

Abstract
Insertion sequences (ISs) are small transposable elements widespread in bacterial genomes, where they play an essential role in chromosome evolution by stimulating recombination and genetic flow. Despite their ubiquity, it is unclear how ISs interact with the host. Here, we report a survey of the orientation patterns of ISs in bacterial chromosomes with the objective of gaining insight into the interplay between ISs and host chromosomal functions. We find that a significant fraction of IS families present a consistent and family-specific orientation bias with respect to chromosomal DNA replication, especially in Firmicutes. Additionally, we find that the transposases of up to nine different IS families with different transposition pathways interact with the β sliding clamp, an essential replication factor, suggesting that this is a widespread mechanism of interaction with the host. Although we find evidence that the interaction with the β sliding clamp is common to all bacterial phyla, it also could explain the observed strong orientation bias found in Firmicutes, because in this group β is asymmetrically distributed during synthesis of the leading or lagging strands. Besides the interaction with the β sliding clamp, other asymmetries also play a role in the biased orientation of some IS families. The utilization of the highly conserved replication sliding clamps suggests a mechanism for host regulation of IS proliferation and also a universal platform for IS dispersal and transmission within bacterial populations and among phylogenetically distant species.
Keywords: transposable elements, insertion sequence, transposase, sliding clamp, DNA replication, bacterial chromosome
Introduction
Insertion sequences (ISs) can be considered autonomous because they encode the enzyme required for their own transposition, the transposase. They can move between genetic elements such as chromosomes, plasmids, and viruses, and across species boundaries (Chandler and Mahillon 2002; Siguier, Filee, et al. 2006). Although their autonomy makes ISs highly promiscuous elements, IS activity is linked to and can be regulated by various host processes (Nagy and Chandler 2004), such as the response to nutritional stress (Twiss et al. 2005), DNA damage (Pasternak et al. 2010), the SOS response, and, generally, chromosomal replication (Chandler 2009). The association between transposition and DNA replication has been documented, among others, for IS1 (Zerbib et al. 1985), IS10 (Roberts et al. 1985), IS50 (Yin et al. 1988), IS903 (Hu and Derbyshire 1998), Tn7 (Wolkow et al. 1996; Parks et al. 2009), Mu (Nakai et al. 2001), or IS608 (Ton-Hoang et al. 2010). Because the known transposition pathways often require host enzymatic functions (Curcio and Derbyshire 2003; Turlan et al. 2004; Parks et al. 2009; Jang et al. 2012), including DNA polymerases and other factors implicated in DNA replication, it is possible that transposition takes place concurrently with chromosomal replication, but no general mechanism linking these processes has been proposed.
The interplay between chromosomal replication and other processes, such as transcription or chromosomal segregation, shapes the organization of bacterial chromosomes (Rocha 2008; Sobetzko et al. 2012). It can result in specific patterns of localization or orientation of genes in the chromosome relative to the origin or replication and the direction of advance of the replication fork. For example, highly expressed genes tend to cluster near the origin of replication in fast-replicating bacteria (Couturier and Rocha 2006), and essential operons like those encoding the highly expressed rrn genes tend to be placed in the leading strand, possibly to prevent the instability caused by head-on clashes between the replication and transcription machineries (Rocha and Danchin 2003; Srivatsan et al. 2010; Paul et al. 2013). ISs are a special class of genetic elements because they could potentially be placed almost anywhere in the chromosome and in any orientation. However, if transposition is mechanistically linked to replication, the interplay between the two processes could be reflected in detectable chromosome-wide patterns. For example, IS608, a member of the IS200/IS605 family, presents an overall orientation bias in the chromosome caused in part by its requirement for single-stranded DNA (ssDNA), present preferentially in the lagging strand (Ton-Hoang et al. 2010).
Despite the documented interplay of ISs with DNA replication, a key to IS ubiquity could be its minimal interaction with the host. Indeed, there are few instances of documented contacts between transposases and host proteins. A recent study showed that TnsE, a Tn7-encoded factor that targets transposition preferentially to replicating conjugative plasmids, interacts with the β sliding clamp (Parks et al. 2009). β is an essential replication factor that provides processivity to DNA polymerases and coordinates numerous enzymatic activities in the replisome (López de Saro et al. 2003; Johnson and O'Donnell 2005). However, Tn7 encodes five proteins, and it was unclear whether interactions with the replisome could be generalized to transposases of less complex mobile elements.
In this study, we first analyzed the orientation of ISs in bacterial genomes with the goal of detecting patterns indicative of an interaction of these elements with chromosomal replication. Our findings suggest that up to 18 IS families show patterns of orientation in chromosomes that are consistent with an interplay between IS transposition and host DNA replication. Further, we show that the orientation biases are IS-family specific and not the result of selection for a specific orientation. Second, we searched for interactions between transposases and the β sliding clamp and find that up to nine different transposases or associated factors interact with this host protein. Our combined results suggest that transposition and replication could be linked and take place coordinately.
Materials and Methods
Genomic Data Set and Computational Pipeline
File collections containing orientation and coordinates of protein coding genes (*.ptt), predicted protein sequences (*.faa), and chromosomal nucleotide sequences (*.fna) of partially and completely sequenced prokaryotic genetic elements were downloaded from the bacterial section of the National Center for Biotechnology Information (NCBI) Genome database, on October 24, 2012, as well as a summary file containing a table that linked accession numbers, replicon type (chromosome, plasmid), and taxonomic name. A computational pipeline written in Perl allowed navigation across the whole collection of files and directed the execution of a number of public domain or in house developed applications to detect, classify, and count IS elements according to their orientation, as described in the following sections. The working, curated data set consisted of 2,074 completely sequenced, circular, bacterial chromosomes, out of which 1,806 contained at least one IS (harbored by 1,685 species or strains).
IS Detection and Classification
The collection of predicted proteins from the genomic data set (6,055,750 sequences) was aligned with HMMER 3.0 against the Pfam 26.0 database (pfam.sanger.ac.uk, last accessed March 19, 2014) of domain profiles (Punta et al. 2012), using domain-specific score thresholds to filter the hits. The output of HMMER was processed with a Perl script to reconstruct protein architectures using a positional competition strategy to assemble the predicted protein domains allowing no overlaps. IS-related proteins were identified by comparing the new annotations against a list of 286 architectures that were considered characteristic of proteins encoded by IS elements and that were composed by a restricted collection of Pfam domains (supplementary table S1, Supplementary Material online). The architecture list was generated by manually extracting IS-encoded protein descriptions from the Pfam database and characterizing the domain structure of IS-encoded proteins from the ISfinder database (www-is.biotoul.fr, last accessed March 19, 2014) (Siguier, Perochon, et al. 2006; Punta et al. 2012). We were able to identify 80,443 IS-associated genes. Once IS-related proteins had been identified in the set of bacterial genomes, IS elements were predicted following a strategy, articulated in four steps, that took into account that ISs can be composed of several genes and that they can appear in chromosomes as tandem insertions, making difficult the definition of their boundaries. In the first step, clusters of consecutive IS genes (separated by intergenic distance ≤ 500 bp) were identified in all genomes to calculate distance distributions for all possible pairs of IS-related gene types (as defined by the architecture of the corresponding gene products). In the second step, cluster detection was repeated, this time restricting the allowed intergenic distances to gene pair-specific distance ranges, deduced from the previous step (mean ± 2 SD). Clusters detected in this step had ten genes at most. In the third step, the resulting collection of clusters was used to manually derive a list of 209 clusters that were accepted as representatives of the genetic organization of complete IS elements, on the basis of their correspondence to described IS structures (supplementary table S2, Supplementary Material online), abundance (assuming that highly abundant and distributed clusters should correspond to complete IS elements), and length (in terms of number of genes). Each of these clusters was classified as belonging to a particular IS family. Accepted clusters had three genes at most. In the fourth step, each IS gene cluster detected in the second step was decomposed into all possible collections of nonoverlapping accepted subclusters to identify the collection that maximized the length of subclusters. Each subcluster from the optimal collection was then assigned to a particular IS family following the correspondences established in the list of accepted clusters (supplementary table S2, Supplementary Material online). A total of 57,515 subclusters were detected, each of them representing a complete IS, that comprised 69,438 (86%) of the IS-related genes. The remaining genes could correspond to chimeric or degenerated IS elements or be composed of protein architectures with ambiguous correspondence to IS families.
To validate the IS detection and classification system of our computational pipeline, we performed two tests. For the first test, we estimated transposase-related gene prediction recall relative to the annotations compiled in PTT files. The total number of genes, in the set of 2,074 completely sequenced, circular, bacterial chromosomes, whose annotation in PTT files contained the string “transpos” was 65,230. A total of 55,800 of them were identified as IS-related genes by the pipeline, which implies an 85% recall rate. Thirty-four percent of the recovered genes had been annotated simply as “transposase” in PTT files. The pipeline identified 24,643 additional IS encoded genes. For the second test, we determined IS family classification accuracy by comparison against the complete set of annotated prokaryotic chromosomes available, in April 2013, from the genomic component of ISfinder (ISbrowser) (Kichenaradja et al. 2010). The comparison involved 866 genes, coming from 33 chromosomes, that had been described as constitutive of IS elements by both ISbrowser and by our computational pipeline. The fraction of genes, considered globally, in which IS family affiliations coincided was 88%. The fraction of genes, by IS family, in which IS family affiliations coincided had average and median values of 79% and 100%, respectively.
Test on the Orientation Distribution of IS elements in Chromosomes
The orientation of each IS element was defined by the orientation of its transposase gene relative to the local GC skew sign. GC skew [(G − C)/(G + C)] reflects an asymmetric nucleotide composition of the leading and lagging strands in Bacteria. The mechanism by which this asymmetry is created is unclear, but it could be related to the mutational or selective pressures on each DNA strand (Rocha et al. 2006). In genomes that have not suffered recent rearrangements, the two replicores have a different GC skew sign. GC skew was taken as a proxy for the direction of movement of the replication fork to correct for the effect of recent genome rearrangements. For each genome, GC skew was calculated with a Perl script over nonoverlapping 3,001-bp-long genome segments. Then, a second script identified genome blocks with a minimal length of 10,000 bp that were composed of consecutive segments having the same GC skew sign but allowing the inclusion of segments with the opposite sign if they were shorter than 10,000 bp. Two blocks with different GC skew sign, corresponding to the replicores defined by the positions of the origin of replication and the termination site, were identified in 60% of the chromosomes. The occurrence of multiple blocks in the remaining genomes can be explained in part as consequence of recent genomic rearrangements. IS element orientation relative to local sign of GC skew was defined as same (s) when the coding strand of the transposase gene had the same sign as that of the container GC skew block and anti (a) when the signs were different. The output of the pipeline consisted of a pair of orientation counts (s, a), for each genome and for each IS family, describing the number of IS elements presenting either of the two possible orientations. To test whether IS elements were distributed randomly in the pair of orientation classes, counts were contrasted against a random binomial distribution with P = 0.5 and a two-tailed P value was calculated (supplementary table S3, Supplementary Material online).
Tests on the Orientation Distribution of IS Elements at Phylum Level
We then set out to determine whether IS families showed a bias at Phylum level by combining information from the chromosomes in each Phylum. The P values obtained in the tests on the orientation distribution of IS elements at chromosomal level (cumulative binomial probabilities, calculated as described earlier) were taken as a measure of the lack of randomness in IS orientation for individual species. To obtain a test statistic representing a combined measure of the lack of orientation randomness for each IS family at Phylum level, we obtained the product of the P values calculated on the corresponding chromosomes, following a strategy similar to that of Bailey and Gribskov (1998). To minimize database bias, we chose randomly only one chromosome per species (1,215 chromosomes; see supplementary table S3, Supplementary Material online, chromosomes labeled red). Because the distribution of such statistic is unknown, statistic values were contrasted against distributions generated after 106 sample, IS family-specific Monte Carlo simulations that assumed the random orientation of IS elements (Besag and Clifford 1991). Simulated data sets had the same IS distribution of the original data in terms of number of chromosomes, number of IS copies per chromosome, and number of IS copies per IS family. Left-tailed P values were calculated as the fraction of simulated samples whose value was equal or lower than the value of the statistic calculated on the original data.
Detection of β-Binding Motifs in Escherichia coli Transposases
To search for the β-binding motif among Escherichia coli transposases, we downloaded a collection of 2,578,009 E. coli protein sequences from the NCBI database (July 9, 2012) from which we identified 53,235 IS-related proteins after assembling Pfam-based architecture descriptions as described earlier. We then used BlastClust (Altschul et al. 1997) to generate a subset consisting of 10,980 unique sequences. Both the nonredundant set of E. coli IS-encoded sequences and the collection of 80,443-associated genes derived from the genomic analysis were analyzed with the application Fuzzpro of the EMBOSS package (Rice et al. 2000) to explore the occurrence of β-binding motifs (QLSLF and selected derivatives). Hits were filtered with a Perl script and then manually curated.
β and MbaPCNA-Binding Assays
See supplementary information, Supplementary Material online, for protein purification methods. β was labeled with Alexa Fluor 350 C5-maleimide (Life Technologies) as recommended by the manufacturer. Maleimide-labeling results in one label per β monomer at Cys-333 and do not alter its interaction with DNA polymerases or its activity in replication assays (Griep and McHenry 1988; López de Saro et al. 2003). Peptides were purchased from Thermo Fisher Scientific GmbH (Ulm, Germany).
We used two assays to determine if the transposase-derived peptides bind to β and the location of the interaction on the surface of β. The binding assay directly tested the interaction of β with the peptide, whereas the second tested the ability of the peptide to compete a preformed complex of β with the little-finger domain of DNA polymerase IV. This domain binds strongly to β, and its three-dimensional structure is known (Bunting et al. 2003).
The binding assay using magnetic beads was performed in a volume of 50 μl using 700 μg of streptavidin-coated magnetic beads (Sigma Aldrich) in Buffer M (50 mM TrisCl, 100 mM NaCl, 5% glycerine, pH 7.5). Biotinylated peptides (440 μM) were mixed with the beads, incubated (30 min, 25 °C), and washed three times before addition of labeled β (4.5 μM) or MbaPCNA (3 μM). After incubation (10 min, 25 °C), unbound β was removed by three washes with the same buffer. The reactions were stopped by addition of 1% SDS, subjected to SDS-PAGE electrophoresis, and β visualized on a UV transilluminator.
The competition assay was performed in 20 μl in Buffer R (40 mM Tris acetate, 1 mM ethylenediaminetetraacetic acid [EDTA], 3% glycerine, 12% DMSO, pH 8.3) supplemented with labeled β (0.55 μM). GST-Pol IVLF (3 μM) and the different peptides (100 μM) were added as indicated. Reactions were incubated (10 min, 25 °C) and loaded on a native gel (7.5% acrylamide/bis 37.5:1, 40 mM Tris acetate, 1 mM EDTA, 10% glycerine, pH 8.3). Electrophoresis (80 min, 16 mA) was performed at 4 °C in TAE buffer. The reaction products were visualized on a UV transilluminator.
Results
Orientation Biases of IS Families in Bacterial Chromosomes
To gain insight into the possible interaction between ISs and the host, we analyzed patterns of orientation of ISs in fully sequenced bacterial chromosomes. ISs were detected and classified following the ISfinder database after producing Pfam-based annotations for all predicted protein sets (see Materials and Methods and supplementary table S2, Supplementary Material online, for IS classification scheme and nomenclature). The orientation of each IS element was defined by the orientation of its transposase. IS orientation patterns were investigated for each chromosome and each IS family by scoring the number of IS elements having either orientation relative to the sense of movement of the replication fork as defined by the local GC skew sign to take into consideration possible recent chromosomal rearrangements. We analyzed the orientation of 57,515 ISs in 1,806 completely sequenced circular bacterial chromosomes (supplementary table S3, Supplementary Material online). We further analyzed only those cases in which six or more copies of a given IS family were found per chromosome and, to avoid database redundancy, in only one strain for each bacterial species (supplementary table S4, Supplementary Material online). We observed 153 cases of significant orientation bias (P < 0.05) of IS families in chromosomes. These could mostly be assigned to a subset of eight IS families for which there was a bias in a large proportion of chromosomes containing six or more copies of the IS. Thus, families IS200 (32% of chromosomes), IS200/IS605 (25%), IS607 (35%), and ISNCYa (20%) tend to be significantly biased for orientation in favor of the sense of advance of the replication fork (i.e., leading strand) in many chromosomes, whereas families IS91 (25%) and ISL3 (16%) show consistent bias for orientation against the sense of movement of the replication fork (i.e., lagging strand). Families IS5a (11%) and IS110 (9%) showed no clear trend: In some chromosomes, the bias was toward a location in the leading, whereas in others, it was toward location in the lagging strand. Mapping of the biased IS families on the chromosomes showed that most IS insertions were well distributed and likely to be the result of independent transposition events (fig. 1A). We found numerous chromosomes in which two different IS families were significantly biased, either in the same or in opposite orientations (fig. 1B), in each case reflecting the pattern of IS bias specific for each family.
Fig. 1.—

IS orientation biases in bacteria. (A) Representative examples for ten IS families biased for their orientation in bacterial chromosomes. P values are given in supplementary table S3, Supplementary Material online, for each IS family and chromosome. The chromosomes are not drawn to scale, and the trace represents the GC skew drawn using the program Artemis of the Sanger Institute (Rutherford et al. 2000) with a window size of 10 kb. Regions of positive GC skew (green) or negative GC skew (magenta) represent the two replichores. The arrows represent individual ISs inserted in the chromosome. For positive GC skew (green), an arrow pointing downward represents an IS oriented in the direction of movement of the replication fork, and an arrow pointing upward represents an IS oriented against the direction of movement of the replication fork. The opposite applies for regions of negative GC skew. (B) Representative examples of chromosomes with two biased IS families of opposite orientations. For Photobacterium profundum, these are IS200 (blue arrows) and IS630 (red); for Streptococcus salivarius, IS200 (blue) and ISL3 (green); and for S. pyogenes, IS3 (gray) and ISAs1 (magenta).
Many of the IS families analyzed (IS1, IS3, IS4a, IS4b, IS4c, IS5b, IS5c, IS5d, IS6, IS30, IS66a, IS66b, IS256, IS481, IS630, IS701, IS982, IS1182, IS1380, IS1595, IS1634, ISAs1, ISAzo13, ISNCYb, Mu, Tn3, and Tn7) showed bias in few or no chromosomes. In some cases, the low number of chromosomes in which the IS was detected (ISAzo13) or the low number of ISs per chromosome (Mu, Tn3, and Tn7) precluded the detection of statistically significant differences. Also, it is likely that bias in IS families that had proliferated quickly and abundantly in chromosomes were easier to detect than in IS families which propagated slowly and in low numbers, as chromosomal reorganizations would blur any original orientation bias. We therefore sought to determine whether orientation biases could be generalized for higher taxonomic levels by considering the counts derived from groups of chromosomes in which a given IS family had been detected. Again, we included only one strain for each bacterial species (supplementary table S3, Supplementary Material online, chromosomes marked in red). We calculated a new statistic for each IS family and compared its value against IS family-specific distributions derived from Monte Carlo simulations (see Materials and Methods). In addition to the families detected by observation of individual chromosomes, we found patterns of statistically significant biased orientation in ten IS families (table 1). Thus, IS66a, IS256, and ISAs1 showed a tendency toward placement in the leading strand; IS3, IS6, IS30, IS481, and ISNCYb showed a tendency to be located in the lagging strand; and IS630 and Tn3 presented a mixed behavior. Further, we found that for many IS families, the biased patterns of orientation were, surprisingly, Phylum dependent. Thus, orientation bias is highly significant (P < 10−2) for ten IS families in Firmicutes but only for three IS families in Proteobacteria and two in Actinobacteria. No biased IS families were found in any other phyla (see supplementary table S6, Supplementary Material online, for Bacteroidetes, Cyanobacteria, and Spirochaeta). Orientation bias in Firmicutes also was particularly strong, with P values < 10−5, for seven IS families. Analysis of the relative abundance of IS families in the different groups revealed that the orientation biases in Firmicutes did not arise from higher numbers of certain IS families in the chromosomes of these organisms (table 1).
Table 1.
Statistical Significance for the Nonrandom Orientation of IS Elements
| Proteobacteria |
Actinobacteria |
Firmicutes |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Chr. | IS | Orient. | Chr. | IS | Orient. | Chr. | IS | Orient. | |
| IS1 | 33 | 1144 | 0.295 | — | — | — | — | — | — |
| IS3 | 358 | 2601 | 0.273 | 100 | 968 | 0.380 | 159 | 1115 | 5.95 × 10−4 |
| IS4a | 43 | 251 | 0.220 | 18 | 83 | 0.260 | — | — | — |
| IS4b | 17 | 82 | 0.116 | — | — | — | 8 | 21 | 0.511 |
| IS4c | 10 | 84 | 0.0903 | — | — | — | — | — | — |
| IS5a | 135 | 735 | 0.749 | — | — | — | 83 | 439 | 5.00 × 10−6 |
| IS5b | 175 | 969 | 0.336 | 55 | 344 | 0.360 | 8 | 27 | 0.375 |
| IS5c | 51 | 192 | 0.684 | — | — | — | — | — | — |
| IS5d | — | — | — | 24 | 100 | 0.466 | 7 | 47 | 0.233 |
| IS6 | 50 | 153 | 0.367 | 9 | 42 | 0.815 | 35 | 104 | 1.21 × 10−3 |
| IS21 | 203 | 787 | 0.0859 | 68 | 218 | 0.0183 | 161 | 509 | 0.380 |
| IS30 | 80 | 289 | 0.241 | 53 | 186 | 0.200 | 91 | 500 | 6.00 × 10−6 |
| IS66a | 135 | 640 | 0.0876 | — | — | — | 33 | 97 | 0.0193 |
| IS66b | 13 | 44 | 0.849 | 4 | 36 | 0.811 | 7 | 26 | 0.667 |
| IS91 | 63 | 174 | 6.53 × 10−3 | — | — | — | 11 | 26 | 0.138 |
| IS110 | 260 | 1442 | 6.10 × 10−5 | 73 | 439 | 0.345 | 112 | 721 | <1 × 10−6 |
| IS200 | 221 | 651 | <1 × 10−6 | — | — | — | 89 | 270 | <1 × 10−6 |
| IS200/IS605 | 27 | 58 | 0.0901 | 11 | 23 | 0.399 | 38 | 90 | 9.40 × 10−5 |
| IS256 | 152 | 711 | 0.0140 | 79 | 578 | 0.510 | 97 | 564 | 0.0161 |
| IS481 | 21 | 260 | 0.0357 | 43 | 147 | 0.493 | — | — | — |
| IS607 | — | — | — | 8 | 30 | 1.00 × 10−6 | 28 | 103 | <1 × 10−6 |
| IS630 | 145 | 1061 | 0.0398 | 39 | 252 | 0.379 | 25 | 120 | 0.565 |
| IS701 | 47 | 234 | 0.223 | 33 | 168 | 0.118 | 6 | 26 | 0.204 |
| IS982 | 30 | 285 | 0.813 | — | — | — | 24 | 145 | 0.653 |
| IS1182 | 86 | 291 | 0.0545 | 26 | 82 | 0.294 | 79 | 386 | 0.312 |
| IS1380 | 28 | 139 | 0.204 | 16 | 39 | 0.339 | 15 | 73 | 0.673 |
| IS1595 | 95 | 333 | 0.340 | 6 | 26 | 0.157 | 7 | 33 | 0.519 |
| IS1634 | 8 | 49 | 0.227 | — | — | 15 | 95 | 0.282 | |
| ISAs1 | 48 | 201 | 0.0142 | 14 | 249 | 0.756 | 8 | 54 | 0.0138 |
| ISL3 | 91 | 336 | 0.343 | 56 | 333 | 0.270 | 84 | 496 | <1 × 10−6 |
| ISNCYa | 68 | 221 | 0.139 | — | — | — | 53 | 203 | <1 × 10−6 |
| ISNCYb | — | — | 10 | 72 | 9.69 × 10−3 | — | — | — | |
| Tn3 | 70 | 138 | 0.0186 | 20 | 43 | 0.237 | — | — | — |
| Tn7 | 46 | 61 | 0.0875 | — | — | — | — | — | — |
Note.—The table presents, for the groups Proteobacteria, Actinobacteria, and Firmicutes, the number of chromosomes in which a particular IS family was detected, the total number of copies, and the orientation test (Orient.), with P values representing the probability of obtaining, in 106 sample Monte Carlo simulations, a value as extreme as the one that was calculated from the observed data (see Tests on the orientation distribution of IS elements at Phylum level under Materials and Methods) are presented in the table. See supplementary table S2, Supplementary Material online, for nomenclature and IS classification scheme. The analyzed genomes are those marked in red in supplementary table S3, Supplementary Material online (one strain per species to avoid database redundancy). P values (P < 0.05 in bold, P < 10−2 shaded gray) indicate an asymmetric distribution of IS elements in chromosomes in terms of their orientation relative to the local GC skew sign. IS families with less than 20 copies detected were omitted.
IS Orientation Biases Are Not Generated by Postinsertion Selection
Our analysis of IS bias in chromosomes revealed that, for ten IS families, the Firmicutes showed a strong bias. Comparative genomics has shown that general gene orientation is nearly neutral for Proteobacteria or Actinobacteria but tends to be highly biased for Firmicutes and Tenericutes, where 78% of genes are co-oriented with the movement of the replication fork (Rocha 2002). It has been speculated that this phenomenon arises to prevent clashes between the DNA replication and transcription machineries, which can eventually lead to genomic instability, and substantial in vitro and in vivo experimental evidence seems to support this model (Wang et al. 2007; Srivatsan et al. 2010; Paul et al. 2013). However, it is not known why the orientation bias is especially strong in Firmicutes and Tenericutes and not in the other groups (see later for biochemical differences between the replisomes of the different groups).
Because ISs are typically very recent additions to chromosomes and do not encode essential or highly expressed genes, it is unlikely that they are subject to selection for any given orientation. However, to determine whether the observed IS orientation biases could just reflect a general preference for insertion in a specific orientation or the effect of selection, we determined global orientation for non-IS encoded and IS-encoded genes in 1,727 bacterial chromosomes (fig. 2). As previously observed (Rocha 2002), we detected a strong trend for non-IS genes to be placed in the leading strand (i.e., direction of movement of the replication fork) in Firmicutes and Tenericutes but not in other groups (fig. 2A). However, we find that IS-related genes (transposases and associated factors) show no orientation bias when considered globally in each chromosome, even in Firmicutes (fig. 2B). Further, there is no correlation between orientation bias for non-IS genes and IS genes in Firmicutes chromosomes (fig. 2C), as it would have been expected if the processes that generate orientation biases were linked. This finding does not present a contradiction with our earlier result of many IS families showing strong orientation bias, as some IS families are consistently oriented in favor and others against replisome movement. In consequence, no net bias is found when considering IS genes globally in chromosomes. Indeed, numerous examples can be found of chromosomes in which different biased IS families show opposite orientation trends (fig. 1B).
Fig. 2.—
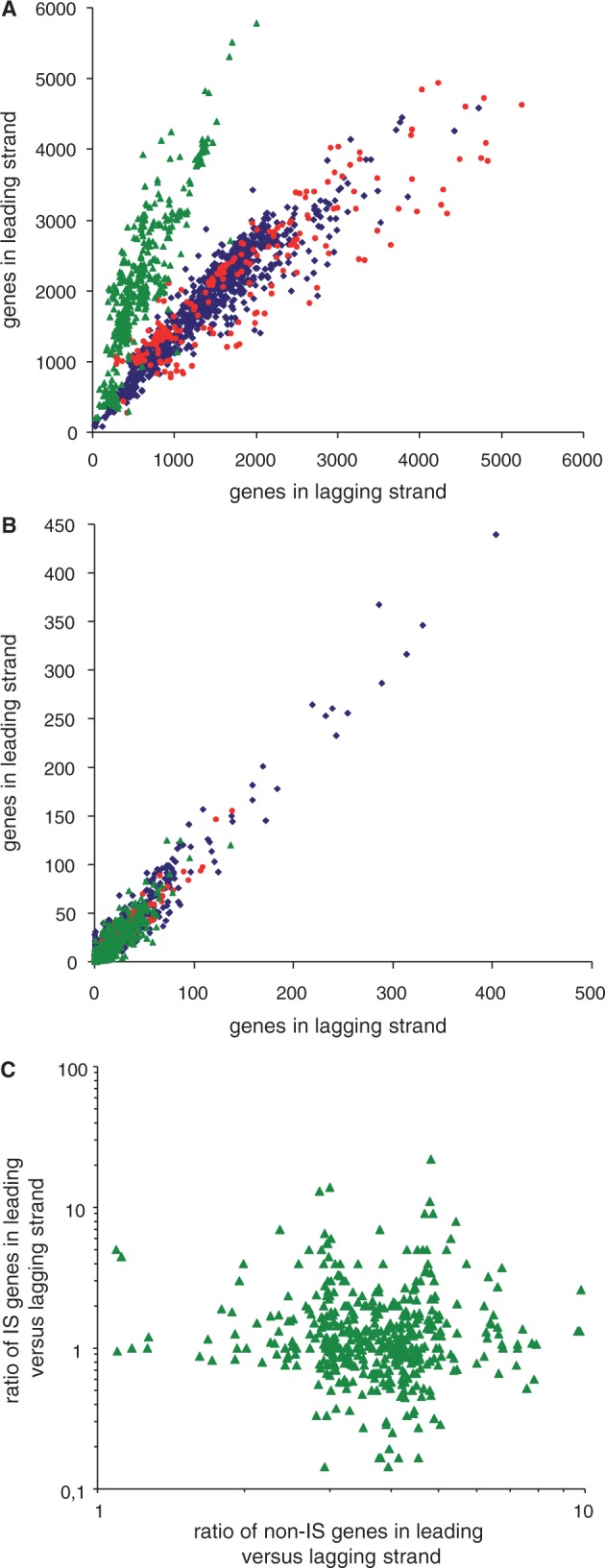
Global gene orientation for 1,727 circular bacterial chromosomes. (A) General gene orientation excluding IS-related and RNA-encoding genes. The chromosomes of four major bacterial phyla are represented: Proteobacteria (blue diamonds, 1,009 chromosomes, 3,067,992 genes, leading/lagging strand ratio = 1.07, R2 = 0.89), Actinobacteria (red circles, 224 chromosomes, 837,682 genes, ratio = 0.79, R2 = 0.82), and Firmicutes and Tenericutes (green triangles, 494 chromosomes, 1,287,236 genes, ratio = 2.58, R2 = 0.76). (B) Orientation of IS-related genes (transposases and associated factors) in genomes for the Proteobacteria (44,504 transposase genes, ratio = 1.05, R2 = 0.94), Actinobacteria (9,202 transposase genes, ratio = 1.00, R2 = 0.92), and Firmicutes and Tenericutes (17,255 transposase genes, ratio = 0.97, R2 = 0.79). (C) Correlation between the ratios of IS-gene orientation and no-IS gene orientation is inexistent in Firmicutes.
Taken together, our data strongly suggest that IS orientation bias for the leading or the lagging strand is unlikely to be the result of selection for co-orientation of IS genes and fork movement but rather specific and intrinsic to the structure and mechanism of transposition of each IS family.
Interaction of Transposases with the β Sliding Clamp
The antiparallel nature of DNA imposes a number of asymmetric features to the DNA replication machinery, its movement, and the synthesis of each strand. The asymmetries in turn lead to various strand-specific differences and biases. For example, IS200 requires ssDNA for transposition, and the excess of ssDNA in the lagging strand could directly explain the strong bias of this IS family in most chromosomes in Proteobacteria and Firmicutes, as described by Ton-Hoang et al. (2010). However, an excess of ssDNA would not account for the orientation bias found for other IS families in Firmicutes but absent in Proteobacteria. Although mechanistically highly similar, the replication fork of Proteobacteria and Firmicutes differ in some relevant ways: In E. coli, only one DNA polymerase (DnaE) is responsible for processive synthesis, whereas in Bacillus subtilis, there are two (PolC and DnaE) (Sanders et al. 2010). Further, recent stoichiometric analyses of the E. coli and B. subtilis replisomes have shown that although in E. coli there are only 3–6 β sliding clamps per fork (Reyes-Lamothe et al. 2010), presumably due to fast recycling of β to new Okazaki fragments, β accumulates during lagging strand synthesis in B. subtilis (up to 200 β/fork, forming “clamp zones”) (Su'etsugu and Errington 2011). Therefore, we reasoned that if transposases or associated factors interacted with β, the difference in β amounts associated with leading and lagging strand synthesis could account for biases in IS insertion in Proteobacteria and Firmicutes. Within the replication fork, the β sliding clamp interacts with a large number of enzymes, which share a short and poorly conserved binding motif (consensus: Q-L-S-L-F/L, Q1, and L4 being the most strongly conserved residues) (Dalrymple et al. 2001). We therefore searched transposases and accessory factors for this specific sequence, focusing on E. coli because of the large number of sequenced genomes and because the interactions of β in this model organism have been extensively analyzed (López de Saro et al. 2003). In contrast to E. coli, no ISs have been detected in the B. subtilis 168 chromosome. Because transposases are frequently toxic and insoluble when overexpressed, we exploited the fact that β-binding motifs are often located on highly flexible, peptide-like, structures at the C-terminus of the protein (Dalrymple et al. 2001; Bunting et al. 2003; López de Saro et al. 2003). Further, unlike most protein–protein interactions, which implicate relatively large surface areas, interactions with β are mostly circumscribed to the motif binding to a hydrophobic pocket on β (Georgescu et al. 2008). We synthesized N-biotinylated peptides (20 aa) derived from sequences of transposases containing putative β-binding motifs (fig. 3A), as well as peptides in which Q1 and L4 had been changed to alanine, and assayed them for β binding by two methodologies. First, we bound the peptides to streptavidin magnetic beads and tested their ability to bind and retain fluorescently labeled β. We find that peptides derived from transposases belonging to nine IS families (IS5a, IS30, IS66a, IS91, IS200, IS1380, ISL3, ISNCYa, and Tn7) bind to β and that their mutated variants do not (fig. 3B).
Fig. 3.—

Interaction between transposases and the β sliding clamp. (A) List of peptides used in the binding assays, aligned at the β-binding motif. The Pol IV peptide was used as a positive control. Peptides derived from transposases found in Escherichia coli chromosomes. Residues corresponding to the consensus β motif are in bold type and those mutated to alanine, underlined. Two peptides (a and b) were designed for different regions of the TnpC protein of IS66a (see E). Two homologous peptides (1 and 2) were designed corresponding to variants of IS200 transposase. NCBI accession numbers for protein sequences are shown. (B) Peptides were coupled to streptavidin-coated paramagnetic beads and used to retain purified Alexa 350-labeled E. coli β. In each panel, the native (wt) and the mutated (mut) peptides were used. (C) Fluorescently labeled β whose mobility was retarded in a native gel by interaction with GST-Pol IVLF was challenged with an excess of transposase-derived peptides, as indicated (See Materials and Methods). (D) N-biotinylated peptides derived from the C-terminus of Methanosarcina barkeri IS200 were bound to streptavidin-coated magnetic beads as in B and used to probe interaction with M. barkeri PCNA (left panel) or E. coli β (right panel). The PCNA consensus motif (bold) and the residues mutated in the mutated peptide (underlined) are marked. (E) Structure of IS66 and diversity in sequence and location of β-binding motifs. The motif can be present in TnpB, TnpC, or in both, and in TnpC, it can be located upstream or downstream of the leuzine zipper domain (LZ). In some Bacilli (Firmicutes), the LZ domain is an independent open reading frame and presents the β-binding motif at its C-terminus (see supplementary table S5, Supplementary Material online).
To determine the interaction site on the surface of β, we tested the peptides in a competitive, native gel mobility-shift assay, using the C-terminal domain (“little finger”) of E. coli DNA polymerase IV (PolIVLF). PolIVLF binds strongly to a hydrophobic pocket on the surface of β that is also the binding site for the other four polymerases in E. coli and for various DNA repair factors (Bunting et al. 2003; López de Saro et al. 2003). Therefore, this competition assay would assure high specificity in the interaction, despite the relative simplicity of the consensus Q-L-S-L-F/L motif. The peptides were tested for their ability to disrupt the β·GST-PolIVLF complex by adding them in a molar excess to the reaction and then separating the products. We find that all peptides that bound to β in the streptavidin-binding assay were also capable of binding β in competition with GST-PolIVLF (fig. 3C), suggesting that they interact with β in the same fashion as other β ligands. The mutant peptide variants, again, were unable to compete with GST-PolIVLF.
The β-binding motifs found in these transposases are conserved (supplementary table S5, Supplementary Material online), suggesting that interaction with β is widespread across phylogenetic groups. Conservation is extensive to Archaeal ISs, but in this case, the PCNA motif is present (consensus Q-x-x-L/I-x-x-F-F) (fig. 3D and IS200 in supplementary table S5, Supplementary Material online). Purified and labeled Methanosarcina barkeri PCNA binds strongly to a peptide derived from MbaIS200 transposase (fig. 3D). Because the PCNA motif is a related variant of the β motif, we tested if MbaIS200 could interact with E. coli β. Indeed, MbaIS200 interacts with E. coli β, and point mutants of this peptide no longer bind to MbaPCNA or β (fig. 3D). Our results suggest that interaction with the replisome would not likely limit the transmission of archaeal ISs to bacterial chromosomes, and few mutations would be required to adapt a bacterial transposase to the archaeal replication machinery.
Discussion
We have performed a general survey on sequenced bacterial genomes to explore patterns of IS insertion that could reveal interactions between transposition and the synthesis of the host chromosome. We find that some IS families reveal orientation patterns that significantly deviate from randomness and that are consistent with an interaction with replication (fig. 1), specifically in Firmicutes (table 1). The data strongly suggest that postinsertion selection is not a general cause of the observed orientation bias among ISs but rather that orientation trends derive from the interplay between the transposition mechanism of each IS family and host chromosomal replication. Independently, we have analyzed a possible interaction between transposases of various IS families and the β sliding clamp, an essential replication factor. We find that up to nine different transposases can bind to β, suggesting that this is a general mechanism of interaction of transposases with the host.
The Source of IS Orientation Biases in Chromosomes
The main hypothesis guiding our analysis of ISs in bacterial genomes was that, if IS insertions are associated with host replication, they could, given some conditions, present orientation patterns in the chromosome. Importantly, our study was severely limited by various factors, namely 1) the heterogeneity present within some IS families (e.g., variability in the orientation of the transposase gene with respect to other elements within the IS); 2) the requirement of a relatively high number of ISs per chromosome to achieve statistical significance (orientation patterns in IS families with low copy number per chromosome could be undetectable); 3) our inability to distinguish between IS insertions resulting from transposition within the chromosome from those incorporated into chromosomes within large blocks of DNA (“genomic islands,” prophages); and 4) the uncertainty derived from using current GC skew as a proxy of replication fork orientation, as any chromosomal rearrangements would tend to randomize any orientation bias.
Despite the mentioned limitations, our analysis of IS orientation in bacterial chromosomes revealed strong orientation bias (P < 10−2) for three IS families in Proteobacteria, two in Actinobacteria, and ten in Firmicutes (table 1). What could be the underlying biological phenomenon generating a biased orientation of ISs in chromosomes? Biases could have been generated by 1) preferred insertion of ISs in nonrandomly oriented sequences in the chromosome, 2) postinsertion selection favoring specific orientation, or 3) by transpososome interaction with an asymmetrical structure within the replication fork. The first possibility, target sequence specificity, has been observed for Tn7, in which the Tn7-encoded protein TnsD directs insertions to a specific location on the chromosome near Ori (attTn7) (Waddell and Craig 1988). Also IS110, a family for which we find strong orientation bias in Proteobacteria and Firmicutes, could possibly reflect oriented insertion into targets such as REP sequences (Tobes and Pareja 2006), the terminal repeats of IS21 (Partridge and Hall 2003), or the recombination sites (attC) of integron gene cassettes (Tetu and Holmes 2008; Post and Hall 2009), which could themselves be biased. However, most ISs show little or weak sequence specificity (Chandler and Mahillon 2002), and the highly distributed placement of most ISs in chromosomes of phylogenetically diverse bacteria renders unlikely the possibility of generalized sequence targeting as a source of bias for most IS families.
The second mechanism, postinsertion selection, could possibly generate a bias if, for example, transcription from upstream genes altered expression of the transposase and this had an effect in viability or if the IS altered regulation of neighboring genes (Plague 2010). However, our analysis of orientation of ISs in bacterial chromosomes does not reveal any global orientation bias of the IS population in chromosomes, even in those with a very strong gene orientation bias, such those of Firmicutes (fig. 2).
Finally, an interaction between transposition and replication could also generate an orientation bias, as it has been recently described in detail for two IS families: T7 (Parks et al. 2009) and IS200 (Ton-Hoang et al. 2010). In the case of T7, an accessory factor, TnsE, interacts physically with the β sliding clamp and targets the T7 transposase preferentially to conjugative plasmids. A study of the orientation of many independent chromosomal insertion events revealed a clear TnsE-dependent, replication-dependent bias (Peters and Craig 2001). On the other hand, IS200 shows a clear orientation bias in bacterial chromosomes, explained by its requirement for ssDNA found mainly in the lagging strand at replication forks (Ton-Hoang et al. 2010). However, it is important to note that interaction with replication does not necessarily impose an orientation bias (see later).
Our observations (fig. 1, table 1) suggest that there is a replication-dependent bias in some IS families and that whether the bias is in favor or against the direction of movement of the replication fork is IS-family dependent. For many families, this bias is strong in Firmicutes but absent in Proteobacteria and Actinobacteria. Because it is unlikely that ISs are mechanistically different in Firmicutes, when compared with the other phyla, this result strongly suggests that ISs insertions in Firmicutes behave differently that in other phyla as direct consequence of distinct chromosomal replication dynamics in this group.
The β Sliding Clamp as a General Link between Transposition and Replication
In our search for host factors that interact with transposition, we performed a systematic search for the β-binding motif in transposases. We then assayed synthetic peptides derived from E. coli transposases by using the E. coli β clamp found in this organism. Our approach was limited by 1) our ability to recognize the canonical β interaction motif in E. coli transposases, as variation within the motif is high, even in well-characterized enzymes (Dalrymple et al. 2001), 2) the sensitivity of the biochemical techniques, and 3) the absence of some IS families in sequenced E. coli genomes. However, the finding of the motif for interaction with β in nine different transposase families suggests a possible general mechanistic link between IS transposition and chromosomal replication.
In addition to providing processivity to DNA polymerases, the main role of sliding clamps in most studied systems, such as Okazaki fragment processing or DNA polymerase switching during lesion bypass, is targeting enzymes to active replication sites to couple and coordinate their activities. However, and given the diversity of transposition mechanisms, it is possible that β is used in distinct ways by the different transposases. It has been proposed that β targets Tn7 to replication in conjugative plasmids as a mechanism for dissemination to new hosts (Peters and Craig 2001; Parks et al. 2009). In the case of IS200, binding to β could help the transposase to localize to sites with increased amounts of ssDNA, such as replication forks or repair sites, thus increasing the efficiency of the excision or insertion processes. If transposition requires DNA synthesis by a DNA polymerase, β would allow polymerase recruitment, because the five DNA polymerases present in E. coli require β (López de Saro et al. 2003). β could be bound by the transposase first to initiate the reaction and then used to target the appropriate polymerase in a subsequent step, as in the case of transposases that use a copying mechanism.
Our results indicate that IS families with diverse transposition mechanisms (DDE, Y1-, Y2-, and S-transposases) could interact with the replisome similarly, suggesting convergent evolution for interaction with the host (supplementary table S5, Supplementary Material online). For example, strong β-binding motifs can be found in TnpB of IS66a, in two different positions within TnpC, or in both proteins (fig. 3E and supplementary table S5, Supplementary Material online). Similarly, a putative motif can be found in the C-terminus of OrfB of IS200/IS605 in Cyanobacteria (supplementary table S5, Supplementary Material online) but seems absent in other phyla at that position and variants of Tn7 harbor β-binding motifs in proteins TnsC or TnsE (Parks et al. 2009). Transposase sequences found in chromosomes show a considerable degree of diversity and degeneracy. This diversity is especially acute near the C-terminus of the proteins. It is tempting to speculate that the C-terminal β motif can be easily not only deleted but also, due to its relative simplicity, regenerated de novo from unrelated sequences. This pattern could explain, for example, the strong β-binding motifs found for ISL3 or for IS200/IS605 (OrfB) in Cyanobacteria (supplementary table S5, Supplementary Material online), which are found within an apparently nonhomologous sequence context. Indeed, we have identified several β-motif sequence alternatives for IS200 transposases (supplementary table S5, Supplementary Material online), two of them present in E. coli (fig. 3A), where the location of the motif within the transposase is very similar but the surrounding sequence context is different. In Archaea, we have identified a clear PCNA motif also at the C-terminus of IS200 (fig. 3D and supplementary table S5, Supplementary Material online). These findings suggest that the ability to interact with sliding clamps is likely to have evolved repeatedly and independently in the different transposase families and even within the different lineages in the same family. Evolutionary convergence has also been proposed for transposase domains and transpososome architecture (Montaño et al. 2012). On the other hand, because sliding clamps are universal and highly conserved, adaptation of transposases to binding β in new organisms could require only subtle sequence changes, facilitating IS exchange among phylogenetically distant organisms.
Although the β-binding motif is short and relatively poorly conserved, the competition assay with the strongly binding ligand GST-PolIVLF (fig. 3C) assured that the proposed peptides bind to β and at the same time mapped the interaction to the canonical hydrophobic pocket where all other enzymes also bind (fig. 4A). However, further work, outside the scope of the global survey presented here but now in progress, will be required to study purified transposases and their functional interaction with β in vitro, as well as the development of in vivo assays of the interaction for each transposition system. The fact that all transposases found interacting with β do so at the same position in competition with replication and DNA repair factors predicts that an excess of ISs in the genome could be disruptive to DNA replication. Competition between ISs within a genome is also a possibility, as suggested by the genome ecosystem hypothesis (Kidwell and Lisch 1997; Brookfield 2005). According to this view, mobile elements in a genome are analogous to an ecological community in which its components have a limited access to host resources (e.g., space in the chromosome, host factors required for transposition). Their fate would be a function of their ability of adaptation and proliferation in a given genomic environment. Future studies will be required to determine if the relative affinity of a transposase for β could alter its chances of success in this ecosystem and, in the process, also determine the fate of its host (Wagner 2009).
Fig. 4.—

Structural and functional asymmetries contributing to biased orientation of ISs in chromosomes. (A) Structure of Escherichia coli β, front (left) and side (right) views (PDB: 2POL). Arrows indicate the hydrophobic pockets on the surface of each monomer of β that are the sites of interaction of all β partners studied and of all the transposase peptides described in this study. (B) The asymmetry of transpososomes (green circles) in their interaction with β could determine the orientation of the transposase gene (orange arrow). The interaction “face” of β is colored red, the other blue. (C) Models of replisomes of E. coli and Bacillus subtilis. β is loaded on DNA by the γ-complex, which for leading strand synthesis positions β facing the direction of movement of the replication fork and in the opposite orientation in the lagging strand. On the left panel, the E. coli the replisome shows a homogeneous concentration of β associated with the synthesis of both strands (Reyes-Lamothe et al. 2010). On the right panel, β accumulates in “clamp zones” as the B. subtilis replisome progresses, possibly due to slow recycling after Okazaki fragment synthesis, creating an asymmetry in the distribution of β associated to the synthesis of leading and lagging strands (Su'etsugu and Errington 2011).
Replisome Composition Could Explain the Orientation Biases in Firmicutes
Although β interacts with transposases involved in various transposition pathways, this alone does not imply the generation of an orientation bias for these IS families in chromosomes. However, the strong orientation bias of some IS families found in Firmicutes (table 1) could be readily explained by three concurring circumstances: first, the interaction of asymmetric transpososomes with β (symmetric transpososomes would not result in chromosomally biased orientations); second, the fact that β is loaded on DNA in a regular, oriented, manner by the replisome and that all factors that interact with it do so on the same face of the ring (López de Saro 2009); and third, differences in the amount of β associated with the synthesis of the leading and lagging strands (fig. 4). In B. subtilis, β is slowly recycled after synthesis of the Okazaki fragments (lagging strand) and tends to accumulates in “clamp zones,” where β is presumably free to interact with other factors (Su'etsugu and Errington 2011). In contrast, β content associated to the synthesis of both strands in the E. coli replisome is homogeneous, possibly because of tighter recycling after Okazaki fragment completion (Reyes-Lamothe et al. 2010). The strongly asymmetric content of β associated with synthesis of the leading versus lagging strands in B. subtilis could explain the orientation bias found for IS families in Firmicutes (fig. 4C). In Proteobacteria, however, we find three strongly biased families, IS91, IS110, and IS200. Although we have found an interaction of IS91 and IS200 with β (fig. 3), other additional mechanisms could add to their orientation pattern. IS91 uses a rolling-circle mechanism that requires DNA synthesis and that, because the IS ends are different, is strongly asymmetric (Garcillán-Barcia et al. 2001; Curcio and Derbyshire 2003; Chandler et al. 2013). IS200 orientation is determined by its use of ssDNA and preferential insertion in the lagging strand (Ton-Hoang et al. 2010). No mechanistic model is available for IS110 that could explain its orientation, but, as mentioned before, specific targeting could also be involved.
Our model relies critically in three levels of asymmetry generating the observed biases: binding to β (fig. 4A), the replication fork (fig. 4C), and the transpososome (fig. 4B). Although asymmetries derived from the first two have been analyzed extensively, only a few transpososomes have been studied in structural detail (reviewed in Dyda et al. 2012). Although transpososomes consist of homomultimeric transposases, major conformational and functional asymmetries (e.g., sequential cleaving of DNA ends) have been found, for example, in the transposition pathways of Tn5 (Reznikoff 2008), Mu (Montaño et al. 2012), IS91 (Garcillán-Barcia et al. 2001), IS3 (Sekine et al. 1999), or IS200 (Ronning et al. 2005). In all cases, if β binds preferentially to one of the transposases, then an orientation bias during insertion on DNA could be the result (fig. 4B). Otherwise, if interaction of β with either transposase is identical, the result would be random orientation with respect to DNA. Both possibilities are plausible, and detailed interaction studies will be required to study what is the case for each transpososome architecture. In the well-studied IS200 transpososome, an ssDNA transposition system, the architecture is inherently asymmetric due to the polarity of ssDNA. According to our peptide data (fig. 3, supplementary table S5, Supplementary Material online), the region of interaction of IS200 TnpA with β would align with αE, an α-helix close to the catalytic tyrosine, as revealed by the ISHp608 crystal structure (Ronning et al. 2005). This C-terminal α-helix is likely to be highly flexible, but it is uncertain to what extent an interaction of either subunit with β would impose an additional asymmetry to the complex.
We have been unable to identify a β motif in 12 IS families that show orientation bias in one or more Phyla. A strong possibility is that we have failed to detect the β motif in sequences from these families, as its conservation is weak and transposases show high variability. Other possibilities are that these transposases bind β using a noncanonical motif or interaction with other replication structures or host factors.
Conclusions
Our study shows a general interaction between transposition and replication mediated by the β sliding clamp, which is revealed differently in the various Phyla as a consequence of distinct chromosome replication dynamics. In Firmicutes, the interplay of transposases with replication could create an orientation bias in many IS families as a consequence of the asymmetry in the distribution of β at the replication fork. We would like to propose that ISs could derive a double benefit from their β-mediated association with replication: first, integration with host chromosomal replication, possibly required for their proliferation within the chromosome, and, second, a universal, highly conserved, platform of dispersal between species. The interaction with β could therefore provide the key to the ubiquitous nature of insertion elements in bacterial genomes, highlighting a remarkable example of extreme molecular adaptability.
Supplementary Material
Supplementary information and tables S1–S6 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Susanna C. Manrubia and Jaime Iranzo (Centro de Astrobiología, Madrid, Spain) for discussions and suggestions for statistical analysis and Miguel Aranda Martín for bioinformatics support. F.J.L.S. gratefully acknowledges support from Ricardo Amils and Víctor Parro (Centro de Astrobiología, Madrid, Spain). This work was funded by the Ministry of Economy and Competitiveness of the Spanish Government grant CGL2010-17384 to F.J.L.S, by Project Consolider Ingenio CSD2007-00005 to M.J.G., by FPI Predoctoral Fellowship from the Spanish Government to H.D.-M., and by an INTA Training Fellowship to E.G.-T.
Literature Cited
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- Besag J, Clifford P. Sequential Monte Carlo p-values. Biometrika. 1991;78:301–304. [Google Scholar]
- Brookfield JFY. The ecology of the genome—mobile DNA elements and their hosts. Nat Rev Genet. 2005;6:128–136. doi: 10.1038/nrg1524. [DOI] [PubMed] [Google Scholar]
- Bunting KA, Roe SM, Pearl LH. Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the β-clamp. EMBO J. 2003;22:5883–5892. doi: 10.1093/emboj/cdg568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler M. Clamping down on transposon targeting. Cell. 2009;138:621–623. doi: 10.1016/j.cell.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Chandler M, Mahillon J. Insertion sequences revisited. In: Craig NL, editor. Mobile DNA II. Washington (DC): ASM Press; 2002. pp. 305–366. [Google Scholar]
- Chandler M, et al. Breaking and joining single-stranded DNA: the HUH endonuclease superfamily. Nat Rev Microbiol. 2013;11:525–538. doi: 10.1038/nrmicro3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier E, Rocha EPC. Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes. Mol Microbiol. 2006;59:1506–1518. doi: 10.1111/j.1365-2958.2006.05046.x. [DOI] [PubMed] [Google Scholar]
- Curcio MJ, Derbyshire KM. The outs and ins of transposition: from mu to kangaroo. Nat Rev Mol Cell Biol. 2003;4: 865–877. doi: 10.1038/nrm1241. [DOI] [PubMed] [Google Scholar]
- Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings P. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc Natl Acad Sci U S A. 2001;98:11627–11632. doi: 10.1073/pnas.191384398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyda F, Chandler M, Hickman AB. The emerging diversity of transpososome architectures. Quart Rev Biophys. 2012;45:493–521. doi: 10.1017/S0033583512000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcillán-Barcia M, Bernales I, Mendiola MV, de la Cruz F. Single-stranded DNA intermediates in IS91 rolling-circle transposition. Mol Microbiol. 2001;39:494–50. doi: 10.1046/j.1365-2958.2001.02261.x. [DOI] [PubMed] [Google Scholar]
- Georgescu RE, et al. Structure of a small-molecule inhibitor of a DNA polymerase sliding clamp. Proc Natl Acad Sci U S A. 2008;105:11116–11121. doi: 10.1073/pnas.0804754105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griep MA, McHenry CS. The dimer of the beta subunit of Escherichia coli DNA polymerase III holoenzyme is dissociated into monomers upon binding magnesium(II) Biochemistry. 1988;27:5210–5215. doi: 10.1021/bi00414a040. [DOI] [PubMed] [Google Scholar]
- Hu WY, Derbyshire KM. Target choice and orientation preference of the insertion sequence IS903. J Bacteriol. 1998;180:3039–3048. doi: 10.1128/jb.180.12.3039-3048.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, Sandler SJ, Harshey RM. Mu insertions are repaired by the double-strand break repair pathway of Escherichia coli. PLoS Genet. 2012;8:e1002642. doi: 10.1371/journal.pgen.1002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A, O'Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- Kichenaradja P, Siguier P, Perochon J, Chandler M. ISbrowser: an extension of ISfinder for visualizing insertion sequences in prokaryotic genomes. Nucleic Acids Res. 2010;38(Database issue):D62–D68. doi: 10.1093/nar/gkp947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG, Lisch D. Transposable elements as sources of variation in animals and plants. Proc Natl Acad Sci U S A. 1997;94:7704–7711. doi: 10.1073/pnas.94.15.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López de Saro FJ. Regulation of interactions with sliding clamps during DNA replication and repair. Curr Genomics. 2009;10:206–215. doi: 10.2174/138920209788185234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López de Saro FJ, Georgescu RE, Goodman MF, O'Donnell M. Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J. 2003;22:6408–6418. doi: 10.1093/emboj/cdg603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaño SP, Pigli YZ, Rice PA. The Mu transpososome structure sheds light on DDE recombinase evolution. Nature. 2012;491:413–417. doi: 10.1038/nature11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Z, Chandler M. Regulation of transposition in bacteria. Res Microbiol. 2004;155:387–398. doi: 10.1016/j.resmic.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Nakai H, Doseeva V, Jones JM. Handoff from recombinase to replisome: insights from transposition. Proc Natl Acad Sci U S A. 2001;98:8247–8254. doi: 10.1073/pnas.111007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AR, et al. Transposition into replicating DNA occurs through interaction with the processivity factor. Cell. 2009;138(4):685–695. doi: 10.1016/j.cell.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge SR, Hall RM. The IS1111 family members IS4321 and IS5075 have subterminal inverted repeats and target the terminalI inverted repeats of Tn21 family transposons. J Bacteriol. 2003;185:6371–6384. doi: 10.1128/JB.185.21.6371-6384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak C, et al. Irradiation-induced Deinococcus radiodurans genome fragmentation triggers transposition of a single resident insertion sequence. PLoS Genet. 2010;6:e1000799. doi: 10.1371/journal.pgen.1000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Million-Weaver S, Chattopadhyay S, Sokurenko E, Merrikh H. Accelerated gene evolution through replication-transcription conflicts. Nature. 2013;495:512–515. doi: 10.1038/nature11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, Craig NL. Tn7 recognizes transposition target structures associated with DNA replication using the DNA-binding protein TnsE. Genes Dev. 2001;15:737–747. doi: 10.1101/gad.870201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plague GR. Intergenic transposable elements are not randomly distributed in bacteria. Genome Biol Evol. 2010;2:584–590. doi: 10.1093/gbe/evq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post V, Hall RM. Insertion sequences in the IS1111 family that target the attC recombination sites of integron-associated gene cassettes. FEMS Microbiol Lett. 2009;290:182–187. doi: 10.1111/j.1574-6968.2008.01412.x. [DOI] [PubMed] [Google Scholar]
- Punta M, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Sherratt DJ, Leake MC. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science. 2010;328:498–501. doi: 10.1126/science.1185757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikoff WS. Transposon Tn5. Annu Rev Genet. 2008;42:269–286. doi: 10.1146/annurev.genet.42.110807.091656. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Roberts D, Hoopes BC, McClure WR, Kleckner N. IS10 transposition is regulated by DNA adenine methylation. Cell. 1985;43:117–130. doi: 10.1016/0092-8674(85)90017-0. [DOI] [PubMed] [Google Scholar]
- Rocha E. Is there a role for replication fork asymmetry in the distribution of genes in bacterial genomes? Trends Microbiol. 2002;10:393–395. doi: 10.1016/s0966-842x(02)02420-4. [DOI] [PubMed] [Google Scholar]
- Rocha EP, Danchin A. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat Genet. 2003;34:377–378. doi: 10.1038/ng1209. [DOI] [PubMed] [Google Scholar]
- Rocha EPC. The organization of the bacterial genome. Annu Rev Genet. 2008;42:211–233. doi: 10.1146/annurev.genet.42.110807.091653. [DOI] [PubMed] [Google Scholar]
- Rocha EPC, Touchon M, Feil EJ. Similar compositional biases are caused by very different mutational effects. Genome Res. 2006;16:1537–1547. doi: 10.1101/gr.5525106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronning DR, et al. Active site sharing and subterminal hairpin recognition in a new class of DNA transposases. Mol Cell. 2005;20: 143–154. doi: 10.1016/j.molcel.2005.07.026. [DOI] [PubMed] [Google Scholar]
- Rutherford K, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Sanders GM, Dallmann G, McHenry CS. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol Cell. 2010;37:273–281. doi: 10.1016/j.molcel.2009.12.025. [DOI] [PubMed] [Google Scholar]
- Sekine Y, Aihara K, Ohtsubo E. Linearization and transposition of circular molecules of insertion sequence IS3. J Mol Biol. 1999;294:21–34. doi: 10.1006/jmbi.1999.3181. [DOI] [PubMed] [Google Scholar]
- Siguier P, Filee J, Chandler M. Insertion sequences in prokaryotic genomes. Curr Opin Microbiol. 2006;9:526–531. doi: 10.1016/j.mib.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobetzko P, Travers A, Muskhelishvili G. Gene order and chromosome dynamics coordinate spatiotemporal gene expression during the bacterial growth cycle. Proc Natl Acad Sci U S A. 2012;109:E42–E50. doi: 10.1073/pnas.1108229109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivatsan A, Tehranchi A, MacAlpine DM, Wang JD. Co-orientation of replication and transcription preserves genome integrity. PLoS Genet. 2010;6(1):e1000810. doi: 10.1371/journal.pgen.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su'etsugu M, Errington J. The replicase sliding clamp dynamically accumulates behind progressing replication forks in Bacillus subtilis cells. Mol Cell. 2011;41(6):720–732. doi: 10.1016/j.molcel.2011.02.024. [DOI] [PubMed] [Google Scholar]
- Tetu SG, Holmes AJ. A family of insertion sequences that impacts integrons by specific targeting of gene cassette recombination sites, the IS1111-attC group. J Bacteriol. 2008;190:4959–4970. doi: 10.1128/JB.00229-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobes R, Pareja E. Bacterial repetitive extragenic palindromic sequences are DNA targets for insertion sequence elements. BMC Genomics. 2006;7:62. doi: 10.1186/1471-2164-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton-Hoang B, et al. Single-stranded DNA transposition is coupled to host replication. Cell. 2010;142:398–408. doi: 10.1016/j.cell.2010.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turlan C, Loot C, Chandler M. IS911 partial transposition products and their processing by the Escherichia coli RecG helicase. Mol Microbiol. 2004;53:1021–1033. doi: 10.1111/j.1365-2958.2004.04165.x. [DOI] [PubMed] [Google Scholar]
- Twiss E, Coros AM, Tavakoli NP, Derbyshire KM. Transposition is modulated by a diverse set of host factors in Escherichia coli and is stimulated by nutritional stress. Mol Microbiol. 2005;57:1593–1607. doi: 10.1111/j.1365-2958.2005.04794.x. [DOI] [PubMed] [Google Scholar]
- Waddell CS, Craig NL. Tn7 transposition: two transposition pathways directed by five Tn7-encoded genes. Genes Dev. 1988;2:137–149. doi: 10.1101/gad.2.2.137. [DOI] [PubMed] [Google Scholar]
- Wagner A. Transposable elements as genomic diseases. Mol Biosyst. 2009;5(1):32–35. doi: 10.1039/b814624c. [DOI] [PubMed] [Google Scholar]
- Wang JD, Berkmen MB, Grossman AD. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc Natl Acad Sci U S A. 2007;104:5608–5613. doi: 10.1073/pnas.0608999104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolkow CA, DeBoy RT, Craig NL. Conjugating plasmids are preferred targets for Tn7. Genes Dev. 1996;10:2145–2157. doi: 10.1101/gad.10.17.2145. [DOI] [PubMed] [Google Scholar]
- Yin JC, Krebs MP, Reznikoff WS. Effect of dam methylation on Tn5 transposition. J Mol Biol. 1988;199:35–45. doi: 10.1016/0022-2836(88)90377-4. [DOI] [PubMed] [Google Scholar]
- Zerbib D, et al. Specificity of insertion of IS1. J Mol Biol. 1985;185:517–524. doi: 10.1016/0022-2836(85)90068-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.