

Abstract
Protein kinase C-α (PKCα) is a member of the conventional family of protein kinase C isoforms (cPKCs) that regulate diverse cellular signaling pathways, share a common activation mechanism, and are linked to multiple pathologies. The cPKC domain structure is modular, consisting of an N-terminal pseudosubstrate peptide, two inhibitory domains (C1A and C1B), a targeting domain (C2), and a kinase domain. Mature, cytoplasmic cPKCs are inactive until they are switched on by a multistep activation reaction that occurs largely on the plasma membrane surface. Often, this activation begins with a cytoplasmic Ca2+ signal that triggers C2 domain targeting to the plasma membrane where it binds phosphatidylserine (PS) and phosphatidylinositol 4,5-bisphosphate (PIP2). Subsequently, the appearance of the signaling lipid diacylglycerol (DAG) activates the membrane-bound enzyme by recruiting the inhibitory pseudosubstrate and one or both C1 domains away from the kinase domain. To further investigate this mechanism, this study has utilized single-molecule total internal reflection fluorescence microscopy (TIRFM) to quantitate the binding and lateral diffusion of full-length PKCα and fragments missing specific domain(s) on supported lipid bilayers. Lipid binding events, and events during which additional protein is inserted into the bilayer, were detected by their effects on the equilibrium bound particle density and the two-dimensional diffusion rate. In addition to the previously proposed activation steps, the findings reveal a major, undescribed, kinase-inactive intermediate. On bilayers containing PS or PS and PIP2, full-length PKCα first docks to the membrane via its C2 domain, and then its C1A domain embeds itself in the bilayer even before DAG appears. The resulting pre-DAG intermediate with membrane-bound C1A and C2 domains is the predominant state of PKCα while it awaits the DAG signal. The newly detected, membrane-embedded C1A domain of this pre-DAG intermediate confers multiple useful features, including enhanced membrane affinity and longer bound state lifetime. The findings also identify the key molecular step in kinase activation: because C1A is already membrane-embedded in the kinase off state, recruitment of C1B to the bilayer by DAG or phorbol ester is the key regulatory event that stabilizes the kinase on state. More broadly, this study illustrates the power of single-molecule methods in elucidating the activation mechanisms and hidden regulatory states of membrane-bound signaling proteins.
The inner leaflet of the plasma membrane serves as a central assembly and diffusion platform on which multiple signaling networks form and conduct their functions as needed. The master kinase protein kinase C-α (PKCα) is targeted by Ca2+ to the plasma membrane where it is activated by a specialized set of lipids and second messengers, thereby triggering its essential functions in an array of signaling pathways.1−7 For example, a local Ca2+ signal at the leading edge of polarized macrophages recruits PKCα to the plasma membrane where it is an essential element of the positive feedback loop that maintains leading edge stability.4 Because PKCα is central to the function of this and many other pathways, its dysfunction or excess activity can trigger diverse pathologies, including inflammation, cancer, diabetes, cardiovascular anomalies, and autoimmune disease.2,8−12
PKCα is a member of the conventional subfamily of protein kinase C isoforms [cPKCs (α, β, and γ)] as reviewed by leading investigators in the field.2,6,13−19 Each cPKC enzyme features an N-terminal pseudosubstrate peptide, two C1 inhibitory domains (C1A and C1B), a C2 targeting domain, and a C-terminal catalytic domain, as illustrated in Figure 1. The individual cPKCs, including PKCα, undergo a maturation process that includes phosphorylation by an upstream kinase (PDK-1) leading to cPKC autophosphorylation, activation, and stabilization.20−22
Figure 1.
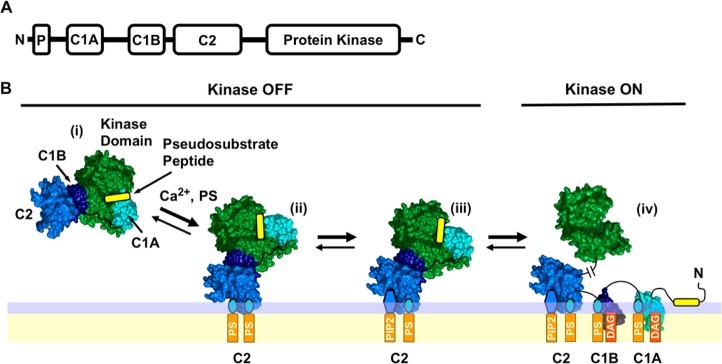
Conventional protein kinase C domain structure and simple activation model. (A) Modular domain organization of conventional protein kinase C isoforms α, β, and γ (cPKCs), consisting of an N-terminal inhibitory pseudosubstrate peptide (P), two inhibitory C1 domains (C1A and C1B), a plasma membrane-targeting C2 domain, and the C-terminal Ser/Thr kinase domain. (B) Simple cPKC activation model based on extensive current data in the field6,7,17,28,33,35,36,39−54 and recent, seminal structural studies.19,28 In response to a Ca2+ signal, the mature but inactive cytoplasmic cPKC (i) docks to plasma membrane PS via its C2 domain, yielding the (PS)2 state (ii). The C2 domain rapidly replaces one PS lipid with the less prevalent but higher-affinity PIP2, triggering a change in the membrane docking geometry because of the large PIP2 headgroup, yielding the (PS-PIP2) state (iii). Later, during a DAG lipid signal the protein, still inhibited by interactions of the kinase domain with the inhibitory elements, encounters and binds two DAG molecules that recruit C1A and C1B to the membrane, thereby relieving inhibition and activating the kinase in state (iv). The indicated protein structures are based on crystal structures of PDB entries 3PFQ(28) and 1PTQ.42 The complex structure is based on ref (28). The lipid binding stoichiometries are from refs (37−39), (100), and (101). The membrane docking geometries are based on refs (28), (42), and (91).
Mature, catalytically competent cPKC resides in the cytoplasm but is held in its inactive state by at least two inhibitory mechanisms. First, the kinase active site is competitively inhibited by the N-terminal region of the protein comprising the pseudosubstrate peptide and the C1A domain.23−26 Second, the C1B domain docks to the kinase domain surface distant from the active site, yielding allosteric inhibition.19,27,28 Kinase activation cannot occur until both parallel types of inhibition are relieved.
Cellular signals can remove these inhibitory constraints, thus switching the cPKC kinase “on” via several routes.29−36 Often, activation begins with a Ca2+ signal that loads the Ca2+ binding site of the C2 domain and triggers targeting to plasma membrane PS and PIP2 lipids. However, the kinase domain remains “off” until both types of inhibition are relieved. Kinase activation is triggered by the appearance of an activating lipid, either diacylglycerol (DAG) or phorbol ester.37−41 The resulting membrane deployment of the C1A and/or C1B domains removes both the competitive and allosteric restraints and thereby activates the enzyme. Figure 1 presents a working activation model based on elegant studies by several laboratories.6,7,28,33,35,36,39−54 In this model, both the C1A and C1B domains bind DAG in the final, active state (iv), as suggested by binding of DAG to both isolated domains, and by the importance of both domains in the targeting of PKCα to the plasma membrane when activating lipid is present.19,28,38,54−56
To test the predictions of the working model, we have employed single-molecule methods to analyze the membrane binding and surface diffusion of full-length PKCα and truncation constructs lacking one or more domains. Single-molecule TIRF microscopy was used to visualize the two-dimensional diffusion tracks of single protein molecules on supported bilayers as previously described.57−59 This approach has been shown to provide extensive information about both tightly bound individual lipids and global protein–bilayer contacts.58−60 Thus, as a protein binds additional lipids or penetrates more deeply into the membrane, the additional frictional drag against the highly viscous bilayer measurably slows its diffusion.58−60 Here such diffusional slowing is used to detect and identify the activation intermediates of PKCα.
The single-molecule studies presented below confirm key features of the working activation model (Figure 1) and also reveal a previously unknown, major pre-DAG intermediate in the PKCα activation reaction. This newly discovered intermediate is the predominant state of the inactive, plasma membrane-bound enzyme as it awaits the appearance of the activating DAG lipid. Furthermore, the molecular features of this predominant intermediate enhance the efficiency of the DAG and phorbol ester activation reactions. The findings yield new insights into the PKCα activation mechanism and show the power of the single-molecule approach as a tool in probing the signaling states of membrane-bound proteins.
Materials and Methods
Reagents
Synthetic dioleolyl phospholipids PC (phosphatidylcholine; 1,2-dioleoyl-sn-glycero-3-phosphocholine), PS (phosphatidylserine; 1,2-dioleoyl-sn-glycero-3-phospho-l-serine), PIP2 (1,2-dioleoyl-sn-glycero-3-phosphoinositol 4,5-bisphosphate), DAG (diacylglycerol; 1,2-dioleoyl-sn-glycerol), and LRB-PE [1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl)] were from Avanti Polar Lipids (Alabaster, AL). Alexa Fluor 555 (AF555) C2-maleimide, Lipofectamine 2000, and CoverWell perfusion chambers were from Invitrogen (Carlsbad, CA). Cover glass was from Ted Pella, Inc. (Redding, CA). 2-Mercaptoethanol was from Fluka (Buchs, Germany). CoA trilithium salt and ultrapure (>99%) BSA and PMA (phorbol 12-myristate 13-acetate) were from Sigma-Aldrich (St. Louis, MO). Anti-HA agarose affinity resin and HA peptide were from Thermo Scientific (Rockford, IL). Amylose affinity resin was from New England Biolabs (Ipswich, MA). Glutathione Sepharose 4B was from GE Healthcare Bio-Sciences (Piscataway, NJ).
Protein Cloning, Expression, and AF555 Labeling
To generate bacterial expression constructs of human PKCα regulatory domains, DNA sequences encoding the C1A domain (residues 26–100), the C1B domain (residues 90–165), the C1 construct (C1A–C1B, residues 26–165), the C2 domain (residues 157–294), C1BC2 (residues 90–294), and the C1C2 construct (C1A–C1B–C2, residues 26–294) were inserted into both pGEX-4T1 and pMAL-c2G bacterial expression vectors. Then, the 11-amino acid recognition sequence for Sfp phospho-pantethienyl-transferase61 was inserted to allow sequence-specific enzymatic labeling with a CoA-linked fluorophore. For each construct, a 5′-term polymerase chain reaction amplification oligonucleotide was synthesized (Integrated DNA Technologies, Coralville, IA) to introduce the labeling tag between the affinity tag and the protein N-terminal sequence. DNA sequencing confirmed the correct full sequence of all final constructs.
To generate a mammalian tissue culture expression construct of full-length, human PKCα, a pair of complementary oligonucleotides encoding the Sfp recognition sequence and flanked by EcoRI restriction sites were digested and ligated into EcoRI sites upstream and downstream of the full-length PKCα sequence in the pHACE plasmid.62 In the former construct, the labeling tag was placed before the N-terminus of the protein; in the latter construct, the labeling tag was placed between the HA affinity tag and the C-terminus of the protein. DNA sequencing confirmed the correct full sequence of all final constructs.
PKCα regulatory domain constructs were expressed in Escherichia coli Rosetta 2(DE3) cells (Novagen). Protein expression of PKCα C2 constructs was induced at 37 °C for 3 h, while the yield and solubility of constructs containing C1 proteins were enhanced by protein induction at 20 °C overnight. GST fusion proteins were retained on glutathione resin and eluted with excess reduced glutathione, except for the C2 domain construct that was released by thrombin cleavage. All MBP fusion proteins were retained on amylose resin and eluted with excess maltose. Each purified domain was ≥90% of the total eluted protein.
Full-length PKCα constructs possessing either the N-terminal or the C-terminal Sfp labeling tag were expressed in HEK 293 mammalian tissue culture cells. After three rounds of passaging, cells were transiently transfected with the appropriate plasmid using lipofectamine. Seventy-two hours after transfection, the cells were scraped and suspended in lysis buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 1% Tween 20, 1 mM EDTA, 2.5 mM EGTA, 10% glycerol, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 0.1 mM PMSF, 1 mM NaF, and 0.1 mM Na3VO4. Cells were homogenized; the cleared lysate was applied to HA resin and washed, and full-length PKCα constructs were eluted with HA peptide. These constructs were found to be fully active (see Results). Using the same approach, we attempted to generate a truncated construct (C1B–C2–kinase) that failed to generate active kinase, presumably because it did not undergo the processing steps essential for intracellular cPKC stability and activity.18
The labeling tag of each construct was covalently modified with AF555 by the Sfp enzyme using a published protocol.58,61 Briefly, ∼2 μM target protein was incubated with 2.5 μM Alexa Fluor 555–CoA conjugate and 0.5 μM Sfp at room temperature for 30 min. Excess fluorophore was removed by buffer exchange using Vivaspin concentrators (Sartorius Stedim, Göttingen, Germany) until the flow-through was not visibly colored by the AF555 fluorophore, and the final flow-through was checked for absorbance at 555 nm to ensure the complete removal of the free label. The concentration of labeled protein and labeling efficiency were determined from the measured absorbances of AF555 and intrinsic aromatic residues.
Supported Lipid Bilayer Preparation
Supported lipid bilayers were prepared from sonicated unilamellar vesicles (SUVs) as described previously,57,58 including a BSA step to block bilayer imperfections.60,63 Control experiments using fluorescent lipid and a lipid-binding protein tested and confirmed this blocking step did not detectably alter the bilayer fluidity or protein–membrane interactions.
Protein Kinase C Assay
Kinase assays were performed at 22 ± 0.5 °C using the PepTag Non-Radioactive Protein Kinase C system (Promega Corp., Madison, WI). The manufacturer’s protocol was followed, except the PKC lipid activator was replaced with the same SUV preparations used to make supported bilayers as described above. Here the total lipid concentration was 200 μg/mL in kinase assay buffer [10 mM MgCl, 26 μM CaCl2, 20 μM EGTA, 1 mM DTT, 1 mM ATP, and 20 mM HEPES (pH 7.4)].
TIRFM Measurements
TIRFM experiments were conducted at 22 ± 0.5 °C on a Nikon A1R TIRF instrument and a home-built, objective-based TIRFM instrument as described previously.57−59 Supported lipid bilayers in physiological buffer [140 mM KCl, 15 mM NaCl, 0.5 mM MgCl, 26 μM CaCl2, 20 μM EGTA, 5 mM reduced l-glutathione, and 25 mM HEPES (pH 7.4)] were imaged before and after sequential additions of buffer, BSA, and fluorescent protein. Typically, only a few dim, rapidly dissociating fluorescent contaminants were observed on the bilayer prior to the addition of protein. After protein addition, samples were allowed to equilibrate for 5 min. Then, to minimize contributions from small numbers of immobile fluorescent particles (presumably inactive protein aggregates), a bleach pulse power ∼30-fold higher than that used for imaging was applied for ∼5 s, and the fluorescence was allowed to recover for 60 s before data were acquired. For each sample, a set of three or four movie streams were acquired at a frame rate of 20 frames/s, and a spatial resolution of 6.3 pixels/μm on the Nikon A1R instrument or 4.2 pixels/μm on the home-built instrument, using NIS Elements Basic Research (Nikon). Subsequent particle tracking analysis was conducted using ImageJ,64 and data processing and fitting were conducted using Mathematica (Wolfram Research) and GraphPad Prism 5 (GraphPad Software, Inc.).
Single-Particle Tracking
As with our previous studies,57−59 diffusion trajectories of single fluorescent lipid and protein molecules were tracked and measured using the Particle Tracker plugin for ImageJ,65 yielding a per-frame quantitation of particle position and brightness. Then resulting data were then imported into Mathematica for further analysis. Only particles possessing fluorescence intensities within a defined range were included in the analysis, thereby eliminating bright protein aggregates and dim, nonprotein contaminants. Additional exclusions removed immobile particles, rapidly dissociating particles, and overlapping tracks for which particle identity is lost. All exclusions were described and validated previously.57−59
Determination of Diffusion Coefficients from Single-Molecule Trajectories
Each data set was analyzed with (i) a one-component fit, (ii) a two-component fit, and (iii) a three-component fit, as described previously.58 Briefly, all trajectories were disassembled into individual steps with a duration of 0.16 s (Δt = 8 frames); the length of each step was calculated, and finally, all step lengths were binned to yield a probability distribution histogram. The histogram was fit either by a linear mean square displacement versus step time function or by a single Rayleigh distribution function,57,66 and for single-component populations, both methods converged and gave the same diffusion constant within error. For heterogeneous populations, the sum of multiple Rayleigh distributions was required for convergence,58 but in all cases, a two-component fit was sufficient and yielded a pair of best-fit diffusion constants.
All-Atom Molecular Dynamics Simulations
Aligned with the C1B nuclear magnetic resonance (NMR) structure (PDB entry 2ELI), the C1A model was generated by the Swiss-Model server.67 This homology model was embedded in a PC/PS bilayer model (120 DOPC and 40 DOPS) in an explicit TIP3P water box with 140 mM NaCl and 10 mM KCl. The membrane model was placed in the X–Y plane. The simulation box had dimensions of ∼80 Å × 80 Å × 80 Å with ∼50000 atoms. All-atom simulations were conducted with the CHARMM force field68 in Desmond version 3.1 (Maestro-Desmond Interoperability Tools, version 3.1, Schrödinger) known to be effective in membrane protein simulations.69 After a 5 ns restrained equilibrium, two semi-isotropic simulations were performed at a constant temperature (296 K) and pressure (1 bar) for 160 and 300 ns. The C1A–C1B–C2 simulation box had dimensions of ∼90 Å × 90 Å × 140 Å with ∼140000 atoms. Embedded in one leaflet of a PC/PS bilayer, each C1A or C1B domain was contacting PS and DAG, while the C2 domain was contacting Ca2+, PS, and PIP2. The C1A–C1B–C2 model was equilibrated for 5 ns and simulated for 10 ns under conditions similar to those used for C1A simulations (above). Data analysis and structure visualization were performed with VMD70 or PyMOL (Schrödinger LLC).
Statistics
Error bars are standard errors of the mean (n = 3–19) except where indicated otherwise. Statistical significance was examined using the appropriate test; most commonly, the single-tailed t test was used to determine whether a given membrane interaction significantly slowed diffusion.
Results
Strategy of Identifying PKCα Activation Intermediates via Single-Molecule Analysis of Two-Dimensional Diffusion Rates
The overall strategy investigates the protein–bilayer contacts formed during the stepwise activation of PKCα on a target membrane, where each activation intermediate is detected and identified by its signature two-dimensional (2D) diffusion kinetics on the bilayer surface. The 2D diffusion constant of a given membrane-contacting domain is inversely proportional to the total frictional drag of its membrane-penetrating surfaces and tightly bound lipids against the highly viscous bilayer;57−60 by contrast, the frictional drag of its aqueous surfaces is negligible because the viscosity of the aqueous phase is ∼100-fold lower.71,72 To a first approximation, when a multidomain protein docks to a supported lipid bilayer,73,74 the frictional drags of the individual domains contribute additively to the total friction, such that the overall diffusion coefficient (DT) of the full protein is57−60,75
| 1 |
where the frictional drag of each domain against the bilayer (frictioni) is operationally defined by the inverse diffusion constant of that isolated domain (1/Di) and is proportional to the coefficient of friction (fi):58−60
| 2 |
It follows that the membrane contacts of PKCα activation intermediates can be probed by quantifying the 2D diffusion properties of the full-length protein and its individual domains, or groups of domains on supported lipid bilayers. The supported bilayer system offers several advantages for such measurements: (i) the ability to utilize controlled lipid mixtures to analyze specific lipid interactions, (ii) approximately 2-fold slower diffusion than free-standing bilayers, allowing more accurate measurement of the fastest-diffusing species,76 and (iii) the convenient additivity of frictional drags on supported bilayers.57−60,75 (Note such additivity may well be a special feature of supported bilayers not shared by free-standing bilayers that are subject to Saffman–Delbruck effects.77,78)
Protein Constructs Employed To Investigate the PKCα Activation Mechanism
The PKCα constructs employed in this analysis are schematically summarized in Figure 2. The single-domain constructs sampled each of the individual PKCα regulatory domains (C1A, C1B, and C2). The multidomain constructs (C1A–C1B, C1A–C1B–C2, and C1B–C2) possessed two or more domains connected by the native interdomain linkers, because they were constructed by truncating the full-length protein from one end or both. Full-length constructs (C1A–C1B–C2–kinase) contained the entire PKCα protein.
Figure 2.

Engineered full-length PKCα and truncation constructs used in this study. Shown are the domain layouts of the human PKCα constructs engineered for this study, and WT PKCα for comparison (native domains defined in Figure 1A). Each engineered construct contained both a fluorescent labeling tag (the 11-residue ybbr target peptide to which the Sfp enzyme covalently couples the fluor) and an affinity tag [the 9-residue HA peptide or the maltose-binding protein (MBP)]. Truncation borders were as follows: C1A–C1B–C2 (residues 26–294), C1B–C2 (residues 90–294), C2 (residues 157–294), C1A–C1B (residues 26–165), C1A (residues 26–100), and C1B (residues 90–165).
Each construct included an N- or C-terminal affinity tag for purification. Because the C1A and C1B domains are notoriously unstable,40,51 maltose-binding protein (MBP) was routinely used as an affinity tag and was retained after purification, because of its ability to enhance the solubility of unstable proteins.79,80 The intrinsically stable PKCα C2 domain was expressed and purified as a GST fusion, and then the GST tag was removed by thrombin cleavage.81 Full-length PKCα was expressed and purified with a C-terminal HA affinity tag (YPYDVPDYA).62
All constructs also possessed an N- or C-terminal, 11-residue labeling tag (DSLEFIASKLA) that was covalently modified with AlexaFluor 555 by a standard enzymatic procedure.61 The resulting fluor-tagged constructs all retained normal membrane binding and, in the case of full-length PKCα, normal Ca2+- and lipid-regulated membrane binding and enzyme regulation (Figure 3 and Figure S1 of the Supporting Information). Notably, full-length constructs labeled at the N- or C-terminus with fluor were indistinguishable from each other and from a corresponding unlabeled construct in four different activity assays, indicating the fluor is unperturbing (Figure S1 of the Supporting Information). For all subsequent studies, the full-length construct labeled at the C-terminus was employed to maintain the native N-terminal regulatory region.
Figure 3.

Dependence of PKCα membrane binding and kinase activation, and domain membrane binding, on lipid composition. (A and D–F) Single-molecule TIRF quantitation of binding of constructs to supported lipid bilayers, normalized to the same total protein concentration (1 pM). A given construct was added to the imaging chamber containing the indicated supported bilayer (Table 1), and then the density of fluorescent protein binding per unit area was quantitated. Each average was determined from 20 temporally isolated frames from three separate movie streams in at least five separate experiments (n ≥ 15). The free Ca2+ concentration was 6 μM in a physiological buffer (Materials and Methods). (B) Total kinase activity of fluorescently labeled PKCα measured by a modified PepTag (Promega) assay (Materials and Methods). The enzyme was activated by membranes containing the same lipid compositions as the binding density measurements, and PKC-specific phosphorylation of the PepTag target peptide was quantitated by electrophoresis. Each condition was repeated twice in duplicate (n = 4) and separate experiments using varying PKCα and/or lipid concentrations confirmed that enzyme saturation did not occur. (C) Specific activity of PKCα determined by the ratio of kinase activity to binding density under each bilayer condition. In all experiments, T = 22 ± 0.5 °C. In this and subsequent figures, an average parameter determined from purely single molecule data is indicated by < parameter >.
Of all the constructs, the full-length PKCα, C1B, C2, and C1B–C2 fluor-tagged constructs were the most stable, as evidenced by the low levels of immobile misfolded or aggregated protein [<10% of the total membrane-bound single particles (see below)]. The C1A, C1A–C1B, and C1A–C1B–C2 constructs exhibited adequate stability but higher levels of immobile particles (<20–30% of the membrane-bound particles), consistent with the tendency of C1 domains to aggregate.40,51 The ability of single-molecule data analysis to filter out immobile and excessively bright particles ensured that misfolded and aggregated proteins were excluded from the study.57−60
Lipid Bilayer Compositions Employed To Investigate the PKCα Activation Mechanism
Table 1 summarizes the lipid compositions of the supported bilayers employed to mimic the native sequence of the acquisition of lipids by PKCα on a target membrane surface (Figure 1B). (i) Following binding of Ca2+ to the C2 domain, the enzyme will typically first associate with PS, which is the major anionic phospholipid (∼25 mol %33,82) of the plasma membrane inner leaflet and thus would virtually always be found in the initial docking footprint (∼14–18 lipids36). To analyze the predominant PS-bound intermediate(s), PKCα and the other constructs were analyzed on PC/PS bilayers. (ii) Next, the membrane-bound enzyme will typically bind PIP2, which is present at ∼10-fold lower densities than PS (∼2 mol %) and thus will often be absent in the initial footprint33,34,83 but binds more tightly.33,34,84,85 This PIP2 binding is rapid: a collision rate calculation86 based on the 2D diffusion constants of individual lipids and the PS-bound C2 domain (Table 2) indicates that a PIP2 collision will occur approximately once per microsecond. The resulting intermediates were investigated on PC/PS/PIP2 bilayers. (iii) Finally, the membrane-bound, inactive enzyme will eventually bind the activating lipid diacylglycerol (DAG) during a transient DAG signaling event when this rare lipid appears at low levels (typically ≪2 mol %). The resulting activated states were studied on PC/PS/PIP2/DAG bilayers. For the sake of symmetry, the PC/PS/DAG lipid mixture was also included (Table 1); in theory, this mixture could be a physiologically relevant mimic for an extreme DAG signal generated by locally high levels of phospholipase C activity that fully hydrolyze local PIP2 to DAG and IP3.87,88
Table 1. Lipid Compositions of Synthetic Bilayers.
| lipid mixturea | lipid mol % |
|---|---|
| PC/PS | 75:25 |
| PC/PS with LRB-PE | 75:25 with 200 ppb |
| PC/PS/PIP2 | 74:24:2 |
| PC/PS/PIP2 with LRB-PE | 74:24:2 with 200 ppb |
| PC/PS/DAG | 74:24:2 |
| PC/PS/DAG with LRB-PE | 74:24:2 with 200 ppb |
| PC/PS/PIP2/DAG | 73:23:2:2 |
| PC/PS/PIP2/DAG with LRB-PE | 73:23:2:2 with 200 ppb |
Abbreviations: PC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; PS, 1,2-dioleoyl-sn-glycero-3-phospho-l-serine; PIP2, 1,2-dioleoyl-sn-glycero-3-phosphoinositol 4,5-bisphosphate; DAG, diacylglycerol or 1,2-dioleoyl-sn-glycerol; LRB-PE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl).
Table 2. Measured Average Single-Molecule Diffusion Constantsa.
| population-weighted
average diffusion constant |
||||
|---|---|---|---|---|
| PC/PS | PC/PS/PIP2 | PC/PS/DAG | PC/PS/PIP2/DAG | |
| lipid | 2.58 ± 0.11 | 2.81 ± 0.12 | 2.63 ± 0.03 | 2.56 ± 0.10 |
| C1A | 0.58 ± 0.07 | 0.73 ± 0.18 | 0.52 ± 0.03 | 0.47 ± 0.10 |
| C1B | 0.48 ± 0.11 | 0.32 ± 0.07 | 0.59 ± 0.18 | 0.41 ± 0.10 |
| C2 | 1.45 ± 0.06 | 1.07 ± 0.02 | 1.41 ± 0.09 | 1.00 ± 0.12 |
| C1A–-C1B | 0.26 ± 0.04 | 0.18 ± 0.03 | 0.21 ± 0.05 | 0.07 ± 0.01 |
| C1A–C1B–C2 | 0.46 ± 0.06 | 0.18 ± 0.03 | 0.21 ± 0.04 | 0.22 ± 0.04 |
| C1B–C2 | 1.02 ± 0.10 | 0.81 ± 0.09 | 1.04 ± 0.04 | 0.50 ± 0.09 |
| FL PKCα | 0.44 ± 0.04 | 0.38 ± 0.07 | 0.41 ± 0.02 | 0.22 ± 0.04 |
Average measured diffusion constants (square micrometers per second) of all constructs except for the lipid and C2 domain were determined by calculating the population-weighted average of single molecule diffusion constants from a two-component Rayleigh best fit of each protein–lipid data set (see Table S2 of the Supporting Information). In the cases of the lipid and C2 domain, a one-component best fit was sufficient (Table S2 of the Supporting Information). Each measurement analyzed at least 2500 diffusion tracks from five experiments (n ≥ 5). In all experiments, the free Ca2+ concentration was 6 μM in a physiological buffer at 22 ± 0.5 °C.
Figure 3 shows that, as predicted by the working PKC activation model (Figure 1B) and its proposed sequence of lipid binding events, the membrane affinity and specific kinase activity of full-length PKCα increased as the bilayer lipid composition was changed from PC/PS to PC/PS/PIP2 to PC/PS/PIP2/DAG. Similarly, on a qualitative level, the engineered fragments displayed the predicted (Figure 1B) relative affinities for their effector lipids; for example, C1A and C1B preferred PS and DAG, while C2 preferred PS and PIP2. In the absence of DAG, addition of PIP2 (2 mol %, the average PIP2 density in the plasma membrane inner leaflet) yielded a small but highly significant (p < 0.001) increase in total PKCα kinase activity, while subsequent addition of DAG generated a much larger increase (Figure 3B). Mass spectrometry115 studies of the lipid mixtures confirmed that no contaminating DAG was present, so the former activity increase was a true PIP2 effect (Table S1 of the Supporting Information). PIP2-triggered kinase activation in the absence of DAG was first described in a previous study utilizing a higher PIP2 density (5 mol %) that fully activated the kinase, suggesting that high PIP2 densities may obviate DAG regulation.85,89
Membrane Binding and Diffusion of Full-Length PKCα and Engineered Constructs
PC/PS Bilayers
When Ca2+-activated PKCα first binds to the target membrane, the working model predicts that the Ca2+-occupied C2 domain will dominate the membrane contacts (Figure 1). As predicted, both full-length PKCα and its isolated C2 domain exhibited stable binding to PC/PS supported bilayers (Figure 3). Notably, however, the equilibrium affinity of full-length PKCα for these bilayers is 2.5-fold higher than that observed for the isolated C2 domain, suggesting that the full-length protein has additional membrane contacts. Furthermore, titration of the PS mole density (Figure 4) yielded sigmoidal binding curves for both full-length PKCα and C2 domain binding, consistent with the cooperative binding of multiple PS lipids, but full-length PKCα exhibited a significantly larger (p < 0.05) Hill coefficient (H values of 4.3 ± 0.2 and 3.5 ± 0.1 for full-length PKCα and the C2 domain, respectively). These Hill coefficients are determined in part by the number of cooperative PS binding sites and in part by the increasing negative charge density of the bilayer surface as the level of PS increases. The larger Hill coefficient observed for full-length PKCα relative to that of its isolated C2 domain is consistent with the binding of at least one additional PS molecule to the full-length protein, or with electrostatic interactions involving additional protein positive charges. Overall, the equilibrium binding data support a model in which full-length PKCα docks to the PC/PS bilayer via its C2 domain, and additional membrane contacts provided by unknown domain(s).
Figure 4.

Dependence of PKCα and C2 domain membrane binding on PS mole density. Membrane binding density was quantified by single-molecule TIRF as described in the legend of Figure 3. A fixed concentration of full-length PKCα or C2 domain (5 pM) was added to supported PC bilayers containing increasing mole fractions of PS, and the density of fluorescent, membrane-bound proteins was measured for 20 temporally isolated frames from three separate movie streams in three separate titration experiments (n = 9). The best fit of the Hill equation yielded the indicated values of K1/2 and the Hill coefficient. In both experiments, the free Ca2+ concentration was 6 μM in a physiological buffer (Materials and Methods) at 22 ± 0.5 °C.
Analysis of the 2D diffusion of full-length PKCα and the isolated C2 domain on PC/PS bilayers provides direct evidence of such additional membrane contacts. The single-molecule, 2D diffusion rates of both constructs on PC/PS supported bilayers were measured by TIRF imaging and quantitation of hundreds of single-particle diffusion tracks as previously described.57,58 Focusing on the initial 20 ms diffusion step of each track (simply the step length in the first full 20 ms frame of that trajectory) revealed that, within error, full-length PKCα and its isolated C2 domain exhibited the same average initial step length, as shown in Figure 5A. This observation fully supported the prediction of the working model that the Ca2+-activated C2 domain of the full-length protein first binds the bilayer surface, yielding a kinase-inactive, membrane-bound intermediate prior to the appearance of the DAG signal.48,49,90
Figure 5.

Single-particle step lengths for freely diffusing full-length PKCα and the C2 domain bound to pre-DAG bilayers. Step sizes were quantitated by TIRF for single-particle tracks on supported bilayers of the indicated lipid composition. (A) Comparison of the average initial step lengths of PKCα and the C2 domain on PC/PS bilayers. The initial step length is the distance traveled per 20 ms frame at the beginning of a single-particle diffusion track (the second frame of the track is quantitated because the first frame often captures a partial step following particle binding), as determined by averaging the initial steps of at least 200 particle tracks (≥30000 steps) per experiment in three separate experiments (n ≥ 600). Data were normalized to the initial step length of FL PKCα (0.40 ± 0.04 μm). (B and C) Time dependence of step length for successive steps during the first 320 ms after the particle had bound to the indicated bilayer. With the second 20 ms step of the diffusion track as a starting point (see the legend of panel A), the length of the indicated step was determined for each diffusion track, and the lengths of corresponding steps were averaged over at least 200 particle tracks in three separate experiments (n ≥ 600) to yield the indicated time courses. Finally, the two time courses in each box were normalized to the initial step length of FL PKC. In all experiments, the free Ca2+ concentration was 6 μM in a physiological buffer (Materials and Methods) at 22 ± 0.5 °C.
After the initial step, the C2 domain retained its original diffusion speed in subsequent steps as would be expected, but surprisingly, the full-length protein slowed dramatically over a period of ∼200 ms as shown in Figure 5B. The exponential decay constant of this slowing on PC/PS bilayers was 54 ± 9 ms. Thus, following binding of the C2 domain to the membrane, the full-length protein formed additional bilayer contacts in a second binding step. These additional contacts increased the membrane residence time of full-length PKCα 3.4-fold relative to that of C2, as determined by their average diffusion track lifetimes (11.8 ± 2.5 and 3.5 ± 0.1 s for PKCα and the C2 domain, respectively). Overall, the average diffusion speed of full-length PKCα was 3.3-fold slower (0.44 ± 0.04 μm2/s) than that of its C2 domain on PC/PS bilayers (1.45 ± 0.06 μm2/s) (Table 2 and Figure 6A). These findings show that after C2 domain docking, full-length PKCα forms a previously unknown, stable, membrane-bound activation intermediate that is not predicted by the working model (Figure 1). Activity measurements found this pre-DAG intermediate possessed little or no kinase activity (Figure 3C).
Figure 6.

Measured and calculated diffusion constants of full-length and truncated PKCα constructs on bilayers mimicking sequential lipid binding and kinase activation. On the plasma membrane, a PKCα molecule will typically encounter and bind PS, then PIP2, and then DAG based on the decreasing mole densities of these lipids (see the text). The indicated supported bilayer compositions (mole percents in Table 1) simulate this sequential binding by allowing protein equilibration first with PS (A), then with PS and PIP2 (B), and then with PS, PIP2, and DAG (C). Black bars are the average experimental diffusion constants of each construct on the indicated bilayer (Table 2 and Table S2 of the Supporting Information), determined by single-molecule TIRF analysis of at least 2500 diffusion tracks in at least five experiments (n ≥ 5). White bars are diffusion constants predicted by the new model (Figures 7 and 10A) for each multidomain construct on the indicated bilayer composition, calculated by combining the experimental diffusion constants of the domains proposed to contact the bilayer (eqs 1 and 2 and Table S2 of the Supporting Information). For contacts involving the C1A and/or C1B domain, calculations included a weighted average over the observed multiple states for these domains (Table S2 of the Supporting Information). Gray bars are diffusion constants predicted by replacing the C1A–bilayer friction of the new model (white bars) with pseudosubstrate peptide–bilayer friction, using the known friction of the tighter binding, more deeply penetrating MARCKS peptide60 as an upper limit on the friction expected for the N-terminal pseudosubstrate region. On each membrane type, this peptide hypothesis could not explain the observed diffusion constant, confirming that a C1 domain–membrane contact is necessary to explain the observed total friction. In all experiments, the free Ca2+ concentration was 6 μM in a physiological buffer at 22 ± 0.5 °C.
Comparison of the diffusion speeds of full-length PKCα and truncation constructs on PC/PS bilayers identified C1A as the domain responsible for the additional membrane contacts in the newly discovered pre-DAG intermediate. Table 2 and Figure 6A show that the isolated C1A and C1B domains each diffused much more slowly than the isolated C2 domain, consistent with their deeper penetration into the bilayer.40,42,49,60,91 Thus, bilayer binding and penetration by either C1A or C1B could, in principle, account for the observed slowing of full-length PKCα following the initial binding of the C2 domain to the PC/PS bilayer (Figure 5B). In contrast, the kinase domain does not contact the membrane and would not be expected to contribute to frictional drag,57,58,60,73 as confirmed by the observation that kinase domain truncation to yield the C1A–C1B–C2 construct had little or no effect on diffusion speed (Figure 6A and Table 2). Strikingly, however, removal of the C1A domain from the C1A–C1B–C2 construct to generate the C1B–C2 construct yielded a dramatic increase in the average diffusion speed to a C2-like level (Figure 6A and Table 2). The simplest interpretation is that the C1A domain is responsible for the extra membrane contacts and frictional drag of full-length PKCα and the C1A–C1B–C2 construct on PC/PS bilayers relative to the isolated C2 domain (Figures 5B and 6A). Consistent with these conclusions, a predicted diffusion constant based on the additive frictional drags of the C1A and C2 domains closely matches the observed diffusion constants of both full-length PKCα and the C1A–C1B–C2 construct (in Figure 6A, compare white and black bars). In this picture, the C1B domain stays bound to the catalytic domain and helps maintain the kinase off state, with no significant membrane contacts.
Further evidence supporting the proposed deployment of C1A, not C1B, to the bilayer after C2 docking was provided by the observation that isolated C1A bound more tightly than C1B to PC/PS bilayers (Figure 3). In addition, previous studies have suggested that C1B of the closely related PKCβ enzyme binds stably to the C2 and kinase domain19,28 while C1A is more mobile and thus should be more available for membrane deployment.28 The new evidence reinforces this picture. Thus, the C1B–C2 construct diffused much like an isolated C2 domain and lacked the large frictional drag observed for the isolated C1B domain (Figure 6A and Table 2), consistent with a C1B–C2 interaction that largely prevented binding of C1B to the bilayer. In contrast, the C1A–C1B construct lacked both the C2 and kinase domains and thus provided no binding partners for C1B, allowing both C1 domains to be embedded in the bilayer. MD simulations (Figure 9) and experimental studies indicate that C1 domains penetrate more deeply than the C2 domain;40,42,49,60,91 thus, the C1A–C1B construct diffused more slowly than full-length PKCα or the C1A–C1B–C2 construct that deployed only C1A and C2 domains on PC/PS bilayers (Figure 6A, Table 2, and eq 1).
Figure 9.
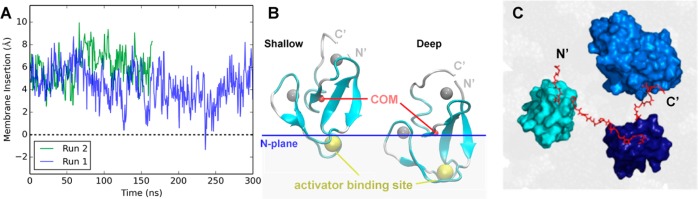
All-atom MD simulations of the isolated C1A domain and the C1A–C1B–C2 construct bound to a PC/PS bilayer. (A) Membrane insertion [measured every 0.54 ns as the distance from the center of mass (COM) of the protein to the bilayer PC average N plane] over time observed in two production runs. (B) Cartoon representation of the C1A domain (cyan) showing the zinc ions (silver spheres), the protein center of mass (COM, red sphere), and the bilayer N plane as a blue line. At the left is a snapshot of the shallow state (8.7 Å) at 59 ns of run 2 and at the right a snapshot of the C1A deep state (0.3 Å) at 245 ns of run 1. A detailed conformational analysis shows that three lysines (K45, K62, and K76) and two arginines (R42 and R77) are the major residues involved in the electrostatic interactions between the C1A domain and the PC/PS headgroups in the shallow state. Four of these residues (R42, K62, K76, and R77) were observed to form a charged “semicircle” around the middle of the C1A domain to stabilize the deep insertion state. It is noteworthy that in the deep state, a stable hydrogen bond is formed between K45 and the backbone oxygen atom of F43 to stabilize the buried charge, while cation−π interactions between side chains of K45 and F43 or F44 may also contribute to the stability of K45 in the deep state. In addition, >60% of the C1A conformers in the deep state were associated with PS, while no shallow state conformers were observed to bind PS. (C) View down the membrane normal toward the bilayer surface of a C1A–C1B–C2 construct (surface representation) on the bilayer (gray) after a 10 ns all-atom MD simulation. The simulation was initiated with the preferred membrane docking geometry of each individual domain (Figure 9B and ref (36)), and with the interdomain linkers (red sticks, H and side-chain atoms hidden) extended with no steric clash. The resulting model was stable in the membrane during the entire simulation. N′ and C′ represent the truncated N- and C-terminal ends of the fragments employed in both experimental studies (Figure 2) and MD simulations.
The N-terminal region of PKCα containing the pseudosubstrate region is known to bind anionic membranes (ΔG = −7 kcal/mol)92 and is likely to be membrane-bound when not occupying the kinase active site;92 thus, the possibility this region contributed significantly to the frictional drag was investigated in two ways. First, the C1A–C1B–C2 construct lacking the region (Figure 2) exhibited the same slow diffusion speed, within error, as full-length PKCα, indicating the pseudosubstrate region made no detectable contribution to the total frictional drag of the full-length enzyme (Figure 6A and Table 2). Second, the known frictional drag of the MARCKS peptide60 was used to estimate the maximal frictional drag that could be expected for the pseudosubstrate region. MARCKS binds anionic membranes with a higher affinity (ΔG = −9 kcal/mol93,94) and possesses more aromatic side chains that penetrate into the bilayer;92−96 thus, MARCKS is expected to generate considerably more bilayer friction than the pseudosubstrate. However, the MARCKS friction is too small (3-fold lower than that of C1A) to account for the observed slowing of PKCα following C2 domain docking (Figures 5B and 6A and eq 1), confirming that C1A must dominate the slowing. This analysis shows that frictional slowing due to pseudosubstrate–bilayer contact would likely be difficult to detect; thus, the approach does not ascertain whether the pseudosubstrate of the new pre-DAG intermediate remains bound in the kinase active site or membrane-deployed.
Overall, the findings provide multiple, strong lines of evidence indicating that the predominant PKCα intermediate formed on PC/PS bilayers possesses membrane-deployed C1A and C2 domains, while C1B does not contact the bilayer but instead helps maintain the observed inhibition of the kinase domain.
PC/PS/PIP2 Bilayers
The equilibrium binding of full-length PKCα and the other constructs was compared on PC/PS and PC/PS/PIP2 bilayers (Figure 3). The results showed that, as expected,32,34−36 the membrane affinities of PKCα and the C2 domain were significantly increased by binding of PIP2 to the C2 domain, while the C1A and C1B domains exhibited no detectable PIP2 affinity (Figure 3). Because PIP2 binding occurs on the microsecond time scale (see above), it was too fast to detect (20 ms per TIRF frame). Thus, as previous models have proposed, most Ca2+-activated PKCα molecules will dock to the target membrane via binding of their C2 domain to PS and then will rapidly bind PIP2 (Figure 1B).32−36 The newly discovered insertion of C1A into the bilayer exhibits a time constant of 43 ± 9 ms on the PC/PS/PIP2 bilayer (Figure 5C) and will generally occur after C2 acquires both its PS and PIP2 target lipids.
Comparing the average 2D diffusion speeds on PC/PS and PC/PS/PIP2 bilayers revealed that the C2 domain exhibited modest slowing when it was bound to PIP2 as previously observed.60 On PC/PS bilayers, this domain is known to bind PS molecules at both its Ca2+ and PIP2 binding sites, while on PC/PS/PIP2 bilayers, the latter PS is replaced with PIP2.33,35,36 Thus, the C2 domain binds the same total number of lipids on the two types of membranes, but electron paramagnetic resonance (EPR) studies of bilayers have shown the larger PIP2 headgroup significantly alters its tilt angle and membrane docking geometry, yielding slightly deeper penetration of the protein into the bilayer.35,36 We have previously shown this deeper penetration and its additional frictional drag can quantitatively account for the observed diffusional slowing of the isolated C2 domain on PC/PS/PIP2 bilayers relative to PC/PS bilayers.60 (Two alternative models of C2 docking geometry based on ATR-IR studies of dehydrated, then rehydrated, C2–lipid multilayers97 or X-ray reflectivity measurements on lipid monolayers116 have been described, but only the EPR studies of bilayers have been confirmed by independent MD simulations.35,36 Moreover, these EPR measurements and MD simulations yield protein penetration depths on membranes lacking and containing PIP2, and thus are most useful in analyzing protein diffusion speed which is highly sensitive to penetration depth.60) Herein, both the isolated C2 domain and the C1B–C2 construct, which diffused much like the C2 domain that dominates its membrane contacts, exhibited significant (p < 0.10 for the C1B–C2 construct, and p < 0.005 for C2) slowing in the PIP2-bound state (Figure 6A,B).
In broader terms, the PIP2-triggered slowing of the C2 domain contributed to a modest slowing of all the multidomain constructs containing the C2 domain on PC/PS/PIP2 bilayers relative to PC/PS bilayers (Figure 6A,B). As anticipated, the effects of PIP2 on the 2D diffusion constant of the C1A–C1B–C2 construct and full-length PKCα were more difficult to quantitate because of (i) the relatively small magnitude of the PIP2-triggered frictional drag and (ii) the inverse relationship between the diffusion constant and the frictional drag, as observed in an extensive study of 17 protein–lipid complexes diffusing on supported bilayers that were well described by eqs 1 and 2.60 At the high-friction limit, this inverse relationship means that additional friction yields monotonically decreasing effects on the diffusion constant as the diffusion speed asymptotically approaches zero. For the C1A–C1B–C2 construct or full-length PKCα bound to PS, the combined membrane insertion of the C1A and C2 domains placed the system near the high-friction limit such that the additional frictional drag triggered by the PIP2-induced tilting of the C2 domain was a small component of the total drag. Thus, the mean diffusion constant of full-length PKCα was reproducibly slower on the PIP2-containing membranes, but the slowing was at the edge of statistical significance (p = 0.20).
These findings provide new insights into the previous observation that binding of PIP2 to the C2 domain increases PKCα kinase activity.89 At 2 mol % PIP2, most of the effect on kinase activity arose from an increased level of membrane binding (Figure 3A,B), though there was also a small increase in the average specific activity of the membrane-bound protein (Figure 3C). At 5 mol % PIP2, near-maximal specific kinase activity is observed even in the absence of DAG.89 Other studies have shown that at sufficiently high PIP2 densities the C2 domain binds two PIP2 molecules and generates a larger domain tilt than one PIP2.36 A simple hypothesis for the PIP2-induced kinase activation proposes that the binding of two successive PIP2 molecules, and the accompanying C2 domain tilt, weakens the C1B–C2 interaction, which could in turn weaken the inhibitory C1B–kinase interaction and facilitate kinase activation. In constructs lacking the kinase domain, the PIP2-induced C1B–bilayer interactions would be amplified because the C1B–kinase interaction is missing. Consistent with this prediction, the C1A–C1B–C2 construct is considerably slower than full-length PKCα on PC/PS/PIP2 bilayers. Moreover, inclusion of weak C1B–bilayer interactions is necessary for the accurate prediction of the diffusion constants of C1B–C2 and C1A–C1B–C2 constructs, while the corresponding prediction for full-length PKCα requires only C1A– and C2–bilayer interactions (Figure 6B, Table S2 of the Supporting Information, and eq 1).
Another interaction that could contribute to PIP2-triggered kinase activation is direct binding of the pseudosubstrate to PIP2 (Figure S2 of the Supporting Information), but as noted for diffusion on PC/PS bilayers (see above), such an interaction would provide too little friction to account for the observed PKCα slowing following docking of the C2 domain to PC/PS/PIP2 bilayers that requires C1A insertion for a quantitative explanation (Figures 5C and 6B). Moreover, pseudosubstrate–bilayer friction would likely be too small to detectably alter total PKCα friction (see above); thus, it is unclear whether the pseudosubstrate is membrane-bound on PC/PS/PIP2 bilayers. If PIP2 does recruit the pseudosubstrate, it would still have to recruit C1B to produce full kinase activation.
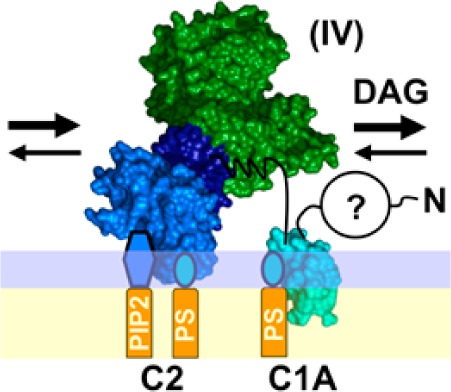
Overall, the findings reveal a previously unknown PKCα pre-DAG intermediate on PC/PS/PIP2 bilayers, analogous to the newly discovered intermediate on PC/PS bilayers (Figure 7). This new state is expected to be the predominant intermediate on unstimulated plasma membranes containing constitutive PS and PIP2, prior to the appearance of a DAG signal. The new intermediate possesses a C2 domain bound to PS and PIP2, as well as a membrane-embedded C1A domain bound to PS (Figures 7, 10A). At moderate PIP2 levels, the C1B domain remains bound to the catalytic domain and helps maintain the kinase off state, but high PIP2 levels are proposed to facilitate binding of C1B and/or the pseudosubstrate to the bilayer.
Figure 7.

Schematic model of the PKCα activation reaction showing interdomain and membrane contacts. Four of the indicated states [(II), (III), (V), and (VI)] are similar to those of a previous model,28 while a new, pre-DAG intermediate [(IV)] is proposed on the basis of new data. The new intermediate (IV) has been directly detected in single-molecule diffusion studies (see Results) and is the predominant intermediate awaiting the appearance of the activating DAG signal. The slow 2D diffusion of this intermediate arises from deployment of its C1A domain to the bilayer as indicated. Binding of DAG to this membrane-deployed C1A domain (IV) is proposed to trigger the formation of a transient state (V), not yet detected, in which binding of DAG to C1A may send a signal through the C1A–C1B linker to help dislodge the inhibitory C1B domain from the kinase domain and/or help guide C1B to the membrane. The resulting kinase on state is stabilized by the binding of a DAG molecule to C1B, thereby trapping its membrane-embedded state (VI). Lines between domains indicate inhibitory contacts (with a crossbar) or other protein–protein contacts; question marks indicate possible but not fully confirmed contacts. Gray contacts indicate a potential weakening of the interaction. A horizontal line represents a bilayer surface.
Figure 10.

Expanded PKC activation model and subpopulations of two PKCα activation states detected by single-molecule diffusion studies. (A) Expanded structural model of PKC activation showing the newly discovered, predominant, pre-DAG intermediate (IV) in which both the C1A and C2 domains contact the bilayer. Four of the indicated states [(II), (III), (V), and (VI)] are similar to those of a previous model,28 while pre-DAG intermediate (IV) is novel. The indicated structures are supported by available evidence, but current data do not rule out alternative structures (Figure S2 of the Supporting Information). (B) Single-molecule TIRF analysis of PKCα single-molecule diffusion shows that states (IV) and (VI) in the expanded activation reaction (see also Figure 7) are each comprised of two major subpopulations as indicated. The relative population sizes were determined by multistate fits of the single-molecule diffusion data on each type of supported bilayer (Table S2 of the Supporting Information). Domain subscripts indicate the relative degree of insertion of the protein into the membrane (s for shallow and d for deep). Question marks indicate that the status of the N-terminal pseudosubstrate region is unresolved in the inactive, pre-DAG intermediate (IV): because the friction of the peptide–bilayer contact is expected to be negligible, the diffusion analysis does not ascertain whether the peptide remains bound to the kinase active site or binds to the bilayer as proposed for the active state.28,92
PC/PS/PIP2/DAG Bilayers
The equilibrium binding of full-length PKCα and its isolated regulatory domains was also measured on PC/PS/PIP2/DAG bilayers. The results showed that, as previously reported,37,38,40,45,56 the membrane affinities of the full-length enzyme and the isolated C1A and C1B domains were significantly increased by the presence of DAG, while the isolated C2 domain was unaffected (Figure 3). Moreover, the specific kinase activity of full-length PKCα was greatly increased by the activating DAG lipid as expected (Figure 3). These findings support a model for the active state (Figure 1B) in which DAG stabilizes both C1 domains in the bilayer, thereby removing kinase inhibition.
While early studies supported a 1:1 stoichiometry for the binding of DAG to full-length PKCα,51,98,99 more recent studies in cells and in vitro, including this study, support a model in which C1A and C1B each deploy to the bilayer and bind one DAG in full-length PKCα as observed for the isolated domains, yielding a 2:1 DAG:protein stoichiometry in DAG-activated PKCα.19,28,38,55,56 The new working model (Figures 7, 10A) proposes that in this DAG-activated state, both C1A and C1B are inserted into the bilayer where their membrane contacts are stabilized by DAG, such that both C1 domains contribute to the observed DAG-triggered increase in membrane affinity (Figure 3). Consistent with this picture, the frictional drags of both C1A and C1B, as well as C2, were needed to quantitatively account for the diffusional slowing of full-length PKCα and the C1A–C1B–C2 construct on PC/PS/PIP2/DAG bilayers (Figure 6C, Table S2 of the Supporting Information, and eq 1). Because of the inverse dependence between the diffusion constant and frictional drag (see above), DAG-triggered slowing of PKCα appears to be relatively small in magnitude but is highly reproducible and significant (p < 0.06). The additional DAG-induced frictional drag was quantitatively consistent with the additive contribution expected for the C1B domain (Figure 6C, Table S2 of the Supporting Information, and eqs 1 and 2). Significant DAG-triggered slowing was also observed for the C1A–C1B (p < 0.05) and C1B–C2 (p < 0.05) constructs. However, quantitative analysis showed the contribution of C1B to this slowing was smaller for the C1B–C2 construct (Figure 6C and Table S2 of the Supporting Information), suggesting that a membrane-anchored C1A domain may assist in C1B membrane association.
The findings identify the PKCα domain that is primarily responsible for triggering kinase activation on a bilayer surface possessing average plasma membrane PS and PIP2 densities (approximately 25 and 2 mol %, respectively). Given the diffusion results described above showing that C1A is already bilayer-associated before addition of DAG (Figure 6A,B), it follows that kinase activation is triggered by the DAG recruitment of C1B to the bilayer where it joins membrane-bound C1A to help generate the large frictional drag observed for the active state (Figures 6C and 7). An alternative possibility, that the active state possesses only one membrane-bound C1 domain (C1A or C1B) together with a membrane-bound pseudosubstrate peptide on the bilayer surface, cannot account for the observed large frictional drag of active PKCα given the small friction expected for a surface-bound peptide [>3-fold less friction than a C1 domain (see above and Figure 6C)].
Testing the New PKCα Activation Model by Comparing the Kinase Activation Efficacies of Diacylglycerol and Phorbol Ester
Previous studies have indicated that the C1A and C1B domains possess contrasting affinities for DAG and phorbol esters such as phorbol 12-myristate 13-acetate (PMA).37−39,56,100,101 Specifically, these studies showed that C1A prefers DAG over PMA while C1B prefers PMA over DAG. Given these preferences, the new model (Figure 7) predicted that PMA would be a significantly better kinase activator than DAG, because PMA would recruit C1B to the membrane and release kinase inhibition more efficiently. When the kinase activating effects of equal bilayer densities (each at 2 mol %) of PMA and DAG were compared, PMA was found to be the superior activator (p < 0.005) as shown in Figure 8, which supports the new model.
Figure 8.

Comparison of the effects of DAG and PMA on the PKCα kinase specific activity. The total PKCα kinase activity and the bound density of PKCα on the bilayer were measured as described in the legend of Figure 3 for membranes containing 3:1 PC/PS bilayers and the indicated levels of activating diacylglycerol and/or phorbol ester PMA (an activating lipid concentration of 48 or 360 nM corresponded to 2.0 or 7.5 mol % in the bilayer, respectively). Specific activity was calculated as the ratio of kinase activity to membrane-bound enzyme density under each bilayer condition. Each bar was calculated from two duplicates for each kinase and binding assay (n = 4). In all experiments, the free Ca2+ concentration was 6 μM in kinase assay buffer (Materials and Methods) at 22 ± 0.5 °C.
Detection of Membrane-Bound Substates for Certain PKCα Activation Intermediates by Diffusional Analysis and Molecular Dynamics
Each experimental diffusion constant presented in Figure 6 was the population average for a given pairing of a protein construct and bilayer composition. When the step size distribution of each construct–bilayer pairing was examined for multiple states, two distinct subpopulations with different diffusion constants were detected for certain pairings, although no third subpopulation of mobile proteins was detected for any case (Table S2 of the Supporting Information). For dominant intermediate (IV) (Figures 7, 10A) and final, fully deployed active kinase (VI), two substates were required to achieve convergence of the Rayleigh best fit in the absence and presence of DAG, respectively (Table S2 of the Supporting Information). The most likely source of this two-state behavior is the membrane-embedded C1A domain, which as an isolated domain possesses two diffusional states on each bilayer type (Table S2 of the Supporting Information).
All-atom MD simulations of the C1A domain bound to a PC/PS bilayer provided a molecular explanation for the two observed C1A diffusional states, as summarized in Figure 9. Specifically, the MD simulations showed that C1A could exist not only in its deeply membrane-embedded state but also in a previously undescribed shallow docking geometry. This shallow docking state is mainly associated with the headgroup layer and is stabilized by a set of basic residues on one face of C1A that interact with PC and PS acidic moieties, indicating the interaction is largely electrostatic and likely serves as an intermediate in the membrane association and dissociation reactions of C1A. Analysis of the deep state yielded an equilibrium membrane docking geometry consistent with previous models based on NMR and single-molecule diffusion results indicating this state penetrates deeply, up to 6 Å into the hydrocarbon region (using the Nagle convention defining the hydrocarbon core boundary as the plane of average atom positions for carbon 2 of the phospholipid acyl chains) into the bilayer hydrocarbon core.60,91,102
Quantitatively Testing the New PKCα Activation Model by Comparing Calculated and Measured Average Diffusion Constants
Finally, the proposed membrane contacts in the new activation model (Figures 7, 10A) were tested by asking whether calculated diffusion constants based on this model would yield reasonable agreement with the observed diffusion constants. Modeling of membrane-bound full-length PKCα confirms that the interdomain linkers are sufficiently long to prevent steric clashes between the individual domains in their normal membrane docking geometries (Figure 9C). Thus, the calculated diffusion constants utilized the frictional drags measured for the individual, isolated regulatory domains (C1A, C1B, and C2) on each membrane type, and the additivity of frictional drags on supported bilayers (eq 1),60 to calculate the total frictional drag and average diffusion constant for each multidomain construct. For cases in which two diffusional substates were observed, both substates were used to calculate the appropriate population-weighted average (Figure 6 and Table S2 of the Supporting Information). Notably, the calculated and measured average diffusion constants were in excellent global agreement (compare white and black bars in Figure 6), providing strong support for the proposed membrane contacts in the new PKCα activation model (Figures 7, 10A).
Discussion
These studies of PKCα directly detect and verify each of the pre-DAG intermediates proposed to occur in the activation reaction that converts inactive, mature cPKC in the cytoplasm into active kinase on the plasma membrane surface (Figure 1B).6,7,28,33,35,36,39−53 In addition, the findings reveal a major new pre-DAG intermediate in the activation reaction. As the now expanded cPKC activation sequence in Figure 7 illustrates, and as shown in the molecular model of Figure 10A, the activation reaction begins with the rapid binding of the C2 domain to two PS molecules at its Ca2+ binding site and basic cluster, respectively. Such rapid PS binding is ensured by the high physiological PS mole density (∼25 mol % in the plasma membrane33,82), such that on average, approximately 4 of the 14–18 lipids in the C2 domain footprint as it docks to the membrane would be PS molecules.36 As confirmation of this picture, full-length PKCα and its isolated C2 domain exhibit the same average diffusion speed when they first bind to the membrane [Figure 5A and (II) in Figures 7 and 10].
Subsequently, the (PS)2 intermediate encounters a PIP2 molecule (∼2 mol % in the plasma membrane) on the microsecond time scale that would rapidly replace PS bound in the C2 domain basic cluster because of its higher PIP2 affinity, generating the (PS-PIP2) intermediate.34−36 PIP2 binding increases membrane affinity, and the resulting change in the membrane docking angle35,36 yields slightly more protein penetration and frictional drag, and modest diffusional slowing [Figures 3 and 6 and (III) in Figures 7 and 10].
After the C2 domain docks to the membrane, the C1A domain inserts into the bilayer with a time constant of ∼50 ms (Figure 5B,C). The deeply penetrating substate of the C1A domain is proposed to bind one or more PS molecules48,49,90,103 to account both for the PS requirement in stable binding of the isolated C1A to the bilayer (Figure 3) and for the higher sensitivity of full-length PKC to the PS density relative to the isolated C2 domain (Figure 4). If C1A is stably contacting the C2 and/or kinase domain in the inactive cPKC, as suggested by some studies,28,49,103 C1A must dissociate from other domain(s) to insert its hydrophobic residues into the bilayer. The resulting, newly discovered pre-DAG intermediate with C1A and C2 contacting the bilayer, and with C1B helping inhibit the catalytic domain [(IV) in Figures 7 and 10], is the predominant inactive intermediate awaiting the appearance of a DAG lipid signal. The membrane-associated C1A domain greatly increases the frictional drag because this domain is embedded more deeply than the C2 domain in the membrane,35,36,42,60,91 thereby slowing the diffusion of the newly discovered intermediate (IV) while increasing its membrane affinity and bound state lifetime, and thus increasing the probability it will remain on the membrane until a DAG signal appears. Moreover, the membrane-bound state of the C1A domain will bind DAG more rapidly and with higher affinity than the aqueous state of C1A, which has no opportunity to collide with the membrane-embedded activating lipid. Finally, the association of C1A with the bilayer is proposed to help guide C1B to the bilayer after the appearance of the DAG signal. It is not yet clear whether the N-terminal pseudosubstrate region of the pre-DAG intermediate remains bound in the kinase active site or deploys to the membrane, because the frictional drag of the resulting peptide–bilayer interaction would likely be negligible relative to the drags of the C1A and C2 domains. Thus, the pre-DAG intermediate could be inhibited either by C1B alone or by both the pseudosubstrate and C1B [compare (IVa,b,c) in Figure S2 of the Supporting Information].
The findings identify the final, essential step in kinase activation. During a DAG signaling event, one DAG molecule is proposed to rapidly occupy the membrane-embedded C1A domain, which is known to bind DAG.14,37,38,44,56,101 This step may also help deploy the pseudosubstrate to the membrane and/or may help dissociate C1B from the kinase domain and if so would contribute to kinase activation [(V) in Figures 7 and 10A]. The transmission of information from C1A to C1B is supported by the highly conserved nature of the 15-residue C1A–C1B linker, which is identical in cow, rat, mouse, and human PKCα or PKCβ and differs at only two positions between the PKCα and PKCβ isoforms. Subsequently, in the final kinase activation step [transition from (V) to (VI) in Figures 7 and 10A], the C1B domain is recruited into the bilayer by PS and DAG, which it is known to bind,40,48,49,103 thereby stabilizing the fully activated kinase (Figures 3 and 6). In the resulting, kinase active state all three regulatory domains, C1A, C1B, and C2, are membrane-associated and all the inhibitory contacts with the kinase domain have been released. The extensive bilayer contacts of the active kinase increase its lifetime on the membrane as it diffuses in two dimensions on the membrane surface, encountering and phosphorylating its substrate receptors and signaling proteins. The diffusion of this active enzyme is ∼10-fold slower than that of a single lipid in the bilayer (Table 2); however, it remains quite mobile, and the 10-fold slowing observed on supported bilayers may overestimate the extent of slowing on the plasma membrane where frictional drag may be less additive.77,78 The recruitment of C1B to the membrane is driven even more strongly by phorbol ester (PMA) than by DAG, because of the known higher affinity of C1B for PMA.37−40,42,48,51,101,104−109 Thus, the model simply explains the known propensity of phorbol esters to activate cPKCs more strongly than DAG, as well as the ability of these plant toxins to stimulate tumor growth.8,98,105,107,110−113 Eventually, unless it is trapped in a complex that maintains its activated state,2,114 the active enzyme will leave the membrane and return to its inactive cytoplasmic state as the Ca2+ and DAG signals return to baseline.
In a population of PKCα molecules, each of the five individual membrane-bound intermediates in the new model (Figures 7 and 10A) can be considered a distinct ensemble. Multistate analysis57,58,66 of the isolated C2 domain on each type of bilayer suggests that the initial intermediates in which membrane contacts are dominated by the C2 domain [(II) and (III)] can be represented as simple, homogeneous populations displaying a single diffusion constant. However, intermediates possessing a membrane-contacting C1A or C1B domain [(IV) and (VI) have been directly observed in this study] can switch between a shallow or deep membrane-penetrating state, yielding ensembles that display multiple diffusion constants: fast and slow arising from shallow and deep penetration of the C1 domain into the bilayer, respectively (Figure 10B and Table S2 of the Supporting Information). Where multiple substates are present, all contribute to the average diffusion constant (Figure 6 and Table 2) with weightings proportional to their population sizes (Table S2 of the Supporting Information).
The molecular model proposed in Figure 10A is based on a SAXS study suggesting the mature, inactive PKCα molecule adopts the indicated compact structure in the cytoplasm, which then docks to the bilayer via its C2 domain during a Ca2+ signal.28 However, it is possible that the membrane-bound enzyme adopts a more extended structure (see multiple possibilities in Figure S2 of the Supporting Information). While further studies are needed to establish the detailed molecular picture of the membrane-bound states, our single-molecule diffusion results firmly define the existence and the domain–bilayer contacts of the observed intermediates [(II), (III), (IV), and (VI)].
In summary, these findings show that the typical activation sequence of a conventional PKC enzyme bound to the plasma membrane involves the previously described rapid acquisition of PS and PIP2 by the C2 domain, followed by a newly observed penetration of C1A into the bilayer that stabilizes the membrane-bound enzyme as it awaits the DAG lipid signal. This C1A-embedded state is the predominant intermediate on the plasma membrane until the DAG signal appears and recruits C1B into the bilayer, thereby releasing the C1B inhibition of the kinase domain and generating the active enzyme [conversion of (IV) to (VI) in Figures 7 and 10A]. Because C1B recruitment by activating lipid is the final trigger for kinase activation, and because C1B binds phorbol esters more tightly than DAG while the reverse is true for C1A, the model explains the superiority of phorbol esters over DAG as lipid activators. Moreover, the findings illustrate the power of single-molecule binding and diffusion studies, in concert with activity studies, to reveal the hidden steps in the activation mechanisms of membrane-bound signaling proteins.
Acknowledgments
We sincerely thank Dr. Thomas Leiker and Prof. Robert C. Murphy (University of Colorado, Denver, CO; support provided by National Institutes of Health Grant U54-GM69338 to Robert C. Murphy) for mass spectrometry analysis of the DAG content of lipid mixtures, Teresa Stines Narheini (University of Colorado Tissue Culture Facility) for training in cell culture, and Profs. James Hurley (University of California at Berkeley, Berkeley, CA) and Alexandra Newton (University of California at San Diego, La Jolla, CA) for helpful discussions and suggestions.
Glossary
Abbreviations
- C1A and C1B
first conserved domains of classical protein kinase C
- C2
second conserved domain of classical protein kinase C
- PKCα
protein kinase C isoform α
- Sfp
phosphopantethienyltransferase
- AF555
Alexa Fluor 555
- PC
phosphatidylcholine or 1,2-dioleoyl-sn-glycero-3-phosphocholine
- PDB
Protein Data Bank
- PS
phosphatidylserine or 1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- PI(4,5)P2 or PIP2
1,2-dioleoyl-sn-glycero-3-phosphoinositol 4,5-bisphosphate
- diacylglycerol or DAG
1,2-dioleoyl-sn-glycerol
- LRB-PE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl)
- SUV
sonicated unilamellar vesicle
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- TIRFM
total internal reflection fluorescence microscopy
- GST
glutathione S-transferase
- MBP
maltose-binding protein
- HA
hemagglutinin
- PMA
phorbol 12-myristate 13-acetate.
Supporting Information Available
SI content includes: Ca2+ and lipid regulation of the full-length PKCα constructs employed here (Figure S1), plausible alternative structural models for the dominant, pre-DAG intermediate (IV) (Figure S2), liquid chromatography–tandem mass spectrometry data showing our lipid mixtures are free of contaminating DAG (Table S1), and the tabulated multistate diffusion analyses carried out for all constructs (Table S2). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ K.E.L.: AvidBiotics Corp., 100 Kimball Way, South San Francisco, CA 94080.
Author Present Address
∥ J.D.K.: Department of Chemistry, University of Colorado, Denver, CO 80217-3364.
Support provided by National Institutes of Health Grants R01 GM-063235 (to J.J.F.) and R01 GM-063796 (to G.A.V.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nishizuka Y. (1995) Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 9, 484–496. [PubMed] [Google Scholar]
- Dempsey E. C.; Newton A. C.; Mochly-Rosen D.; Fields A. P.; Reyland M. E.; Insel P. A.; Messing R. O. (2000) Protein kinase C isozymes and the regulation of diverse cell responses. Am. J. Physiol. 279, L429–L438. [DOI] [PubMed] [Google Scholar]
- Griner E. M.; Kazanietz M. G. (2007) Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 7, 281–294. [DOI] [PubMed] [Google Scholar]
- Evans J. H.; Falke J. J. (2007) Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proc. Natl. Acad. Sci. U.S.A. 104, 16176–16181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos L. L.; Newton A. C. (2008) Spatiotemporal dynamics of lipid signaling: Protein kinase C as a paradigm. IUBMB Life 60, 782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton A. C. (2009) Lipid activation of protein kinases. J. Lipid Res. 50(Suppl.), S266–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oancea E.; Meyer T. (1998) Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell 95, 307–318. [DOI] [PubMed] [Google Scholar]
- Cataisson C.; Joseloff E.; Murillas R.; Wang A.; Atwell C.; Torgerson S.; Gerdes M.; Subleski J.; Gao J. L.; Murphy P. M.; Wiltrout R. H.; Vinson C.; Yuspa S. H. (2003) Activation of cutaneous protein kinase C α induces keratinocyte apoptosis and intraepidermal inflammation by independent signaling pathways. J. Immunol. 171, 2703–2713. [DOI] [PubMed] [Google Scholar]
- Spitaler M.; Cantrell D. A. (2004) Protein kinase C and beyond. Nat. Immunol. 5, 785–790. [DOI] [PubMed] [Google Scholar]
- Haughian J. M.; Reno E. M.; Thorne A. M.; Bradford A. P. (2009) Protein kinase C α-dependent signaling mediates endometrial cancer cell growth and tumorigenesis. Int. J. Cancer 125, 2556–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughian J. M.; Bradford A. P. (2009) Protein kinase C α (PKCα) regulates growth and invasion of endometrial cancer cells. J. Cell. Physiol. 220, 112–118. [DOI] [PubMed] [Google Scholar]
- Pearce L. R.; Komander D.; Alessi D. R. (2010) The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 11, 9–22. [DOI] [PubMed] [Google Scholar]
- Dekker L. V.; Palmer R. H.; Parker P. J. (1995) The protein kinase C and protein kinase C related gene families. Curr. Opin. Struct. Biol. 5, 396–402. [DOI] [PubMed] [Google Scholar]
- Hurley J. H.; Newton A. C.; Parker P. J.; Blumberg P. M.; Nishizuka Y. (1997) Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 6, 477–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W.; Stahelin R. V. (2005) Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 34, 119–151. [DOI] [PubMed] [Google Scholar]
- Kheifets V.; Mochly-Rosen D. (2007) Insight into intra- and inter-molecular interactions of PKC: Design of specific modulators of kinase function. Pharmacol. Res. 55, 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbalan-Garcia S.; Gomez-Fernandez J. C. (2010) The C2 domains of classical and novel PKCs as versatile decoders of membrane signals. Biofactors 36, 1–7. [DOI] [PubMed] [Google Scholar]
- Newton A. C. (2010) Protein kinase C: Poised to signal. Am. J. Physiol. 298, E395–E402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard T. A.; Hurley J. H. (2011) Regulation of protein kinases by lipids. Curr. Opin. Struct. Biol. 21, 785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutil E. M.; Toker A.; Newton A. C. (1998) Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1). Curr. Biol. 8, 1366–1375. [DOI] [PubMed] [Google Scholar]
- Sonnenburg E. D.; Gao T.; Newton A. C. (2001) The phosphoinositide-dependent kinase, PDK-1, phosphorylates conventional protein kinase C isozymes by a mechanism that is independent of phosphoinositide 3-kinase. J. Biol. Chem. 276, 45289–45297. [DOI] [PubMed] [Google Scholar]
- Newton A. C. (2003) Regulation of the ABC kinases by phosphorylation: Protein kinase C as a paradigm. Biochem. J. 370, 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House C.; Kemp B. E. (1987) Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science 238, 1726–1728. [DOI] [PubMed] [Google Scholar]
- Pears C. J.; Kour G.; House C.; Kemp B. E.; Parker P. J. (1990) Mutagenesis of the pseudosubstrate site of protein kinase C leads to activation. Eur. J. Biochem. 194, 89–94. [DOI] [PubMed] [Google Scholar]
- Orr J. W.; Keranen L. M.; Newton A. C. (1992) Reversible exposure of the pseudosubstrate domain of protein kinase C by phosphatidylserine and diacylglycerol. J. Biol. Chem. 267, 15263–15266. [PubMed] [Google Scholar]
- Orr J. W.; Newton A. C. (1994) Intrapeptide regulation of protein kinase C. J. Biol. Chem. 269, 8383–8387. [PubMed] [Google Scholar]
- Grodsky N.; Li Y.; Bouzida D.; Love R.; Jensen J.; Nodes B.; Nonomiya J.; Grant S. (2006) Structure of the catalytic domain of human protein kinase C β II complexed with a bisindolylmaleimide inhibitor. Biochemistry 45, 13970–13981. [DOI] [PubMed] [Google Scholar]
- Leonard T. A.; Rozycki B.; Saidi L. F.; Hummer G.; Hurley J. H. (2011) Crystal structure and allosteric activation of protein kinase C βII. Cell 144, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D.; Luo J.; Mochly-Rosen D. (1995) C2 region-derived peptides inhibit translocation and function of β protein kinase C in vivo. J. Biol. Chem. 270, 24180–24187. [DOI] [PubMed] [Google Scholar]
- Ron D.; Mochly-Rosen D. (1995) An autoregulatory region in protein kinase C: The pseudoanchoring site. Proc. Natl. Acad. Sci. U.S.A. 92, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedovitzky M.; Mochly-Rosen D.; Johnson J. A.; Gray M. O.; Ron D.; Abramovitch E.; Cerasi E.; Nesher R. (1997) Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic β-cells. J. Biol. Chem. 272, 1417–1420. [DOI] [PubMed] [Google Scholar]
- Corbalan-Garcia S.; Guerrero-Valero M.; Marin-Vicente C.; Gomez-Fernandez J. C. (2007) The C2 domains of classical/conventional PKCs are specific PtdIns(4,5)P(2)-sensing domains. Biochem. Soc. Trans. 35, 1046–1048. [DOI] [PubMed] [Google Scholar]
- Corbin J. A.; Evans J. H.; Landgraf K. E.; Falke J. J. (2007) Mechanism of specific membrane targeting by C2 domains: Localized pools of target lipids enhance Ca2+ affinity. Biochemistry 46, 4322–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna D.; Bhardwaj N.; Vora M. S.; Stahelin R. V.; Lu H.; Cho W. (2008) Differential roles of phosphatidylserine, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 in plasma membrane targeting of C2 domains. Molecular dynamics simulation, membrane binding, and cell translocation studies of the PKCα C2 domain. J. Biol. Chem. 283, 26047–26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf K. E.; Malmberg N. J.; Falke J. J. (2008) Effect of PIP2 binding on the membrane docking geometry of PKC α C2 domain: An EPR site-directed spin-labeling and relaxation study. Biochemistry 47, 8301–8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C. L.; Landgraf K. E.; Voth G. A.; Falke J. J. (2010) Membrane docking geometry and target lipid stoichiometry of membrane-bound PKCα C2 domain: A combined molecular dynamics and experimental study. J. Mol. Biol. 402, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater S. J.; Kelly M. B.; Taddeo F. J.; Rubin E.; Stubbs C. D. (1994) Evidence for discrete diacylglycerol and phorbol ester activator sites on protein kinase C. Differences in effects of 1-alkanol inhibition, activation by phosphatidylethanolamine and calcium chelation. J. Biol. Chem. 269, 17160–17165. [PubMed] [Google Scholar]
- Slater S. J.; Ho C.; Kelly M. B.; Larkin J. D.; Taddeo F. J.; Yeager M. D.; Stubbs C. D. (1996) Protein kinase Cα contains two activator binding sites that bind phorbol esters and diacylglycerols with opposite affinities. J. Biol. Chem. 271, 4627–4631. [DOI] [PubMed] [Google Scholar]
- Slater S. J.; Milano S. K.; Stagliano B. A.; Gergich K. J.; Ho C.; Mazurek A.; Taddeo F. J.; Kelly M. B.; Yeager M. D.; Stubbs C. D. (1999) Synergistic activation of protein kinase Cα, -βI, and -γ isoforms induced by diacylglycerol and phorbol ester: Roles of membrane association and activating conformational changes. Biochemistry 38, 3804–3815. [DOI] [PubMed] [Google Scholar]
- Medkova M.; Cho W. (1999) Interplay of C1 and C2 domains of protein kinase C-α in its membrane binding and activation. J. Biol. Chem. 274, 19852–19861. [DOI] [PubMed] [Google Scholar]
- Slater S. J.; Seiz J. L.; Cook A. C.; Buzas C. J.; Malinowski S. A.; Kershner J. L.; Stagliano B. A.; Stubbs C. D. (2002) Regulation of PKC α activity by C1-C2 domain interactions. J. Biol. Chem. 277, 15277–15285. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Kazanietz M. G.; Blumberg P. M.; Hurley J. H. (1995) Crystal structure of the cys2 activator-binding domain of protein kinase C δ in complex with phorbol ester. Cell 81, 917–924. [DOI] [PubMed] [Google Scholar]
- Edwards A. S.; Newton A. C. (1997) Regulation of protein kinase C βII by its C2 domain. Biochemistry 36, 15615–15623. [DOI] [PubMed] [Google Scholar]
- Newton A. C. (1997) Regulation of protein kinase C. Curr. Opin. Cell Biol. 9, 161–167. [DOI] [PubMed] [Google Scholar]
- Medkova M.; Cho W. (1998) Differential membrane-binding and activation mechanisms of protein kinase C-α and -ε. Biochemistry 37, 4892–4900. [DOI] [PubMed] [Google Scholar]
- Sutton R. B.; Sprang S. R. (1998) Structure of the protein kinase Cβ phospholipid-binding C2 domain complexed with Ca2+. Structure 6, 1395–1405. [DOI] [PubMed] [Google Scholar]
- Verdaguer N.; Corbalan-Garcia S.; Ochoa W. F.; Fita I.; Gomez-Fernandez J. C. (1999) Ca2+ bridges the C2 membrane-binding domain of protein kinase Cα directly to phosphatidylserine. EMBO J. 18, 6329–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. E.; Giorgione J.; Newton A. C. (2000) The C1 and C2 domains of protein kinase C are independent membrane targeting modules, with specificity for phosphatidylserine conferred by the C1 domain. Biochemistry 39, 11360–11369. [DOI] [PubMed] [Google Scholar]
- Bittova L.; Stahelin R. V.; Cho W. (2001) Roles of ionic residues of the C1 domain in protein kinase C-α activation and the origin of phosphatidylserine specificity. J. Biol. Chem. 276, 4218–4226. [DOI] [PubMed] [Google Scholar]
- Corbalan-Garcia S.; Garcia-Garcia J.; Rodriguez-Alfaro J. A.; Gomez-Fernandez J. C. (2003) A new phosphatidylinositol 4,5-bisphosphate-binding site located in the C2 domain of protein kinase Cα. J. Biol. Chem. 278, 4972–4980. [DOI] [PubMed] [Google Scholar]
- Giorgione J.; Hysell M.; Harvey D. F.; Newton A. C. (2003) Contribution of the C1A and C1B domains to the membrane interaction of protein kinase C. Biochemistry 42, 11194–11202. [DOI] [PubMed] [Google Scholar]
- Colon-Gonzalez F.; Kazanietz M. G. (2006) C1 domains exposed: From diacylglycerol binding to protein-protein interactions. Biochim. Biophys. Acta 1761, 827–837. [DOI] [PubMed] [Google Scholar]
- Evans J. H.; Murray D.; Leslie C. C.; Falke J. J. (2006) Specific translocation of protein kinase Cα to the plasma membrane requires both Ca2+ and PIP2 recognition by its C2 domain. Mol. Biol. Cell 17, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dries D. R.; Newton A. C. (2008) Kinetic analysis of the interaction of the C1 domain of protein kinase C with lipid membranes by stopped-flow spectroscopy. J. Biol. Chem. 283, 7885–7893. [DOI] [PubMed] [Google Scholar]
- Bogi K.; Lorenzo P. S.; Szallasi Z.; Acs P.; Wagner G. S.; Blumberg P. M. (1998) Differential selectivity of ligands for the C1a and C1b phorbol ester binding domains of protein kinase Cδ: Possible correlation with tumor-promoting activity. Cancer Res. 58, 1423–1428. [PubMed] [Google Scholar]
- Ananthanarayanan B.; Stahelin R. V.; Digman M. A.; Cho W. (2003) Activation mechanisms of conventional protein kinase C isoforms are determined by the ligand affinity and conformational flexibility of their C1 domains. J. Biol. Chem. 278, 46886–46894. [DOI] [PubMed] [Google Scholar]
- Knight J. D.; Falke J. J. (2009) Single-molecule fluorescence studies of a PH domain: New insights into the membrane docking reaction. Biophys. J. 96, 566–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J. D.; Lerner M. G.; Marcano-Velazquez J. G.; Pastor R. W.; Falke J. J. (2010) Single molecule diffusion of membrane-bound proteins: Window into lipid contacts and bilayer dynamics. Biophys. J. 99, 2879–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemba B. P.; Knight J. D.; Falke J. J. (2012) Assembly of membrane-bound protein complexes: Detection and analysis by single molecule diffusion. Biochemistry 51, 1638–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemba B. P.; Falke J. J. (2013) Lateral diffusion of peripheral membrane proteins on supported lipid bilayers is controlled by the additive frictional drags of (1) bound lipids and (2) protein domains penetrating into the bilayer hydrocarbon core. Chem. Phys. Lipids 172–173, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.; Lin A. J.; Golan D. E.; Walsh C. T. (2006) Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 1, 280–285. [DOI] [PubMed] [Google Scholar]
- Soh J. W.; Weinstein I. B. (2003) Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J. Biol. Chem. 278, 34709–34716. [DOI] [PubMed] [Google Scholar]
- Nair P. M.; Salaita K.; Petit R. S.; Groves J. T. (2011) Using patterned supported lipid membranes to investigate the role of receptor organization in intercellular signaling. Nat. Protoc. 6, 523–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbalzarini I. F.; Koumoutsakos P. (2005) Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Biol. 151, 182–195. [DOI] [PubMed] [Google Scholar]
- Schutz G. J.; Schindler H.; Schmidt T. (1997) Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophys. J. 73, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. (2006) The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. III; Mackerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caflisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. (2009) CHARMM: The biomolecular simulation program. J. Comput. Chem. 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jonsson A. L.; Beuming T.; Shelley J. C.; Voth G. A. (2013) Ligand-dependent activation and deactivation of the human adenosine A(2A) receptor. J. Am. Chem. Soc. 135, 8749–8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14, 27–38. [DOI] [PubMed] [Google Scholar]
- Saffman P. G.; Delbruck M. (1975) Brownian motion in biological membranes. Proc. Natl. Acad. Sci. U.S.A. 72, 3111–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters R.; Cherry R. J. (1982) Lateral and rotational diffusion of bacteriorhodopsin in lipid bilayers: Experimental test of the Saffman-Delbruck equations. Proc. Natl. Acad. Sci. U.S.A. 79, 4317–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackmann E. (1996) Supported membranes: Scientific and practical applications. Science 271, 43–48. [DOI] [PubMed] [Google Scholar]
- Tamm L. K.; Groves J. T. (2009) Supported membranes in structural biology. J. Struct. Biol. 168, 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm L. K. (1988) Lateral diffusion and fluorescence microscope studies on a monoclonal antibody specifically bound to supported phospholipid bilayers. Biochemistry 27, 1450–1457. [DOI] [PubMed] [Google Scholar]
- Przybylo M.; Sykora J.; Humpolickova J.; Benda A.; Zan A.; Hof M. (2006) Lipid diffusion in giant unilamellar vesicles is more than 2 times faster than in supported phospholipid bilayers under identical conditions. Langmuir 22, 9096–9099. [DOI] [PubMed] [Google Scholar]
- Camley B. A.; Brown F. L. (2012) Contributions to membrane-embedded-protein diffusion beyond hydrodynamic theories. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 85, 061921. [DOI] [PubMed] [Google Scholar]
- Camley B. A.; Brown F. L. (2013) Diffusion of complex objects embedded in free and supported lipid bilayer membranes: Role of shape anisotropy and leaflet structure. Soft Matter 9, 4767–4779. [Google Scholar]
- Sun P.; Tropea J. E.; Waugh D. S. (2011) Enhancing the solubility of recombinant proteins in Escherichia coli by using hexahistidine-tagged maltose-binding protein as a fusion partner. Methods Mol. Biol. 705, 259–274. [DOI] [PubMed] [Google Scholar]
- Bell M. R.; Engleka M. J.; Malik A.; Strickler J. E. (2013) To fuse or not to fuse: What is your purpose?. Protein Sci. 22, 1466–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohout S. C.; Corbalan-Garcia S.; Torrecillas A.; Gomez-Fernandez J. C.; Falke J. J. (2002) C2 domains of protein kinase C isoforms α, β, and γ: Activation parameters and calcium stoichiometries of the membrane-bound state. Biochemistry 41, 11411–11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventis P. A.; Grinstein S. (2010) The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39, 407–427. [DOI] [PubMed] [Google Scholar]
- McLaughlin S.; Wang J.; Gambhir A.; Murray D. (2002) PIP2 and proteins: Interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175. [DOI] [PubMed] [Google Scholar]
- Sanchez-Bautista S.; Marin-Vicente C.; Gomez-Fernandez J. C.; Corbalan-Garcia S. (2006) The C2 domain of PKCα is a Ca2+-dependent PtdIns(4,5)P2 sensing domain: A new insight into an old pathway. J. Mol. Biol. 362, 901–914. [DOI] [PubMed] [Google Scholar]
- Perez-Lara A.; Egea-Jimenez A. L.; Ausili A.; Corbalan-Garcia S.; Gomez-Fernandez J. C. (2012) The membrane binding kinetics of full-length PKCα is determined by membrane lipid composition. Biochim. Biophys. Acta 1821, 1434–1442. [DOI] [PubMed] [Google Scholar]
- Hardt S. L. (1979) Rates of diffusion controlled reactions in one, two and three dimensions. Biophys. Chem. 10, 239–243. [DOI] [PubMed] [Google Scholar]
- Allen V.; Swigart P.; Cheung R.; Cockcroft S.; Katan M. (1997) Regulation of phospholipase Cδ by changes in Ca2+ ion concentrations. Biochem. J. 327(Part 2), 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wal J.; Habets R.; Varnai P.; Balla T.; Jalink K. (2001) Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J. Biol. Chem. 276, 15337–15344. [DOI] [PubMed] [Google Scholar]
- Egea-Jimenez A. L.; Perez-Lara A.; Corbalan-Garcia S.; Gomez-Fernandez J. C. (2013) Phosphatidylinositol 4,5-bisphosphate decreases the concentration of Ca2+, phosphatidylserine and diacylglycerol required for protein kinase C α to reach maximum activity. PLoS One 8, e69041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr J. W.; Newton A. C. (1992) Interaction of protein kinase C with phosphatidylserine. 2. Specificity and regulation. Biochemistry 31, 4667–4673. [DOI] [PubMed] [Google Scholar]
- Xu R. X.; Pawelczyk T.; Xia T. H.; Brown S. C. (1997) NMR structure of a protein kinase C-γ phorbol-binding domain and study of protein-lipid micelle interactions. Biochemistry 36, 10709–10717. [DOI] [PubMed] [Google Scholar]
- Mosior M.; McLaughlin S. (1991) Peptides that mimic the pseudosubstrate region of protein kinase C bind to acidic lipids in membranes. Biophys. J. 60, 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Gambhir A.; McLaughlin S.; Murray D. (2004) A computational model for the electrostatic sequestration of PI(4,5)P2 by membrane-adsorbed basic peptides. Biophys. J. 86, 1969–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Gambhir A.; Hangyas-Mihalyne G.; Murray D.; Golebiewska U.; McLaughlin S. (2002) Lateral sequestration of phosphatidylinositol 4,5-bisphosphate by the basic effector domain of myristoylated alanine-rich C kinase substrate is due to nonspecific electrostatic interactions. J. Biol. Chem. 277, 34401–34412. [DOI] [PubMed] [Google Scholar]
- Gambhir A.; Hangyas-Mihalyne G.; Zaitseva I.; Cafiso D. S.; Wang J.; Murray D.; Pentyala S. N.; Smith S. O.; McLaughlin S. (2004) Electrostatic sequestration of PIP2 on phospholipid membranes by basic/aromatic regions of proteins. Biophys. J. 86, 2188–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch M. E.; Ferguson C. G.; Prestwich G. D.; Cafiso D. S. (2002) Myristoylated alanine-rich C kinase substrate (MARCKS) sequesters spin-labeled phosphatidylinositol 4,5-bisphosphate in lipid bilayers. J. Biol. Chem. 277, 14068–14076. [DOI] [PubMed] [Google Scholar]
- Ausili A.; Corbalan-Garcia S.; Gomez-Fernandez J. C.; Marsh D. (2011) Membrane docking of the C2 domain from protein kinase Cα as seen by polarized ATR-IR. The role of PIP2. Biochim. Biophys. Acta 1808, 684–695. [DOI] [PubMed] [Google Scholar]
- Kikkawa U.; Takai Y.; Tanaka Y.; Miyake R.; Nishizuka Y. (1983) Protein kinase C as a possible receptor protein of tumor-promoting phorbol esters. J. Biol. Chem. 258, 11442–11445. [PubMed] [Google Scholar]
- Konig B.; DiNitto P. A.; Blumberg P. M. (1985) Stoichiometric binding of diacylglycerol to the phorbol ester receptor. J. Cell. Biochem. 29, 37–44. [DOI] [PubMed] [Google Scholar]
- Ho C.; Slater S. J.; Stagliano B. A.; Stubbs C. D. (1999) Conformation of the C1 phorbol-ester-binding domain participates in the activating conformational change of protein kinase C. Biochem. J. 344(Part 2), 451–460. [PMC free article] [PubMed] [Google Scholar]
- Slater S. J.; Seiz J. L.; Stagliano B. A.; Cook A. C.; Milano S. K.; Ho C.; Stubbs C. D. (2001) Low- and high-affinity phorbol ester and diglyceride interactions with protein kinase C: 1-O-Alkyl-2-acyl-sn-glycerol enhances phorbol ester- and diacylglycerol-induced activity but alone does not induce activity. Biochemistry 40, 6085–6092. [DOI] [PubMed] [Google Scholar]
- Nagle J. F.; Tristram-Nagle S. (2000) Structure of lipid bilayers. Biochim. Biophys. Acta 1469, 159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahelin R. V.; Wang J.; Blatner N. R.; Rafter J. D.; Murray D.; Cho W. (2005) The origin of C1A-C2 interdomain interactions in protein kinase Cα. J. Biol. Chem. 280, 36452–36463. [DOI] [PubMed] [Google Scholar]
- Quest A. F.; Bardes E. S.; Bell R. M. (1994) A phorbol ester binding domain of protein kinase C γ. High affinity binding to a glutathione-S-transferase/Cys2 fusion protein. J. Biol. Chem. 269, 2953–2960. [PubMed] [Google Scholar]
- Kozhemiakina O. V.; Iaguzhinskii L. S. (1990) [Complexes of phorbol tumor promoters with protein kinase C]. Biokhimiia 55, 595–607. [PubMed] [Google Scholar]
- Sharkey N. A.; Blumberg P. M. (1985) Highly lipophilic phorbol esters as inhibitors of specific [3H]phorbol 12,13-dibutyrate binding. Cancer Res. 45, 19–24. [PubMed] [Google Scholar]
- Leach K. L.; Blumberg P. M. (1985) Modulation of protein kinase C activity and [3H]phorbol 12,13-dibutyrate binding by tumor promoters in mouse brain cytosol. Cancer Res. 45, 1958–1963. [PubMed] [Google Scholar]
- Arcoleo J. P.; Weinstein I. B. (1985) Activation of protein kinase C by tumor promoting phorbol esters, teleocidin and aplysiatoxin in the absence of added calcium. Carcinogenesis 6, 213–217. [DOI] [PubMed] [Google Scholar]
- Blumberg P. M.; Delclos K. B.; Dunphy W. G.; Jaken S. (1982) Specific binding of phorbol ester tumor promoters to mouse tissues and cultured cells. Carcinog.—Compr. Surv. 7, 519–535. [PubMed] [Google Scholar]
- Castagna M.; Takai Y.; Kaibuchi K.; Sano K.; Kikkawa U.; Nishizuka Y. (1982) Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 257, 7847–7851. [PubMed] [Google Scholar]
- Anderson W. B.; Estival A.; Tapiovaara H.; Gopalakrishna R. (1985) Altered subcellular distribution of protein kinase C (a phorbol ester receptor). Possible role in tumor promotion and the regulation of cell growth: Relationship to changes in adenylate cyclase activity. Adv. Cyclic Nucleotide Protein Phosphorylation Res. 19, 287–306. [PubMed] [Google Scholar]
- Tapley P. M.; Murray A. W. (1984) Modulation of Ca2+-activated, phospholipid-dependent protein kinase in platelets treated with a tumor-promoting phorbol ester. Biochem. Biophys. Res. Commun. 122, 158–164. [DOI] [PubMed] [Google Scholar]
- Hubinont C. J.; Best L.; Sener A.; Malaisse W. J. (1984) Activation of protein kinase C by a tumor-promoting phorbol ester in pancreatic islets. FEBS Lett. 170, 247–253. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D. (1995) Localization of protein kinases by anchoring proteins: A theme in signal transduction. Science 268, 247–251. [DOI] [PubMed] [Google Scholar]
- Leiker T. J.; Barkley R. M.; Murphy R. C. (2011) Analysis of Diacylglycerol Molecular Species in Cellular Lipid Extracts by Normal-Phase LC-Electrospray Mass Spectrometry. Int. J. Mass Spectrom. 305, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. H.; Málková S.; Pingali S. V.; Long F.; Garde S.; Cho W.; Schlossman M. (2009) Configuration of PKCα-C2 domain bound to mixed SOPC/SOPS lipid monolayers. Biophys. J. 97, 2794–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.