

Abstract
High cholesterol levels are an established risk factor for cardiovascular disease (CVD), the world’s leading cause of death. Inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (statins) are prescribed to lower serum cholesterol levels and reduce the risk of CVD. Despite the success of statins, many patients abandon treatment owing to neuromuscular adverse drug reactions (ADRs). Genome-wide association studies have identified the single-nucleotide polymorphism (SNP) rs4149056 in the SLCO1B1 gene as being associated with an increased risk for statin-induced ADRs. By studying slow-channel syndrome transgenic mouse models, we determined that statins trigger ADRs in mice expressing the mutant allele of the rs137852808 SNP in the nicotinic acetylcholine receptor (nAChR) α-subunit gene CHRNA1. Mice expressing this allele show a remarkable contamination of end-plates with caveolin-1 and develop early signs of neuromuscular degeneration upon statin treatment. This study demonstrates that genes coding for nAChR subunits may contain variants associated with statin-induced ADRs.
Introduction
According to the World Health Organization (WHO), cardiovascular disease (CVD) is the world’s leading cause of death. It has been estimated that 17.3 million people died from CVD in 2008 alone, representing 30% of all global deaths. High levels of cholesterol carried by low-density lipoprotein, colloquially known as ‘bad cholesterol’, is a firmly established independent risk factor for CVD.1, 2, 3 Naturally, as CVD claims so many lives, cholesterol-lowering medications represent an essential strategy to reduce CVD mortality rates.4 Inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase—collectively named statins—inhibit the rate-limiting step in the biosynthesis of cholesterol, thus reducing its availability and effectively lowering plasma low-density lipoprotein levels. Despite its demonstrated safety, a fraction of those treated with statins suffer adverse drug reactions (ADRs), mostly neuromuscular symptoms, which eventually force patients to discontinue treatment. Because the number of patients on statins is so high (more than 17 million people are prescribed Lipitor™), the actual number of patients that abandon treatment due to ADRs can be substantial. Many are left with an increased risk for CVD as there is a linear dose–response relationship between increasing adherence to statin treatment and decreasing coronary mortality.5
The most common statin ADRs are neuromuscular problems involving muscle pain or weakness, and range in severity from myalgia to rhabdomyolysis. Myalgia is defined as muscle weakness or pain without an elevation in serum creatine kinase (CK) levels. In clinical trials, the incidence of myalgia is 1–5%, although observational studies reveal that myalgia is more frequent (9–20%) than expected.6 Very rare muscle-related ADRs include myositis, which refers to muscular symptoms with serum creatine kinase elevation, and the life-threatening rhabdomyolysis, which is characterized by a marked creatine kinase elevation and can cause severe muscle pain, renal failure, disseminated intravascular coagulation, and death.7, 8 Genetic factors have been suspected to have a role in the etiology of ADRs, thus prompting genome-wide association studies (GWAS), and candidate gene studies looking for genes associated with increased risk for ADRs.9, 10 The SLCO1B1 gene has been shown in several studies to be associated with statin ADRs, exceeding even the stringent P-value thresholds of GWAS.11 Candidate genes have been selected on the basis of their hypothesized role in the etiology of statin-induced ADRs, such as genes coding for proteins involved in pain perception,12 vascular homeostasis,13 statin transport into hepatocytes,14 and drug metabolism,15 among others. However, while neuromuscular problems are among the most common ADRs, genes coding for proteins expressed in the neuromuscular junction (NMJ) are absent in candidate gene studies.
The nicotinic acetylcholine receptor (nAChR) is a transmembrane glycoprotein highly expressed in skeletal muscle NMJs that transduces the chemical signal of acetylcholine released by nerve endings into an electrical signal and subsequent muscle contraction.16, 17, 18 Owing to its fundamental role in the transmission of nerve impulses across the NMJ, mutation-induced structural changes in nAChRs can substantially alter its function and affect nerve transmission across the synapse, resulting in muscle weakness and pain. For instance, slow-channel congenital myasthenic syndromes (SCS), characterized by generalized muscle weakness and fatigability, result from point mutations in nAChRs that extend channel open time.19, 20 Previous experiments showed that nAChR mutations can also dramatically modify cholesterol-dependent regulation of receptor function.21, 22, 23 Therefore, it is hypothesized that statin sensitivity is merely a manifestation of the cholesterol sensitivity of nAChR genetic variants.
Using transgenic mouse models expressing different SCS mutations, we demonstrated that the αC418W mutation produced a myopathy-like picture upon statin treatment resembling statin-induced ADRs. The nonsynonymous single-nucleotide polymorphism (SNP) rs137852808 (αC418W), responsible for a mild SCS, was found to be cholesterol-sensitive, as its macroscopic response to agonist stimulus increased significantly upon cholesterol depletion.24 The results presented herein demonstrate that genetic variants of genes coding for nAChR subunits could be related to an increased risk for statin-induced ADRs, and suggest that detailed, statistically powered candidate gene studies, including the nAChR genes and perhaps other genes coding for proteins expressed in the NMJ, are likely to result in the identification of variants related to statin-induced ADRs and are therefore warranted.
Materials and Methods
See Supplementary Information for details. All fluorescence microscopy experiments were carried out as described in the Supplementary Information Materials and Methods; NMJs were stained using α-BuTx Alexa Fluor 488 conjugate and size measurements done on z-stacks collected with a confocal microscope using a × 40 objective. The Cav-1 immunohistochemistry was also imaged using a confocal microscope with a × 63 oil immersion objective.
Results
αC418W neuromuscular transmission is significantly impaired after 3 days of statin treatment
To screen for statin ADRs, we studied three different transgenic lines expressing SCS mutations (αC418W, δS262T, and αV249F) for impairment in neuromuscular transmission using repetitive stimulation electromyograms. This technique compares the amplitude of a series of compound muscle action potentials (CMAP) repetitively evoked by sciatic nerve stimulation for evidence of decrement in the CMAP, given that the decremental response is a typical pattern of myasthenic disorders.25 After 3 days of statin treatment, which significantly lowered total cholesterol levels (see Supplementary Figure S1), the αC418W compound muscle action potential amplitude decrement increased significantly (1.497±0.136 fold, P<0.01, n=4). In contrast, the statin treatment had no significant effect on the wild type (WT), δS262T, and αV249F mice (Figure 1a).
Figure 1.
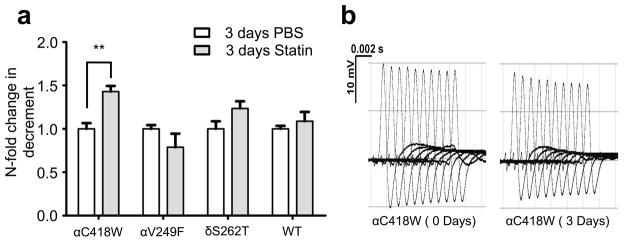
Statin treatment impairs neuromuscular transmission in αC418W mice. (a) The decrement in CMAP in αC418W mice, as a response to train like stimuli, increased upon 3 days of statin treatment (**P<0.01), whereas αV249F, δS262T, and WT had no response to the treatment (n=5, 3, 5, 4 and 4, 3, 5, 4 for αC418W, αV249F, δS262T, and WT mice in PBS and statin treatment, respectively). (b) Representative recordings of αC418W CMAP recorded over the flexor digitorum brevis and evoked by repetitive stimulation of the sciatic nerve showed the decrement after 3 days of statin treatment.
Statin treatment induces decreased locomotor activity in αC418W mice after 18 days of atorvastatin treatment
In order to determine whether the impairment in neuromuscular transmission in αC418W mice is associated with a change in physical activity levels, we measured voluntary locomotor activity during a 24-h period in αC418W and WT mice. We assessed the ability of mice to run for prolonged periods of time in a cage containing an activity wheel that recorded the instantaneous velocity at a frequency of 1Hz over a 24-h period (see Supplementary Figure S3). To understand the progression of statin-induced ADRs as a function of time, the activity measurements were obtained over a 36-day period at 3, 7, 18 and 36 days and the results plotted in heat maps (Figure 2). The most frequent activity is seen at low values of both velocity (X-axis) and duration (Y-axis), because this represents the periods when the mice ran for short intervals of time at low velocities. A second peak of activity occurs when the values of both velocity and duration are high. Because the high velocity- or long-duration peak represents periods when the mice are engaged in considerable physical activity, it is of particular interest. The initial differences between the WT and αC418W strains are negligible, but over the course of the statin treatment, the high velocity or long duration peak in αC418W is observed to progressively decrease in frequency, particularly at 18 and 36 days, in αC418W mice. These results suggest that αC418W mice are engaging in some physical activity, but it is being abandoned after short periods of time, causing a decrease in the frequency of higher-activity periods. WT animals treated over the same period with atorvastatin (Figure 2) as well as placebo-treated αC418W and WT mice remained unchanged (see Supplementary Figure S2). This is consistent with a higher predisposition to suffer from statin-induced ADRs.
Figure 2.

Statin treatment reduces the capacity of αC418W transgenic mice to run at high velocities for long periods of time. The locomotor activity was assessed by means of detailed measurement of time spent running (non-stop) in the activity wheel, and the corresponding velocity in a 24-h period. WT and αC418W mice were treated with statin daily, and on days 0, 3, 7, 18, and 36, mice were placed in cages with activity wheels (n=5, 5, 5, 4, 5 for WT and n=4, 4, 3, 3, 4 for αC418W in 0, 3, 7, 18, and 36 days of treatment, respectively). Time running and corresponding velocities were recorded and plotted in heat maps. The Y-axis corresponds to log10 time spent running (non-stop) and the X-axis is the corresponding velocity. Color corresponds to number of occasions the mice ran for a particular period of time and velocity. The heat maps reveal that after 18 days of statin treatment, the αC418W transgenic mice ran less frequently at high velocities for long periods of time when compared with WT mice.
Statin-induced NMJ calcium overload and caspase activation
To further explore the effects of cholesterol sensitivity associated with the rs137852808 SNP, we performed histochemical studies to look for an effect on calcium overload of end-plates. Unlike other SCS mice and SCS patients, motor end-plates in αC418W mice had a low level of calcium overload. Serial cryosections of tibialis anterior muscle from statin-treated αC418W and WT mice were stained for calcium deposits (via GBHA) and also for cholinesterase to localize end-plates. Looking at these in adjacent sections, we were able to measure the percent of calcium-positive NMJs as previously described.26 In statin-treated αC418W mice, this calcium overload was observed to increase as a function of time, reaching nearly a 3-fold increase over control levels at 36 days (Figure 3b). The statin treatment did not cause NMJ calcium overload in WT mice (Figure 3a). We previously showed that increased end-plate calcium in the muscle of SCS patients and mice is associated with increased levels of activated caspases, proteases known to mediate apoptosis.27, 28 To test for similar increases resulting from statin-induced end-plate calcium overload, we analyzed muscle homogenates using a luminometric assay for caspase-3, the downstream mediator of apoptosis, after 3, 7, 18, and 36 days of statin treatment. In αC418W mice, after 18 and 36 days of treatment, caspase-3 activity was increased 1.475- and 1.986-fold, respectively, as compared to controls (Figure 3c) (P<0.001, n=5). As expected, in statin-treated WT mice, caspase-3 activity remained unchanged when compared with the placebo-treated control group. These findings of increased caspase-3 activity in statin-treated αC418W mice are consistent with the previous electrophysiological, behavioral, and histological findings.
Figure 3.

Statin treatment results in NMJ Ca2+ overload and caspase-3 activation in NMJ transgenic but not WT mice. (a) The tibialis anterior muscles of WT and αC418W mice were serially sectioned (10μm thick), so that end-plates could be visualized in at least two consecutive slides. Consecutive sections of WT mice muscle display acetylcholinesterase staining (counterstained with eosin), yet no GBHA staining (counterstained with methylene blue) is seen. Conversely, consecutive sections of αC418W muscle reveal GBHA staining on end-plates, indicating the presence of Ca2+ overloading in the transgenic end-plate upon statin treatment. (b) GBHA staining in the transgenic end-plate is most frequent upon 36 days of statin treatment (***P<0.001) (n=5 for all αC418W mice treated with statins, n=5, 5, 5, 3 for αC418W mice treated with PBS in 0, 3, 7, 18, and 36 days of treatment, respectively). End-plates in WT mice showed no GBHA labeling regardless of the treatment type. Using an enzyme-based activity kit in which a proluminescent substrate for caspase-3 is cleaved to produce aminoluciferin, a substrate of luciferase, the activity of caspase-3 was assayed in αC418W and WT mice as a function of statin treatment. (c and d) Caspase-3 activity significantly increased with the duration of statin treatment in αC418W mice after 18 days of treatment, whereas not in the WT mice (***P<0.001, n=5 in WT and αC418W mice in 0, 3, 7, 18, and 36 days of PBS and statin treatment, respectively).
The distribution of end-plate size is altered in transgenic mice after 36 days of statin treatment
SCS is associated with distinct changes in end-plate morphology including simplification and shrinkage of end-plates, a finding that has proven to be reproducible in SCS mice.29 To investigate the effect of statin treatment on NMJ structure in the αC418W mice, we measured NMJ size using confocal fluorescence microscopy after labeling nAChRs with Alexa Fluor 488-conjugated α-bungarotoxin. This was done at day 36 of treatment, the time at which a maximum effect on locomotor-activity loss, calcium overload, and increased caspase activity was seen. After 36 days of statin treatment, WT NMJ size distribution remained unchanged (Figure 4a). PBS treatment had no effect on either WT or αC418W end-plates. However, statin treatment caused an appreciable change in the size distribution of αC418W NMJs (Figure 4b); it should be noted that the resulting distribution of statin-treated αC418W NMJs was not dissimilar from that seen in PBS-treated WT mice (see Supplementary Figure S4).
Figure 4.
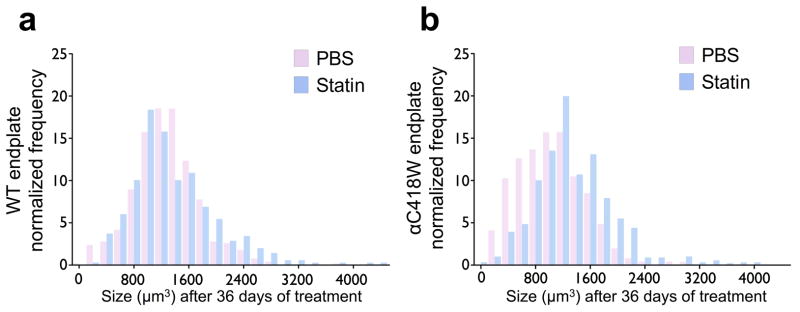
The distribution of end-plate size is altered in transgenic mice after 36 days of statin treatment. (a) The end-plate size distribution of statin-treated WT mice remained similar to the PBS-treated group (n=3 for PBS and statin treatment). (b) Conversely, the αC418W mice NMJ size distribution is altered upon statin treatment (n=3 for PBS and statin treatment). 345, 418, 348, and 502 αC418W and WT end-plates in statin- and PBS-treated mice were analyzed, respectively.
Cav-1-positive NMJs are more numerous and sensitive to statins in αC418W mice
As previously reported, the αC418W mutation creates a caveolin-binding motif in nAChRs expressed in vitro.30 Immunohistochemistry was used to test the effect of the αC418W caveolin-binding motif on the distribution of Cav-1 by comparing the percentage of Cav-1-positive end-plates along the tibialis anterior muscle sections of αC418W and WT mice. After measuring the percentage of Cav-1-positive NMJs, we found increased immunolabeling of NMJs with anti-Cav-1 antibody in αC418W mice relative to WT mice (16.14±1.41% versus 2.90±0.83%, P<0.01, n=3) (Figure 5a), suggesting that this mutation increases the presence of Cav-1 within the αC418W NMJ. Finally, we explored the effect of 36 days of statin treatment on Cav-1 localization in αC418W mice. We found that the percentage of Cav-1 positive NMJs in αC418W mice was significantly reduced in statin-treated αC418W mice (from 16.14±1.41% to 10.84±1.21%, P<0.05, n=3), whereas the WT mice remained unchanged (from 2.90±0.83% to 2.69±0.66%) (Figure 5b), suggesting that retention of Cav-1 in αC418W end-plates is sensitive to cholesterol concentration.
Figure 5.
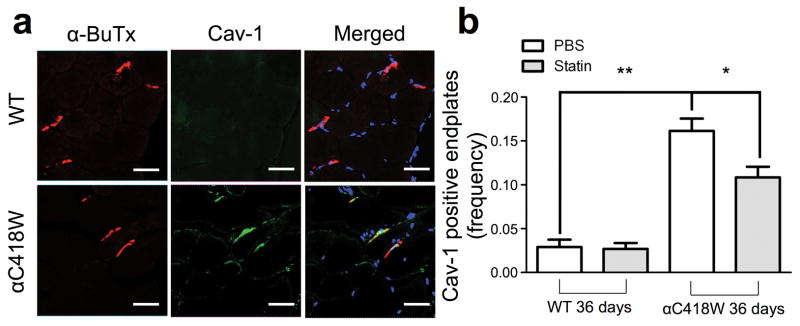
αC418W mice display a higher percentage of Cav-1 positive end-plates, which is in turn reduced by statin treatment. Tibialis anterior muscle cryo-sections were obtained from αC418W and WT mice. These were stained for nAChRs, Cav-1 and nuclei with α-bungarotoxin, immunohistochemistry using anti-Cav-1 antibodies and DAPI, respectively. (a) αC418W mice showed a significant amount of Cav-1 colocalizing with end-plates, as identified by nAChR staining, whereas WT mice displayed a significant lower amount (**P<0.01, n=3 for αC418W and WT mice on PBS treatment) (scale bars represent 36μm). (b) After 36 days of treatment, αC418W mice showed a significant reduction in the percentage of Cav-1-positive end-plates, whereas the WT percentages remained unchanged (*P<0.05, n=3 for αC418W and WT mice on PBS and statin treatment).
Disscussion
CVD is the leading cause of death and a major cause of disability. High cholesterol levels are a major risk factor in the development of CVD.1, 2, 3 Medications that reduce cholesterol levels, such as statins, have been shown to decrease the incidence of cardiovascular events by 20–30% per mmol l−1 reduction in low-density lipoprotein.4 However, although proper use of statins may substantially decrease the likelihood of suffering from CVD, many at risk choose not to continue treatment. Indeed, nonadherence can be as high as 75% after 5 years of treatment.31 A significant reason for nonadherence to statins is the fear of suffering ADRs.31, 32 Despite great efforts, the causes of statin-induced ADRs have remained elusive and unpredictable, causing excessive concerns among patients and leading to nonadherence. Unfortunately, nonadherence may be more dangerous than ADRs. For instance, nonadherence is associated with an 85% increase in mortality.31 In this study, we provide evidence that some ADRs may be related to genetic variants of membrane proteins, such as the nAChR, that result in an abnormal level of cholesterol sensitivity that affects the NMJ.
Genetic factors have been suspected to have a role in the etiology of ADRs, prompting GWAS and candidate gene studies to look for genes associated with increased risk for ADRs.9, 10 GWAS, which seek to find common genetic variants statistically more prevalent in patients affected by disease or ADRs, have been demonstrated to work.33 However, because GWAS examine the whole genome, true signals can be overshadowed with statistical noise from variants not associated with ADRs. The P-value threshold to reach ‘genome-wide significance’ is therefore very low (5 × 10−8) to avoid false positives, and sample sizes are often in the thousands in order to have adequate power to detect associations.34 GWAS necessary stringent P-values also mean that potentially important genetic variants may not reach genome-wide significance if the sample size is not large enough.33 By focusing on a small number of genes rather than examining the whole genome, the candidate gene approach may have a higher statistical power, identifying genes as associated with risk for ADRs in studies with smaller sample sizes.35, 36 In addition, rare variants of individually large effect can be identified in candidate gene-resequencing studies.37 However, the candidate gene approach is hypothesis-driven, and thus limited by how much is known about the underlying biology of the disease mechanism.38 The underlying mechanism for statin ADRs is not completely understood, thus the selection of candidate genes can be challenging. Genes selected as candidates on the basis of their hypothesized role in the etiology of statin-induced ADRs include genes encoding the organic anion-transporter polypeptide member 1B1, which is expressed in the hepatocyte basolateral membrane and is responsible for the hepatocellular uptake of endogenous and foreign substances, including statins;14, 39, 40 serotonin receptors and transporters involved in pain perception;12 angiotensin II Type 1 receptors and nitric oxide synthase 3, which are involved in vascular homeostasis;13 and cytochrome P450 drug metabolizing enzymes,40 among other proteins. Nevertheless, although neuromuscular problems are among the most common ADRs, candidate gene studies focusing on genes coding for proteins expressed in the NMJ are lacking.
The nAChR has a pivotal role in neuromuscular transmission.16, 17, 18 Indeed, point mutations in the four subunits making up the nAChR are responsible for SCS, which are disorders of neuromuscular transmission characterized by muscle weakness and fatigability.19, 20 To study the potential role of the nAChR in the etiology of statin-induced ADRs, we screened three different transgenic animals expressing SCS-causing mutations—including the cholesterol-sensitive αC418W mouse—for impaired neuromuscular transmission upon atorvastatin treatment by means of EMG experiments. We hypothesized that, among the SCS transgenic mice studied, only the αC418W mouse would display impaired neuromuscular transmission following a drop in membrane cholesterol concentration achieved through statin treatment, consistent with previous studies that established the cholesterol-sensitive nature of the αC418W nAChR.24, 30 As hypothesized, the transgenic mouse model expressing the cholesterol-sensitive allele of the nonsynonymous SNP rs137852808 (αC418W) was the only strain that developed impaired neuromuscular transmission upon atorvastatin treatment (Figure 1a).
To further characterize the atorvastatin sensitivity displayed by this nAChR genetic variant, we devised a novel technique to study the voluntary locomotor activity of these mice using a running wheel. This technique permitted us to measure the frequency at which the mice ran for a specific period of time and at a specific velocity. Using this method, we demonstrated that atorvastatin treatment transforms otherwise seemingly normal mice into weakened mice, as determined by their unwillingness or inability to run at relatively high velocities for long periods of time (Figure 2).
Previous studies have shown that SCS mutant mice, including αC418W mice, develop calcium overload of NMJs.26 Here we found that upon prolonged atorvastatin treatment, the proportion of NMJs overloaded with calcium in αC418W transgenic mice increased when compared with the placebo-treated αC418W mice (Figure 3b). In contrast, statin treatment did not cause calcium overload at any NMJ in WT mice (Figure 3a). The activity of mutant nAChRs and local disturbance of the ionic milieu, such as calcium overload, have been presumed to result in the eventual activation of caspases.28 As expected, examination of caspase-3 activity showed that αC418W mice had increased levels at 18 and 36 days of statin treatment, whereas WT mice showed no response. This phenomenon was exclusive to αC418W mice, as δS262T and αV249F mice did not show an increase in caspase-3 activity (see Supplementary Figure S5).
We used confocal microscopy to examine transgenic mice end-plates, fluorescently labeled with Alexa Fluor 488-conjugated α-bungarotoxin, to gain insight into the effects of atorvastatin treatment on end-plate integrity. Our results demonstrate that the distribution of end-plate size is altered in the αC418W mice upon statin treatment but not in WT mice. In addition, careful examination revealed that upon 36 days of atorvastatin treatment, the size distribution of transgenic mice end-plates overlapped the distribution of WT mice end-plates (see Supplementary Figure S4). Our transgenic mice were created by microinjection of single-cell mouse embryos and the expression of the transgene was highly variable among fibers.41 Such variations may affect the proportion of mutant nAChRs in a given end-plate.26 In light of this, our results suggest that the proportion of end-plates with a higher expression of the nAChR transgene is reduced upon statin treatment. These end-plates may be more susceptible to the concomitant cholesterol depletion of statin treatment, which is consistent with the previously reported cholesterol sensitivity of the αC418W nAChR.24
We found that there was a significantly higher proportion of Cav-1-positive end-plates in αC418W mice compared with WT mice. Upon 36 days of statin treatment, the proportion of Cav-1 in αC418W mice end-plates was significantly reduced (Figure 5b). Caveolins are structural proteins that are indispensable components of the cholesterol-rich membrane raft domains, known as caveolae, and exist in three isoforms, namely Cav-1, Cav-2 and Cav-3.42, 43 Cav-3 is generally regarded as the caveolin isoform expressed in muscle; therefore, the unexpected presence of Cav-1 in the transgenic mice end-plates may be associated with sensitivity to atorvastatin treatment. Caveolins bind cholesterol in a 1:1 ratio,44 and thus its expression in end plates could be increasing their cholesterol levels. Presumably, contamination of end-plates with Cav-1 (and Cav-1-positive membrane domains), which are very dependent on cholesterol concentration,43, 45 confers upon αC418W-expressing end-plates, a susceptibility to cholesterol reductions, a phenomenon that is not present in WT end-plates. This contamination could contribute to changes in end-plate plasticity and have a prominent role in the etiology of the concomitant statin-induced ADRs that lead to end-plate myopathy.
Our results demonstrate that genetic variants of the nAChR could be related to the onset of statin-induced ADRs. This is exemplified by the nonsynonymous SNP rs137852808 of the α subunit of the nAChR (αC418W), which showed a remarkable increase in risk for suffering an atorvastatin-induced ADR. The mechanism appears to relate to an effect of the variant rendering the NMJ sensitive to changes in cholesterol and, therefore, the action of statins. An alternative hypothesis could be that the prolonged gating kinetics of αC418W,26, 46 rather than its cholesterol-sensitive nature, renders it sensitive to blockade by atorvastatin and hence produces an increased risk for statin-induced ADRs. This possibility is unlikely based on the finding that other slow-channel transgenic mice expressing δS262T and αV249F mutations have no impairment of synaptic transmission or caspase activation after atorvastatin treatment (see Supplementary Figure S5). These experiments demonstrate that slow channels are not necessarily risk factors for statin-induced ADRs.
Clinical trials have demonstrated that statin-induced ADRs affect a relatively modest fraction of those who were prescribed the medication; however, in clinical practice the incidence of ADRs is greater than in controlled trials.6 Furthermore, the prevalence of statin-induced ADRs dramatically increases to 25% in patients who exercise and to >75% in professional athletes.47 This raises the paradoxical situation where exercise appears to be contraindicated in statin-treated patients. Nevertheless, despite the inherent dangers of statin-induced ADRs, the real danger is the discontinuation of statin treatment, as adherence may be a matter of life or death for CVD patients. In a recent survey, a 26% rate of nonadherence in patients with coronary artery disease was associated with an alarming 85% increase in overall mortality.31 Along this line, this article should not be viewed as one to foster unjustified concerns about statin treatment and the fear of statin-induced ADRs, or as providing additional reasons to disregard physicians’ advise to consider the use of statins. On the contrary, this study contributes to the wealth of knowledge regarding statin-induced ADR etiology and could lead to strategies to assess the risks of and/or alleviate common statin-induced ADRs. Because patients will be in a better position to make an informed decision of whether or not to initiate or continue statin treatment, such mechanistic studies are expected to increase medical adherence, and therefore result in better outcomes in the treatment of CVD.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) grants 2R01GM56371-12 and SNRP U54NS0430311 to J.A. Lasalde-Dominicci and NIH grant R01NS033202 to C. Gómez. We acknowledge the contribution to this study by grants ISI0 RR-13705-01 and DBI-0923132 to establish and upgrade the Confocal Microscopy Facility at the University of Puerto Rico (CIF-UPR). G. Grajales-Reyes, J.G. Grajales-Reyes, and W.F. García-Beltrán were supported by the Research Initiative for Scientific Enhancement (RISE) Program Grant R25GM61151 and the Minority Access to Research Careers (MARC) Program Grant T34GM007821. M. Delgado-Vélez was supported by the Research Initiative for Scientific Enhancement (RISE) Program Grant R25GM61151 and the PR-LSAMP Program Grant HRD0601843. We thank Andrew Loza for his excellent assistance with the MATLAB heat map analysis.
Footnotes
Supplementary Information accompanies the paper on The Pharmacogenomics Journal website
Conflict of interest
GEGR, CABP, CAL, OQ and JALD have filed a provisional patent application for materials related to this manuscript. Myo ADR Technologies, Corp. was founded by CABP, CAL, OQ and JALD to commercialize intellectual property published in this manuscript.
References
- 1.Kannel WB, Dawber TR, Kagan A, Revotskie N, Stokes J., III Factors of risk in the development of coronary heart disease—six year follow-up experience. The Framingham Study. Ann Intern Med. 1961;55:33–50. doi: 10.7326/0003-4819-55-1-33. [DOI] [PubMed] [Google Scholar]
- 2.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brautbar A, Ballantyne CM. Pharmacological strategies for lowering LDL cholesterol: statins and beyond. Nat Rev Cardiol. 2011;8:253–265. doi: 10.1038/nrcardio.2011.2. [DOI] [PubMed] [Google Scholar]
- 4.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 903056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 5.Haukka J, Niskanen L, Partonen T, Lönnqvist J, Tiihonen J. Statin usage and all-cause and disease-specific mortality in a nationwide study. Pharmacoepidemiology and Drug Safety. 2012;21:61–69. doi: 10.1002/pds.2255. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez G, Spatz ES, Jablecki C, Phillips PS. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med. 2011;78:393–403. doi: 10.3949/ccjm.78a.10073. [DOI] [PubMed] [Google Scholar]
- 7.Tomaszewski M, Stêpieñ KM, Tomaszewska J, Czuczwar SJ. Statin-induced myopathies. Pharmacol Rep. 2011;63:859–866. doi: 10.1016/s1734-1140(11)70601-6. [DOI] [PubMed] [Google Scholar]
- 8.Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med. 2002;346:539–540. doi: 10.1056/NEJM200202143460721. [DOI] [PubMed] [Google Scholar]
- 9.Vladutiu GD. Genetic predisposition to statin myopathy. Curr Opin Rheumatol. 2008;20:648–655. doi: 10.1097/BOR.0b013e328314b7b4. [DOI] [PubMed] [Google Scholar]
- 10.Mangravite LM, Thorn CF, Krauss RM. Clinical implications of pharmacogenomics of statin treatment. Pharmacogenomics J. 2006;6:360–374. doi: 10.1038/sj.tpj.6500384. [DOI] [PubMed] [Google Scholar]
- 11.Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 12.Ruaño G, Thompson PD, Windemuth A, Seip RL, Dande A, Sorokin A, et al. Physiogenomic association of statin-related myalgia to serotonin receptors. Muscle Nerve. 2007;36:329–335. doi: 10.1002/mus.20871. [DOI] [PubMed] [Google Scholar]
- 13.Ruaño G, Thompson PD, Windemuth A, Smith A, Kocherla M, Holford TR, et al. Physiogenomic analysis links serum creatine kinase activities during statin therapy to vascular smooth muscle homeostasis. Pharmacogenomics. 2005;6:865–872. doi: 10.2217/14622416.6.8.865. [DOI] [PubMed] [Google Scholar]
- 14.Brunham LR, Lansberg PJ, Zhang L, Miao F, Carter C, Hovingh GK, et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. The Pharmacogenomics J. doi: 10.1038/tpj.2010.92. advance online publication, 18 January 2011. [DOI] [PubMed] [Google Scholar]
- 15.Marciante KD, Durda JP, Heckbert SR, Lumley T, Rice K, McKnight B, et al. Cerivastatin, genetic variants, and the risk of rhabomyolysis. Pharmacogenet Genomics. 2011;21:280–288. doi: 10.1097/FPC.0b013e328343dd7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karlin A. Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci. 2002;3:102–114. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- 17.Corringer PJ, Le Novère N, Changeux JP. Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- 18.Le Novère N, Corringer P-J, Changeux J-P. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol. 2002;53:447–456. doi: 10.1002/neu.10153. [DOI] [PubMed] [Google Scholar]
- 19.Engel AG, Shen X-M, Selcen D, Sine SM. What have we learned from the congenital myasthenic syndromes. J Mol Neurosci. 2010;40:143–153. doi: 10.1007/s12031-009-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otero-Cruz JD, Báez-Pagán CA, Dorna-Pérez L, Grajales-Reyes GE, Ramírez-Ordoñez RT, Luciano CA, et al. Decoding pathogenesis of slow-channel congenital myasthenic syndromes using recombinant expression and mice models. P R Health Sci J. 2010;29:4–17. [PMC free article] [PubMed] [Google Scholar]
- 21.Burger K, Gimpl G, Fahrenholz F. Regulation of receptor function by cholesterol. Cell Mol Life Sci. 2000;57:1577–1592. doi: 10.1007/PL00000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fong TM, McNamee MG. Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry. 1986;25:830–840. doi: 10.1021/bi00352a015. [DOI] [PubMed] [Google Scholar]
- 23.Guzmán GM, Ortiz-Acevedo A, Santiago J, Rojas LV, Lasalde-Dominicci JA. Recent Res Devel Membrane Biol. Research Signpost; India: 2002. Regulation of acetylcholine receptor function by cholesterol; pp. 127–146. [Google Scholar]
- 24.Santiago J, Guzmàn GR, Rojas LV, Marti R, Asmar-Rovira GA, Santana LF, et al. Probing the effects of membrane cholesterol in the Torpedo californica acetylcholine receptor and the novel lipid-exposed mutation alpha C418W in Xenopus oocytes. J Biol Chem. 2001;276:46523–46532. doi: 10.1074/jbc.M104563200. [DOI] [PubMed] [Google Scholar]
- 25.Gomez CM, Maselli R, Gundeck JE, Chao M, Day JW, Tamamizu S, et al. Slow-channel transgenic mice: a model of postsynaptic organellar degeneration at the neuromuscular junction. J Neurosci. 1997;17:4170–4179. doi: 10.1523/JNEUROSCI.17-11-04170.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomez CM, Maselli RA, Groshong J, Zayas R, Wollmann RL, Cens T, et al. Active calcium accumulation underlies severe weakness in a panel of mice with slow-channel syndrome. J Neurosci. 2002;22:6447–6457. doi: 10.1523/JNEUROSCI.22-15-06447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vohra BPS, Groshong JS, Maselli RA, Verity MA, Wollmann RL, Gomez CM. Focal caspase activation underlies the endplate myopathy in slow-channel syndrome. Ann Neurol. 2004;55:347–352. doi: 10.1002/ana.10823. [DOI] [PubMed] [Google Scholar]
- 28.Vohra BPS, Groshong JS, Zayas R, Wollmann RL, Gomez CM. Activation of apoptotic pathways at muscle fiber synapses is circumscribed and reversible in a slow-channel syndrome model. Neurobiol Dis. 2006;23:462–470. doi: 10.1016/j.nbd.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 29.Groshong JS, Spencer MJ, Bhattacharyya BJ, Kudryashova E, Vohra BPS, Zayas R, et al. Calpain activation impairs neuromuscular transmission in a mouse model of the slow-channel myasthenic syndrome. J Clin Invest. 2007;117:2903–2912. doi: 10.1172/JCI30383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Báez-Pagán CA, Martínez-Ortiz Y, Otero-Cruz JD, Salgado-Villanueva IK, Velázquez G, Ortiz-Acevedo A, et al. Potential role of caveolin-1-positive domains in the regulation of the acetylcholine receptor’s activatable pool: implications in the pathogenesis of a novel congenital myasthenic syndrome. Channels (Austin) 2008;2:180–190. doi: 10.4161/chan.2.3.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lardizabal JA, Deedwania PC. Benefits of statin therapy and compliance in high risk cardiovascular patients. Vasc Health Risk Manag. 2010;6:843–853. doi: 10.2147/VHRM.S9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobson TA. Toward ‘pain-free’ statin prescribing: clinical algorithm for diagnosis and management of myalgia. Mayo Clin Proc. 2008;83:687–700. doi: 10.4065/83.6.687. [DOI] [PubMed] [Google Scholar]
- 33.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirschhorn JN, Gajdos ZKZ. Genome-wide association studies: results from the first few years and potential implications for clinical medicine. Annu Rev Med. 2011;62:11–24. doi: 10.1146/annurev.med.091708.162036. [DOI] [PubMed] [Google Scholar]
- 35.Cannon TD. Candidate gene studies in the GWAS era: the MET proto-oncogene, neurocognition, and schizophrenia. Am J Psychiatry. 2010;167:369–372. doi: 10.1176/appi.ajp.2010.10010082. [DOI] [PubMed] [Google Scholar]
- 36.Amos W, Driscoll E, Hoffman JI. Candidate genes versus genome-wide associations: which are better for detecting genetic susceptibility to infectious disease? Proc Biol Sci. 2011;278:1183–1188. doi: 10.1098/rspb.2010.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johansen CT, Wang J, Lanktree MB, Cao H, McIntyre AD, Ban MR, et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon JM, Goate AM. The candidate gene approach. Alcohol Res Health. 2000;24:164–168. [PMC free article] [PubMed] [Google Scholar]
- 39.Couvert P, Chapman MJ, Carrié A. Impact of genetic variation in the SLCO1B1 gene on statin efficacy in low-density lipoprotein cholesterol-lowering therapy. Pharmacogenomics. 2011;12:137–139. doi: 10.2217/pgs.10.214. [DOI] [PubMed] [Google Scholar]
- 40.Voora D, Shah SH, Spasojevic I, Ali S, Reed CR, Salisbury BA, et al. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol. 2009;54:1609–1616. doi: 10.1016/j.jacc.2009.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomez CM, Bhattacharyya BB, Charnet P, Day JW, Labarca C, Wollmann RL, et al. A transgenic mouse model of the slow-channel syndrome. Muscle Nerve. 1996;19:79–87. doi: 10.1002/(SICI)1097-4598(199601)19:1<79::AID-MUS11>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 42.Williams TM, Lisanti MP. The caveolin proteins. Genome Biol. 2004;5:214. doi: 10.1186/gb-2004-5-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lajoie P, Nabi IR. Lipid rafts, caveolae, and their endocytosis. Int Rev Cell Mol Biol. 2010;282:135–163. doi: 10.1016/S1937-6448(10)82003-9. [DOI] [PubMed] [Google Scholar]
- 44.Murata M, Peränen J, Schreiner R, Wieland F, Kurzchalia TV, Simons K. VIP21/caveolin is a cholesterol-binding protein. Proc Natl Acad Sci USA. 1995;92:10339–10343. doi: 10.1073/pnas.92.22.10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang WJ, Rothberg KG, Kamen BA, Anderson RG. Lowering the cholesterol content of MA104 cells inhibits receptor-mediated transport of folate. J Cell Biol. 1992;118:63–69. doi: 10.1083/jcb.118.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen X-M, Deymeer F, Sine SM, Engel AG. Slow-channel mutation in acetylcholine receptor alphaM4 domain and its efficient knockdown. Ann Neurol. 2006;60:128–136. doi: 10.1002/ana.20861. [DOI] [PubMed] [Google Scholar]
- 47.Meador BM, Huey KA. Statin-associated myopathy and its exacerbation with exercise. Muscle Nerve. 2010;42:469–479. doi: 10.1002/mus.21817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.