as noncompetitive inhibitors for AKR1D1 [89]. Their potency towards AKR1D1 is in the low micromolar range, similar to the potency for AKR1C1 and AKR1C3 but much lower than the potency for AKR1C2, which is a known human bile acid binding protein and binds bile acids with nanomolar affinity. [129]. AKR1D1 inhibition by bile acids may play a role in the feedback inhibition of bile-acid biosynthesis.
d) Structural Biology
Numerous crystal structures of AKR1D1 have been reported (Table 1). The enzyme possesses the same triose-phosphate isomerase (TIM) barrel core structure composed of eight alternating α-helices and β-strands shared by the other aldo-keto reductase family members. The steroid cavity is situated at the C-terminal of the β-strands surrounded by three long flexible loops. In the productive binding mode, the cofactor NADPH and steroid bind perpendicular to each other (Figure 5A) [120]. The steroid 3-ketone group is anchored to the oxyanion site through hydrogen bonding to Tyr58 and Glu120. The β-face of the steroid is presented to the cofactor with the aid of Trp230 and Tyr132. Thus the Δ4-double bond is positioned within an appropriate distance and orientation from the cofactor to permit 4-pro-R hydride transfer from the nicotinamide ring to the 5β-position of the steroid. The end of the steroid cavity is open to solvent, which also explains the promiscuity of AKR1D1 for substrates with varied C-17 side-chains. Smaller steroid substrates like testosterone and progesterone can be tethered by Tyr132, Ser225, and Asn227 to a non-productive binding mode in which the steroid lies parallel with the cofactor (Figure 5B). This non-productive binding mode provides an explanation for substrate inhibition. This is supported by studies with the Val309Phe mutant. Val309 resides in the non-productive cavity and when it is mutated to the bulkier Phe, substrate inhibition is eliminated [130].
Table 1.
Crystal structures of AKR1D1.
| PDB ID | AKR1D1 | Cofactor/steroid | Reference |
|---|---|---|---|
| 3BV7 | WT | NADP+ | [120] |
| 3BUV | WT | NADP+ | [120] |
| 3BUR | WT | NADP+, testosterone | [120] |
| 3CMF | WT | NADP+, cortisone | [120] |
| 3COT | WT | NADP+, progesterone | [120] |
| 3CAQ | WT | NADPH | [132] |
| 3CAV | WT | NADP+, 5β-pregnane-3,20-dione | [130] |
| 3CAS | WT | NADP+, Δ4-androstenedione | [130] |
| 3DOP | WT | NADP+, 5β-dihydrotestosterone | [133] |
| 3G1R | WT | NADP+, finasteride | [128] |
| 3UZW | Glu120His | NADP+ | [131] |
| 3UZX | Glu120His | NADP+, epiandrosterone, 3α-androstanediol | [131] |
| 3UZY | Glu120His | NADP+, 5β-dihydrotestosterone | [131] |
| 3UZZ | Glu120His | NADP+, Δ4-androstenedione, testosterone | [131] |
Figure 5.

Crystal Structure of AKR1D1 showing normal (A, PDB: 3CMF) and non-productive (B, PDB: 3BUR) binding modes. The steroids and NADP+ are colored in black. All the other atoms are color-coded as follows: carbon, green; oxygen, red; and nitrogen, blue; phosphor, orange. Hydrogen bonds are indicated by red dashes. All structural figures are prepared using The PyMOL Molecular Graphics System, Version 1.20 Schrödinger, LLC.
The active site of AKR1D1 is fine tuned for 5β-reduction and its function is precisely controlled by a single residue, Glu120. Glu120 is also the most prominent residue that distinguishes 5β-reductase from the other AKR family members in sequence alignment. A Glu120His mutation abolishes 5β-reductase activity and perfectly converts the enzyme into a highly efficient 3β-hydroxysteroid dehydrogenase [131]. Crystal structures of the Glu120His mutant reveal that the residue controls enzyme function by manipulating the relative position between the steroid and cofactor. With the smaller glutamate in position, the steroid can reach “deeper” into the active site to bring the steroid C5 carbon within hydride transfer distance to the cofactor (Figure 6A). Once substituted by histidine, the bulky imidazole side chain pushes the steroid away so that only the steroid C3 carbon can be reduced. In this new position the steroid C3 ketone group in the Glu120His mutant superimposes on the corresponding group in ketosteroids bound in the oxyanion hole in the AKR1C enzymes (Figure 6B). This Glu120His mutation sets an excellent example of how a single amino acid governs enzyme function and emphasizes that the presence of the glutamate residue is essential when assigning a newly identified AKR gene as a 5β-reductase.
Figure 6.

Crystal Structure of the AKR1D1-Glu120His mutant (magenta) in superposition with WT AKR1D1 (A, green, PDB: 3CMF) and 3α-hydroxysteroid dehydrogenase AKR1C9 (B, blue, PDB: 1AFS). In the AKR1D1 Glu120His mutant structure, the face of the steroid that presents to the cofactor is flipped and the C3 ketone of the steroid does not penetrate as deeply into the active site permitting 3β-hydroxysteroid dehydrogenase activity.
e) Chemical mechanism
5β-Reduction is an irreversible double bond reduction of Δ4-3-ketosteroids. During the chemical event, the hydride at the 4-pro-R position on the nicotinamide ring is stereospecifically transferred to C5 position of the steroid on the β-face, which is facilitated by a highly conserved Tyr58 that acts as a general acid and Glu120 that acts as a “superacid” to enolize the α,β-unsaturated ketone group of the steroid (Figure 7) [120]. In the mechanism of rat 3α-hydroxysteroid dehydrogenase (AKR1C9), Asp53, Tyr58, Lys87, and His120 (AKR1D1 numbering) are collectively called the catalytic tetrad. The mechanism depicts Tyr58 as the general acid/base, where His120 facilitates proton donation from tyrosine in the reduction direction, and Lys87 and Asp53 facilitates proton removal by the tyrosine in the oxidation direction. In the structure of AKR1D1, Lys87 and Asp53 remain in hydrogen bond network with Tyr58 and Glu120. Faucher et al. proposed that Lys87 and Asp53 play roles in the proton relay by shuttling protons from Tyr58 [132]. However, this mechanism has yet to be supported by pH-rate studies.
Figure 7.

The chemical mechanism of AKR1D1. Note the steroid is drawn with the α-face towards the viewer and the hydride is transferred to the β-face. The tetrad residues and steroid are colored in black. All the other atoms are color-coded as in Figure 5. Hydrogen bonds are indicated by red dashes.
4. Future directions
The physiological functions of 5β-reduced steroids other than bile acids still remain to be fully elucidated along with their modes of action. Wild type AKR1D1 can be used as a synthon to catalyze the formation of A/B cis ring fusions in steroid substrates, which is difficult to perform chemically. AKR1D1 mutations have been associated with bile acid deficiency. With the onset of whole exome sequencing, SNPs are likely to be revealed in AKR1D1 that can be mapped to the existing crystal structure. Examination of these mutants by site-directed mutagenesis will determine whether they affect enzyme function or folding and could be deleterious to health. Defects in AKR1D1 activity may also compromise glucocorticoid metabolism and as a result the loss of enzyme activity could play a role in obesity. The basis of the tissue specific expression of 5β-reductase needs to be elucidated to determine whether this is related to tissue specific transcription factors or epigenetic imprinting.
Highlight Points.
5β-reduced steroids are metabolites of all Δ4-3-ketosteroids
5β-reduced androstanes, pregnanes, and cholanes perform biological functions
All mammalian bile-acids are 5β-cholanes
Aldo-keto reductase (AKR) 1D1 is the only human 5β-reductase
Structure-function studies on AKR1D1 provide a basis for bile-acid deficiency
Acknowledgement
This work was supported by R01-DK47015 and P30-ES13508 awarded from the National Institutes of Health to TMP and by grant F32 (F32-DK089827) awarded by the National Institutes of Health to MC.
Abbreviations
- AKR
aldo-keto reductase
- AKR1C1-AKR1C4
human hydroxysteroid dehydrogenase
- AKR1D1
human steroid 5β-reductase
- AKR1D2
rat steroid 5β-reductase
- CAR
constitutive androstane receptor
- ERK
extracellular-signal regulated kinase
- FXR
farnesoid X receptor
- JNK
c-Jun NH2-terminal kinase
- LXR
liver X receptor
- PXR
pregnane X receptor
- SREBP
sterol regulatory element binding protein
- SHP
short heterodimer partner
- TIM
triose phosphate isomerase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Langlois VS, Zhang D, Cooke GM, Trudeau VL. Evolution of steroid-5α-reductases and comparison of their function with 5β-reductase. Gen Comp Endocrinol. 2010;166:489–497. doi: 10.1016/j.ygcen.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Russell DW, Wilson JD. Steroid 5α-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- 3.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 4.Agrawal AA, Petschenka G, Bingham RA, Weber MG, Rasmann S. Toxic cardenolides: chemical ecology and coevolution of specialized plant-herbivore interactions. New Phytol. 2012;194:28–45. doi: 10.1111/j.1469-8137.2011.04049.x. [DOI] [PubMed] [Google Scholar]
- 5.Huang X, Warren JT, Gilbert LI. New players in the regulation of ecdysone biosynthesis. J Genet Genomics. 2008;35:1–10. doi: 10.1016/S1673-8527(08)60001-6. [DOI] [PubMed] [Google Scholar]
- 6.Thorn A, Egerer-Sieber C, Jager CM, Herl V, Muller-Uri F, Kreis W, et al. The crystal structure of progesterone 5β-reductase from Digitalis lanata defines a novel class of short chain dehydrogenases/reductases. J Biol Chem. 2008;283:17260–17269. doi: 10.1074/jbc.M706185200. [DOI] [PubMed] [Google Scholar]
- 7.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 8.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roda A, Hofmann AF, Mysels KJ. The influence of bile salt structure on self-association in aqueous solutions. J Biol Chem. 1983;258:6362–6370. [PubMed] [Google Scholar]
- 10.Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65:2461–2483. doi: 10.1007/s00018-008-7568-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allouche M, Castano S, Colin D, Desbat B, Kerfelec B. Structure and orientation of pancreatic colipase in a lipid environment: PM-IRRAS and Brewster angle microscopy studies. Biochemistry. 2007;46:15188–15197. doi: 10.1021/bi701831f. [DOI] [PubMed] [Google Scholar]
- 12.Wang CS, Hartsuck JA. Bile salt-activated lipase. A multiple function lipolytic enzyme. Biochim Biophys Acta. 1993;1166:1–19. doi: 10.1016/0005-2760(93)90277-g. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Wang CS, Tang J, Dyda F, Zhang XC. The crystal structure of bovine bile salt activated lipase: insights into the bile salt activation mechanism. Structure. 1997;5:1209–1218. doi: 10.1016/s0969-2126(97)00271-2. [DOI] [PubMed] [Google Scholar]
- 14.Fontbonne H, Brisson L, Vérine A, Puigserver A, Lombardo D, Ajandouz EH. Human bile salt-dependent lipase efficiency on medium-chain acyl-containing substrates: control by sodium taurocholate. J Biochem (Tokyo) 2011;149:145–151. doi: 10.1093/jb/mvq132. [DOI] [PubMed] [Google Scholar]
- 15.Hofmann AF, Hagey LR, Krasowski MD. Bile salts of vertebrates: structural variation and possible evolutionary significance. J Lipid Res. 2009;51:226–246. doi: 10.1194/jlr.R000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 17.Chiang JYL. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peyer A-K, Jung D, Beer M, Gnerre C, Keogh A, Stroka D, et al. Regulation of human liver δ-aminolevulinic acid synthase by bile acids. Hepatology. 2007;46:1960–1970. doi: 10.1002/hep.21879. [DOI] [PubMed] [Google Scholar]
- 20.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han S, Li T, Ellis E, Strom S, Chiang JYL. A novel bile acid-activated vitamin D receptor signaling in human hepatocytes. Mol Endocrinol. 2010;24:1151–1164. doi: 10.1210/me.2009-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka H, Makino Y, Miura T, Hirano F, Okamoto K, Komura K, et al. Ligand-independent activation of the glucocorticoid receptor by ursodeoxycholic acid. Repression of IFN-gamma-induced MHC class II gene expression via a glucocorticoid receptor-dependent pathway. The Journal of Immunology. 1996;156:1601–1608. [PubMed] [Google Scholar]
- 24.Miura T, Ouchida R, Yoshikawa N, Okamoto K, Makino Y, Nakamura T, et al. Functional modulation of the glucocorticoid receptor and suppression of NF-κB-dependent transcription by ursodeoxycholic Acid. J Biol Chem. 2001;276:47371–47378. doi: 10.1074/jbc.M107098200. [DOI] [PubMed] [Google Scholar]
- 25.Weitzel C, Stark D, Kullmann F, Scholmerich J, Holstege A, Falk W. Ursodeoxycholic acid induced activation of the glucocorticoid receptor in primary rat hepatocytes. Eur J Gastroenterol Hepatol. 2005;17:169–177. doi: 10.1097/00042737-200502000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen A, Bouscarel B. Bile acids and signal transduction: role in glucose homeostasis. Cell Signal. 2008;20:2180–2197. doi: 10.1016/j.cellsig.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiao L, Studer E, Leach K, McKinstry R, Gupta S, Decker R, et al. Deoxycholic acid (DCA) causes ligand-independent activation of epidermal growth factor receptor (EGFR) and FAS receptor in primary hepatocytes: inhibition of EGFR/mitogen-activated protein kinase-signaling module enhances DCA-induced apoptosis. Mol Biol Cell. 2001;12:2629–2645. doi: 10.1091/mbc.12.9.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amaral JD, Viana RJ, Ramalho RM, Steer CJ, Rodrigues CM. Bile acids: regulation of apoptosis by ursodeoxycholic acid. J Lipid Res. 2009;50:1721–1734. doi: 10.1194/jlr.R900011-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raimondi F, Santoro P, Barone MV, Pappacoda S, Barretta ML, Nanayakkara M, et al. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G906–G913. doi: 10.1152/ajpgi.00043.2007. [DOI] [PubMed] [Google Scholar]
- 31.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 33.Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, et al. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE. 2011;6:e25637. doi: 10.1371/journal.pone.0025637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84. doi: 10.1016/j.bbrc.2008.04.171. [DOI] [PubMed] [Google Scholar]
- 35.Li T, Holmstrom SR, Kir S, Umetani M, Schmidt DR, Kliewer SA, et al. The G protein-coupled bile acid receptor, TGR5, stimulates gallbladder filling. Mol Endocrinol. 2011;25:1066–1071. doi: 10.1210/me.2010-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khurana S, Raufman J-P, Pallone TL. Bile acids regulate cardiovascular function. Clin Transl Sci. 2011;4:210–218. doi: 10.1111/j.1752-8062.2011.00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nonappa, Maitra U. Unlocking the potential of bile acids in synthesis, supramolecular/materials chemistry and nanoscience. Org Biomol Chem. 2008;6:657–669. doi: 10.1039/b714475j. [DOI] [PubMed] [Google Scholar]
- 38.Navarro JF. In the erythropoietin era, can we forget alternative or adjunctive therapies for renal anaemia management? The androgen example. Nephrol Dial Transplant. 2003;18:2222–2226. doi: 10.1093/ndt/gfg370. [DOI] [PubMed] [Google Scholar]
- 39.Incefy GS, Kappas A. Enhancement of RNA synthesis in avian liver cell cultures by a 5β-steroid metabolite during induction of δ-aminolevulinate synthase. Proc Natl Acad Sci U S A. 1974;71:2290–2294. doi: 10.1073/pnas.71.6.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levere RD, Kappas A, Granick S. Stimulation of hemoglobin synthesis in chick blastoderms by certain 5β-androstane and 5β-pregnane steroids. Proc Natl Acad Sci U S A. 1967;58:985–990. doi: 10.1073/pnas.58.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Urabe A, Sassa S, Kappas A. The influence of steroid hormone metabolites on the in vitro development of erythroid colonies derived from human bone marrow. J Exp Med. 1979;149:1314–1325. doi: 10.1084/jem.149.6.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dillard GM, Bodel P. Studies on steroid fever. II. Pyrogenic and anti-pyrogenic activity in vitro of some endogenous steroids of man. J Clin Invest. 1970;49:2418–2426. doi: 10.1172/JCI106461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bodel P, Dillard M. Studies on steroid fever: I. Production of leukocyte pyrogen in vitro by etiocholanolone. J Clin Invest. 1968;47:107–117. doi: 10.1172/JCI105701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimball HR, Wolff SM, Vogel JM, Perry S. Experimental etiocholanolone fever: febrile reactivity in men and women. J Clin Endocrinol Metab. 1966;26:222–224. doi: 10.1210/jcem-26-2-222. [DOI] [PubMed] [Google Scholar]
- 45.Bondy PK, Cohn GL, Gregory PB. Etiocholanolone fever. Medicine (Baltim) 1965;44:249–262. doi: 10.1097/00005792-196505000-00003. [DOI] [PubMed] [Google Scholar]
- 46.George JM, Wolff SM, Diller E, Bartter FC. Recurrent fever of unknown etiology: failure to demonstrate association between fever and plasma unconjugated etiocholanolone. J Clin Invest. 1969;48:558–563. doi: 10.1172/JCI106014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sieghart W, Fuchs K, Tretter V, Ebert V, Jechlinger M, Höger H, et al. Structure and subunit composition of GABAA receptors. Neurochem Int. 1999;34:379–385. doi: 10.1016/s0197-0186(99)00045-5. [DOI] [PubMed] [Google Scholar]
- 48.Callachan H, Cottrell GA, Hather NY, Lambert JJ, Nooney JM, Peters JA. Modulation of the GABAA receptor by progesterone metabolites. Proc R Soc Lond B Biol Sci. 1987;231:359–369. doi: 10.1098/rspb.1987.0049. [DOI] [PubMed] [Google Scholar]
- 49.Wang M, He Y, Eisenman LN, Fields C, Zeng C-M, Mathews J, et al. 3β-Hydroxypregnane steroids are pregnenolone sulfate-like GABAA receptor antagonists. J Neurosci. 2002;22:3366–3375. doi: 10.1523/JNEUROSCI.22-09-03366.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benarroch EE. NMDA receptors: recent insights and clinical correlations. Neurology. 2011;76:1750–1757. doi: 10.1212/WNL.0b013e31821b7cc9. [DOI] [PubMed] [Google Scholar]
- 51.Kussius CL, Kaur N, Popescu GK. Pregnanolone sulfate promotes desensitization of activated NMDA receptors. J Neurosci. 2009;29:6819–6827. doi: 10.1523/JNEUROSCI.0281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheehan PM. A possible role for progesterone metabolites in human parturition. Aust N Z J Obstet Gynaecol. 2006;46:159–163. doi: 10.1111/j.1479-828X.2006.00548.x. [DOI] [PubMed] [Google Scholar]
- 53.Sheehan PM, Rice GE, Moses EK, Brennecke SP. 5β-Dihydroprogesterone and steroid 5β-reductase decrease in association with human parturition at term. Mol Hum Reprod. 2005;11:495–501. doi: 10.1093/molehr/gah201. [DOI] [PubMed] [Google Scholar]
- 54.Thornton S, Terzidou V, Clark A, Blanks A. Progesterone metabolite and spontaneous myometrial contractions in vitro. Lancet. 1999;353:1327–1329. doi: 10.1016/S0140-6736(98)05247-7. [DOI] [PubMed] [Google Scholar]
- 55.Kubli-Garfias C, Medrano-Conde L, Beyer C, Bondani A. In vitro inhibition of rat uterine contractility induced by 5α and 5β progestins. Steroids. 1979;34:609–617. doi: 10.1016/0039-128x(79)90131-4. [DOI] [PubMed] [Google Scholar]
- 56.Hill M, Cibula D, Havlíková H, Kancheva L, Fait T, Kancheva R, et al. Circulating levels of pregnanolone isomers during the third trimester of human pregnancy. J Steroid Biochem Mol Biol. 2007;105:166–175. doi: 10.1016/j.jsbmb.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 57.Gilbert Evans SE, Ross LE, Sellers EM, Purdy RH, Romach MK. 3α-Reduced neuroactive steroids and their precursors during pregnancy and the postpartum period. Gynecol Endocrinol. 2005;21:268–279. doi: 10.1080/09513590500361747. [DOI] [PubMed] [Google Scholar]
- 58.Mitchell BF, Mitchell JM, Chowdhury J, Tougas M, Engelen SME, Senff N, et al. Metabolites of progesterone and the pregnane X receptor: A novel pathway regulating uterine contractility in pregnancy? Am J Obstet Gynecol. 2005;192:1304–1313. doi: 10.1016/j.ajog.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 59.Putnam CD, Brann DW, Kolbeck RC, Mahesh VB. Inhibition of uterine contractility by progesterone and progesterone metabolites: mediation by progesterone and gamma amino butyric acidA receptor systems. Biol Reprod. 1991;45:266–272. doi: 10.1095/biolreprod45.2.266. [DOI] [PubMed] [Google Scholar]
- 60.Burger K, Fahrenholz F, Gimpl G. Non-genomic effects of progesterone on the signaling function of G protein-coupled receptors. FEBS Lett. 1999;464:25–29. doi: 10.1016/s0014-5793(99)01668-3. [DOI] [PubMed] [Google Scholar]
- 61.Perusquia M, Villalón CM. The relaxant effect of sex steroids in rat myometrium is independent of the gamma-amino butyric acid system. Life Sci. 1996;58:913–926. doi: 10.1016/0024-3205(96)00034-3. [DOI] [PubMed] [Google Scholar]
- 62.Grazzini E, Guillon G, Mouillac B, Zingg HH. Inhibition of oxytocin receptor function by direct binding of progesterone. Nature. 1998;392:509–512. doi: 10.1038/33176. [DOI] [PubMed] [Google Scholar]
- 63.Astle S, Khan RN, Thornton S. The effects of a progesterone metabolite, 5β-dihydroprogesterone, on oxytocin receptor binding in human myometrial membranes. BJOG. 2003;110:589–592. doi: 10.1046/j.1471-0528.2003.02041.x. [DOI] [PubMed] [Google Scholar]
- 64.Gorshein D, Gardner FH. Erythropoietic activity of steroid metabolites in mice. Proc Natl Acad Sci U S A. 1970;65:564–568. doi: 10.1073/pnas.65.3.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Besa EC, Gorshein D, Hait WA, Gardner FH. Effective erythropoiesis induced by 5β-pregnane-3β-hydroxy-20-one in squirrel monkeys. J Clin Invest. 1973;52:2278–2282. doi: 10.1172/JCI107415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gorshein D, Reisner EH, Jr, gardner FH. Tissue culture of bone marrow. V. Effect of 5β(H) steroids and cyclic AMP on heme synthesis. Am J Physiol. 1975;228:1024–1028. doi: 10.1152/ajplegacy.1975.228.4.1024. [DOI] [PubMed] [Google Scholar]
- 67.Swales K, Negishi M. CAR, driving into the future. Mol Endocrinol. 2004;18:1589–1598. doi: 10.1210/me.2003-0397. [DOI] [PubMed] [Google Scholar]
- 68.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, et al. Orphan nuclear receptors constitutive androstane receptor and pregnane X receptor share xenobiotic and steroid ligands. J Biol Chem. 2000;275:15122–15127. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 69.Bertilsson G, Heidrich J, Svensson K, Åsman M, Jendeberg L, Sydow-Bäckman M, et al. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci U S A. 1998;95:12208–12213. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Southren AL, Gordon GG, Munnangi PR, Vittek J, Schwartz J, Monder C, et al. Altered cortisol metabolism in cells cultured from trabecular meshwork specimens obtained from patients with primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 1983;24:1413–1417. [PubMed] [Google Scholar]
- 71.McInnes KJ, Kenyon CJ, Chapman KE, Livingstone DEW, Macdonald LJ, Walker BR, et al. 5α-Reduced glucocorticoids, novel endogenous activators of the glucocorticoid receptor. J Biol Chem. 2004;279:22908–22912. doi: 10.1074/jbc.M402822200. [DOI] [PubMed] [Google Scholar]
- 72.Weinstein BI, Gordon GG, Southren AL. Potentiation of glucocorticoid activity by 5β-dihydrocortisol: Its role in glaucoma. Science. 1983;222:172–173. doi: 10.1126/science.6623065. [DOI] [PubMed] [Google Scholar]
- 73.Clark AF, Lane D, Wilson K, Miggans ST, McCartney MD. Inhibition of dexamethasone-induced cytoskeletal changes in cultured human trabecular meshwork cells by tetrahydrocortisol. Invest Ophthalmol Vis Sci. 1996;37:805–813. [PubMed] [Google Scholar]
- 74.Penland SN, Morrow AL. 3α,5β-Reduced cortisol exhibits antagonist properties on cerebral cortical GABAA receptors. Eur J Pharmacol. 2004;506:129–132. doi: 10.1016/j.ejphar.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 75.Kawamura A, Guo J, Itagaki Y, Bell C, Wang Y, Haupert GT, Jr, et al. On the structure of endogenous ouabain. Proc Natl Acad Sci U S A. 1999;96:6654–6659. doi: 10.1073/pnas.96.12.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferrandi M, Manunta P, Balzan S, Hamlyn JM, Bianchi G, Ferrari P. Ouabain-like factor quantification in mammalian tissues and plasma: comparison of two independent assays. Hypertension. 1997;30:886–896. doi: 10.1161/01.hyp.30.4.886. [DOI] [PubMed] [Google Scholar]
- 77.Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, et al. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci U S A. 1991;88:6259–6263. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lichtstein D, Gati I, Samuelov S, Berson D, Rozenman Y, Landau L, et al. Identification of digitalis-like compounds in human cataractous lenses. Eur J Biochem. 1993;216:261–268. doi: 10.1111/j.1432-1033.1993.tb18141.x. [DOI] [PubMed] [Google Scholar]
- 79.Bagrov AY, Fedorova OV, Dmitrieva RI, Howald WN, Hunter AP, Kuznetsova EA, et al. Characterization of a urinary bufodienolide Na+,K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension. 1998;31:1097–1103. doi: 10.1161/01.hyp.31.5.1097. [DOI] [PubMed] [Google Scholar]
- 80.Qazzaz HM, Cao Z, Bolanowski DD, Clark BJ, Valdes R., Jr De novo biosynthesis and radiolabeling of mammalian digitalis-like factors. Clin Chem. 2004;50:612–620. doi: 10.1373/clinchem.2003.022715. [DOI] [PubMed] [Google Scholar]
- 81.Jortani SA, Valdes R., Jr Mammalian cardenolides as biomarkers in congestive heart failure. Cardiovasc Toxicol. 2001;1:165–170. doi: 10.1385/ct:1:2:165. [DOI] [PubMed] [Google Scholar]
- 82.Báthori M, Tóth N, Hunyadi A, Márki A, Zádor E. Phytoecdysteroids and anabolic-androgenic steroids--structure and effects on humans. Curr Med Chem. 2008;15:75–91. doi: 10.2174/092986708783330674. [DOI] [PubMed] [Google Scholar]
- 83.Schwedes CC, Carney GE. Ecdysone signaling in adult Drosophila melanogaster. J Insect Physiol. 2012;58:293–302. doi: 10.1016/j.jinsphys.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 84.Tomkins GM. The enzymatic reduction of Δ4-3-ketosteroids. J Biol Chem. 1957;225:13–24. [PubMed] [Google Scholar]
- 85.Okuda A, Okuda K. Purification and characterization of Δ4-3-ketosteroid 5β-reductase. J Biol Chem. 1984;259:7519–7524. [PubMed] [Google Scholar]
- 86.Onishi Y, Noshiro M, Shimosato T, Okuda K. Molecular cloning and sequence analysis of cDNA encoding Δ4-3-ketosteroid 5β-reductase of rat liver. FEBS Lett. 1991;283:215–218. doi: 10.1016/0014-5793(91)80591-p. [DOI] [PubMed] [Google Scholar]
- 87.Kondo KH, Kai MH, Setoguchi Y, Sjöblom P, Setoguchi T, Okuda KI, et al. Cloning and expression of cDNA of human Δ4-3-oxosteroid 5β-reductase and substrate specificity of the expressed enzyme. Eur J Biochem. 1994;219:357–363. doi: 10.1111/j.1432-1033.1994.tb19947.x. [DOI] [PubMed] [Google Scholar]
- 88.Drury J, Penning T. Delta4-3-ketosteroid 5beta-reductase (AKR1D1): Properties and role in bile acid synthesis. Enzymology and Molecular Biology of Carbonyl Metabolism. 2007:332–340. [Google Scholar]
- 89.Chen M, Drury JE, Penning TM. Substrate specificity and inhibitor analyses of human steroid 5β-reductase (AKR1D1) Steroids. 2011;76:484–490. doi: 10.1016/j.steroids.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Palermo M, Marazzi MG, Hughes BA, Stewart PM, Clayton PT, Shackleton CH. Human Δ4-3-oxosteroid 5beta-reductase (AKR1D1) deficiency and steroid metabolism. Steroids. 2008;73:417–423. doi: 10.1016/j.steroids.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 91.Penning TM, Drury JE. Human aldo-keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch Biochem Biophys. 2007;464:241–250. doi: 10.1016/j.abb.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Charbonneau A. The V-L. Genomic organization of a human 5β-reductase and its pseudogene and substrate selectivity of the expressed enzyme. BBA-Gene Struct Expr. 2001;1517:228–235. doi: 10.1016/s0167-4781(00)00278-5. [DOI] [PubMed] [Google Scholar]
- 93.Pircher PC, Kitto JL, Petrowski ML, Tangirala RK, Bischoff ED, Schulman IG, et al. Farnesoid X receptor regulates bile acid-amino acid conjugation. J Biol Chem. 2003;278:27703–27711. doi: 10.1074/jbc.M302128200. [DOI] [PubMed] [Google Scholar]
- 94.Tsuji M, Terada N, Yabumoto H, Takeyama M, Matsumoto K. Hormonal regulation of activities of 4-ene-5β and 5α-reductases and 17β-ol-dehydrogenase in immature golden hamster ovary. J Steroid Biochem. 1983;18:777–781. doi: 10.1016/0022-4731(83)90259-5. [DOI] [PubMed] [Google Scholar]
- 95.Hutchison JB, Steimer T. Brain 5β-reductase: a correlate of behavioral sensitivity to androgen. Science. 1981;213:244–246. doi: 10.1126/science.7244635. [DOI] [PubMed] [Google Scholar]
- 96.Ghraf R, Lax ER, Schriefers H. The hypophysis in the regulation of androgen and oestrogen dependent enzyme activities of steroid hormone metabolism in rat liver cytosol. Hoppe Seylers Z Physiol Chem. 1975;356:127–134. doi: 10.1515/bchm2.1975.356.1.127. [DOI] [PubMed] [Google Scholar]
- 97.MacLeod AK, Kelly VP, Higgins LG, Kelleher MO, Price SA, Bigley AL, et al. Expression and localization of rat aldo-keto reductases and induction of the 1B13 and 1D2 isoforms by phenolic antioxidants. Drug Metab Dispos. 2009;38:341–346. doi: 10.1124/dmd.109.030544. [DOI] [PubMed] [Google Scholar]
- 98.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lisboa BP, Strassner M, Wulff C, Hoffmann U. 5β-Reductase in human fetal brain. Acta Endocrinol (Copenh) 1974;77:S156. [Google Scholar]
- 101.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kolker E, Higdon R, Haynes W, Welch D, Broomall W, Lancet D, et al. MOPED: Model Organism Protein Expression Database. Nucleic Acids Res. 2012;40:D1093–D1099. doi: 10.1093/nar/gkr1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang Y, Nadeau Ml, Faucher Fdr, Lescelleur O, Biron S, Daris M, et al. Progesterone metabolism in adipose cells. Mol Cell Endocrinol. 2009;298:76–83. doi: 10.1016/j.mce.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 104.Baudrand R, DomÃnguez JM, Carvajal CA, Riquelme A, Campino C, Macchiavello S, et al. Overexpression of hepatic 5α-reductase and 11β-hydroxysteroid dehydrogenase type 1 in visceral adipose tissue is associated with hyperinsulinemia in morbidly obese patients. Metabolism. 2011;60:1775–1780. doi: 10.1016/j.metabol.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 105.Finken MJJ, Andrews RC, Andrew R, Walker BR. Cortisol metabolism in healthy young adults: sexual dimorphism in activities of A-ring reductases, but not 11β-hydroxysteroid dehydrogenases. J Clin Endocrinol Metab. 1999;84:3316–3321. doi: 10.1210/jcem.84.9.6009. [DOI] [PubMed] [Google Scholar]
- 106.Barrett T, Troup DB, Wilhite SE, Ledoux P, Evangelista C, Kim IF, et al. NCBI GEO: archive for functional genomics data sets-10 years on. Nucleic Acids Res. 2011;39:D1005–D1010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Soma KK, Schlinger BA, Wingfield JC, Saldanha CJ. Brain aromatase 5 alpha-reductase, and 5 beta-reductase change seasonally in wild male song sparrows: relationship to aggressive and sexual behavior. J Neurobiol. 2003;56:209–221. doi: 10.1002/neu.10225. [DOI] [PubMed] [Google Scholar]
- 108.Mode A, Rafter I. The sexually differentiated Δ4-3-ketosteroid 5β-reductase of rat liver. Purification, characterization, and quantitation. J Biol Chem. 1985;260:7137–7141. [PubMed] [Google Scholar]
- 109.Barski OA, Mindnich R, Penning TM. Alternative splicing in the aldo-keto reductase superfamily: Implications for protein nomenclature. Chem Biol Interact. doi: 10.1016/j.cbi.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Setchell KD, Suchy FJ, Welsh MB, Zimmer-Nechemias L, Heubi J, Balistreri WF. Δ4-3-oxosteroid 5β-reductase deficiency described in identical twins with neonatal hepatitis. A new inborn error in bile acid synthesis. The Journal of Clinical Investigation. 1988;82:2148–2157. doi: 10.1172/JCI113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Clayton PT. Disorders of bile acid synthesis. J Inherit Metab Dis. 2011;34:593–604. doi: 10.1007/s10545-010-9259-3. [DOI] [PubMed] [Google Scholar]
- 112.Heubi JE, Setchell KDR, Bove KE. Inborn Errors of Bile Acid Metabolism. Semin Liver Dis. 2007;27:282–294. doi: 10.1055/s-2007-985073. [DOI] [PubMed] [Google Scholar]
- 113.Clayton PT, Mills KA, Johnson AW, Barabino A, Marazzi MG. Δ4-3-oxosteroid 5β-reductase deficiency: failure of ursodeoxycholic acid treatment and response to chenodeoxycholic acid plus cholic acid. Gut. 1996;38:623–628. doi: 10.1136/gut.38.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Muto A, Takei H, Unno A, Murai T, Kurosawa T, Ogawa S, et al. Detection of Δ4-3-oxo-steroid 5β-reductase deficiency by LC-ESI-MS/MS measurement of urinary bile acids. J Chromatogr B. 900:24–31. doi: 10.1016/j.jchromb.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 115.Zhao J, Fang LJ, Setchell KD, Chen R, Li LT, Wang JS. Primary Δ4-3-oxosteroid 5β-reductase deficiency: Two cases in China. World J Gastroenterol. 2012;18:7113–7117. doi: 10.3748/wjg.v18.i47.7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ueki I, Kimura A, Chen H-L, Yorifuji T, Mori J, Itoh S, et al. SRD5B1gene analysis needed for the accurate diagnosis of primary 3-oxo-Δ4-steroid 5β-reductase deficiency. J Gastroenterol Hepatol. 2009;24:776–785. doi: 10.1111/j.1440-1746.2008.05669.x. [DOI] [PubMed] [Google Scholar]
- 117.Lemonde HA, Custard EJ, Bouquet J, Duran M, Overmars H, Scambler PJ, et al. Mutations in SRD5B1 (AKR1D1), the gene encoding Δ4-3-oxosteroid 5β-reductase, in hepatitis and liver failure in infancy. Gut. 2003;52:1494–1499. doi: 10.1136/gut.52.10.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gonzales E, Cresteil D, Baussan C, Dabadie A, Gerhardt M-F, Jacquemin E. SRD5B1 (AKR1D1) gene analysis in Δ4-3-oxosteroid 5β-reductase deficiency: evidence for primary genetic defect. J Hepatol. 2004;40:716–718. doi: 10.1016/j.jhep.2003.12.024. [DOI] [PubMed] [Google Scholar]
- 119.Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Di Costanzo L, Drury JE, Penning TM, Christianson DW. Crystal structure of human liver Δ4-3-ketosteroid 5β-reductase (AKR1D1) and implications for substrate binding and catalysis. J Biol Chem. 2008;283:16830–16839. doi: 10.1074/jbc.M801778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Drury JE, Mindnich R, Penning TM. Characterization of disease-related 5β-reductase (AKR1D1) mutations reveals their potential to cause bile acid deficiency. J Biol Chem. 2010;285:24529–24537. doi: 10.1074/jbc.M110.127779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Clayton PT. Δ4-3-Oxosteroid 5β-reductase deficiency and neonatal hemochromatosis. The Journal of pediatrics. 1994;125:845–846. [PubMed] [Google Scholar]
- 123.Cooper WC, Jin Y, Penning TM. Elucidation of a complete kinetic mechanism for a mammalian hydroxysteroid dehydrogenase (HSD) and identification of all enzyme forms on the reaction coordinate. J Biol Chem. 2007;282:33484–33493. doi: 10.1074/jbc.M703414200. [DOI] [PubMed] [Google Scholar]
- 124.Jin Y, Penning TM. Multiple steps determine the overall rate of the reduction of 5α-dihydrotestosterone catalyzed by human type 3 3α-hydroxysteroid dehydrogenase: Implications for the elimination of androgens. Biochemistry. 2006;45:13054–13063. doi: 10.1021/bi060591r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Heredia VV, Cooper WC, Kruger RG, Jin Y, Penning TM. Alanine scanning mutagenesis of the testosterone binding site of rat 3α-hydroxysteroid dehydrogenase demonstrates contact residues influence the rate-determining step. Biochemistry. 2004;43:5832–5841. doi: 10.1021/bi0499563. [DOI] [PubMed] [Google Scholar]
- 126.Burda E, Kraußer M, Fischer G, Hummel W, Müller-Uri F, Kreis W, et al. Recombinant Δ4,5-steroid 5β-reductases as biocatalysts for the reduction of activated C=C-double bonds in monocyclic and acyclic molecules. Advanced Synthesis & Catalysis. 2009;351:2787–2790. [Google Scholar]
- 127.Adeniji AO, Chen M, Penning TM. AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol. 2013 doi: 10.1016/j.jsbmb.2013.05.012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Drury JE, Di Costanzo L, Penning TM, Christianson DW. Inhibition of human steroid 5β-reductase (AKR1D1) by finasteride and structure of the enzyme-inhibitor complex. J Biol Chem. 2009;284:19786–19790. doi: 10.1074/jbc.C109.016931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bauman DR, Rudnick SI, Szewczuk LM, Jin Y, Gopishetty S, Penning TM. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67:60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 130.Faucher F, Cantin L, Luu-The V, Labrie F, Breton R. Crystal structures of human Δ4-3-ketosteroid 5β-reductase (AKR1D1) reveal the presence of an alternative binding site responsible for substrate inhibition. Biochemistry. 2008;47:13537–13546. doi: 10.1021/bi801276h. [DOI] [PubMed] [Google Scholar]
- 131.Chen M, Drury JE, Christianson DW, Penning TM. Conversion of human steroid 5β-reductase (AKR1D1) into a 3β-hydroxysteroid dehydrogenase by a single-point mutation E120H; an example of perfect enzyme-engineering. J Biol Chem. 2012;287:16609–16622. doi: 10.1074/jbc.M111.338780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Faucher F, Cantin L, Luu-The V, Labrie F, Breton R. Crystal structures of human Δ4-3-ketosteroid 5β-reductase defines the functional role of the residues of the catalytic tetrad in the steroid double bond reduction mechanism. Biochemistry. 2008;47:8261–8270. doi: 10.1021/bi800572s. [DOI] [PubMed] [Google Scholar]
- 133.Di Costanzo L, Drury JE, Christianson DW, Penning TM. Structure and catalytic mechanism of human steroid 5β-reductase (AKR1D1) Mol Cell Endocrinol. 2009;301:191–198. doi: 10.1016/j.mce.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]