

Abstract
The cytokine thrombopoietin (Tpo) plays a critical role in hematopoiesis by binding to the extracellular domain and inducing homodimerization of the intracellular signaling domain of its receptor, c-Mpl. Mpl homodimerization can also be accomplished by binding of a synthetic ligand to a constitutively expressed fusion protein F36VMpl consisting of a ligand binding domain (F36V) and the intracellular signaling domain of Mpl. Unexpectedly, in contrast to Tpo stimulation, robust erythropoiesis is induced after dimerization of F36VMpl in human CD34+ progenitor cells. The goal of this study was to define the hematopoietic progenitor stages at which dimerization of intracellular Mpl induces erythropoiesis and the downstream molecular events that mediate this unanticipated effect. Dimerization (in the absence of erythropoietin and other cytokines) in human common myeloid progenitors and megakaryocytic erythroid progenitors caused a significant increase in CD34+ cells (p < .01) and induced all stages of erythropoiesis including production of enucleated red blood cells. In contrast, erythropoiesis was not seen with Tpo stimulation. CD34+ cell expansion was the result of increased cell cycling and survival (p < .05). Microarray profiling of CD34+ cells demonstrated that a unique transcriptional pattern is activated in progenitors by F36VMpl dimerization. Ligand-inducible dimerization of intracellular Mpl in human myeloerythroid progenitors induces progenitor expansion and erythropoiesis through molecular mechanisms that are not shared by Tpo stimulation of endogenous Mpl.
Keywords: humans, myeloid progenitor cells, erythropoiesis, c-Mpl
INTRODUCTION
The cytokine thrombopoietin (Tpo) is a megakaryocytic development factor that also plays a critical role in hematopoietic stem cell (HSC) self-renewal and expansion [1]. The actions of Tpo are mediated through its cell surface receptor c-Mpl, which is expressed by HSC, myeloerythroid progenitors, megakaryocytes, and platelets [2-4]. The binding of Tpo to the extracellular domain of c-Mpl results in homodimerization of its intracellular domain leading to activation of proliferation, survival and differentiation pathways [4, 5].
Although Tpo produces little expansion of progenitor cells when used alone, it is commonly used in combination with other cytokines for ex vivo maintenance and expansion of progenitor cells [1]. However, c-Mpl is internalized after binding to Tpo and its expression is downregulated during differentiation, resulting in a limited window of development during which Tpo can act. To directly regulate Mpl signaling, an alternative approach has been used, involving the constitutive intracellular expression of a fusion protein (referred to as F36VMpl) that consists of a ligand-binding domain (F36V) fused to the intracellular signaling domain of c-Mpl. The addition of a synthetic diffusible ligand (chemical inducer of dimerization; CID) to cells results in homodimerization of F36VMpl and signaling is induced.
Our laboratory has previously used a lentiviral vector to express F36VMpl in cord blood (CB) HSC. The addition of CID to transduced HSC resulted in significant expansion of CD34+ cells with multilineage potential, making this a potential gene therapy approach for primitive hematopoietic progenitor expansion [2]. The progenitor expansion seen after F36VMpl dimerization of HSC was not seen in a previous study, which targeted a more heterogeneous and differentiated progenitor population [6]. However, both aforementioned studies showed significant erythroid differentiation with F36VMpl dimerization. The unexpected finding of erythroid differentiation with F36VMpl dimerization formed the basis of the studies described here, the goal of which was to define the mechanisms by which F36VMpl dimerization induces erythropoiesis and progenitor expansion. Our approach was to identify the hematopoietic myeloerythroid progenitor cell stages at which F36VMpl dimerization acts and to examine the downstream molecular targets of dimerization of intracellular Mpl to understand more clearly how these compared with Tpo stimulation. Defining these mechanisms is critical for understanding how F36VMpl dimerization could be applied for clinical translational uses.
During hematopoiesis, primitive lymphomyeloid progenitor cells give rise to the more lineage-restricted common myeloid [3] and common lymphoid progenitors [7-9]. The myeloerythroid lineages arise from a common myeloid progenitor (CMP), which gives rise to the more lineage restricted megakaryocytic erythroid progenitors (MEPs) and granulocytic monocytic progenitors (GMPs). MEPs give rise to megakaryocytes and erythrocytes, whereas GMPs give rise to granulocytes and monocytes [3]. As both CMP and MEP normally express c-Mpl [3], we focused on these specific progenitor stages to directly compare the functional and molecular effects of F36VMpl dimerization with those caused by binding of the natural ligand Tpo to its endogenous receptor. Our data show that ligand inducible dimerization of intracellular Mpl activates functional and molecular programs in CMP and MEP that are markedly different from those activated by Tpo.
MATERIALS AND METHODS
Isolation of Human Progenitor Populations
Umbilical CB was collected from normal deliveries, according to guidelines approved by the University of California Los Angeles Investigational Review Board. Enrichment of CD34+ cells was performed using the magnetic-activated cell sorting system (Miltenyi Biotec, Auburn, CA, http://www.miltenyibiotec.com/en/default.aspx) or RoboSep human CD34 selection system (StemCell Technologies, Vancouver, BC, Canada, http://www.stemcell.com/). CD34+ enriched cells were incubated with the following anti-human–specific monoclonal antibodies: CD34 PerCP-Cy5.5, CD38 PE-Cy7, CD123 (interleukin-3 receptor alpha) PE, CD 45RA PE-Cy5, FITC-labeled lineage-specific antibodies: CD2, CD7, CD10, and CD20 and the lineage allophycocyanin (APC)-labeled antibodies: CD3, CD4, CD8, CD11b, CD14, CD19, CD56, and glycophorin A (Gly A; all from Becton Dickinson, San Jose, CA, http://www.bdbiosciences.com/). An unstained (no antibody) control was used to define negative gates. The following published immunophenotypic definitions were used to isolate myeloid progenitors from thawed CB CD34+ enriched cells by fluorescence-activated cell-sorting (FACS): CD34+CD38+lin-CD45RA-CD123lo (CMP), CD34+CD38+lin-CD45RA-CD123- (MEP), CD34+CD38+lin-CD45RA+CD123lo for granulocyte monocyte progenitors (GMP) [3], and CD34+ 38-lin- primitive lymphomyeloid progenitors (HSC) [10]. Sorting was performed on a FACSAria (Becton Dickinson) equipped with five lasers (355, 405, 488, 561, and 633 nm). Isolated populations were analyzed by FACS to assess post sort purity.
Lentiviral Vector Expression of F36VMpl
A F36VMpl plasmid (a generous gift of Dr C.A.Blau, University of Washington, Seattle, WA) was used to produce lentiviral (HIV-1) vectors that express the fusion protein F36VMpl under control of the murine stem cell leukemia virus promoter and a marker gene (green fluorescent protein [GFP] for in vitro and luciferase [LUC] for in vivo studies) expressed under control of the hPGK (human phosphoglycerate kinase) promoter (Fig. 1D). Control vectors coexpressed F36V (no c-MPL) and GFP. Vectors were pseudotyped using vesicular stomatitis virus (VSV) envelope or Gibbon Ape leukemia virus (GALV) envelope by transient transfection into 293T cells and concentrated by ultrafiltration and ultracentrifugation [11, 12]. CB progenitors were incubated in 100 μl per well of serum free transduction medium on recombinant fibronectin fragment CH-296 (50 μg/ml; Takara Bio Inc, Otsu, Japan, http://www.takara-bio.com/) coated non-tissue culture 48-well plates (100,000 cells per well) for 16 hours. The transduction medium consisted of X-vivo15 (BioWhittaker, Walkersville, MD, http://www.lonzabio.com/resources/product-instructions/biowhittaker-cell-culture/); stem cell factor (50 ng/ml); Flt 3 ligand (50 ng/ml), and Tpo (50 ng/ml), (all cytokines from R&D Systems, Minneapolis, MN, http://www.rndsystems.com/). Viral supernantant (2 × 107 units/ml) was then added along with an additional 100 μl of transduction medium per well to make the total volume 200 μl/well. A second dose of viral supernatant was added 4 hours later. After 30 hours of total exposure to vector, cells were washed with 10 times the volume of Dulbecco phosphate-buffered saline (DPBS; Mediatech, Herndon, VA, http://www.cellgro.com/). Cells were transduced in a common pool and then divided for culture or for transplantation with or without CID.
Figure 1.

Isolation and transduction of progenitors. (A): CD34+lin– cells were sorted to obtain common myeloid progenitor (CMP), megakaryocytic–erythroid progenitor (MEP), and granulocytic monocytic progenitor (GMP). (B): Relative colony-forming unit output from CMP, MEP, GMP, and total CD34+ cells (500 cells per population plated). (C): Relative B-lymphoid output in lymphoid cultures initiated with populations shown. (D): Lentiviral F36VMpl vector. Abbreviations: CFU, colony-forming unit; CMP, common myeloid progenitor; F36VMpl, fusion protein; GFP, green fluorescent protein; GMP, granulocytic monocytic progenitor; HSC: hematopoietic stem cell; MEP, megakaryocytic erythroid progenitor; MSCV LTR, murine stem cell leukemia virus long-term repeat promoter; PGK, human phosphoglycerate kinase promoter.
Dimerization
Lyophylized CID (AP20187), (ARIAD Pharmaceuticals, Cambridge, MA, http://www.ariad.com/; regulation kit) was solubilized in 100% ethanol to produce 1 mM stock solution (for in vitro use) or 62.5 mg/ml (for in vivo use) and stored at −20°C. For in vitro experiments, CID was diluted fresh in the culture medium to a final concentration of 100 nM and added to cultures every 48 hours. For in vivo administration, stock solution was diluted fresh on the day of injection. The final solution for injection contained 10% polyethelene glycol 400, 2% Tween 80, and 2.5 mg/ml of CID.
CMP, MEP, GMP, and Serum Free Cultures
CMP, MEP, and GMP were transduced with either F36V-Mpl or F36V control vectors and cultured in the presence or absence of CID (100 nM) and in the absence of cytokines. Cells were plated in bulk in 96-well tissue-culture plates onto established MS5 (murine marrow stromal) cell line in lymphoid medium (RPMI 1,640 [Irvine Scientific, Santa Ana, CA, http://www.irvinesci.com/], 5% fetal calf serum (FCS) [screened for B-cell cultures], 50 mM 2-ME, 50 U/ml penicillin/50 μg/ml streptomycin, and 200 mM L-glutamine). Cells were recovered for analysis at different time points by trypsinizing wells. Viable cells were counted using trypan blue. Fold increase in the total number of cells compared with the original number of cells plated was calculated. Myeloid lineage potential of progenitors was assessed using a colony-forming unit cell (CFU-C) assay by plating cells in methylcellulose using METHOCULT GF_H4435 (StemCell Technologies) and counting colonies after 14 days. Lymphoid potential of progenitors was tested by culturing them (cultures initiated with 1,000 cells per progenitor) on MS-5 stroma in lymphoid medium with FLT-3 (5 ng/ml), Tpo (5 ng/ml), and IL-7 (5 ng/ml) for 4 weeks; and using FACS to detect CD19+ B lymphocytes. Erythroid differentiation in serum free conditions was tested by transducing CB CD34+ cells with F36VMpl and culturing the transduced cells in X-vivo 15 medium on recombinant fibronectin fragment CH-296 coated non-tissue culture 48-well plates in the presence of CID, Tpo (50 ng/ml), Epo (5 units/ml), Epo+CID, or no growth factors.
Immunophenotypic Analysis of Cultured and Transplanted Cells
FACS analysis of cultured cells, as well as bone marrow and spleen cells harvested from mice, was performed on LSR II (Becton Dickinson) by direct immunofluorescence staining with human specific monoclonal antibodies and detection of GFP expression after incubation in 1.2% human intravenous immunoglobulin (IVIG; Cutter, Berkley, CA, http://www.bayer.com/). Analysis used FlowJo (Tree Star, Ashland, OR, http://www.treestar.com/). Lineage-specific human antigen expression was determined using the following antibodies: CD45-APC Cy7, CD34-PE Cy7, CD41a-PE Cy5, CD66-PE, Gly A-APC or -PE, CD19-APC or Percp, CD56-PE, and CD14 FTIC (all from Becton Dickinson). The following immunophenotypes were used to identify myeloid progenitors from culture: CMP (CD34+CD38+CD123loCD45RA− or CD34+lin-CD123loCD45RA−), MEP (CD34+CD38+CD123-CD45RA− or CD34+lin-CD123-CD45RA−), and GMP (CD34+CD38+CD123loCD45RA+ or CD34+lin-CD123 loCD45RA+).
Cell Cycle and Apoptosis Analyses
Transduced cells cultured for 5–7 days with or without CID were harvested and stained with Hoechst (Becton Dickinson) for cell cycle analysis and Annexin-APC (Becton Dickinson) for apoptosis analysis. Unstained cells were used to set negative gates. In a separate experiment, transduced cells were cultured for 7 days with or without CID and then incubated with bromodeoxyuridine (BrDU) for 30 or 120 minutes before harvest and analysis. Harvested cells were fixed, permeabilized, and stained with 7-AAD and APC conjugated antibody to BrDU. Unstained cells were used to set negative gates and the BrDU positive cells were scored as cells in the S-phase of cell cycle.
Cell Morphology Analysis
F36vMpl transduced CMP were cultured for 35 days, harvested, and stained with Giemsa-Wright. The stained cells were scanned with Scanscope XT (Aperio technologies, Vista, CA, http://www.aperio.com/) to acquire digital images, which were analyzed using Imagescope software (Aperio technologies, Vista, CA).
Transplantation Assays
Nonobese diabetic/severe combined immunodeficiency interleukin-2 receptor gamma chain knock out (NSG) mice (Jackson Laboratories, Bar Harbor, ME, http://www.jax.org/) were used for in vivo experiments according to protocols approved by the Institutional Animal Care and Use Committee of University of California Los Angeles. Transduced human cells were suspended in DPBS and inoculated (30–50 microliters per animal) into the right tibia of sublethally irradiated (250 cGy 4–6 hours before transplantation) mice. Engraftment and differentiation of human cells were assessed by in vivo bioluminescence imaging and/or FACS from harvested organs after preincubation in 1.2% human IVIG (Cutter) and purified rat anti-mouse CD16/CD32 (Fc III/II receptor; Becton Dickinson) to prevent nonspecific antibody binding. The transplantation of relatively low numbers of cells (30,000–48,000 MEP cells per mouse) was designed to provide minimal baseline engraftment from unstimulated cells. CID of 10 mg/kg (or control vehicle) was administered by intraperitoneal (IP) injection daily starting 1-day before transplantation (day 1) until the time of killing.
Bioluminescence Imaging
In vivo bioluminescence imaging was performed under general anesthesia with 2% isoflurane. Fifteen minutes before imaging, each mouse was given 125 mg/kg IP luciferin (Promega, Madison, WI, http://www.promega.com/). In vivo optical imaging was performed with a prototype IVIS 3D bioluminescence/fluorescence optical imaging system (Xenogen, Alameda, CA, http://www.caliperls.com/products/preclinical-imaging/) at different time points. Regions were manually drawn around the bodies of the mice to assess signal intensity emitted. Ventral and dorsal images were acquired and total signal was calculated for each time point [13].
Quantitative RT-PCR
CMP transduced with F36VMpl-GFP or F36V-GFP were cultured on MS-5 stroma in lymphoid medium with or without CID. CD34+GFP+ cells were isolated from culture on day 7 by FACS and RNA was extracted using a Qiagen micro kit (Qiagen, Valencia, CA, http://www.qiagen.com/default.aspx). An omniscript reverse transcriptase kit was used to make cDNA, which was subjected to quantitative polymerase chain reaction (qPCR) using TaqMan probe-based gene expression analysis assays (Applied Biosystems, Carlsbad, California, http://www.appliedbiosystems.com/absite/us/en/home.html) for VEGF (Hs00900055_m1), E-cadherin (Hs01023894_m1), GATA1 (Hs01085823_m1), TAL1 (Hs01097987_m1), EPOR (Hs00959427_m1), VEGFR1 (Hs01052961_m1), and VEGFR2 (Hs00911700_m1). The qPCR was done using a 7500 Real time PCR system Applied Biosystems. To identify appropriate housekeeping genes, CB CD34+ cells were transduced with F36VMpl-GFP or F36V-GFP and cultured for 7 days on MS-5 stroma with or without CID. RNA was extracted from equal numbers of CD34+GFP+ cells isolated from culture and subjected to qPCR for 12 housekeeping genes (18SrRNA, ACTB, B2M, GAPDH, GUSB, HPRT1, PPIA, HSP90AB1, RPL13A, RPLP0, TFRC, and UBC) on a human housekeeping gene RT profiler PCR array (SA Biosciences, Frederick, MD, http://www.sabiosciences.com/). Based on the results of the housekeeping array (Supporting Information Table S1); RPL13a (Hs01926559_g1), HSP90 (Hs00607336_gH), and beta actin (Hs99999903_m1) were chosen as housekeeping genes because their expression values were similar in dimerized and control cells (Supporting Information Table S1). The data were analyzed using the comparative C (T) method [14].
Microarray Analysis
CB CD34+ cells transduced with F36VMpl-GFP were cultured in lymphoid medium on MS-5 stroma either with CID alone, with Epo (5 international units/ml) alone, or with Tpo (50 ng/ml) alone. Transduced cells cultured on MS-5 stroma in the absence of either growth factors or CID served as controls. Three biologically independent experiments were performed. CD34+GFP+ cells were isolated by FACS after 7 days of culture and subjected to RNA extraction using a Qiagen micro kit. Microarray analysis was performed using an affymetrix U133 plus 2.0 platform (Affymetrix, Santa Clara, California, http://www.affymetrix.com) and dCHIP software (Department of Biostatistics, Harvard School of Public Health, Boston, MA, http://biosun1.harvard.edu/complab/dchip/). The quantile method [15] was used to normalize gene expression values in a PM/MM model [16]. Gene expression values from control cultures served as a baseline for normalization. Genes were considered differentially expressed if they were greater than or equal to twofold upregulated or downregulated and significant at a p <0.05 on a paired t-test when compared with control cells over all three independent experiments. The Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov) was used to derive functional gene class enrichment data. Ingenuity software (Ingenuity systems, Redwood City, California, http://www.ingenuity.com/) was used to generate functional gene network data. The microarray data were deposited in NCBI’s Gene Expression Omnibus and is accessible through the following link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=tzkxvssqeseoors&acc=GSE30585.
Statistical Analysis
Analysis of variance (ANOVA) and linear regression were performed to compare the lymphoid potential of the four different cell types (CMP, MEP, GMP, and HSC). The Generalized Cochran–Mantel–Haenszel test was used to determine whether or not a difference exists in the frequency of colony type (burst forming unit erythroid (BFU-E), colony-forming unit granulocyte macrophage (CFU-GM), and colony-forming unit granulocyte macrophage (CFU-GEMM)) among the four cell types (CMP, MEP, GMP, and CD34). The analysis of the differences in cell numbers between the F36VMpl+CID, F36VMpl without CID, and F36V+CID arms was based on linear regression that adjusted for the time of cell counts. A nested ANOVA was performed to examine the association between the proportions of cycling or apoptotic cells among the three arms. QPCR data were analyzed with a two-way ANOVA model, which included a Benjamini and Hochberg adjustment to correct for the false discovery rates due to multiple comparisons. Statistical analyses were carried out using SAS and R software.
RESULTS
F36VMpl Dimerization in CMP and MEP Increases Total and CD34+ Cell Numbers and Induces Erythropoiesis
Lineage potentials of the CMP, GMP, and MEP immunophenotypes were initially confirmed through in vitro myeloerythroid and lymphoid assays of populations isolated by FACS from CB (Fig. 1A). As expected, CMP possessed both granulocyte–monocyte and erythroid clonogenic potential, as shown by robust production of BFU-E, CFU-GM, and CFU-GEMM (Fig. 1B). Consistent with more lineage-restriction, MEP produced predominantly erythroid colonies and GMP produced predominantly granulocytic-monocytic colonies (Fig. 1B, p < .05 for difference in frequency of types of colonies for the four cell types- CMP, MEP, GMP, and unfractionated CD34+ cells, n = 3 independent experiments). In keeping with commitment to the myeloid pathway, CMP, MEP, and GMP were almost devoid of lymphoid potential when compared with control CD34+CD38− progenitor cells (Fig. 1C, p < .05, n = 2 independent experiments).
The effects of F36VMpl dimerization on erythropoiesis were first investigated in MEP, which are progenitors enriched in erythroid potential. MEP cells were transduced to express F36VMpl (Fig. 1D; or control vector) and cultured in the presence or absence of CID on the murine stromal line MS5 in the absence of exogenous growth factors. CID-treated MEP that expressed F36VMpl produced significantly higher numbers of total CD45+ cells from and CD34+ progenitors than cultures from control cells (Fig. 2A, p < .01, n = 5 independent experiments). Of note, Gly A+ (erythroid) cells comprised the predominant cell type produced in dimerized cultures but were not seen in control cultures (Fig. 2A, 2B, p < .01, n = 5 independent experiments). In view of the significant progenitor expansion seen in vitro, we also explored the effects of delivering the dimerizing ligand in vivo after transplantation of MEP into NSG mice. Flow cytometry of bone marrow harvested from mice euthanized on day 14 showed the presence of GFP+ Gly A+ human cells in CID-treated mice and no human cells in control vehicle-treated mice (Supporting Information Fig. S2). In further transplantation experiments, the GFP marker was replaced in the vector with firefly LUC allowing dynamic tracking of engraftment. Bioluminescent imaging of mice that received CID in vivo from day 1 to day 14 showed higher short-term engraftment than control mice in three of four independent experiments (Figs. 2C, 2D; one representative experiment out of a total of four experiments, and Supporting Information Fig. S2).
Figure 2.

Fusion protein (F36VMpl) dimerization in megakaryocytic erythroid progenitor (MEP) expands enhanced green fluorescent protein (GFP) positive progenitors and induces erythropoiesis. Transduced MEP cultured ± chemical inducer of dimerization (CID) on stroma without growth factors (starting cell number = 3,300–8,666 cells per arm, n = 5 independent experiments, equal cell numbers were used for each arm in a given experiment) were analyzed for (A) the output of cell types shown and (B) immunophenotype (day 7). (C): Nonobese diabetic/severe combined immunodeficiency interleukin-2 receptor gamma chain knock out (NSG) mice transplanted with transduced MEP and administered CID or control vehicle were imaged (day 14). (D): Longitudinal quantification of bioluminescence. Abbreviations: Gly A, glycophorin A.  , F36VMpl+CID;
, F36VMpl+CID;  , F36VMpl, no CID;
, F36VMpl, no CID; , F36V+CID
, F36V+CID
In contrast to MEP, no significant effect on cell growth or differentiation was seen when transduced GMP were cultured in the presence of CID (data not shown). The effects of inducing Mpl dimerization in CMP, a more primitive myeloid progenitor that gives rise to MEP and GMP, were next tested. Similar to MEP, transduced CMP stimulated with CID gave rise to significantly higher numbers of total CD45+ (p < .01) and CD34+ (p < .01) cells than nondimerized cultures (Fig. 3A, n = 5 independent experiments). Gly A expressing cells were again the predominant population (Fig. 3A, 3B, p < .01, n = 5 independent experiments). Morphological analysis of cultures generated from CMP demonstrated that F36VMpl dimerization induced full erythroid differentiation from erythroid progenitors to mature enucleated red blood cells (Fig. 3D, day 35 of culture) despite the absence of Epo. Flow cytometry confirmed that dimerized CMP generated enucleated red blood cells (shown as Gly A+ cells that were Hoechst negative, Fig. 3E). In addition to erythroid cells, significantly higher numbers of megakaryocytic (CD41 bright Gly A−) cells were produced in dimerized cultures than in control cultures (p < .05 for five independent experiments for MEP and four independent experiments for CMP, Supporting Information Fig. S3A, S3B). Morphologic analysis of cultures also demonstrated generation of monocytes, megakaryocytes, and neutrophils (Supporting Information Fig. S3C).
Figure 3.

Dimerization in common myeloid progenitor (CMP) induces expansion of CD45+, CD34+, and erythroid cells and enhances cell cycling and survival. Transduced CMP cultured ± chemical inducer of dimerization without growth factors (starting cell number = 7,000–53,000 cells per arm, n = 5 independent experiments, equal cell numbers were used for each arm in a given experiment) were analyzed for (A) cell output, (B) immunophenotype (day7), as well as (C) cell cycle and apoptosis (day 7). (D): Giemsa-Wright staining ( ×10 magnification; red arrows, erythrocytes; green arrows, erythroid progenitors) and (E) flow cytometry for enucleated erythrocytes in dimerized cultures (day 35). Abbreviations: CID, chemical inducer of dimerization; GFP, green fluorescent protein; Gly A, glycophorin A.  , F36VMpl+CID;
, F36VMpl+CID;  , F36VMpl, no CID;
, F36VMpl, no CID;  , F36V+CID.
, F36V+CID.
Most of the CD34+ cells generated during Mpl dimerization expressed relatively low levels of CD34 irrespective of whether the starting population was unfractionated CB CD34+ cells (Supporting Information Fig. S4A), MEP (Fig. 2B), or CMP (Fig. 3B). The CD34 dim population contained clonogenic cells (particularly erythroid colonies; Supporting Information Fig. S4B). Immunophenotypic analyses of cultures generated from CMP showed that dimerized cultures generated all three types of progenitors (CMP, MEP, and GMP). In comparison, control cultures contained GMP and CMP but few or no MEP (Fig. 4A, 4B, one representative experiment of three). These data suggest that F36VMpl dimerization of CMP promotes the in vitro generation of MEP but not GMP.
Figure 4.

Dimerization in common myeloid progenitor (CMP) results in selective generation of vector-expressing megakaryocytic erythroid progenitor (MEP) and CMP. Transduced CMP were cultured ± chemical inducer of dimerization for 7 days (starting cell number = 9,000 cells/arm) and analyzed for (A) CMP, MEP and granulocytic monocytic progenitor immunophenotypes (gated out of CD34+ lin- cells), (B) cell counts of populations in (A), (C) green fluorescent protein expression of populations in (A). (One representative experiment of three depicted). Abbreviations: CID, chemical inducer of dimerization; CMP, common myeloid progenitor; MEP, megakaryocytic erythroid progenitor; GMP, granulocytic monocytic progenitor; GFP, green fluorescent protein.
F36VMPL dimerization generated a significantly higher proportion of GFP+ cells when compared with control cells (Figs. 2B, 3B, p < .05), indicating that cells expressing the F36VMpl fusion protein were selectively expanded without a bystander effect on nontransduced cells. The selection advantage provided by dimerization was seen specifically in the CMP and MEP (but not GMP) generated from CMP (Fig. 4C) again demonstrating that dimerization results in selective proliferation of CMP and MEP but not GMP.
F36VMpl Dimerization Enhances Cell Cycling and Survival
To further elucidate the mechanism underlying the F36VMpl dimerization induced increase in cell output from progenitors, cell cycle status and apoptosis were assayed in cultures initiated with transduced CMP and stimulated for 7 days in the presence or absence of CID. F36VMpl dimerization of CMP significantly increased the proportion of CD34+ and total cells in cell cycle by Hoechst uptake (p < .05, n = 3 independent experiments; and confirmed with BrDU uptake; Fig. 3C). In addition, dimerization produced a significantly lower proportion of apoptotic cells (Fig. 3C, p < .05, n = 3 independent experiments).
F36VMpl Dimerization Induces Erythroid Gene Expression in CD34+ Progenitors
In view of the expansion of CD34+ progenitor numbers in association with induction of erythropoiesis, we next investigated whether F36VMpl dimerization upregulates erythroid gene expression at the CD34+ progenitor stage. CD34+GFP+ cells generated from CMP were isolated on day 7 from cultures with or without CID, and subjected to quantitative PCR. Mpl dimerization significantly upregulated expression of the erythroid transcription factors SCL and GATA1 [17], E-cadherin (CDH1) (a downstream target of erythropoietin) [18], and the erythropoietin receptor (EPOR) (Fig. 5, p < .05, n = 3 independent experiments). Interestingly, dimerization also significantly upregulated expression of VEGF (p < .05, n = 3 independent experiments), an anti-apoptotic factor for hematopoietic and erythroid progenitors [19, 20].
Figure 5.

F36VMpl dimerization induces erythroid gene expression in CD34+ progenitors. Transduced common myeloid progenitor were cultured ± chemical inducer of dimerization (CID) for 7 days. CD34+ GFP+ cells were then sorted and subjected to qPCR. mRNA expression relative to the no CID arm is depicted. Beta actin, RPL13a and HSP90 were used as housekeeping genes.*p < .05 for F36VMpl + CID versus controls. Abbreviation: CID, chemical inducer of dimerization.
Erythropoiesis Is Induced by F36VMpl Dimerization but Not Tpo, and Is Independent of Epo
Tpo induces erythropoiesis in serum containing culture systems [21]. This erythroid effect of Tpo is dependent on the erythropoietin present in serum and is not seen in serum free culture conditions [21]. Since gene expression analysis demonstrated that F36VMpl dimerization results in upregulation of the erythropoietin receptor gene (Fig. 5), we explored whether the erythropoiesis induced by Mpl dimerization was secondary to an increased sensitivity of dimerized cells to erythropoietin present in bovine fetal calf serum. Dimerization in serum-free conditions in the absence of stroma still resulted in erythroid differentiation (Fig. 6), proving that the erythropoiesis induced by F36VMpl dimerization occurs in the absence of erythropoietin. The effects of F36VMpl dimerization on CD34+ progenitors were next directly compared with those of Tpo (the natural ligand of full-length c-Mpl) in serum-free, stroma-free conditions. In contrast to F36VMpl dimerization, Tpo stimulation did not induce erythropoiesis (Fig. 6, n = 2 independent experiments).
Figure 6.
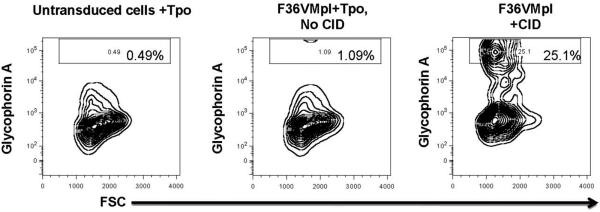
F36VMpl dimerization, and not Thrombopoietin, induces erythropoiesis in the absence of Erythropoietin. Transduced CD34+ cells were cultured in serum-free medium in the absence of erythropoietin, either with chemical inducer of dimerization or thrombopoietin (Tpo). Untransduced CD34+ cells were cultured in the presence of Tpo. Flow cytometry shows generation of erythroid cells (glycophorin A +) at day7. Abbreviations: CID, chemical inducer of dimerization; Tpo, thrombopoietin.
When F36VMpl dimerization was compared with stimulation with Epo, no significant difference was seen in the frequency or number of Gly A+ cells in the CID, Epo, and CID+Epo arms (Supporting Information Fig. S5, n = 2 independent experiments).
F36VMpl Dimerization in CD34+ Progenitors Activates Novel Gene Networks Not Seen with Tpo Stimulation
Microarray analysis was performed to explore further the downstream gene networks activated by stimulation of progenitors through F36VMpl dimerization. CD34+GFP+ cells were isolated after 7 days of culture in either CID, Epo, or Tpo. Cells grown in the absence of either CID or growth factors served as negative controls (n = 3 independent experiments). Distinct expression profiles were noted in each of the three experimental arms (Fig. 7A). Specifically, of the genes differentially expressed relative to the negative control (p < .05), 675, 349, and 455 genes were unique to CID-, Epo-, and Tpo-treated cells, respectively (Fig. 7B).
Figure 7.

F36VMpl dimerization induces a distinct gene expression profile. Transduced CD34+ cells were cultured in the presence of chemical inducer of dimerization (CID), thrombopoietin, Epo or no treatment. CD34+ GFP+ cells were isolated on day 7 for microarray. (A) Selected genes up-regulated >2-fold with CID. ( B) Numbers of genes differentially expressed in each condition compared with no treatment. (C) Network analysis of genes differentially expressed with CID. Abbreviations: CID, chemical inducer of dimerization; Epo, erythropoietin; Tpo, thrombopoietin
In keeping with the enhanced cycling status of dimerized cells seen in culture (Fig. 3C), dimerization of CD34+ progenitors induced upregulation of cyclin D1 (CCND1). Consistent with the decreased apoptosis of dimerized cells, CID-treated cells showed upregulation of survival genes (Fig. 7A). In concordance with the erythroid differentiation seen in culture, Mpl dimerization upregulated several genes associated with erythropoiesis (Fig. 7A), including spectrin genes (SPTB and SPTA1), pyruvate kinase liver and red-cell (PKLR), and hemoglobin subunits beta (HBB), delta (HBD), γ-1 (HBG1).
Both Epo and CID upregulated genes involved in erythroid development (Fig. 7A). In contrast, CID-treated cells but not Epo-treated cells, showed upregulation of platelet associated genes. Interestingly, CID but not Epo-treated cells showed upregulation of TIMP1. In addition to being a platelet associated gene, TIMP1 is an erythroid potentiating factor that acts on early erythroid progenitors and potentiates the erythropoietic effects of Epo [22-24]. Consistent with the lack of erythroid induction seen in culture, Tpo-treated CD34+ cells did not show upregulation of erythroid genes (Fig. 7A). Interestingly, genes associated with megakaryopoiesis, including TIMP1, were also not upregulated by Tpo at the CD34+ progenitor stage.
A network analysis of the differentially expressed genes in the dimerized progenitors showed enrichment for expression of genes involved in myelopoiesis, erythropoiesis, proliferation of blood cells, and red-cell apoptosis (Fig. 7C, p < .05). Network analyses of Tpo stimulated cells did not show enrichment for these networks, providing further evidence that mechanisms underlying the effects of F36VMpl dimerization are distinct from those activated by Tpo (Supporting Information Fig. S6).
DISCUSSION
We have previously shown that F36VMpl dimerization of primitive CD34+CD38- cells with lymphomyeloid potential expanded CD34+ progenitor cells and enhanced engraftment [2, 4], making it a potential strategy to improve hematopoietic reconstitution post-HSC transplantation. Of note, this study also showed erythroid differentiation with F36VMpl dimerization. Studies before our own that used the F36VMpl approach with unfractionated CD34+ cells noted erythroid differentiation with no progenitor expansion [6]. The goal of the current study was to understand at which stage of differentiation F36VMpl dimerization acts to promote progenitor expansion and the mechanisms by which dimerization induces erythropoiesis. We conclude that F36VMpl dimerization in progenitors with myeloerythroid potential (CMP and MEP) induces both CD34+ progenitor expansion and erythropoiesis. Further we show that the erythropoiesis induced by dimerization is independent of the presence of erythropoietin and reflects molecular mechanisms that are distinct from stimulation of full-length c-Mpl by its natural ligand Tpo.
F36VMpl dimerization studies by Richard et al. [6] demonstrated erythroid differentiation without progenitor expansion. Even though Epo was not added exogenously, these studies [6] were performed in the presence of serum and thus did not show that the erythroid effect of dimerization was independent of Epo. Also, molecular mechanisms induced by dimerization in progenitors were not investigated. The contrasting effects on progenitor expansion in our studies and those by Richard et al. are unlikely to be explained by the stage of differentiation at which dimerization was induced. The former study [6] used the total population of unfractionated CD34+ cells, which contains the CMP and MEP subpopulations. It is more likely that differences in the conditions in which dimerization were induced in the former study, particularly the absence of stroma, reduced progenitor survival, and promoted differentiation.
Unlike Tpo, F36VMpl dimerization does not induce detectable phosphorylation of the signaling molecules STAT5 (signal transduction and transcriptional activation five) or JAK2 (Janus Kinase two) [25]. We did not detect phosphorylation of STA5 or JAK2 in western blot assays of BaF3 (murine pro B cell leukemia cell line) cells that had been transduced with F36VMpl and cultured in the presence of CID (data not shown). In view of functional and signal transduction pathway differences between Tpo stimulation and F36VMpl dimerization, we used microarray to investigate differences in downstream gene expression between CD34+ progenitors undergoing Tpo stimulation or F36VMpl dimerization.
The current study demonstrated that F36VMpl dimerization affects multiple molecular pathways associated with proliferation, survival, and erythroid differentiation and that these pathways are activated at the CD34+ progenitor stage. Importantly, there were marked functional and molecular differences between the effects of dimerizing the constitutively expressed intracellular domain of Mpl (through F36VMpl) and exposing cells that express endogenous Mpl to the native ligand Tpo. Several studies have demonstrated that Tpo enhances erythropoiesis [26-28]. However, all such studies examined the effect of Tpo in the presence of Epo or serum, which contains bovine Epo [21]. Consistent with studies from Kobayashi et al. [26], and Liu et al. [21], our experiments did not reveal any functional or molecular evidence that Tpo induces erythroid differentiation in the absence of Epo or in serum free conditions.
Interestingly, although Tpo is well known to be critical for megakaryocytopoiesis, platelet associated genes were upregulated in F36VMpl dimerized, but not Tpo stimulated CD34+ cells. However, it should be noted that studies showing upregulation of megakaryocytic genes in the presence of Tpo involved gene analysis studies in differentiated CD41+ megakaryocytes rather than in their CD34+ progenitors [29, 30]. However, Tpo stimulation of CD34+ progenitors did result in upregulation of certain known downstream targets of c-Mpl signaling, for example, PIM1 and lipid synthesis genes [29, 30].
At least two possibilities may explain the disparate effects of Tpo and F36VMpl dimerization on erythropoiesis. The constitutive expression of intracellular Mpl in progenitors may allow signaling at progenitor stages that do not normally express the endogenous Mpl receptor and thus cannot respond to Tpo. Retroviral over expression of c-mpl in mouse hematopoietic progenitors led to enhanced erythroid differentiation [31]. However, the expression of c-Mpl in CMP and MEP [3] make this a less likely explanation.
Alternatively, CID mediated dimerization of the intracellular domain of Mpl may result in conformational changes of the receptor that lead to signaling pathways that are different to those activated by Tpo induced dimerization of full-length Mpl. Native Tpo and Epo receptors contain extracellular, transmembrane and intracellular domains [32]. The intracellular part of c-Mpl and the Epo receptor contain conserved box1 and box2 domains, which are important for signaling and proliferation [5, 33]. The distal region (50 aminoacids) of the intracellular part of c-Mpl is different from that of other cytokine receptors, and contains a domain that is important for differentiation effects [5]. In the case of Epo, binding to the two extracellular domains of the receptor induces specific conformational changes, which are then transmitted through the transmembrane domains to the cystosolic domains that mediate signaling pathways [34]. Differences in the efficiency and degree of downstream signaling pathway activation have been noted between the native full-length EpoR and mutant receptors that have an altered extracellular or transmembrane domain conformation [34]. In addition, synthetic extracellular domain binding ligands that induce conformational changes different from those induced by native Epo, result in differing degrees of signal transduction [35]. Similar conformation-dependent signaling mechanisms have been postulated for Tpo [32], but to our knowledge these have not yet been proven. The fusion protein F36VMpl contains 121 amino acids from the intracellular domain of c-Mpl, including the box1 and box2 domains, which are important for signaling [5]. However, it does not contain the extracellular or transmembrane domains of Mpl. Thus, we propose that differences between the conformations of F36VMpl and the full-length native Tpo receptor may account for the differences between the effects of F36VMpl dimerization and Tpo.
SUMMARY
In summary, this study demonstrates that intracellular Mpl dimerization induces progenitor expansion and erythropoiesis in human myeloerythroid progenitors through molecular mechanisms that are not shared by binding of Tpo to native Mpl. The ability to regulate these unique functional effects in human hematopoietic progenitors could be potentially exploited for the enhancement of myeloerythropoiesis in the cell therapy setting. Delineating these effects provides valuable insights into the effects of dimerization in specific hematopoietic progenitor populations. These data also emphasize how constitutive activation of a cytokine signaling domain can produce effects that are not predictable based on stimulation of the endogenous full-length receptor. Defining the mechanisms underlying these differences is essential for the development, characterization, and clinical translation of cytokine signaling domain based gene therapy strategies.
Supplementary Material
ACKNOWLEDGMENTS
We thank ARIAD Pharmaceuticals (Cambridge, MA) for providing CID, Richard Sposto (Childrens Hospital Los Angeles, Los Angeles, CA) and David Gjertson (University of California Los Angeles [UCLA] David Geffen School of Medicine, Department of Pathology and Laboratory Medicine, CA) for guidance regarding statistical analyses, Felicia Codrea (UCLA Broad Stem Cell Research Center) for assistance with flow cytometry, Shantha Seenadheera for assistance with viral vectors, Batul Suterwala for assistance with cell morphology analysis, and Gautam Dravid and Lisa Kohn for critical reading of the manuscript. This work was made possible by the following core facilities at UCLA: Broad Stem Cell Research Center flow cytometry, Clinical Microarray, Jonsson Comprehensive Cancer Center Small Animal Imaging facility, DLAM. This work was supported by a program project grant from the National Heart Lung and Blood Institute (NHLBI) Grant Number 1P01–HL073104 (G.M.C. and D.B.K.). C.P. was the recipient of a St. Baldrick fellowship award and a California Institute for Regenerative Medicine Training Grant (T-0032).
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors indicate no potential conflicts of interest.
REFERENCES
- 1.Piacibello W, Sanavio F, Garetto L, et al. Extensive amplification and self-renewal of human primitive hematopoietic stem cells from cord blood. Blood. 1997;89:2644–2653. [PubMed] [Google Scholar]
- 2.Abdel-Azim H, Zhu Y, Hollis R, et al. Expansion of multipotent and lymphoid-committed human progenitors through intracellular dimerization of Mpl. Blood. 2008;111:4064–4074. doi: 10.1182/blood-2007-08-107466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manz MG, Miyamoto T, Akashi K, et al. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci USA. 2002;99:11872–11877. doi: 10.1073/pnas.172384399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deutsch VR, Tomer A. Megakaryocyte development and platelet production. Br J Haematol. 2006;134:453–466. doi: 10.1111/j.1365-2141.2006.06215.x. [DOI] [PubMed] [Google Scholar]
- 5.Drachman JG, Kaushansky K. Dissecting the thrombopoietin receptor: functional elements of the Mpl cytoplasmic domain. Proc Natl Acad Sci USA. 1997;94:2350–2355. doi: 10.1073/pnas.94.6.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richard RE, Wood B, Zeng H, et al. Expansion of genetically modified primary human hemopoietic cells using chemical inducers of dimerization. Blood. 2000;95:430–436. [PubMed] [Google Scholar]
- 7.Galy A, Travis M, Cen D, et al. Human T, B, natural killer, and dendritic cells arise from a common bone marrow progenitor cell subset. Immunity. 1995;3:459–473. doi: 10.1016/1074-7613(95)90175-2. [DOI] [PubMed] [Google Scholar]
- 8.Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91:661–672. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 9.Hao QL, Zhu J, Price MA, et al. Identification of a novel, human multilymphoid progenitor in cord blood. Blood. 2001;97:3683–3690. doi: 10.1182/blood.v97.12.3683. [DOI] [PubMed] [Google Scholar]
- 10.Hao QL, Smogorzewska EM, Barsky LW, et al. In vitro identification of single CD34+CD38- cells with both lymphoid and myeloid potential. Blood. 1998;91:4145–4151. [PubMed] [Google Scholar]
- 11.Dull T, Zufferey R, Kelly M, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ailles L, Schmidt M, Santoni de Sio FR, et al. Molecular evidence of lentiviral vector-mediated gene transfer into human self-renewing, multi-potent, long-term NOD/SCID repopulating hematopoietic cells. Mol Ther. 2002;6:615–626. [PubMed] [Google Scholar]
- 13.Wang X, Rosol M, Ge S, et al. Dynamic tracking of human hematopoietic stem cell engraftment using in vivo bioluminescence imaging. Blood. 2003;102:3478–3482. doi: 10.1182/blood-2003-05-1432. [DOI] [PubMed] [Google Scholar]
- 14.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 15.Bolstad BM, Irizarry RA, Astrand M, et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 16.Rajagopalan D. A comparison of statistical methods for analysis of high density oligonucleotide array data. Bioinformatics. 2003;19:1469–1476. doi: 10.1093/bioinformatics/btg202. [DOI] [PubMed] [Google Scholar]
- 17.Tsiftsoglou AS, Vizirianakis IS, Strouboulis J. Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life. 2009;61:800–830. doi: 10.1002/iub.226. [DOI] [PubMed] [Google Scholar]
- 18.Sivertsen EA, Hystad ME, Gutzkow KB, et al. PI3K/Akt-dependent Epo-induced signalling and target genes in human early erythroid progenitor cells. Br J Haematol. 2006;135:117–128. doi: 10.1111/j.1365-2141.2006.06252.x. [DOI] [PubMed] [Google Scholar]
- 19.Gerber HP, Ferrara N. The role of VEGF in normal and neoplastic hematopoiesis. J Mol Med. 2003;81:20–31. doi: 10.1007/s00109-002-0397-4. [DOI] [PubMed] [Google Scholar]
- 20.Martin R, Lahlil R, Damert A, et al. SCL interacts with VEGF to suppress apoptosis at the onset of hematopoiesis. Development. 2004;131:693–702. doi: 10.1242/dev.00968. [DOI] [PubMed] [Google Scholar]
- 21.Liu W, Wang M, Tang DC, et al. Thrombopoietin has a differentiative effect on late-stage human erythropoiesis. Br J Haematol. 1999;105:459–469. [PubMed] [Google Scholar]
- 22.Golde DW, Bersch N, Quan SG, et al. Production of erythroid-potentiating activity by a human T-lymphoblast cell line. Proc Natl Acad Sci USA. 1980;77:593–596. doi: 10.1073/pnas.77.1.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gasson JC, Golde DW, Kaufman SE, et al. Molecular characterization and expression of the gene encoding human erythroid-potentiating activity. Nature. 1985;315:768–771. doi: 10.1038/315768a0. [DOI] [PubMed] [Google Scholar]
- 24.Hayakawa T, Yamashita K, Tanzawa K, et al. Growth-promoting activity of tissue inhibitor of metalloproteinases-1 (TIMP-1) for a wide range of cells. A possible new growth factor in serum. FEBS Lett. 1992;298:29–32. doi: 10.1016/0014-5793(92)80015-9. [DOI] [PubMed] [Google Scholar]
- 25.Otto KG, Broudy VC, Lin NL, et al. Membrane localization is not required for Mpl function in normal hematopoietic cells. Blood. 2001;98:2077–2083. doi: 10.1182/blood.v98.7.2077. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi M, Laver JH, Kato T, et al. Recombinant human thrombopoietin (Mpl ligand) enhances proliferation of erythroid progenitors. Blood. 1995;86:2494–2499. [PubMed] [Google Scholar]
- 27.Kaushansky K, Broudy VC, Grossmann A, et al. Thrombopoietin expands erythroid progenitors, increases red cell production, and enhances erythroid recovery after myelosuppressive therapy. J Clin Invest. 1995;96:1683–1687. doi: 10.1172/JCI118210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ratajczak MZ, Ratajczak J, Marlicz W, et al. Recombinant human thrombopoietin (TPO) stimulates erythropoiesis by inhibiting erythroid progenitor cell apoptosis. Br J Haematol. 1997;98:8–17. doi: 10.1046/j.1365-2141.1997.1802997.x. [DOI] [PubMed] [Google Scholar]
- 29.Lim CK, Hwang WY, Aw SE, et al. Study of gene expression profile during cord blood-associated megakaryopoiesis. Eur J Haematol. 2008;81:196–208. doi: 10.1111/j.1600-0609.2008.01104.x. [DOI] [PubMed] [Google Scholar]
- 30.Kim JA, Jung YJ, Seoh JY, et al. Gene expression profile of megakaryocytes from human cord blood CD34(+) cells ex vivo expanded by thrombopoietin. Stem Cells. 2002;20:402–416. doi: 10.1634/stemcells.20-5-402. [DOI] [PubMed] [Google Scholar]
- 31.Yan XQ, Lacey DL, Saris C, et al. Ectopic overexpression of c-mpl by retroviral-mediated gene transfer suppressed megakaryopoiesis but enhanced erythropoiesis in mice. Exp Hematol. 1999;27:1409–1417. doi: 10.1016/s0301-472x(99)00069-7. [DOI] [PubMed] [Google Scholar]
- 32.Kaushansky K. Molecular mechanisms of thrombopoietin signaling. J Thromb Haemost. 2009;7(suppl 1):235–238. doi: 10.1111/j.1538-7836.2009.03419.x. [DOI] [PubMed] [Google Scholar]
- 33.Miller CP, Liu ZY, Noguchi CT, et al. A minimal cytoplasmic subdomain of the erythropoietin receptor mediates erythroid and megakaryocytic cell development. Blood. 1999;94:3381–3387. [PubMed] [Google Scholar]
- 34.Kubatzky KF, Liu W, Goldgraben K, et al. Structural requirements of the extracellular to transmembrane domain junction for erythropoietin receptor function. J Biol Chem. 2005;280:14844–14854. doi: 10.1074/jbc.M411251200. [DOI] [PubMed] [Google Scholar]
- 35.Constantinescu SN, Huang LJ, Nam H, et al. The erythropoietin receptor cytosolic juxtamembrane domain contains an essential, precisely oriented, hydrophobic motif. Mol Cell. 2001;7:377–385. doi: 10.1016/s1097-2765(01)00185-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.