

Abstract
SUMMARY
We present a comprehensive overview of the hierarchical network of intracellular processes revolving around central nitrogen metabolism in Escherichia coli. The hierarchy intertwines transport, metabolism, signaling leading to posttranslational modification, and transcription. The protein components of the network include an ammonium transporter (AmtB), a glutamine transporter (GlnHPQ), two ammonium assimilation pathways (glutamine synthetase [GS]-glutamate synthase [glutamine 2-oxoglutarate amidotransferase {GOGAT}] and glutamate dehydrogenase [GDH]), the two bifunctional enzymes adenylyl transferase/adenylyl-removing enzyme (ATase) and uridylyl transferase/uridylyl-removing enzyme (UTase), the two trimeric signal transduction proteins (GlnB and GlnK), the two-component regulatory system composed of the histidine protein kinase nitrogen regulator II (NRII) and the response nitrogen regulator I (NRI), three global transcriptional regulators called nitrogen assimilation control (Nac) protein, leucine-responsive regulatory protein (Lrp), and cyclic AMP (cAMP) receptor protein (Crp), the glutaminases, and the nitrogen-phosphotransferase system. First, the structural and molecular knowledge on these proteins is reviewed. Thereafter, the activities of the components as they engage together in transport, metabolism, signal transduction, and transcription and their regulation are discussed. Next, old and new molecular data and physiological data are put into a common perspective on integral cellular functioning, especially with the aim of resolving counterintuitive or paradoxical processes featured in nitrogen assimilation. Finally, we articulate what still remains to be discovered and what general lessons can be learned from the vast amounts of data that are available now.
INTRODUCTION
A fact of life that many microorganisms are confronted with is the unreliable environmental availability of nutrients, including N-containing compounds. Some microorganisms have to deal with virtually every possible nutritional state between feast and famine; periods of nutrient excess may well be followed by periods of extreme starvation, or they may not be or not always be. As they can grow fast and often exponentially, many microorganisms readily deplete their own environment of the nutrient that is least in excess and thereby have to endure nutrient limitation of some sort much more frequently than they can enjoy full nutritional affluence. Adaptive physiological responses to feast or famine conditions are the result of a highly regulated system consisting of signal transduction coupled to transport, metabolism, and genetic circuits.
A general challenge for every microorganism is how to choose to respond to changing environmental conditions if it has more than one possibility to react at its disposal. This challenge is well illustrated by Escherichia coli when it is confronted with a change in the nitrogen supply in its surroundings. Since it possesses two central nitrogen-assimilatory routes, it has to make a decision as to what extent either pathway or both pathways should be adapted most.
The ammonium assimilation network of E. coli comprises a complex and hierarchical regulatory network that involves transport, signaling, metabolism, posttranslational modification, and transcription. At the protein level, the network includes an ammonium transporter, two ammonium assimilation pathways, two bifunctional protein modification enzymes, two trimeric signal transduction proteins, and a two-component regulatory system composed of a histidine protein kinase and the corresponding response regulator (1). At the metabolic level, the amino acids glutamine and glutamate, the free energy currency molecules ADP and ATP, the redox currency molecules NADPH/NADP, and the tricarboxylic acid (TCA) cycle intermediate 2-oxoglutarate are relevant metabolites in the central ammonium assimilation network; all of these metabolites rank among the top 10 key metabolites in cellular metabolism, based on connectivity statistics (2, 3). After uptake, ammonium is incorporated directly only into the amino acids glutamate and glutamine, which subsequently function as nitrogen donors in transamination and transamidation reactions. These lead to other amino acids and to precursors for the biosynthesis of purines and pyrimidines (4, 5).
Ammonium assimilation by the glutamine synthetase (GS)-glutamate synthase (glutamine 2-oxoglutarate amidotransferase [GOGAT]) pathway may account for a substantial percentage of the cell's ATP requirement when it is growing on glucose minimal medium (6), even though the alternative of ammonium assimilation through glutamate dehydrogenase (GDH) alone accomplishes the same chemistry at the cost of less ATP. The former pathway appears to be used, even though expression of the operons encoding the proteins GS and GOGAT, as well as the activity of GS, can be regulated in multiple ways (6–9). On the other hand, for many other carbon and energy sources used for growth of E. coli, the ATP requirement of ammonium assimilation through GS-GOGAT as a percentage of the total ATP turnover is smaller than that for glucose, suggesting that the extra ATP requirement of the GS-GOGAT pathway would be tolerable if the GS-GOGAT pathway would serve functions other than the mere assimilation of ammonium. Indeed, GS-GOGAT is only the tip of the iceberg of a complex regulatory network, which might contribute in other ways to maximum fitness of E. coli. Accordingly, our research should be dedicated not only to characterizing the properties of the components in isolation but also to a full systemic understanding of the role that each and every component plays in bringing about the behavior and function of the network. The science that studies how interactions and networks contribute to biological function has been called systems biology (10, 11), and this systems biology is needed to understand ammonium assimilation. Studying just any single molecule, such as GS, in the network will not suffice, as the properties of that molecule will be affected by the redox state, the energy state, signal transduction, gene expression, and the intracellular levels of ammonium, glutamine, and glutamate, which are also determined by the other macromolecules in the network. An understanding of the network without understanding the molecules will not suffice either, as it is the dynamic response of the macromolecules through the levels of the micromolecules that determines network functioning (12). One needs to study the molecules and their networking to understand biological function (10).
In this review, we focus on the assimilatory response of E. coli to changes in the availability of nitrogen-containing compounds, in particular ammonium and glutamine. Our strategy is to first describe all the components of this network one by one, paying attention to their interactive properties. We then discuss the different (sub)networks that involve these components, with the aim of showing what their functional roles are in nitrogen assimilation in E. coli. Finally, we distill some general biology lessons from this particular subject. This review will thereby focus on (i) the molecular data regarding the structures and functions of the proteins involved in the central nitrogen assimilation network, most notably on the GS regulatory cascade and (ii) the systemic view that puts the molecular data in a systems biological perspective. (Note that the term “ammonium” is used in this review if the exact nature of the molecular species is not relevant [or not known], whereas the chemical formulas “NH4+” and “NH3” are used when these particular species are at stake.)
Central Nitrogen Assimilation in a Nutshell
This review focuses on E. coli, but the closely related Enterobacteriaceae Salmonella enterica serovar Typhimurium (13) and Klebsiella pneumoniae (Klebsiella aerogenes) are not excluded. The genus Shigella, which also belongs to the highly diverse species E. coli (14), will not be discussed because of a lack of data on nitrogen assimilation. Other enterobacteria, proteobacteria, and even archaea are discussed only when important for an understanding of E. coli function by comparison.
Nitrogen is an essential element for all organisms. Like other enteric bacteria, E. coli is able to use a host of organic nitrogen-containing compounds as sole nitrogen sources (15, 16). Ammonium is considered the preferred nitrogen source, as it supports the highest growth rate (7). In batch culture, E. coli cannot grow on any inorganic compound other than ammonium (6, 7), although in C-limited chemostats at low specific growth rates, nitrate can be used as the single N source (17).
The assimilation of ammonium into glutamate is the process where the element nitrogen is assimilated by carbon metabolism. For this incorporation of ammonium into 2-oxoglutarate, E. coli (and other enteric bacteria) possesses two pathways (Fig. 1 and Table 1), i.e., the GDH and the GS-GOGAT pathways (6, 18). GDH catalyzes the reductive amination of 2-oxoglutarate to glutamate. GS catalyzes the amidation of glutamate to glutamine at the cost of the hydrolysis of one molecule of ATP. Both enzymes use ammonium as the nitrogen source. The reductive transfer of the glutamine amide group to the 2-position of 2-oxoglutarate, thereby forming two molecules of glutamate, is catalyzed by GOGAT. Both GDH and GOGAT of enterobacteria are specific for NADPH over NADH (18–20). Net glutamate production from 2-oxoglutarate can hereby be achieved not only by GDH alone but also by GS-GOGAT coupled together. The latter process expends one ATP.
Fig 1.

Two pathways for glutamate synthesis. The left panel shows the GDH pathway, and the right panel shows the cyclic GS-GOGAT pathway. The basic characteristics of the two pathways are shown in the box below the scheme. The GDH pathway consists of a single enzyme, glutamate dehydrogenase (GDH). One net glutamate is produced from the reductive amination of 2-oxoglutarate; no ATP is involved, and GDH had a relatively low affinity for ammonium. The cyclic GS-GOGAT pathway consists of two enzymes, i.e., glutamine synthetase (GS) and glutamate synthase (GOGAT), which, in a cyclic configuration in effect, also produces one net glutamate by reductive amination of 2-oxoglutarate. However, one ATP is invested for every amino group assimilated, and GS has a relatively high affinity for ammonium.
Table 1.
Biosynthetic reactions catalyzed by GDH, GS, GOGAT, and GS plus GOGAT

Glutamine is the main amide donor for nucleotide biosynthesis and, hence, DNA and RNA synthesis (4). Net glutamine production from 2-oxoglutarate can be achieved by the combined activity of GDH and GS or of GOGAT and GS twice, at the cost of one or two ATP molecules, respectively. However, DNA and RNA make up a smaller fraction of biomass than protein, and consequently, the flux into glutamate is much higher than the flux into glutamine other than for the purpose of producing glutamate. We shall therefore focus on the process of synthesis of glutamate from ammonium and 2-oxoglutarate.
A salient feature of the two central nitrogen-assimilating enzymes of E. coli is the difference in their respective Km values for ammonium. Purified GDH has a relatively high Km value for ammonium (∼1 mM) (19, 21), whereas purified GS has a 10-fold-lower Km for ammonium, i.e., ∼0.1 mM (22–24). Therefore, the received view is that the “cheap and low-affinity” enzyme GDH takes care of nitrogen assimilation during growth in high-ammonium and low-carbon/energy media, while the “free energy-expensive and high-affinity” enzyme system GS-GOGAT functions during growth in low-ammonium and high-carbon/energy media.
GS activity is regulated in multiple ways. Already metabolically, flow through GS is regulated 4-fold, i.e., via substrate levels (intracellular ammonium and glutamate), product inhibition (glutamine), the redox state of the cell (NADPH), and the free energy state of the cell (ATP). The catalytic rate is also regulated through signal transduction, which determines the reversible covalent modification state of the enzyme. The latter mode of regulation is connected to transcription regulation by a common sensor and signal transduction system (1, 6, 25–31). Generally, in the absence of ammonium, the global nitrogen response regulator I (NRI) is phosphorylated by its cognate sensor (NRII) at a low-ammonium assimilation state of the cell. NRI-phosphate (NRI-P) then stimulates the expression of some 75 genes (32). Most of these genes have σ54-dependent promoters. One such gene codes for the nitrogen assimilation control (Nac) protein. Nac regulates the expression of genes involved in nitrogen metabolism that have σ70-dependent promoters (some 25 genes) (32).
Metabolic, signal transduction, and gene expression regulations each involve entire pathways, e.g., of free energy metabolism, of uridylylation and adenylylation, and of transcription and translation, respectively. In addition, the three types of pathways are intertwined, already around GS itself. The functioning of this hierarchical network depends on the interaction of its components. Below, we therefore discuss molecules, interactions, networks, and function in more detail, with a focus on the dynamic integration of the former into the latter.
THE PROTEINS IN ISOLATION
At least 14 proteins are engaged in assimilation of extracellular ammonium in intracellular biochemistry (Table 2): 5 enzymes, 3 regulatory proteins, 4 transcription factors, and 2 transporters, that is, the enzymes GS, GOGAT, GDH, adenylyltransferase (ATase), and uridylyltransferase (UTase); the regulatory proteins GlnB, GlnK, and NRII; the transcription factors NRI, Nac, cyclic AMP (cAMP) receptor protein (Crp), and leucine-responsive regulatory protein (Lrp); and the ammonium carrier (AmtB) and glutamine carrier (GlnHPQ) transporters. In addition, six enzymes (NAD synthetase, carbamoyl phosphate synthetase, asparagine synthetases A and B, and glutaminases A and B) and the nitrogen-phosphotransferase system (N-PTS) are possibly involved and are reviewed below. The first four of the latter enzymes are discussed because they have the ability to assimilate ammonium, although they normally engage in other reactions. The glutaminases are potentially important as they may be involved in controlling the glutamine pool. The N-PTS is a recently discovered system that may or may not turn out to be relevant for nitrogen assimilation. All genes, proteins (abbreviations and full names), and activities discussed in this review are shown in Table 2. In this section, we introduce each of these >20 components in terms of their structural and interactive properties, as determined for the components in isolation.
Table 2.
Components of the nitrogen assimilation network
| Genea | Proteina,b | Full protein name and/or activity(ies) |
|---|---|---|
| glnA | GS | Glutamine synthetase; catalyzes the synthesis of glutamine |
| glnB | GlnB (PII)c | GlnB protein, signal-transducing protein; GlnB stimulates adenylylation of GS, GlnB-UMP stimulates deadenylylation of GS-AMP, and GlnB stimulates phosphatase activity of NRII |
| glnD | UTased | Uridylyltransferase/uridylyl-removing protein; uridylylates GlnB and GlnK and deuridylylates GlnB-UMP and GlnK-UMP |
| glnE | ATased | Adenylyltransferase/adenylyl-removing protein; adenylylates GS and deadenylylates GS-AMP |
| glnF (ntrA; rpoN) | σ54 (σN) | RNA polymerase sigma factor 54 |
| glnG (ntrC) | NRI (NtrC)e | Nitrogen regulator I/NtrC; NRI-P activates glnALG transcription at the glnAp2 promoter, and NRI represses glnALG transcription at the glnAp1 and glnLp promoters |
| glnL (ntrB) | NRII (NtrB)e | Nitrogen regulator II/NtrB; NRII phosphorylates NRI, and NRII + GlnB or GlnK dephosphorylates NRI-P |
| glnK | GlnKc | GlnK protein, signal-transducing protein; GlnK stimulates adenylylation of GS, GlnK-UMP stimulates deadenylylation of GS-AMP, GlnK stimulates phosphatase activity of NRII, and GlnK inhibits AmtB activity |
| amtB | AmtB | Ammonium transporter AmtB; conducts ammonium across the cytoplasmic membrane and functions as a sensor for extracellular ammonium |
| glnHPQ | GlnHPQ | Glutamine transporter; high-affinity ABC transporter |
| nac | Nac | Nitrogen assimilation control protein |
| crp | Crp | cAMP receptor protein |
| lrp | Lrp | Leucine-responsive regulatory protein |
| gltBDF | GOGAT | Glutamate synthase |
| gdhA | GDH | Glutamate dehydrogenase |
Alternative name(s) is in parentheses.
Historical nomenclature is as follows: PI, ATase; PIIA, GlnB; PIID, GlnB-UMP.
PII and GlnK are PII paralogs, but for clarity, both proteins are named after their gene name throughout this review; thus, PII is called GlnB.
UTase instead of UTase/UR and ATase instead of ATase/AR are used throughout this review to denote these bifunctional/ambiguous enzymes.
We propose the use of NRI and NRII, because they are a complete set of glnA-specific expression regulators (there is no NRIII), which is not suggested by the naming of NtrB and NtrC, because NtrA is a sigma factor that regulates multiple operons, including non-nitrogen-related ones, and is itself not part of the glnALG operon.
Glutamine Synthetase
GS lies at the heart of the nitrogen assimilation network. GS is a dodecamer of identical monomers of 52 kDa encoded by the glnA gene (33, 34).
Electron microscopic (35) and X-ray crystallographic analyses of completely unadenylylated GS of S. Typhimurium have shown that the 12 subunits in each complex are arranged in two rings of six subunits each, with the second hexagon inverted on top of the first. The two layers of subunits are held together largely by the apolar carboxyl terminus of each subunit inserting into a hydrophobic pocket formed by two neighboring subunits on the opposite ring (36, 37). Within each layer, each of the six active sites is located at the interface of a pair of subunits. A cylindrical active site is formed by six antiparallel β-strands of one subunit and two strands of the neighboring subunit and holds two divalent cations (Mn2+ ions) as cofactors (36) necessary for catalysis (37). The substrate binding sites of glutamate, ATP, and ammonium are located within this active site (36, 38–41). GS has 12 active sites that may well act cooperatively (see below).
The various steps in the synthesis of glutamine by fully unadenylylated GS have been visualized beautifully as a series of molecular interactions in time and space by X-ray crystallography of crystal structures of enzyme-substrate complexes (38, 41). GS prefers an ordered catalytic cycle: ATP first binds to GS, glutamate then binds and attacks ATP to form γ-glutamyl phosphate and ADP, and, finally, ammonium binds to GS and loses a proton to form ammonia, which attacks the γ-glutamyl phosphate to yield glutamine (38).
The rate equation for νGS is as follows:
with the apparent maximal rate constant = θGS · VGS = θGS · [GS] · kcatGS. The affinity constants are as follows: KATP is 0.4 mM, KGlu is 4 mM, KNH4+ is 0.1 mM, KADP is 0.06 mM, KPi is 4 mM, and KGln is 6 mM (determined at an average adenylylation state of ~2.5 AMP groups [out of a maximum of 12] covalently attached to dodecameric GS). The equilibrium constant (Keq) is 460 and the maximum capacity of GS (VGS) is 600 mM/min under ammonium-limited conditions. As the enzyme concentration ([GS]) is ~14 µM with glutamine as the single N source, kcatGS may amount to ~720 s−1 (see references 1, 24, 42, and 43). This value is comparable to the turnover number of ~1,400 s−1 determined for GS purified from cells grown with glutamate as the N source (677), another N-limited growth condition. θGS is a function of the adenylylation state of the enzyme (44, 45).
Glutamate Synthase
GOGAT is an abbreviation of glutamine 2-oxoglutarate amidotransferase (EC 1.4.1.13). However, it should be noted that GOGAT is classified by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (IUBMB) as an oxidoreductase but not as a transferase. Its systematic name is glutamate:NADP+ oxidoreductase (transaminating).
E. coli GOGAT is a heterodimer with subunits with molecular masses of 166 and 52 kDa, which can aggregate further to form an octamer. The two different subunits are encoded by the gltB and gltD genes, respectively. It has been suggested that GOGAT has two active forms (19, 46).
E. coli GOGAT is a member of the class of NADPH-dependent glutamate synthases. In general, the class can be characterized as follows: the large α- and small β-subunits form a catalytically active αβ-heterodimer, which contains one flavin adenine dinucleotide (FAD), one flavin mononucleotide (FMN), and three different Fe-S clusters. The GltB protein comprises an amidotransferase domain coupled to a synthase domain through an intramolecular ammonium tunnel. The GltD protein delivers the reducing equivalents from NADPH to the active site. The first glutamate molecule is released at the “glutaminase” site, and the second glutamate is released from the “synthase” site of the α-subunit (47).
In general, glutamine-dependent amidotransferases generate NH3 by glutamine hydrolysis, followed by NH3 transport via an intramolecular tunnel and further reaction by an enzyme-specific synthase activity (48). Under some conditions, these enzymes use cytosolic ammonium instead of glutamine and, by doing so, function as ammonium-assimilating enzymes. For instance, native GOGAT (as well as the apoglutamate synthase) can use ammonium instead of glutamine as the amino donor to 2-oxoglutarate; the former activity is only 5 to 7% of the latter activity, however (49).
On the basis of data obtained with a polar gltF mutant, GltF was assigned a regulatory role in nitrogen catabolism and ammonium transport (50, 51), but this observation could not be confirmed when a nonpolar mutant was used (52). GltF may be translocated to the periplasmic space, which may make a direct regulatory function in GOGAT activity unlikely (52). Thus, until now, its precise function remains unclear (50–53).
The rate equation of GOGAT (νGOGAT) may read
The dissociation constants are as follows: KGln is 0.18 mM, KKG is 0.007 mM (where KG denotes 2-oxoglutarate), KNADPH is 0.0015 mM, KGlu is 11 mM, KNADP is 0.0037 mM (54), and KMetGlu is 0.7 mM (manual optimization, referring to inhibition by metabolites derived from glutamate). The maximum capacity of GOGAT (VGOGAT) is 85 mM/min under ammonium-limited growth conditions (44).
Glutamate Dehydrogenase
GDH of E. coli is a hexamer (275 to 300 kDa) of identical polypeptides (45 to 50 kDa) (19, 55, 56). The monomer is encoded by the gdhA gene, which translates to a polypeptide of 447 amino acids (48.4 kDa) (57, 58). So-called biosynthetic GDH catalyzes the reductive amination of 2-oxoglutarate to glutamate using NADPH as the single reducing agent (19, 20). The production of glutamate is favored, since the apparent equilibrium constant for the biosynthetic reaction amounts to 2,850 mM−1 at an ionic strength (I) of 0 and a temperature of 27°C, although it decreases with increasing ionic strength to a value of 285 mM−1 at an I of 0.5 and goes down to 90 mM−1 at an I of 0.5 and a temperature of 37°C (59). The latter value means that at equal concentrations of NADPH and NADP and of glutamate and 2-oxoglutarate, the reaction will run in the direction of ammonium assimilation whenever the intracellular concentration of ammonium exceeds 11 μM. S. Typhimurium GDH features an ordered ter-bi (three substrates, two products) mechanism, where NADPH first binds and ammonium and, finally, 2-oxoglutarate then bind; first glutamate and then NADP dissociate (60).
Confusing results have been presented with respect to the stability of the enzyme. Purified GDH of E. coli was heat stable (19, 20) and remained active when stored for several months at room temperature, while it was inactivated by freezing; potassium protected against freezing damage (19). In other hands, purified GDH was found to lose activity within minutes at room temperature (21). Purified GDH displayed an unusual resistance to high concentrations of the protein denaturant urea (20) and guanidine HCl (19). The same was true for purified S. Typhimurium GDH (55). Purified GDH has a single tight binding site for NADPH (61). Binding of NADPH in the absence of other ligands destabilizes the enzyme, except when tri- and dicarboxylic acids, including 2-oxoglutarate, or nucleoside di- and triphosphates, including ATP and GTP, are present. Native GDH was stable in exponentially growing cells but was degraded by ATP-dependent proteases under conditions of carbon or nitrogen starvation (61).
The Km values for the substrates and products of purified GDH of E. coli as measured by six different groups differed considerably: the ranges of Km values were 0.2 to 6.0 mM for 2-oxoglutarate, 1 to 36 mM for ammonium, and 12 to 83 μM for NADPH (21). The Km values for the substrates and products of purified GDH of S. Typhimurium lie in the respective ranges (60). Purified GDH showed substantial substrate inhibition with 2-oxoglutarate (>10 mM) and glutamate (>250 mM) (21). Purified GDH showed a pH optimum at 8.0 for the reductive amination reaction (20, 21), while that of S. Typhimurium showed a pH optimum at 8.6 (55). Interestingly, GDH has been shown to be phosphorylated at a histidine residue in vitro (62) and in vivo (63) in the exponential growth phase. Low ATP (up to 2 mM) concentrations stimulated GDH activity, while high (>8 mM) concentrations were inhibitory (60). It is unknown whether ATP is the direct phosphoryl donor for the histidine phosphorylation of GDH. There is evidence for an allosteric binding site for GTP, and perhaps also for ppGpp, in GDH (61).
For a rate equation for GDH (νGDH), Bruggeman et al. (44) used the following equation:
Here KKG is 0.32 mm, KNADPH is 0.04 mm, KNH4+ is 1.1 mM, KNADP is 0.04 mM, KGlu is 10 mM, and Keq is 1,300 mM−1.
Other Enzymes Capable of Direct Ammonium Assimilation
To date, besides GS and GDH (and GOGAT to some extent), E. coli possesses four more enzymes capable of directly incorporating ammonium into organic biomolecules, i.e., NAD synthetase, carbamoyl phosphate synthetase, and two asparagine synthetases, AsnA and AsnB. In this section, we briefly describe these enzymes (Table 3), even though these enzymes are not featured any further in this review.
Table 3.
Reactions catalyzed by enzymes capable of direct ammonium assimilationa

Direct ammonium assimilation occurs in reactions 1, 2b, 3, and 4b. Reactions 2a and 4a show the reactions with the physiological amino donor glutamine. Reaction 1 includes ammonium, as the specific molecular species is so far unknown.
NAD synthetase.
The gene efg (nadE) was found to code for an ammonium-dependent NAD synthetase (64). Purified NAD synthetase is homodimer with a subunit molecular mass of 30.6 kDa (65). There is strong evidence that ammonium is the sole physiological donor of the amino group for NadE in E. coli and S. Typhimurium (64, 66, 67), which is in contrast to the eukaryotic enzymes, which are glutamine dependent. If true, enterobacterial NAD synthetase is a second essential ammonium-dependent enzyme; GS is the other (66). Indeed, the enzyme has been shown to be essential for growth (68, 69). S. Typhimurium nit mutants are defective in nitrogen assimilation, in spite of having normal levels of GS, GOGAT, and GDH (66, 70). It has been shown that nit is nadE (66).
Recently, an interesting link between carbon and nitrogen metabolism has been observed to involve CyaR, a so-called small noncoding regulatory RNA (71). The expression of nadE was found to be negatively affected by CyaR. Probably, because NadE is an essential enzyme, downregulation was less than that with other targets. In turn, CyaR was positively regulated by the global transcriptional regulator Crp under conditions where cAMP levels were high, i.e., under glucose-limiting conditions, thereby establishing a linkage between C and N metabolisms (71).
It is unknown what the ammonium assimilation rate of this enzyme is, but it is not expected to be substantial, since the cofactors NAD and NADH are not consumed but are recycled in redox reactions: at any significant growth rate, the net rate of NAD(H) synthesis is expected to be much lower than the net rate of ammonium assimilation into biomass synthesis.
Carbamoyl phosphate synthetase.
The carAB operon codes for the two subunits of carbamoyl phosphate synthetase (72). Carbamoyl phosphate is synthesized from bicarbonate, ATP, and glutamine (73). The transfer of the amido-N of glutamine in the form of NH3 takes place via a long hydrophobic tunnel made up from the small subunit (CarA) and the large subunit (CarB). The latter subunit carries the binding sites for bicarbonate and ATP. The native heterodimer as well as the purified large subunit have been shown to be able to produce carbamoyl phosphate from bicarbonate, ATP, and ammonium; NH3, and not NH4+, is the substrate (74–76). Thus, in principle, carbamoyl phosphate synthetase is another enzyme capable of direct ammonium assimilation. However, the low affinity of either CarAB (75) or CarB (76) for NH3 (Km = 5 mM) renders it unlikely that this reaction occurs at a substantial physiological rate. CarB is essential, while CarA is not, and cells lacking CarA grow albeit slowly (77).
Asparagine synthetases A and B.
Two asparagine synthetases exist, i.e., an ammonium- and a glutamine-dependent enzyme, encoded by asnA and asnB, respectively (78, 79). Mutant strains in which the genes for both asparagine synthetase A and asparagine synthetase B are deleted require asparagine, whereas either single mutant strain grows at normal rates without asparagine (79).
The asnA gene codes for a minor ammonium-assimilating enzyme that catalyzes the amination of aspartate with ammonium to produce asparagine (80). This gene is downregulated under N-limiting conditions; NRI-P stimulates the synthesis of the transcription factor Nac, and Nac represses asnC, the product of which is required to activate transcription of asnA (80). Thus, asnA expression is repressed under N-limiting conditions. AsnB catalyzes the amination of aspartate with glutamine to produce asparagine (78). Glutamine hydrolysis to yield NH3 takes place in the N-terminal domain; thereafter, the NH3 released is channeled to the active site at the C terminus, where aspartate is converted into asparagine (81). Remarkably, AsnB has also been shown to be able to use cytosolic NH3 instead of glutamine as the N donor (81). Thus, both asparagine synthetases are, in principle, capable of assimilating ammonium. Because it is the amide and not the amino group of asparagine that is involved, their contribution to direct ammonium assimilation is expected to be of minor importance, although the in vivo fluxes remain to be determined.
Adenylyltransferase/Adenylyl-Removing Enzyme
Structural aspects.
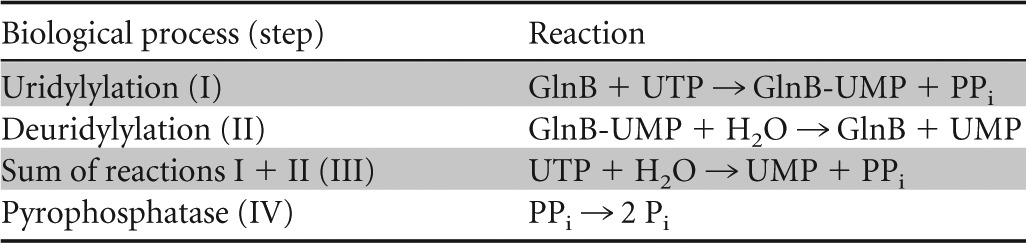
The term ATase is used to represent the bifunctional enzyme that has adenylyltransferase (AT) and adenylyl-removing (AR) activities. ATase functioning as an adenylyltransferase catalyzes the addition of AMP to GS, using ATP as the substrate and with the release of pyrophosphate (PPi) (82–85). Pyrophosphatase catalyzes the hydrolysis of PPi into phosphate. When ATase functions as an adenylyl-removing enzyme, it catalyzes the removal of AMP from GS in a phosphate-dependent reaction, which results in the release of ADP (83, 86). The reactions catalyzed by ATase and pyrophosphatase are shown in Table 4.
Table 4.
Reactions catalyzed by ATase and pyrophosphatase

The glnE gene is located at position 3254 of the E. coli chromosome (66.75 min on the genetic map) (87). It codes for ATase, a protein of 945 amino acids (102.4 kDa) (88). In native gel electrophoresis and in sedimentation equilibrium experiments, ATase behaved as a monomer of ∼115 kDa (89).
Biochemical analyses (with partly purified enzyme preparations) suggested that both the adenylylation and the deadenylylation reactions are catalyzed by the same enzyme, ATase (89–91). Indeed, mutants lacking glnE had neither adenylylation nor deadenylylation activity (92, 93). The presence of these two activities within ATase was also confirmed by showing that highly purified ATase, expressed from the cloned E. coli glnE gene (94), contained both adenylylation and deadenylylation activity in vitro (95).
Although reactions I and II are functionally the opposite of one another, chemically, they are not the reversal of each other (95, 96). The sum of the adenylylation reaction and the deadenylylation reaction (reaction III) equals the conversion of ATP and phosphate to ADP and pyrophosphate. The standard free energy of hydrolysis (ΔG°′) of ATP (to ADP) and PPi (to Pi) are −33 and −22 kJ/mol, respectively. Free energy estimates of ATP and PPi hydrolysis under cytosolic conditions at pH 7.6 and 3 mM Mg2+ are −50 and −20 kJ/mol, respectively (97), and therefore, free energy to an amount of 30 kJ/mol is dissipated by the sum of the two reactions (reaction III) (Table 4), but 50 kJ/mol if the product pyrophosphate would be hydrolyzed further to inorganic phosphate (reaction IV) (Table 4). Here the ATase is special biochemically not only in that it is bifunctional (i.e., catalyzing two different reactions) but also in that its two activities are antagonistic: the enzyme is “ambiguous” (95). Its preference for either reaction might be toggled by the binding of allosteric effectors or covalent modification. Such ambiguity is not unique; the UTase and certain protein kinases may also be ambiguous.
Comparison of the amino acid sequence of ATase, a bifunctional nucleotidyltransferase, with itself reveals a two-domain structure (88). Each domain is specialized in one of the two reactions catalyzed by ATase. The N-terminal domain catalyzes the deadenylylation reaction, while the C-terminal domain catalyzes the adenylylation reaction (95, 98, 99). A central, regulatory “R” domain of some 200 amino acids is located between the N- and C-terminal domains (98, 100). The regulatory R domain, which may engage in intramolecular signal transduction, perhaps prevents one domain from catalyzing adenylylation and the other from deadenylylation at the same time (95). The three domains are separated from each other by two Q linkers, small peptides of 15 to 25 amino acids that are relatively rich in glutamine (hence Q linker), arginine, glutamate, serine, and proline (101). Thus, the overall domain structure of ATase is N-Q1-R-Q2-C.
Each catalytic domain contains the 24-amino-acid-long conserved motif observed in enzymes catalyzing nucleotidyltransferase reactions (98, 100, 102, 103). This motif includes two aspartate residues, separated from each other by one amino acid, which may be involved in positioning of the Mg2+ ion(s) (103, 104) required for catalysis (96). Indeed, replacement of one or two of these aspartate residues with Ala, Glu, or Asn individually or in pairs in each domain abolished the corresponding domain activities completely (105). X-ray crystallographic analyses of the N-terminal (deadenylylation) domain (104) and of the central plus C-terminal (adenylylation) domains together (103) revealed that the three-dimensional structure of the active site of the two activity domains is highly conserved. In addition, it showed that the overall shape of the former domain is discoidal and that the overall shape of the latter is oblate spheroid. A compilation of the complete ATase protein built from the two X-ray structures (including the two activity domains plus the regulatory domain) revealed that (i) the superhelical regulatory domain is bridging the two catalytic domains and (ii) the two catalytic active sites are located on opposite sides of the molecule and face away from each other (103).
By extensive fitting to in vitro kinetic data from the literature, Bruggeman et al. (44) proposed the following rate equations for the adenylylation and deadenylylation reactions, respectively:
where ϑAD is a complex regulatory function of 2-oxoglutarate containing GlnB and glutamine, and ϑdead is such a function of 2-oxoglurate containing GlnB, GlnB-UMP, and glutamine. It is likely that these expressions are incomplete, however, as GS and GS-AMP should inhibit deadenylylation and adenylylation, respectively (see reference 106). Indeed, the deadenylylation reaction might compete with the adenylylation reaction, as the enzyme is “ambiguous” (95). The above-described equations reflect neither this possibility nor the regulatory role of the R domain explicitly (see above), which may limit this competition by keeping the two reactions separate, in the N and C domains, respectively.
Interactions with GS and GlnB.
The above-mentioned structural model of ATase was used to construct a more speculative model of ATase docked onto GS showing that when the adenylylation domain is bound to GS, the deadenylylation domain is pointing away from GS and vice versa (103). In this way, the activity of the two domains would have less steric hindrance from each other, and the two opposed activities would not interfere with each other. However, an opposing view on the interactions between ATase and GS has also been presented, which was not based on structural but on biochemical data; here it was suggested that the binding of one ATase domain to a GS monomer has a strong cooperative effect on the binding of the other domain to an adjacent GS monomer (107). In contrast to the above-mentioned model, the two domains of the same ATase would both point toward GS.
ATase reduces the activity of GS by covalent modification, i.e., by the covalent linkage of an AMP group to each GS subunit, using ATP as an AMP donor and with the release of pyrophosphate (PPi) (82, 108–110) (Table 4). Each subunit of GS can be adenylylated so that one molecule of GS12 can have 12 AMP groups covalently attached (GS12-AMP12). The activity of GS decreases as the average number of adenylylated subunits per enzyme molecule increases (111). The modified amino acid of GS is tyrosine 397 (112–115), which is located at the outer diameter of the hexagonal ring structure (116). The decrease in activity upon adenylylation of a GS monomer is due to a decrease of both the Vmax and the affinity for glutamate (111). However, the exact conformation change upon full adenylylation is not known because, so far, no nuclear magnetic resonance (NMR) or X-ray crystallographic analysis has been performed with fully adenylylated GS, although crystals with partially adenylylated GS have been analyzed (117–119). Partial adenylylation had no effect on the quaternary structure but might have small effects on the tertiary structure (118). Finally, two other changes of GS become apparent upon adenylylation; i.e., (i) the metal ion cofactor requirement changes from Mg2+ to Mn2+ (45, 120) and (ii) the pH optimum in the γ-glutamyl transferase assay becomes lower, at least for K. pneumoniae (K. aerogenes) (121) and E. coli (122).
As described above, ATase regulates the activity of GS by adenylylation or deadenylylation. Direct regulation of ATase itself occurs by its binding of glutamine and uridylylated and unmodified forms of GlnB, while indirect regulation takes place through the modulation of the activity of GlnB species binding 2-oxoglutarate. As expected, the adenylylation domain contains the binding site for glutamine because glutamine is known to activate the adenylylation activity (98, 100). However, as glutamine also inhibits the deadenylylation reaction, “intramolecular signal transduction” between the AT and AR domains appears to be involved (123). It was suggested that both GlnB and GlnB-UMP bind in the central regulatory domain R (100), but more specific allocations for the binding sites have also been proposed, i.e., at the junctions between the central region and the AR and AT domains (123). Paradoxically, the binding site of GlnB and GlnB-UMP is located on the deadenylylation and the adenylylation domains, respectively. This is unexpected because GlnB stimulates the adenylylation reaction, while GlnB-UMP stimulates the deadenylylation reaction. Thus, the two signal-transducing proteins bind to the domain with the opposing activity (98, 123), again a feature of intramolecular signal transduction. However, regarding inhibition of the two ATase activities, either signal transducer protein binds to the ATase domain with inhibited activity (98). This implies that the two activity domains need to communicate with each other for activation of the opposed activity. Indeed, the adenylylation activity becomes independent of GlnB after deletion of the deadenylylation domain (98, 100, 123).
Uridylyltransferase/Uridylyl-Removing Enzyme
Structural aspects.
UTase is encoded by the glnD gene, which translates to a protein of 890 amino acids (102 kDa) (88, 124, 125). This gene product migrated as an 89-kDa (88) or a 95-kDa (126) protein in SDS-PAGE gels and eluted as a protein of ∼100 kDa from a gel filtration column (124), suggesting that UTase is a monomer.
Comparison of the GlnD amino acid sequence with itself did not reveal a two-domain structure as observed for ATase (88). In addition, the conserved motif observed in enzymes catalyzing nucleotidyl transferase reactions (102), which is supposed to be located at the active site, is present only once in UTase, while in ATase, this motif is present twice (see above). These findings are unexpected, because UTase and ATase are both ambiguous nucleotidyl transferases. UTase has uridylyltransferase (UT) and uridylyl-removing (UR) activities. The discrepancy between UTase and ATase may be taken to suggest that the two catalytic activities of UTase reside at the same site or that the catalytic site of one of the two activities is not homologous to the putative consensus sequence for the nucleotidyl transferases. The former suggestion is consistent with a kinetic analysis of UTase that indicated that both reactions catalyzed by UTase may occur at a single active center (127). The latter suggestion, however, is consistent with a recent analysis of the four distinct domains of E. coli GlnD (128). The site of GlnD homologous to the catalytic domains of nucleotidyl transferases, which is approximately 100 amino acids in length and located at the N terminus of GlnD (102, 128), is essential for the uridylyl transferase activity (128). By implication, the remaining part of the protein (∼800 amino acids), or a subdomain of it, may be involved in the deuridylylation reaction. Indeed, this remaining part of GlnD contains three other domains: one HD domain (128, 129) and, further downstream, two ACT domains (128, 130), in the middle and at the C terminus of GlnD, respectively (128, 131). The HD domain and the ACT domain are named after the conserved His and Asp residues and after three enzymes in which this domain is found, respectively (128). The HD and ACT domains harbor the uridylyl-removing activity and the binding site for glutamine, respectively, as determined by in vitro assays of GlnD variant proteins (128). The uridylyl-removing activity is a hydrolytic reaction (reaction II) (Table 5). This agrees with the observation that the HD domain is present in proteins with phosphohydrolase activity (128, 129). The ACT domain has been found by amino acid sequence analysis in proteins that bind amino acids or small effector molecules (128, 130). This agrees with the regulation of GlnD activity by glutamine (127).
Table 5.
Reactions catalyzed by UTase and pyrophosphatase
Rate equations for the uridylylation and deuridylylation activities have been developed by Bruggeman et al. (44). These activities are highly complex in terms of their regulation by glutamine and unmodified and uridylylated forms of GlnB.
Interactions with GlnB and GlnK.
GlnB can be modified covalently by uridylyltransferase (UTase), resulting in GlnB-UMP and PPi while starting from GlnB and UTP (Table 5). UTase can also use other nucleotides, like CTP, GTP, and ATP, to modify GlnB (127). The preferences for these substrates can be estimated by calculating the so-called specificity constants (kcat/Km) (132). The (calculated) constants are 2,200, 170, 21, and 16 mM · min−1, respectively (127), and thus, UTP is by far the preferred substrate. In addition, Jiang and coworkers (127) showed that GlnB-AMP, and GlnB-CMP could activate the deadenylylation reaction of GS-AMP catalyzed by ATase but not as well as GlnB-UMP, which is known to be an essential activator for this reaction (133). In addition, GlnB-AMP and GlnB-CMP did not, like GlnB-UMP, activate the NRII phosphatase activity (127).
Like ATase, UTase is an ambiguous enzyme: it catalyzes both uridylylation and deuridylylation but as two distinct reactions. However, both the uridylylation and deuridylylation reactions of UTase are (indirectly) stimulated by 2-oxoglutarate and ATP. The latter molecules affect the reactions by binding to GlnB and GlnB-UMP (and not to UTase itself) (127, 134). This is in contrast to glutamine, which stimulates the deuridylylation reaction but inhibits the uridylylation reaction (127). As the activation constants of the UT and UR activities of UTase for 2-oxoglutarate (5 to 30 μM) are at least 10-fold lower than the 2-oxoglutarate concentrations in intact cells (0.5 to 10 mM) (135), the in vivo uridylylation state of GlnB may well be independent of variations in the 2-oxoglutarate concentration. It was therefore concluded that the UTase-GlnB monocycle may function as a glutamine rather than a 2-oxoglutarate sensor (127, 136).
Mg2+ is the metal ion cofactor for both activities of UTase (127). Deuridylylation of GlnB-UMP is a hydrolytic reaction that results in the release of UMP (127, 137). Because this is not a phosphorolytic reaction, like the deadenylylation reaction (Table 4), but rather a hydrolytic reaction (Table 5), the sum of the two UTase reactions is energetically more costly than that of the two ATase reactions. As with ATase, the uridylylation reaction is not the reverse of the deuridylylation reaction (for a comprehensive analysis of the kinetics of the two activities of UTase with GlnB as the substrate, see reference 127).
Like GlnB, GlnK can be modified by UTase (138–140). However, the products (and substrates) of both the uridylylation and the deuridylylation reactions are still unknown, but they would probably be the same as those for the reactions with GlnB as the substrate (Table 5), because of the homology of GlnK with GlnB.
GlnB
It was observed by Stadtman and coworkers that the deadenylylation reaction of GS-AMP was catalyzed by the combined action of two protein components (PI and PII) (141, 142). Those authors concluded that the PI component was the catalyst (i.e., the enzyme adenylyltransferase [see above] [i.e., PI = ATase]) and that the PII component was probably involved in regulating the activity of PI. The PII protein is encoded by the glnB gene. The gene product (GlnB) consists of 112 amino acids (143–146), and here we refer to PII as GlnB. Tyrosine 51 of GlnB is modified by the uridylylation reaction, as has been shown by biochemical analysis and site-directed mutagenesis (145, 147, 148). From results based on gel filtration (124) and combined native and SDS-polyacrylamide gel electrophoresis, it was concluded that GlnB consists of four identical subunits (149). However, after sedimentation equilibrium analysis (150) and preliminary X-ray diffraction analysis (150, 151), it was concluded that GlnB was a homotrimer instead of a homotetramer. This was confirmed by resolving the structure of nonuridylylated GlnB at a high resolution through X-ray diffraction analyses (152, 153). Most of the residues in the three monomers pack into a rigid, squat barrel, approximately 5 nm (50 Å) in diameter and 3 nm (30 Å) high, consisting of three tightly interlocking β-sheets surrounded by α-helices (an interlocking double βαβ-fold). The most distinctive features of the trimer are the three large (flexible) 20-residue loops (T loops), which protrude from the compact main body of the molecule. Tyrosine 51 is located at the apex of each of the T loops, placing the hydroxyl group of tyrosine 51 approximately 1.3 nm (13 Å) above the flat surfaces of the barrel, making this residue readily accessible for uridylylation. In the trimer, all three subunits may be uridylylated, and thus, in the cell, GlnB may exist in at least 4 different forms (GlnB3-UMP0–3), depending on the nitrogen status of the cell. In addition, each monomer has small B and C loops. A cleft is formed between neighboring monomers, i.e., between the B, C, and T loops (152). The importance of the T-loop structure for the interaction with ATase, UTase, or NRII was shown by reducing the length of the T loop by 7 amino acids (the apex of the T loop including Tyr51). This completely abolished the interaction of the resulting GlnBΔ47–53 variant with all three target proteins, as was concluded from activity measurements in vitro (148). This result suggested that (at least one of) the deleted amino acids, or the presence of the intact T loop, are necessary for these interactions. However, the deleted amino acids of the T loop were not necessary for the binding of ATP or 2-oxoglutarate (148). Single-amino-acid substitutions within the T loop resulted generally in a reduced interaction of the GlnB variants with ATase, UTase, or NRII (147, 148).
GlnK
GlnK is the most recently discovered member of the GS regulatory system (139, 154, 155). The glnK gene forms an operon together with the amtB gene. glnK, like glnB, codes for a product of 112 amino acids (12.2 kDa), and GlnK is 67% identical to GlnB (139, 155). In addition, GlnK, like GlnB, contains tyrosine 51. Indeed, it has been concluded from the absence of uridylylation of GlnKY51N (a GlnK variant in which Tyr51 is replaced by Asn) and its analogy with GlnB that Tyr51 may be the site of modification (138). The amino acid sequence of GlnK is highly conserved among known GlnB(-like) proteins, including those present in nonenteric bacteria (25, 139). GlnK, like GlnB, is a homotrimer, and its three-dimensional structure is similar to that of GlnB, including the B and C loops and the three large protruding T loops, as deduced by X-ray crystallographic analysis (156). An X-ray diffraction analysis of the E. coli GlnK-ATP cocrystal revealed binding of ATP in a cleft on the side of the molecule to the Walker A consensus motif present in the B loop. This binding could influence the structure of the flexible T loop (156). The binding site for 2-oxoglutarate, which is expected to be present in all GlnB(-like) proteins, has been difficult to identify.
Recently, however, it was convincingly shown by X-ray crystallographic analysis (157) that 2-oxoglutarate binds at a site near the ATP binding site of GlnZ, which is a GlnK-like protein (25, 158) present in the proteobacterium Azospirillum brasilense. This makes sense, because binding of ATP and 2-oxoglutarate is synergistic, as has been shown for GlnB (29, 127, 134), and this may also be the case for GlnK.
Although the base and ribose of ATP bind in a similar way to the E. coli GlnB and GlnK proteins, the binding of ATP to the two paralogs differs in the way in which the phosphates interact with the proteins (159; see references 26 and 160 for reviews about novel structural insights of GlnB-like proteins). The different way of binding ATP may be an explanation for the difference in the activities between GlnK and GlnB. Indeed, GlnK and GlnK-UMP are less potent than GlnB and GlnB-UMP in activating the adenylylation and deadenylylation reactions, respectively (138, 139, 161). In addition, GlnK is less potent than GlnB in stimulating the phosphatase activity of NRII (138). Thus, in regulating GS activity, GlnB is the principal signal transducer and regulator. As described above, the UTase-GlnB couple can be perceived as an efficient glutamine-sensing device. Although GlnK is almost as readily uridylylated as GlnB, deuridylylation of GlnK-UMP is about 10 times slower than that of GlnB-UMP (138), implying that the UTase-GlnK couple might be a worse device in sensing glutamine. However, on the basis of in vivo experiments, Atkinson and Ninfa suggested that the reversible modification of GlnK was rapid (162). Also, in vivo complex formation between AmtB and GlnK, which is dependent on the uridylylation status of GlnK, is reversibly and rapidly adjusted to the internal glutamine pool (163).
Nitrogen Regulator II
Structural aspects.
NRII and NRI together constitute a two-component system consisting of a sensor/transmitter and a receiver/response regulator protein (for reviews concerning two-component systems, see references 164–166). NRII alone can act as a kinase or a phosphatase of NRI, i.e., a positive or negative regulator for glnA expression, respectively, while it is a phosphatase in the presence of GlnB or GlnK (133, 138, 167–169). The glnL (ntrB) gene translates to a protein of 349 amino acids (34). It has a molecular mass of approximately 36 kDa per monomer and forms homodimers in solution (170). NRII consists of three domains (171), i.e., a nonconserved amino-terminal domain of approximately 120 amino acids (the sensor module); a middle domain, which contains the dimerization determinants, consisting of two helices; and a carboxy-terminal domain. The latter two domains together, also called the kinase/phosphatase domain (the transmitter module), are approximately 230 amino acids and possess three regions that are highly conserved among transmitter proteins of two-component systems (171, 172). The helix in the middle domain close to the N-terminal domain includes histidine 139, the site of autophosphorylation (173). The C-terminal domain includes the putative nucleotide binding site (174–176). After dimerization, the two helices of each monomer may together form a four-helix bundle (176). Analysis of many NRII variants in vivo revealed that NRII variants that could not activate glnA expression also behaved as phosphatases in the absence of GlnB (174). Because of this strong correlation, it was proposed that the mechanism of regulation by GlnB may be to shift the equilibrium between the kinase and phosphatase conformations of NRII toward the latter; thus, GlnB is essential for the phosphatase activity of NRII but not for the kinase activity (174). The decreased kinase activity of some other NRII variants (174) was shown to be the result of a disabled autokinase activity (177). It was suggested that the sensor module controls the switch between the kinase and phosphatase activities of the transmitter module by influencing the interaction between the middle domain and the C-terminal domain in the transmitter module (172).
Interactions with GlnB (GlnK).
An X-ray crystallographic analysis of the C-terminal domain of NRII revealed that its three-dimensional structure shares characteristics with other sensor proteins, except for a novel β-hairpin (176). This structural element may be involved in binding of GlnB to NRII because substitutions in the vicinity of this β-hairpin reduced the binding of GlnB to NRII, as has been shown by cross-linking studies (178, 179). The binding of GlnB to the kinase domain of NRII was shown to occur in an ATP- and 2-oxoglutarate-dependent manner (179). In analogy with GlnB, GlnK may also bind to the β-hairpin of NRII.
Nitrogen Regulator I
The glnG (ntrC) gene translates to a protein that has been named NRI. It consists of 468 amino acids (34), has a molecular mass of approximately 55 kDa per monomer, and forms dimers in solution (180). NRI harbors three domains (181). The first domain is an amino-terminal domain, also called the receiver domain, of approximately 120 amino acids, which is conserved among receiver proteins of two-component systems and contains the conserved Asp54, the site of phosphorylation (182). This domain is essential for transcription activation (183). The second is a central domain that contains the activation plus oligomerization determinants (184). The third domain is a carboxy-terminal domain that harbors a helix-turn-helix DNA binding motif and dimerization determinants (185, 186). Besides binding to DNA and dimerization, a third function for the C-terminal domain might be phosphorylation-independent oligomerization (187). The central activation domain has an ATPase activity that is essential for activation of transcription (188, 189). The amino-terminal and central domains are linked together by a flexible Q linker (101). Transcription activation by wild-type NRI-P in vitro (190) and phosphorylation of wild-type NRI at D54 in vitro require Mg2+ (168, 191). Replacement of aspartate 54 of NRI by various other amino acids abolished phosphorylation of NRI, confirming that D54 is the sole site of phosphorylation (192, 193). Replacement of D54 by glutamate activated the protein, presumably by mimicking the aspartyl-phosphate entity (192, 193); perhaps, the negative charge of the carboxy group of glutamate may be at the same distance from the α-carbon as the negative charge of the phosphate group of aspartyl-phosphate. However, the corresponding receiver domain (D54E) fails to bind Mg2+. Thus, because full-length NRI (D54E) can activate transcription, it has been concluded that Mg2+ does not change the structure of the receiver domain necessary for intramolecular signal transduction and activation of NRI (194). Similar to NRI (D54E), an adduct of NRI with beryllofluoride (BeF3), prepared in situ, could mimic phosphorylation of NRI and activate transcription (195).
NMR spectroscopy of the nonphosphorylated receiver domain of NRI of S. Typhimurium, without Mg2+, revealed a structure comprised of a central five-stranded parallel β-sheet surrounded by five α-helices, a (β/α)5 topology (196). NMR spectroscopy of the nonphosphorylated receiver domain (D54E) of NRI of S. Typhimurium and those of additional constitutive mutants (194), selected as suppressors of growth defects caused by glnL-negative mutants (184, 197), revealed huge structural changes compared to the wild-type nonphosphorylated receiver domain, in a region from α-helix 3 to β-strand 5, the so-called “3445 face” (for α3, β4/α4, and β5) (194). Thus, the 3445 face and the active site appear to interact (194). The importance of this 3445 face has been confirmed by NMR spectroscopy of (i) a transiently phosphorylated switch in the receiver domain of NRI using a high concentration of a low-molecular-mass phosphoryl molecule (198) and (ii) the beryllofluoride adduct of the receiver domain of NRI (199).
Constitutive E. coli mutants, which were isolated in a way similar to that described above for S. Typhimurium, had amino acid substitutions in the 3445 face and in α-helix 5 (200). Both types of E. coli NRI variants exhibited ATPase activity, could bind specifically to the promoter without phosphorylation, and could activate transcription in vitro. Moreover, apart from the involvement of the 3445 face in interdomain signal transduction, a second interaction was detected, where α-helix 5 of the receiver domain interacts with the central domain (200).
As mentioned above, the C-terminal domain contains the helix-turn-helix DNA binding motif. Replacement of three hydrophilic residues in the second helix of the helix-turn-helix motif by alanines resulted in a variant of S. Typhimurium NRI that did not bind DNA but which was structurally and functionally intact (186). Surprisingly, at concentrations of 100 nM or higher, this phosphorylated variant activated transcription from the glnAp2 promoter. This activation was similar by using templates with or without an enhancer. Thus, the roles of the enhancer can be bypassed if the protein is present at high concentrations in solution (201).
Nitrogen Assimilation Control Protein
Nac is a transcriptional regulator. It is present in the enteric bacteria E. coli (NacE) and K. pneumoniae (K. aerogenes) (NacK) but is absent from S. Typhimurium (202). So far, the main focus of research has been on the Klebsiella Nac system (for reviews, see references 203 and 204).
The translated gene product of both nacE and nacK counts 305 amino acids (202, 205). The molecular mass of NacE (33 kDa) (202), expressed from a temperature-inducible phage λ pL promoter located on a vector, is similar to that of purified NacK (32 kDa) (206), as assessed by SDS-polyacrylamide gel electrophoresis. NacE could not be purified with the same methods used for NacK, because it was insoluble under all conditions tested; furthermore, it was degraded rather fast, with a half-life (t1/2) of only 15 min at 30°C (202). NacK is a homodimer in solution at a concentration below 5 to 7 μM (203, 207). However, at higher concentrations, Nac eluted as a tetramer, as determined by fast protein liquid chromatography (FPLC)-mediated gel filtration (207), suggesting that the tetramer is a complex of two dimers. NacE was insoluble. However, using FPLC, a maltose binding protein (MBP)-NacE fusion was monomeric, but NacE was dimeric after cleavage of MBP (208).
Although NacE and NacK have exactly 305 amino acids, the amino acid sequence identity was only 79%, with most of the divergence in the C-terminal two-thirds of the protein (202). The N-terminal 100 residues of NacE and NacK, which may contain a helix-turn-helix motif (see below), turned out to harbor many known properties of Nac in vivo and in vitro, such as (i) the activation of hut; (ii) the repression of gdh; (iii) the ability to bind DNA, as assessed by a gel mobility shift assay; and probably (iv) carrying determinants of dimerization (208). Indeed, NacK variants with the C-terminal domain deleted were still dimers in solution, as assessed by FPLC (207). In addition, some Nac proteins having a point substitution in the C-terminal domain, e.g., NacL111K, were dimers in solution instead of tetramers, suggesting that the tetramerization determinant is located in the C-terminal domain (207).
Nac is a member of a large family of highly conserved LysR-type global transcription regulators, acting as either activators or repressors of single or cistronic genes (for reviews concerning LysR-type regulators, see references 209 and 210). Structurally, they contain a coeffector binding domain at the C terminus and a helix-turn-helix motif at the N terminus, which enables binding to promoter DNA (209, 210). The minimal binding consensus sequence of NacK, as determined by DNase I footprint and mobility shift assays, seems to be the 15-nucleotide consensus DNA binding site 5′-ATA-n9-TAT-3′ (the first and last 3 nucleotides form a dyad symmetry) (206, 211). When NacK was cloned downstream of an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter, the activity of hut and gdh, genes regulated by NacK, depended only on IPTG, even when cells were grown with excess ammonium (212). The authors of that study suggested that apparently no coeffector or modification of NacK was required for regulation (212).
Indeed, binding of partially purified NacK from cells grown under conditions of nitrogen limitation or nitrogen excess shifted hutP DNA equally (206). In addition, potential nitrogen coeffectors, like glutamine, glutamate, 2-oxoglutarate, and ammonium at 10 mM, had no effect on shifting hutP DNA by NacK (206). Using gel mobility shift assays, it was shown that wild-type NacK (NacWTK) and NacL111KK shifted the Nac-activated promoters phutU and pureD (for histidine and urea utilization, respectively) as dimers, with protected regions in DNase I footprint analyses of 28 bp (206, 207). However, NacWTK and NacL111KK bound to the repressed promoters pnac and pgdhA as a tetramer and a dimer, respectively (207). DNase I footprint analysis of NacWTK bound to the pnac promoter showed that it protected two adjacent regions of approximately 26 bp, each with a region of hypersensitivity between them, while NacL111KK protected only one region (207). The region of hypersensitivity may be the result of bending of the DNA by bound Nac, as has been determined by gel mobility shift assays (207, 213). In addition, NacL111KK (dimer) failed to repress gdhA, while NacWTK (tetramer) strongly repressed gdhA (207). Like most LysR-type transcriptional regulators (LTTRs), tetrameric NacK may adopt a compact or an extended conformation that recognizes short or long DNA binding sites, respectively. Unlike most members of the LysR family, the conformation is not determined by the intervention of a coeffector (a small metabolite) but by the nature of the DNA site (214).
cAMP Receptor Protein
Crp (also known as CAP [catabolite activator protein] or CGAP [catabolite gene activator protein]) is a global transcriptional regulator that controls approximately 400 mono- or multicistronic operons in E. coli (215, 216; for reviews about Crp, see references 217 and 218). Crp plays a regulatory role in carbon source utilization. There is some evidence that it might also be involved in the regulation of nitrogen metabolism. This is described in the section below about transcription regulation of glnALG. Here general and structural knowledge is presented.
Crp is encoded by the gene crp, which yields a product of 209 amino acids (219) or 210 amino acids (220). This gene product has a calculated molecular mass of 23.6 kDa (219). The molecular mass of Crp, as determined by denaturing gel electrophoresis, was determined to be 23.0 kDa (219) or 22.0 kDa (221). However, Crp eluted from a G-100 Sephadex column as a 45-kDa protein (222), and thus, Crp is considered a homodimer (221, 223). Each subunit may bind one cAMP (223, 224), but Crp exhibits negative cooperativity in binding cAMP (225).
After enzymatic proteolysis of Crp in the presence of cAMP, a peptide of ∼12.5 kDa was observed, while wild-type Crp (22 to 23 kDa) disappeared. This was taken to suggest that a Crp monomer consists of two domains (226, 227). On the basis of a lack of proteolysis of Crp in the absence of cAMP by the same proteolytic enzymes, it was concluded that binding of cAMP induced a conformational change in Crp (226, 227). From the high level of similarity between the amino acid sequences of a two-alpha-helix motif at the C-terminal domain of Crp and a region at the N-terminal domain of the lac and gal repressors, it was inferred that this motif may be involved in binding DNA (228). X-ray diffraction analysis of Crp-cAMP complexes (223, 224, 229) confirmed (i) the two-domain structure of a Crp subunit and (ii) the two-alpha-helix motif as the DNA binding unit. Furthermore, the structures showed that the large N-terminal domain forms the contacts between the two Crp subunits of the dimer and binds cAMP. The small C-terminal domain contains a helix-turn-helix DNA binding motif, and both C-terminal domains are involved in binding to DNA (223).
The DNA binding consensus sequence is indeed palindromic and covers 22 bp, 11 bp on either side of the axis of symmetry. It consists of two core motifs, i.e., a 5-bp sequence, TGTGA (one motif on either side of the symmetry axis), separated by a 6-bp spacer (for an elaborate study concerning the spacer length, see reference 230). In the absence of cAMP, Crp binds nonspecifically to DNA; its affinity and specificity for the binding site are greatly enhanced by cAMP (221, 231). The crystal structure of Crp-cAMP and especially that of its two DNA binding domains fit perfectly in the major groove of left-handed B DNA (223). It has been suggested that cAMP exerts its allosteric effect by changing the relative orientation of the two subunits that allows them to bind to the DNA (223, 229). Crp requires cAMP both for specific binding to DNA and for activation of transcription, as has been shown in an in vitro transcription assay (222, 232). The Crp-cAMP1 complex is considered the relevant active form of Crp under physiological conditions (233).
Besides binding to DNA, Crp-cAMP is also able to bend DNA, as determined by nondenaturing gel electrophoresis mobility shift analyses (234, 235). Bending of the DNA in a DNA-Crp-cAMP complex has been confirmed by X-ray diffraction analyses of the complex (236, 237). Bending may increase the rate of transcription in vivo, as was suggested by the observed higher in vitro transcription rate using synthetic DNA-bending sequences at the E. coli lac promoter (238). This can then either activate or repress transcription. Activation or repression occurs if Crp-cAMP is correctly positioned with respect to the transcription start site, i.e., at bp −40 to −75 (239), or if the binding site overlaps that of the RNA polymerase or the transcription start site, respectively (e.g., see reference 240).
Leucine-Responsive Regulatory Protein
The leucine-responsive regulatory protein of E. coli is the best-studied member of the Lrp family (241) or the FFRP (feast/famine regulatory protein) family (242). Because many reviews (6, 241–244) dealing with Lrp can be found in the literature, we only summarize here.
Lrp is a small protein (∼19 kDa) (244). It consists of an N-terminal DNA binding domain, harboring the canonical helix-turn-helix motif, connected with a flexible hinge to a C-terminal ligand binding RAM (regulator of amino acid metabolism) domain (241, 242). As for Crp and IHF (integration host factor), an important function of Lrp may be to bend DNA (243). It affects transcription of at least 10% of the genes of E. coli (241). Lrp regulates the expression of genes involved in catabolism and anabolism of amino acids via repression or activation (244). In general, it regulates genes that function during famine and feast positively and negatively, respectively. Lrp often works together with other global regulators like Crp, IHF, and H-NS (the histone-like nucleoid-structuring protein) (241). Although lrp is autogenously regulated, Lrp is a moderately abundant DNA binding protein, since some 3,000 Lrp dimers are present per cell (241, 243). Also, other multimeric states are found, from tetramers and octamers up to hexadecamers (241). Determination of the structure of Lrp revealed that the octamer, which is a tetramer of dimers, is basically an open, linear structure, in contrast to the closed, octameric rings formed by Lrp homologs (245). Interconversions of the multimeric states are possibly affected by leucine, and the multimeric state determines the affinity for DNA (241, 242). Leucine indicates amino acid sufficiency, and it sometimes strongly affects the regulation of transcription of operons by Lrp (243), but in most cases, control by Lrp is leucine independent (241). Alanine is almost as effective with Lrp as leucine (243, 246). However, Lrp responds to a broader range of amino acids than was previously appreciated: responsiveness to methionine was comparable to that to leucine, and Lrp was also fairly responsive to isoleucine, histidine, and threonine (247). In relation to nitrogen metabolism, unliganded Lrp has been demonstrated to activate gltBDF and pyridine nucleotide transhydrogenase (6). Also, the operon glnALG was claimed to be positively affected, but this may be only an indirect effect due to the effect of Lrp on gltBDF transcription (243).
Nitrogen-Phosphotransferase System
E. coli possesses a so-called nitrogen-phosphotransferase system (nitrogen-PTS or PTSNtr) (248), in addition to the well-known sugar uptake system PTS (sugar-PTS) (249, 250). The nitrogen-PTS consists of a cascade of three phosphoryl-transferring proteins. The proteins are homologous to their equivalents of the sugar-PTS. Initially, the nitrogen-PTS seemed to provide a link between carbon and nitrogen assimilation, and the name “nitrogen-PTS” or “PTSNtr” appeared to be appropriate. However, in light of recent evidence (see below), it may be inappropriately named after all (251, 252), because the link of the nitrogen-PTS with the nitrogen-regulated system is limited, if present at all. However, we maintain Ntr in superscript to indicate this system (PTSNtr) and two of its components (EIIANtr and EINtr).
The rpoN gene, also known as ntrA (197) and glnF (190), codes for the σ54 factor. RNA polymerase associated with σ54 (σ54-RNA polymerase) binds to the DNA consensus sequence, with the most distinct feature of two doublets, GG (position −25/−24) and GC (position −13/−12), relative to the transcription start site (190, 253, 254). The first and second doublets are conserved in ∼100% and 96% of the sequences, respectively (253). In contrast to the housekeeping σ70-RNA polymerase, σ54-RNA polymerase requires an activator protein, such as NRI (see “Transcription regulation of glnALG,” below), to initiate transcription (254, 255).
σ54 has also been denoted σN, where the superscript N illustrates the fact that σ54 functions in transcription of nitrogen assimilation and nitrogen fixation genes such as glnA and nifLA, respectively (for a review, see reference 255). Recently, however, a comparative analysis of all sequenced prokaryotes revealed that σ54 (σN) not only engages in transcription of N-related genes but also is a central player in the control over the very many processes that involve the interaction of a microbe with its environment (256). This implies that the superscript N in σN no longer does justice to the many disparate activities in which σ54 is engaged.
The rpoN gene appeared to have a few neighboring genes that altogether constitute an operon. The genes within the rpoN operon were discovered on the basis of suppression of a conditionally lethal temperature-sensitive era [era(Ts)] mutant (257). The era gene codes for an essential GTPase, which may be important for cell division and ribosome function (for a review, see reference 258). It was concluded from an analysis of the various suppressors and other kinds of mutants which prevented the expression of ptsN that the defect caused by era was relieved by the absence of EIIANtr (nitrogen-related enzyme IIA) (257). This rpoN operon contains five genes (248), two of which code for proteins with high amino acid sequence similarity to the phosphoenolpyruvate (PEP):carbohydrate PTS proteins. The latter two proteins were designated EIIANtr (ptsN) and NPr (nitrogen-related HPr) (npr) (257). The npr gene has been renamed ptsO (259). The genes in the rpoN operon are expressed from two promoters, i.e., a strong promoter upstream of rpoN (257, 260) and a weak promoter upstream of the ptsNO genes (257). Transcription of the rpoN gene (operon) was found not to be regulated by nitrogen (257, 261, 262), which, in hindsight, is not so remarkable anymore in light of the involvement of the sigma factor in all kinds of bacterium-environment interactions (256). A third gene that coded for a protein with homology to a PTS component was ptsP. It was not part of the rpoN operon (257) but has been localized elsewhere on the E. coli chromosome by sequence analysis (259). The ptsP gene encodes a protein with homology to the EI protein and has been named EINtr, making the set of three PTS homologs complete (nitrogen-PTS).
The nitrogen-PTS, like the sugar-PTS, uses PEP as the high-energy phosphate input substrate. Phosphoryl transfer in the nitrogen-PTS has been shown to proceed in vitro (263) and in vivo (264) from PEP to EINtr to NPr to EIIANtr, i.e., just like the phosphorelay observed for the sugar-PTS. His73 is the single site phosphorylated in EIIANtr in vivo (264). In strong contrast to the sugar-PTS, however, phosphoryl transfer from EIIANtr-P to a specific small-molecule target has not been reported so far (248). However, dephosphorylation of EIIANtr-P is expected to occur, and perhaps, the exchange of the phosphoryl group via cross talk with the sugar-PTS may have to be considered in this respect.
Cross talk between the nitrogen-PTS and the sugar-PTS has been shown to be possible in principle. In vitro, phosphoryl transfer from HPr-P to EIIANtr (257, 265) and from EAII-P to Npr (257) has been demonstrated. However, phosphoryl transfer in vitro between the first two proteins of the two systems is strictly phosphorelay dependent: EINtr-P prefers NPr as an acceptor, and likewise, EI-P has a preference for HPr, and the two pairs have little cross-reactivity (257, 263). In vivo, EI and Hpr as such are each separately capable of phosphorylating EIIANtr (264).
Ammonium Transporter
AmtB belongs to the Amt/MEP/Rh (rhesus) membrane superfamily of ammonium transporters, members of which are widespread in organisms of all domains of life (266). Its ubiquity has given rise to quite a vivid field of research. The AmtB/MEP/Rh proteins have been studied extensively using a variety of methods: site-directed mutagenesis, whole-cell and vesicle uptake assays, crystallography, electron microscopy, atomic force microscopy, and molecular dynamics simulation have shed light on the mechanistic, structural, and kinetic behaviors of the transporter proteins. It is striking that in spite of this wealth of experimental data, no consensus view on several important aspects of ammonium transport has emerged yet. This may become understandable by acknowledging the observation that highly similar transport proteins of the same family can display fundamental mechanistic differences (267).
Membrane topology.
The amtB gene encodes a preprotein with a signal peptide that is cleaved off upon membrane insertion. Deletion of the signal peptide results in lower levels of AmtB accumulating in the membrane (268). The AmtB monomer consists of 11 trans-membrane-spanning α-helices, with the N-terminal and C-terminal domains located in the periplasm and cytoplasm, respectively (269, 270). The AmtB protein consists of 428 amino acids (139), but in E. coli, the 22 N-terminal residues are removed upon insertion of the protein into the membrane (271). The first and second five trans-membrane segments form structurally homologous domains, whereas the 11th segment is not part of this domain structure (272). Upon purification, the protein is obtained as a homotrimer, even in a detergent solution (273). The trimeric configuration of the protein was confirmed by electron and atomic force microscopy (274) and X-ray diffraction analysis (271). AmtB is a trimer of three channels, each with 11 trans-membrane-spanning α-helices forming a right-handed helical bundle around its channel (271, 275). Each channel consists of an extracellular vestibule, a narrow hydrophobic region including two in-line histidines, and an intracellular exit pore (271, 275).
A single file of NH3 molecules was suggested to be present in the hydrophobic core but only when the crystals were formed in the presence of ammonium (271). However, electronic density was also observed in the pore in the absence of ammonium (275). Furthermore, the hydrophobic pore was able to stabilize a file of water molecules (276). The presence of water in the channel could challenge the dominant view that NH3 is conducted through the pore (277).
There is general agreement on the presence of four distinguished binding sites on AmtB, i.e., site 1 (S1), S2, S3, and S4. S1 most probably constitutes the binding site for NH4+. S2 is the first site in the pore after passage of the so-called “Phe gate.” S3 and S4 are sites lower down in the pore. Furthermore, two more sites were defined, S5 and S6; they are both located in the cytoplasmic vestibule and have been suggested to play a role in the protonation state of NH3 (278–280).
Recruitment of ammonium.
AmtB is expressed only if no or very little ammonium is available in the environment (139, 281–283). By inference, a high-affinity binding site for ammonium would seem to a prerequisite for AmtB functioning under these conditions. S1 is located at the bottom of the periplasmic vestibule. It is the putative binding site for ammonium, although the NH4+ ion itself has not been observed on the spot yet; only thallium (Tl+) has been observed (284). W148, F107, and S219 are envisioned to form a highly selective cage for ammonium, which would not readily be accessible to NH3, CO2, K+, Na+, or water (284–287). Methylammonium, imidazole, and Tl+, however, are considered to be able to bind to S1 (284). Residue W148 is thought to be an important part of an aromatic cage around the NH4+ ion (284, 285). However, tryptophan was not essential for recruitment of NH4+ and could be replaced with leucine (288). Also, aspartate (D160) has been considered the most important amino acid for recruiting NH4+ (287), but evidence contrary to this finding has also been presented (285).
The dynamic Phe gate and the hydrophobic pore.
Two phenylalanines are considered to separate the periplasmic vestibule from the hydrophobic pore. Together, they form a dynamic intramolecular gate through which ammonium might gain access to the hydrophobic pore (284). The phenyl rings of residues F107 and F215 block the entrance of the pore. The partially stacked phenyl rings flip open and closed synchronously at a high frequency (285). F215, but not F107, was found to be absolutely required for AmtB function, and removal of the gate produced an open but inactive channel (284). In complete contrast, individual or simultaneous replacement of the phenylalanines by aliphatic residues was found not to cause a loss of AmtB function (289).
Fong et al. assigned a particular role to W148, which, besides being part of the aromatic collar, would also restrict the entry of (methyl)ammonium into the channel, probably together with the Phe “flap” (288).
Below the Phe gate, a narrow hydrophobic pore, some 20 Å long, is present (271, 290). Because of the hydrophobicity of the pore, NH4+ is not usually considered to be the permeating species. A lower pKa for NH3 (<6) would prevail inside the channel at S3 and S4, thereby stabilizing the movement of the NH3 released inward to and through the channel (271). In addition, two histidine residues in line with the channel, but with their imidazole rings protruding into the channel, might help to conduct NH3 through the channel (271, 290). They would be absolutely required for optimum conductance (290). Fourteen variants of the twin histidines were shown to be inactive, except one. In the latter case, substituting the first histidine for glutamate decreased AmtB activity to 25% (290). However, in another study, replacement of one or two of the conserved histidine residues with acidic amino acids did not result in a significant loss of AmtB activity (289). It was concluded that the twin histidines are not required for AmtB function but instead serve to optimize its performance. Also, changing the first histidine to a glutamate caused an upshift of 2 pH units of the optimal pH, from 4 to 6, for methylammonium uptake in cells (267). In a molecular dynamics study of (methyl)ammonium transport, it was concluded that conduction through the pore is facilitated by the hydrogen bond network comprised of both histidines and Tyr32 in coordination with π interactions with Trp212. In particular, Trp212 would be important for removing NH3 from its strong hydrogen bonding to His168 (291).
Deprotonation of ammonium.
If NH4+ binds to AmtB and NH3 is the species that is conducted through the pore, an important issue is where exactly along the pathway after ammonium binding the proton would be released and what the proton acceptor and the final destination of the proton would be. This is because the answers have strong consequences for the intracellular ammonium level and for the energetics of the transporter (see “Passive versus active transport,” below).
Unfortunately, to date, no unambiguous answer can be found in the literature. Amino acids at different positions in the monomer have been implicated to play a role in deprotonation. Some argue that deprotonation might take place in the periplasmic vestibule via small basic molecules such as carbonates or phosphates (283) or at S219 (292). Conflicting suggestions with respect to the involvement of the aspartate residue (D160) in deprotonation of ammonium have been uttered. Some argue for a direct involvement (293, 294), while others suggest no involvement at all (279, 285). Also, the twin histidines have been considered to play a role in deprotonation (290). Nevertheless, the site most frequently mentioned in this respect is the phenylalanine gate (278, 279, 284, 285, 294–297). Thus, currently, a preference for the phenylalanine gate as the site of deprotonation seems to develop. On the other hand, (de)protonation reactions are fast in almost any environment, such that no catalytic group may be needed.
However, accepting this as the consensus view, it is still not clear what happens with the released proton. Is it (i) transferred to a residue close to the gate (His168) and from there moved “downwards” (i.e., toward the cytoplasm) on the electric gradient powered by the transmembrane electric potential difference in live E. coli cells, (ii) translocated forward into the pore lumen, or (iii) extruded back into the periplasmic vestibule? At present, there is no agreement on this point: authors in the field are in favor of the first (e.g., see reference 284), the second (e.g., see reference 276), or the third (e.g., see reference 285) possibility. Still others disagree with the above-described general view and claim that deprotonation is not at stake, because NH4+ is the species that permeates the hydrophobic channel (288). We note that all the evidence is based on inference from structural information. There is no direct evidence where NH4+ rather than NH3 has actually been observed to reside in the channel or to move through the channel.
Passive versus active transport.
The topic of passive versus active transport of ammonium by the AmtB/MEP/Rh transporter family is vividly debated in the literature (e.g., see reference 298), but here we focus on the controversy in E. coli (see transport modes in Fig. 2).
Fig 2.

Four modes of ammonium transport. The beige rounded rectangle represents the AmtB carrier. Four modes (mechanisms) of transport are shown. (A) Transport via passive diffusion of NH3 (Sdif); (B) facilitated transport of NH3 (Sfac); (C) active transport of NH3, corresponding to facilitated transport of NH4+ (SH+); (D) active transport of NH3 where NH3 and the symported H+ follow separate routes (S/H+). In panels A and B, H+ shown at the vertical arrows denotes production or consumption of scalar protons.
If the released proton ends up in the periplasm (mode Sfac), transport would be characterized as diffusion of NH3 facilitated by AmtB, but if the proton is symported somehow along with NH3 to the cytoplasm (modes SH+ and S/H+), it would count as active transport. In the former case, no accumulation of NH3 inside the cell could ever occur, while in the latter case, substantial intracellular accumulation of NH3 could be accomplished. In all transport mechanisms, the issue should not be merely to come with a mechanism of NH3 or NH4+ transport but also to come with a mechanism that prevents the transport of protons or hydroxyl ions independent of the movement of ammonium. Such uncoupled transport would cause dissipation of the proton motive force, a most important free energy potential in E. coli.
On the basis of uptake studies with AmtB-containing liposomes, it was concluded that the gas NH3, instead of the ion NH4+, is conducted through the channels (271), but the experimental results could not be reproduced, despite extensive efforts (299). Although movement of NH4+ as such through the hydrophobic pore of E. coli AmtB seems to be excluded by most scientists (see above), and conductance of the uncharged NH3 through the channels of AmtB may be the consensus view, this does not necessarily imply that AmtB is a passive NH3 facilitator and that the NH3 gradient is the sole driving force for uptake. NH3 transport might be coupled energetically to the migration of a proton to the cytoplasm via a microscopically distinct but mechanistically coupled route (276, 277, 300). Potentially, this could take place via the two conserved histidine residues or via a file of water molecules in the pore. Alternatively, the movement of NH3 might be coupled to the movement of another base, i.e., hydroxylate in the opposite direction. Because of their affinity for protons, binding sites for hydroxylate and NH3 might be similar. In either case, ammonium uptake would be active and driven by the chemical potential difference for NH3 and the electrochemical potential difference for protons (277, 300). A systems biology approach has been used to suggest that AmtB-mediated ammonium transport must be active (301). The authors of that study combined the observation that AmtB is expressed only in cells growing under nitrogen-limited conditions with the fact that GS has a relatively high Km for NH4+ to infer that AmtB-mediated passive diffusion of NH3, no matter how much stimulated by AmtB, would not result in sufficiently high intracellular NH4+ concentrations to enable GS to reach a flux compatible with growth of E. coli at the observed growth rates. Here the limitation was essentially provided by the thermodynamic point that the intracellular concentration of NH3 could never exceed its extracellular concentration when the nitrogen flux should be inward. This argument becomes particularly strong with low extracellular ammonium concentrations and acidic extracellular pH, where E. coli can still grow with ammonium as the nitrogen source.
Cytoplasmic vestibule and cytoplasmic C-terminal region.
The cytoplasmic vestibule and cytoplasmic C-terminal region (CTR) of AmtB have been studied the least, and much remains to be elucidated. If mode Sfac or mode S/H+ (Fig. 2) is correct, NH3 may be protonated to form NH4+ in the cytoplasmic vestibule. Where exactly this happens is unknown so far. Several options are available, however. The proton could be delivered by water molecules present in the cytoplasmic vestibule or by some amino acid residues that form part of the exit pore. Indeed, Ser263 has been implicated in the translocation of NH3 from the exit gate into the cytoplasm by hydrogen bond interactions (291). Alternatively, in the case of the S/H+ mechanism, one idea is that the cotransported proton rejoins with the NH3 molecule just after passage through the pore at His318.
Based on genetic evidence, an interesting idea was put forward that the C-terminal tail of AmtB facilitates an oscillation of transmembrane segment 5 (TM5) that controls the opening of both the periplasmic gate and the cytoplasmic exit (302). It is tempting to speculate that the synchronized flipping of the Phe gate (285) is related to TM5 oscillation.
The role of this region of some 25 residues is not known precisely. The structure of the CTR was not resolved in the X-ray structures of E. coli AmtB (271, 275). Deletion of the entire region substantially reduces AmtB activity to an intermediate (25 to 30%) level (269, 302, 303) and prevents association with GlnK (303). The addition of PhoA or LacZ peptides to the C terminus completely abolishes methylamine (MA) transport activity (270). Site-directed mutagenesis was used to study the role of the cytoplasmic C-terminal region. Nearly all mutations significantly impaired AmtB activity. The mutants fell into two phenotypic classes: one that had a phenotype comparable to that of the deletion of the entire region and another that exhibited virtually no AmtB activity (<7%). It was concluded that the CTR plays a significant role in AmtB activity and possibly mediates cooperativity between the three AmtB subunits (269, 302). Notably, complete inactivation of AmtB was shown to require participation of the HflB protein, which leads to incorrect folding of mutated AmtB (676).
Inhibitors of ammonium transport.
Uptake of MA (at 20 μM) in unwashed assay mixtures has been shown to be very sensitive to ammonium, already at submicromolar concentrations of the latter (Ki = 0.5 μM [304] or ∼0.8 μM [305]). Also, Tl+ was shown to be a relatively effective inhibitor (Ki of 5.3 μM [304] and full inhibition at 0.5 mM [284]). Cs+ and imidazole are weak inhibitors (10 to 100 mM) (284). All of these inhibitors may compete for the binding site for ammonium (S1) in the periplasmic vestibule of AmtB.
Several other ions and compounds that have been shown to inhibit MA uptake may not compete for the ammonium binding site. Conflicting results were presented for K+, which would be a noncompetitive inhibitor (Ki = 1 mM [304]) or not an inhibitor at all (at 50 mM [284]). Na+ (50 mM) was not inhibitory (284). l-Glutamine (Ki = 18 μM) (but not d-glutamine or glutamate), glycylglycine (Ki = 43 μM), and the glutamine analogs γ-l-glutamyl hydroxamate (Ki = 0.4 mM) and γ-l-glutamyl hydrazide (Ki = 0.8 mM) all showed noncompetitive inhibition of MA transport (306). Using washed assays, i.e., uptake assays with a washing step, 10 to 25 μM ammonium was shown to partially inhibit MA uptake in E. coli (307) and 50 μM ammonium or 50 μM l-glutamine was found to inhibit MA uptake completely (308).
The fact that glutamine inhibits MA uptake, although at higher concentrations than those required for inhibition with ammonium, deserves some more explanation. In contrast with ammonium, glutamine did not instantaneously inhibit MA uptake, as the t1/2 of the rate of onset of inhibition was 3 min. Furthermore, inhibition did not occur when a mutant with a defective glutamine transporter was used. The conclusion then was that ammonium transport was regulated by the internal glutamine pool via feedback inhibition (306).
AmtA is not involved in ammonium transport.
A mutant with <10% of the ammonium transport activity of the wild type has been isolated and characterized (309). The corresponding gene (amtA) had been cloned, sequenced, and localized (310, 311). First, the gene product was suggested to be a periplasmic component of an ammonium transport system (309), but later, it was predicted to be a cytoplasmic component (310). However, it has been convincingly shown that the cysQ gene is identical to this amtA gene and that CysQ helps to prevent accumulation of the toxic metabolite 3′-phosphoadenoside 5′-phosphosulfate (PAPS), which is an intermediate in the conversion of sulfate into sulfite (312). Purified CysQ was shown to exhibit NADP(H) phosphatase and fructose-1,6-bisphosphatase activities as well (313), the reactions of which bear no relation with ammonium transport. Therefore, most probably, amtA (cysQ) has no role in ammonium uptake.
Glutamine Transporter
There is firm, direct evidence for the existence of a high-affinity inward ABC transporter for glutamine. The transport system has been relatively well characterized, especially when it concerns the structure of the periplasmic binding protein and the dynamics of physiological (see “Growth with excess glutamine as the N source,” below) and transcription [see “Transcription regulation by and of other nitrogen assimilation-associated genes. (v) The glnHPQ operon,” below] regulation. There is also some indirect evidence for the presence of a low-affinity transporter (e.g., see references 314 and 315). However, little is known about the latter transporter, and even more than one such low-affinity permease may exist. We further focus on the high-affinity glutamine transporter.
In the 1970s, evidence had already accrued for the presence of an osmotic shock-sensitive periplasmic binding protein for glutamine. Both the specificity and the binding affinity of glutamine for the purified protein were high (315–317). It was demonstrated that glnH was the structural gene for the periplasmic binding protein (318) and that the N-terminal 22 residues constitute a signal peptide (319). GlnH is a monomeric protein with a molecular mass of 24 kDa (315, 319). The glnH gene was shown to belong to the glnHPQ operon (319). The glnP gene codes for the membrane-bound glutamine permease, and the glnQ gene codes for the ATP-hydrolyzing component of the ABC transporter system (320). Together, they enable active transport of glutamine into cells with a high specificity.
Whereas the periplasmic binding protein has been quite well characterized, virtually nothing is known about the other two components. The structure of the GlnH (also called GlnBP) protein and the GlnBP-Gln complex has been studied extensively, but we address only some of these data. The binding protein exhibits open unliganded and closed liganded states (321). GlnH possesses two functional domains (termed large and small) connected by a linker region; a solvent-accessible cleft between the two domains provides the glutamine binding pocket (321, 322), which is a general feature of periplasmic binding proteins (323). Two tryptophan residues at positions 32 and 220 are involved in interactions with membrane-bound components of the glutamine transporter system and are both required for optimal transport of glutamine but are not essential for its binding (324).
Restoration of glutamine uptake by the purified binding protein was demonstrated both in membrane vesicles prepared from a binding protein mutant (325) and in spheroplasts made from wild-type cells (326). With the vesicles, binding of glutamine was insensitive to variations in ionic strength and pH (22, 65, 154, 327) and required neither potassium nor phosphate ions, whereas glutamine transport activity was inhibited by increasing ionic strength, had a narrow pH optimum, and required potassium and phosphate ions (328). Glutamine uptake into cells was inhibited by osmotic stress produced by NaCl but was stimulated when sucrose was used instead (329). Uptake in vesicles (325, 328) and in spheroplasts (326) was concluded to be energy dependent and was found to be driven by ATP in cells (330, 331). There is no evidence that it would belong to the so-called TRAP (tripartite ATP-independent periplasmic) transporters (332) that employ a periplasmic binding protein but where the driving force is the proton-motive force rather than ATP hydrolysis. The transporter is now considered an ABC transporter (320, 333). In this light, earlier observations that ATP was insufficient to drive glutamine uptake in vesicles (328) or that ATP and the transmembrane electric potential were both required for uptake (334) are difficult to interpret.
Glutaminases A and B
Glutaminases catalyze the hydrolytic deamidation of glutamine to glutamate and ammonium. It should be noted that this reaction is not the reverse of the reaction catalyzed by glutamine synthetase. If both operate simultaneously, these two reactions constitute a so-called futile cycle (see “GS plus glutaminase wasting ATP,” below). Here we discuss glutaminases as separate enzymes and not as a subunit of an amidotransferase possessing glutaminase activity (335), like carbamoyl-phosphate synthetase or asparagine synthetase B, which are both discussed in another paragraph above. The ammonium formed as a result of glutaminase catalysis is released into the cytosol.
In 1968, a major glutaminase of E. coli, in hindsight most likely glutaminase A, was isolated (336). A few years later, a second glutaminase, named glutaminase B, comprising a minute fraction of the total protein, was isolated (337). Glutaminase A is a tetrameric protein, and glutaminase B is a dimeric protein (338–340). Glutaminase B is active under neutral to mildly alkaline pH conditions, while glutaminase A remains active under mildly acidic conditions (337–339). Glutaminase B has an isoelectric point of 5.4, as determined by isoelectric focusing (340). Both glutaminases A and B are highly specific for l-glutamine. d-Glutamine, isoglutamine, l-asparagine, or d-asparagine is not deamidated (336, 340). Both enzymes have a relatively low affinity for glutamine, with the Km values being 7.3 and 31 mM, respectively. Thus, like other glutaminases, the glutaminases from E. coli combine low affinity with high selectivity (338).
Glutaminase A is particularly expressed in the stationary phase, and glutaminase B is constitutively expressed albeit at a low level, since this level was not affected at all when tested under a variety of culture conditions (337, 341). It has been suggested that both glutaminases play a significant role in glutamine metabolism (338), although the high Km value (31 mM) of glutaminase B for glutamine seems to preclude a substantial flux in E. coli at steady state, in view of recent measurements of the glutamine pool under conditions of ammonium excess, which amount to 3 to 12 mM (135, 342, 343). In transient states, glutamine may reach concentrations of up to 50 mM, however (135), and both glutaminases may then show substantial fluxes. Glutaminase A may be involved in a new system for acid resistance, while glutaminase B may be required for growth with glutamine as the sole carbon and nitrogen source (338).
Glutaminase B is a so-called cold-labile enzyme (337, 340), a feature that shows up with many regulatory enzymes. Indeed, glutaminase B activity is modulated by nucleotides and divalent cations: ATP and ADP inhibit glutaminase B, while AMP and divalent cations activate it (344). The modulation by energy charge has been implicated in preventing a futile cycle constituted by GS and glutaminase B (344).
Thirty years later, Brown and coworkers cloned two genes of E. coli predicted to be glutaminases. The genes have the systematic names ybaS and yneH, and the amino acid sequences of the two translated gene products share 38% sequence identity (338). They have been overexpressed, purified, kinetically characterized, and crystallized. The gene products of ybaS and yneH have calculated molecular masses of 32.8 and 33.4 kDa, respectively, and calculated isoelectric points of 4.6 and 6.0, respectively, according to the Colibri database (345). As the previously measured molecular masses and the isoelectric points are similar to those calculated, and the pH profiles are also similar, YbaS and YneH are most likely glutaminases A and B, respectively (338).
The two glutaminases were inhibited by low concentrations of the divalent metal cations Mg2+ and Mn2+. For YneH, the 50% inhibitory concentrations (IC50s) were 2.0 mM and 1.0 mM, respectively. However, YbaS was approximately 10 times more sensitive to these cations; i.e., the IC50s for Mg2+ and Mn2+ were 0.2 mM and 0.1 mM, respectively (338). In strong contrast, however, Prusiner and Stadtman observed activation of glutaminase B by Mg2+ and Mn2+ at concentrations similar to those that were found to be inhibitory by Brown et al. (344). The reason for this discrepancy is unknown. Either way, both glutaminases are likely affected by Mg2+, since the free intracellular Mg2+ concentration is estimated to be in the range of 1 to 5 mM (327), whereas only YbaS may be modulated by Mn2+, since the free intracellular Mn2+ concentration is probably not >0.1 mM (346).
ACTIVITIES, REGULATION, AND LIMITATIONS OF THE COMPONENTS OF THE NITROGEN ASSIMILATION NETWORK
As discussed above, protein structures and homologies have suggested binding sites for the various small and large molecules engaged in nitrogen assimilation. Together with biochemical assays in vitro and genetic experiments, this has elucidated the catalytic mechanisms of the enzymes in the network. This type of information does not immediately demonstrate, however, that the proposed mechanisms suffice to explain the in vivo flux of nitrogen assimilation required for rapid growth, nor does the information show how the organism adjusts the assimilation rate to its various needs. In this section, we address the above-mentioned questions for a number of processes in the network and for their regulation by the network.
We distinguish four aspects, i.e., gene expression, signal transduction, kinetics, and thermodynamics. We do this for both the rates of processes and their regulation, on the basis of the following rationale: the rate of the reaction catalyzed by an enzyme can almost always be written as follows (347):
where ΔG is the Gibbs free energy of reaction, [e] is the concentration of the enzyme, RT is the product of the gas constant and the temperature, kcat is the catalytic rate constant of the forward reaction, KS is the Michaelis constant for substrate S, and Σ is a sum of products of ratios of concentrations of metabolites and their Michaelis constants. The S/KS ratio represents the product of corresponding ratios for all reaction substrates, i.e., substances that are consumed by the reaction. If the same enzyme can be in either of two states, at mole fractions φ1 and φ2, the total catalyzed reaction rate becomes
with the kinetic factor
representing the dependence of the kinetic activity of the enzyme on substrate concentrations and the enzyme's catalytic properties when in state 1 and with an analogous equation defining f2. Using the coordinate transformation and f = f1 + f2, the above-described expression for the rate can be rewritten as follows:
Here we have simplified to the case where the second state of the enzyme is inactive. The prime then drops, and the subscript 1 can be replaced by “active.”
The equation is a concretization of the concept that four aspects contribute to the rate of a biological process, i.e., thermodynamics, enzyme kinetics, gene expression, and signal transduction: the rate of a process is equal to the multiplication of (i) the thermodynamic driving force; (ii) the catalytic activity; (iii) the fraction of the enzyme in the active state, as determined by signal transduction, resulting in covalent modification of the enzyme (such as adenylylation of GS); and (iv) the enzyme concentration, as determined by gene expression. The first two terms were grouped together previously as reflecting the influence of metabolism. Each of these four factors is determined by the network around the enzyme and is thereby a systems biology issue.
When conditions change, living organisms tend to adapt their activities, i.e., fluxes. They can do this in various ways, and these ways can be distinguished in a similar way: by measuring changes in the various factors of the above-described equation, as well as the change in flux through any process involved in nitrogen assimilation, and by dividing the logarithm of the changes of the former by that of the latter, one finds that total regulation of flux should equal 1, as follows: 1 = ρth + ρki +ρst + ρge. This equation is a generalization of an expression used in hierarchical regulation analysis (348). It quantifies the intuition that there are four types of regulation, i.e., thermodynamic, kinetic (these two together have also been called metabolic regulation), signal transduction, and gene expression regulation. The four types add up to a total of 1. In quantitative terms, the thermodynamic regulation coefficient measures regulation by adjustments of the thermodynamic driving force:
The second regulation coefficient refers to kinetic regulation:
The third regulation coefficient in this equation refers to signal transduction that leads to covalent modification of the enzyme, such as in adenylylation of GS. It is defined as
The final regulation coefficient deals with gene expression regulation:
This coefficient may be subdivided further into regulation at the level of mRNA (transcription and mRNA stability) and regulation at the level of protein (translation and protein stability) (349).
For growth to become possible at a certain rate, the ammonium assimilation flux should exceed that growth rate divided by a positive number representing the theoretical growth yield on nitrogen. Once the ammonium assimilation rate predicted by the above-described equation is positive, the organism may increase it to that value, for instance, by increasing the expression levels of the genes encoding the relevant enzymes. Alternatively, it may bring the existing enzymes into a more active state by covalent modification, or it may increase the catalytic rate constant or decrease the Michaelis constant for a substrate. None of these measures would help, however, if the rate predicted by the above-described rate equation was negative. In this case, the Gibbs free energy difference across the reaction would be positive; in other words, the product-over-substrate concentration ratio would exceed the equilibrium constant. Such a thermodynamic problem cannot be catalyzed away.
Accordingly, for each reaction residing in the presumed pathway of nitrogen assimilation of E. coli, three issues arise. First, is there a Gibbs free energy drop across the reaction, or does the Gibbs free energy in the products exceed that in the substrates of the reaction? Such thermodynamic issues surround the supposed transporter of ammonia (NH3) at low external concentrations of NH3 and acidic external pH and the GDH reaction, which may well begin to work in the direction of ammonium production (as it does in mammalian liver) when the intracellular ammonium concentration becomes low.
Second, are the enzyme concentration, the activity of the enzyme in the modification state it is in, the catalytic efficacy, and the magnitude of the thermodynamic driving force high enough collectively to sustain the flux rate needed for the observed growth rate? For some reactions, the driving force and the catalytic efficacy may be high, e.g., because it is a chemically simple reaction to catalyze (e.g., a hydratase), and the organism may need to expend less biosynthetic capacity to establish a high enzyme concentration. For others (e.g., GS), the reaction is complex in the sense of coupling an ATPase and a synthetase reaction together, potentially leading to a low kcat and requiring higher enzyme concentrations.
Third, there is the issue of regulation, i.e., of how the organism adjusts its fluxes of nitrogen assimilation to changes under conditions leading to nitrogen limitation or nitrogen excess. Above, we have reviewed that E. coli GS uses the former two regulatory mechanisms, i.e., gene expression and signal transduction. Ammonium assimilation can effectively employ the thermodynamic mechanism to regulate by substituting GS plus GOGAT for GDH: this effectively increases the Gibbs free energy dissipation accompanying the reductive synthesis of glutamate from 2-oxoglutarate and ammonium by coupling it to the hydrolysis of a molecule of ATP. The binding of an additional protein such as GlnK could regulate kinetically by reducing the kcat, which is what we discuss below for the putative ammonium transporter. Below, we examine which of these regulatory strategies the organism actually employs when confronted with ammonium limitation.
In this section, we discuss the four different aspects sequentially, for example, in the central ammonium assimilation pathway of E. coli.
Thermodynamic Regulation and Constraints: Transport
Ammonium transport could be a challenge.
If developments in science would give credence to the idea that science is perhaps never completely finished, the recent history of research into bacterial ammonium uptake could be a telling example. In short, ammonium carriers were first deemed unnecessary; NH4+ (“SH+”) transporters were then shown to be present under some conditions, and these were claimed to be membrane potential dependent; AmtB permeases were next proposed to function as NH3 (S) channels instead; and active transport may finally be back on stage but now perhaps in the form of NH3 transport mechanistically coupled to the transport of a proton (S + H+) (Fig. 2). For ammonium transport, the issue is one of thermodynamics; i.e., under some conditions, the driving force across the proposed transport mechanism may point in the wrong direction or might be too small to accumulate ammonium to intracellular concentrations sufficient for its subsequent assimilation into glutamine by GS to proceed fast enough.
As described above, structural evidence has been taken to demonstrate that AmtB is a passive transporter of ammonia (NH3). For this transport mechanism, the thermodynamic driving force expressed in the concentration of the dominant ammonium ion equals
For physiological and biochemical reasons, the intracellular pH will have to remain close to 7.2, so the above-described equation implies that during growth that depends on the influx of ammonium, the intracellular ammonium concentration is lower than the extracellular concentration, especially when the extracellular pH is <7.2: [NH4+in] < [NH4+out] · 10pH−7.2.
For instance, at an extracellular pH of 6.2 and an extracellular ammonium concentration of 0.1 mM, the intracellular ammonium concentration cannot exceed 10 μM, which is low compared to the Michaelis constants of both GDH and GS for ammonium.
The data described above show that in pH-neutral environments where >0.5 mM ammonium is continuously available, the NH3 transport mechanism alone will have sufficient inward driving force. Under this condition, not only is unmediated NH3 diffusion rapid enough to fulfill the biosynthetic ammonium flux, the resulting intracellular ammonium concentration will also be high enough so as to saturate ammonium-assimilating enzymes to a sufficient degree (301). Indeed, Boogerd et al. calculated that under such conditions, no transporters should be needed, as NH3 diffusion alone through a lipid bilayer would have sufficient flux-carrying capacity. Thus, in the majority of cases of bacterial growth with ammonium as a nitrogen source studied in the laboratory, the availability of nitrogen as an intracellular ammonium ion for the biochemistry leading to glutamate is not problematic for bacteria.
However, it is worthwhile to briefly discuss the condition of unlimited ammonium supply. Since even water can be administered at levels that are toxic to organisms, might excess ammonium not be a problem for bacteria? Would ammonium concentrations of more than, say, 500 mM not be toxic to bacteria? After all, it is known that ammonium is rather toxic for humans (low μM range), plants (low mM range), and yeast (mM range) (350). However, several studies indicate that it might not be toxic for bacteria up to rather high concentrations, except perhaps for the cyanobacteria (351). The growth rates of Corynebacterium glutamicum, Bacillus subtilis, and E. coli were not affected by up to 500 mM ammonium in batch culture (352). Assuming that the extracellular and intracellular pH was 7.5, the intracellular ammonia (NH3) concentrations would have amounted to some 9 mM. In a fed-batch reactor in which the ammonium level was controlled at 5 to 200 mM, inhibition of growth by ammonium occurred at >170 mM extracellularly (353). As common batch culture media with excess ammonium have concentrations ranging from 10 to 100 mM, negative effects on growth should not be expected.
A related issue is what happens if cells that have been growing under N-limited conditions are confronted with a sudden increase in the ammonium concentration. There is a wide range of external ammonium concentrations that are described as “ammonium shocks,” e.g., from 0.05 mM (163) to 0.2 mM (354), 10 mM (135), 20 mM (355), and up to 30 mM (139, 303, 356). In natural environments, perturbations in the high mM range (10 to 30 mM) are unlikely. If a high mM ammonium shock is applied, cells would have to cope with a rapid internal alkalinization due to the instantaneous diffusion of NH3 into the cell and the immediate consumption of 1 proton per NH3 molecule to yield NH4+. In view of a buffering capacity of some 30 mM protons/pH unit (357), internal pH may increase up to 1 full unit upon a 30 mM upshift. It remains to be seen if this occurs, but in principle, this could be assessed experimentally, as substantial (1.5 units) and rapid (within seconds) internal pH changes upon addition of HCl or KOH have been demonstrated using green fluorescent protein (GFP) fluorimetry (358, 359).
Kleiner gathered data on intracellular and extracellular ammonium concentrations as observed for a host of different eubacteria (including some enterobacteria but not E. coli) grown with a variety of N sources in batch or continuous cultures (360). The single N source was N2, nitrate, glutamine, or ammonium. If ammonium is the N source, it will have to be taken up prior to assimilation. The situation is different for the first two N sources mentioned (glutamine as an N source represents a special case and is discussed below). After uptake, these N sources (N2 and nitrate) will invariably have to be converted into ammonium in the cytoplasm through reduction to serve as the N donor for nitrogenous metabolites. Hence, part of the intracellular NH4+ formed in this way will inevitably leak out of the cell as NH3 and instantaneously be converted into extracellular NH4+. Kleiner argued that reuptake of NH4+ is then required to compensate for the loss. His collection of data showed that for cells of several bacterial species (grown with N2, nitrate, or ammonium), quite substantial outward ammonium gradients had indeed been observed: from 1 to 2 orders of magnitude for eubacteria to even up to 3 orders of magnitude for cyanobacteria (360). Thus, intracellular ammonium concentrations would seem to be much higher in many bacterial species, although the intracellular production of ammonium from N2 and nitrate was a confounding factor.
Methylammonium uptake by whole cells.
Similar gradients had been found for the ammonium analog methylamine (MA) (CH3NH2 and/or CH3NH3+), however (360), where production of the compound intracellularly should be absent. The gradient could result only from active uptake of the neutral form of MA or the passive uptake of the MA cation (but see below). This presents a challenge of the notion that ammonium uptake proceeds through an NH3 transport mechanism. Indeed, in a review on ammonium transporters in prokaryotes, fungi, and plants, corroborating evidence was presented that bacterial (methyl)ammonium transport takes place via electrogenic (CH3NH3+) NH4+ uniporters (361).
However, this view changed radically in the 1990s upon the discovery of the amtB gene and the ensuing (direct) structural characterization of wild-type and variant transport proteins. Structural evidence was presented for the view that AmtB functioned as channel for ammonia (NH3), as discussed above.
To date, no reliable direct assay of rapid cellular ammonium transport via AmtB has been reported. The major reason for this is that a quick, sensitive, and accurate method is required to measure ammonium concentrations, but such a method has yet to be developed. NH3 is a highly permeable molecule that will quickly equilibrate between cells and the surrounding fluid, and this fact will compound the assay, in particular when intracellular pools are to be measured. The use of MA carrying radioactively labeled 14C as a substitute for ammonium has overcome most of these difficulties, although fast equilibration of the neutral form of MA between cells and medium remains a problem (see below). Transport of [14C]methylamine is generally considered to represent AmtB-mediated uptake of ammonium by whole cells, but we note that this is an assumption that would itself require validation. Indeed, MA uptake has been used as a measure for the activity of wild-type AmtB in wild-type and mutant (but not AmtB mutant) cells (e.g., see references 281 and 303) as well as of mutated AmtB derivatives (e.g., see references 269 and 288–290) and of His-tagged AmtB (273). In view of the high degree of specificity of binding of NH4+ to the periplasmic vestibule and of the proposed transfer of NH3 specifically through the hydrophobic channel, it is quite remarkable that MA is transported via AmtB at all: MA been considered a poor analog for ammonium biochemically and physiologically (289, 305).
In contrast to ammonium, methylammonium is not metabolized any further after uptake (MA cannot be used as an N source), except for its conversion to methylglutamine by GS (281, 362). Because the conversion of MA to methylglutamine is ATP dependent, as is incorporation of ammonium into glutamine, a major issue is whether uptake per se is being measured in transport assays or uptake together with (limited) metabolism (281). A solution to this problem is the use of mutants that lack GS, but using the commonly used washed assay, Soupene et al. then did not observe any uptake at all (281; but see reference 288). However, an explanation for this negative result was presented by Javelle et al. (305), who resuscitated the unwashed assay described previously (362) and carefully corrected for nonspecific binding of MA to biomass that resulted from omitting the washing steps. This unwashed assay has shed light on the confusing and sometimes conflicting results regarding MA uptake experiments, in which a phase featuring rapid initial uptake of MA followed by a phase with a much slower further uptake of MA were observed (305). Javelle and colleagues concluded that MA uptake occurs in two phases, the first of which is AmtB specific and the second of which is a consequence of GS activity.
Washing of cells has been shown to remove all of the free and unmetabolized MA from the cytoplasm (305, 362), with only the metabolized product of MA, i.e., methylglutamine, remaining. Actually, full advantage of washing has been taken, because it enabled the determination of the in vivo GS activity with MA as a substrate for both AmtB and GS. With this method, the effective in vivo Km value of GS for MA was some 200-fold lower than the in vitro-determined value. The authors of that study suggested that an explanation might be that the function of GS could be closely coupled to that of AmtB (305). Because GS was found to not be membrane bound in an AmtB-dependent fashion, we suggest the alternative interpretation of a considerable accumulation of CH3NH3+ through active transport.
Although both Jayakumar et al. and Javelle et al. used unwashed assays, they claimed very different accumulation factors for MA: 100-fold (304) versus 4-fold (305). The latter authors considered their 4-fold accumulation close enough to 1.0 to be taken as evidence for uptake of the neutral species CH3NH2, while Jayakumar et al. perceived their factor and other experiments with K+-depleted and -repleted cells as evidence for an NH4+/K+ antiport transport system.
In unwashed assays, the fast phase does not take much longer than 30 s, and the issue can be raised regarding what causes this leveling off. If transport of MA would be active, then an answer might be that after some time, active uptake of the cation MA just equals the loss of the neutral CH3NH2 to the medium by diffusion through the membrane. Thereafter, only conversion of MA by GS to the impermeable methylglutamine will lead to a steady but slow further increase in cytoplasmic radioactivity.
Energetics of ammonium uptake by proteoliposomes and vesicles.
It has been claimed that reconstitution of AmtB into proteoliposomes showed that AmtB conducts uncharged NH3 (271, 363). The basic premise of this experimental setup is that uptake of NH3, either through the lipid membrane or via AmtB, will subsequently result in protonation of NH3 in the intraliposomal compartment, whereas uptake of NH4+ would not. Khademi et al. observed that the increase in intraliposomal pH was 10-fold faster than in control experiments with liposomes without AmtB. Thus, AmtB strongly stimulated the transfer of NH3, and this finding was taken to support the passage of NH3 through the hydrophobic channel, as concluded from analysis of the crystallographic structure of AmtB in the same article. However, the stimulation could not be reproduced despite extensive efforts (299). Also, uptake studies with right-side vesicles prepared from whole cells overexpressing AmtB did not show an increased uptake rate compared to control vesicles harboring a defective AmtB (299). Moreover, it was argued that the permeability assay used by Khademi et al. (271) is inconclusive with respect to the nature of the transported species, NH3 or NH4+ (301): (i) the above-mentioned factor of 10 was recalculated to amount to just 2, and (ii) the formation of a transmembrane electric potential that would quickly result from the uptake of NH4+ and would drive proton extrusion or hydroxyl uptake and cause intraliposomal alkalinization, as predicted by nonequilibrium thermodynamic analysis (364), could not be ruled out, as no permeant counterions had been added to the assay mixture.
We conclude that although AmtB functioning as a channel for NH3 was the widely accepted view in the 2000s, anomalous observations have continued to appear in more recent years. In the end, it may well turn out to be that AmtB-mediated transport is electrogenic after all, with either NH4+ as such being transported “passively by facilitated/protein-catalyzed diffusion,” which would correspond to active uptake of NH3 or symport of a molecule of NH3 and (at least) one proton or antiport of NH3 and OH−. After all, transport is a product of the concentration in the membrane and the rate of transfer across the membrane and can be rapid if the intramembrane concentrations are so low that any one binding site is <10% saturated and, hence, invisible as such in any structural determination, but the flux rate is fast.
A mechanism in which ammonium ion is transported or one in which NH3 is symported with a proton has a much more favorable inward driving force, due to the contribution of the transmembrane electric potential:
The membrane potential-dependent term is approximately −18 kJ/mol in living cells, which is able to drive an ammonium accumulation ratio of 1,000 (347).
AmtB-GlnK interaction and kinetic regulation.
On the basis of the strong conservation of the glnK and amtB genes as gene pairs in many prokaryotic genomes, it has been argued that the gene products may interact physically (365). Indeed, both in vitro and in vivo, GlnK binds to AmtB, and the binding then inhibits ammonium transport (Fig. 3). This interaction is dependent on the uridylylation state of GlnK. It is GlnK in its deuridylylated conformation that binds to AmtB (163, 303, 356). Trimeric GlnK binds trimeric AmtB in a 1:1 ratio (356, 366). The stability of the complex does not require other proteins (356). GlnK binds AmtB at the highly conserved cytosolic C-terminal domain of AmtB (303). Complex formation or dissociation in vitro was sensitive to the small effector molecules 2-oxoglutarate, Mg2+, ATP, and ADP and depended on whether these were present separately or combined (356). The key features promoting the AmtB-GlnK association in vitro and in vivo turned out to be the rapid decline in the 2-oxoglutarate concentration and a change in the ATP/ADP ratio upon a small ammonium upshift (354), conditions pertaining to a reduced requirement of AmtB transport activity (not flux). GlnK may cause kinetic regulation of the ammonium transported by reducing the kcat of the latter upon binding. X-ray diffraction analysis of a complex of E. coli AmtB and nonuridylylated GlnK suggested a mechanism of inhibition of AmtB by native GlnK. The extended T loop of three GlnK monomers inserts deeply into the cytoplasmic vestibule of the three AmtB monomers, possibly blocking the exit of ammonium into the cytoplasm sterically (366–368). These structures may also clarify the inability of uridylylated GlnK to bind AmtB. The space in the vestibule is too narrow to accommodate the uridylyl group at the apex of the T loop (367).
Fig 3.

Regulation of AmtB activity by GlnK. The trimeric ammonium transporter AmtB and the trimeric regulatory protein GlnK are induced only under nitrogen-limited conditions. The bottom right part of the figure shows that each of the three channels of AmtB is able to translocate ammonium from the outside to the inside. Each of the monomers of GlnK harbors a T loop that protrudes from the top of the barrel-like structure (to the left of the green arrow). The three loops each fit deeply into the cytoplasmic vestibule of each of the channels of the trimeric AmtB transporter (to the right of the green arrow). Hereby, transport of ammonium is blocked. The top left part of the figure displays that under conditions of high (dark blue) or low (light blue) intracellular glutamine concentrations, the bifunctional enzyme UTase catalyzes the deuridylylation or uridylylation (i.e., the covalent removal or coupling of UMP groups) of the tips of the T loops of a GlnK trimer, respectively. Binding of GlnK-UMP to AmtB is no longer possible, since the bulky UMP groups preclude insertion of the T loops into the cytoplasmic vestibules of the AmtB channels. The lower middle part of the figure shows that complex formation between AmtB and GlnK is affected by the metabolites 2-oxoglutarate, ATP, and ADP and by the cation Mg2+.
Complex formation is fast (within seconds) and reversible in response to the extracellular ammonium concentration (163). When the latter is low, AmtB would conduct ammonium, which is then converted to glutamine by GS. The concentration of glutamine is sensed by UTase, leading to an adjustment of the uridylylation state of GlnK (and GlnB), which may in turn regulate the activity of AmtB. Thus, AmtB can be perceived not only as a transporter but also as a sensor of extracellular ammonium and should be considered a full member of the signal transduction cascade (163), producing signal at the level of the intracellular ammonium concentration and, as such, being regulated by the N status of the cell.
AmtB may also bind GlnB and thereby antagonize GlnB signaling through NRII. As amtB and glnK are cotranscribed, GlnK may prevent titration of GlnB by AmtB (369).
In view of the possible function of GlnK as an inhibitor of AmtB activity, it is important to establish the amounts of these two proteins in the cell. Quantitative measurements of AmtB are rare: for cells grown with glucose and glutamine, Western blotting quantification gave a value of some 1,500 AmtB trimers per cell (354). Unfortunately, there is no consensus about the level of GlnK in cells grown under the same conditions. The intracellular concentration of GlnK was estimated to amount to 1 μM (161). Radchenko et al. reported a value of 50 μM (354). The latter value would imply a ratio of GlnK over AmtB of ∼8. This seems to be at variance, however, with data presented previously (163, 356) for the extent of sequestration of GlnK to the membrane upon an ammonium upshift: maximally, 10% of cytoplasmic GlnK could then be sequestered to AmtB in the membrane fraction, leaving minimally 90% in the cytoplasmic fraction. However, the experimental results (163, 356) showed that no GlnK whatsoever remained in the cytoplasmic fraction. Also, a GlnK concentration of 50 μM is much too high for a regulatory protein, as it would be even 4 times higher than the GS concentration in nitrogen-limited cells (around 12 μM), where GS is a highly active enzyme. We therefore hold the GlnK concentration of around 1 μM, as measured by van Heeswijk et al., to be more likely than 50 μM.
Kinetic Regulation
Feedback inhibition of GS.
Quite remarkably, direct inhibition of GS by its own product glutamine is limited (44). This may be due to the fact that its product inhibition constant lies at around 6 mM and the Michaelis constant for glutamate is around 4 mM, whereas the concentration of glutamate readily exceeds that of glutamine by a factor of 20 or 50 under conditions of excess or limited ammonium, respectively (see references 135 and 342): the concentration term in the thermodynamic driving force is in the forward direction, and the substrate competes the product inhibitor away from its binding site, which reduces kinetic limitations. Bruggeman et al. (44) found glutamine to be the third important short-term regulator, but this was based on a lower intracellular glutamate concentration of 5 to 20 mM, which may therefore have overestimated the regulatory role of glutamine product inhibition.
Much of the longer-term kinetic regulation of GS may, however, depend on a mechanism that has been referred to as “cumulative feedback inhibition” (31). Purified GS has been shown to be inhibited by many metabolites: alanine, glycine, serine, histidine, tryptophan, carbamoyl-phosphate, glucosamine-6-phosphate, AMP, and CTP (370–372). Each of these compounds caused only partial inhibition at relatively high concentrations (3 to 20 mM). However, various combinations of them give rise to a so-called “cumulative inhibition” of GS, where the total inhibition is determined by the product of the fractional activities (which is what would occur with noncompetitive inhibitors) and not by their sum (which would happen if all inhibitors would be competing for the same site) (371, 372). The inhibitors were divided into two classes. Alanine, glycine, and serine constitute class I, and the others constitute class II (373). Except for the class I inhibitors, each metabolite is an end product of a pathway that utilizes glutamine as the donor of an amide group (4). On the basis of the cumulative nature of the feedback inhibition, it was suggested that each compound had its own binding site (371, 372). To obtain the binding sites of the inhibitors of GS by X-ray diffraction analysis, crystals of unadenylylated GS from S. Typhimurium were used (38, 374). Although this is perhaps not the physiologically relevant form of GS (see below), the results give an indication of the binding sites of the inhibitors at GS-AMP. The class I inhibitors were all found in the binding pocket of glutamate of unadenylylated GS and to be competitive inhibitors with respect to glutamate (374), which means that their inhibition should not be additive/cumulative with respect to each other. AMP was observed to bind at the ATP binding site of unadenylylated GS and to be a competitive inhibitor with respect to ATP (38), but it should be noncompetitive with respect to glutamate and, hence, cumulative with respect to, for instance, alanine. Class II inhibitors would all bind to separate allosteric sites (373). In view of the limited possibilities of having many different binding sites on the altogether not extraordinarily large GS monomer (52 kDa), we accept the proposed cumulative inhibition between the amino acids and the adenine nucleotides but find additive inhibition within these groups more likely. More detailed enzyme kinetic studies under in vivo conditions (375) are warranted here. The idea of cumulative inhibition of the class II compounds that depend on amide donation by glutamine for their biosynthesis, and thereby each “booking” their part of glutamine biosynthesis, independent of the others, remains interesting, however.
Stadtman and colleagues (109, 120, 376) and Holzer et al. (377) observed that adenylylated GS is generally (much) more sensitive to feedback inhibitors than unadenylylated GS. In hindsight, early results obtained regarding feedback inhibition were confounded by the fact that the cultivation method and the growth phase of cells used to obtain GS preparations affected the adenylylation state of GS and, hence, the level of inhibition (120, 377).
Reitzer ascribed physiological significance to the observed differential sensitivity of adenylylated and unadenylylated GS to the feedback inhibitors. Adenylylated GS is not primarily engaged in ammonium assimilation (373) but is engaged more in the synthesis of glutamine as one of the 20 amino acids required for protein synthesis. For cells growing in a low-ammonium medium, the primary function of the then deadenylylated GS (together with GOGAT) is that of assimilating ammonium. i.e., incorporating nitrogen into biomass. Reitzer concluded from this that feedback inhibition would not make sense for the deadenylylated GS, which is involved in this assimilation of ammonium. This is not necessarily convincing to us, as massive flux of ammonia assimilation might also have to be regulated to prevent its expensive overflow. However, indeed, when the primary function of GS is to synthesize glutamine and hence its class II inhibitors (see above), glutamine and those inhibitors might accumulate and should then inhibit glutamine synthesis either by the fully adenylylated GS or by the residual activity of the fraction of GS that is still unadenylylated. Complete feedback inhibition of the residual activity of adenylylated GS would have to be prevented for cells growing in the presence of excess ammonium, since GS is an essential enzyme (77), i.e., the only enzyme capable of glutamine synthesis. It is questionable whether in vivo the activity of GS-AMP will show up, due to the effects of two divalent cations: Mn2+ is required at 0.3 mM for activity (122), but intracellular levels are probably <0.1 mM (346), and Mg2+ inhibits the GS-AMP activity, while its intracellular levels are in the 1 to 5 mM range. In this respect, it seems easier to secure the necessary glutamine-synthesizing activity through incomplete, instead of full, adenylylation of GS. The more recent finding that the adenylylation state of GS is dynamically determined by the simultaneous actions of both ATase activities is consistent with this view. In addition, this mechanism would allow fine-tuning of GS activity to the needs of E. coli (107). Finally, the cellular concentrations of most of the inhibitors (excluding glycine and carbamoyl-phosphate) have been determined for cells grown with glucose, glycerol, or acetate as the C source and excess ammonium as the N source (342), and only alanine and CTP are present in a concentration range (1 to 3 mM) that could give rise to some inhibition of GS-AMP activity. Alanine has to compete with the substrate glutamate, which is invariably present in high concentrations (>50 mM) in vivo.
Signal Transduction-Mediated Regulation
Signal transduction onto GS.
The nitrogen status of the cell, as signaled by glutamine and 2-oxoglutarate, regulates the activity of GS. Although glutamine does not inhibit E. coli GS activity directly (see above), it exerts indirect control via signal transduction through the enzyme adenylyltransferase (ATase): it reduces the activity of GS by causing covalent modification, i.e., a covalent linkage of an AMP group to each subunit of GS (109, 115, 378). ATase is also able to deadenylylate GS-AMP (83, 90) (Fig. 4). Thus, ATase is a bifunctional, “ambiguous” enzyme. Ambiguous enzymes need careful regulation to prevent their wasting of Gibbs free energy, in this case, of the ATP that provides the AMP group. Indeed, both reactions are strongly regulated. The adenylylation reaction is stimulated by the regulatory protein GlnB as well as by glutamine, whereas the deadenylylation reaction is stimulated by a modified form of GlnB, i.e., GlnB-UMP, and by 2-oxoglutarate (95, 96, 379). The conversion of GlnB to GlnB-UMP and vice versa is catalyzed by the enzyme UTase, which is again an ambiguous enzyme: it can both uridylylate GlnB and deuridylylate GlnB-UMP (124, 142, 149, 380). If the intracellular nitrogen concentration is low, UTase converts GlnB and UTP to GlnB-UMP and pyrophosphate. On the other hand, if the intracellular nitrogen concentration is high, UTase deuridylylates GlnB-UMP to form GlnB plus UMP. Therefore, GlnB signals cellular nitrogen affluence, while GlnB-UMP signals nitrogen poverty. In sum, the covalent modification of GS is regulated by two nucleotidyltransferases, UTase and ATase, forming a bicyclic cascade (Fig. 5).
Fig 4.

The covalent modification cycle of GS. Glutamine synthetase consists of 12 monomers, each of which can be adenylylated or deadenylylated by the bifunctional enzyme ATase. The adenylylation state (nAMP) of GS can be any integer between 0 and 12. The catalytic activity of GS decreases continuously upon increasing adenylylation. The gradient bars at the top of the figure indicate the sensitivity of the enzyme ATase for glutamine (GLN), fully uridylylated GlnB carrying 3 2-oxoglutarate molecules (GlnB-UMP3∼OG3), and unmodified GlnB carrying a single 2-oxoglutarate molecule (GlnB∼OG1). The latter two GlnB species, out of 16 possible GlnB species, represent the two molecules that give the highest stimulation of the deadenylylating and the adenylylating activities of ATase, respectively. GS itself is insensitive to these three molecular species.
Fig 5.

The full regulatory cascade of GS. The heart of the figure shows that GS is covalently (de)adenylylated (denoted −AMP) by the action of ATase. The two opposing activities of ATase are affected directly by glutamine (∼GLN). These activities are also affected by binding two specific species of the regulatory protein GlnB and the corresponding GlnK species. Covalent uridylylation (denoted −UMP) of both GlnB (top) and GlnK (bottom) is shown to be catalyzed by the enzyme UTase, which is sensitive to glutamine. Differential binding of 2-oxoglutarate (∼OG) to the unmodified and uridylylated GlnB/GlnK species is also shown. Solid blue arrows denote positive effects and red arrows denote negative effects on ATase and UTase. The two dashed blue arrows indicate that the GlnK species are (much) less potent, at least in vitro, in activating the ATase activity than are the corresponding GlnB species.
Under some conditions, E. coli has a GlnB-like protein called GlnK at its disposal (139, 154, 155). GlnK, like GlnB, can be uridylylated by UTase (138, 139), while its product, GlnK-UMP, can be deuridylylated by UTase (138). Like GlnB, GlnK is able to stimulate the adenylylation reaction (138, 139), while GlnK-UMP can stimulate the deadenylylation of modified GS (140, 162). Accordingly, GS modification is regulated by two parallel bicyclic cascades (Fig. 5). The different proteins involved in the covalent modification of GS are discussed in more detail in the following sections.
UTase activity.
From the results of a kinetic analysis of UTase (127) and of a reconstitution of the UTase-GlnB monocycle (136), it was concluded that the monocycle functions as a glutamine-sensing device: at a fixed 2-oxoglutarate concentration, a low or high glutamine concentration resulted in an almost completely uridylylated GlnB or deuridylylated GlnB, respectively (136). If the in vitro experiments reflect the in vivo situation, the physiological function of UTase may be one of sensing the glutamine concentration and transducing this information to the GlnB protein by adjusting the degree of uridylylation of the latter. Indeed, neither E. coli nor S. Typhimurium glnD mutants lacking both activities of UTase (which is encoded by glnD) sense the nitrogen status of the cell: they exhibit a high-adenylylation state of GS independent of the absence or presence of ammonium in the growth medium (92, 133, 381, 382).
For wild-type E. coli under conditions of nitrogen limitation, this implies that if the glutamine concentration were to drop to below the Km of GOGAT for glutamine (which is approximately 0.2 mM in vitro [46, 54]), GlnB will be highly uridylylated (see reference 136), and as a consequence, GS will be completely deadenylylated and, thus, operate at its maximum activity. This may then lead to an increase in the intracellular glutamine concentration such that the increased thermodynamic driving force for GOGAT will push the enzyme to produce glutamate again. However, the intracellular glutamine concentration is some 2 mM during nitrogen limitation (135), which is far above the Km value of GOGAT. The competitive inhibitors of glutamine are aspartate (Ki = 0.35 mM) and glutamate (Ki = 28 mM) (46), both of which, however, are present at concentrations (Asp = 2 mM; Glu = 70 mM) above their Ki values (135), increasing the effective Km for glutamine and making the above-described kinetic regulation relevant again: the competition for active sites is physiologically important (see below). For nitrogen-limited S. Typhimurium, the glutamine concentrations are lower, roughly 0.5 mM (calculated from data in reference 383).
In the literature, the ranges of intracellular glutamine (0.2 to 0.5 mM) and 2-oxoglutarate (0.1 to 0.9 mM) concentrations presented in a seminal paper by Senior (384) are often referred to. It should be noted, however, that these concentration ranges were determined by using chemostat cultures at different growth rates for glucose-limited cells, i.e., for cells that were growing with excess ammonium.
GlnB activity.
By constructing heterotrimers in vitro consisting of two GlnBΔ47–53 subunits and one wild-type subunit, Jiang and coworkers (385) demonstrated that a single T loop within a GlnB trimer suffices in vitro to interact productively with all three target proteins (UTase, ATase, and NRII). Those authors suggested, therefore, that the functional unit of GlnB is the individual monomer. These measurements showed in addition that the heterotrimer, containing one unmutated T loop, could be even better uridylylated than wild-type GlnB and activated NRII better than, and ATase equally as well as, the wild-type GlnB trimer (385). A reason why GlnB is a homotrimer instead of a monomer has been proposed by Thomas et al. (365). Based on a phylogenetic analysis, they concluded that trimeric GlnB may have evolved from trimeric GlnK, which is part of the conserved GlnK-AmtB couple (organized as an operon). In addition, a trimer may exhibit more regulability than a monomer. Indeed, several findings point in this direction. The clefts between the loops of the monomers are important for binding of ATP, ADP, and 2-oxoglutarate (see below) and, hence, for regulation of the activity of the GlnB trimer. Furthermore, it has been shown that the multiple binding sites for ATP, ADP, and 2-oxoglutarate within the trimer cooperate to regulate the functioning of a single T loop (386). Also, the number of 2-oxoglutarate molecules bound to trimeric GlnB affects the ability of the latter to regulate its targets ATase (387) and NRII (388). In contrast, uridylylation of GlnB is a noncooperative reaction (161, 389), suggesting that uridylylation of one subunit neither stabilizes nor destabilizes the other nonuridylylated loops.
As described above, 2-oxoglutarate and ATP can bind to GlnB with high affinity, each with a Kd (dissociation constant) in the μM range, and binding of ATP stimulates the binding of 2-oxoglutarate and vice versa (127, 134). However, ADP acted strongly antagonistically to 2-oxoglutarate (390). Moreover, the binding of 2-oxoglutarate and ATP/ADP requires cooperation of the three ligand binding sites within the GlnB trimer (386).
Binding of 2-oxoglutarate to GlnB is necessary for its uridylylation (127, 134), which suggests that binding of 2-oxoglutarate (and possibly also ATP) to GlnB is important for the presentation of the correct loop structure to the active site of UTase (147). It should be noted that although 2-oxoglutarate controls the ability of GlnB to regulate the activities of NRII and ATase, it does not control the binding of GlnB and GlnB-UMP to these receptors of the nitrogen status of the cell (96, 387, 388).
It has been noted (151) that GlnB contains an amino acid sequence similar to the “Walker A consensus motif” (391) involved in the binding of ATP to a number of proteins. This sequence in GlnB may be G84-X-X-X-X-G89-K90. The site is located within a small loop structure (B loop), located between an α-helix and a β-sheet at the exterior surface of the trimer (152, 153). Indeed, substitutions within this motif resulted in GlnB variants to which the binding of ATP (and 2-oxoglutarate) was strongly reduced and which did not interact with either UTase, ATase, or NRII (392). Binding of ATP to this site changed the conformation of the B loop of GlnB, as was observed by X-ray diffraction analysis of a crystal of GlnB bound with ATP (159). In addition, the structure of the latter compared to the structure of uncomplexed GlnB (152, 153) revealed that the T loop may block the binding site of ATP.
Studies with reconstituted signal transduction systems have shown that the action of ADP is targeted to GlnB and GlnB-UMP and that the interactions of GlnB with ATase, UTase, and NRII are affected by the presence of ADP (390). ATP and ADP bind to the same nucleotide binding site on GlnB (159). The binding of ATP was strongly synergistic with 2-oxoglutarate, while the binding of ADP was not (390). ATP concentrations measured for E. coli cells grown under a variety of batch and chemostat conditions varied from 2 to 10 mM (342, 343, 393–397). ATP/ADP ratios were observed to range from 3 to 12 (343, 398, 399). Recent metabolome data indicate that 2-oxoglutarate concentrations are in the range of 0.2 to 0.6 mM in cells growing with excess ammonium (342, 343) and around 10 mM when nitrogen is limiting (135). Therefore, under most circumstances, ATP is expected to dominate at the binding site on GlnB, and regulation of GlnB by the ATP/ADP ratio or the energy charge might be of physiological significance when both the ATP/ADP ratio is low and nitrogen is not limiting.
In sum, GlnB can bind 3 molecules of ATP or ADP and 3 molecules of 2-oxoglutarate noncovalently and 3 UMP groups covalently. The ATP/ADP ratio may represent a Gibbs free energy signal, 2-oxoglutarate being an intermediate of the TCA cycle may represent a carbon source signal, and uridylylation of GlnB may represent a nitrogen source signal. Altogether, GlnB may adopt many different signaling states (386). Thus, instead of simply transducing a signal, GlnB may function as an integrator of the three signals mentioned above and transduce the integrated signal to its receptor proteins NRII and ATase (for reviews, see references 28, 29, and 400).
GlnK activity.
GlnB and GlnK are structurally similar but functionally distinct regulatory proteins. First and foremost, glnB is constitutively expressed at a low level (88, 144), while the expression of glnK is induced under conditions of nitrogen limitation or starvation (139, 401). Second, the two paralogous proteins regulate the degree of NRI phosphorylation via their interaction with NRII differently during different stages of nitrogen supply, i.e., under nitrogen excess, nitrogen limitation, and nitrogen starvation conditions. Together, they indirectly control the expression of the nitrogen-regulated genes (glnALG, nac, glnK, and perhaps other nitrogen-regulated genes) under a variety of environmental conditions (401, 402). The differences in the physiological roles may not be due to differences between the proteins per se but to the timing of expression of their genes and the levels of accumulation (403).
When it concerns GlnK alone, a variety of alternative metabolic functions for GlnK has been suggested. GlnK would function as follows. First, GlnK would function as a regulatory protein acting in the nitrogen-regulated regulon much like GlnB does (138, 139, 162, 403). However, GlnK was found to be a less potent actor than GlnB, at least for some of its activities (138–140). Second, GlnK may function as a “memory protein,” enabling the cell to keep track of its growth history (139). The cell might remember the condition to which it had been adapted through the concentration of the GlnK protein, and this might prevent the cell from responding to each and every transient change in the environment. Third, It may function as a regulator of the expression level of NRI-P under conditions of severe nitrogen starvation (162, 402). In the absence of GlnK, rather high expression levels of the glnK and nac promoters occurred in nitrogen-starved cells. Apparently, GlnB is unable to control nac and glnK expression, and GlnK is required to limit expression of these two promoters during nitrogen starvation. Fourth, GlnK may be a fine-tuning protein for GlnB action via heterotrimer formation (140, 161, 404). Indeed, GlnK and GlnB can form heterotrimers between them in vivo (140, 404) and in vitro (140). The activity in stimulating the deadenylylation reaction decreases from uridylylated GlnB homotrimers and uridylylated GlnB-GlnK heterotrimers to uridylylated GlnK homotrimers in vivo (161) as well as in vitro (140). The formation of heterotrimers with both GlnK and GlnB is not surprising, because GlnB heterotrimers can also be formed in vitro from wild-type and mutant monomers (385). Actually, considering the fact that the glnK gene is more ancient than the glnB gene (365), GlnB may be, in terms of differences in its amino acid sequence relative to that of GlnK, regarded as a variant form of GlnK. (v) GlnK may be a factor needed to survive nitrogen starvation and to recover rapidly when fed ammonium afterwards (402). In contrast to the wild type, a glnK mutant failed to recover rapidly when starved for nitrogen for 10 h or more. Also, these cells showed faster loss of viability than the wild type. (vi) GlnK may function as a protein needed to prevent titration of GlnB when AmtB is highly expressed (369). Cotranscription of glnK with amtB ensures that both proteins are expressed in similar quantities, and thereby, the GlnB protein, which is capable of binding to AmtB, would not be lost for signaling by AmtB-mediated sequestration to the cytoplasmic membrane. (vii) GlnK may be an inhibitory regulator of the transport activity of AmtB by direct binding (163, 303, 356). Building on this GlnK activity, it was hypothesized that it serves to minimize AmtB-mediated futile cycling of ammonium across the membrane while maintaining an intracellular ammonium level sufficient for growth (301). These functions are discussed in “AmtB-GlnK interaction and kinetic regulation,” above, and “AmtB-mediated futile cycling of ammonium,” below.
Although it may seem that there are too many functions to all be true, the concepts of conditional (405) and quantitative (406) phenotypes make it conceivable that most, if not all, of the above-described functions might indeed be physiologically relevant. On the other hand, the most straightforward function of GlnK seems to be the regulation of AmtB transport activity. The observation that GlnK deuridylylation is dependent on AmtB activity under physiological conditions may be taken to suggest that this indeed might be its major activity (163, 407). Indeed, it has been argued that part of the evidence for the other functions of GlnK has been collected by studying glnK mutants in which the absence of GlnK causes GlnB to be unnaturally sequestered to the membrane by AmtB (163).
ATase activity.
The physiological function of the adenylylation and inactivation of GS by ATase may be 4-fold. First, it may prevent depletion of the glutamate pool and enable the resumption of growth after an ammonium upshift of cells that had been growing on a poor nitrogen source, a condition that results in a high glnA expression level and, thus, in a high concentration of its unmodified gene product, GS (408, 409). Second, as glutamate has been regarded as the major counterion for free potassium, ATase may indirectly prevent depletion of the potassium pool in the situation described above (410–412). Third, ATase may control the activity of GS homeostatically. When glnA is overexpressed or upregulated, adenylylation of unmodified GS may balance the activity of GS (161, 413). Conversely, a glnE mutant, without adenylylation activity and thus with high GS activity, might decrease the synthesis of GS to compensate for its increased activity (92, 408, 413). Fourth, at steady-state growth, the adenylylation and deadenylylation reactions may set the adenylylation state of GS to the optimal number of adenylylated GS subunits (414, 415).
Autophosphorylation of NRII.
NRII can be autophosphorylated at histidine 139 (173). The reaction requires Mg2+ and ATP but no other protein (hence the prefix “auto”) (reaction I) (Table 6) (191, 416). The putative ATP binding site is involved in the autokinase reaction because a substitution in this site impaired ATP binding and resulted in a much lower degree of autophosphorylation (177). The autokinase reaction of NRII proceeds by binding of ATP to one subunit followed by intermonomer phosphorylation in trans within the NRII dimer (177, 417). Consequently, the dimerization reaction of NRII monomers may have control of NRII phosphorylation.
Table 6.
Reactions catalyzed by the NRI-NRII two-component system
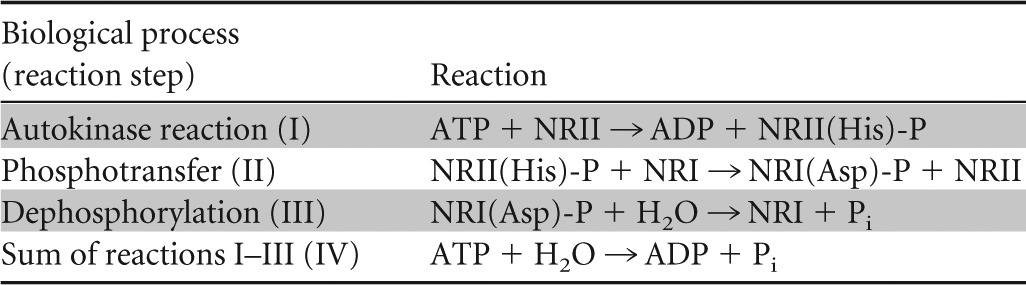
As shown in Fig. 6, the signal transduction occurs through the transfer of a phosphoryl group from ATP via NRII (His139) to NRI (Asp54) (182; for reviews, see references 164–166): NRII is a protein kinase of NRI. Any of the three reactions may contribute to the regulation and control of the level of NRI-P, the transcriptional activator in its active form. The autophosphorylation of NRII is inhibited by unmodified GlnB complexed with ATP and 2-oxoglutarate (178, 179). However, GlnB can bind and inhibit the autokinase reaction of NRII only when the trimer is complexed with three ATP molecules and only one 2-oxoglutarate molecule (Kd of ∼5 μM) (134, 178, 388). A complex of GlnB with ATP and without any or with two additional molecules of 2-oxoglutarate (Kd of ∼150 μM) cannot bind NRII (178, 388). If these in vitro-determined dissociation constants are also valid under in vivo conditions, a paradox arises for the received view of regulation under conditions of nitrogen excess. According to this view, a reduced concentration of 2-oxoglutarate would make GlnB inhibit the kinase of NRI and lead to a lack of activation of glnA expression and, hence, to lower levels of GS. However, the physiological 2-oxoglutarate concentrations under excess-nitrogen conditions are mostly higher than the above-mentioned second Kd. For instance, in E. coli cells growing in a chemostat with a glucose-limited excess-ammonium medium or in batch culture with excess glucose and ammonium, the cellular 2-oxoglutarate concentration varied between 0.1 and 0.9 mM depending on the growth rate (384) or was 0.5 to 0.6 mM (135, 342), respectively. Hence, cells would not be able to make a switch from inactive GlnB–2-oxoglutarate3 to active GlnB–2-oxoglutarate and inhibit the autophosphorylation of NRII as a response to the changes in the 2-oxoglutarate concentration. It is noteworthy that it would be no problem for the recent view that GS together with GOGAT is also the primary producer of glutamate under excess-ammonium conditions (see “GS-GOGAT Activity under Conditions of Internal Nitrogen Sufficiency?” below). Alternatively, one should accommodate the likelihood that glutamate competes for the binding of 2-oxoglutarate to GlnB and thereby greatly increases the corresponding effective binding constant.
Fig 6.
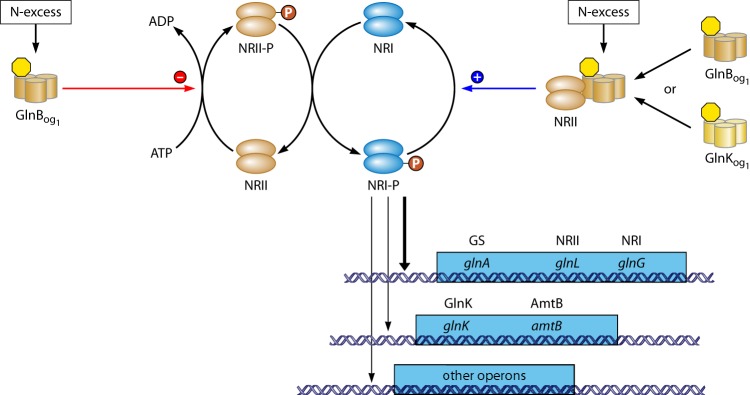
Transcription regulation of nitrogen-regulated genes by NRI-NRII. The heart of the figure displays the phosphorylation cycle of the NRI-NRII two-component system. At the bottom right part of the figure, the glnALG and the glnK-amtB operons (and other operons) that are regulated by the response regulator NRI-P are shown. The transmitter protein NRII features autokinase properties and can be phosphorylated by ATP to yield NRII-P. The autokinase activity is inhibited by unmodified GlnB carrying a single 2-oxoglutarate molecule. When the same molecular species forms a complex with NRII, it stimulates the dephosphorylation of NRI-P to yield NRI. By analogy, the corresponding GlnK species might do the same. A blue arrow denotes a positive effect, and a red arrow indicates a negative effect.
The autophosphorylation reaction is reversible when purified NRII-P is incubated with ADP (191). It seems, therefore, that the cellular ATP/ADP ratio may have control over the extent of autophosphorylation of NRII (418) and indirectly over the phosphorylation of NRI.
Phosphotransfer from NRII-phosphate to NRI.
Phosphorylation of NRI can occur via two different physiological routes. The major route is a phosphotransfer from NRII-phosphate (NRII-P) (reaction II) (Table 6). Isolated NRII-P, free of ATP, phosphorylates NRI in the presence of Mg2+ (191, 416). This suggests that NRII does not catalyze a direct transfer of γ-phosphate of ATP to NRI but engages more intricately in the reaction through the intermediate formation of NRII-P. Incubation of NRI-P with ADP results in the formation of ATP and disappearance of NRI-P and requires NRII and Mg2+ (191), suggesting that the phosphotransfer of NRII-P to NRI is a thermodynamically reversible reaction. In addition, it seems that the phosphotransfer from NRII-P to NRI is much faster than the autophosphorylation of NRII, because when NRI was phosphorylated with [γ-32P]ATP in the presence of NRII, in vitro, label appeared almost solely in NRI and hardly in NRII under the experimental conditions (124, 168). The (initial) rate of phosphotransfer from NRII-P to NRI is not influenced by GlnB (124, 168).
The second route to the synthesis of NRI-P is a phosphotransfer from small phosphate donors like acetyl-phosphate, the transfer of which also requires Mg2+ (419, 420). This reaction might be relevant physiologically for the activation of NRI in wild-type cells to facilitate the shift from nitrogen-rich growth conditions to nitrogen-poor growth conditions (419, 420). Under the former growth condition, the cellular concentration of NRII (and NRI) is very low, because the level of the two regulators themselves is nitrogen regulated. It is this activity, i.e., the phosphotransfer from a small phosphate donor to the signal receiver protein NRI, that may be taken to suggest that NRI itself is the enzyme that catalyzes its own phosphorylation, rather than the sensor protein kinase NRII, as has been proposed (182, 418, 421). NRII-P may serve as the substrate only for the phosphorylation of NRI, and NRI may catalyze the phosphoryl transfer. On the other hand, a kinase is defined as a phosphotransferase with ATP as the substrate, and NRI does not readily accept a phosphoryl group from ATP without the help of NRII.
Dephosphorylation of NRI-phosphate.
The acyl phosphate bond of NRI-P is naturally unstable and hydrolyzes at pH 7.0, with a t1/2 of about 6 min (168, 191). This reaction may be referred to as the autophosphatase activity of NRI-P (reaction III) (Table 6 and Fig. 6) (168, 191). Because histidine 139 of NRII is not required for the dephosphorylation of NRI-P, this phosphatase activity is not the reversal of the phosphotransfer reaction (174, 422). Based on a high-resolution NMR structure of the beryllofluoride-activated receiver domain of NRI, it has been suggested that the His84 of NRI may be involved in the catalysis of autodephosphorylation of NRI-phosphate (199).
Presumably, to enable a quick response to a changing environment, nonuridylylated GlnB complexed with one 2-oxoglutarate molecule (indicator of a nitrogen-rich state [see above]) stimulates an NRI-P phosphatase activity of NRII, which is known as the regulated phosphatase activity of NRII (168). The paradox discussed above in “Autophosphorylation of NRII,” i.e., the difficulty to lose two of the three 2-oxoglutarate molecules complexed to GlnB because the 2-oxoglutarate concentration remains saturating after an ammonium upshift, also seems to apply here. However, glutamate might solve the paradox in this case, since it enables GlnB to stimulate the dephosphorylation of NRI-P in vitro, provided that the glutamate concentration is >5 mM (134, 169). Probably, GlnB may also be bound with ATP and glutamate instead of ATP and 2-oxoglutarate, although the stoichiometry of the complex is unknown. As in cells growing under excess-ammonium conditions, the measured intracellular glutamate concentrations are no less than 30 to 40 mM (343, 410, 423), and even in the range of 70 to 150 mM (135, 342), this backup control by glutamate might be operative. On the other hand, the binding of glutamate to GlnB must be seen in its competition with the binding of 2-oxoglutarate; the presence of the latter in a 10-fold excess over its binding constant should increase the apparent binding constant for glutamate by a factor of 10.
NRII homodimers containing a single substitution in one of the three domains, including the ATP and GlnB binding site, showed a decreased GlnB-stimulated NRI-P phosphatase activity; heterodimers containing these variant subunits were formed in various combinations, and with some of them, complementation of the GlnB-activated phosphatase activity was observed in vitro (424). On the basis of these results, it was suggested that the three domains collaborate to function as a phosphatase (424). However, it was concluded from additional (de)phosphorylation experiments in vitro by NRII with or without GlnB using variants of NRI containing a single-amino-acid substitution or deletion in the N-terminal domain close to the site of phosphorylation that the regulated dephosphorylation of NRI-P is a result of a collaboration between the NRII-GlnB complex and NRI-P and, thus, that NRII is not a true phosphatase (425). The argument may not be quite convincing, however, as any specific phosphatase requires some involvement of its substrate.
GlnB-UMP does not interact with NRII; therefore, uridylylation of GlnB reduces only the concentration of unmodified GlnB capable of interacting with NRII (389). Both GlnB and GlnK are potent activators of the NRI-P phosphatase activity of NRII (138).
A strain with an NRII variant, which is disabled in terms of reducing the degree of phosphorylation of NRI-P, features a so-called “GlnC phenotype”: it is unable to reduce the expression of glnA and other nitrogen-regulated operons in response to ammonium in the growth medium (167). NRII variants resulting in strains having a GlnC phenotype may have a substitution located near His139, the site of autophosphorylation (426).
Signal transduction by the nitrogen-PTS?
It has been argued that functionally related proteins are likely to evolve in parallel and often are coded in a single operon (365, 427). Because rpoN (σ54), ptsN, and ptsO are within the same operon (257, 262), and this has been shown to be the case for many more bacteria (248, 257), it seemed plausible that ptsN and ptsO might regulate σ54-dependent promoters. Indeed, in comparison with wild-type K. pneumoniae, ptsN and ptsO mutants showed increased (2- to 3-fold) and decreased (2-fold) σ54-dependent activities of the glnAp2 promoter, respectively (262, 265). Neither the promoter of σ54 itself (rpoNp) nor σ70-dependent promoters were affected, though (262, 265). In contrast, a ptsN mutant showed a slightly decreased glnAp2 promoter activity (1.5-fold) in comparison to wild-type E. coli (257). Transcription of the Pseudomonas putida xylS operon was decreased (2- to 3-fold) by ptsN in E. coli (428). Thus, only modest effects of the nitrogen-PTS on σ54-dependent transcription of nitrogen-related genes have been observed.
Although the nitrogen-PTS has been proposed to play a general role in nitrogen assimilation and to provide a link between carbon and nitrogen assimilation (257, 259, 429, 430), recent evidence does not corroborate this view, at least not for E. coli. In particular, the phenotypes associated with a lack of ptsN were all shown to be due to the presence of a nonfunctional version of the ilvG gene encoding acetohydroxy acid synthase II (AHASII) (431). Indeed, various other roles, not related to nitrogen assimilation, for the components of the nitrogen-PTS have been put forward for EINtr and Npr but especially for EIIANtr. These roles are consecutively discussed below.
The EINtr protein possesses a GAF domain (named after some of the proteins in which it is found) at its N-terminal domain (432, 433). The GAF domain of Azotobacter vinelandii NifA has been shown to bind a small molecule (2-oxoglutarate) that regulates its activity (434). Similarly, the N-terminal GAF domain in EINtr may regulate the catalytic activity of EINtr (or another protein) upon binding of a small molecule (432, 434). Remarkably, in spite of the lack of a GAF domain (259), EI involved in the glucose-dependent PTS has been shown to bind 2-oxoglutarate (435).
The dephosphorylated protein Npr decreases lipid A synthesis of the lipopolysaccharide layer by inhibiting the enzyme LpxD (436), the N-acyltransferase that catalyzes the third step of lipid A biosynthesis (437).
The IIANtr protein, unlike the IIAGlc protein, is involved in neither sugar transport nor catabolite repression (428). To date, four different activities have been observed for IIANtr. First, the dephosphorylated protein EIIANtr has been shown to bind the K+ transport protein TrkA and to inhibit TrkA-mediated transport (438). TrkA is an essential subunit of the Trk potassium transport systems (439, 440), which have a low affinity for potassium and are constitutively expressed (441). In addition, the dephosphorylated protein EIIANtr was shown to bind the sensor kinase KdpD, stimulating its kinase activity (442). The latter results in an increased level of the phosphorylated transcriptional regulator KdpE, which stimulates the expression of the kdpFABC operon. This operon encodes the KdpFABC K+ transport system (442), which has a high affinity for potassium (441). Thus, the nonmodified EIIANtr protein regulates both the activity of the low-affinity potassium uptake systems and the expression of the high-affinity potassium uptake systems, indicating the importance of protein EIIANtr for potassium homeostasis. Second, it has been shown that IIANtr modulates the pho regulon, which comprises over 30 genes. Comparison of mutants lacking EIIANtr with mutants deficient for phosphorylation suggested that nonphosphorylated EIIANtr modulates the expression of pho. In addition, EIIANtr was found to bind directly to the sensor kinase PhoR and, through this interaction, to increase the amount of the phosphorylated response regulator PhoB (443). Importantly, the phosphate starvation response was controlled by nitrogen-PTS independent from its effects on K+ uptake. Third, EIIANtr has also been shown to be a member of the σE regulon and to play a role in maintaining cell envelope integrity upon stress (444). Fourth, for Salmonella, EIIANtr has been demonstrated to interact directly with SsrB, the response regulator of the SsrA/SsrB two-component system that mediates survival and replication within host cells (445).
In sum, the early proposal that the nitrogen-PTS connects carbon to nitrogen assimilation seems to have lost most of its credit; instead, a variety of other seemingly unrelated activities and functions have surfaced for the nitrogen-PTS components.
Signal transduction toward lysine-acetylated proteins?
Recently, acetylation of lysine residues has been shown to function as a reversible posttranslational modification of proteins that plays an important role in regulation of eukaryotic metabolism (446, 447). Protein lysine acetylation also occurs in Salmonella enterica and coordinates carbon source utilization (448). For E. coli, 85 proteins (449) or 91 proteins (450) were identified as having acetylated sites, but although both sets included enzymes in amino acid metabolism, none of the proteins of the central nitrogen assimilation metabolism were part of the set. Thus, it seems likely that regulation by protein acetylation does not affect this part of N metabolism. Proteins discussed in this review that are acetylated are Crp, glutaminase, pyrophosphatase, and carbamoyl phosphate synthetase. However, with enhancing proteomics, we may be in for the discovery of more covalent modification states of proteins, and the potential importance of this remains open.
Gene Expression Regulation
In this section, we focus on the fourth aspect of regulation, i.e., that of regulation by adjusting gene expression. In principle, this issue involves the operon organization of genes, transcription, stability, as well as translation, but the focus here rests on the former two aspects, as less is known of the latter two. Overviews of all of the transcriptional regulators that pass in review in this section are shown in Fig. 7.
Fig 7.

Nitrogen-related transcription factors. A summary of the nitrogen-related transcription factors is presented. The transcription factors that have well-known effects on the genes/operons displayed in the figure (Nac, ArgR, IHF, Lrp, CRP-cAMP, and NRI-P) are shown in gray boxes with solid lines. GadE and FNR are shown in a dotted box, because the evidence for transcription regulation of the gltBDF operon by these factors is incomplete. Positive effects are indicated by blue circles, and negative effects are indicated by red circles. NRI-P and CRP-cAMP may have positive or negative effects on the expression of the glnALG operon, depending on the actual conditions.
Transcription regulation of glnALG.
The glnA gene (33, 34), encoding GS, is part of a complex operon, the glnALG (451) or glnA-ntrBC operon (452; for reviews concerning two-component systems, see references 164–166).
(i) DNA elements in cis of the glnALG operon.
There are two promoters upstream of the glnALG operon (453–455), a distal weak glnAp1 promoter containing the Eσ70 recognition site and a proximal strong glnAp2 promoter containing the Eσ54 recognition site (197, 253, 254, 453, 455, 456) (Fig. 8A). A Crp binding site is located upstream of the glnAp1 promoter. In addition, upstream of the glnAp2 promoter, five binding sites for NRI are present, which NRI binds unequally (197). DNase I footprint analysis has revealed the order of apparent affinity of NRI to these five binding sites. NRI has the highest affinity for the two most upstream binding sites, sites 1 and 2; a lower affinity for sites 3 and 4, in the middle of the upstream region of glnA; and the lowest affinity for site 5, proximal to the glnAp2 promoter (197) (Fig. 8B). The numbers of these NRI binding sites follow (197). Binding sites 1 and 2 are separated from each other by 3 turns of a helix, and these together have been called the enhancer of glnALG (457, 458). NRI binding sites 3 and 4, between the enhancer and the glnAp2 promoter, have been called the “governor” of the glnAp2 promoter (459).
Fig 8.
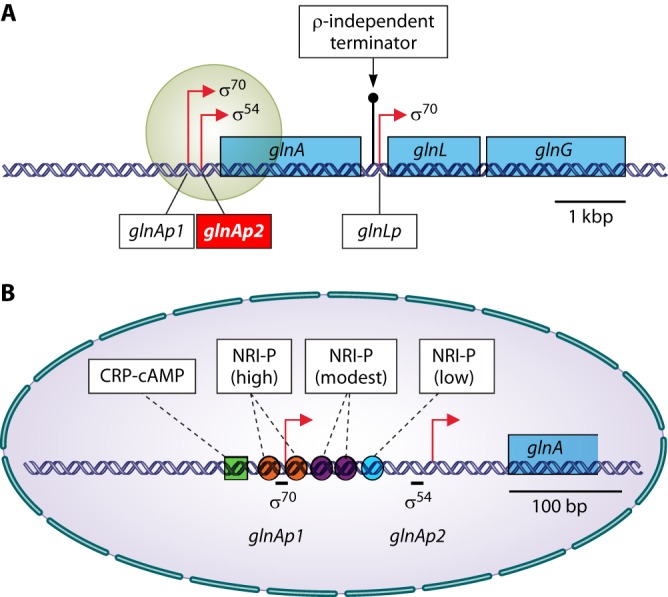
Transcription regulation of glnALG by NRI-P. (A) The glnALG region. The glnA, glnL, and glnG genes constitute the structural genes of the operon. Thin arrows indicate the transcription start sites of the glnAp1 (σ70), glnAp2 (σ54), and glnLp (σ70) promoters. The black-dotted arrow denotes the position of the ρ-independent transcription terminator site. The green circle indicates the upstream region of the operon that is presented in panel B. (B) The glnA upstream region is shown enlarged. This region contains a σ54 promoter, a σ70 promoter, five NRI-P binding sites (circles), and one CRP-cAMP binding site (square). The five NRI-P binding sites have been qualified from left to right as high-, high-, modest-, modest-, and low-affinity sites, respectively.
There is an additional Eσ70 promoter between glnA and glnL (glnLp), which, like glnAp1 (454, 455), has an overlapping binding site for NRI, as assessed by DNase I footprint analysis (460). Moreover, the intercistronic glnA-glnL region contains a Rho-independent terminator (460) and one (461) or two (462) repetitive extragenic palindromic (REP) sequences. A REP sequence possesses an imperfect dyad symmetry that has the potential to form a stem-loop structure in the DNA (461). REP and Rho-independent terminator sequences have been shown to bind DNA gyrase (463) and DNA polymerases I (464) and IV (465) (see references 466–469 for more information concerning such DNA elements). Each of these two different DNA elements (REP and Rho-independent terminator) may contribute to reduce, although to different extents, the readthrough of the transcription initiated from the glnAp2 promoter and, thus, decrease the expression of glnLG. Expression initiated at the glnAp1 and glnLp promoters results in a housekeeping level of the encoded proteins.
(ii) Initiation of glnALG transcription.
The consensus DNA binding site of NRI-P counts 17 nucleotides (TGCACCAnnnTGGTGCA). The first and last 7 nucleotides (in uppercase type) have dyad symmetry (470). Although phosphorylation of NRI affects neither dimerization of wild-type NRI (185) nor binding to a single DNA binding site (470), upon phosphorylation, preexisting NRI dimers form oligomers, and this oligomerization rather than dimerization is required for activation (471, 472). The oligomers bind tightly to enhancer sequences and contact the σ54 holoenzyme form of RNA polymerase, bound to the promoter, by means of a DNA loop (457, 473–475). This complex hydrolyzes ATP to catalyze open promoter complex formation by σ54-RNA polymerase (189; for a review, see reference 476).
(iii) Transcription activation by NRI.
Phosphorylation of NRI at Asp54 resulted in a rearrangement of the α-helices and the β-strands located in a region called the “3445 face” in the N-terminal domain, especially an axial rotation of helix 4. This event created an exposed hydrophobic surface, as has been shown by NMR spectroscopy of transient phosphorylation of NRI using a high concentration of a low-molecular-mass phosphoryl molecule (198). This helix, upon phosphorylation, contacts the beginning of the central domain within the same monomer or the partner molecule of the dimer, as has been elegantly shown by cleavage of the dimers by iron chelators tethered to cysteines engineered into the N-terminal domain within this helix (477). This contact then makes the dimer ready for oligomerization by moving a residue that prevented oligomerization but was also needed for catalysis into the active site, as was shown by X-ray crystallographic analysis of (i) the central domain and (ii) the amino-terminal and central domains together (478). Oligomerization (actually heptamerization) completes the active site of ATPase (478). Every monomer in the ATPase domain contains a novel loop. The seven monomers form a ring structure with a hole in the center of the heptamer. The loops together, which point toward the center of the pore, may form an extended structure that could contact σ54 and stimulate open complex formation (478). Open complex formation by the σ54-RNA polymerase holoenzyme has been reviewed previously (479).
NRI-P binding sites 1 and 2 are located normally approximately 140 and 100 bp upstream of the transcription start site, respectively (197). However, when the two binding sites 1 and 2 together were moved more than 1 kb from the transcription start site, transcription activation of glnAp2 by NRI-P did not decrease in vivo (458). Similarly, moving binding sites 1 and 2 together approximately 1 kb upstream or downstream of glnAp2 on linear DNA templates resulted in efficient open complex formation in vitro (457). Thus, these two binding sites together are like the transcription enhancers in eukaryotic cells (458), hence clarifying their name enhancer. Two NRI-P dimers strongly bind the enhancer in a cooperative way (470, 471). The function of the enhancer is to increase the local concentration of NRI-P (201, 457, 458, 471, 473, 480, 481). This facilitates oligomerization (472). The enhancer–NRI-P complex may move to the closed complex, comprising the glnAp2 promoter and bound σ54-RNA polymerase, through a DNA loop (475, 481) and then catalyze open complex formation (480) (Fig. 9).
Fig 9.

Transcription activation at the glnAp2 promoter. (A) Two NRI-P dimers bind the enhancer, which is composed of two binding sites for dimers. The enhancer is located at positions −108 and −140 with respect to the transcription start site. The RNA polymerase holoenzyme containing sigma factor 54 (Eσ54) binds to the glnAp2 promoter to form a closed complex. (B) Two NRI-P dimers contact the Eσ54 holoenzyme by means of DNA loop formation (intermediate complex). (C) Hydrolysis of ATP by NRI-P results in the formation of an open complex, i.e., DNA strand separation at the promoter. (Adapted from reference 181.)
The function of the governor is to limit the maximal expression of glnA. Expression of the glnALG operon not only results in an increase in the cellular GS concentration but also increases the cellular concentration of NRI and NRII. The latter two may again stimulate the glnALG operon, which results in a positive-feedback loop and, consequently, a potentially huge cellular concentration of GS. Binding of NRI-P to the governor brings the expression of the glnALG operon to a halt. This may possibly occur by preventing productive contact between enhancer-bound NRI-P and promoter-bound σ54-RNA polymerase (459). However, this mechanism awaits experimental verification. Intricate mechanisms keeping positive-feedback loops that are important for starting up new activities in check appear to be important in biology (482). Previously, it was observed that hyperexpression of NRI-P reduced the activity of the glnAp2 promoter, but in hindsight, it can be concluded that this apparently happened in a manner independent of the governor binding sites 3 and 4 (483).
NRI-P can stimulate the expression of genes only when their promoter contains an Eσ54 binding site. This binding site is different from that of the housekeeping Eσ70 binding site and consists of the highly conserved doublets GG and GC, 24 and 12 bp upstream of the transcription initiation point, respectively (253, 456).
When E. coli is growing on a nitrogen-rich source, unmodified GlnB stimulates the phosphatase activity of NRII, which dephosphorylates NRI-P (reaction III) (Table 6). The glnALG operon is then transcribed at the basal level by σ70-RNA polymerase holoenzyme from the glnAp1 and glnLp promoters, which may be repressed by nonphosphorylated NRI (1). In addition, the latter is unable to stimulate gene expression.
(iv) Transcription regulation of glnALG by Crp.
The enhancer sequence upstream of glnA overlaps the binding site of Crp, and thus, NRI-P and Crp compete for the same site, enabling an integrated transcription response to alterations in N and C metabolism. Indeed, Crp has direct and indirect effects on the expression of glnALG, and this is discussed below.
Increased levels of intracellular cAMP correspond to a hunger signal, referring to hunger for energy or carbon. Physiologically, it may make sense that such a signal represses other activities that may no longer be necessary because energy or carbon that is required for growth is missing. cAMP added to a culture medium containing glucose as a carbon source and excess ammonium as the nitrogen source increased the level of GS and GDH and decreased the level of GOGAT in wild-type cells, but cAMP had no effect on these activities in a crp mutant (484). Thus, Crp-cAMP played a role in regulation of nitrogen metabolism. More recently, Crp was shown to moderately activate transcription of the σ70-dependent glnAp1 housekeeping promoter but to strongly (21-fold) repress the activity of the σ54-dependent glnAp2 promoter, as assessed by comparing β-galactosidase activities of various glnAp1p2::lacZYA promoter constructs in wild-type, cya single, and cya crp double mutants (455). The overall effect of Crp on glnA is a 4-fold expression reduction, which was in agreement with a similar reduction of GS activity in vivo. Therefore, it was proposed that a physiological role of Crp activation of glnAp1 is to compensate for its repression of glnAp2, allowing a low but significant housekeeping expression level of the glnALG genes (455). The glnAp2 promoter is activated by two NRI-P dimers bound strongly, in a cooperative way (470, 471), to the enhancer (457, 458, 470). Binding of Crp may result in bending of the DNA (see “cAMP Receptor Protein,” above), which prevents NRI-P from contacting the σ54-RNA polymerase holoenzyme and inhibits transcription from glnAp2 (453).
The second, indirect, type of regulation of glnALG by Crp is realized via the glutamine ABC transporter. The transcription of the glnHPQ operon is regulated by both NRI-P and Crp [see “Transcription regulation by and of other nitrogen assimilation-associated genes. (v) The glnHPQ operon,” below]. Crp activates expression of glnHPQ, resulting in an increased carbon source-dependent uptake capacity of glutamine. When glutamine is present in the growth medium, the imported glutamine increases the cellular glutamine concentration, which stimulates UTase to deuridylylate GlnB-UMP toward unmodified GlnB. The latter stimulates NRII to dephosphorylate NRI-P, resulting in abolishment of the expression of glnALG to around the housekeeping level of glnA (as described above) (453, 485, 486).
Transcription regulation of the GOGAT genes gltB and gltD.
The two GOGAT genes (gltB and gltD) are part of the gltBDF operon (50, 53). The product of the third gene of this operon, GltF, has a molecular mass of 26 to 30 kDa (50, 51). Expression of gltBDF is regulated by an unusually large number of global and local transcription regulators (487). It is activated by Lrp (488–491), by IHF (490, 492), and by GadE, a transcriptional regulator of genes involved in pH homeostasis (493) (see below). In addition, transcription of gltBDF is repressed by Crp-cAMP (484, 490), by the fumarate nitrate reductase regulator (FNR) (494), by Nac (32, 53), and by ArgR, the arginine repressor (490) (see sections on Nac and Crp-cAMP below).
(i) Activators.
(a) Lrp.
Lrp regulates the expression of many genes whose gene products are involved in the synthesis of amino acids (for reviews concerning Lrp, see references 241, 243, 495, and 496). Lrp is a positive regulator of GOGAT, and regulation is relatively insensitive to exogenous leucine (497), at least for growth in minimal media compared to rich media (498). Lrp dimers bind cooperatively to three sites of the gltBDF promoter, with the centers of the binding sites located at bp −152, −215, and −246 relative to the transcription start site, as assessed by DNase I footprint analysis in vitro (490, 491). Thus, the region protected by Lrp is large, approximately 100 bp. The distance between the proximal Lrp binding site and the −35 element of the Eσ70 promoter is approximately 120 bp: the three Lrp binding sites are unusually far upstream of the transcriptional start site compared to other Eσ70 promoters (499). Moreover, this analysis detected a phased hypersensitivity covering the three Lrp binding sites, suggesting phased DNA bending. DNase I footprinting in vitro also revealed that the three sites must be in phase with each other to be protected from DNase I digestion (491). By measuring β-galactosidase activity of a strain containing a chromosomal gltBp::lacZYA fusion, maximal β-galactosidase activity was observed when the transcription start site was in phase with all three Lrp binding sites (491). The results from in vivo and in vitro analyses together suggested that the three Lrp dimers may wrap the gltBDF promoter DNA like a nucleoprotein complex (491).
(b) IHF.
It was shown by gel mobility shift and DNase I footprinting assays that between this Lrp-gltB promoter DNA complex and the Eσ70 promoter, IHF could bind to the DNA, thereby covering the two overlapping 13-bp consensus binding sites centered at bp −85 and −89 relative to the transcription start site (492). In addition, DNase I footprinting revealed hypersensitivity when IHF bound the gltB promoter, indicative of DNA bending by IHF (492). Gel mobility shift analyses showed that Lrp and IHF alone could shift the promoter DNA independently from each other, but together, an extra shifted band was observed, perhaps representing a ternary complex (492). Moreover, binding of IHF was required for transcriptional activation of gltBDF, as transcription of gltBp::lacZYA was abolished in an IHF-negative mutant (492). This result suggests that IHF may activate transcription by bringing the Lrp complex in the vicinity of RNA polymerase (492).
(c) GadE.
The transcriptional regulator GadE (systematic name YhiE) has been implicated as a regulator of the expression of genes encoding proteins involved in maintenance of pH homeostasis (500) (see “GOGAT functions,” below). Induction of gltD was detected by microarray analysis of a strain overexpressing GadE versus the wild type growing in glucose minimal medium at pH 5.5 (493). This regulation of GadE may be direct, as the gltB promoter DNA could be gel band shifted by purified GadE. In addition, the binding capacity of GadE to the gltB promoter was increased in the presence of glutamate (493). However, the location of the GadE binding site at the promoter region of gltBDF remains to be determined, as is the regulation of the gltBp::lacZYA reporter fusion by a gadE-negative mutant versus the wild type.
(ii) Repressors.
(a) Crp-cAMP.
Prusiner et al. concluded from growth experiments with the wild-type and cya and crp mutant strains growing on glucose minimal medium with or without cAMP that GOGAT activity is regulated negatively by Crp-cAMP (484). As Crp-cAMP has a regulon of some 400 operons, this negative regulation of GOGAT activity by Crp-cAMP could have been indirect. However, Paul et al. (490) have convincingly shown that the repression of gltBD by Crp-cAMP is direct, as follows. First, a gel mobility shift analysis showed binding of purified Crp-cAMP to the wild-type gltB promoter DNA, but binding to the same promoter without the Crp consensus binding site was absent. Second, a DNase I footprint analysis of Crp-cAMP binding to the gltB promoter DNA showed a protected region covering the 23-bp Crp consensus binding site plus hypersensitivity at this site, indicative of bending by Crp-cAMP. The center of the Crp binding site is at approximately bp −65 relative to transcription start site, between the IHF binding sites and the −35 hexamer promoter element. Third, a comparison was made of the β-galactosidase activity between a crp+ strain possessing a chromosomal gltBp::lacZYA reporter fusion containing the wild-type promoter and a similar strain carrying a promoter with a mutated Crp binding site. Both strains were grown in glucose-rich medium with or without dibutyryl-cAMP. The former strain showed a lower β-galactosidase activity with dibutyryl-cAMP than without. The β-galactosidase activity of the latter strain was similar with or without dibutyryl-cAMP in this medium. Thus, the gltBDF operon may be negatively regulated by Crp-cAMP, confirming the results of Prusiner et al. (484). In addition, gel mobility shift and DNase I footprint analyses have shown that Lrp, IHF, and Crp-cAMP could bind the gltB-promoter DNA independently of one another (490). As Crp-cAMP is normally an activator, the mechanism of negative regulation of gltBDF expression by Crp-cAMP remains to be determined. However, one interesting possibility has been proposed without yet testing it: Crp may bend the DNA in an opposite direction compared to that of IHF (490).
(b) Nac.
Two lines of evidence have been presented that expression of the gltBDF operon is repressed by Nac [see “Transcription regulation by and of other nitrogen assimilation-associated genes. (i) Transcription regulation of and by nitrogen assimilation control protein,” below]. First, a comparison of GOGAT activity of a wild-type strain grown on glucose-arginine medium (which is regarded as being severely nitrogen limiting, resulting in a high cellular concentration of Nac) to that of the same strain grown on glucose-nitrogen-rich medium showed a 4-fold decrease of GOGAT activity. However, a nac deletion strain compared to the wild type grown on glucose-arginine medium showed an approximately 2-fold increase in GOGAT activity (53). Second, a 3-fold or higher repression of the gltBDF operon by Nac was observed by determination via microarray analysis of the NRI regulon, which includes the Nac regulon (32). However, regulation of the gltBp::lacZYA reporter fusion by a nac-negative mutant versus the wild type should confirm this indirect evidence by the microarray analysis. Also, direct binding of Nac to, and location at, the promoter region of gltBDF remains to be determined.
(c) FNR.
A microarray analysis of an fnr-negative strain versus the wild type revealed gltBDF as a candidate operon to be regulated by FNR (501). The center of the putative 14-bp FNR binding site in the promoter of gltB has been mapped at 26.5 bp upstream the transcription start site by regulon DB version 7.0 (215), which compared automatically the promoter sequence of gltB with the consensus binding sequence of FNR. The binding site overlaps partly the −35 hexamer, the binding site of RNA polymerase. Thus, the negative regulation of gltBD by FNR may be due to steric hindrance of binding of RNA polymerase to the gltB promoter by FNR. However, the location of the FNR binding site remains to be confirmed experimentally.
(d) ArgR.
Surface-enhanced laser desorption/ionization (SELDI) is a technique by which one fishes in a crude cell extract for proteins that bind to specific (here gltB) promoter DNA followed by a determination of the mass of the bound protein(s) by mass spectrometry (MS). By using SELDI, besides Lrp, Crp, and subunits α and β of IHF, a new protein was also detected, with a mass corresponding to that of ArgR (490). The detection of ArgR as a repressor was confirmed 4-fold: (i) the β-galactosidase activity of a strain carrying the chromosomal gltBp::lacZYA fusion growing on glucose minimal medium decreased by approximately 50% when arginine, the corepressor of ArgR, was added to the medium; (ii) the β-galactosidase activity of gltBp::lacZYA in an ΔargR deletion strain was increased twice compared to the wild-type background, and thus, ArgR may repress gltBDF; (iii) ArgR-Arg could bind to the gltB promoter containing the 18-bp ArgR-Arg binding site, as assessed by gel mobility shift analysis; and (iv) DNase I footprint analysis revealed that ArgR-Arg bound to two 18-bp consensus binding sites, centered at bp −332 and −353 relative to the transcription start site, upstream of the most distantly bound Lrp (490). The third and fourth points show that repression of gltBDF expression by ArgR is direct. Although the position of the two ArgR binding sites is far upstream, ArgR could still repress gltB transcription. However, its mechanism remains to be determined.
Finally, binding of RNA polymerase alone to the gltB promoter DNA was unstable, as indicated by an unclear DNase I footprint in vitro. Binding of Crp-cAMP alone or Lrp plus IHF together may stabilize the binding of RNA polymerase, as the two combinations revealed clear footprints of RNA polymerase to the gltB promoter DNA in vitro, although the footprints were different from each other (490).
Transcription regulation of gdhA.
As described above, strong repression of the K. pneumoniae gdhA gene required NacK to be a tetramer (207). Indeed, for this strong repression, >50-fold (observed when cells are growing in glucose glutamate medium, which is regarded as a severely nitrogen-limiting condition), the two dimers of the NacK tetramer bind to the consensus binding sites, the center nucleotides of which are located at bp −89 and +57 relative to the transcription start site (502). This location of the two NacK binding sites, i.e., up- and downstream of the transcriptional start site, suggests that binding of the NacK tetramer may block the binding of σ70-RNA polymerase. In addition, the long DNA distance between the two NacK binding sites requires and allows loop formation (203, 502). However, when one of these two binding sites (or both) was absent, strong repression of gdhA was lost in vivo (502). This is in agreement with the observation, as described above, that NacL11KK (which is a dimer) fails to repress gdhA strongly, in contrast to NacWT (tetramer) (207). In addition, changing the face of the two binding sites, e.g., by deleting 5 or 15 bp or inserting 5 bp, resulted in a significant reduction in the repression of gdhA in vivo, while insertion or deletion of 10 bp between these two binding sites, thus leaving the face between them intact, had only little effect (502). Thus, for strong repression of gdhA by NacKWT, the two dimers bound to each binding site may contact each other via loop formation (203, 502).
The expression of gdhA in E. coli is also repressed by Nac (503). However, whether the mechanism of repression of gdhA in E. coli by NacE is similar to that of NacK remains to be determined. The expression of gdhA may also be affected by the global transcriptional regulator FNR during growth in high-osmolarity media, but the underlying mechanism is unknown (494). Finally, gdhA transcription is regulated by ArgP. ArgP is a nucleoid-associated protein and also a LysR-type transcriptional regulator (504, 505). K. pneumoniae as well as E. coli gdhA genes have been shown to feature lysine-sensitive activation by ArgP (504, 506).
Transcription regulation by and of other nitrogen assimilation-associated genes.
(i) Transcription regulation of and by nitrogen assimilation control protein.
Components involved in regulation may surface at two positions, (i) where they regulate and (ii) where they are regulated. We first discuss the role of Nac as a regulator. In a subsequent section, we discuss its role as a component that is being regulated.
(a) Transcription regulation by nitrogen assimilation control protein.
While the LysR family harbors activators or repressors (209), NacK can be either. In general, it activates the transcription of genes whose products are involved in ammonium production (e.g., hut and ure), whereas it represses genes whose products use ammonium (e.g., gdhA) (203, 507). Also, NacE appears to function as a repressor and activator altogether (32). NacE has been shown to complement NacK in vivo (202). Both Nac proteins control genes that have σ70-dependent promoters but are themselves transcribed by the σ54-RNA polymerase holoenzyme when activated by NRI-P under nitrogen-limited conditions (32, 401, 508, 509). Therefore, it has been suggested that Nac may function to connect NRI-activated transcription by σ54-RNA polymerase holoenzyme to the expression of genes having σ70-dependent promoters (204).
To date, the Nac regulon in E. coli counts 9 operons, as detected by microarray analysis (32). All of the gene products of these operons are involved in nitrogen metabolism. In K. pneumoniae, 99 unique DNA regions were detected by using the chromatin immunoprecipitation (ChIP) technique (211), a technique by which (promoter) DNA bound to a transcriptional regulator can be detected in vivo. For 84 of the 99 regions, the association with purified Nac was confirmed by electrophoretic mobility shift assays in vitro. Regulation of 15 of the ChIP-identified fragments was tested further by primer extension analysis: 9 were activated, and 6 were repressed (211). As in E. coli, many detected genes of the Nac regulon in K. pneumoniae are involved in nitrogen metabolism. However, unlike in E. coli, in K. pneumoniae, this regulon also contains genes involved in carbon and energy metabolism. Thus, not only does the Nac regulon of K. pneumoniae cover more metabolic functions than that of E. coli, it also has more genes in its regulon (211).
One explanation for this difference is that K. pneumoniae has Nac-regulated operons which are lacking in E. coli, like ure, encoding urease (211). Another explanation is that some Nac-regulated genes or operons, present in both organisms, have a point mutation in the Nac consensus binding site in the promoter of the gene/operon in E. coli (203, 510), e.g., the dadAX operon in E. coli compared to the dadAB operon in K. pneumoniae, coding for the smaller subunit of d-amino acid dehydrogenase and the catabolic alanine racemase, respectively (510). The point mutation in the Nac consensus binding site in the promoter of dadAX resulted in a loss of binding of Nac to the promoter DNA, as assessed by gel mobility assays. This resulted in a loss of regulation of this operon by Nac, as assessed by a d-amino acid dehydrogenase activity assay (510).
An observation with potential physiological relevance is the following. When NacK was cloned downstream of an IPTG-inducible promoter, the activity of hut and gdh, genes regulated by NacK, was dependent only on IPTG, even when cells were grown with excess ammonium (212). The authors of that study suggested that apparently no coeffector or modification of NacK was required for regulation (212). Indeed, binding of partially purified NacK from cells grown under conditions of nitrogen limitation or nitrogen excess, gel band shifted hutP DNA (the regulatory region of hut) equally well (206). In addition, potential nitrogen coeffectors, like glutamine, glutamate, 2-oxoglutarate, or ammonium at 10 mM, had no effect on the shifting of hutP DNA by NacK (206). Thus, NacK, in contrast to other LTTRs, is capable of transcriptional regulation without binding of a coeffector.
Nac downregulates its own synthesis (203). Goss and Bender have implicitly given three reasons for this negative autoregulation: (i) Nac is always in its active form, as there are no metabolite effectors controlling the activity of Nac (206); (ii) Nac is a promiscuous regulator, as it will bind to almost any AT-rich DNA to some extent; and (iii) growth is hampered by overproduction of Nac (511).
The concentration of NRI-P necessary for expression of nacE is higher than that for the glnALG operon (401) (see also below). This creates a hierarchy of gene transcription: first those regulated by NRI-P and then those regulated by Nac. Indeed, the hierarchy seems to be activation of transcription by NRI-P of genes encoding transporters for nitrogen-containing compounds first (32), followed by activation by Nac of genes whose protein products degrade poor nitrogen-containing compounds for the synthesis of ammonium and by repression by Nac of genes whose protein products use ammonium for biosynthesis (32) (see “Nitrogen Assimilation Control Protein,” above).
However, this NRI-P–Nac system seems to include an oddity. When cells are growing in a nitrogen-poor medium, NRI becomes phosphorylated and stimulates transcription of some 75 genes, including nac (32). However, when these cells experience a sudden ammonium shock, NRI-P is immediately dephosphorylated by the NRII-GlnB complex, and transcription of these genes ceases almost simultaneously. However, the Nac protein will still be present and active in stimulating transcription, even when these cells are exposed to a nitrogen-rich medium. Perhaps, binding of a coeffector to Nac, which is a LysR-type protein after all, may switch off the activity of Nac. However, such a coeffector has not yet been described.
(b) Transcription regulation of nitrogen assimilation control protein.
As described above, the expression of both nacE (401) and nacK (509) is activated by NRI-P, but in both organisms, nac was less efficiently expressed than glnA; i.e., its expression required a higher concentration of NRI-P, both in vivo and in vitro (401, 509). DNase I footprint analysis showed that two elements were responsible for this lower efficiency of nacK than of E. coli glnA (509): (i) the upstream enhancer of nacK possesses both a high- and a low-affinity binding site for NRI-P, while the upstream enhancer of glnAp2 possesses two adjacent high-affinity NRI-P binding sites, and (ii) the promoter of glnA bound σ54-RNA polymerase with a higher affinity than the nacK promoter.
Nac is both a transcription activator and a repressor. Indeed, one of the repressed genes is its own gene (213). As described above, NacK binds to its promoter as a tetramer, which includes a region from directly downstream of the σ54-RNA polymerase binding site toward the upstream high-affinity NRI-P binding site and partially overlapping the low-affinity NRI-P binding site (213). In spite of the observation that binding of NacK induces bending of the nacK upstream region (207, 213), the latter resulted neither in the loss of binding of σ54-RNA polymerase nor in the loss of binding of NRI-P to the high- and low-affinity binding sites, as assessed by DNase I footprinting (213). Despite an intact binding of these two proteins, NacK repressed its own promoter. However, in vitro transcription analysis showed that when increasing the distance between the NacK and σ54-RNA polymerase binding sites by 4 to 34 bp, without changing the distance between NacK and the two NRI-P binding sites, transcription increased (i.e., repression decreased) compared to that of the wild-type nacK promoter (213). It was hypothesized from this work that NacK represses its own gene by preventing the interaction between NRI-P and σ54-RNA polymerase. The authors of that study called this mechanism an antiactivation mechanism, which may be a result of the possibility of Nac to bend DNA (213). Thus, a possible overexpression of nacK by NRI-P is controlled by a negative-feedback loop of NacK. There is some evidence that this is also the case for the expression of nacE (202).
(ii) The glnE, glnB, and glnD genes.
(a) glnE.
The glnE gene, which encodes ATase, is located at around 66.8 min on the E. coli chromosome (87). The open reading frames upstream of glnE (orfXE) and glnE are cotranscribed, as observed by RNase protection assays (88). In addition, the intergenic sequence between orfXE and glnE is very small (24 bp), and no Rho-independent terminator-like structure was found in this region (88). The transcription start site of this operon is located upstream of orfXE (88), but it has not been mapped precisely. Transcription of the orfXE-glnE operon is neither regulated in response to nitrogen status nor dependent on σ54-RNA polymerase, as established by an RNase protection assay (88).
(b) glnB.
The glnB gene is located between 54.4 and 55.4 min on the E. coli chromosome, near glyA (144) and downstream of hmpA (144, 146). It encodes the regulatory protein GlnB. The two open reading frames upstream of glnB are transcribed in the same direction (orf1-orf2 [orfXB-glnB]) (144, 146).
Four transcription start sites of E. coli glnB have been described. The proximal transcription start site (P1) has been mapped by three studies at 33 bp upstream of the translation start codon of glnB, i.e., in the intergenic region between orfXB and glnB (144, 146, 512). This promoter accounts for the majority of the glnB transcripts (144, 512). Two additional glnB transcripts initiate at bp −95/−99 (P2) and bp −115/−118 (P3) upstream of the translation start, i.e., in the orfXB coding region (144, 512). Compared to the amount of P2 transcript, the amount of P3 transcript was very small. No −35 sequences for an Eσ70 promoter were located upstream of any the three transcript start sites, nor did the DNA upstream of the transcription start site P3 have a −10 consensus sequence for an Eσ70 promoter (144, 512). Consequently, perhaps, the observed P3-mRNA products are not physiologically relevant. The fourth promoter is located upstream of orf1 but is approximately 20% as efficient as the three other promoters together (144). Although the transcripts initiated from this promoter are perhaps not relevant for glnB physiologically, they are necessary for orf1 and orf2. This promoter has not been mapped.
None of the four promoters are regulated in response to nitrogen status, nor are they dependent on σ54-RNA polymerase, as established by RNase protection assays (146), primer extension analyses (144), and β-galactosidase measurements of a glnB-lacZ fusion (133, 144, 512). Thus, transcription of glnB is not regulated by nitrogen. In addition, none of the proteins ORF1, ORF2, GlnB, and HmpA are required for glnB expression (144).
He et al. (512) detected a binding sequence for the PurR repressor between the transcription start sites P2 and P1. PurR is responsible for repression of genes encoding enzymes required for purine biosynthesis (e.g., purF, encoding glutamine phosphoribosylpyrophosphate amidotransferase [513]), which is a pathway dependent on glutamine synthesis (5). Binding of PurR to promoter-operator sequences is increased when PurR is complexed with one of the corepressors hypoxanthine and guanine or both (514, 515). Although the promoter sequence of glnB could bind purified PurR complexed with the corepressor guanine, as shown by a gel retardation assay (512), its affinity was rather low, 33-fold lower than that for the promoter-operator sequence of purF (512). In addition, its binding site was upstream of P1, the major transcription start site. Indeed, expression of glnB-lacZ was only 2-fold repressed relative to the expression in a purR mutant, and this reduction of cellular GlnB influences neither the glnA expression level nor GS activity, independent of whether cells were growing under nitrogen-rich or nitrogen-limiting conditions (512). Therefore, the physiological relevance of the binding of PurR to the glnB promoter region remains uncertain.
(c) glnD.
The glnD gene, encoding UTase, is located between the map and dapD genes at position 199 of the E. coli chromosome (4.0 min on the genetic map) (88, 125, 381, 516). The intergenic sequence between map and glnD contains a Rho-independent terminator-like structure (517), which terminates map transcription before progressing into glnD (88). Using primer extension analysis, the transcription start of glnD was mapped at 5 bp (88), 7 bp (518), and 40 bp (124) upstream of the translation start codon. The former two measurements imply that the mRNA has a very short leader, while the latter means that the start site is located within the Rho-independent terminator-like structure. An Eσ70 factor binding site (519) may be present upstream of the former two transcription start sites, while an Eσ54 factor binding site (253, 456), although imperfect, could be detected upstream of the latter transcription start site, suggesting that transcription of glnD could be partly regulated by the nitrogen status of the cell (124). An imperfect Eσ54 factor binding site (124) is consistent with the observation of similar mRNA levels of glnD in an rpoN mutant (encoding the σ54 factor) and in the isogenic wild type, suggesting that the expression of glnD should not be regulated much by the nitrogen status (88).
Three studies, one using primer extension analysis (124), a second using an RNA protection assay (88), and a third one using a glnD-luc transcription fusion (the luc gene, encoding luciferase) (518), have shown that the expression of glnD is not regulated by nitrogen: the mRNA levels were similar for cells grown in minimal glucose medium plus glutamine as the N source (N-poor medium) and for cells grown in the same medium plus ammonium as the N source (N-rich medium). When mRNA from cells grown in glucose-arginine medium (“very N poor”) was compared to that of cells grown in Luria-Bertani (LB) medium (“very N rich”), Kamberov et al. (124) reported some regulation of glnD expression. However, LB medium is an undefined medium and may affect gene expression patterns through a plethora of regulatory routes.
In the sequence upstream of glnD, Kamberov and coworkers (124) identified two putative binding sites for the transcriptional regulatory protein Nac (203). There is no evidence, however, that Nac binds to these sites.
(iii) The glutaminase genes ybaS and yneH.
(a) ybaS.
The level of glutaminase A (YbaS) increases 10-fold when cells enter the stationary phase (339, 341). Addition of 5 mM cAMP to the culture medium containing glucose as the carbon source and ammonium as the nitrogen source decreased the level of glutaminase A approximately 5-fold in wild-type cells, but cAMP had no effect on the activity of glutaminase A in a crp mutant, indicating that transcription of ybaS is under the control of Crp (484). The former result may suggest that expression of ybaS is under the control of RpoS, the sigma factor of RNA polymerase, σs, controlling expression of genes when cells enter the stationary phase. Indeed, a global gene expression profile of rpoS versus the wild type revealed expression of ybaS to be RpoS dependent (520).
Brown and coworkers (338) noted that according to the EcoCyc database (487), the ybaS gene is located upstream of and cotranscribed with ybaT, encoding an uncharacterized amino acid transporter.
(b) yneH.
Glutaminase B (YneH) appears to be a constitutive enzyme, since its level did not change (i) by altering the nutritional conditions of the culture medium (340, 341), (ii) when cells enter the stationary phase (339, 341), or (iii) after addition of 5 mM cAMP to the culture medium containing glucose as the carbon source (484).
The gene upstream of yneH is yneG, the function of which is unknown. The last nucleotide of yneG is the same as the first nucleotide of yneH, and therefore, the two open reading frames may constitute an operon (Colibri [345] and EcoCyc [487] databases).
(iv) The glnK-amtB operon.
In a diverse range of Archaea and Bacteria, including E. coli and other enterobacteria, glnK is linked to amtB in an operon (365, 407). There is no invariant coupling, however, as the association is absent in many other species, e.g., in all analyzed Lactobacillaceae (521). The glnK-amtB operon is located between the mdl and tesB genes, at kb 475.5 to 481.7 on the E. coli chromosome (154). The glnK-amtB operon is not constitutively expressed. The upstream region of the operon contains a perfect Eσ54 binding site (139). The glnK promoter has been shown to be regulated by NRI-P (32, 139, 401). The glnK promoter is activated by NRI-P binding to an upstream enhancer (401). Unlike the glnA enhancer, the glnK enhancer consists of a high-affinity NRI-P binding site adjacent to a low-affinity NRI-P binding site (139). As a consequence, the glnK promoter is activated only when the NRI-P concentration is near its physiological maximum. However, when activated, the promoter is strong (401, 402).
In general, induction of the operon has been shown to occur, directly (by demonstrating the presence of the AmtB and/or GlnK protein) and/or indirectly (by observing methylammonium uptake), in culture media that are considered to yield internal N limitation. AmtB is absent in cells that are grown in batch cultures in media with excess ammonium (say, >1 mM), whereas it is present in media containing glucose as the carbon source and glutamine (305, 308, 522), glutamate (304, 306), arginine (282), or low levels (say, <0.5 mM) of ammonium (308) as the N source (the references given are just examples). Remarkably, though, in batch culture with “poor” C sources (glycerol, galactose, and succinate) and glutamine as the N source, expression of glnK was not observed (486). Thus, during batch growth in minimal media with glutamine as the N source, expression of the glnK-amtB operon seems to be the exception rather than the rule (see also “Growth with excess glutamine as the N source,” below), possibly because growth in batch cultures with poor C sources is effectively C limited. In batch cultures with various amounts of ammonium, the glnK promoter was silent during the entire exponential growth phase but was expressed at the transition to the stationary phase if this was caused by ammonium depletion. No glnK promoter activity was observed, however, when cells entered the stationary phase while ammonium was still in excess (401). Even in rich LB medium, the operon was expressed when the first group of amino acids was depleted after some 4 h of growth (523). Also, in ammonium-limited (from undetectable to low μM residual ammonium levels) chemostat cultures of S. Typhimurium, AmtB is present (282). All in all, if present, AmtB levels and/or transport activities may vary to a substantial degree.
Like glnD, the regulatory genes glnB and glnE are also constitutively expressed in E. coli, i.e., expressed irrespective of the nitrogen status (88). However, the expression of the regulatory gene glnK is regulated by the nitrogen status (139, 401). Because GlnK is 40 times less potent than GlnB in activation of the adenylylation activity of ATase (138), and GlnK-UMP is almost inactive in stimulating the deadenylylation activity of ATase (140), GlnK has no significant role in the regulation of the two activities of ATase (138), at least in vitro. Therefore, the functioning of the GS adenylylation cascade is regulated mainly by modulations of the activities of GlnB, UTase, and ATase rather than by changes in the expression levels of these proteins (88).
An interesting observation is that GOGAT mutants do not seem to express AmtB. GOGAT-negative mutants grown with glutamine as the N source did not show methylammonium uptake activity (308), and it expressed AmtB poorly (281). Most likely, gltB mutants cannot synthesize the AmtB protein. Perhaps, such mutant cells, in contrast to wild-type cells, accumulate glutamine to such a level that NRI-P becomes dephosphorylated and no longer activates the transcription of the glnK-amtB operon. Indeed, a GOGAT mutant of E. coli growing under ammonium-limited conditions accumulated glutamine (12 mM) relative to the wild type (2 mM) (135). Induction of AmtB from a plasmid by IPTG (in order to increase the intracellular ammonium concentration so that GDH would be active) did not have the anticipated positive effect on growth of the gltB mutant, however (281).
(v) The glnHPQ operon.
E. coli can use glutamine not only as a sole source of nitrogen (7, 16, 338) but also as a sole source of carbon and nitrogen (338). This suggests that expression of glnHPQ, which encodes the glutamine transporter, may be regulated by signals of both nitrogen and carbon. Indeed, it has been shown that transcription of glnHPQ is regulated by both NRI-P (453, 524, 525) and Crp (453). Similarly, in S. Typhimurium, transcription of the glnHPQ operon depended on NRI-P and σ54 (526). The glnHPQ operon is located at kb 847.2 on the E. coli chromosome (18.3 min on the genetic map) (Colibri database [345]). It has been cloned and sequenced by Nohno and colleagues (319). The first codon of glnQ overlaps the termination codon of glnP. This indicates that there is no untranslated intercistronic region between these two genes, suggesting that the two genes are transcribed to approximately the same level (319). The intercistronic region between the glnH and glnP genes harbors a palindromic sequence, which seems to play a regulatory role in reducing the expression of the glnPQ genes relative to glnH (319). This region also lacks a promoter. Therefore, transcription of the three genes is initiated at one of the two promoters upstream of glnH, depending on the growth conditions. From a deletion analysis of the glnH promoter region of a glnH′-lacZ fusion, it was apparent that the promoter region possesses two different types of promoters, i.e., glnHp1 and glnHp2 (453), which is in agreement with the detection of two transcription start sites by primer extension analysis (453, 527).
The upstream glnHp1 promoter is involved in constitutive expression of the operon by the σ70-RNA polymerase holoenzyme, as deduced from the presence of the −35 and −10 recognition sequences (319) and DNase I footprint analysis of Eσ70 binding to glnHp1 (453). The downstream glnHp2 promoter is involved in the inducible expression of the operon under glutamine-deprived conditions (319) or in the absence of ammonium (525). Initiation of transcription may be performed by the σ54-RNA polymerase holoenzyme, as deduced from the presence of the two conserved doublets, GG (position −25/−24) and GC (position −13/−12), relative to the transcription start site (319). Two strong and two weak NRI binding sites have been detected by DNase I footprint analysis upstream of the Eσ54 binding site (525), and induction of the glnHPQ operon by NRI-P was observed by microarray analysis (32).
Also, IHF plays a role in transcription of glnHPQ. A binding site for IHF was detected between the proximal weak NRI site and the Eσ54 binding site by DNase I footprint analysis (525). Binding of IHF results in bending of the DNA, which enhances open complex formation by NRI-P. On linear DNA, IHF is required for activation by NRI-P, while on supercoiled DNA, IHF is not essential but still enhances the activation of transcription by NRI-P (524). When bound to a site between the promoter and enhancer (as in the upstream region of glnHPQ), IHF may increase the specificity of activation of the glnHp2 promoter by the enhancer-bound activator (such as NRI-P) (528). This capacity of IHF to increase the specificity of activation may be due its ability to bend the DNA. Indeed, a crystal structure of an IHF-DNA complex showed a huge bend of >160°, like a U-turn (529).
As described above, the glnHp1 promoter is initiated by the σ70-RNA polymerase holoenzyme. DNase I footprint analysis detected a DNA binding site for Crp upstream of the binding site of σ70-RNA polymerase, even though there is no obvious Crp consensus binding sequence in the DNA sequence upstream of glnHp1, nor did Crp shift the glnHp1 promoter DNA in a gel band shift assay (453). DNase I footprint analysis revealed that Crp could bind the glnHp1 promoter DNA only in the presence of σ70-RNA polymerase. Thus, the two proteins may bind to the promoter DNA synergistically. Indeed, an in vitro transcription assay using σ70-RNA polymerase and Crp demonstrated an increase in glnHp1 transcription with increasing Crp levels (453). E. coli possessing a glnHp1p2′-lacZ fusion and growing on minimal medium with glycerol as the carbon source and glutamine as the nitrogen source showed 3-fold induction by wild-type Crp in the presence of exogenous cAMP (453). In addition, it was shown by real-time reverse transcription-PCR from transcription of chromosomal glnHPQ that Crp activates glnHp1 but represses glnHp2 (453).
Two observations may bear relevance to the expression of the glutamine transporter. The glnHPQ operon was upregulated by H2, and cells grown with H2 showed increased glutamine uptake (530). The nucleoid-associated protein Fis has been shown to repress transcription of glnQ 3-fold (531).
INTEGRAL FUNCTIONING OF THE NITROGEN ASSIMILATION NETWORK
Up to this point, we first reviewed the properties of the individual components of the nitrogen assimilation network of E. coli, also taking into account their potential interactive properties. We then reviewed how each component engaged in activities and regulation related to thermodynamics, kinetics, signal transduction, or gene expression. In this section, we discuss a number of examples where multiple network components function together to produce functional properties that are more than the sum of the properties of the components. In all these cases, it is the network rather than a molecule that is responsible for ultimate biological function, which is itself a property of the network of 4,000 genes and their expression products.
The Metabolic Nitrogen State of Cells
First, we should be conceptually as clear as possible about the “nitrogen state” of bacterial cells. In the literature, several terms are employed to reflect the nitrogen state, such as nitrogen starvation, severe nitrogen starvation, nitrogen limitation, nitrogen deficient, nitrogen poor, very nitrogen poor, low nitrogen, nitrogen sufficient, high nitrogen, nitrogen rich, and nitrogen excess. These terms are supposed to abstract away from the specific experimental conditions used and to infer more general statements. A problem is that the N state as described by the above-mentioned terms is mostly not defined explicitly. The N state terms may allude to relative specific growth rates, relative growth yields, the N source available in the environment, the nitrogen-related intracellular processes, or a combination of some of these factors. Not only does this introduce ambiguity, it also introduces vagueness, for even if the meaning is unambiguous, it is not always clear what the domain of application of the term is. Also, even if the N state is further sketched, this is done mostly in qualitative terms only and not so much in quantitative terms. For example, incipient external nitrogen limitation has been shown to be associated with a drop in internal glutamine pools (383), and since then, many scientists have adopted this finding as the general definition for nitrogen-limited growth, but exactly how fast intracellular glutamine should decrease and from what level to what level have not been explicated.
The same problem, mutatis mutandis, applies to the definition of the carbon state of cells. As an increasing number of papers actually demonstrate the links between N and C metabolisms (see, e.g., references 435, 453, 486, 532, and 533), it would be good practice to always mention the carbon and nitrogen sources in experimental procedures.
The underlying problem is that the N state of the cell is a state function in a multidimensional space of thousands of metabolites and gene products (347). The underlying history is that of reductionistic oversimplification, i.e., the desire to see an organism like E. coli encoded by more than 300 essential genes (534) or 600 essential genes (535) in terms of a few so-called key enzymes. This explains the persistent urge to do the impossible, i.e., to try to capture something as complex as the nitrogen state of the cell in terms of a single qualifier. Metabolic control analysis has shown experimentally that control in intracellular networks tends to be distributed (536), that some of the key proteins and genes in E. coli failed to exercise control of the growth rate (161, 398), and that such control may be distributed even among various levels in an intracellular hierarchy (537), and a combined experimental and modeling approach showed that all of this also seems to apply to ammonium assimilation (44, 161). Living organisms are irreducibly (though not infinitely) complex (538, 539); their analysis should be made as simple as possible but not simpler.
We advocate the use of in silico replicas of E. coli nitrogen assimilation as qualifiers of the nitrogen status of the real cell, as was tried first by Bruggeman et al. (44). These true-to-life replicas may then be analyzed in terms of reduced dimensionality under some conditions, which should also lead to the identification of a manageable number of quantifiers of the state function. Short of this, we tend to quantify the nitrogen state of a cell by the combination of the intracellular concentrations of glutamine, glutamate, 2-oxoglutarate, ammonium ion, and also, for its dynamics, GS, GOGAT, and GDH.
Experimental Ambiguities
Growth with limiting ammonium as the N source.
Also, experimentally, ambiguity/vagueness cannot always be avoided. In batch cultures, growth with excess ammonium cannot be compared with growth with a limiting amount of ammonium, because in the latter case, no (or hardly any) growth will be observed. For this reason, it is common practice to compare cells grown with a single amino acid as the N source with cells grown on the same medium plus excess ammonium (e.g., see references 140, 281, 401, 455, 483, 486, and 540). Usually, glutamine is used as the amino acid. The comparison of the regulation of nitrogen assimilation under these two conditions in batch culture could be flawed for several reasons: (i) conceptually, growth with two completely different N sources is compared (an amino acid versus ammonium), which also may invoke very different physiologies for reasons not related to nitrogen limitation (e.g., glutamine contains carbon, whereas ammonium does not); (ii) growth proceeds under unrestricted conditions since the nitrogen source is available in excess; (iii) when the C source is consumed, in the case of glutamine, growth will continue, with the remaining N source as the C and N sources; (iv) N-diauxie could occur in media with ammonium and glutamine; and (v) the growth history of the cells may affect the outcome of the experiments, since memory and learning can have strong effects on the state of pregrown cells (161).
It is worthwhile to note that the above-mentioned problem of comparing growth on two different nitrogen sources can be avoided by using chemostat cultures. In chemostat cultures, unlike in batch cultures, ammonium-limited growth can be easily established and compared to growth in the presence of excess ammonium, albeit in the latter case, some other limitation must be put in place to establish the same growth rate.
Are cells in a batch culture in steady state?
Batch cultures are used almost universally to study nitrogen metabolism and for obvious and good reasons. However, the very fact that growth conditions inherently change during growth in a batch culture is a major caveat, and it calls for caution in interpreting results from batch growth experiments. First of all, growing populations of cells may be in different states of growth: steady-state growth, balanced growth, and exponential growth ought to be distinguished (541, 542). In short, steady-state growth implies balanced growth, and because this usually includes cell mass, it also implies exponential growth. A culture is said to be in steady state only if every intensive variable (e.g., single-cell size, metabolite concentration, intracellular ionic strength, and pH) is time invariant and every extensive property (e.g., total protein, RNA, or DNA or cell mass of the population) increases by the same constant factor over a small time interval. Cell size is correlated directly with the nutrient source and growth rate in S. Typhimurium and E. coli (543). Therefore, a sensitive indicator of steady-state growth is the constancy of the cell size or, better, the time invariance of the distribution of cell sizes (544). Although it is usually implicitly presumed that cells in the exponential phase of growth of a batch culture also feature completely balanced growth and steady-state growth, there are some experimental observations that raise doubts about the general applicability of this assumption, elaborated here for E. coli. First, it has been shown that steady-state growth in batch cultures requires large dilutions and incubation for many (at least 8 to 10) generations at low cell densities (544). Experimentally, this can be achieved by diluting cells serially several times or by diluting the preculture (>105-fold) to a very small number of cells at the start of the batch culture. The latter is preferred above the former, notably for growth in rich media. Second, even so, it has been observed that such a pseudo-steady state did not hold with respect to the redox state of the quinone pool (545). Third, even in the uniform environment provided by a (glutamate-limited) chemostat culture, an E. coli strain harboring a tac promoter-controlled fusion between glnA and gfp showed large fluctuations in the GlnA fusion protein level (2 orders of magnitude of variation in protein numbers), as measured by flow cytometry (546). It is our expectation that many more examples of heterogeneity of intracellular states at the single-cell level caused by stochasticity will be demonstrated in due time. Fourth, a rule of thumb for growth in continuous cultures is that the culture is in steady state after 6 to 10 generations of growth. However, even for this standard cultivation method, it is not always a safe assumption: in glucose-limited chemostats of K. pneumoniae, constancy of the biomass was indeed established within some 10 generations, but it took 40 to 60 generations to arrive at a constant residual glucose concentration (547), and in glucose-limited chemostat cultures of E. coli, a similar observation was made, but after the initial physiological adaptation phase, a genetic (long-term) adaptation phase started (548). Fifth, during growth of E. coli with glucose and glutamine as the sole N sources, the growth rate decreased at a certain point in the growth curve. Before this point, glnA was partially activated and glnK and nac were not activated, while afterwards, all three genes were strongly activated (401). Sixth, the main carbon sources in the commonly used LB broth (casein digest, yeast extract, and NaCl) are catabolizable amino acids and not sugars (549). LB broth supports growth to an optical density at 600 nm (OD600) of 7, but exponential growth already slows down at an OD600 of 0.3. In the first short phase, the sugars are used for growth, while thereafter, the various catabolizable amino acids present in LB broth are consumed (549). The growth pattern in LB medium is complex, as a switch occurs from a sequential mode to a simultaneous mode of substrate utilization (550). Also, during growth on a mixture of amino acids (serine, aspartate, tryptophan, glutamate, glycine, threonine, and alanine) as C and N sources, a characteristic pattern of sequential and simultaneous consumption was observed (551). Similarly, a complex usage pattern of different carbon sources (sugars and organic acids) when present simultaneously in a batch culture was observed with ammonium as the N source (552). Indeed, in a batch culture with ample glucose (10 mM) plus a mixture of amino acids (LB broth or Casamino Acids) as the N source, cell size, which is a sensitive indicator of steady state, was shown to vary even within the exponential growth phase. In fact, it started to decrease already at rather low cell concentrations (OD550 = 0.02), whereas exponential growth continued until the OD550 reached 0.1 to 0.2 (544). Thereafter, growth continued up to an OD550 of 1.0, but it was no longer exponential. In contrast, cell size remained more or less constant, and the growth rate was exponential up to an OD550 of 0.7 in a glucose-plus-ammonium minimal medium. In conclusion, in rich media, no steady-state growth takes place whatsoever, because the physiological state of the cells varies constantly due to the sequential/simultaneous consumption of the C and N sources. In minimal medium, the occurrence of steady-state growth depends on whether particular precautions had been taken. However, a general screening of growth curves in the literature shows that much more often than not, growth does not proceed exponentially, which, by implication, means that growth cannot be in the steady state either.
Growth with excess glutamine as the N source.
It has been known since the 1930s (see reference 553 and references therein) that glutamine decomposes spontaneously; in culture media, glutamine spontaneously hydrolyzes to pyroglutamate and ammonia. The first-order rate constant observed for chemical degradation was determined at various pHs, temperatures, medium compositions, and glutamine concentrations (64, 554–556). Based on these data, it can be calculated that under the conditions often used to study growth with glutamine as the sole N source (37°C, neutral pH, and 14 mM glutamine), approximately 0.5 mM ammonium would be formed from glutamine in the absence of bacteria within 8 h. Therefore, during batch growth in culture media with excess glutamine, small amounts of ammonium will be produced continuously and will probably be used as a nitrogen source. If the latter holds true, growth proceeds with the simultaneous consumption of two N sources. How this will affect nitrogen assimilation is not known.
In an influential paper by Ikeda et al., it was observed for S. Typhimurium that when growth was slowed by nitrogen limitation, the intracellular glutamine pool was lowered by a factor of up to 10 (383). The phenomenon was seen with wild-type strains and a variety of N sources (except with alanine) in batch cultures, with a host of mutants that showed impaired growth with excess ammonium, and also with the wild type in ammonium-limited chemostat cultures. The results were similar when glycerol, citrate, or glucose was used as the single C source. For this reason, it was postulated that external nitrogen limitation is perceived as a drop in the internal glutamine pool.
Also, growth with alanine as the N source represents a special case. As noted by Ikeda et al. (383), alanine was the single exception among the N sources tested that was not compatible with the general observation that external N limitation is perceived as internal glutamine limitation. Alanine has been shown to perturb growth of S. Typhimurium (383) and of K. pneumoniae (K. aerogenes) (510) on ammonium.
Although growth in batch culture with glucose as the C source and glutamine as the N source was not tested by Ikeda et al., many authors have studied N metabolism in cells grown under this condition, and it is generally considered an example of N-limited growth. However, growth under this condition represents a metabolic oddity, because these cells exhibit several paradoxical features: (i) even though ample external glutamine is available and is supporting rapid growth (557), the cells display a nitrogen-deficient response, as judged from a high expression level of NRI (180), an increased amount of GS (486), and a high expression level of the glnK-amtB operon (281); (ii) this apparent nitrogen deficiency shows up in spite of the fact that several glutamine transporters are known to exist, not only one or more low-affinity transport systems but also a high-affinity ATP-dependent ABC transporter (GlnHPQ); (iii) the exponential phase of the growth curve is biphasic, where high-level glnA, glnK, and nac promoter activities show up only in the second part (401); (iv) the GDH capacity, which is expected to be low, turns out to be as high as that in cells grown with excess ammonium; and (v) with glutamine as the only nitrogen source, Nac did not repress gdhA, whereas with glutamate or low ammonium concentrations, such repression was observed (503). In light of the consensus that external nitrogen limitation is perceived as a drop in the internal glutamine pool (383), one might suggest that this pool is apparently small enough for cells to exhibit a nitrogen deficiency response when growing in a glucose-glutamine medium, but this being the case while excess glutamine is abundant in the environment remains an intriguing paradox.
A clue to understanding the first two odd paradoxes was found when carbon sources other than glucose were tested in combination with glutamine as the nitrogen source. With the C source gluconate (558), glucose-6-phosphate (455), or xylose (486), expression levels of GS were high and similar to those with glucose. However, the use of the carbon source fructose (455), galactose (486), glycerol (455, 486), lactate (558), or succinate (486, 558) resulted in low expression levels of GS. Moreover, cells grown in a glucose medium supplemented with exogenous cAMP also had low GS expression levels (486). These results were interpreted to suggest that cAMP, produced internally or added externally, enhances the transport of glutamine into the cell and thereby increases the glutamine pool to such a level that GlnB-UMP becomes deuridylylated. As a consequence, NRI-P will become dephosphorylated, and transcription of glnA from the glnAp2 promoter will no longer be activated (453, 486). Thus, the carbon source/cAMP effect is mediated through GlnB deuridylylation and NRI dephosphorylation. Second, apart from this explanation at the metabolic level, an explanation at the transcriptional level via Crp-cAMP has also been offered: the Crp-cAMP complex would strongly inhibit glnAp2 promoter activity [see “Transcription regulation of glnALG. (iv) Transcription regulation of glnALG by Crp,” above].
In summary, (i) the nitrogen-limited behavior of cells grown in the frequently used glucose/glutamine medium should perhaps be considered an exception rather than the rule and not be held representative of N limitation more in general, and (ii) the carbon source may influence the “nitrogen state” (453, 486), and therefore, further investigations into the integration of carbon and nitrogen metabolism seem urgent. Inspired by the works of Mandelstam (533) and Magasanik (559), a working hypothesis could be that cells slowly growing with a poor C source and glutamine as the N source experience internal C limitation and therefore do not feature nitrogen-limited behavior, whereas the converse situation would apply to fast growth with rich C sources and glutamine.
All in all, theoretical and experimental ambiguities exist when defining the metabolic nitrogen state of cells. To avoid unnecessary confusion, one should explicitly declare what is meant by terms and concepts related to the cellular nitrogen status, at least qualitatively and, if possible, quantitatively, or one should accept much more comprehensive descriptions.
GDH Activity under Conditions of Internal Nitrogen Limitation?
Although according to the received view GDH is absent or inactive under internal nitrogen-limited conditions, there are various types of direct and indirect data indicating the presence/activity of GDH in wild-type E. coli strains under conditions that are supposed to lead to intracellular nitrogen limitation and, thus, to the primacy of the GS-GOGAT route in nitrogen assimilation. First, in ammonium-limited chemostats of E. coli, GDH was also present at lower growth rates. Its maximal capacity strongly (8-fold) increased with the specific growth rate (384, 560). Second, in ammonium-limited chemostat cultures, K. pneumoniae GDH was completely repressed (384). Under low-ammonium conditions, GDH of E. coli was only moderately (2-fold) repressed by the nitrogen-regulated system-dependent Nac, in contrast to the strong repression (20-fold) of GDH of K. pneumoniae (K. aerogenes) (202, 207). Third, an additional Nac-independent mechanism responsible for overcoming nac repression during nitrogen-limited growth is likely to be present in E. coli (202, 503). Fourth, growth conditions that result in internal N limitation may or may not lead to suppression of GDH. GDH is repressed during growth with excess arginine or glutamate as the N source, but its level is elevated during growth with glutamine (503). In glutamate-limited chemostat cultures, GDH was highly expressed, while it was repressed in proline-limited cultures (384). Fifth, in glucose-glutamine medium, considered to yield intracellular nitrogen-limited conditions, GDH was hyperinduced in a GOGAT mutant (53). Sixth, no growth of a GOGAT mutant was observed in a glucose medium with a low (0.4 mM) ammonium concentration, but growth did occur when poorer carbon sources (such as maltose, glycerol, or succinate) were used instead (494). It was argued that C limitation might lead to accumulation of ppGpp, which in its turn might stimulate GDH expression. Seventh, the growth defect of a GOGAT S. Typhimurium mutant growing in an ammonium-limited chemostat could be completely rescued by acquiring two suppressor mutations that resulted in increased GDH expression and lowered GS expression levels (411). Thus, these cells grew at a wild-type growth rate even without an intact GS-GOGAT system, while the residual ammonium concentration was <20 μM. Similarly, GOGAT mutants of E. coli growing in chemostats under conditions of N limitation showed increased levels of mRNA of gdhA and increased GDH capacity compared to growth under N-rich conditions (561). Finally, external ammonium limitation in a chemostat culture perhaps cannot be automatically considered to result in internal ammonium limitation. It depends on whether ammonium transport is passive or active (see “Passive versus active transport,” above). In the latter case, the intracellular ammonium concentration might be 10- or even 100-fold higher than the extracellular ammonium concentration. The implications would then be that so-called ammonium-limited chemostat cells do not experience internal ammonium limitation and that GDH may then indeed substantially contribute to ammonium assimilation and glutamate production.
Estimating Fluxes In Vivo
Flux methods.
There are four methods for measuring/calculating cellular metabolic fluxes: flux balance analysis (FBA), metabolic flux analysis (MFA), kinetic flux profiling (KFP), and kinetic modeling (KM). The merits and drawbacks of the first three methods were discussed previously (562). The pros and cons of various types of models, including kinetic models, were dealt with previously (563, 564). FBA relies on an objective function, which is mostly not validated (565). Also, FBA cannot predict metabolite concentrations, because it does not use kinetic parameters (566). MFA is limited to carbon metabolism, because only feeding cells with a 13C-labeled compound yields a spectrum of labeling patterns of metabolites that is rich enough to allow for flux deconvolution. Labeling by other elements rarely produces a rich-enough spectrum. KFP enables the quantification of metabolic fluxes in live cells by measuring the kinetics of cellular incorporation of stable isotopes from nutrients into downstream metabolites (562, 567). KM employs the kinetic parameters of the enzymes, such as maximal capacities and affinities for metabolites, to construct rate equations that, together with mass balances for every metabolite, enable the calculation of steady-state fluxes (as well as metabolite concentrations) in an enzyme network.
Kinetic and flux balance models and kinetic flux profiling.
Bruggeman et al. made a detailed kinetic model for central ammonium metabolism based on all of the kinetic data on the components that were available (44). Steady-state and transient behavior was calculated with the model for the wild type and various knockout mutants with internal ammonium concentrations in the range of 0.05 to 1.0 mM. It was confirmed that based on the information then available, the adenylylation state of GS should be a major regulator in the network but also that the interplay between GS-GOGAT and GDH critically should depend on the signaling cascade composed of ATase, UTase, and GlnB. Moreover, a method was developed to quantify the relative importance of the various regulators in the network. Adenylylation of GS accounted for 60% of the regulation. The model has been extended to include GlnK-regulated AmtB-mediated ammonium transport (568). The main message of the paper together with the corrigendum was that only if ammonium would be actively transported into the cell could a substantial growth rate be established, while at the same time, back diffusion of NH3 was inevitable. Maximal enzyme capacities are vital parameters of kinetic models, and it has been argued that to obtain realistic Vmax values, traditional biochemical assays tailored to a particular enzyme could better be replaced with a single in vivo-like assay mimicking the cytoplasmic conditions as much as possible (375). Another simplified kinetic model of the GS, GOGAT, and GDH subnetwork allowed the glutamine, 2-oxoglutarate, glutamate, and ammonium concentrations to vary. The presence/absence of GS regulation and of GOGAT was analyzed. GS regulation provided a greater robustness of the fluxes against perturbations of enzyme or ammonium concentrations, in particular when the ammonium supply fluctuated (569). A flux balance model for nitrogen metabolism was developed (570) and verified by experiments carried out for the wild-type and GDH and GOGAT knockout strains (135, 567). Furthermore, by employing the kinetic equations built for GS, GDH, and GOGAT (44), Yuan et al. were able to predict maximal capacities of the three enzymes in wild-type and mutant strains as a function of the ammonium availability. The results suggested that among the three central enzymes, GS would be the preferred regulation point.
Another model of the GS cascade arrived at the conclusion that the bicyclic cascade is suboptimal for homeostatically controlling the glutamine pool (571). It was hypothesized that a slow inactivation of GS by adenylylation allows GS to transiently deal with ammonium toxicity imposed on cells by a sudden internal ammonium upshift from 0.1 to 100 mM. The authors of that study assumed that 100 mM ammonium is toxic, although there are data showing that neither NH4+ nor NH3 is toxic per se for bacteria (see “Ammonium transport could be a challenge,” above). A dynamic mathematical model was developed to analyze and understand biological design principles of the complex but highly structured feedback modules in ammonium assimilation (572, 573). The former was the first model for central ammonium assimilation that linked the metabolite and protein levels to the DNA level, while the latter presented an extended and improved version. The GS activity control module was unified with a GS synthesis module, and the robustness of the full model toward internal and external perturbations was studied. Several differences surfaced between the biological ammonium assimilation model and classical engineering systems. In another application of the method, it was shown that the redundant feedback loops generate robust properties to multiple gene deletions (574). A steady-state analysis of a network consisting of the GS cascade, the NRI/NRII system, and the glnALG operon indicated that the output of the operon should be switch-like, while the activation of the transcription factor NRI should be graded (575).
An elegant experimental setup that enables a quick change of the nutrient environment is a filter culture technique developed by Rabinowitz and coworkers (562, 576): cells are growing on a membrane filter on top of agarose plates loaded with media. The cells are fed by nutrient diffusion from the underlying medium up through the filter. This technique allows quick medium switching by transferring the filter between agarose plates of different compositions. The filter methodology was nicely employed in the kinetics flux profiling protocol; by switching from unlabeled to isotope-labeled forms of nutrients, metabolic fluxes can be quantified, based on the kinetics of cellular incorporation of stable isotopes from the nutrient into downstream metabolites. An important result obtained with kinetic flux profiling is discussed in the next section.
GS-GOGAT Activity under Conditions of Internal Nitrogen Sufficiency?
The textbook view on nitrogen assimilation in E. coli states that the free energy-intensive GS-GOGAT pathway for glutamate synthesis is tuned down and that the ATP-independent GDH takes over the production of most of the glutamate whenever ammonium is available in sufficient amounts. This picture is based mainly on in vitro knowledge gathered in biochemical experiments with purified enzymes and by simplifications, typically neglecting the smaller of two parallel activities that might well be active simultaneously, as more often, Occam's razor may be deceptive in biology (538, 539): that a simple explanation at first glance looks plausible enough to be real is insufficient for living organisms with their networks of irreducible core complexity.
Indeed, data obtained with the new in vivo method (562) for measuring metabolic fluxes in live cells, KFP, have seriously challenged this view. The fluxes predicted by FBA to allow optimal biomass production agreed well with fluxes measured with the KFP method on the basis of the kinetics of nitrogen assimilation in exponentially growing cells (567). However, there was one notable exception: in the presence of glucose and ample ammonium, KFP indicated that glutamate was synthesized largely via the GS-GOGAT route and much less via the GDH route. This unexpected result was further put to the test by applying kinetic flux profiling to mutant strains lacking either GDH or GOGAT. The results confirmed the previous conclusion, and over 85% of the total glutamate biosynthetic flux in the wild type was assigned to GS-GOGAT activity. This conclusion was corroborated by the finding that the 2-oxoglutarate pool in both the wild type and a GDH mutant hardly responded to ammonium depletion down to 0.1 mM, while the pool in a GOGAT mutant increased by a factor of 4 (577). Finally, in 1975, Senior had already observed that GS was deadenylylated under conditions of ammonium excess at high growth rates in glucose-limited chemostats (384).
Measuring Metabolites In Vivo
Introduction to metabolomics.
In the past, at most, a few to a dozen metabolites were painstakingly determined at a time (e.g., see references 355 and 384), but this situation has changed dramatically by the spectacular development of high-throughput techniques for measuring intracellular metabolites. Via intermediate metabolome analysis covering only high-abundance metabolites (>0.5 mM) (e.g., see reference 578), the technique has been further developed such that currently, out of 800 to 2,000 metabolites, about 200 metabolites can be qualitatively identified down to the micromolar level (579), and some 100 (342) metabolites reach levels sufficiently high to be identified and quantified in vivo. Ultimately, the new science of metabolomics aims at the qualitative and quantitative analysis of the complete set of all low-molecular-mass metabolites present in and around growing cells at a given time (580, 581).
Various experimental methods required in several different steps of the analysis are available or are being developed in this field of research. The first steps concern rapid sample collection, instant quenching of metabolic activity, separation of extracellular medium, and extraction of intracellular metabolites (e.g., see references 582–584). Each of these steps faces its own problems and pitfalls, e.g., incomplete inactivation of metabolism, leakage of metabolites, and incomplete extraction. A survey of comparisons of seven extraction procedures for metabolome analysis in microorganisms convincingly showed that the authors' evaluation of the different extraction techniques used by them did not go together very well (585). The last, and perhaps the least troublesome, step involves the quantification of the metabolites through enzyme-based analytical methods or different chromatographic techniques (liquid or gas chromatography and capillary electrophoresis) coupled to mass spectrometry (MS) or NMR analysis (580).
It has been estimated that the maximum total metabolite concentration, as constrained by the mechanical strength of the cell wall, is some 500 mM (586). Since about 800 to 2,000 different metabolites are present in a cell (579), one might expect an average intracellular metabolite concentration of roughly 0.5 mM. For stochastic and diffusion reasons, submicromolar concentrations may be hard to operate by cells. Therefore, most concentrations may not be too far away from the average, and low intracellular metabolite concentrations seem to be a general property of any cell (586). The compatible solutes glutamate, K+, trehalose, glycine betaine, and proline (587) seem to be important exceptions to the rule, however. Also, for E. coli cells grown with glucose, glycerol, or acetate as the C source and excess ammonium as the N source, the top 20 metabolites are all present at concentrations that are significantly higher than this 0.5 mM, i.e., in the range of 2 to 17 mM (342).
Glutamine and 2-oxoglutarate signal nitrogen availability.
For enterobacteria, external nitrogen limitation may be perceived as a drop in the internal glutamine pool under both aerobic (383) and anaerobic (588) conditions. Glutamate was found to remain reasonably constant, although its concentration may change by a factor of 2.5 (135, 383). In a more recent metabolomics study, these observations were confirmed and extended. Here the filter culture technique was used to determine the metabolic response to a sudden removal of ammonium from the medium (576). Two dominant starvation responses were observed. A generic response accounted for 42% of the metabolite concentration changes and typically included depletion of biosynthetic intermediates. An ammonium-specific response was responsible for another 30%. Ammonium deprivation was characterized by a 16-fold increase in the 2-oxoglutarate concentration and a 64-fold decrease in the glutamine concentration, while the glutamate concentration was decreased by only 2-fold. Thus, besides a markedly decreased glutamine level, an increased 2-oxoglutarate level is also indicative of internal nitrogen limitation. A rapid increase in the 2-oxoglutarate concentration was also observed when cells from batch cultures were filtered and washed with solutions containing less ammonium than the growth medium at filtration (577).
In another study, the integrated regulation of central nitrogen metabolism through metabolomics and modeling was examined by shifting filter cultures from limiting to excess ammonium conditions (135). The metabolome response upon the ammonium upshift (10 mM), covering 59 metabolites, was found to boil down to just two characteristic response patterns. The first one accounted for 63% of the available data, and 2-oxoglutarate contributed most strongly to the pattern. The second pattern accounted for 23%, and glutamine was the dominant molecule. Other affected compounds were proposed to be closely related to either 2-oxoglutarate (TCA cycle compounds) or glutamine (amino acids). Still many other compounds retained concentration homeostasis, including ATP and NAD+ (135). A rapid drop in the 2-oxoglutarate pool was also observed with a 0.2 mM ammonium upshift (354).
It should be noted that data from some articles (107, 572, 573) are based on the assumption that the 2-oxoglutarate-over-glutamine ratio would be maintained homeostatically, which, in our opinion, might not be valid under all conditions (see below). The ratio was found to be constant at 1.8 for cells growing in chemostat cultures under conditions of glucose limitation and excess ammonium but only at relatively high specific growth rates (>0.4 h−1). Below specific growth rates of 0.4 h−1, it gradually declined to 0.4 (384). Thus, the measured ratio has been shown to be robust only under conditions of ammonium excess for a limited range of growth rates. Furthermore, from more recently determined concentrations in other experimental setups (135, 342, 343), the ratio can be calculated to vary from approximately 10 under conditions of N limitation to approximately 0.04 under conditions of N excess, i.e., a 250-fold change of the ratio. We think that the ratio of 2-oxoglutarate over glutamine might actually be the cell's robust monitor for a changing cellular N state. The ratio should be more sensitive than the concentrations of glutamine and 2-oxoglutarate taken separately as N-state indicators. In addition, some sites that either 2-oxoglutarate or glutamine interacts with may bind both compounds competitively because of their structural relatedness.
Active-site competition in central nitrogen metabolism?
Absolute intracellular concentrations of some 100 metabolites were determined via liquid chromatography-tandem MS (LC-MS/MS) (562) in aerobic, exponentially growing cells in filter cultures with glucose, glycerol, or acetate as the carbon source and ammonium as the nitrogen source (342). The total observed metabolite pool was about 300 mM, while the intracellular concentrations ranged widely from 0.001 to 20 mM, with glutamate as the notable exception (50 to 150 mM). The 10 most abundant compounds comprised 77% of the total molar concentration of the measured metabolome.
Interestingly, the metabolite concentrations exceeded the Kms for most substrate-enzyme pairs (342). As a consequence, fluxes could be insensitive to the substrate concentration, and this might potentially lead to large variations in metabolite concentrations. On the other hand, competition by products or other compounds for the substrate binding site will increase the effective Km for the substrate and contribute to kinetic regulation of its activity; the authors of that study proposed such active-site competition as a new way to regulate fluxes and, thereby, metabolite levels in general (342). Other ways of flux regulation could also be operative, such as enzyme activity regulation by covalent modification subsequent to signal transduction and enzyme concentration control by transcription regulation (see “Signal Transduction-Mediated Regulation” and “Gene Expression Regulation,” above).
As a matter of fact, introducing active-site competition of glutamine, glutamate, and aspartate for the glutamine binding site of GOGAT was necessary for the ordinary differential equation model of central nitrogen metabolism to properly simulate all of the experimental ammonium upshift data (135). More specifically, insertion of modest competitive inhibition by aspartate increased the elasticity coefficient of GOGAT flux to glutamine and rendered the elasticity coefficient of GOGAT flux to glutamate less negative.
However, two arguments can be mounted against the model: (i) the fitted Vmax value for GS is rather high (9,120 mM/min), far (15-fold) above the values measured in ammonium-limited chemostats at a D of 0.3 h−1 (600 mM/min [44]), and (ii) the term NH3 appears to denote NH3 in the diffusion equation but NH4+ in the rate equations for GS and GDH, which of course cannot be true at the same time. For these reasons, the calculated intracellular ammonium (ammonia?) concentration of 1 μM in the nitrogen-limited wild type becomes questionable. Probably, this computed concentration was extraordinarily low because the Vmax of GS was taken to be extraordinarily high.
Reliability of the metabolome?
An important issue is how one can be sure that the metabolome data constitute a reliable snapshot of the metabolic state of cells. Reliability is necessary to compare results under different growth conditions gathered by one and the same laboratory but also to enable comparison of data sets obtained by different research groups. For example, there are differences between the data set for cells growing in microcolonies on filters with ample glucose and ammonium (342) and for cells growing in high-density glucose-limited chemostat cultures (D = 0.1 h−1) with ample ammonium (343); most metabolite concentrations are (sometimes substantially) lower in the latter case, although some are equal or higher. A meaningful biological interpretation of the apparent differences requires that they should be attributable only to microbial behavior and growth conditions. Therefore, much effort is and should be spent to find ways to standardize and optimize protocols for metabolomics (e.g., see references 343, 585, and 589).
Considering the major pitfalls in determining intracellular metabolite concentrations, an important issue is how the overall quality of metabolome data can be checked. Two simple checks for healthy cells are recommended by Taymaz-Nikerel and coworkers (343): (i) the value of the adenylate energy charge (590) should be between 0.80 and 0.95 (591), and (ii) the calculated mass action ratio of known near-equilibrium reactions should be close to the equilibrium constant. Bolten has shown that some literature data do not conform to the expected range of the adenylate energy charge (582). Application of the checks to a relatively recent data set (342) shows that with glucose, glycerol, or acetate as the carbon source, the adenylate energy charges (all three approximately 0.95) are indeed within the range but that the adenylate kinase reactions (9, 64, and 11, respectively, compared with a Keq of 0.6 to 1.1) as well as the reaction catalyzed by fumarase (all three approximately 0.08, compared with a Keq of 0.23) appeared to be away from equilibrium. This observation raises some doubt about the reliability of the above-mentioned measurements.
Taymaz-Nikerel et al. (343) put much effort into avoiding practical problems when analyzing the metabolome. They found that the commonly applied quenching method of cold aqueous methanol was not suitable for E. coli because of leakage of a major part of the metabolites. They arrived at the so-called differential method as the best option to obtain reliable metabolome data, at least for E. coli; the method requires metabolite measurements in total broth and filtrate for each measurement.
GS, GOGAT, and GDH Functions
GS functions.
In E. coli and other enteric bacteria, the reaction catalyzed by GS is the only known biosynthetic route for the synthesis of glutamine. Deletion of the glnA gene results in an absolute requirement of glutamine (592). The prime function of GS is to provide cells with glutamine for protein synthesis and as a donor of amide groups (not to be confused with the amino groups provided by glutamate in transaminase reactions) for other N-containing compounds. Therefore, with ammonium as the sole nitrogen source, GS activity should never be completely shut down, despite the existence of extensive mechanisms that regulate its activity. Indeed, in the presence of ammonia, the expression of glnA is not completely shut down (133), and adenylylated GS still has some activity (371, 372) (see “Feedback inhibition of GS,” above).
GOGAT functions.
E. coli mutants lacking GOGAT represent an interesting case for two reasons: (i) they cannot deplete glutamine except by biosynthetic reactions, and (ii) they cannot accumulate glutamate under intracellular N-limiting conditions. Because of the latter reason, it was envisaged that they can grow only in minimal medium containing ammonium as the only nitrogen source when the ammonium concentration is relatively high (>1 mM) (373). Likewise, it was argued that GOGAT mutants grow slowly on poor organic nitrogen sources, because they are starved for glutamate (53). Later, however, it was demonstrated that with ammonium as the N source, it holds true only when glucose is the carbon source; a minimal medium with poorer C sources (such as glycerol, succinate, or maltose) did support growth at a low ammonium concentration (0.4 mM), where glucose failed to do so (494). Mutants lacking both GOGAT and GDH require externally added glutamate for growth, even in the presence of ample ammonium (593).
GDH functions.
At first sight, GDH seems to be redundant. In principle, the high-ammonium-affinity GS-GOGAT system should suffice for enteric bacteria to assimilate ammonium, independent of the extracellular ammonium concentration. Indeed, a deletion in gdhA conferred no overt phenotype and did not induce a glutamate requirement (593–595). However, the phenotype of “silent” genes that show no effect on growth rates or other fluxes can be made visible by studying the metabolome of such mutants (406). Moreover, some bacteria that harbor GS and GOGAT (e.g., Erwinia carotovora and Bacillus megaterium) seem to lack GDH naturally (596, 597).
Nevertheless, GDH seems to be invariably present during growth in all kinds of different media, and it is probably also active under most growth conditions. We put forward the following (possible) functions of GDH and its biosynthetic product glutamate, mostly on the basis of the following suggestions made by other authors.
(i) Enabling rapid growth.
GDH activity increases linearly with the growth rate in ammonium-limited chemostats of E. coli (384). Perhaps, GDH enables rapid growth.
(ii) Producing glutamate to maintain the steady-state K+ pool.
Under normal growth conditions, K+ is by far the most abundant cation in E. coli (598), and glutamate is the most abundant amino acid anion (599). Not glutamate but trehalose is the most abundant metabolite only at low growth rates (578). Indeed, it had already been shown in 1972 that these two ions have some correlation in growing bacteria (599). Studies of GOGAT mutant strains of S. Typhimurium grown at low ammonium concentrations and subjected to an ammonium upshift indicated that glutamate is specifically required to maintain the K+ pool and that K+-glutamate is required for optimal growth (411, 412).
(iii) Producing the osmoprotectant glutamate.
Addition of NaCl (2 to 4%) to K. pneumoniae (and E. coli) effected a dramatic increase in the glutamate pool; this effect was irrespective of the nature of the growth limitation in chemostat (599). Under excess-ammonium conditions, GDH may synthesize glutamate when enterobacteria are hyperosmotically stressed (but GS-GOGAT might also do so [see “GS-GOGAT Activity under Conditions of Internal Nitrogen Sufficiency?,” above]); apparently, only glutamate can serve as the counterion for K+ that is accumulated upon osmostress (410, 599, 600). Eventually, however, the K+-glutamate couple is replaced by the compatible osmoprotectants trehalose, glycine betaine, and proline (601). Trehalose is produced from glucose-6-phosphate and UDP-glucose in a two-step reaction, catalyzed by OtsA and OtsB (131).
(iv) Producing glutamate under conditions of free energy limitation.
E. coli might possess and use GDH in case free energy limits growth and ample ammonium is present, because it costs less ATP equivalents than GS plus GOGAT (602).
(v) Catabolizing glutamate.
Mammalian GDH operates in the catabolic direction, oxidizing glutamate (603), but is coupled to NAD(H) rather than NADP(H). In contrast, bacterial GDH usually operates in the biosynthetic direction. Because of the relatively low Keq (90 mM−1 at an I of 0.5 and a T of 37°C) (59), the reaction catalyzed by E. coli GDH may in principle be reversed under some conditions. Recently, absolute intracellular metabolite concentrations in E. coli (some 100 metabolites) growing on filters with excess glucose and ammonium were measured (342); from the concentrations determined for NADP+ (0.002 mM), NADPH (0.12 mM), glutamate (96 mM), and 2-oxoglutarate (0.4 mM), it can be calculated that if the intracellular ammonium concentration is <44 μM, oxidation of glutamate becomes thermodynamically feasible. Since such a low internal ammonium concentration is not likely when external ammonium is present in excess, biosynthesis of glutamate takes place, even at very high internal glutamate concentrations. When cells are growing under ammonium-limited conditions, the glutamate concentration remains rather high, at ∼70 mM, but the 2-oxoglutarate concentration increases up to 12 mM (135). Based on the assumption that the NADPH/NADP ratio is the same as that during growth with excess ammonium, biosynthesis of glutamate is then possible whenever the internal ammonium concentration exceeds 1 μM. Thus, degradation of glutamate is not expected to occur under this condition either.
We note, however, that this expectation depends on the very high NADPH/NADP ratio (604) measured, which may be lower under more oxidized conditions or if the effective proton motive force-driven transhydrogenase reaction falters because of a reduction in the energy state of the cell. Indeed, ratios of 3.0 (605) and 0.8 (606) have also been measured. If the ratio is 3.0, the internal ammonium concentration below which glutamate catabolism becomes thermodynamically possible becomes 880 or 20 μM when the external ammonium concentration is in excess or limiting, respectively. For a ratio of 0.8, the threshold intracellular ammonium concentration increases even further to 3,330 or 75 μM, respectively. Hence, these calculations at least show that under some conditions, GDH might possibly catalyze glutamate degradation instead of biosynthesis.
Senior (384) noted that when E. coli is growing on minimal medium containing excess glutamate and limiting glucose in a chemostat, NH4+ was excreted. Also, in batch culture with glutamate as the single nitrogen source, ammonium was found to be excreted in the late phase of growth (377). In both cases, the carbon skeleton of glutamate probably serves as an extra C source producing excess ammonium. As suggested by Senior (384), externally added glutamate might become deaminated by GDH, although rather low NADPH/NADP and 2-oxoglutarate/glutamate ratios would seem to be required to reach the observed high level of ammonium (20 mM). Alternatively, degradation via aspartate aminotransferase and aspartate ammonia-lyase might occur instead (607).
(vi) pH homeostasis.
The hallmark of E. coli cells growing on glucose minimal medium appears to be the formation and excretion of acetate and subsequent metabolism of the acetate and protection of the cells from acid stress (608). Recovery of normal internal pH occurs via glutamate synthesis (609) and through proton-consuming decarboxylation of glutamate by GadB and secretion of the product γ-aminobutyrate by GadC (500). GadE seems to regulate the three acid resistance systems of E. coli, of which the GadAB system is the most important and most regulated one (493, 500).
(vii) Producing glutamate to enhance protein-DNA interactions.
GDH may produce glutamate to enhance protein-DNA interactions (e.g., see reference 610) or, together with potassium, to act differentially on cellular promoters so as to activate some or inhibit others (611).
(viii) GDH functionality.
Because enzymes are embedded in networks, it can be difficult to identify all their functionalities. For GDH, functionality tends to be discussed in terms of its production of glutamate. Additional functions, however, might reside in keeping the 2-oxoglutarate concentration at an adequate level. Indeed, for K. pneumoniae (K. aerogenes), it has been hypothesized that GDH might play a role in controlling the size of the 2-oxoglutarate pool at such a level that both GDH and the TCA cycle enzyme 2-oxoglutarate dehydrogenase, and thereby the TCA cycle itself, remain active (612, 613). 2-Oxoglutarate has also been shown to provide a link between C and N metabolisms by virtue of its ability to block glucose uptake by inhibiting enzyme I of the sugar-phosphoenolpyruvate phosphotransferase system (435).
Signal Transduction Cascades
Protein activity can be regulated by covalent modification: a chemical group is covalently attached to the target protein, the process of which is catalyzed by a converter enzyme, while another converter enzyme catalyzes the removal of the same chemical group. The interconversion of the target protein is reversible through the action of this cycle. Kinase/phosphatase couples of converter enzymes that modify a protein by phosphorylation are the prime examples of such monocyclic cascades. Eukaryotic glycogen phosphorylase was the first enzyme reported to undergo reversible phosphorylation-dephosphorylation (614, 615). A bacterial example is isocitrate dehydrogenase, which is (de)phosphorylated by a bifunctional converter enzyme (616–618). Bicyclic and multicyclic cascades can be formed by concatenating monocycles. For instance, the eukaryotic mitogen-activated protein (MAP) cascade is an illustrious example of a tricyclic phosphorylation cascade (619). With respect to ammonium metabolism in E. coli, covalent modification is known to play a prime role in its regulation. Adenylylation of GS, uridylylation of GlnB and of GlnK, and phosphorylation of NRI and of NRII are the three relevant covalent modification reactions that have been studied in great detail. The GS regulatory cascade at large (UTase, ATase, GlnB, GlnK, and GS) has been implicated as a promising candidate for featuring ultrasensitivity.
Definitions and terminology.
Speaking in general terms, signals elicit responses. This statement applies to single enzymes as well as to mono- or multicyclic cascades. The input-output relations may show less (subsensitivity) or greater (ultrasensitivity) responsiveness than the usual hyperbolic (Michaelian) sensitivity (620). Ultrasensitivity is associated with a sigmoidal stimulus-response curve. It has received much attention, and we therefore focus on this form of signal amplification.
The amplification of a signal generally takes two forms: magnitude and sensitivity amplification (620). Magnitude amplification occurs when target molecules are produced in greater numbers than the stimulus molecules. Magnitude amplification can be readily achieved in single steps; in principle, there is no need for monocyclic, let alone multicyclic, cascades. Sensitivity amplification is about the degree of change in a response in comparison to the degree of change in the stimulus. Sensitivity magnification can be realized in single steps by positive cooperativity, but monocyclic and multicyclic cascades may further increase sensitivity compared to individual steps (620–622). Although a cascade structure does not necessarily result in ultrasensitivity, even a monocyclic cascade can readily generate a substantial response coefficient (622). Multicyclic cascades have the potential to feature even higher degrees of ultrasensitivity (in the absence of feedback loops), since the total response coefficient equals the product of all the local response coefficients, one for each level of the cascade (623–625).
The classical definition of sensitivity amplification takes the so-called “amplification factor” expressed in finite terms: the percent change in response to the percent change in stimulus (620, 626). In later studies, the precise vocabulary and analysis tools of metabolic control analysis (MCA) were introduced to deal with sensitivity amplification. By using MCA, it is possible to pinpoint exactly which processes should be affected and to what degree they should be affected to obtain ultrasensitivity (627). The theorems of MCA have been adapted to include covalent modification cycles (624, 627, 628). The “response coefficient,” which corresponds to the infinitesimal form of the amplification factor, is the variable that is relevant in MCA (622, 629). It has the advantage that the mechanism underlying sensitivity need not be known (622, 627). The maximum response coefficient derived from a sigmoidal input-output curve equals the Hill coefficient, another measure of ultrasensitivity used with single enzymes (630). Response coefficients (R = ε.C) of cooperative and allosteric mechanisms will generally be lower than the corresponding Hill coefficients, because elasticities are at most equal to Hill coefficients, and control coefficients maximally equal 1.0 (631). In the case of zero-order ultrasensitivity that can be obtained in cascades, the maximum response coefficient is infinite (624, 628). By definition, a response coefficient less than, equal to, or greater than 1.0 indicates a subsensitive, hyperbolic, or ultrasensitive response, respectively. For ultrasensitivity, it is noteworthy that the definition implies that the term is applicable to moderate and strong cases alike. Also note that early on, the term response coefficient was defined differently as the ratio of concentrations of a factor required to change the response from a 10% to a 90% response (620, 632, 633). For a Michaelis enzyme, the response coefficient amounts to 81, and the smaller this ratio, the more sensitive the response. We use the MCA meaning of the term response coefficient.
Application of MCA has shown that the conditions are determined by a limited number of elasticities: the elasticities of the modifier enzymes for their substrates (the modifiable target enzymes) should be minimized, and the elasticities of the modifier enzymes for their effectors should be maximized (627). Ortega et al. (106) showed that considering that, in many cases, the modification and the demodification enzymes are identical, one should take product inhibition and the consequent increase in elasticities into account, largely removing the ultrasensitivity of such cascades. The sensitivity of the response to signal can be expressed in terms that are equivalent to Hill coefficients or in MCA terms as response coefficients.
Functions of ultrasensitivity.
An intriguing general question is why the regulation of protein activity occurs by interconversion via a mono- or multicyclic cascade of covalent modifications rather than by allosteric effectors acting on single enzymes. In other words, what are the functional advantages of the former over the latter? Several functions may be put forward on the basis of modeling studies. The first function is more extended regulation: in a cascade, more proteins are present, and thus, more allosteric effectors can influence regulation than with a single protein. The second function may be stronger sensitivity amplification; for allosteric proteins, Hill coefficients of >4 are rarely observed (620). Although a monocyclic cascade per se is no guarantee of high sensitivity and typically even results in subsensitivity (106, 634), it may nevertheless exhibit enormous sensitivities; Hill coefficients as high as 800 can be achieved, although this value was not considered of any real physiological significance (629, 631). Third, in a multicyclic cascade, additional sensitivity can be achieved, because the response coefficient of the entire cascade equals the product of the local coefficients of the individual cycles (623–625).
Several characteristics of strong ultrasensitivity per se that may be considered useful for the cell are as follows (635): (i) generating all-or-none decisions, (ii) filtering out noise, (iii) delaying responses, (iv) displaying oscillations when combined with negative feedback, and (v) displaying bistability in combination with positive feedback.
Mechanisms for ultrasensitivity.
Several mechanisms for generating an ultrasensitive response may be distinguished (620, 626, 632, 633, 636, 637): cooperative, multistep, multisite, zero-order, stoichiometric inhibitor, and branch-point mechanisms. Importantly, more than one mechanism may occur in one and the same system. If so, the respective sensitivities all contribute to the overall ultrasensitivity (633). The zero-order variant has been studied most extensively, especially in theoretical studies. We discuss this form of ultrasensitivity in more detail.
Conditions for zero-order ultrasensitivity.
The conditions for maximizing zero-order ultrasensitivity turned out to be quite stringent (106, 629, 632, 633). They can best be illustrated with the aid of a simple monocyclic enzyme cascade (Fig. 10). The conditions can then be summarized as follows.
Fig 10.

Monocyclic interconvertible enzyme cascade. A target enzyme may exist in two states, an active state and an inactive state. These can be interconverted by the action of the modifier enzymes, enzyme1 and enzyme2. An effector regulates the activity of the target enzyme indirectly by activating modifying enzyme1, inhibiting modifying enzyme2, or both. The two modification reactions are not each other's precise reversal.
First, either one or both modifier enzymes should operate under conditions approaching saturation by their substrate (the target enzyme). In other words, the concentration of the modified enzyme should exceed the effective Michaelis constant prevailing in the presence of the product of the modification reaction (106). This is the basic condition that the phrase zero order alludes to (632). If the enzymes operate in the region of first-order kinetics, hyperbolic or subsensitive responses are found instead. Although zero order suggests that the enzymes should be nearly saturated, a 36% degree of saturation of both converter enzymes has been shown to suffice for a Hill coefficient of 10 (629).
Second, the effectors must act on both modifier enzymes in opposite direction (627, 629).
Third, the effectors should have catalytic (kcat) rather than specific (Km) effects on the modifier enzymes (627, 629).
Fourth, the modifier enzyme subject to activation must respond to lower effector concentrations than the modifier enzyme subject to inhibition (627, 629).
Fifth, the covalent modification reactions of the target enzyme must be virtually irreversible. In practice, this boils down to reactions with a high (effective) equilibrium constant; if the reaction is reversible, zero-order ultrasensitivity disappears (106).
Sixth, the conversion reactions must also be product insensitive. The product inhibition constant should be lower than the Michaelis constant; if the conversion reactions are saturated by their product, zero-order ultrasensitivity disappears (106).
Seventh, the interconversion of the target enzyme should be catalyzed by two independent modifier enzymes. Covalent modification cycles that are catalyzed by one bifunctional “ambiguous” enzyme rather than by two independent converter proteins cannot have high-level responses, because high substrate concentrations and low product concentrations for both reactions of the cycle are inconsistent (106). Paradoxically, in another theoretical study, bifunctionality (among other factors) was considered essential for ultrasensitivity (638).
Eighth, the sum of modified and unmodified target proteins must be constant.
Ninth, target enzymes should not be sequestered by the converter enzymes. Zero-order ultrasensitivity requires that target proteins are present in excess over the converter enzymes of the modification cycle, i.e., free target protein pools virtually equal total target protein concentrations. However, for most signaling cascades, converting enzymes and target proteins are present in comparable concentrations, and consequently, large fractions of the target protein may be sequestered by the modifying enzymes. This sequestration will result in a decrease in ultrasensitivity (635).
Finally, random fluctuations should be negligible. All of the above-described conditions were recognized by macroscopic modeling, which implicitly assumes that cells are infinitely large and fluctuations are negligible. Using mesoscopic modeling, which takes finite cell size and stochastic aspects into account, it was found that zero-order ultrasensitivity was much more gradual than in macroscopic models (639). The general message is that the demonstration of ultrasensitivity in macroscopic models does not guarantee it will prevail under the same conditions in vivo.
Ultrasensitivity in models of the GS cascade.
A steady-state analysis of adenylylation of GS with glutamine as the input in a model of the GS bicyclic cascade (UTase-ATase-GS) demonstrated an ultrasensitive response; it was concluded to be brought about by allosteric interactions of glutamine and 2-oxoglutarate, bifunctionality of converter enzymes, and a closed-loop bicyclic cascade structure (638). In a follow-up study, a steady-state modular analysis of a network made up of the above-mentioned GS bicyclic closed-loop cascade, together with the NRII-NRI two-component system and an autoregulated glnALG operon, was presented. The simulation indicated that the inactivation of GS is moderately ultrasensitive to input stimulus by glutamine (575). A mathematical model has been made to study to what extent simple feedback regulation could account for the dynamic behavior as observed for nitrogen assimilation. It was found that it could lead to dangerously large metabolite pools but also that the generation of such pools could be prevented when multiple regulatory mechanisms were working in concert to produce ultrasensitive feedback (640).
On the basis of the laws of hierarchical control analysis (624), it was shown that the sum of the control of the adenylylation and the deadenylylation reactions on steady-state ammonium assimilation should be nil, whereas the sum of their control of the dynamics of adenylylation and, hence, of ammonium assimilation should be quite significant (−1 for the total control on duration and “area under the curve,” making the deadenylylation potentially more important than the adenylylation reaction) (44, 641, 642). Also, a ligand, such as perhaps glutamine, adversely stimulating the adenylylation reaction and inhibiting the deadenylylation reaction should readily obtain a highly sensitive response (642).
Experimental ultrasensitivity in the GS cascade.
In contrast to the number of theoretical studies on ultrasensitivity in general, only a few experimental studies have dealt with ultrasensitivity in the GS cascade. In a reconstituted UTase-GlnB-ATase-GS bicyclic system, a modest ultrasensitive response (Hill coefficient of 1.5) of the GS adenylylation state to the glutamine concentration was already demonstrated in 1978 (643). The same system was studied 20 years later (387) and some 30 years later (390, 644) by Jiang and coworkers. The reconstituted system now showed a sensitivity to glutamine that was equivalent to a Hill coefficient of 5.2. Since UTase and ATase are both bifunctional enzymes, zero-order ultrasensitivity is not likely (106, 644). More recently, the source of ultrasensitivity has been explored for this in vitro system, and it was found that a fully deuridylylated trimeric GlnB was required for the activation of the AT activity of ATase (644). Its composite UTase-GlnB and ATase-GS cycles displayed only moderate glutamine sensitivities when examined separately. Both the trimeric nature of GlnB and the completely unmodified form of GlnB were important factors in the glutamine response of the bicyclic in vitro system (644).
In another experimental study, GlnB expression was modulated in vivo around the wild-type level, and MCA was used to study the function of the GS signal transduction chain (161). Under two relevant physiological conditions, i.e., nitrogen limitation (glutamine) and nitrogen sufficiency (high ammonium), neither the steady-state level of adenylylation of GS nor the rate at which GS-AMP became deadenylylated upon ammonium deprivation depended on glnB gene expression or on the concentration of GlnB plus GlnK. Thus, the pivotal regulatory proteins GlnB and GlnK had no control over the steady-state GS level or activity. This in vivo test refuted ultrasensitivity to GlnB and GlnK for the steady-state function of the GS cascade, and instead, a subtle, quantitative, and context-dependent adjustment of regulation was put forward as the principal steady-state function of the cascade.
Possible subtleties include that high sensitivities toward glutamine do not have to be accompanied by high sensitivities toward GlnB, especially in view of the ambiguity of GlnB (stimulating both adenylylation and deadenylylation depending on its own modification state): this paradoxical phenomenon is due to the fact that the steady-state adenylylation state of GS is determined not merely by the adenylylation rate but also by the deadenylylation rate and, in fact, just as much (624, 642; see reference 641 for experimental demonstrations for the MAP kinase pathway). Indeed, in 1974, evidence was already presented to support the conclusion that the constant states of adenylylation at steady state reflect a dynamic state in which the rates of adenylylation and deadenylylation are equal (415). In line with this reasoning is the more recent finding that the AR activity of ATase is mandatory to counterbalance its AT activity during steady-state growth under both N-limiting and excess-N conditions (414). A graded response of GS instead of a switch-like, all-or-none response also fits nicely with the obligatory presence of active GS under all conditions (for it is the single enzyme capable of glutamine synthesis) and also with the observation that GS-GOGAT (not GDH) is the predominant producer of glutamate during growth under conditions of ammonium excess (135, 567). Finally, with an in vitro-reconstituted nitrogen assimilation subsystem consisting of purified UTase and GlnB, it has been shown that in steady state, sequestration of GlnB by NRII (a downstream target) may alter a moderately ultrasensitive response observed in the absence of NRII into a subsensitive response in its presence (645). This phenomenon has been termed “stoichiometric retroactivity.” In a follow-up study with the above-described in vitro experimental system, it was shown that even when steady-state properties were unaffected by the downstream target (no stoichiometric retroactivity), the dynamic behavior of the system may be altered by so-called “load-induced modulation,” i.e., changes in response to time-varying input stimuli (646). It was noted, however, that there existed an apparent discrepancy in the model used, as it did not reflect the bifunctional, ambiguous character of UTase (647). The authors of that study responded that only in this way did the ultrasensitive response obtained in the experiments show up in the model (648).
In conclusion, the multitude of theoretical conditions that should be fulfilled simultaneously and the relative paucity of experimental support render signal transduction cascades, i.e., the GS signal transduction cascade, featuring zero-order ultrasensitivity in vivo rather unlikely. Although further research is needed to examine other forms of ultrasensitivity in detail, in our opinion, it may turn out to be rather difficult to find conditions where GS will or should be shut down completely in a switch-like manner in response to just a small change in the internal glutamine concentration (strong ultrasensitivity) in vivo. Regulatory cascades overall might perhaps serve the function of subtlety of regulation rather than high signal amplification (161). Accordingly, it has been argued that not so much ultrasensitivity but relatively (and adjustably) slow dynamics may have an evolutionary advantage under conditions of fluctuating ammonium concentrations (649).
So-Called Futile Cycles
In this section, we describe and discuss cyclic processes in the nitrogen assimilation network which at first sight seem to behave as futile, i.e., useless, cycles but which upon closer inspection are in most cases useful, if not necessary, investments that serve a clear purpose after all. In some cases, they are not likely to occur under most physiological conditions.
ATase consuming ATP.
The sum of the adenylylation reaction and the deadenylylation reaction equals the conversion of ATP and phosphate to ADP and pyrophosphate (reaction III) (Table 4). Because of the presence of pyrophosphatase activity in the cell, free energy is dissipated by the sum of the three reactions. Since both adenylylation and deadenylylation reactions are catalyzed by the same ambiguous enzyme, appropriate control of the two activities may seem necessary to prevent futile cycling. Indeed, such control is executed by the action of the regulatory protein GlnB in combination with the small effector molecules glutamine and 2-oxoglutarate (for a comprehensive kinetic analysis of the regulation of the two activities of ATase by GlnB, GlnB-UMP, and small effector molecules, see reference 96). On the other hand, it was already argued in 1974 that the final state of adenylylation represents a dynamic steady state in which the rates of adenylylation and deadenylylation of GS are equal but not necessarily zero (415). This conclusion was confirmed in a recent study of adenylylation of GS in the wild type and an E. coli mutant (414). Here it was shown that the adenylyl-removing activity of ATase was required to counterbalance its adenylyl-transferase activity during steady-state growth under both excess-nitrogen and nitrogen-limiting conditions. Both processes take place at the same time, and although at first glance it may seem that free energy is wasted, the actual ATP costs are only some 0.003% of the total rate of ATP consumption for cells growing at a growth rate of 0.66 h−1 (414, 415). Although Goldbeter and Koshland in general concluded that, “a significant fraction of the total energy expenditure of an organism is required for the large number of reactions which involve covalent modification of proteins,” it was also acknowledged that the fraction was much lower for microorganisms (650). This is mainly because the ATP turnover rate of microbial cells is on average 2 orders of magnitude higher than the turnover rate of human cells (say, 200 to 1,000 versus 2 to 10 mM/min, respectively). In this particular case, the benefit at low ATP costs for the microbial cell might be that by sustaining intermediate states of adenylylation, the activity of GS is carefully and rapidly adjusted to the level needed by the cell and rapid changes therein.
“Futile” cycling, such as through the simultaneous activity of adenylyl transferase and deadenylylase, offers the advantage of allowing metabolic activities such as that of GS to be regulated directly by “key intermediates” of intracellular metabolism, such as 2-oxoglutarate and glutamine. Since key regulatory metabolites correspond to nodes with connections to multiple central metabolic processes, a paradox arises: on the one hand, the changes in their concentrations should be so small that they do not compromise all the processes that depend on them, while on the other hand, the changes in their concentrations should be large enough to serve as signals for adaptations of metabolism. The very presence of both the adenylyltransferase stimulated by glutamine and the deadenylylase stimulated by 2-oxoglutarate makes some degree of futile cycling inescapable, for if an increase in glutamine levels is necessary to decrease the activity of GS through adenylylation of the latter, the glutamine cannot increase from zero to some value but only from its preexisting physiological value to a value not too different from that. This implies that in the physiological steady state, both glutamine and 2-oxoglutarate are already present at concentrations that activate the adenylyltransferase and the deadenylylase, respectively; i.e., both activities must already be present at the steady state before perturbation. Consequently, the issue is not whether or not there will be futile cycling but, rather, how much. If the turnover time of the adenylylation/deadenylylation cycle at the steady state is τA seconds, then a 25% increase in the glutamine level will decrease the adenylylation state of GS by some 50% in approximately 2τA seconds. If GS were inactivated because of an increase in ammonium, which would make GDH energetically cheaper than GS plus GOGAT, then any retardation in the response time would cost ATP, and the 2τA seconds would cost on the order of 2τA/τGS molecules of ATP, where τGS is the turnover time of GS in terms of glutamine synthesis. If a switch between nitrogen-poor and nitrogen-rich conditions would occur on average once every τS seconds, then the total cost would amount to , which is very large at both very slow futile cycling (high τA) and very fast futile cycling (low τA) and which has a minimum in between at approximately . In other words, the optimal futile cycling rate would be between the turnover rate of GS (perhaps 100 s−1) and the geometric average switching time between nitrogen-rich and nitrogen-poor conditions (perhaps 10−4 s−1), hence perhaps once every 10 s. In the example, the futile cycle would be present but not futile. Also, it would not require zero-order ultrasensitivity to be relevant.
It is tempting to speculate that, mutatis mutandis, a similar reasoning as that given above for ATase might be applied to the other bifunctional enzyme UTase, which would seem to “waste” free energy (UTP) when the uridylyl-transferase and the uridylyl-removing activity would occur simultaneously to achieve a steady-state level of uridylylation of GlnB or GlnK. In both cases, it may be relevant that the cycles waste some ATP so as to prevent the wasting of ATP by inappropriate activity of GS plus GOGAT when GDH suffices.
GS-GOGAT plus GDH wasting ATP.
As discussed above (see “GDH functions”), it is possible that under some circumstances, GDH operates in the catabolic direction, and if GS-GOGAT is active at the same time, a futile cycle of ammonium assimilation and production will be generated. Although such a cycle has not yet been demonstrated, a sudden ammonium downshift may invoke a futile cycle; when cells growing with excess ammonium are confronted with a sudden downshift in ammonium availability, it is possible that GDH starts operating in the catabolic direction when the threshold level of intracellular ammonium is passed. If GS-GOGAT is still active at the same time at this lower intracellular ammonium level, one ATP will be wasted per futile cycle of ammonium. Under this particular condition, GDH should be turned off, but no such rapid metabolic regulation of GDH activity is known so far, nor is gene expression regulation strong enough (see “Transcription regulation of gdhA,” above). This issue certainly warrants further experimental study. There is a possible and paradoxical aspect to this; if under such conditions, the ammonium transporter becomes expressed, this would increase the intracellular ammonium concentration quite significantly and possibly to above the threshold concentration for GDH reversal. A tradeoff would arise between the futile cycle of ammonium across the membrane and the GDH/GS-GOGAT cycle.
GS plus glutaminase wasting ATP.
Glutaminase B, which is formed during growth, would, together with GS, constitute a futile cycle wasting ATP (651). However, two factors may prevent futile cycle: (i) glutaminase B is inhibited by ATP (and ADP) (344), and this characteristic may limit the activity of the enzyme under conditions where the ATP-demanding GS is also active, and (ii) the high Km of glutaminase B for glutamine (31 mM) and the usual glutamine concentration range of 3 to 12 mM (135, 342, 343) seem to preclude enzyme activity, except for transient states, however, where glutamine may reach concentrations of up to 50 mM (135).
AmtB-mediated futile cycling of ammonium.
If ammonium uptake is a form of active transport, accumulation of ammonium takes place in order to sustain nitrogen assimilation required for growth under nitrogen-limited conditions (301). In this case, the obvious benefit comes at a cost, since NH3 will inevitably flow out of the cell down its concentration gradient. Building on this hypothesis, it has been further hypothesized that the inhibition of AmtB activity by GlnK serves the purpose of minimizing the cost of futile cycling by adjusting the uptake rate of ammonium such that the intracellular ammonium concentration becomes just sufficient for growth. Recently, results obtained from elegant experiments in a microfluidic chemostat (652) were consistent with both of these hypotheses (301). Here it was demonstrated that cells growing with glycerol, glucose, or glucose-6-phosphate plus gluconate abruptly activate ammonium transport when the ambient ammonium concentration is reduced to some 30, 40, or 80 μM, respectively, and from there on maintain the internal ammonium concentration (as deduced by calculation) at 15, 20, or 40 μM, respectively, when the external ammonium concentration was decreased further down to 4 μM in all cases. Thus, a 4-, 5-, or 10-fold outward NH4+ gradient was maintained. Although transport of ammonium as such was not studied, Kim et al. (652) assumed it to be active transport of NH3, corresponding to passive transport of NH4+. We suggest that maintenance of these carbon source-dependent ammonium gradients is required for growth at the respective growth rates and that the costs of the accompanying futile cycling through efflux of NH3 and active reuptake of NH4+ via AmtB are minimized by fine-tuning AmtB-mediated transport by reversible binding of the inhibitor GlnK to AmtB.
Remarkably, methylamine, unlike ammonium, does not promote deuridylylation of GlnK (305). Thus, if true, AmtB-mediated MA transport would not be modulated by GlnK, and therefore, more futile cycling should be expected with MA than with ammonium.
Already in the 1980s, Kleiner argued that futile cycling of ammonium will occur when single N sources other than ammonium are used for growth (360, 361). Cells growing under such conditions will have to produce ammonium internally for biosynthetic purposes, and consequently, NH3 will leak out of the cell. Since AmtB is expressed under these conditions, ammonium will be transported back into the cell, which together then generates a futile cycle of ammonium.
In the above-described two cases (ammonium-limited growth and growth with an N source other than ammonium), futile cycling inevitably takes place, but this is then again a negative consequence of the cellular need for a biosynthetically sufficient level of internal ammonium. This is what it takes to be able to grow under internally ammonium-limited conditions.
Kdp-mediated futile cycling of ammonium.
Ammonium futile cycling has been shown to occur via the high-affinity K+ transporter Kdp under conditions of K+ limitation in the presence of high ammonium concentrations (50 to 75 mM) (653). Ammonium and K+ have similar ionic radii and can to some extent functionally replace each other (654). Ammonium will also be taken up actively via the Kdp transporter, and as a consequence, NH3 will flow out the cell, constituting a futile cycle. In this particular case, apparently no advantage can be coupled to this process, but the conditions under which it takes place are somewhat artificial. The combination of low levels of K+ and high levels of ammonium will not be encountered frequently in the natural environment.
Growth History and Intelligence-Like Behavior
The growth history of cells is a neglected and underestimated area of research. Growth in batch cultures is normally assumed to be in a quasi-steady state when cells have entered the exponential phase. However, this might very well be the exception rather than the rule, as we discuss above (see “Are cells in a batch culture in steady state?”). Here we briefly discuss the possible relevance of the concepts of memory, learning, cross talk, and bistability, all in relation to central N metabolism. We do not discuss earlier evidence of C metabolism (but see reference 655 and references therein).
The life (growth) history is also relevant for central N metabolism. An example is the finding that various phosphorylated NRI levels are necessary for subsequent transcriptional activation of the various nitrogen-regulated genes. In other words, NRI-P has a differential effect on the transcription of the nitrogen-regulated genes: the glnALG operon itself becomes already activated at submicromolar NRI-P concentrations, whereas for transcription of the glnK-amtB operon or the nac gene, a higher (some 10-fold) NRI-P concentration is required (400, 401). Because transcription and translation have a delayed response at the level of protein activity and metabolism, this has the effect that E. coli remembers its previous state, with memory times that differ between different components in the network. Another example is the observation that GlnK is required to resume rapid growth when fed ammonium after a relatively long period of nitrogen starvation (402). Here GlnK would effectively function as a “memory” protein (but see reference 301 for an alternative explanation). In the same vein, it has been argued that the inductive protein GlnK, in contrast to the constitutive protein GlnB, serves as a memory protein enabling the cell to keep track of its growth history (139).
It has been hypothesized that the various two-component systems in a bacterial cell may constitute a phospho-neural network (656) and that primitive forms of memory and learning may occur in the corresponding signal transduction systems (657–659). Learning and memory are different from physiological adaptations and should be understood in the sense of the “immunological” meanings of the terms: they give rise to a boosted response upon a repeated stimulus. Several two-component regulatory systems are subject to autoamplification, including the NRI/NRII system. As described above, the couple NRI/NRII forms a nitrogen-sensitive two-component system, where NRII is the sensor and NRI is the response regulator. They are both involved in the activation of transcription of their own operon (glnALG). The autocatalytic expression of the transcription factor NRI is a process that possibly allows for primitive forms of intelligence to occur in a bacterium. Indeed, by comparing two core models, one with and another without NRI-P-controlled transcription of glnALG, it has been shown that both conditioning and learning took place after several brief periods (pulses) of ammonium shortage (657). The concentration of and flux through GS increased with each of the four initial pulses in the inductive model compared to the constitutive model (conditioning), but at the fifth pulse, increased conditioning (learning) was observed. The concentrations of NRI/NRII and of GS at any moment might be dependent on the life history of E. coli. This might also be valid for the rate at which NRI becomes phosphorylated. Experimental evidence for this in the area of ammonium assimilation in E. coli is still limited, because of the complexity of the corresponding experiments. In yeast subjected to a pulse of glucose, such learning behavior has been demonstrated (660).
Cross talk sensu stricto has been defined as the transfer of phosphoryl groups from a sensor kinase to a noncognate response regulator (656). Cross talk can relatively easily be demonstrated in vitro by using purified components. For instance, the protein kinases of chemotaxis (CheA) and the nitrogen-regulated system (NRII) have cross-specificities: CheA can phosphorylate NRI, and NRII can phosphorylate CheY (416). In vivo, this has been much more difficult to demonstrate. In a study focusing on the interactions between four two-component systems (Uhp, Pho, Arc, and NR sensing the availability of phosphorylated sugars, phosphate, oxygen, and nitrogen, respectively), no significant cross talk was detectable in wild-type cells. Only in an NRII deletion strain did cross talk toward NRI occur upon joint activation of Uhp, Pho, and the nitrogen-regulated system (540), suggesting that cross talk is prevented by loud straight talk. In organisms under conditions such as stationary phase where multiple limitations may whisper simultaneously, the cross talk may be more active.
At the end of the 1970s and at the beginning of the 1980s, a number of papers were published by Müller and coworkers (e.g., see references 560 and 661–664). They were reported to have observed bistability (two stable stationary states) in ammonium-limited chemostat cultures of E. coli (strain ML30) at low specific growth rates (0.15 < μ < 0.20 h−1). The bistability in ammonium metabolism was expressed in GS and GDH activity and also in the residual ammonium concentration. Which stationary state was actually attained depended on the cultivation history of the cells, i.e., by whether the cells were grown at a high or a low μ before.
WHAT REMAINS TO BE DISCOVERED
Thinking about the secret of life, Westerhoff et al. argued that it is not so much a single macromolecule hidden in the cellular jungle but more the jungle of interactions among macromolecules itself (665). The term jungle should not be taken to imply that it would be an impossible task to find one's way in the jungle, as the number of interactions is large yet finite (666), and sets of interactions can be taken together in a modular approach. Consequently, it has been argued that Occam's razor as such (complexity should not be assumed unnecessarily) is not an appropriate paradigm for studying living organism (539). Indeed, it is not only in psychology that Occam's razor is all too often turned into Occam's chainsaw (clear-cutting the forest until just one tree is left standing) or into Occam's Swiss army knife (switching back and forth among multiple terms) (667). At least considerable complexity always goes with any (micro)organism, and high degrees of complexity seem to be the rule rather than the exception. To keep to the metaphor, perhaps Occam's safety razor (668) would do the job properly in systems biology. Emergent functional properties will be visible only at higher levels of complexity, and they will be lost at the level of the isolated components of a cell (669). A mechanistic explanation of the “live” state requires a pluralist research program that should apply multiple intralevel and interlevel theories and methodologies (670). Especially, the ever-changing interlevel relations (e.g., between biochemistry and physiology) grow out of the developing sciences, and these are better not discussed as relations between “completed” sciences (671). After all, the grand challenges to biology are to understand how organisms function and to discover how function arises in dynamic interactions. Systems biology endeavors to find the corresponding mechanistic links between molecules and physiology (564, 604, 655, 672, 673). Taking a systems biological perspective all the way from molecules to cell function, this review has provided a comprehensive survey of the many molecular and systems biological data that have been gathered on ammonium assimilation in E. coli regarding signal transduction, transcription, translation, metabolism, and transport. Its aim is to show how networking affected molecular activities and how some functional aspects emerged from the interactions.
Although the influence of a systems biological approach filling the gap between the holism of microbial physiology and the reductionism of molecular microbiology is strongly on the increase since the beginning of the 21st century, the reductionist molecular biological approach is still quite dominant (539, 674, 675). The fact that the molecular biology articles referenced in this review outnumber the systems biology papers testifies to the above-mentioned statement. We expect that substantial progress in understanding ammonium assimilation in E. coli will be realized when the focus shifts from the molecular to the systemic approach. If it comes down to understanding the parts or understanding the whole, we agree with Cornish-Bowden and Cárdenas that, “the emphasis ought to be on the needs of the system as a whole for understanding the components, not the converse” (674). If, indeed, the jungle of interactions constitutes the secret of life, we need to find the Rosetta stones of the common functioning of the biological languages of gene expression, signal transduction, metabolism, and transport (665).
WHAT WE LEARNED FROM NITROGEN ASSIMILATION ABOUT BIOLOGY IN GENERAL
Here a number of general statements are made point by point, which are then illustrated by back reference to the sections above.
It is a common understanding that biology engages in either one mechanism or a different mechanism, i.e., that it is binary or black/white. An enzyme is the rate-limiting step, or it is not. Cells follow one pathway or a different pathway. The concept of continuously variable transmission, i.e., not one gear or another but both at the same time but to various extents, may be more appropriate for biology. A prime example is the central nitrogen assimilation network of E. coli. Contrary to the textbook view on ammonium assimilation, GDH and GS-GOGAT may also be active under low- and high-ammonium conditions, respectively (see “GDH Activity under Conditions of Internal Nitrogen Limitation?” and “GS-GOGAT Activity under Conditions of Internal Nitrogen Sufficiency?,” above).
In biology, different from physics, details matter; the devil is in the details. The regulation of GS by ATase (see “ATase activity,” above) might be seen as a detail, but it is likely to be essential for survival. The implication is that reduction to simple descriptions may forego the essence of life.
Biology cannot be understood by first understanding components and then adding these understandings. For instance, both GDH and GS can fix ammonia, but when both are active, GDH should not work in the reverse direction. Synergy is important. Considering the equilibrium constant of the reaction catalyzed by GDH and the actual concentrations of its substrates and products, the cell has to face the problem of a potential reversal of the biosynthetic GDH reaction. Since the internal concentration of the product glutamate has been shown to be rather high (ranging from 50 to 300 mM) under a variety of growth conditions, reversal of the reaction should become thermodynamically possible when the intracellular ammonium concentrations would be in the low μM range (see “GDH functions,” above).
Biology can be understood only by considering components together, i.e., when they engage in interactions. This may be done in vivo (but difficult), in vitro (but hard to get the conditions right), or in silico. Examples of this statement are given throughout this review.
Functions always engage and are controlled by multiple components (see section on MCA above), and at the same time, most components have multiple functions. For instance, GS has the function of ammonium assimilation but also of its regulation and as a regulator of protein synthesis and distribution of nitrogen between glutamine and glutamate, etc.
It is essential for biology that issues and concepts are defined quantitatively; e.g., when enterobacteria would perceive external nitrogen limitation as internal glutamine limitation or 2-oxoglutarate excess, the qualifications of “low” glutamine or “high” 2-oxoglutarate concentrations in the cell are not enough: it should be quantitatively specified how low or high these levels should be (see “Glutamine and 2-oxoglutarate signal nitrogen availability,” above).
Physical-chemical and thermodynamic limitations should be incorporated. This is not common practice in the cartoons of biology, which may thereby be impossible. For example, growth in ammonium-limited chemostat cultures at substantial rates would not be possible if AmtB would only facilitate the diffusion of NH3 across the cytoplasmic membrane (see “Ammonium transport could be a challenge,” above).
Structural information is not decisive (see the paragraph above and “Ammonium transport could be a challenge,” above); systems biochemistry information may be more so. However, an understanding of the biochemistry information on the basis of nonobvious structural aspects then becomes important and fascinating.
Cells do not care only about free energy/ATP. They may well “waste” ATP if other things are important. Loss of NH3 from the cell via diffusion is inevitable if the internal ammonium concentration has to be maintained at a sufficiently high level and active transport of NH4+ is required to sustain growth (see “Passive versus active transport,” above). Still other examples can be found (see “So-Called Futile Cycles,” above).
Neither metabolism nor genes are important; it is their integrated activity that decides about cell function and survival when challenged. Examples can be found throughout the review.
It is impossible to understand any biology intuitively; the amount of detail required is too much to fit into a human head. Solutions are needed, such as silicon cells, i.e., dynamic models with data attached (see “Estimating Fluxes In Vivo” and “Measuring Metabolites In Vivo,” above).
ATP costs are important but are not often made explicit. For instance, textbooks and reviews often mention 15 to 18% as the extra ATP costs for ammonium assimilation if GS-GOGAT would do the job alone instead of just GDH; actually, it can be made plausible by calculation that the value is ±10% and furthermore that it decreases when C sources poorer than glucose are used for growth.
Redundancy may be explained by reference to subtle function differences, e.g., GDH versus GOGAT plus GS. See the example of GDH and GS-GOGAT activities above.
Functional roles of compounds and phenomena may differ. For example, glutamine is considered a poor N source when present outside the cell but is also perceived as a signal for nitrogen sufficiency when available inside the cell. Also, ammonium upshift experiments have been carried out with nitrogen-limited cells, which resulted in a wide range of final ammonium concentrations after the shift (from 0.2 to 30 mM, i.e., a 150-fold range); the cellular effects of such a wide range of ammonium upshifts are likely to yield considerably different results.
To understand biology, both models and experiments are needed: models alone do not work because actual detail matters, and experiments alone do not work because the data cannot be integrated. For instance, the cellular functioning of the intricate network of all transcription factors involved in nitrogen assimilation cannot be understood solely from the properties of each of the factors separately; a model that integrates all these properties is needed to describe, analyze, and understand the emergent behavior arising from the direct and indirect interactions among a multitude of factors. Another example where modeling and experimentation should go hand in hand surfaces when one wishes to fully understand the plethora of metabolic regulatory processes going on altogether in ammonium assimilation. Ultimately, our ambition should be to unite all our knowledge of time- and space-dependent processes in transport, signal transduction, transcription, translation, and metabolism in one grand model of central nitrogen assimilation. Such a full in silico model constituting a replica of nitrogen-related cellular functioning would not entail the end of modeling and/or experimentation; instead, it would allow not only for the testing of a variety of predictions of the model but also for carrying out “dry in silico experiments” that would be impossible through wet experimentation. As in computational physics, ultimate biological understanding may require such close-to-reality replica models.
ACKNOWLEDGMENTS
We thank Martijn J. Moné, Jeantine E. Lunshof, and Klaas Krab for support and discussions. We are grateful to the three reviewers for appraising the huge manuscript in great detail and for providing excellent comments, most of which we used to improve this review.
H.V.W. thanks various funding programs of the NWO, BBSRC, EPSRC, EU-FP6 and -7, and VUA.
Wally C. van Heeswijk passed away during the preparation of this review. He considered this review, which is primarily his work, his final and major contribution to science. We miss him and his unflagging optimism in all aspects of life. His work constituted a pillar under the systems understanding of biology that we require so much.
Biographies

Wally C. van Heeswijk graduated in Chemistry in 1989 and obtained his Ph.D. in 1998, both at the University of Amsterdam. He did his Ph.D. research (on the glutamine synthetase adenylylation cascade) in four different laboratories: the Netherlands Cancer Institute, EC Slater Institute, and Molecular Cell Physiology, all in Amsterdam, and CNRS-INRA in Castanet-Tolosan, France (with Daniel Kahn). He worked from 1998 to 2001 as a postdoc at the James Cook University, Australia (with Subhash Vasudevan) and the University of California (with Sidney Kustu). He received a PULS grant from the Netherlands Organization for Scientific Research (2001 to 2004) to study regulation of nitrogen metabolism in Bacillus subtilis. After falling ill in 2005, he devoted as much time as possible to his magnum opus, this first understanding of a crucial biological function in terms of all its molecules. He persevered until 3 weeks before he died of cancer on 12 January 2012.

Hans V. Westerhoff received his Ph.D. cum laude from the University of Amsterdam on the topics of mosaic nonequilibrium thermodynamics and (the control of) biological free energy transduction (1983). After a brief postdoc at the University of Padua, he became Visiting Scientist at the National Institutes of Health (United States) to work on DNA supercoiling, regulation, and control and antimicrobial peptides. After 6 subsequent years at the Netherlands Cancer Institute, he became full professor of Microbial Physiology at the VU University Amsterdam. From 2005, he also held the AstraZeneca Chair for Systems Biology at the University of Manchester, as director of the Manchester Centre for Integrative Systems Biology and the Doctoral Training Centre Integrative Systems Biology. Also, as Director of the biennial FEBS Advanced Lecture Course Systems Biology, as Professor of Synthetic Systems Biology at the University of Amsterdam (since 2011), and as one of the drivers of the Infrastructure of Systems Biology Europe (ISBE), he actively promotes Systems Biology. His research interests include integrated experimental and computational systems biology of microorganisms; systems biology of cancer; carbon, nitrogen, and energy metabolism; signal transduction; integral regulation of cell function; the silicon human; systems biology; multifactorial disease; and personalized medicine.

Fred C. Boogerd is assistant professor in the Molecular Cell Physiology research group at VU University Amsterdam. He graduated in Chemistry (cum laude) in 1979 and obtained his Ph.D. in the field of microbiology in 1984, both at VU University. He worked for 7 years as a postdoctoral researcher at Leiden University and Delft University of Technology, and he was on sabbatical at the Technical University of Denmark (Copenhagen). He is interested in microbiology and studied in particular denitrification, manganese oxidation, microbial desulfurization of coal, free-living and symbiotic nitrogen fixation, and succinate transport. As of 2000, he became interested in the philosophy of (systems) biology, especially in the topics of reductionism, emergence, mechanism, and explanation. His current research interest is in systems microbiology in general and ammonium transport and metabolism in E. coli in particular.
REFERENCES
- 1.van Heeswijk WC. 1998. The glutamine synthetase adenylylation cascade. A search for its control and regulation. University of Amsterdam, Amsterdam, the Netherlands [Google Scholar]
- 2.Fell DA, Wagner A. 2000. The small world of metabolism. Nat. Biotechnol. 18:1121–1122. 10.1038/81025 [DOI] [PubMed] [Google Scholar]
- 3.Wagner A, Fell DA. 2001. The small world inside large metabolic networks. Proc. Biol. Sci. 268:1803–1810. 10.1098/rspb.2001.1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prusiner S, Stadtman ER. 1973. The enzymes of glutamine metabolism. Academic Press, Inc, New York, NY [Google Scholar]
- 5.Zalkin H, Smith JL. 1998. Enzymes utilizing glutamine as an amide donor. Adv. Enzymol. Relat. Areas Mol. Biol. 72:87–144 [DOI] [PubMed] [Google Scholar]
- 6.Reitzer L. 2003. Nitrogen assimilation and global regulation in Escherichia coli. Annu. Rev. Microbiol. 57:155–176. 10.1146/annurev.micro.57.030502.090820 [DOI] [PubMed] [Google Scholar]
- 7.Magasanik B. 1996. Regulation of nitrogen utilization, p 1344–1356 In Neidhardt FC, Curtiss R, III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC [Google Scholar]
- 8.Merrick MJ, Edwards RA. 1995. Nitrogen control in bacteria. Microbiol. Rev. 59:604–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reitzer L, Schneider BL. 2001. Metabolic context and possible physiological themes of σ54-dependent genes in Escherichia coli. Microbiol. Mol. Biol. Rev. 65:422–444. 10.1128/MMBR.65.3.422-444.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boogerd FC, Bruggeman FJ, Hofmeyr J-HS, Westerhoff HV. (ed). 2007. Systems biology (philosophical foundations), 1st ed. Elsevier, Amsterdam, the Netherlands [Google Scholar]
- 11.Westerhoff HV, Palsson BØ. 2004. The evolution of molecular biology into systems biology. Nat. Biotechnol. 22:1249–1252. 10.1038/nbt1020 [DOI] [PubMed] [Google Scholar]
- 12.Westerhoff HV, Kolodkin A, Conradie R, Wilkinson SJ, Bruggeman FJ, Krab K, van Schuppen JH, Hardin H, Bakker BM, Moné MJ, Rybakova KN, Eijken M, van Leeuwen HJP, Snoep JL. 2009. Systems biology towards life in silico: mathematics of the control of living cells. J. Math. Biol. 58:7–34. 10.1007/s00285-008-0160-8 [DOI] [PubMed] [Google Scholar]
- 13.Brenner FW, Villar RG, Angulo FJ, Tauxe R, Swaminathan B. 2000. Salmonella nomenclature. J. Clin. Microbiol. 38:2465–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lan R, Reeves PR. 2002. Escherichia coli in disguise: molecular origins of Shigella. Microbes Infect. 4:1125–1132. 10.1016/S1286-4579(02)01637-4 [DOI] [PubMed] [Google Scholar]
- 15.Gutnick D, Calvo JM, Klopotowski T, Ames BN. 1969. Compounds which serve as the sole source of carbon or nitrogen for Salmonella typhimurium LT-2. J. Bacteriol. 100:215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyler B. 1978. Regulation of the assimilation of nitrogen compounds. Annu. Rev. Biochem. 47:1127–1162. 10.1146/annurev.bi.47.070178.005403 [DOI] [PubMed] [Google Scholar]
- 17.Brons HJ, Zehnder AJ. 1990. Aerobic nitrate and nitrite reduction in continuous cultures of Escherichia coli E4. Arch. Microbiol. 153:531–536. 10.1007/BF00245261 [DOI] [PubMed] [Google Scholar]
- 18.Tempest DW, Meers JL, Brown CM. 1970. Synthesis of glutamate in Aerobacter aerogenes by a hitherto unknown route. Biochem. J. 117:405–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakamoto N, Kotre AM, Savageau MA. 1975. Glutamate dehydrogenase from Escherichia coli: purification and properties. J. Bacteriol. 124:775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veronese FM, Boccu E, Conventi L. 1975. Glutamate dehydrogenase from Escherichia coli: induction, purification and properties of the enzyme. Biochim. Biophys. Acta 377:217–228. 10.1016/0005-2744(75)90304-6 [DOI] [PubMed] [Google Scholar]
- 21.Sharkey MA, Engel PC. 2008. Apparent negative co-operativity and substrate inhibition in overexpressed glutamate dehydrogenase from Escherichia coli. FEMS Microbiol. Lett. 281:132–139. 10.1111/j.1574-6968.2008.01086.x [DOI] [PubMed] [Google Scholar]
- 22.Alibhai M, Villafranca JJ. 1994. Kinetic and mutagenic studies of the role of the active site residues Asp-50 and Glu-327 of Escherichia coli glutamine synthetase. Biochemistry 33:682–686. 10.1021/bi00169a008 [DOI] [PubMed] [Google Scholar]
- 23.Colanduoni J, Nissan R, Villafranca JJ. 1987. Studies of the mechanism of glutamine synthetase utilizing pH-dependent behavior in catalysis and binding. J. Biol. Chem. 262:3037–3043 [PubMed] [Google Scholar]
- 24.Meek TD, Villafranca JJ. 1980. Kinetic mechanism of Escherichia coli glutamine synthetase. Biochemistry 19:5513–5519. 10.1021/bi00565a008 [DOI] [PubMed] [Google Scholar]
- 25.Arcondéguy T, Jack R, Merrick M. 2001. PII signal transduction proteins, pivotal players in microbial nitrogen control. Microbiol. Mol. Biol. Rev. 65:80–105. 10.1128/MMBR.65.1.80-105.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forchhammer K. 2008. PII signal transducers: novel functional and structural insights. Trends Microbiol. 16:65–72. 10.1016/j.tim.2007.11.004 [DOI] [PubMed] [Google Scholar]
- 27.Leigh JA, Dodsworth JA. 2007. Nitrogen regulation in bacteria and archaea. Annu. Rev. Microbiol. 61:349–377. 10.1146/annurev.micro.61.080706.093409 [DOI] [PubMed] [Google Scholar]
- 28.Ninfa AJ, Atkinson MR. 2000. PII signal transduction proteins. Trends Microbiol. 8:172–179. 10.1016/S0966-842X(00)01709-1 [DOI] [PubMed] [Google Scholar]
- 29.Ninfa AJ, Jiang P. 2005. PII signal transduction proteins: sensors of alpha-ketoglutarate that regulate nitrogen metabolism. Curr. Opin. Microbiol. 8:168–173. 10.1016/j.mib.2005.02.011 [DOI] [PubMed] [Google Scholar]
- 30.Rhee SG, Chock PB, Stadtman ER. 1989. Regulation of Escherichia coli glutamine synthetase. Adv. Enzymol. Relat. Areas Mol. Biol. 62:37–92 [DOI] [PubMed] [Google Scholar]
- 31.Stadtman ER. 2001. The story of glutamine synthetase regulation. J. Biol. Chem. 48:44357–44364 [DOI] [PubMed] [Google Scholar]
- 32.Zimmer DP, Soupene E, Lee HL, Wendisch VF, Khodursky AB, Peter BJ, Bender RA, Kustu S. 2000. Nitrogen regulatory protein C-controlled genes of Escherichia coli: scavenging as a defense against nitrogen limitation. Proc. Natl. Acad. Sci. U. S. A. 97:14674–14679. 10.1073/pnas.97.26.14674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colombo G, Villafranca JJ. 1986. Amino acid sequence of Escherichia coli glutamine synthetase deduced from the DNA nucleotide sequence. J. Biol. Chem. 261:10587–10591 [PubMed] [Google Scholar]
- 34.Miranda-Rios J, Sanchez-Pescador R, Urdea M, Covarrubias AA. 1987. The complete nucleotide sequence of the glnALG operon of Escherichia coli K12. Nucleic Acids Res. 15:2757–2770. 10.1093/nar/15.6.2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valentine RC, Shapiro BM, Stadtman ER. 1968. Regulation of glutamine synthetase. XII. Electron microscopy of the enzyme from Escherichia coli. Biochemistry 7:2143–2152 [DOI] [PubMed] [Google Scholar]
- 36.Almassy RJ, Janson CA, Hamlin R, Xuong NH, Eisenberg D. 1986. Novel subunit-subunit interactions in the structure of glutamine synthetase. Nature 323:304–309. 10.1038/323304a0 [DOI] [PubMed] [Google Scholar]
- 37.Yamashita MM, Almassy RJ, Janson CA, Cascio D, Eisenberg D. 1989. Refined atomic model of glutamine synthetase at 3.5 Å resolution. J. Biol. Chem. 264:17681–17690 [DOI] [PubMed] [Google Scholar]
- 38.Liaw SH, Eisenberg D. 1994. Structural model for the reaction mechanism of glutamine synthetase, based on five crystal structures of enzyme-substrate complexes. Biochemistry 33:675–681. 10.1021/bi00169a007 [DOI] [PubMed] [Google Scholar]
- 39.Liaw SH, Jun G, Eisenberg D. 1993. Extending the diffraction limit of protein crystals: the example of glutamine synthetase from Salmonella typhimurium in the presence of its cofactor ATP. Protein Sci. 2:470–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liaw SH, Jun G, Eisenberg D. 1994. Interactions of nucleotides with fully unadenylylated glutamine synthetase from Salmonella typhimurium. Biochemistry 33:11184–11188. 10.1021/bi00203a014 [DOI] [PubMed] [Google Scholar]
- 41.Liaw SH, Kuo I, Eisenberg D. 1995. Discovery of the ammonium substrate site on glutamine synthetase, a third cation binding site. Protein Sci. 4:2358–2365. 10.1002/pro.5560041114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruggeman F. 2005. Of molecules and cells. Emergent mechnisms. VU University Amsterdam, Amsterdam, the Netherlands [Google Scholar]
- 43.Rhee SG, Chock PB, Stadtman ER. 1985. Glutamine synthetase from Escherichia coli. Methods Enzymol. 113:213–241. 10.1016/S0076-6879(85)13032-6 [DOI] [PubMed] [Google Scholar]
- 44.Bruggeman FJ, Boogerd FC, Westerhoff HV. 2005. The multifarious short-term regulation of ammonium assimilation of Escherichia coli: dissection using an in silico replica. FEBS J. 272:1965–1985. 10.1111/j.1742-4658.2005.04626.x [DOI] [PubMed] [Google Scholar]
- 45.Ginsburg A, Yeh J, Hennig SB, Denton MD. 1970. Some effects of adenylylation on the biosynthetic properties of the glutamine synthetase from Escherichia coli. Biochemistry 9:633–649. 10.1021/bi00805a025 [DOI] [PubMed] [Google Scholar]
- 46.Miller RE, Miller RE, Stadtman ER, Stadtman ER. 1972. Glutamate synthase from Escherichia coli. An iron-sulfide flavoprotein. J. Biol. Chem. 247:7407–7419 [PubMed] [Google Scholar]
- 47.Vanoni MA, Curti B. 2008. Structure-function studies of glutamate synthases: a class of self-regulated iron-sulfur flavoenzymes essential for nitrogen assimilation. IUBMB Life 60:287–300. 10.1002/iub.52 [DOI] [PubMed] [Google Scholar]
- 48.List F, Vega MC, Razeto A, Häger MC, Sterner R, Wilmanns M. 2012. Catalysis uncoupling in a glutamine amidotransferase bienzyme by unblocking the glutaminase active site. Chem. Biol. 19:1589–1599. 10.1016/j.chembiol.2012.10.012 [DOI] [PubMed] [Google Scholar]
- 49.Mantsala P, Zalkin H. 1976. Properties of apoglutamate synthase and comparison with glutamate dehydrogenase. J. Biol. Chem. 251:3300–3305 [PubMed] [Google Scholar]
- 50.Castano I, Bastarrachea F, Covarrubias AA. 1988. gltBDF operon of Escherichia coli. J. Bacteriol. 170:821–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castano I, Flores N, Valle F, Covarrubias AA, Bolivar F. 1992. gltF, a member of the gltBDF operon of Escherichia coli, is involved in nitrogen-regulated gene expression. Mol. Microbiol. 6:2733–2741. 10.1111/j.1365-2958.1992.tb01450.x [DOI] [PubMed] [Google Scholar]
- 52.Grassl G, Bufe B, Muller B, Rosel M, Kleiner D. 1999. Characterization of the gltF gene product of Escherichia coli. FEMS Microbiol. Lett. 179:79–84. 10.1111/j.1574-6968.1999.tb08711.x [DOI] [PubMed] [Google Scholar]
- 53.Goss TJ, Perez-Matos A, Bender RA. 2001. Roles of glutamate synthase, gltBD, and gltF in nitrogen metabolism of Escherichia coli and Klebsiella aerogenes. J. Bacteriol. 183:6607–6619. 10.1128/JB.183.22.6607-6619.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rendina AR, Orme-Johnson WH. 1978. Glutamate synthase: on the kinetic mechanism of the enzyme from Escherichia coli W. Biochemistry 17:5388–5393. 10.1021/bi00618a011 [DOI] [PubMed] [Google Scholar]
- 55.Coulton JW, Kapoor M. 1973. Purification and some properties of the glutamate dehydrogenase of Salmonella typhimurium. Can. J. Microbiol. 19:427–438. 10.1139/m73-071 [DOI] [PubMed] [Google Scholar]
- 56.Lin HP, Reeves HC. 1991. Purification and characterization of NADP+-specific glutamate dehydrogenase from Escherichia coli. Curr. Microbiol. 22:371–376. 10.1007/BF02092157 [DOI] [Google Scholar]
- 57.McPherson MJ, Wootton JC. 1983. Complete nucleotide sequence of the Escherichia coli gdhA gene. Nucleic Acids Res. 11:5257–5266. 10.1093/nar/11.15.5257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valle F, Becerril B, Chen E, Seeburg P, Heyneker H, Bolivar F. 1984. Complete nucleotide sequence of the glutamate dehydrogenase gene from Escherichia coli K-12. Gene 27:193–199. 10.1016/0378-1119(84)90140-9 [DOI] [PubMed] [Google Scholar]
- 59.Engel PC, Dalziel K. 1967. The equilibrium constants of the glutamate dehydrogenase systems. Biochem. J. 105:691–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coulton JW, Kapoor M. 1973. Studies on the kinetics and regulation of glutamate dehydrogenase of Salmonella typhimurium. Can. J. Microbiol. 19:439–450. 10.1139/m73-072 [DOI] [PubMed] [Google Scholar]
- 61.Maurizi MR, Rasulova F. 2002. Degradation of glutamate dehydrogenase from Escherichia coli: allosteric regulation of enzyme stability. Arch. Biochem. Biophys. 397:206–216. 10.1006/abbi.2001.2703 [DOI] [PubMed] [Google Scholar]
- 62.Lin HP, Reeves HC. 1992. Properties of phosphorylated NADP+-specific glutamate dehydrogenase from Escherichia coli. Curr. Microbiol. 24:73–79. 10.1007/BF01570901 [DOI] [Google Scholar]
- 63.Lin HP, Reeves HC. 1994. In vivo phosphorylation of NADP+ glutamate dehydrogenase in Escherichia coli. Curr. Microbiol. 28:63–65. 10.1007/BF01569047 [DOI] [Google Scholar]
- 64.Willison JC, Tissot G. 1994. The Escherichia coli efg gene and the Rhodobacter capsulatus adgA gene code for NH3-dependent NAD synthetase. J. Bacteriol. 176:3400–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allibert P, Willison JC, Vignais PM. 1987. Complementation of nitrogen-regulatory (ntr-like) mutations in Rhodobacter capsulatus by an Escherichia coli gene: cloning and sequencing of the gene and characterization of the gene product. J. Bacteriol. 169:260–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneider BL, Reitzer LJ. 1998. Salmonella typhimurium nit is nadE: defective nitrogen utilization and ammonia-dependent NAD synthetase. J. Bacteriol. 180:4739–4741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spencer RL, Preiss J. 1967. Biosynthesis of diphosphopyridine nucleotide. The purification and the properties of diphospyridine nucleotide synthetase from Escherichia coli B. J. Biol. Chem. 242:385–392 [PubMed] [Google Scholar]
- 68.Hughes KT, Olivera BM, Roth JR. 1988. Structural gene for NAD synthetase in Salmonella typhimurium. J. Bacteriol. 170:2113–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willison JC. 1992. An essential gene (efg) located at 38.1 minutes on the Escherichia coli chromosome. J. Bacteriol. 174:5765–5766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Broach J, Neumann C, Kustu S. 1976. Mutant strains (nit) of Salmonella typhimurium with a pleiotropic defect in nitrogen metabolism. J. Bacteriol. 128:86–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Lay N, Gottesman S. 2009. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J. Bacteriol. 191:461–476. 10.1128/JB.01157-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mergeay M, Gigot D, Beckmann J, Glansdorff N, Pierard A. 1974. Physiology and genetics of carbamoylphosphate synthesis in Escherichia coli K12. Mol. Gen. Genet. 133:299–316. 10.1007/BF00332706 [DOI] [PubMed] [Google Scholar]
- 73.Holden HM, Thoden JB, Raushel FM. 1999. Carbamoyl phosphate synthetase: an amazing biochemical odyssey from substrate to product. Cell. Mol. Life Sci. 56:507–522. 10.1007/s000180050448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guy HI, Rotgeri A, Evans DR. 1997. Activation by fusion of the glutaminase and synthetase subunits of Escherichia coli carbamyl-phosphate synthetase. J. Biol. Chem. 272:19913–19918. 10.1074/jbc.272.32.19913 [DOI] [PubMed] [Google Scholar]
- 75.Rubino SD, Nyunoya H, Lusty CJ. 1986. Catalytic domains of carbamyl phosphate synthetase. Glutamine-hydrolyzing site of Escherichia coli carbamyl phosphate synthetase. J. Biol. Chem. 261:11320–11327 [PubMed] [Google Scholar]
- 76.Rubino SD, Nyunoya H, Lusty CJ. 1987. In vivo synthesis of carbamyl phosphate from NH3 by the large subunit of Escherichia coli carbamyl phosphate synthetase. J. Biol. Chem. 262:4382–4386 [PubMed] [Google Scholar]
- 77.Kim J, Copley SD. 2007. Why metabolic enzymes are essential or nonessential for growth of Escherichia coli K12 on glucose. Biochemistry 46:12501–12511. 10.1021/bi7014629 [DOI] [PubMed] [Google Scholar]
- 78.Felton J, Michaelis S, Wright A. 1980. Mutations in two unlinked genes are required to produce asparagine auxotrophy in Escherichia coli. J. Bacteriol. 142:221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Humbert R, Simoni R. 1980. Genetic and biochemical studies demonstrating a second gene coding for asparagine synthetase in Escherichia coli. J. Bacteriol. 142:212–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Poggio S, Domeinzain C, Osorio A, Camarena L. 2002. The nitrogen assimilation control (Nac) protein represses asnC and asnA transcription in Escherichia coli. FEMS Microbiol. Lett. 206:151–156. 10.1111/j.1574-6968.2002.tb11001.x [DOI] [PubMed] [Google Scholar]
- 81.Li KK, Beeson WT, Ghiviriga I, Richards NG. 2007. A convenient gHMQC-based NMR assay for investigating ammonia channeling in glutamine-dependent amidotransferases: studies of Escherichia coli asparagine synthetase B. Biochemistry 46:4840–4849. 10.1021/bi700145t [DOI] [PubMed] [Google Scholar]
- 82.Ebner E, Wolf D, Gancedo C, Elsasser S, Holzer H. 1970. ATP:glutamine synthetase adenylyltransferase from Escherichia coli B. Purification and properties. Eur. J. Biochem. 14:535–544 [DOI] [PubMed] [Google Scholar]
- 83.Heilmeyer L, Jr, Battig F, Holzer H. 1969. Characterization of a glutamine synthetase b activating (deadenylylating) enzyme system in Escherichia coli. Eur. J. Biochem. 9:259–262. 10.1111/j.1432-1033.1969.tb00603.x [DOI] [PubMed] [Google Scholar]
- 84.Stadtman ER, Shapiro BM, Kingdon HS, Woolfolk CA, Hubbard JS. 1968. Cellular regulation of glutamine synthetase activity in Escherichia coli. Adv. Enzyme Regul. 6:257–289. 10.1016/0065-2571(68)90017-4 [DOI] [PubMed] [Google Scholar]
- 85.Wohlhueter RM, Ebner E, Wolf DH. 1972. Studies on the reaction mechanism of adenosine triphosphate:glutamine synthetase adenylyltransferase from Escherichia coli B. Evidence for an ordered mechanism. J. Biol. Chem. 247:4213–4218 [PubMed] [Google Scholar]
- 86.Anderson WB, Stadtman ER. 1970. Glutamine synthetase deadenylation: a phosphorolytic reaction yielding ADP as nucleotide product. Biochem. Biophys. Res. Commun. 41:704–709. 10.1016/0006-291X(70)90070-7 [DOI] [PubMed] [Google Scholar]
- 87.Muse WB, Bender RA. 1992. Map position of the glnE gene from Escherichia coli. J. Bacteriol. 174:7876–7877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Heeswijk WC, Rabenberg M, Westerhoff HV, Kahn D. 1993. The genes of the glutamine synthetase adenylylation cascade are not regulated by nitrogen in Escherichia coli. Mol. Microbiol. 9:443–457. 10.1111/j.1365-2958.1993.tb01706.x [DOI] [PubMed] [Google Scholar]
- 89.Caban CE, Ginsburg A. 1976. Glutamine synthetase adenylyltransferase from Escherichia coli: purification and physical and chemical properties. Biochemistry 15:1569–1580. 10.1021/bi00652a030 [DOI] [PubMed] [Google Scholar]
- 90.Anderson WB, Hennig SB, Ginsburg A, Stadtman ER. 1970. Association of ATP:glutamine synthetase adenylyltransferase activity with the P1 component of the glutamine synthetase deadenylylation system. Proc. Natl. Acad. Sci. U. S. A. 67:1417–1424. 10.1073/pnas.67.3.1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Engleman EG, Francis SH. 1978. Cascade control of E. coli glutamine synthetase. II. Metabolite regulation of the enzymes in the cascade. Arch. Biochem. Biophys. 191:602–612 [DOI] [PubMed] [Google Scholar]
- 92.Bancroft S, Rhee SG, Neumann C, Kustu S. 1978. Mutations that alter the covalent modification of glutamine synthetase in Salmonella typhimurium. J. Bacteriol. 134:1046–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reuveny Z, Foor F, Magasanik B. 1981. Regulation of glutamine synthetase by regulatory protein PII in Klebsiella aerogenes mutants lacking adenylyltransferase. J. Bacteriol. 146:740–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rhee SG, Park SC, Koo JH. 1985. The role of adenylyltransferase and uridylyltransferase in the regulation of glutamine synthetase in Escherichia coli. Curr. Top. Cell. Regul. 27:221–232 [DOI] [PubMed] [Google Scholar]
- 95.Jaggi R, van Heeswijk WC, Westerhoff HV, Ollis DL, Vasudevan SG. 1997. The two opposing activities of adenylyl transferase reside in distinct homologous domains, with intramolecular signal transduction. EMBO J. 16:5562–5571. 10.1093/emboj/16.18.5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiang P, Mayo AE, Ninfa AJ. 2007. Escherichia coli glutamine synthetase adenylyltransferase (ATase, EC 2.7.7.49): kinetic characterization of regulation by PII, PII-UMP, glutamine, and alpha-ketoglutarate. Biochemistry 46:4133–4146. 10.1021/bi0620510 [DOI] [PubMed] [Google Scholar]
- 97.Davies JM, Poole RJ, Sanders D. 1993. The computed free energy change of hydrolysis of inorganic pyrophosphate and ATP: apparent significance for inorganic-pyrophosphate-driven reactions of intermediary metabolism. Biochim. Biophys. Acta 1141:29–36. 10.1016/0005-2728(93)90185-I [DOI] [Google Scholar]
- 98.Jiang P, Pioszak AA, Ninfa AJ. 2007. Structure-function analysis of glutamine synthetase adenylyltransferase (ATase, EC 2.7.7.49) of Escherichia coli. Biochemistry 46:4117–4132. 10.1021/bi0620508 [DOI] [PubMed] [Google Scholar]
- 99.Xu Y, Wen D, Clancy P, Carr PD, Ollis DL, Vasudevan SG. 2004. Expression, purification, crystallization, and preliminary X-ray analysis of the N-terminal domain of Escherichia coli adenylyl transferase. Protein Expr. Purif. 34:142–146. 10.1016/j.pep.2003.11.001 [DOI] [PubMed] [Google Scholar]
- 100.Clancy P, Xu Y, van Heeswijk WC, Vasudevan SG, Ollis DL. 2007. The domains carrying the opposing activities in adenylyltransferase are separated by a central regulatory domain. FEBS J. 274:2865–2877. 10.1111/j.1742-4658.2007.05820.x [DOI] [PubMed] [Google Scholar]
- 101.Wootton JC, Drummond MH. 1989. The Q-linker: a class of interdomain sequences found in bacterial multidomain regulatory proteins. Protein Eng. 2:535–543. 10.1093/protein/2.7.535 [DOI] [PubMed] [Google Scholar]
- 102.Holm L, Sander C. 1995. DNA polymerase β belongs to an ancient nucleotidyltransferase superfamily. Trends Biochem. Sci. 20:345–347. 10.1016/S0968-0004(00)89071-4 [DOI] [PubMed] [Google Scholar]
- 103.Xu Y, Carr PD, Vasudevan SG, Ollis DL. 2010. Structure of the adenylylation domain of E. coli glutamine synthetase adenylyl transferase: evidence for gene duplication and evolution of a new active site. J. Mol. Biol. 396:773–784. 10.1016/j.jmb.2009.12.011 [DOI] [PubMed] [Google Scholar]
- 104.Xu Y, Zhang R, Joachimiak A, Carr PD, Huber T, Vasudevan SG, Ollis DL. 2004. Structure of the N-terminal domain of Escherichia coli glutamine synthetase adenylyltransferase. Structure 12:861–869. 10.1016/j.str.2004.02.029 [DOI] [PubMed] [Google Scholar]
- 105.Clancy P. 2004. Studies of protein complexes involved in the adenylylation cascade of the nitrogen signalling pathway of Escherichia coli. Ph.D. thesis James Cook University, Townsville, Australia [Google Scholar]
- 106.Ortega F, Acerenza L, Westerhoff HV, Mas F, Cascante M. 2002. Product dependence and bifunctionality compromise the ultrasensitivity of signal transduction cascades. Proc. Natl. Acad. Sci. U. S. A. 99:1170–1175. 10.1073/pnas.022267399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hart Y, Madar D, Yuan J, Bren A, Mayo AE, Rabinowitz JD, Alon U. 2011. Robust control of nitrogen assimilation by a bifunctional enzyme in E. coli. Mol. Cell 41:117–127. 10.1016/j.molcel.2010.12.023 [DOI] [PubMed] [Google Scholar]
- 108.Heinrich CP, Battig FA, Mantel M, Holzer H. 1970. Formation of pyrophosphate during ATP:glutamine synthetase-adenylyltransferase-reaction in E. coli. Arch. Mikrobiol. 73:104–110 [DOI] [PubMed] [Google Scholar]
- 109.Kingdon HS, Shapiro BM, Stadtman ER. 1967. Regulation of glutamine synthetase. 8. ATP:glutamine synthetase adenylyltransferase, an enzyme that catalyzes alterations in the regulatory properties of glutamine synthetase. Proc. Natl. Acad. Sci. U. S. A. 58:1703–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wulff K, Mecke D, Holzer H. 1967. Mechanism of the enzymatic inactivation of glutamine synthetase from E. coli. Biochem. Biophys. Res. Commun. 28:740–745. 10.1016/0006-291X(67)90378-6 [DOI] [PubMed] [Google Scholar]
- 111.Denton MD, Ginsburg A. 1970. Some characteristics of the binding of substrates of glutamine synthetase from Escherichia coli. Biochemistry 9:617–632. 10.1021/bi00805a024 [DOI] [PubMed] [Google Scholar]
- 112.Heinrikson RL, Kingdon HS. 1971. Primary structure of Escherichia coli glutamine synthetase. II. The complete amino acid sequence of a tryptic heneicosapeptide containing covalently bound adenylic acid. J. Biol. Chem. 246:1099–1106 [PubMed] [Google Scholar]
- 113.Heinrikson RL, Kingdon HS. 1970. The amino acid sequence in the vicinity of the covalently bound adenylic acid in glutamine synthetase from Escherichia coli. J. Biol. Chem. 245:138–142 [PubMed] [Google Scholar]
- 114.Rhee SG. 1984. 5′-Nucleotidyl-O-tyrosine bond in glutamine synthetase. Methods Enzymol. 107:183–200. 10.1016/0076-6879(84)07012-9 [DOI] [PubMed] [Google Scholar]
- 115.Shapiro BM, Stadtman ER. 1968. 5′-Adenylyl-O-tyrosine. The novel phosphodiester residue of adenylylated glutamine synthetase from Escherichia coli. J. Biol. Chem. 243:3769–3771 [PubMed] [Google Scholar]
- 116.Frink RJ, Eisenberg D, Glitz DG. 1978. Localization of the site of adenylylation of glutamine synthetase by electron microscopy of an enzyme-antibody complex. Proc. Natl. Acad. Sci. U. S. A. 75:5778–5782. 10.1073/pnas.75.12.5778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bywater RP, Carlisle CH, Jackson RB, Mackay AL, Timmins PA. 1975. Crystals of glutamine synthetase from Escherichia coli. J. Mol. Biol. 91:293–300. 10.1016/0022-2836(75)90381-2 [DOI] [PubMed] [Google Scholar]
- 118.Heidner EG, Frey TG, Held U, Weissman LJ, Fenna RE, Lei M, Harel M, Kabsch H, Sweet RM, Eisenberg D. 1978. New crystal forms of glutamine synthetase and implications for the molecular structure. J. Mol. Biol. 122:163–173. 10.1016/0022-2836(78)90033-5 [DOI] [PubMed] [Google Scholar]
- 119.Kabsch W, Kabsch H, Eisenberg D. 1976. Packing in a new crystalline form of glutamine synthetase from Escherichia coli. J. Mol. Biol. 100:283–291. 10.1016/S0022-2836(76)80064-2 [DOI] [PubMed] [Google Scholar]
- 120.Shapiro BM, Kingdon HS, Stadtman ER. 1967. Regulation of glutamine synthetase. VII. Adenylyl glutamine synthetase: a new form of the enzyme with altered regulatory and kinetic properties. Proc. Natl. Acad. Sci. U. S. A. 58:642–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bender RA, Janssen KA, Resnick AD, Blumenberg M, Foor F, Magasanik B. 1977. Biochemical parameters of glutamine synthetase from Klebsiella aerogenes. J. Bacteriol. 129:1001–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stadtman ER, Smyrniotis PZ, Davis JN, Wittenberger ME. 1979. Enzymic procedures for determining the average state of adenylylation of Escherichia coli glutamine synthetase. Anal. Biochem. 95:275–285. 10.1016/0003-2697(79)90217-3 [DOI] [PubMed] [Google Scholar]
- 123.Jiang P, Ninfa AJ. 2009. Reconstitution of Escherichia coli glutamine synthetase adenylyltransferase from N-terminal and C-terminal fragments of the enzyme. Biochemistry 48:415–423. 10.1021/bi801775b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kamberov ES, Atkinson MR, Feng J, Chandran P, Ninfa AJ. 1994. Sensory components controlling bacterial nitrogen assimilation. Cell. Mol. Biol. Res. 40:175–191 [PubMed] [Google Scholar]
- 125.Kim IH, Park SC. 1990. Organization and nucleotide sequence of genes in the four-minute region of Escherichia coli chromosome map locates between rpsB and glnD. Mol. Cells 1:49–56 [Google Scholar]
- 126.Park SC. 1989. Optimum conditions for overproduction of uridylyltransferase-uridylyl removing enzyme via glnD overexpression vector in Escherichia coli. Korean J. Biochem. 21:103–111 [Google Scholar]
- 127.Jiang P, Peliska JA, Ninfa AJ. 1998. Enzymological characterization of the signal-transducing uridylyltransferase/uridylyl-removing enzyme (EC 2.7.7.59) of Escherichia coli and its interaction with the PII protein. Biochemistry 37:12782–12794. 10.1021/bi980667m [DOI] [PubMed] [Google Scholar]
- 128.Zhang Y, Pohlmann EL, Serate J, Conrad MC, Roberts GP. 2010. Mutagenesis and functional characterization of the four domains of GlnD, a bifunctional nitrogen sensor protein. J. Bacteriol. 192:2711–2721. 10.1128/JB.01674-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aravind L, Koonin EV. 1998. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 23:469–472. 10.1016/S0968-0004(98)01293-6 [DOI] [PubMed] [Google Scholar]
- 130.Aravind L, Koonin EV. 1999. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 287:1023–1040. 10.1006/jmbi.1999.2653 [DOI] [PubMed] [Google Scholar]
- 131.Tondervik A, Torgersen HR, Botnmark HK, Strom AR. 2006. Transposon mutations in the 5′ end of glnD, the gene for a nitrogen regulatory sensor, that suppress the osmosensitive phenotype caused by otsBA lesions in Escherichia coli. J. Bacteriol. 188:4218–4226. 10.1128/JB.00513-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Eisenthal R, Danson MJ, Hough DW. 2007. Catalytic efficiency and kcat/KM: a useful comparator? Trends Biotechnol. 25:247–249. 10.1016/j.tibtech.2007.03.010 [DOI] [PubMed] [Google Scholar]
- 133.Bueno R, Pahel G, Magasanik B. 1985. Role of glnB and glnD gene products in regulation of the glnALG operon of Escherichia coli. J. Bacteriol. 164:816–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kamberov ES, Atkinson MR, Ninfa AJ. 1995. The Escherichia coli PII signal transduction protein is activated upon binding 2-ketoglutarate and ATP. J. Biol. Chem. 270:17797–17807. 10.1074/jbc.270.30.17797 [DOI] [PubMed] [Google Scholar]
- 135.Yuan J, Doucette CD, Fowler WU, Feng XJ, Piazza M, Rabitz HA, Wingreen NS, Rabinowitz JD. 2009. Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol. Syst. Biol. 5:302. 10.1038/msb.2009.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jiang P, Peliska JA, Ninfa AJ. 1998. Reconstitution of the signal-transduction bicyclic cascade responsible for the regulation of Ntr gene transcription in Escherichia coli. Biochemistry 37:12795–12801. 10.1021/bi9802420 [DOI] [PubMed] [Google Scholar]
- 137.Mangum JH, Magni G, Stadtman ER. 1973. Regulation of glutamine synthetase adenylylation and deadenylylation by the enzymatic uridylylation and deuridylylation of the PII regulatory protein. Arch. Biochem. Biophys. 158:514–525. 10.1016/0003-9861(73)90543-2 [DOI] [PubMed] [Google Scholar]
- 138.Atkinson MR, Ninfa AJ. 1999. Characterization of the GlnK protein of Escherichia coli. Mol. Microbiol. 32:301–313. 10.1046/j.1365-2958.1999.01349.x [DOI] [PubMed] [Google Scholar]
- 139.van Heeswijk WC, Hoving S, Molenaar D, Stegeman B, Kahn D, Westerhoff HV. 1996. An alternative PII protein in the regulation of glutamine synthetase in Escherichia coli. Mol. Microbiol. 21:133–146. 10.1046/j.1365-2958.1996.6281349.x [DOI] [PubMed] [Google Scholar]
- 140.van Heeswijk WC, Wen D, Clancy P, Jaggi R, Ollis DL, Westerhoff HV, Vasudevan SG. 2000. The Escherichia coli signal transducers PII (GlnB) and GlnK form heterotrimers in vivo: fine tuning the nitrogen signal cascade. Proc. Natl. Acad. Sci. U. S. A. 97:3942–3947. 10.1073/pnas.97.8.3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Anderson WB, Stadtman ER. 1971. Purification and functional roles of the PI and PII components of Escherichia coli glutamine synthetase deadenylylation system. Arch. Biochem. Biophys. 143:428–443. 10.1016/0003-9861(71)90229-3 [DOI] [PubMed] [Google Scholar]
- 142.Brown MS, Segal A, Stadtman ER. 1971. Modulation of glutamine synthetase adenylylation and deadenylylation is mediated by metabolic transformation of the PII-regulatory protein. Proc. Natl. Acad. Sci. U. S. A. 68:2949–2953. 10.1073/pnas.68.12.2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Holtel A, Merrick M. 1988. Identification of the Klebsiella pneumoniae glnB gene: nucleotide sequence of wild-type and mutant alleles. Mol. Gen. Genet. 215:134–138. 10.1007/BF00331314 [DOI] [PubMed] [Google Scholar]
- 144.Liu J, Magasanik B. 1993. The glnB region of the Escherichia coli chromosome. J. Bacteriol. 175:7441–7449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Son HS, Rhee SG. 1987. Cascade control of Escherichia coli glutamine synthetase. Purification and properties of PII protein and nucleotide sequence of its structural gene. J. Biol. Chem. 262:8690–8695 [PubMed] [Google Scholar]
- 146.Vasudevan SG, Armarego WLF, Shaw DC, Lilley PE, Dixon NE, Poole RK. 1991. Isolation and nucleotide sequence of the hmp gene that encodes a haemoglobin-like protein in Escherichia coli K-12. Mol. Gen. Genet. 226:49–58 [DOI] [PubMed] [Google Scholar]
- 147.Jaggi R, Ybarlucea W, Cheah E, Carr PD, Edwards KJ, Ollis DL, Vasudevan SG. 1996. The role of the T-loop of the signal transducing protein PII from Escherichia coli. FEBS Lett. 391:223–228. 10.1016/0014-5793(96)00737-5 [DOI] [PubMed] [Google Scholar]
- 148.Jiang P, Zucker P, Atkinson MR, Kamberov ES, Tirasophon W, Chandran P, Schefke BR, Ninfa AJ. 1997. Structure/function analysis of the PII signal transduction protein of Escherichia coli: genetic separation of interactions with protein receptors. J. Bacteriol. 179:4342–4353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Adler SP, Purich D, Stadtman ER. 1975. Cascade control of Escherichia coli glutamine synthetase. Properties of the PII regulatory protein and the uridylyltransferase-uridylyl-removing enzyme. J. Biol. Chem. 250:6264–6272 [PubMed] [Google Scholar]
- 150.Vasudevan SG, Gedye C, Dixon NE, Cheah E, Carr PD, Suffolk PM, Jeffrey PD, Ollis DL. 1994. Escherichia coli PII protein: purification, crystallization and oligomeric structure. FEBS Lett. 337:255–258. 10.1016/0014-5793(94)80203-3 [DOI] [PubMed] [Google Scholar]
- 151.de Mel VS, Kamberov ES, Martin PD, Zhang J, Ninfa AJ, Edwards BF. 1994. Preliminary X-ray diffraction analysis of crystals of the PII protein from Escherichia coli. J. Mol. Biol. 243:796–798. 10.1016/0022-2836(94)90049-3 [DOI] [PubMed] [Google Scholar]
- 152.Carr PD, Cheah E, Suffolk PM, Vasudevan SG, Dixon NE, Ollis DL. 1996. X-ray structure of the signal transduction protein PII from Escherichia coli at 1.9 Å. Acta Crystallogr. D Biol. Crystallogr. 52:93–104. 10.1107/S0907444995007293 [DOI] [PubMed] [Google Scholar]
- 153.Cheah E, Carr PD, Suffolk PM, Vasudevan SG, Dixon NE, Ollis DL. 1994. Structure of the Escherichia coli signal transducing protein PII. Structure 2:981–990. 10.1016/S0969-2126(94)00100-6 [DOI] [PubMed] [Google Scholar]
- 154.Allikmets R, Gerrard B, Court D, Dean M. 1993. Cloning and organization of the abc and mdl genes of Escherichia coli: relationship to eukaryotic multidrug resistance. Gene 136:231–236. 10.1016/0378-1119(93)90470-N [DOI] [PubMed] [Google Scholar]
- 155.van Heeswijk WC, Stegeman B, Hoving S, Molenaar D, Kahn D, Westerhoff HV. 1995. An additional PII in Escherichia coli: a new regulatory protein in the glutamine synthetase cascade. FEMS Microbiol. Lett. 132:153–157 [DOI] [PubMed] [Google Scholar]
- 156.Xu Y, Cheah E, Carr PD, van Heeswijk WC, Westerhoff HV, Vasudevan SG, Ollis DL. 1998. GlnK, a PII-homologue: structure reveals ATP binding site and indicates how the T-loops may be involved in molecular recognition. J. Mol. Biol. 282:149–165. 10.1006/jmbi.1998.1979 [DOI] [PubMed] [Google Scholar]
- 157.Truan D, Huergo LF, Chubatsu LS, Merrick M, Li XD, Winkler FK. 2010. A new P(II) protein structure identifies the 2-oxoglutarate binding site. J. Mol. Biol. 400:531–539. 10.1016/j.jmb.2010.05.036 [DOI] [PubMed] [Google Scholar]
- 158.Sant'Anna FH, Trentini DB, de Souto WS, Cecagno R, da Silva SC, Schrank IS. 2009. The PII superfamily revised: a novel group and evolutionary insights. J. Mol. Evol. 68:322–336. 10.1007/s00239-009-9209-6 [DOI] [PubMed] [Google Scholar]
- 159.Xu Y, Carr PD, Huber T, Vasudevan SG, Ollis DL. 2001. The structure of the PII-ATP complex. Eur. J. Biochem. 268:2028–2037. 10.1046/j.1432-1327.2001.02074.x [DOI] [PubMed] [Google Scholar]
- 160.Huergo LF, Chandra G, Merrick M. 2013. PII signal transduction proteins: nitrogen regulation and beyond. FEMS Microbiol. Rev. 37:251–283 [DOI] [PubMed] [Google Scholar]
- 161.van Heeswijk WC, Molenaar D, Hoving S, Westerhoff HV. 2009. The pivotal regulator GlnB of Escherichia coli is engaged in subtle and context-dependent control. FEBS J. 276:3324–3340. 10.1111/j.1742-4658.2009.07058.x [DOI] [PubMed] [Google Scholar]
- 162.Atkinson MR, Ninfa AJ. 1998. Role of the GlnK signal transduction protein in the regulation of nitrogen assimilation in Escherichia coli. Mol. Microbiol. 29:431–447. 10.1046/j.1365-2958.1998.00932.x [DOI] [PubMed] [Google Scholar]
- 163.Javelle A, Severi E, Thornton J, Merrick M. 2004. Ammonium sensing in Escherichia coli. Role of the ammonium transporter AmtB and AmtB-GlnK complex formation. J. Biol. Chem. 279:8530–8538 [DOI] [PubMed] [Google Scholar]
- 164.Ninfa AJ. 1991. Protein phosphorylation and the regulation of cellular processes by the homologous two-component regulatory systems of bacteria. Genet. Eng. 13:39–72 [DOI] [PubMed] [Google Scholar]
- 165.Parkinson JS, Kofoid EC. 1992. Communication modules in bacterial signaling proteins. Annu. Rev. Genet. 26:71–112. 10.1146/annurev.ge.26.120192.000443 [DOI] [PubMed] [Google Scholar]
- 166.Stock JB, Ninfa AJ, Stock AM. 1989. Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiol. Rev. 53:450–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen YM, Backman K, Magasanik B. 1982. Characterization of a gene, glnL, the product of which is involved in the regulation of nitrogen utilization in Escherichia coli. J. Bacteriol. 150:214–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Keener J, Kustu S. 1988. Protein kinase and phosphoprotein phosphatase activities of nitrogen regulatory proteins NTRB and NTRC of enteric bacteria: roles of the conserved amino-terminal domain of NTRC. Proc. Natl. Acad. Sci. U. S. A. 85:4976–4980. 10.1073/pnas.85.14.4976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liu J, Magasanik B. 1995. Activation of the dephosphorylation of nitrogen regulator I-phosphate of Escherichia coli. J. Bacteriol. 177:926–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ninfa AJ, Ueno-Nishio S, Hunt TP, Robustell B, Magasanik B. 1986. Purification of nitrogen regulator II, the product of the glnL (ntrB) gene of Escherichia coli. J. Bacteriol. 168:1002–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ninfa AJ, Atkinson MR, Kamberov ES, Feng J, Ninfa EG. 1995. Control of nitrogen assimilation by the NRI-NRII two-component system of enteric bacteria, p 67–88 In Hoch JA, Silhavy TJ. (ed), Two-component signal transduction. ASM Press, Washington, DC [Google Scholar]
- 172.Kramer G, Weiss V. 1999. Functional dissection of the transmitter module of the histidine kinase NtrB in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 96:604–609. 10.1073/pnas.96.2.604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ninfa AJ, Bennett RL. 1991. Identification of the site of autophosphorylation of the bacterial protein kinase/phosphatase NRII. J. Biol. Chem. 266:6888–6893 [PubMed] [Google Scholar]
- 174.Atkinson MR, Ninfa AJ. 1993. Mutational analysis of the bacterial signal-transducing protein kinase/phosphatase nitrogen regulator II (NRII or NtrB). J. Bacteriol. 175:7016–7023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jiang P, Atkinson MR, Srisawat C, Sun Q, Ninfa AJ. 2000. Functional dissection of the dimerization and enzymatic activities of Escherichia coli nitrogen regulator II and their regulation by the PII protein. Biochemistry 39:13433–13449. 10.1021/bi000794u [DOI] [PubMed] [Google Scholar]
- 176.Song Y, Peisach D, Pioszak AA, Xu Z, Ninfa AJ. 2004. Crystal structure of the C-terminal domain of the two-component system transmitter protein nitrogen regulator II (NRII; NtrB), regulator of nitrogen assimilation in Escherichia coli. Biochemistry 43:6670–6678. 10.1021/bi049474r [DOI] [PubMed] [Google Scholar]
- 177.Ninfa EG, Atkinson MR, Kamberov ES, Ninfa AJ. 1993. Mechanism of autophosphorylation of Escherichia coli nitrogen regulator II (NRII or NtrB): trans-phosphorylation between subunits. J. Bacteriol. 175:7024–7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jiang P, Ninfa AJ. 1999. Regulation of autophosphorylation of Escherichia coli nitrogen regulator II by the PII signal transduction protein. J. Bacteriol. 181:1906–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pioszak AA, Jiang P, Ninfa AJ. 2000. The Escherichia coli PII signal transduction protein regulates the activities of the two-component system transmitter protein NRII by direct interaction with the kinase domain of the transmitter module. Biochemistry 39:13450–13461. 10.1021/bi000795m [DOI] [PubMed] [Google Scholar]
- 180.Reitzer LJ, Magasanik B. 1983. Isolation of the nitrogen assimilation regulator NRI, the product of the glnG gene of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 80:5554–5558. 10.1073/pnas.80.18.5554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Porter S, North A, Kustu S. 1995. Mechanism of transcriptional activation by NtrC, p 147–158 In Hoch JA, Silhavy TJ. (ed), Two-component signal transduction. ASM Press, Washington, DC [Google Scholar]
- 182.Sanders DA, Gillece-Castro BL, Burlingame AL, Koshland DEJ. 1992. Phosphorylation site of NtrC, a protein phosphatase whose covalent intermediate activates transcription. J. Bacteriol. 174:5117–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Drummond MH, Contreras A, Mitchenall LA. 1990. The function of isolated domains and chimaeric proteins constructed from the transcriptional activators NifA and NtrC of Klebsiella pneumoniae. Mol. Microbiol. 4:29–37. 10.1111/j.1365-2958.1990.tb02012.x [DOI] [PubMed] [Google Scholar]
- 184.Flashner Y, Weiss DS, Keener J, Kustu S. 1995. Constitutive forms of the enhancer-binding protein NtrC: evidence that essential oligomerization determinants lie in the central activation domain. J. Mol. Biol. 249:700–713. 10.1006/jmbi.1995.0330 [DOI] [PubMed] [Google Scholar]
- 185.Klose KE, North AK, Stedman KM, Kustu S. 1994. The major dimerization determinants of the nitrogen regulatory protein NTRC from enteric bacteria lie in its carboxy-terminal domain. J. Mol. Biol. 241:233–245. 10.1006/jmbi.1994.1492 [DOI] [PubMed] [Google Scholar]
- 186.Pelton JG, Kustu S, Wemmer DE. 1999. Solution structure of the DNA-binding domain of NtrC with three alanine substitutions. J. Mol. Biol. 292:1095–1110. 10.1006/jmbi.1999.3140 [DOI] [PubMed] [Google Scholar]
- 187.Yang XF, Ji Y, Schneider BL, Reitzer L. 2004. Phosphorylation-independent dimer-dimer interactions by the enhancer-binding activator NtrC of Escherichia coli: a third function for the C-terminal domain. J. Biol. Chem. 279:36708–36714. 10.1074/jbc.M405205200 [DOI] [PubMed] [Google Scholar]
- 188.Mettke I, Fiedler U, Weiss V. 1995. Mechanism of activation of a response regulator: interaction of NtrC-P dimers induces ATPase activity. J. Bacteriol. 177:5056–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Weiss DS, Batut J, Klose KE, Keener J, Kustu S. 1991. The phosphorylated form of the enhancer-binding protein NTRC has an ATPase activity that is essential for activation of transcription. Cell 67:155–167. 10.1016/0092-8674(91)90579-N [DOI] [PubMed] [Google Scholar]
- 190.Hunt TP, Magasanik B. 1985. Transcription of glnA by purified Escherichia coli components: core RNA polymerase and the products of glnF, glnG, and glnL. Proc. Natl. Acad. Sci. U. S. A. 82:8453–8457. 10.1073/pnas.82.24.8453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Weiss V, Magasanik B. 1988. Phosphorylation of nitrogen regulator I (NRI) of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 85:8919–8923. 10.1073/pnas.85.23.8919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Klose KE, Weiss DS, Kustu S. 1993. Glutamate at the site of phosphorylation of nitrogen-regulatory protein NTRC mimics aspartyl-phosphate and activates the protein. J. Mol. Biol. 232:67–78. 10.1006/jmbi.1993.1370 [DOI] [PubMed] [Google Scholar]
- 193.Moore JB, Shiau SP, Reitzer LJ. 1993. Alterations of highly conserved residues in the regulatory domain of nitrogen regulator I (NtrC) of Escherichia coli. J. Bacteriol. 175:2692–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nohaile M, Kern D, Wemmer D, Stedman K, Kustu S. 1997. Structural and functional analyses of activating amino acid substitutions in the receiver domain of NtrC: evidence for an activating surface. J. Mol. Biol. 273:299–316. 10.1006/jmbi.1997.1296 [DOI] [PubMed] [Google Scholar]
- 195.Yan D, Cho HS, Hastings CA, Igo MM, Lee SY, Pelton JG, Stewart V, Wemmer DE, Kustu S. 1999. Beryllofluoride mimics phosphorylation of NtrC and other bacterial response regulators. Proc. Natl. Acad. Sci. U. S. A. 96:14789–14794. 10.1073/pnas.96.26.14789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Volkman BF, Nohaile MJ, Amy NK, Kustu S, Wemmer DE. 1995. Three-dimensional solution structure of the N-terminal receiver domain of NTRC. Biochemistry 34:1413–1424. 10.1021/bi00004a036 [DOI] [PubMed] [Google Scholar]
- 197.Hirschman J, Wong PK, Sei K, Keener J, Kustu S. 1985. Products of nitrogen regulatory genes ntrA and ntrC of enteric bacteria activate glnA transcription in vitro: evidence that the ntrA product is a sigma factor. Proc. Natl. Acad. Sci. U. S. A. 82:7525–7529. 10.1073/pnas.82.22.7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kern D, Volkman BF, Luginbuhl P, Nohaile MJ, Kustu S, Wemmer DE. 1999. Structure of a transiently phosphorylated switch in bacterial signal transduction. Nature 402:894–898 [DOI] [PubMed] [Google Scholar]
- 199.Hastings CA, Lee SY, Cho HS, Yan D, Kustu S, Wemmer DE. 2003. High-resolution solution structure of the beryllofluoride-activated NtrC receiver domain. Biochemistry 42:9081–9090. 10.1021/bi0273866 [DOI] [PubMed] [Google Scholar]
- 200.Harrod AC, Yang X, Junker M, Reitzer L. 2004. Evidence for a second interaction between the regulatory amino-terminal and central output domains of the response regulator NtrC (nitrogen regulator I) in Escherichia coli. J. Biol. Chem. 279:2350–2359 [DOI] [PubMed] [Google Scholar]
- 201.North AK, Kustu S. 1997. Mutant forms of the enhancer-binding protein NtrC can activate transcription from solution. J. Mol. Biol. 267:17–36. 10.1006/jmbi.1996.0838 [DOI] [PubMed] [Google Scholar]
- 202.Muse WB, Bender RA. 1998. The nac (nitrogen assimilation control) gene from Escherichia coli. J. Bacteriol. 180:1166–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bender RA. 2010. A NAC for regulating metabolism: the nitrogen assimilation control protein (NAC) from Klebsiella pneumoniae. J. Bacteriol. 192:4801–4811. 10.1128/JB.00266-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bender RA. 1991. The role of the NAC protein in the nitrogen regulation of Klebsiella aerogenes. Mol. Microbiol. 5:2575–2580. 10.1111/j.1365-2958.1991.tb01965.x [DOI] [PubMed] [Google Scholar]
- 205.Schwacha A, Bender RA. 1993. The nac (nitrogen assimilation control) gene from Klebsiella aerogenes. J. Bacteriol. 175:2107–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Goss TJ, Bender RA. 1995. The nitrogen assimilation control protein, NAC, is a DNA binding transcription activator in Klebsiella aerogenes. J. Bacteriol. 177:3546–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rosario CJ, Bender RA. 2005. Importance of tetramer formation by the nitrogen assimilation control protein for strong repression of glutamate dehydrogenase formation in Klebsiella pneumoniae. J. Bacteriol. 187:8291–8299. 10.1128/JB.187.24.8291-8299.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Muse WB, Bender RA. 1999. The amino-terminal 100 residues of the nitrogen assimilation control protein (NAC) encode all known properties of NAC from Klebsiella aerogenes and Escherichia coli. J. Bacteriol. 181:934–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Maddocks SE, Oyston PC. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154:3609–3623. 10.1099/mic.0.2008/022772-0 [DOI] [PubMed] [Google Scholar]
- 210.Schell MA. 1993. Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47:597–626. 10.1146/annurev.mi.47.100193.003121 [DOI] [PubMed] [Google Scholar]
- 211.Frisch RL, Bender RA. 2010. Expanded role for the nitrogen assimilation control protein in the response of Klebsiella pneumoniae to nitrogen stress. J. Bacteriol. 192:4812–4820. 10.1128/JB.00931-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schwacha A, Bender RA. 1993. The product of the Klebsiella aerogenes nac (nitrogen assimilation control) gene is sufficient for activation of the hut operons and repression of the gdh operon. J. Bacteriol. 175:2116–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Feng J, Goss TJ, Bender RA, Ninfa AJ. 1995. Repression of the Klebsiella aerogenes nac promoter. J. Bacteriol. 177:5535–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rosario CJ, Frisch RL, Bender RA. 2010. The LysR-type nitrogen assimilation control protein forms complexes with both long and short DNA binding sites in the absence of coeffectors. J. Bacteriol. 192:4827–4833. 10.1128/JB.00968-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Gama-Castro S, Salgado H, Peralta-Gil M, Santos-Zavaleta A, Muniz-Rascado L, Solano-Lira H, Jimenez-Jacinto V, Weiss V, Garcia-Sotelo JS, Lopez-Fuentes A, Porron-Sotelo L, Alquicira-Hernandez S, Medina-Rivera A, Martinez-Flores I, Alquicira-Hernandez K, Martinez-Adame R, Bonavides-Martinez C, Miranda-Rios J, Huerta AM, Mendoza-Vargas A, Collado-Torres L, Taboada B, Vega-Alvarado L, Olvera M, Olvera L, Grande R, Morett E, Collado-Vides J. 2011. RegulonDB version 7.0: transcriptional regulation of Escherichia coli K-12 integrated within genetic sensory response units (Gensor units). Nucleic Acids Res. 39:D98–D105. 10.1093/nar/gkq1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Shimada T, Fujita N, Yamamoto K, Ishihama A. 2011. Novel roles of cAMP receptor protein (CRP) in regulation of transport and metabolism of carbon sources. PLoS One 6:e20081. 10.1371/journal.pone.0020081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kolb A, Busby S, Buc H, Garges S, Adhya S. 1993. Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 62:749–795. 10.1146/annurev.bi.62.070193.003533 [DOI] [PubMed] [Google Scholar]
- 218.Lawson CL, Swigon D, Murakami KS, Darst SA, Berman HM, Ebright RH. 2004. Catabolite activator protein:DNA binding and transcription activation. Curr. Opin. Struct. Biol. 14:10–20. 10.1016/j.sbi.2004.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Aiba H, Fujimoto S, Ozaki N. 1982. Molecular cloning and nucleotide sequencing of the gene for E. coli cAMP receptor protein. Nucleic Acids Res. 10:1345–1361. 10.1093/nar/10.4.1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Cossart P, Gicquel-Sanzey B. 1982. Cloning and sequence of the crp gene of Escherichia coli K 12. Nucleic Acids Res. 10:1363–1378. 10.1093/nar/10.4.1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Riggs AD, Reiness G, Zubay G. 1971. Purification and DNA-binding properties of the catabolite gene activator protein. Proc. Natl. Acad. Sci. U. S. A. 68:1222–1225. 10.1073/pnas.68.6.1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zubay G, Schwartz D, Beckwith J. 1970. Mechanism of activation of catabolite-sensitive genes: a positive control system. Proc. Natl. Acad. Sci. U. S. A. 66:104–110. 10.1073/pnas.66.1.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.McKay DB, Steitz TA. 1981. Structure of catabolite gene activator protein at 2.9 Å resolution suggests binding to left-handed B-DNA. Nature 290:744–749. 10.1038/290744a0 [DOI] [PubMed] [Google Scholar]
- 224.Weber IT, Steitz TA. 1987. Structure of a complex of catabolite gene activator protein and cyclic AMP refined at 2.5 Å resolution. J. Mol. Biol. 198:311–326. 10.1016/0022-2836(87)90315-9 [DOI] [PubMed] [Google Scholar]
- 225.Harman JG. 2001. Allosteric regulation of the cAMP receptor protein. Biochim. Biophys. Acta 1547:1–17. 10.1016/S0167-4838(01)00187-X [DOI] [PubMed] [Google Scholar]
- 226.Eilen E, Pampeno C, Krakow JS. 1978. Production and properties of the alpha core derived from the cyclic adenosine monophosphate receptor protein of Escherichia coli. Biochemistry 17:2469–2473. 10.1021/bi00606a001 [DOI] [PubMed] [Google Scholar]
- 227.Krakow JS, Pastan I. 1973. Cyclic adenosine monophosphate receptor: loss of cAMP-dependent DNA binding activity after proteolysis in the presence of cyclic adenosine monophosphate. Proc. Natl. Acad. Sci. U. S. A. 70:2529–2533. 10.1073/pnas.70.9.2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Weber IT, McKay DB, Steitz TA. 1982. Two helix DNA binding motif of CAP found in lac repressor and gal repressor. Nucleic Acids Res. 10:5085–5102. 10.1093/nar/10.16.5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.McKay DB, Weber IT, Steitz TA. 1982. Structure of catabolite gene activator protein at 2.9 Å resolution. Incorporation of amino acid sequence and interactions with cyclic AMP. J. Biol. Chem. 257:9518–9524 [PubMed] [Google Scholar]
- 230.Barber AM, Zhurkin VB, Adhya S. 1993. CRP-binding sites: evidence for two structural classes with 6-bp and 8-bp spacers. Gene 130:1–8. 10.1016/0378-1119(93)90339-5 [DOI] [PubMed] [Google Scholar]
- 231.Majors J. 1975. Specific binding of CAP factor to lac promoter DNA. Nature 256:672–674. 10.1038/256672a0 [DOI] [PubMed] [Google Scholar]
- 232.Eron L, Arditti R, Zubay G, Connaway S, Beckwith JR. 1971. An adenosine 3′:5′ cyclic monophosphate-binding protein that acts on the transcription process. Proc. Natl. Acad. Sci. U. S. A. 68:215–218. 10.1073/pnas.68.1.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Takahashi M, Blazy B, Baudras A, Hillen W. 1989. Ligand-modulated binding of a gene regulatory protein to DNA. Quantitative analysis of cyclic-AMP induced binding of CRP from Escherichia coli to non-specific and specific DNA targets. J. Mol. Biol. 207:783–796 [DOI] [PubMed] [Google Scholar]
- 234.Gartenberg MR, Crothers DM. 1988. DNA sequence determinants of CAP-induced bending and protein binding affinity. Nature 333:824–829. 10.1038/333824a0 [DOI] [PubMed] [Google Scholar]
- 235.Liu-Johnson HN, Gartenberg MR, Crothers DM. 1986. The DNA binding domain and bending angle of E. coli CAP protein. Cell 47:995–1005. 10.1016/0092-8674(86)90814-7 [DOI] [PubMed] [Google Scholar]
- 236.Parkinson G, Wilson C, Gunasekera A, Ebright YW, Ebright RE, Berman HM. 1996. Structure of the CAP-DNA complex at 2.5 Å resolution: a complete picture of the protein-DNA interface. J. Mol. Biol. 260:395–408. 10.1006/jmbi.1996.0409 [DOI] [PubMed] [Google Scholar]
- 237.Schultz SC, Shields GC, Steitz TA. 1991. Crystal structure of a CAP-DNA complex: the DNA is bent by 90 degrees. Science 253:1001–1007. 10.1126/science.1653449 [DOI] [PubMed] [Google Scholar]
- 238.Gartenberg MR, Crothers DM. 1991. Synthetic DNA bending sequences increase the rate of in vitro transcription initiation at the Escherichia coli lac promoter. J. Mol. Biol. 219:217–230. 10.1016/0022-2836(91)90563-L [DOI] [PubMed] [Google Scholar]
- 239.Gaston K, Bell A, Kolb A, Buc H, Busby S. 1990. Stringent spacing requirements for transcription activation by CRP. Cell 62:733–743. 10.1016/0092-8674(90)90118-X [DOI] [PubMed] [Google Scholar]
- 240.Fandl JP, Thorner LK, Artz SW. 1990. Mutations that affect transcription and cyclic AMP-CRP regulation of the adenylate cyclase gene (cya) of Salmonella typhimurium. Genetics 125:719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Brinkman AB, Ettema TJ, de Vos WM, van der Oost J. 2003. The Lrp family of transcriptional regulators. Mol. Microbiol. 48:287–294. 10.1046/j.1365-2958.2003.03442.x [DOI] [PubMed] [Google Scholar]
- 242.Yokoyama K, Ishijima SA, Clowney L, Koike H, Aramaki H, Tanaka C, Makino K, Suzuki M. 2006. Feast/famine regulatory proteins (FFRPs): Escherichia coli Lrp, AsnC and related archaeal transcription factors. FEMS Microbiol. Rev. 30:89–108. 10.1111/j.1574-6976.2005.00005.x [DOI] [PubMed] [Google Scholar]
- 243.Calvo JM, Matthews RG. 1994. The leucine-responsive regulatory protein, a global regulator of metabolism in Escherichia coli. Microbiol. Rev. 58:466–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Newman EB, D'Ari R, Lin RT. 1992. The leucine-Lrp regulon in E. coli: a global response in search of a raison d'être. Cell 68:617–619. 10.1016/0092-8674(92)90135-Y [DOI] [PubMed] [Google Scholar]
- 245.de los Rios S, Perona JJ. 2007. Structure of the Escherichia coli leucine-responsive regulatory protein Lrp reveals a novel octameric assembly. J. Mol. Biol. 366:1589–1602. 10.1016/j.jmb.2006.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Janes BK, Bender RA. 1999. Two roles for the leucine-responsive regulatory protein in expression of the alanine catabolic operon (dadAB) in Klebsiella aerogenes. J. Bacteriol. 181:1054–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hart BR, Mishra PK, Lintner RE, Hinerman JM, Herr AB, Blumenthal RM. 2011. Recognition of DNA by the helix-turn-helix global regulatory protein Lrp is modulated by the amino terminus. J. Bacteriol. 193:3794–3803. 10.1128/JB.00191-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Pflüger-Grau K, Görke B. 2010. Regulatory roles of the bacterial nitrogen-related phosphotransferase system. Trends Microbiol. 18:205–214. 10.1016/j.tim.2010.02.003 [DOI] [PubMed] [Google Scholar]
- 249.Deutscher J, Francke C, Postma PW. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 70:939–1031. 10.1128/MMBR.00024-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Postma PW, Lengeler JW, Jacobson GR. 1993. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57:543–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Ninfa AJ. 2007. Regulation of carbon and nitrogen metabolism: adding regulation of ion channels and another second messenger to the mix. Proc. Natl. Acad. Sci. U. S. A. 104:4243–4244. 10.1073/pnas.0700325104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Ninfa AJ. 2011. Unnecessary signaling: poorly named? J. Bacteriol. 193:4571–4573. 10.1128/JB.05682-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Barrios H, Valderrama B, Morett E. 1999. Compilation and analysis of sigma(54)-dependent promoter sequences. Nucleic Acids Res. 27:4305–4313. 10.1093/nar/27.22.4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kustu S, Santero E, Keener J, Popham D, Weiss D. 1989. Expression of sigma54 (ntrA)-dependent genes is probably united by a common mechanism. Microbiol. Rev. 53:367–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Merrick MJ. 1993. In a class of its own—the RNA polymerase sigma factor σ54 (σN). Mol. Microbiol. 10:903–909. 10.1111/j.1365-2958.1993.tb00961.x [DOI] [PubMed] [Google Scholar]
- 256.Francke C, Kormelink TG, Hagemeijer Y, Overmars L, Sluijter V, Moezelaar R, Siezen RJ. 2011. Comparative analyses imply that the enigmatic sigma factor 54 is a central controller of the bacterial exterior. BMC Genomics 12:385. 10.1186/1471-2164-12-385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Powell BS, Court DL, Inada T, Nakamura Y, Michotey V, Cui X, Reizer A, Saier MHJ, Reizer J. 1995. Novel proteins of the phosphotransferase system encoded within the rpoN operon of Escherichia coli. Enzyme IIANtr affects growth on organic nitrogen and the conditional lethality of an erats mutant. J. Biol. Chem. 270:4822–4839 [DOI] [PubMed] [Google Scholar]
- 258.Caldon CE, March PE. 2003. Function of the universally conserved bacterial GTPases. Curr. Opin. Microbiol. 6:135–139. 10.1016/S1369-5274(03)00037-7 [DOI] [PubMed] [Google Scholar]
- 259.Reizer J, Reizer A, Merrick MJ, Plunkett GI, Rose DJ, Saier MHJ. 1996. Novel phosphotransferase-encoding genes revealed by analysis of the Escherichia coli genome: a chimeric gene encoding an enzyme I homologue that possesses a putative sensory transduction domain. Gene 181:103–108. 10.1016/S0378-1119(96)00481-7 [DOI] [PubMed] [Google Scholar]
- 260.Jones DHA, Franklin FC, Thomas CM. 1994. Molecular analysis of the operon which encodes the RNA polymerase sigma factors 54 of Escherichia coli. Microbiology 140:1035–1043. 10.1099/13500872-140-5-1035 [DOI] [PubMed] [Google Scholar]
- 261.Castano I, Bastarrachea F. 1984. glnF-lacZ fusions in Escherichia coli: studies on glnF expression and its chromosomal orientation. Mol. Gen. Genet. 195:228–233. 10.1007/BF00332751 [DOI] [PubMed] [Google Scholar]
- 262.Merrick MJ, Coppard JR. 1989. Mutations in genes downstream of the rpoN gene (encoding σ54) of Klebsiella pneumoniae affect expression from σ54-dependent promoters. Mol. Microbiol. 3:1765–1775. 10.1111/j.1365-2958.1989.tb00162.x [DOI] [PubMed] [Google Scholar]
- 263.Rabus R, Reizer J, Paulsen I, Saier MHJ. 1999. Enzyme I (Ntr) from Escherichia coli. A novel enzyme of the phosphoenolpyruvate-dependent phosphotransferase system exhibiting strict specificity for its phosphoryl acceptor, NPr. J. Biol. Chem. 274:26185–26191 [DOI] [PubMed] [Google Scholar]
- 264.Zimmer B, Hillmann A, Görke B. 2008. Requirements for the phosphorylation of the Escherichia coli EIIANtr protein in vivo. FEMS Microbiol. Lett. 286:96–102. 10.1111/j.1574-6968.2008.01262.x [DOI] [PubMed] [Google Scholar]
- 265.Merrick JM, Taylor M, Saier MHJ, Reizer A. 1995. The role of genes downstream of the σN structural gene rpoN in Klebsiella pneumoniae, p 189–194 In Tikhonovich IA, Provorov NA, Romanov VI, Newton WE. (ed), Nitrogen fixation: fundamentals and applications. Kluwer Academic Publishers, Dordrecht, the Netherlands [Google Scholar]
- 266.Wirén NV, Merrick M. 2004. Regulation and function of ammonium carriers in bacteria, fungi, and plants, p 95–120 In Boles E, Kramer R. (ed), Topics in current genetics. Molecular mechanisms controlling transmembrane transport. Springer, Berlin, Germany [Google Scholar]
- 267.Boeckstaens M, Andre B, Marini AM. 2008. Distinct transport mechanisms in yeast ammonium transport/sensor proteins of the Mep/Amt/Rh family and impact on filamentation. J. Biol. Chem. 283:21362–21370. 10.1074/jbc.M801467200 [DOI] [PubMed] [Google Scholar]
- 268.Thornton J, Blakey D, Scanlon E, Merrick M. 2006. The ammonia channel protein AmtB from Escherichia coli is a polytopic membrane protein with a cleavable signal peptide. FEMS Microbiol. Lett. 258:114–120. 10.1111/j.1574-6968.2006.00202.x [DOI] [PubMed] [Google Scholar]
- 269.Severi E, Javelle A, Merrick M. 2007. The conserved carboxy-terminal region of the ammonia channel AmtB plays a critical role in channel function. Mol. Membr. Biol. 24:161–171. 10.1080/09687860601129420 [DOI] [PubMed] [Google Scholar]
- 270.Thomas GH, Mullins JG, Merrick M. 2000. Membrane topology of the Mep/Amt family of ammonium transporters. Mol. Microbiol. 37:331–344. 10.1046/j.1365-2958.2000.01994.x [DOI] [PubMed] [Google Scholar]
- 271.Khademi S, O'Connell JI, Remis J, Robles-Colmenares Y, Miercke LJ, Stroud RM. 2004. Mechanism of ammonia transport by Amt/MEP/Rh: structure of AmtB at 1.35 Å. Science 305:1587–1594. 10.1126/science.1101952 [DOI] [PubMed] [Google Scholar]
- 272.Sobczak I, Lolkema JS. 2005. Structural and mechanistic diversity of secondary transporters. Curr. Opin. Microbiol. 8:161–167. 10.1016/j.mib.2005.02.005 [DOI] [PubMed] [Google Scholar]
- 273.Blakey D, Leech A, Thomas GH, Coutts G, Findlay K, Merrick M. 2002. Purification of the Escherichia coli ammonium transporter AmtB reveals a trimeric stoichiometry. Biochem. J. 364:527–535. 10.1042/BJ20011761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Conroy MJ, Jamieson SJ, Blakey D, Kaufmann T, Engel A, Fotiadis D, Merrick M, Bullough PA. 2004. Electron and atomic force microscopy of the trimeric ammonium transporter AmtB. EMBO Rep. 5:1153–1158. 10.1038/sj.embor.7400296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Zheng L, Kostrewa D, Bernèche S, Winkler FK, Li X-D. 2004. The mechanism of ammonia transport based on the crystal structure of AmtB of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 101:17090–17095. 10.1073/pnas.0406475101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Lamoureux G, Klein ML, Bernèche S. 2007. A stable water chain in the hydrophobic pore of the AmtB ammonium transporter. Biophys. J. 92:L82–L84. 10.1529/biophysj.106.102756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Lamoureux G, Javelle A, Baday S, Wang S, Berneche S. 2010. Transport mechanisms in the ammonium transporter family. Transfus. Clin. Biol. 17:168–175. 10.1016/j.tracli.2010.06.004 [DOI] [PubMed] [Google Scholar]
- 278.Bostick DL, Brooks CL. 2007. Deprotonation by dehydration: the origin of ammonium sensing in the AmtB channel. PLoS Comput. Biol. 3:e22. 10.1371/journal.pcbi.0030022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Bostick DL, Brooks CL. 2007. On the equivalence point for ammonium (de)protonation during its transport through the AmtB channel. Biophys. J. 92:L103–L105. 10.1529/biophysj.107.109165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Ishikita H. 2007. Modulation of the protein environment in the hydrophilic pore of the ammonia transporter protein AmtB upon GlnK protein binding. FEBS Lett. 581:4293–4297. 10.1016/j.febslet.2007.07.085 [DOI] [PubMed] [Google Scholar]
- 281.Soupene E, He L, Yan D, Kustu S. 1998. Ammonia acquisition in enteric bacteria: physiological role of the ammonium/methylammonium transport B (AmtB) protein. Proc. Natl. Acad. Sci. U. S. A. 95:7030–7034. 10.1073/pnas.95.12.7030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Soupene E, Lee H, Kustu S. 2002. Ammonium/methylammonium transport (Amt) proteins facilitate diffusion of NH3 bidirectionally. Proc. Natl. Acad. Sci. U. S. A. 99:3926–3931. 10.1073/pnas.062043799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Winkler FK. 2006. Amt/MEP/Rh proteins conduct ammonia. Pflugers Arch. 451:701–707. 10.1007/s00424-005-1511-6 [DOI] [PubMed] [Google Scholar]
- 284.Javelle A, Lupo D, Ripoche P, Fulford T, Merrick M, Winkler FK. 2008. Substrate binding, deprotonation, and selectivity at the periplasmic entrance of the Escherichia coli ammonia channel AmtB. Proc. Natl. Acad. Sci. U. S. A. 105:5040–5045. 10.1073/pnas.0711742105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Akgun U, Khademi S. 2011. Periplasmic vestibule plays an important role for solute recruitment, selectivity, and gating in the Rh/Amt/MEP superfamily. Proc. Natl. Acad. Sci. U. S. A. 108:3970–3975. 10.1073/pnas.1007240108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Liu Y, Hu X. 2006. Molecular determinants for binding of ammonium ion in the ammonia transporter AmtB—a quantum chemical analysis. J. Phys. Chem. A 110:1375–1381. 10.1021/jp054261c [DOI] [PubMed] [Google Scholar]
- 287.Nygaard TP, Alfonso-Prieto M, Peters GH, Jensen MO, Rovira C. 2010. Substrate recognition in the Escherichia coli ammonia channel AmtB: a QM/MM investigation. J. Phys. Chem. B 114:11859–11865. 10.1021/jp102338h [DOI] [PubMed] [Google Scholar]
- 288.Fong RN, Kim KS, Yoshihara C, Inwood WB, Kustu S. 2007. The W148L substitution in the Escherichia coli ammonium channel AmtB increases flux and indicates that the substrate is an ion. Proc. Natl. Acad. Sci. U. S. A. 104:18706–18711. 10.1073/pnas.0709267104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Hall JA, Kustu S. 2011. The pivotal twin histidines and aromatic triad of the Escherichia coli ammonium channel AmtB can be replaced. Proc. Natl. Acad. Sci. U. S. A. 108:13270–13274. 10.1073/pnas.1108451108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Javelle A, Lupo D, Zheng L, Li XD, Winkler FK, Merrick M. 2006. An unusual twin-His arrangement in the pore of ammonia channels is essential for substrate conductance. J. Biol. Chem. 281:39492–39498. 10.1074/jbc.M608325200 [DOI] [PubMed] [Google Scholar]
- 291.Wang J, Yang H, Zuo Z, Yan X, Wang Y, Luo X, Jiang H, Chen K, Zhu W. 2010. Molecular dynamics simulations on the mechanism of transporting methylamine and ammonia by ammonium transporter AmtB. J. Phys. Chem. B 114:15172–15179. 10.1021/jp104508k [DOI] [PubMed] [Google Scholar]
- 292.Ishikita H, Knapp E-W. 2007. Protonation states of ammonia/ammonium in the hydrophobic pore of ammonia transporter protein AmtB. J. Am. Chem. Soc. 129:1210–1215. 10.1021/ja066208n [DOI] [PubMed] [Google Scholar]
- 293.Lin Y, Cao Z, Mo Y. 2009. Functional role of Asp160 and the deprotonation mechanism of ammonium in the Escherichia coli ammonia channel protein AmtB. J. Phys. Chem. B 113:4922–4929. 10.1021/jp810651m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Lin Y, Cao Z, Mo Y. 2006. Molecular dynamics simulations on the Escherichia coli ammonia channel protein AmtB: mechanism of ammonia/ammonium transport. J. Am. Chem. Soc. 128:10876–10884. 10.1021/ja0631549 [DOI] [PubMed] [Google Scholar]
- 295.Luzhkov VB, Almlöf M, Nervall M, Aqvist J. 2006. Computational study of the binding affinity and selectivity of the bacterial ammonium transporter AmtB. Biochemistry 45:10807–10814. 10.1021/bi0610799 [DOI] [PubMed] [Google Scholar]
- 296.Nygaard TP, Rovira C, Peters GH, Jensen MØ. 2006. Ammonium recruitment and ammonia transport by E. coli ammonia channel AmtB. Biophys. J. 91:4401–4412. 10.1529/biophysj.106.089714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Yang H, Xu Y, Zhu W, Chen K, Jiang H. 2007. Detailed mechanism for AmtB conducting NH4+/NH3: molecular dynamics simulations. Biophys. J. 92:877–885. 10.1529/biophysj.106.090191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Andrade SLA, Einsle O. 2007. The Amt/Mep/Rh family of ammonium transport proteins. Mol. Membr. Biol. 24:357–365. 10.1080/09687680701388423 [DOI] [PubMed] [Google Scholar]
- 299.Javelle A, Lupo D, Li XD, Merrick M, Chami M, Ripoche P, Winkler FK. 2007. Structural and mechanistic aspects of Amt/Rh proteins. J. Struct. Biol. 158:472–481. 10.1016/j.jsb.2007.01.004 [DOI] [PubMed] [Google Scholar]
- 300.Wang S, Orabi EA, Baday S, Bernèche S, Lamoureux G. 2012. Ammonium transporters achieve charge transfer by fragmenting their substrate. J. Am. Chem. Soc. 134:10419–10427. 10.1021/ja300129x [DOI] [PubMed] [Google Scholar]
- 301.Boogerd FC, Ma H, Bruggeman FJ, van Heeswijk WC, García-Contreras R, Molenaar D, Krab K, Westerhoff HV. 2011. AmtB-mediated NH3 transport in prokaryotes must be active and as a consequence regulation of transport by GlnK is mandatory to limit futile cycling of NH4+/NH3. FEBS Lett. 585:23–28. 10.1016/j.febslet.2010.11.055 [DOI] [PubMed] [Google Scholar]
- 302.Inwood WB, Hall JA, Kim KS, Fong R, Kustu S. 2009. Genetic evidence for an essential oscillation of transmembrane-spanning segment 5 in the Escherichia coli ammonium channel AmtB. Genetics 183:1341–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Coutts G, Thomas G, Blakey D, Merrick M. 2002. Membrane sequestration of the signal transduction protein GlnK by the ammonium transporter AmtB. EMBO J. 21:536–545. 10.1093/emboj/21.4.536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Jayakumar A, Epstein W, Barnes EMJ. 1985. Characterization of ammonium (methylammonium)/potassium antiport in Escherichia coli. J. Biol. Chem. 260:7528–7532 [PubMed] [Google Scholar]
- 305.Javelle A, Thomas G, Marini AM, Kramer R, Merrick M. 2005. In vivo functional characterization of the Escherichia coli ammonium channel AmtB: evidence for metabolic coupling of AmtB to glutamine synthetase. Biochem. J. 390:215–222. 10.1042/BJ20042094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Jayakumar A, Hong JS, Barnes EMJ. 1987. Feedback inhibition of ammonium (methylammonium) ion transport in Escherichia coli by glutamine and glutamine analogs. J. Bacteriol. 169:553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Stevenson R, Silver S. 1977. Methylammonium uptake by Escherichia coli: evidence for a bacterial NH4+ transport system. Biochem. Biophys. Res. Commun. 75:1133–1139. 10.1016/0006-291X(77)91501-7 [DOI] [PubMed] [Google Scholar]
- 308.Servín-González L, Bastarrachea F. 1984. Nitrogen regulation of synthesis of the high affinity methylammonium transport system of Escherichia coli. J. Gen. Microbiol. 130:3071–3077 [DOI] [PubMed] [Google Scholar]
- 309.Jayakumar A, Hwang SJ, Fabiny JM, Chinault AC, Barnes EMJ. 1989. Isolation of an ammonium or methylammonium ion transport mutant of Escherichia coli and complementation by the cloned gene. J. Bacteriol. 171:996–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Fabiny JM, Jayakumar A, Chinault AC, Barnes EMJ. 1991. Ammonium transport in Escherichia coli: localization and nucleotide sequence of the amtA gene. J. Gen. Microbiol. 137:983–989. 10.1099/00221287-137-4-983 [DOI] [PubMed] [Google Scholar]
- 311.Jayakumar A, Rudd KE, Fabiny JM, Barnes EMJ. 1991. Localization of the Escherichia coli amtA gene to 95.8 minutes. J. Bacteriol. 173:1572–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Neuwald AF, Krishnan BR, Brikun I, Kulakauskas S, Suziedelis K, Tomcsanyi T, Leyh TS, Berg DE. 1992. cysQ, a gene needed for cysteine synthesis in Escherichia coli K-12 only during aerobic growth. J. Bacteriol. 174:415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Fukuda C, Kawai S, Murata K. 2007. NADP(H) phosphatase activities of archaeal inositol monophosphatase and eubacterial 3′-phosphoadenosine 5′-phosphate phosphatase. Appl. Environ. Microbiol. 73:5447–5452. 10.1128/AEM.02703-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Masters PS, Hong JS. 1981. Genetics of the glutamine transport system in Escherichia coli. J. Bacteriol. 147:805–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Weiner JH, Heppel LA. 1971. A binding protein for glutamine and its relation to active transport in Escherichia coli. J. Biol. Chem. 246:6933–6941 [DOI] [PubMed] [Google Scholar]
- 316.Weiner JH, Furlong CE, Heppel LA. 1971. A binding protein for L-glutamine and its relation to active transport in E. coli. Arch. Biochem. Biophys. 142:715–717. 10.1016/0003-9861(71)90538-8 [DOI] [PubMed] [Google Scholar]
- 317.Willis RC, Iwata KK, Furlong CE. 1975. Regulation of glutamine transport in Escherichia coli. J. Bacteriol. 122:1032–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Kustu SG, McFarland NC, Hui SP, Esmon B, Ames GF-L. 1979. Nitrogen control of Salmonella typhimurium: co-regulation of synthesis of glutamine synthetase and amino acid transport systems. J. Bacteriol. 138:218–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Nohno T, Saito T, Hong JS. 1986. Cloning and complete nucleotide sequence of the Escherichia coli glutamine permease operon (glnHPQ). Mol. Gen. Genet. 205:260–269. 10.1007/BF00430437 [DOI] [PubMed] [Google Scholar]
- 320.Linton KJ, Higgins CF. 1998. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol. Microbiol. 28:5–13 [DOI] [PubMed] [Google Scholar]
- 321.Bermejo GA, Strub M-P, Ho C, Tjandra N. 2010. Ligand-free open-closed transitions of periplasmic binding proteins: the case of glutamine-binding protein. Biochemistry 49:1893–1902. 10.1021/bi902045p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Hsiao CD, Sun YJ, Rose J, Wang BC. 1996. The crystal structure of glutamine-binding protein from Escherichia coli. J. Mol. Biol. 262:225–242. 10.1006/jmbi.1996.0509 [DOI] [PubMed] [Google Scholar]
- 323.Davidson AL, Dassa E, Orelle C, Chen J. 2008. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 72:317–364. 10.1128/MMBR.00031-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Shen QC, Simplaceanu V, Cottam PF, Wu JL, Hong JS, Ho C. 1989. Molecular genetic, biochemical and nuclear magnetic resonance studies on the role of the tryptophan residues of glutamine-binding protein from Escherichia coli. J. Mol. Biol. 210:859–867. 10.1016/0022-2836(89)90113-7 [DOI] [PubMed] [Google Scholar]
- 325.Hunt AG, Hong J. 1981. The reconstitution of binding protein-dependent active transport of glutamine in isolated membrane vesicles from Escherichia coli. J. Biol. Chem. 256:11988–11991 [PubMed] [Google Scholar]
- 326.Masters PS, Hong J. 1981. Reconstitution of binding protein dependent active transport of glutamine in spheroplasts of Escherichia coli. Biochemistry 20:4900–4904. 10.1021/bi00520a015 [DOI] [PubMed] [Google Scholar]
- 327.Alatossava T, Jütte H, Kuhn A, Kellenberger E. 1985. Manipulation of intracellular magnesium content in polymyxin B nonapeptide-sensitized Escherichia coli by ionophore A23187. J. Bacteriol. 162:413–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Hunt AG, Hong JS. 1983. Involvement of histidine and tryptophan residues of glutamine binding protein in the interaction with membrane-bound components of the glutamine transport system of Escherichia coli. Biochemistry 22:851–854. 10.1021/bi00273a022 [DOI] [PubMed] [Google Scholar]
- 329.Gehring K, Hofnung M, Nikaido H. 1990. Stimulation of glutamine transport by osmotic stress in Escherichia coli K-12. J. Bacteriol. 172:4741–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Berger EA. 1973. Different mechanisms of energy coupling for the active transport of proline and glutamine in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 70:1514–1518. 10.1073/pnas.70.5.1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Berger EA, Heppel LA. 1974. Different mechanisms of energy coupling for the shock-sensitive and shock-resistant amino acid permeases of Escherichia coli. J. Biol. Chem. 249:7747–7755 [PubMed] [Google Scholar]
- 332.Kelly DJ, Thomas GH. 2001. The tripartite ATP-independent periplasmic (TRAP) transporters of bacteria and archaea. FEMS Microbiol. Rev. 25:405–424. 10.1111/j.1574-6976.2001.tb00584.x [DOI] [PubMed] [Google Scholar]
- 333.Higgins CF. 1992. ABC transporters—from microorganisms to man. Annu. Rev. Cell Biol. 8:67–113. 10.1146/annurev.cb.08.110192.000435 [DOI] [PubMed] [Google Scholar]
- 334.Plate CA. 1979. Requirement for membrane potential in active transport of glutamine by Escherichia coli. J. Bacteriol. 137:221–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Raushel FM, Thoden JB, Holden HM. 1999. The amidotransferase family of enzymes: molecular machines for the production and delivery of ammonia. Biochemistry 38:7891–7899. 10.1021/bi990871p [DOI] [PubMed] [Google Scholar]
- 336.Hartman SC. 1968. Glutaminase of Escherichia coli. I. Purification and general catalytic properties. J. Biol. Chem. 243:853–863 [PubMed] [Google Scholar]
- 337.Prusiner S, Stadtman ER. 1971. On the regulation of glutaminase in E. coli: metabolite control. Biochem. Biophys. Res. Commun. 45:1474–1481. 10.1016/0006-291X(71)90186-0 [DOI] [PubMed] [Google Scholar]
- 338.Brown G, Singer A, Proudfoot M, Skarina T, Kim Y, Chang C, Dementieva I, Kuznetsova E, Gonzalez CF, Joachimiak A, Savchenko A, Yakunin AF. 2008. Functional and structural characterization of four glutaminases from Escherichia coli and Bacillus subtilis. Biochemistry 47:5724–5735. 10.1021/bi800097h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Hartman SC, Stochaj EM. 1973. Glutaminase A of Escherichia coli. Subunit structure and cooperative behavior. J. Biol. Chem. 248:8511–8517 [PubMed] [Google Scholar]
- 340.Prusiner S, Davis JN, Stadtman ER. 1976. Regulation of glutaminase B in Escherichia coli. I. Purification, properties, and cold lability. J. Biol. Chem. 251:3447–3456 [PubMed] [Google Scholar]
- 341.Prusiner S. 1975. Regulation of glutaminase levels in Escherichia coli. J. Bacteriol. 123:992–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. 2009. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5:593–599. 10.1038/nchembio.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Taymaz-Nikerel H, De Mey M, Ras C, ten Pierick A, Seifar RM, van Dam JC, Heijnen JJ, van Gulik WM. 2009. Development and application of a differential method for reliable metabolome analysis in Escherichia coli. Anal. Biochem. 386:9–19. 10.1016/j.ab.2008.11.018 [DOI] [PubMed] [Google Scholar]
- 344.Prusiner S, Stadtman ER. 1976. Regulation of glutaminase B in Escherichia coli. III. Control by nucleotides and divalent cations. J. Biol. Chem. 251:3463–3469 [PubMed] [Google Scholar]
- 345.Médigue C, Viari A, Hénaut A, Danchin A. 1993. Colibri: a functional data base for the Escherichia coli genome. Microbiol. Rev. 57:623–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Kehres DG, Maguire ME. 2003. Emerging themes in manganese transport, biochemistry and pathogenesis in bacteria. FEMS Microbiol. Rev. 27:263–290. 10.1016/S0168-6445(03)00052-4 [DOI] [PubMed] [Google Scholar]
- 347.Westerhoff HV, van Dam K. 1987. Thermodynamics and control of biological free-energy transduction. Elsevier Science Ltd, Amsterdam, the Netherlands [Google Scholar]
- 348.ter Kuile BH, Westerhoff HV. 2001. Transcriptome meets metabolome: hierarchical and metabolic regulation of the glycolytic pathway. FEBS Lett. 500:169–171. 10.1016/S0014-5793(01)02613-8 [DOI] [PubMed] [Google Scholar]
- 349.Daran-Lapujade P, Rossell S, van Gulik WM, Luttik MAH, de Groot MJL, Slijper M, Heck AJR, Daran J-M, de Winde JH, Westerhoff HV, Pronk JT, Bakker BM. 2007. The fluxes through glycolytic enzymes in Saccharomyces cerevisiae are predominantly regulated at posttranscriptional levels. Proc. Natl. Acad. Sci. U. S. A. 104:15753–15758. 10.1073/pnas.0707476104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Lorenz MC. 2006. A marriage of old and new: chemostats and microarrays identify a new model system for ammonium toxicity. PLoS Biol. 4:e388. 10.1371/journal.pbio.0040388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Kleiner D, Traglauer A, Domm S. 1998. Does ammonia production by Klebsiella contribute to pathogenesis? Bull. Inst. Pasteur 96:257–260. 10.1016/S0020-2452(99)80006-2 [DOI] [Google Scholar]
- 352.Muller T, Walter B, Wirtz A, Burkovski A. 2006. Ammonium toxicity in bacteria. Curr. Microbiol. 52:400–406. 10.1007/s00284-005-0370-x [DOI] [PubMed] [Google Scholar]
- 353.Thompson BG, Kole M, Gerson DF. 1985. Control of ammonium concentration in Escherichia coli fermentations. Biotechnol. Bioeng. 27:818–824. 10.1002/bit.260270610 [DOI] [PubMed] [Google Scholar]
- 354.Radchenko MV, Thornton J, Merrick M. 2010. Control of AmtB-GlnK complex formation by intracellular levels of ATP, ADP and 2-oxoglutarate. J. Biol. Chem. 285:31037–31045. 10.1074/jbc.M110.153908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Lowry OH, Carter J, Ward JB, Glaser L. 1971. The effect of carbon and nitrogen sources on the level of metabolic intermediates in Escherichia coli. J. Biol. Chem. 246:6511–6521 [PubMed] [Google Scholar]
- 356.Durand A, Merrick M. 2006. In vitro analysis of the Escherichia coli AmtB-GlnK complex reveals a stoichiometric interaction and sensitivity to ATP and 2-oxoglutarate. J. Biol. Chem. 281:29558–29567. 10.1074/jbc.M602477200 [DOI] [PubMed] [Google Scholar]
- 357.Slonczewski JL, Fujisawa M, Dopson M, Krulwich TA. 2009. Cytoplasmic pH measurement and homeostasis in bacteria and archaea. Adv. Microb. Physiol. 55:1–79, 317. 10.1016/S0065-2911(09)05501-5 [DOI] [PubMed] [Google Scholar]
- 358.Kitko RD, Wilks JC, Garduque GM, Slonczewski JL. 2010. Osmolytes contribute to pH homeostasis of Escherichia coli. PLoS One 5:e10078. 10.1371/journal.pone.0010078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Wilks JC, Slonczewski JL. 2007. pH of the cytoplasm and periplasm of Escherichia coli: rapid measurement by green fluorescent protein fluorimetry. J. Bacteriol. 189:5601–5607. 10.1128/JB.00615-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Kleiner D. 1985. Bacterial ammonium transport. FEMS Microbiol. Rev. 32:87–100. 10.1111/j.1574-6968.1985.tb01185.x [DOI] [Google Scholar]
- 361.Kleiner D. 1981. The transport of NH3 and NH4+ across biological membranes. Biochim. Biophys. Acta 639:41–52. 10.1016/0304-4173(81)90004-5 [DOI] [PubMed] [Google Scholar]
- 362.Jayakumar A, Barnes EM. 1983. A filtration method for measuring cellular uptake of [14C]methylamine and other highly permeant solutes. Anal. Biochem. 135:475–478. 10.1016/0003-2697(83)90715-7 [DOI] [PubMed] [Google Scholar]
- 363.Khademi S, Stroud RM. 2006. The Amt/MEP/Rh family: structure of AmtB and the mechanism of ammonia gas conduction. Physiology 21:419–429. 10.1152/physiol.00051.2005 [DOI] [PubMed] [Google Scholar]
- 364.Westerhoff HV, Arents JC, Hellingwerf KJ. 1981. The effect of ionophores and light intensity on the initial rate of proton uptake into bacteriorhodopsin liposomes can be quantitatively described by mosaic non-equilibrium thermodynamics. Biochim. Biophys. Acta 637:69–79. 10.1016/0005-2728(81)90211-5 [DOI] [Google Scholar]
- 365.Thomas G, Coutts G, Merrick M. 2000. The glnKamtB operon. A conserved gene pair in prokaryotes. Trends Genet. 16:11–14 [DOI] [PubMed] [Google Scholar]
- 366.Gruswitz F, O'Connell JI, Stroud RM. 2007. Inhibitory complex of the transmembrane ammonia channel, AmtB, and the cytosolic regulatory protein, GlnK, at 1.96 Å. Proc. Natl. Acad. Sci. U. S. A. 104:42–47. 10.1073/pnas.0609796104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Conroy MJ, Durand A, Lupo D, Li XD, Bullough PA, Winkler FK, Merrick M. 2007. The crystal structure of the Escherichia coli AmtB-GlnK complex reveals how GlnK regulates the ammonia channel. Proc. Natl. Acad. Sci. U. S. A. 104:1213–1218. 10.1073/pnas.0610348104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Radchenko M, Merrick M. 2011. The role of effector molecules in signal transduction by PII proteins. Biochem. Soc. Trans. 39:189–194. 10.1042/BST0390189 [DOI] [PubMed] [Google Scholar]
- 369.Blauwkamp TA, Ninfa AJ. 2003. Antagonism of PII signalling by the AmtB protein of Escherichia coli. Mol. Microbiol. 48:1017–1028. 10.1046/j.1365-2958.2003.03479.x [DOI] [PubMed] [Google Scholar]
- 370.Holzer H, Mecke D, Wulff K, Liess K, Heilmeyer LJ. 1967. Metabolite-induced enzymatic inactivation of glutamine synthetase in E. coli. Adv. Enzyme Regul. 5:211–225. 10.1016/0065-2571(67)90017-9 [DOI] [PubMed] [Google Scholar]
- 371.Woolfolk CA, Stadtman ER. 1964. Cumulative feedback inhibition in the multiple end product regulation of glutamine synthetase activity in Escherichia coli. Biochem. Biophys. Res. Commun. 17:313–319. 10.1016/0006-291X(64)90003-8 [DOI] [Google Scholar]
- 372.Woolfolk CA, Stadtman ER. 1967. Regulation of glutamine synthetase. 3. Cumulative feedback inhibition of glutamine synthetase from Escherichia coli. Arch. Biochem. Biophys. 118:736–755 [DOI] [PubMed] [Google Scholar]
- 373.Reitzer LJ. 1996. Ammonia assimilation and the biosynthesis of glutamine, glutamate, aspartate, asparagine, L-alanine, and D-alanine, p 391–407 In Neidhardt FC, Curtiss R, III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC [Google Scholar]
- 374.Liaw SH, Pan C, Eisenberg D. 1993. Feedback inhibition of fully unadenylylated glutamine synthetase from Salmonella typhimurium by glycine, alanine, and serine. Proc. Natl. Acad. Sci. U. S. A. 90:4996–5000. 10.1073/pnas.90.11.4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.García-Contreras R, Vos P, Westerhoff HV, Boogerd FC. 2012. Why in vivo may not equal in vitro—new effectors revealed by measurement of enzymatic activities under the same in vivo-like assay conditions. FEBS J. 279:4145–4159. 10.1111/febs.12007 [DOI] [PubMed] [Google Scholar]
- 376.Kingdon HS, Stadtman ER. 1967. Two E. coli glutamine synthetases with different sensitivities to feedback effectors. Biochem. Biophys. Res. Commun. 27:470–473. 10.1016/S0006-291X(67)80008-1 [DOI] [PubMed] [Google Scholar]
- 377.Holzer H, Schutt H, Masek Z, Mecke D. 1968. Regulation of two forms of glutamine synthetase in Escherichia coli by the ammonia content of the growth medium. Proc. Natl. Acad. Sci. U. S. A. 60:721–724. 10.1073/pnas.60.2.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Gancedo C, Holzer H. 1968. Enzymatic inactivation of glutamine synthetase in Enterobacteriaceae. Eur. J. Biochem. 4:190–192. 10.1111/j.1432-1033.1968.tb00192.x [DOI] [PubMed] [Google Scholar]
- 379.Shapiro BM. 1969. The glutamine synthetase deadenylylating enzyme system from Escherichia coli. Resolution into two components, specific nucleotide stimulation, and cofactor requirements. Biochemistry 8:659–670 [DOI] [PubMed] [Google Scholar]
- 380.Garcia E, Rhee SG. 1983. Cascade control of Escherichia coli glutamine synthetase. Purification and properties of PII uridylyltransferase and uridylyl-removing enzyme. J. Biol. Chem. 258:2246–2253 [PubMed] [Google Scholar]
- 381.Bloom FR, Levin MS, Foor F, Tyler B. 1978. Regulation of glutamine synthetase formation in Escherichia coli: characterization of mutants lacking the uridylyltransferase. J. Bacteriol. 134:569–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Foor F, Cedergren RJ, Streicher SL, Rhee SG, Magasanik B. 1978. Glutamine synthetase of Klebsiella aerogenes: properties of glnD mutants lacking uridylyltransferase. J. Bacteriol. 134:562–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Ikeda TP, Shauger AE, Kustu S. 1996. Salmonella typhimurium apparently perceives external nitrogen limitation as internal glutamine limitation. J. Mol. Biol. 259:589–607. 10.1006/jmbi.1996.0342 [DOI] [PubMed] [Google Scholar]
- 384.Senior PJ. 1975. Regulation of nitrogen metabolism in Escherichia coli and Klebsiella aerogenes: studies with the continuous-culture technique. J. Bacteriol. 123:407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Jiang P, Zucker P, Ninfa AJ. 1997. Probing interactions of the homotrimeric PII signal transduction protein with its receptors by use of PII heterotrimers formed in vitro from wild-type and mutant subunits. J. Bacteriol. 179:4354–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Jiang P, Ninfa AJ. 2009. Sensation and signaling of alpha-ketoglutarate and adenylylate energy charge by the Escherichia coli PII signal transduction protein require cooperation of the three ligand-binding sites within the PII trimer. Biochemistry 48:11522–11531. 10.1021/bi9011594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Jiang P, Peliska JA, Ninfa AJ. 1998. The regulation of Escherichia coli glutamine synthetase revisited: role of 2-ketoglutarate in the regulation of glutamine synthetase adenylylation state. Biochemistry 37:12802–12810. 10.1021/bi980666u [DOI] [PubMed] [Google Scholar]
- 388.Jiang P, Ninfa AJ. 2009. Alpha-ketoglutarate controls the ability of the Escherichia coli PII signal transduction protein to regulate the activities of NRII (NtrB) but does not control the binding of PII to NRII. Biochemistry 48:11514–11521. 10.1021/bi901158h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Atkinson MR, Kamberov ES, Weiss RL, Ninfa AJ. 1994. Reversible uridylylation of the Escherichia coli PII signal transduction protein regulates its ability to stimulate the dephosphorylation of the transcription factor nitrogen regulator I (NRI or NtrC). J. Biol. Chem. 269:28288–28293 [PubMed] [Google Scholar]
- 390.Jiang P, Ninfa AJ. 2007. Escherichia coli PII signal transduction protein controlling nitrogen assimilation acts as a sensor of adenylate energy charge in vitro. Biochemistry 46:12979–12996. 10.1021/bi701062t [DOI] [PubMed] [Google Scholar]
- 391.Walker JE, Saraste M, Runswick MJ, Gay NJ. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1:945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Magasanik B. 1996. Regulation of nitrogen assimilation, p 281–290 In Lin ECC, Lynch AS. (ed), Regulation of gene expression in Escherichia coli. Chapman & Hall, London, United Kingdom [Google Scholar]
- 393.Holm AK, Blank LM, Oldiges M, Schmid A, Solem C, Jensen PR, Vemuri GN. 2010. Metabolic and transcriptional response to cofactor perturbations in Escherichia coli. J. Biol. Chem. 285:17498–17506. 10.1074/jbc.M109.095570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Jensen PR, Michelsen O. 1992. Carbon and energy metabolism of atp mutants of Escherichia coli. J. Bacteriol. 174:7635–7641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Koebmann BJ, Westerhoff HV, Snoep JL, Nilsson D, Jensen PR. 2002. The glycolytic flux in Escherichia coli is controlled by the demand for ATP. J. Bacteriol. 184:3909–3916. 10.1128/JB.184.14.3909-3916.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Rohwer JM. 1997. Interaction of functional units in metabolism. Control and regulation of the bacterial phosphoenolpyruvate-dependent phosphotransferase system. University of Amsterdam, Amsterdam, the Netherlands [Google Scholar]
- 397.Tran QH, Unden G. 1998. Changes in the proton potential and the cellular energetics of Escherichia coli during growth by aerobic and anaerobic respiration or by fermentation. Eur. J. Biochem. 251:538–543. 10.1046/j.1432-1327.1998.2510538.x [DOI] [PubMed] [Google Scholar]
- 398.Jensen PR, Westerhoff HV, Michelsen O. 1993. Excess capacity of H+-ATPase and inverse respiratory control in Escherichia coli. EMBO J. 12:1277–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Koefoed S. 2003. Is ATP in charge? E. coli as a test case. VU University Amsterdam, Amsterdam, the Netherlands [Google Scholar]
- 400.Ninfa AJ, Jiang P, Atkinson MR, Peliska JA. 2000. Integration of antagonistic signals in the regulation of nitrogen assimilation in Escherichia coli. Curr. Top. Cell. Regul. 36:31–75 [DOI] [PubMed] [Google Scholar]
- 401.Atkinson MR, Blauwkamp TA, Bondarenko V, Studitsky V, Ninfa AJ. 2002. Activation of the glnA, glnK, and nac promoters as Escherichia coli undergoes the transition from nitrogen excess growth to nitrogen starvation. J. Bacteriol. 184:5358–5363. 10.1128/JB.184.19.5358-5363.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Blauwkamp TA, Ninfa AJ. 2002. Physiological role of the GlnK signal transduction protein of Escherichia coli: survival of nitrogen starvation. Mol. Microbiol. 46:203–214. 10.1046/j.1365-2958.2002.03153.x [DOI] [PubMed] [Google Scholar]
- 403.Atkinson MR, Blauwkamp TA, Ninfa AJ. 2002. Context-dependent functions of the PII and GlnK signal transduction proteins in Escherichia coli. J. Bacteriol. 184:5364–5375. 10.1128/JB.184.19.5364-5375.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Forchhammer K, Hedler A, Strobel H, Weiss V. 1999. Heterotrimerization of PII-like signalling proteins: implications for PII-mediated signal transduction systems. Mol. Microbiol. 33:338–349. 10.1046/j.1365-2958.1999.01477.x [DOI] [PubMed] [Google Scholar]
- 405.Oliver SG. 1996. From DNA sequence to biological function. Nature 379:597–600. 10.1038/379597a0 [DOI] [PubMed] [Google Scholar]
- 406.Raamsdonk LM, Teusink B, Broadhurst D, Zhang N, Hayes A, Walsh MC, Berden JA, Brindle KM, Kell DB, Rowland JJ, Westerhoff HV, van Dam K, Oliver SG. 2001. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 19:45–50. 10.1038/83496 [DOI] [PubMed] [Google Scholar]
- 407.Javelle A, Merrick M. 2005. Complex formation between AmtB and GlnK: an ancestral role in prokaryotic nitrogen control. Biochem. Soc. Trans. 33:170–172. 10.1042/BST0330170 [DOI] [PubMed] [Google Scholar]
- 408.Kustu S, Hirschman J, Burton D, Jelesko J, Meeks JC. 1984. Covalent modification of bacterial glutamine synthetase: physiological significance. Mol. Gen. Genet. 197:309–317. 10.1007/BF00330979 [DOI] [PubMed] [Google Scholar]
- 409.Kustu S, Hirschman J, Meeks JC. 1985. Adenylylation of bacterial glutamine synthetase: physiological significance. Curr. Top. Cell Regul. 27:201–213 [DOI] [PubMed] [Google Scholar]
- 410.McLaggan D, Naprstek J, Buurman ET, Epstein W. 1994. Interdependence of K+ and glutamate accumulation during osmotic adaptation of Escherichia coli. J. Biol. Chem. 269:1911–1917 [PubMed] [Google Scholar]
- 411.Yan D. 2007. Protection of the glutamate pool concentration in enteric bacteria. Proc. Natl. Acad. Sci. U. S. A. 104:9475–9480. 10.1073/pnas.0703360104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Yan D, Ikeda TP, Shauger AE, Kustu S. 1996. Glutamate is required to maintain the steady-state potassium pool in Salmonella typhimurium. Proc. Natl. Acad. Sci. U. S. A. 93:6527–6531. 10.1073/pnas.93.13.6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Foor F, Janssen KA, Magasanik B. 1975. Regulation of synthesis of glutamine synthetase by adenylylated glutamine synthetase. Proc. Natl. Acad. Sci. U. S. A. 72:4844–4848. 10.1073/pnas.72.12.4844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Okano H, Hwa T, Lenz P, Yan D. 2010. Reversible adenylylation of glutamine synthetase is dynamically counterbalanced during steady-state growth of Escherichia coli. J. Mol. Biol. 404:522–536. 10.1016/j.jmb.2010.09.046 [DOI] [PubMed] [Google Scholar]
- 415.Segal A, Brown MS, Stadtman ER. 1974. Metabolite regulation of the state of adenylylation of glutamine synthetase. Arch. Biochem. Biophys. 161:319–327. 10.1016/0003-9861(74)90267-7 [DOI] [Google Scholar]
- 416.Ninfa AJ, Ninfa EG, Lupas AN, Stock A, Magasanik B, Stock J. 1988. Crosstalk between bacterial chemotaxis signal transduction proteins and regulators of transcription of the Ntr regulon: evidence that nitrogen assimilation and chemotaxis are controlled by a common phosphotransfer mechanism. Proc. Natl. Acad. Sci. U. S. A. 85:5492–5496. 10.1073/pnas.85.15.5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Jiang P, Peliska JA, Ninfa AJ. 2000. Asymmetry in the autophosphorylation of the two-component regulatory system transmitter protein nitrogen regulator II of Escherichia coli. Biochemistry 39:5057–5065. 10.1021/bi992921w [DOI] [PubMed] [Google Scholar]
- 418.Stock JB, Surette MG, Levit M, Park P. 1995. Two-component signal transduction systems: structure-function relationships and mechanisms of catalysis, p 25–51 In Hoch JA, Silhavy TJ. (ed), Two-component signal transduction. ASM Press, Washington, DC [Google Scholar]
- 419.Feng J, Atkinson MR, McCleary W, Stock JB, Wanner BL, Ninfa AJ. 1992. Role of phosphorylated metabolic intermediates in the regulation of glutamine synthetase synthesis in Escherichia coli. J. Bacteriol. 174:6061–6070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.McCleary WR, Stock JB. 1994. Acetyl phosphate and the activation of two-component response regulators. J. Biol. Chem. 269:31567–31572 [PubMed] [Google Scholar]
- 421.McCleary WR, Stock JB, Ninfa AJ. 1993. Is acetyl phosphate a global signal in Escherichia coli? J. Bacteriol. 175:2793–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Kamberov ES, Atkinson MR, Chandran P, Ninfa AJ. 1994. Effect of mutations in Escherichia coli glnL (ntrB), encoding nitrogen regulator II (NRII or NtrB), on the phosphatase activity involved in bacterial nitrogen regulation. J. Biol. Chem. 269:28294–28299 [PubMed] [Google Scholar]
- 423.Cayley S, Lewis BA, Guttman HJ, Record MTJ. 1991. Characterization of the cytoplasm of Escherichia coli K-12 as a function of external osmolarity. Implications for protein-DNA interactions in vivo. J. Mol. Biol. 222:281–300 [DOI] [PubMed] [Google Scholar]
- 424.Pioszak AA, Ninfa AJ. 2003. Mechanism of the PII-activated phosphatase activity of Escherichia coli NRII (NtrB): how the different domains of NRII collaborate to act as a phosphatase. Biochemistry 42:8885–8899. 10.1021/bi030065p [DOI] [PubMed] [Google Scholar]
- 425.Pioszak AA, Ninfa AJ. 2004. Mutations altering the N-terminal receiver domain of NRI (NtrC) that prevent dephosphorylation by the NRII-PII complex in Escherichia coli. J. Bacteriol. 186:5730–5740. 10.1128/JB.186.17.5730-5740.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Atkinson MR, Ninfa AJ. 1992. Characterization of Escherichia coli glnL mutations affecting nitrogen regulation. J. Bacteriol. 174:4538–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. 1999. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. U. S. A. 96:4285–4288. 10.1073/pnas.96.8.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Du Y, Holtel A, Reizer J, Saier MHJ. 1996. Sigma54-dependent transcription of the Pseudomonas putida xylS operon is influenced by the IIANtr protein of the phosphotransferase system in Escherichia coli. Res. Microbiol. 147:129–132. 10.1016/0923-2508(96)80212-9 [DOI] [PubMed] [Google Scholar]
- 429.Commichau FM, Forchhammer K, Stülke J. 2006. Regulatory links between carbon and nitrogen metabolism. Curr. Opin. Microbiol. 9:167–172. 10.1016/j.mib.2006.01.001 [DOI] [PubMed] [Google Scholar]
- 430.Lee CR, Koo BM, Cho SH, Kim YJ, Yoon MJ, Peterkofsky A, Seok YJ. 2005. Requirement of the dephospho-form of enzyme IIANtr for derepression of Escherichia coli K12 ilvBN expression. Mol. Microbiol. 58:334–344. 10.1111/j.1365-2958.2005.04834.x [DOI] [PubMed] [Google Scholar]
- 431.Reaves ML, Rabinowitz JD. 2011. Characteristic phenotypes associated with ptsN-null mutants in Escherichia coli K-12 are absent in strains with functional ilvG. J. Bacteriol. 193:4576–4581. 10.1128/JB.00325-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Aravind L, Ponting CP. 1997. The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem. Sci. 22:458–459. 10.1016/S0968-0004(97)01148-1 [DOI] [PubMed] [Google Scholar]
- 433.Peterkofsky A, Wang G, Seok Y-J. 2006. Parallel PTS systems. Arch. Biochem. Biophys. 453:101–107. 10.1016/j.abb.2006.01.004 [DOI] [PubMed] [Google Scholar]
- 434.Little R, Dixon R. 2003. The amino-terminal GAF domain of Azotobacter vinelandii NifA binds 2-oxoglutarate to resist inhibition by NifL under nitrogen-limiting conditions. J. Biol. Chem. 278:28711–28718. 10.1074/jbc.M301992200 [DOI] [PubMed] [Google Scholar]
- 435.Doucette CD, Schwab DJ, Wingreen NS, Rabinowitz JD. 2011. α-Ketoglutarate coordinates carbon and nitrogen utilization via enzyme I inhibition. Nat. Chem. Biol. 7:894–901. 10.1038/nchembio.685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Kim H-J, Lee C-R, Kim M, Peterkofsky A, Seok Y-J. 2011. Dephosphorylated NPr of the nitrogen PTS regulates lipid A biosynthesis by direct interaction with LpxD. Biochem. Biophys. Res. Commun. 409:556–561. 10.1016/j.bbrc.2011.05.044 [DOI] [PubMed] [Google Scholar]
- 437.Bartling CM, Raetz CRH. 2008. Steady-state kinetics and mechanism of LpxD, the N-acyltransferase of lipid A biosynthesis. Biochemistry 47:5290–5302. 10.1021/bi800240r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Lee CR, Cho SH, Yoon MJ, Peterkofsky A, Seok YJ. 2007. Escherichia coli enzyme IIANtr regulates the K+ transporter TrkA. Proc. Natl. Acad. Sci. U. S. A. 104:4124–4129. 10.1073/pnas.0609897104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Bossemeyer D, Borchard A, Dosch DC, Helmer GC, Epstein W, Booth IR, Bakker EP. 1989. K+-transport protein TrkA of Escherichia coli is a peripheral membrane protein that requires other trk gene products for attachment to the cytoplasmic membrane. J. Biol. Chem. 264:16403–16410 [PubMed] [Google Scholar]
- 440.Dosch DC, Helmer GL, Sutton SH, Salvacion FF, Epstein W. 1991. Genetic analysis of potassium transport loci in Escherichia coli: evidence for three constitutive systems mediating uptake potassium. J. Bacteriol. 173:687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Rhoads DB, Waters FB, Epstein W. 1976. Cation transport in Escherichia coli. VIII. Potassium transport mutants. J. Gen. Physiol. 67:325–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Lüttmann D, Heermann R, Zimmer B, Hillmann A, Rampp IS, Jung K, Görke B. 2009. Stimulation of the potassium sensor KdpD kinase activity by interaction with the phosphotransferase protein IIANtr in Escherichia coli. Mol. Microbiol. 72:978–994. 10.1111/j.1365-2958.2009.06704.x [DOI] [PubMed] [Google Scholar]
- 443.Lüttmann D, Göpel Y, Görke B. 2012. The phosphotransferase protein EIIANtr modulates the phosphate starvation response through interaction with histidine kinase PhoR in Escherichia coli. Mol. Microbiol. 86:96–110. 10.1111/j.1365-2958.2012.08176.x [DOI] [PubMed] [Google Scholar]
- 444.Hayden JD, Ades SE. 2008. The extracytoplasmic stress factor, sigmaE, is required to maintain cell envelope integrity in Escherichia coli. PLoS One 3:e1573. 10.1371/journal.pone.0001573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Choi J, Shin D, Yoon H, Kim J, Lee C-R, Kim M, Seok Y-J, Ryu S. 2010. Salmonella pathogenicity island 2 expression negatively controlled by EIIANtr-SsrB interaction is required for Salmonella virulence. Proc. Natl. Acad. Sci. U. S. A. 107:20506–20511. 10.1073/pnas.1000759107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. 2009. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840. 10.1126/science.1175371 [DOI] [PubMed] [Google Scholar]
- 447.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan K-L. 2010. Regulation of cellular metabolism by protein lysine acetylation. Science 327:1000–1004. 10.1126/science.1179689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning Z-B, Zeng R, Xiong Y, Guan K-L, Zhao S, Zhao G-P. 2010. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327:1004–1007. 10.1126/science.1179687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Yu BJ, Kim JA, Moon JH, Ryu SE, Pan J-G. 2008. The diversity of lysine-acetylated proteins in Escherichia coli. J. Microbiol. Biotechnol. 18:1529–1536 [PubMed] [Google Scholar]
- 450.Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu C-F, Grishin NV, Zhao Y. 2009. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics 8:215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Pahel G, Rothstein DM, Magasanik B. 1982. Complex glnA-glnL-glnG operon of Escherichia coli. J. Bacteriol. 150:202–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.McFarland N, McCarter L, Artz S, Kustu S. 1981. Nitrogen regulatory locus glnR of enteric bacteria is composed of cistrons ntrB and ntrC: identification of their protein products. Proc. Natl. Acad. Sci. U. S. A. 78:2135–2139. 10.1073/pnas.78.4.2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Mao XJ, Huo YX, Buck M, Kolb A, Wang YP. 2007. Interplay between CRP-cAMP and PII-Ntr systems forms novel regulatory network between carbon metabolism and nitrogen assimilation in Escherichia coli. Nucleic Acids Res. 35:1432–1440. 10.1093/nar/gkl1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Reitzer LJ, Magasanik B. 1985. Expression of glnA in Escherichia coli is regulated at tandem promoters. Proc. Natl. Acad. Sci. U. S. A. 82:1979–1983. 10.1073/pnas.82.7.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Tian ZX, Li QS, Buck M, Kolb A, Wang YP. 2001. The CRP-cAMP complex and downregulation of the glnAp2 promoter provides a novel regulatory linkage between carbon metabolism and nitrogen assimilation in Escherichia coli. Mol. Microbiol. 41:911–924 [DOI] [PubMed] [Google Scholar]
- 456.Merrick M, Chambers S. 1992. The helix-turn-helix motif of σ54 is involved in recognition of the −13 promoter region. J. Bacteriol. 174:7221–7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Ninfa AJ, Reitzer LJ, Magasanik B. 1987. Initiation of transcription at the bacterial glnAp2 promoter by purified E. coli components is facilitated by enhancers. Cell 50:1039–1046. 10.1016/0092-8674(87)90170-X [DOI] [PubMed] [Google Scholar]
- 458.Reitzer LJ, Magasanik B. 1986. Transcription of glnA in E. coli is stimulated by activator bound to sites far from the promoter. Cell 45:785–792. 10.1016/0092-8674(86)90553-2 [DOI] [PubMed] [Google Scholar]
- 459.Atkinson MR, Pattaramanon N, Ninfa AJ. 2002. Governor of the glnAp2 promoter of Escherichia coli. Mol. Microbiol. 46:1247–1257. 10.1046/j.1365-2958.2002.03211.x [DOI] [PubMed] [Google Scholar]
- 460.Ueno-Nishio S, Mango S, Reitzer LJ, Magasanik B. 1984. Identification and regulation of the glnL operator-promoter of the complex glnALG operon of Escherichia coli. J. Bacteriol. 160:379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Dimri GP, Rudd KE, Morgan MK, Bayat H, Ames GF. 1992. Physical mapping of repetitive extragenic palindromic sequences in Escherichia coli and phylogenetic distribution among Escherichia coli strains and other enteric bacteria. J. Bacteriol. 174:4583–4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Rocha M, Vazquez M, Garciarrubio A, Covarrubias AA. 1985. Nucleotide sequence of the glnA-glnL intercistronic region of Escherichia coli. Gene 37:91–99. 10.1016/0378-1119(85)90261-6 [DOI] [PubMed] [Google Scholar]
- 463.Yang Y, Ames GF-L. 1988. DNA gyrase binds to the family of prokaryotic repetitive extragenic palindromic sequences. Proc. Natl. Acad. Sci. U. S. A. 85:8850–8854. 10.1073/pnas.85.23.8850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Gilson E, Perrin D, Hofnung M. 1990. DNA polymerase I and a protein complex bind specifically to E. coli palindromic unit highly repetitive DNA: implications for bacterial chromosome organization. Nucleic Acids Res. 18:3941–3952. 10.1093/nar/18.13.3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Sladewski TE, Hetrick KM, Foster PL. 2011. Escherichia coli Rep DNA helicase and error-prone DNA polymerase IV interact physically and functionally. Mol. Microbiol. 80:524–541. 10.1111/j.1365-2958.2011.07590.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Gilson E, Saurin W, Perrin D, Bachellier S, Hofnung M. 1991. The BIME family of bacterial highly repetitive sequences. Res. Microbiol. 142:217–222. 10.1016/0923-2508(91)90033-7 [DOI] [PubMed] [Google Scholar]
- 467.Higgins CF, Ames GF, Barnes WM, Clement JM, Hofnung M. 1982. A novel intercistronic regulatory element of prokaryotic operons. Nature 298:760–762. 10.1038/298760a0 [DOI] [PubMed] [Google Scholar]
- 468.Hulton CS, Higgins CF, Sharp PM. 1991. ERIC sequences: a novel family of repetitive elements in the genomes of Escherichia coli, Salmonella typhimurium and other enterobacteria. Mol. Microbiol. 5:825–834. 10.1111/j.1365-2958.1991.tb00755.x [DOI] [PubMed] [Google Scholar]
- 469.Stern MJ, Ames GF-L, Smith NH, Robinson EC, Higgins CF. 1984. Repetitive extragenic palindromic sequences: a major component of the bacterial genome. Cell 37:1015–1026. 10.1016/0092-8674(84)90436-7 [DOI] [PubMed] [Google Scholar]
- 470.Weiss V, Claverie-Martin F, Magasanik B. 1992. Phosphorylation of nitrogen regulator I of Escherichia coli induces strong cooperative binding to DNA essential for activation of transcription. Proc. Natl. Acad. Sci. U. S. A. 89:5088–5092. 10.1073/pnas.89.11.5088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Porter SC, North AK, Wedel AB, Kustu S. 1993. Oligomerization of NTRC at the glnA enhancer is required for transcriptional activation. Genes Dev. 7:2258–2273. 10.1101/gad.7.11.2258 [DOI] [PubMed] [Google Scholar]
- 472.Wyman C, Rombel I, North AK, Bustamante C, Kustu S. 1997. Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science 275:1658–1661. 10.1126/science.275.5306.1658 [DOI] [PubMed] [Google Scholar]
- 473.Popham DL, Szeto D, Keener J, Kustu S. 1989. Function of a bacterial activator protein that binds to transcriptional enhancers. Science 243:629–635. 10.1126/science.2563595 [DOI] [PubMed] [Google Scholar]
- 474.Reitzer LJ, Movsas B, Magasanik B. 1989. Activation of glnA transcription by nitrogen regulator I (NRI)-phosphate in Escherichia coli: evidence for a long-range physical interaction between NRI-phosphate and RNA polymerase. J. Bacteriol. 171:5512–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Su W, Porter S, Kustu S, Echols H. 1990. DNA-looping and enhancer activity: association between DNA-bound NtrC activator and RNA polymerase at the bacterial glnA promoter. Proc. Natl. Acad. Sci. U. S. A. 87:5504–5508. 10.1073/pnas.87.14.5504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Rombel I, North A, Hwang I, Wyman C, Kustu S. 1998. The bacterial enhancer-binding protein NtrC as a molecular machine. Cold Spring Harb. Symp. Quant. Biol. 63:157–166. 10.1101/sqb.1998.63.157 [DOI] [PubMed] [Google Scholar]
- 477.Lee J, Owens JT, Hwang I, Meares C, Kustu S. 2000. Phosphorylation-induced signal propagation in the response regulator ntrC. J. Bacteriol. 182:5188–5195. 10.1128/JB.182.18.5188-5195.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Lee SY, De La Torre A, Yan D, Kustu S, Nixon BT, Wemmer DE. 2003. Regulation of the transcriptional activator NtrC1: structural studies of the regulatory and AAA+ ATPase domains. Genes Dev. 17:2552–2563. 10.1101/gad.1125603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Buck M, Gallegos MT, Studholme DJ, Guo Y, Gralla JD. 2000. The bacterial enhancer-dependent sigma54 (sigmaN) transcription factor. J. Bacteriol. 182:4129–4136. 10.1128/JB.182.15.4129-4136.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Wedel A, Kustu S. 1995. The bacterial enhancer-binding protein NTRC is a molecular machine: ATP hydrolysis is coupled to transcriptional activation. Genes Dev. 9:2042–2052. 10.1101/gad.9.16.2042 [DOI] [PubMed] [Google Scholar]
- 481.Wedel A, Weiss DS, Popham D, Droge P, Kustu S. 1990. A bacterial enhancer functions to tether a transcriptional activator near a promoter. Science 248:486–490. 10.1126/science.1970441 [DOI] [PubMed] [Google Scholar]
- 482.Teusink B, Walsh MC, van Dam K, Westerhoff HV. 1998. The danger of metabolic pathways with turbo design. Trends Biochem. Sci. 23:162–169. 10.1016/S0968-0004(98)01205-5 [DOI] [PubMed] [Google Scholar]
- 483.Shiau SP, Schneider BL, Gu W, Reitzer LJ. 1992. Role of nitrogen regulator I (NtrC), the transcriptional activator of glnA in enteric bacteria, in reducing expression of glnA during nitrogen-limited growth. J. Bacteriol. 174:179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Prusiner S, Miller RE, Valentine RC. 1972. Adenosine 3′:5′-cyclic monophosphate control of the enzymes of glutamine metabolism in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 69:2922–2926. 10.1073/pnas.69.10.2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Forchhammer K. 2007. Glutamine signalling in bacteria. Front. Biosci. 12:358–370. 10.2741/2069 [DOI] [PubMed] [Google Scholar]
- 486.Maheswaran M, Forchhammer K. 2003. Carbon-source-dependent nitrogen regulation in Escherichia coli is mediated through glutamine-dependent GlnB signalling. Microbiology 149:2163–2172. 10.1099/mic.0.26449-0 [DOI] [PubMed] [Google Scholar]
- 487.Keseler IM, Collado-Vides J, Santos-Zavaleta A, Peralta-Gil M, Gama-Castro S, Muniz-Rascado L, Bonavides-Martinez C, Paley S, Krummenacker M, Altman T, Kaipa P, Spaulding A, Pacheco J, Latendresse M, Fulcher C, Sarker M, Shearer AG, Mackie A, Paulsen I, Gunsalus RP, Karp PD. 2011. EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 39:D583–D590. 10.1093/nar/gkq1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Borst DW, Blumenthal RM, Matthews RG. 1996. Use of an in vivo titration method to study a global regulator: effect of varying Lrp levels on expression of gltBDF in Escherichia coli. J. Bacteriol. 178:6904–6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Chen C, Newman EB. 1998. Comparison of the sensitivities of two Escherichia coli genes to in vivo variation of Lrp concentration. J. Bacteriol. 180:655–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Paul L, Mishra PK, Blumenthal RM, Matthews RG. 2007. Integration of regulatory signals through involvement of multiple global regulators: control of the Escherichia coli gltBDF operon by Lrp, IHF, Crp, and ArgR. BMC Microbiol. 7:2. 10.1186/1471-2180-7-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Wiese DE, Ernsting BR, Blumenthal RM, Matthews RG. 1997. A nucleoprotein activation complex between the leucine-responsive regulatory protein and DNA upstream of the gltBDF operon in Escherichia coli. J. Mol. Biol. 270:152–168. 10.1006/jmbi.1997.1057 [DOI] [PubMed] [Google Scholar]
- 492.Paul L, Blumenthal RM, Matthews RG. 2001. Activation from a distance: roles of Lrp and integration host factor in transcriptional activation of gltBDF. J. Bacteriol. 183:3910–3918. 10.1128/JB.183.13.3910-3918.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Hommais F, Krin E, Coppée JY, Lacroix C, Yeramian E, Danchin A, Bertin P. 2004. GadE (YhiE): a novel activator involved in the response to acid environment in Escherichia coli. Microbiology 150:61–72. 10.1099/mic.0.26659-0 [DOI] [PubMed] [Google Scholar]
- 494.Saroja GN, Gowrishankar J. 1996. Roles of SpoT and FNR in NH4+ assimilation and osmoregulation in GOGAT (glutamate synthase)-deficient mutants of Escherichia coli. J. Bacteriol. 178:4105–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.D'Ari R, Lin RT, Newman EB. 1993. The leucine-responsive regulatory protein: more than a regulator? Trends Biochem. Sci. 18:260–263. 10.1016/0968-0004(93)90177-O [DOI] [PubMed] [Google Scholar]
- 496.Newman EB, Lin R. 1995. Leucine-responsive regulatory protein: a global regulator of gene expression in E. coli. Annu. Rev. Microbiol. 49:747–775. 10.1146/annurev.mi.49.100195.003531 [DOI] [PubMed] [Google Scholar]
- 497.Ernsting BR, Atkinson MR, Ninfa AJ, Matthews RG. 1992. Characterization of the regulon controlled by the leucine-responsive regulatory protein in Escherichia coli. J. Bacteriol. 174:1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Ernsting BR, Denninger JW, Blumenthal RM, Matthews RG. 1993. Regulation of the gltBDF operon of Escherichia coli: how is a leucine-insensitive operon regulated by the leucine-responsive regulatory protein? J. Bacteriol. 175:7160–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Collado-Vides J, Magasanik B, Gralla JD. 1991. Control site location and transcriptional regulation in Escherichia coli. Microbiol. Rev. 55:371–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Ma Z, Gong S, Richard H, Tucker DL, Conway T, Foster JW. 2003. GadE (YhiE) activates glutamate decarboxylase-dependent acid resistance in Escherichia coli K-12. Mol. Microbiol. 49:1309–1320. 10.1046/j.1365-2958.2003.03633.x [DOI] [PubMed] [Google Scholar]
- 501.Constantinidou C, Hobman JL, Griffiths L, Patel MD, Penn CW, Cole JA, Overton TW. 2006. A reassessment of the FNR regulon and transcriptomic analysis of the effects of nitrate, nitrite, NarXL, and NarQP as Escherichia coli K12 adapts from aerobic to anaerobic growth. J. Biol. Chem. 281:4802–4815 [DOI] [PubMed] [Google Scholar]
- 502.Goss TJ, Janes BK, Bender RA. 2002. Repression of glutamate dehydrogenase formation in Klebsiella aerogenes requires two binding sites for the nitrogen assimilation control protein, NAC. J. Bacteriol. 184:6966–6975. 10.1128/JB.184.24.6966-6975.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Camarena L, Poggio S, Garcia N, Osorio A. 1998. Transcriptional repression of gdhA in Escherichia coli is mediated by the Nac protein. FEMS Microbiol. Lett. 167:51–56. 10.1111/j.1574-6968.1998.tb13206.x [DOI] [PubMed] [Google Scholar]
- 504.Marbaniang CN, Gowrishankar J. 2011. Role of ArgP (IciA) in lysine-mediated repression in Escherichia coli. J. Bacteriol. 193:5985–5996. 10.1128/JB.05869-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Ruiz J, Haneburger I, Jung K. 2011. Identification of ArgP and Lrp as transcriptional regulators of lysP, the gene encoding the specific lysine permease of Escherichia coli. J. Bacteriol. 193:2536–2548. 10.1128/JB.00815-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Goss TJ. 2008. The ArgP protein stimulates the Klebsiella pneumoniae gdhA promoter in a lysine-sensitive manner. J. Bacteriol. 190:4351–4359. 10.1128/JB.00295-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Best EA, Bender RA. 1990. Cloning of the Klebsiella aerogenes nac gene, which encodes a factor required for nitrogen regulation of the histidine utilization (hut) operons in Salmonella typhimurium. J. Bacteriol. 172:7043–7048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Bender RA, Snyder PM, Bueno R, Quinto M, Magasanik B. 1983. Nitrogen regulation system of Klebsiella aerogenes: the nac gene. J. Bacteriol. 156:444–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Feng J, Goss TJ, Bender RA, Ninfa AJ. 1995. Activation of transcription initiation from the nac promoter of Klebsiella aerogenes. J. Bacteriol. 177:5523–5534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Janes BK, Bender RA. 1998. Alanine catabolism in Klebsiella aerogenes: molecular characterization of the dadAB operon and its regulation by the nitrogen assimilation control protein. J. Bacteriol. 180:563–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Blauwkamp TA, Ninfa AJ. 2002. Nac-mediated repression of the serA promoter of Escherichia coli. Mol. Microbiol. 45:351–363. 10.1046/j.1365-2958.2002.02994.x [DOI] [PubMed] [Google Scholar]
- 512.He B, Choi KY, Zalkin H. 1993. Regulation of Escherichia coli glnB, prsA, and speA by the purine repressor. J. Bacteriol. 175:3598–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Tso JY, Hermodson MA, Zalkin H. 1982. Glutamine phosphoribosylpyrophosphate amidotransferase from cloned Escherichia coli purF. NH2-terminal amino acid sequence, identification of the glutamine site, and trace metal analysis. J. Biol. Chem. 257:3532–3536 [PubMed] [Google Scholar]
- 514.Choi KY, Zalkin H. 1992. Structural characterization and corepressor binding of the Escherichia coli purine repressor. J. Bacteriol. 174:6207–6214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Rolfes RJ, Zalkin H. 1990. Purification of the Escherichia coli purine regulon repressor and identification of corepressors. J. Bacteriol. 172:5637–5642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.van Heeswijk WC, Kuppinger O, Merrick M, Kahn D. 1992. Localization of the glnD gene on a revised map of the 200-kilobase region of the Escherichia coli chromosome. J. Bacteriol. 174:1702–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Christie GE, Farnham PJ, Platt T. 1981. Synthetic sites for transcription termination and a functional comparison with tryptophan operon termination sites in vitro. Proc. Natl. Acad. Sci. U. S. A. 78:4180–4184. 10.1073/pnas.78.7.4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Kim IH, Kwak SJ, Kang J, Park SC. 1998. Transcriptional control of the glnD gene is not dependent on nitrogen availability in Escherichia coli. Mol. Cells 8:483–490 [PubMed] [Google Scholar]
- 519.Mendoza-Vargas A, Olvera L, Olvera M, Grande R, Vega-Alvarado L, Taboada B, Jimenez-Jacinto V, Salgado H, Juarez K, Contreras-Moreira B, Huerta AM, Collado-Vides J, Morett E. 2009. Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLoS One 4:e7526. 10.1371/journal.pone.0007526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Dong T, Schellhorn HE. 2009. Control of RpoS in global gene expression of Escherichia coli in minimal media. Mol. Genet. Genomics 281:19–33. 10.1007/s00438-008-0389-3 [DOI] [PubMed] [Google Scholar]
- 521.Groot Kormelink T, Koenders E, Hagemeijer Y, Overmars L, Siezen RJ, de Vos WM, Francke C. 2012. Comparative genome analysis of central nitrogen metabolism and its control by GlnR in the class Bacilli. BMC Genomics 13:191. 10.1186/1471-2164-13-191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Jayakumar A, Schulman I, MacNeil D, Barnes EMJ. 1986. Role of the Escherichia coli glnALG operon in regulation of ammonium transport. J. Bacteriol. 166:281–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Baev MV, Baev D, Radek AJ, Campbell JW. 2006. Growth of Escherichia coli MG1655 on LB medium: monitoring utilization of amino acids, peptides, and nucleotides with transcriptional microarrays. Appl. Microbiol. Biotechnol. 71:317–322. 10.1007/s00253-005-0310-5 [DOI] [PubMed] [Google Scholar]
- 524.Claverie-Martin F, Magasanik B. 1992. Positive and negative effects of DNA bending on activation of transcription from a distant site. J. Mol. Biol. 227:996–1008. 10.1016/0022-2836(92)90516-M [DOI] [PubMed] [Google Scholar]
- 525.Claverie-Martin F, Magasanik B. 1991. Role of integration host factor in the regulation of the glnHp2 promoter of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 88:1631–1635. 10.1073/pnas.88.5.1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Klose KE, Mekalanos JJ. 1997. Simultaneous prevention of glutamine synthesis and high-affinity transport attenuates Salmonella typhimurium virulence. Infect. Immun. 65:587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Nohno T, Saito T. 1987. Two transcriptional start sites found in the promoter region of Escherichia coli glutamine permease operon, glnHPQ. Nucleic Acids Res. 15:2777. 10.1093/nar/15.6.2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Dworkin J, Ninfa AJ, Model P. 1998. A protein-induced DNA bend increases the specificity of a prokaryotic enhancer-binding protein. Genes Dev. 12:894–900. 10.1101/gad.12.6.894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Rice PA, Yang S, Mizuuchi K, Nash HA. 1996. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell 87:1295–1306. 10.1016/S0092-8674(00)81824-3 [DOI] [PubMed] [Google Scholar]
- 530.Lamichhane-Khadka R, Frye JG, Porwollik S, McClelland M, Maier RJ. 2011. Hydrogen-stimulated carbon acquisition and conservation in Salmonella enterica serovar Typhimurium. J. Bacteriol. 193:5824–5832. 10.1128/JB.05456-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Xu J, Johnson RC. 1995. Identification of genes negatively regulated by Fis: Fis and RpoS comodulate growth-phase-dependent gene expression in Escherichia coli. J. Bacteriol. 177:938–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Kumar R, Shimizu K. 2011. Transcriptional regulation of main metabolic pathways of cyoA, cydB, fnr, and fur gene knockout Escherichia coli in C-limited and N-limited aerobic continuous cultures. Microb. Cell Fact. 10:3. 10.1186/1475-2859-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Mandelstam J. 1962. The repression of constitutive β-galactosidase in Escherichia coli by glucose and other carbon sources. Biochem. J. 82:489–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Gerdes SY, Scholle MD, Campbell JW, Balázsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gel'fand MS, Bhattacharya A, Kapatral V, D'Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabási A-L, Oltvai ZN, Osterman AL. 2003. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J. Bacteriol. 185:5673–5684. 10.1128/JB.185.19.5673-5684.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Groen AK, Wanders RJ, Westerhoff HV, van der Meer R, Tager JM. 1982. Quantification of the contribution of various steps to the control of mitochondrial respiration. J. Biol. Chem. 257:2754–2757 [PubMed] [Google Scholar]
- 537.Jensen PR, van der Weijden CC, Jensen LB, Westerhoff HV, Snoep JL. 1999. Extensive regulation compromises the extent to which DNA gyrase controls DNA supercoiling and growth rate of Escherichia coli. Eur. J. Biochem. 266:865–877. 10.1046/j.1432-1327.1999.00921.x [DOI] [PubMed] [Google Scholar]
- 538.Kolodkin A, Simeonidis E, Westerhoff HV. 2013. Computing life: add logos to biology and bios to physics. Prog. Biophys. Mol. Biol. 111:69–74. 10.1016/j.pbiomolbio.2012.10.003 [DOI] [PubMed] [Google Scholar]
- 539.Westerhoff HV, Winder C, Messiha H, Simeonidis E, Adamczyk M, Verma M, Bruggeman FJ, Dunn W. 2009. Systems biology: the elements and principles of life. FEBS Lett. 583:3882–3890. 10.1016/j.febslet.2009.11.018 [DOI] [PubMed] [Google Scholar]
- 540.Verhamme DT, Arents JC, Postma PW, Crielaard W, Hellingwerf KJ. 2002. Investigation of in vivo cross-talk between key two-component systems of Escherichia coli. Microbiology 148:69–78 [DOI] [PubMed] [Google Scholar]
- 541.Fishov I, Zaritsky A, Grover NB. 1995. On microbial states of growth. Mol. Microbiol. 15:789–794. 10.1111/j.1365-2958.1995.tb02349.x [DOI] [PubMed] [Google Scholar]
- 542.Kell DB, Ryder HM, Kaprelyants AS, Westerhoff HV. 1991. Quantifying heterogeneity: flow cytometry of bacterial cultures. Antonie Van Leeuwenhoek 60:145–158. 10.1007/BF00430362 [DOI] [PubMed] [Google Scholar]
- 543.Chien A-C, Hill NS, Levin PA. 2012. Cell size control in bacteria. Curr. Biol. 22:R340–R349. 10.1016/j.cub.2012.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Akerlund T, Nordstrom K, Bernander R. 1995. Analysis of cell size and DNA content in exponentially growing and stationary-phase batch cultures of Escherichia coli. J. Bacteriol. 177:6791–6797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Bekker M, Kramer G, Hartog AF, Wagner MJ, de Koster CG, Hellingwerf KJ, de Mattos MJT. 2007. Changes in the redox state and composition of the quinone pool of Escherichia coli during aerobic batch-culture growth. Microbiology 153:1974–1980. 10.1099/mic.0.2007/006098-0 [DOI] [PubMed] [Google Scholar]
- 546.Yamada T, Furusawa C, Nagahisa K, Kashiwagi A, Yomo T, Shimizu H. 2007. Analysis of fluctuation in protein abundance without promoter regulation based on Escherichia coli continuous culture. Biosystems 90:614–622. 10.1016/j.biosystems.2007.02.001 [DOI] [PubMed] [Google Scholar]
- 547.Rutgers M, Teixeira de Mattos MJ, Postma PW, van Dam K. 1987. Establishment of the steady state in glucose-limited chemostat cultures of Klebsiella pneumoniae. J. Gen. Microbiol. 133:445–451 [DOI] [PubMed] [Google Scholar]
- 548.Franchini AG, Egli T. 2006. Global gene expression in Escherichia coli K-12 during short-term and long-term adaptation to glucose-limited continuous culture conditions. Microbiology 152:2111–2127. 10.1099/mic.0.28939-0 [DOI] [PubMed] [Google Scholar]
- 549.Sezonov G, Joseleau-Petit D, D'Ari R. 2007. Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 189:8746–8749. 10.1128/JB.01368-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Baev MV, Baev D, Radek AJ, Campbell JW. 2006. Growth of Escherichia coli MG1655 on LB medium: monitoring utilization of sugars, alcohols, and organic acids with transcriptional microarrays. Appl. Microbiol. Biotechnol. 71:310–316. 10.1007/s00253-006-0317-6 [DOI] [PubMed] [Google Scholar]
- 551.Prüß BM, Nelms JM, Park C, Wolfe AJ. 1994. Mutations in NADH:ubiquinone oxidoreductase of Escherichia coli affect growth on mixed amino acids. J. Bacteriol. 176:2143–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Beg QK, Vazquez A, Ernst J, de Menezes MA, Bar-Joseph Z, Barabási A-L, Oltvai ZN. 2007. Intracellular crowding defines the mode and sequence of substrate uptake by Escherichia coli and constrains its metabolic activity. Proc. Natl. Acad. Sci. U. S. A. 104:12663–12668. 10.1073/pnas.0609845104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Tritsch GL, Moore GE. 1962. Spontaneous decomposition of glutamine in cell culture media. Exp. Cell Res. 28:360–364. 10.1016/0014-4827(62)90290-2 [DOI] [PubMed] [Google Scholar]
- 554.Heeneman S, Deutz NE, Buurman WA. 1993. The concentrations of glutamine and ammonia in commercially available cell culture media. J. Immunol. Methods 166:85–91. 10.1016/0022-1759(93)90331-Z [DOI] [PubMed] [Google Scholar]
- 555.Mena FV, Baab PJ, Zielke CL, Huang Y, Zielke HR. 2005. Formation of extracellular glutamate from glutamine: exclusion of pyroglutamate as an intermediate. Brain Res. 1052:88–96. 10.1016/j.brainres.2005.06.014 [DOI] [PubMed] [Google Scholar]
- 556.Ozturk SS, Palsson BO. 1990. Chemical decomposition of glutamine in cell culture media: effect of media type, pH, and serum concentration. Biotechnol. Prog. 6:121–128. 10.1021/bp00002a005 [DOI] [PubMed] [Google Scholar]
- 557.Reitzer LJ. 1996. Sources of nitrogen and their utilization, p 380–390 In Neidhardt FC, Curtiss R, III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC [Google Scholar]
- 558.van West M, van Heeswijk WC, Westerhoff HV, Postma PW. 1996. Ntr: a signal transduction system with neural network characteristics? BioThermoKinetics Press, Amsterdam, the Netherlands [Google Scholar]
- 559.Magasanik B. 1970. Glucose effects: inducer exclusion and repression, p 189–221 In Beckwith JR, Zipser D. (ed), The lactose operon; Cold Spring Harbor Symposium. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 560.Müller PJ, von Frommannshausen B, Schutz H. 1981. Regulation of ammonia assimilation in ammonia-limited chemostat cultures of Escherichia coli ML 30: evidence of bistability. Z. Allg. Mikrobiol. 21:361–372. 10.1002/jobm.3630210503 [DOI] [PubMed] [Google Scholar]
- 561.Kumar R, Shimizu K. 2010. Metabolic regulation of Escherichia coli and its gdhA, glnL, gltB,D mutants under different carbon and nitrogen limitations in the continuous culture. Microb. Cell Fact. 9:8. 10.1186/1475-2859-9-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Yuan J, Bennett BD, Rabinowitz JD. 2008. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat. Protoc. 3:1328–1340. 10.1038/nprot.2008.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Bruggeman FJ. 2007. Systems biology: at last an integrative wet and dry biology! Biol. Theory 2:183–188 [Google Scholar]
- 564.Bruggeman FJ, Westerhoff HV. 2007. The nature of systems biology. Trends Microbiol. 15:45–50. 10.1016/j.tim.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 565.Simeonidis E, Murabito E, Smallbone K, Westerhoff HV. 2010. Why does yeast ferment? A flux balance analysis study. Biochem. Soc. Trans. 38:1225–1229 [DOI] [PubMed] [Google Scholar]
- 566.Orth JD, Thiele I, Palsson BØ 2010. What is flux balance analysis? Nat. Biotechnol. 28:245–248. 10.1038/nbt.1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Yuan J, Fowler WU, Kimball E, Lu W, Rabinowitz JD. 2006. Kinetic flux profiling of nitrogen assimilation in Escherichia coli. Nat. Chem. Biol. 2:529–530. 10.1038/nchembio816 [DOI] [PubMed] [Google Scholar]
- 568.Ma H, Boogerd FC, Goryanin I. 2009. Modelling nitrogen assimilation of Escherichia coli at low ammonium concentration. J. Biotechnol. 144:175–183. (Erratum, 150:207, 2010) [DOI] [PubMed] [Google Scholar]
- 569.Lodeiro A, Melgarejo A. 2008. Robustness in Escherichia coli glutamate and glutamine synthesis studied by a kinetic model. J. Biol. Phys. 34:91–106. 10.1007/s10867-008-9109-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Wang L, Lai L, Ouyang Q, Tang C. 2011. Flux balance analysis of ammonia assimilation network in E. coli predicts preferred regulation point. PLoS One 6:e16362. 10.1371/journal.pone.0016362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Kidd PB, Wingreen NS.2010. Modeling the role of covalent enzyme modification in Escherichia coli nitrogen metabolism. Phys. Biol. 7:016006. 10.1088/1478-3975/55/1/016006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Kurata H, Masaki K, Sumida Y, Iwasaki R. 2005. CADLIVE dynamic simulator: direct link of biochemical networks to dynamic models. Genome Res. 15:590–600. 10.1101/gr.3463705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Masaki K, Maeda K, Kurata H. 2012. Biological design principles of complex feedback modules in the E. coli ammonia assimilation system. Artif. Life 18:53–90 [DOI] [PubMed] [Google Scholar]
- 574.Kurata H, Inoue K, Maeda K, Masaki K, Shimokawa Y, Zhao Q. 2007. Extended CADLIVE: a novel graphical notation for design of biochemical network maps and computational pathway analysis. Nucleic Acids Res. 35:e134. 10.1093/nar/gkm769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Mutalik VK, Venkatesh KV. 2007. A theoretical steady state analysis indicates that induction of Escherichia coli glnALG operon can display all-or-none behavior. Biosystems 90:1–19. 10.1016/j.biosystems.2006.06.003 [DOI] [PubMed] [Google Scholar]
- 576.Brauer MJ, Yuan J, Bennett BD, Lu W, Kimball E, Botstein D, Rabinowitz JD. 2006. Conservation of the metabolomic response to starvation across two divergent microbes. Proc. Natl. Acad. Sci. U. S. A. 103:19302–19307. 10.1073/pnas.0609508103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Yan D, Lenz P, Hwa T. 2011. Overcoming fluctuation and leakage problems in the quantification of intracellular 2-oxoglutarate levels in Escherichia coli. Appl. Environ. Microbiol. 77:6763–6771. 10.1128/AEM.05257-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Tweeddale H, Notley-McRobb L, Ferenci T. 1998. Effect of slow growth on metabolism of Escherichia coli, as revealed by global metabolite pool (metabolome) analysis. J. Bacteriol. 180:5109–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.van der Werf MJ, Overkamp KM, Muilwijk B, Coulier L, Hankemeier T. 2007. Microbial metabolomics: toward a platform with full metabolome coverage. Anal. Biochem. 370:17–25. 10.1016/j.ab.2007.07.022 [DOI] [PubMed] [Google Scholar]
- 580.Mashego MR, Rumbold K, De Mey M, Vandamme E, Soetaert W, Heijnen JJ. 2007. Microbial metabolomics: past, present and future methodologies. Biotechnol. Lett. 29:1–16 [DOI] [PubMed] [Google Scholar]
- 581.Reaves ML, Rabinowitz JD. 2011. Metabolomics in systems microbiology. Curr. Opin. Biotechnol. 22:17–25. 10.1016/j.copbio.2010.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Bolten CJ, Kiefer P, Letisse F, Portais J-C, Wittmann C. 2007. Sampling for metabolome analysis of microorganisms. Anal. Chem. 79:3843–3849. 10.1021/ac0623888 [DOI] [PubMed] [Google Scholar]
- 583.Maharjan R, Ferenci T. 2003. Global metabolite analysis: the influence of extraction methodology on metabolome profiles of Escherichia coli. Anal. Biochem. 313:145–154. 10.1016/S0003-2697(02)00536-5 [DOI] [PubMed] [Google Scholar]
- 584.van Gulik WM. 2010. Fast sampling for quantitative microbial metabolomics. Curr. Opin. Biotechnol. 21:27–34. 10.1016/j.copbio.2010.01.008 [DOI] [PubMed] [Google Scholar]
- 585.Canelas AB, ten Pierick A, Ras C, Seifar RM, van Dam JC, van Gulik WM, Heijnen JJ. 2009. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal. Chem. 81:7379–7389. 10.1021/ac900999t [DOI] [PubMed] [Google Scholar]
- 586.Heijnen JJ. 2010. Impact of thermodynamic principles in systems biology. Adv. Biochem. Eng. Biotechnol. 121:139–162 [DOI] [PubMed] [Google Scholar]
- 587.Csonka LN, Hanson AD. 1991. Prokaryotic osmoregulation: genetics and physiology. Annu. Rev. Microbiol. 45:569–606. 10.1146/annurev.mi.45.100191.003033 [DOI] [PubMed] [Google Scholar]
- 588.Schmitz RA. 2000. Internal glutamine and glutamate pools in Klebsiella pneumoniae grown under different conditions of nitrogen availability. Curr. Microbiol. 41:357–362. 10.1007/s002840010149 [DOI] [PubMed] [Google Scholar]
- 589.Winder CL, Dunn WB, Schuler S, Broadhurst D, Jarvis R, Stephens GM, Goodacre R. 2008. Global metabolic profiling of Escherichia coli cultures: an evaluation of methods for quenching and extraction of intracellular metabolites. Anal. Chem. 80:2939–2948. 10.1021/ac7023409 [DOI] [PubMed] [Google Scholar]
- 590.Atkinson DE. 1968. The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 7:4030–4034 [DOI] [PubMed] [Google Scholar]
- 591.Chapman AG, Fall L, Atkinson DE. 1971. Adenylate energy charge in Escherichia coli during growth and starvation. J. Bacteriol. 108:1072–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Mayer EP, Smith OH, Fredricks WW, McKinney MA. 1975. The isolation and characterization of glutamine-requiring strains of Escherichia coli K12. Mol. Gen. Genet. 137:131–142. 10.1007/BF00341679 [DOI] [PubMed] [Google Scholar]
- 593.Berberich MA. 1972. A glutamate-dependent phenotype in E. coli K12: the result of two mutations. Biochem. Biophys. Res. Commun. 47:1498–1503. 10.1016/0006-291X(72)90242-2 [DOI] [PubMed] [Google Scholar]
- 594.Brenchley JE, Magasanik B. 1974. Mutants of Klebsiella aerogenes lacking glutamate dehydrogenase. J. Bacteriol. 117:544–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Brenchley JE, Prival MJ, Magasanik B. 1973. Regulation of the synthesis of enzymes responsible for glutamate formation in Klebsiella aerogenes. J. Biol. Chem. 248:6122–6128 [PubMed] [Google Scholar]
- 596.Elmerich C, Aubert JP. 1971. Synthesis of glutamate by a glutamine:2-oxo-glutarate amidotransferase (NADP oxidoreductase) in Bacillus megaterium. Biochem. Biophys. Res. Commun. 42:371–376. 10.1016/0006-291X(71)90380-9 [DOI] [PubMed] [Google Scholar]
- 597.Meers JL, Tempest DW, Brown CM. 1970. “Glutamine(amide):2-oxoglutarate amino transferase oxido-reductase (NADP),” an enzyme involved in the synthesis of glutamate by some bacteria. J. Gen. Microbiol. 64:187–194. 10.1099/00221287-64-2-187 [DOI] [PubMed] [Google Scholar]
- 598.Christian JHB, Waltho JA. 1961. The sodium and potassium content of non-halophilic bacteria in relation to salt tolerance. J. Gen. Microbiol. 25:97–102. 10.1099/00221287-25-1-97 [DOI] [PubMed] [Google Scholar]
- 599.Tempest DW, Meers JL, Brown CM. 1970. Influence of environment on the content and composition of microbial free amino acid pools. J. Gen. Microbiol. 64:171–185. 10.1099/00221287-64-2-171 [DOI] [PubMed] [Google Scholar]
- 600.Csonka LN, Ikeda TP, Fletcher SA, Kustu S. 1994. The accumulation of glutamate is necessary for optimal growth of Salmonella typhimurium in media of high osmolality but not induction of the proU operon. J. Bacteriol. 176:6324–6333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Booth IR, Higgins CF. 1990. Enteric bacteria and osmotic stress: intracellular potassium glutamate as a secondary signal of osmotic stress? FEMS Microbiol. Rev. 6:239–246 [DOI] [PubMed] [Google Scholar]
- 602.Helling RB. 1994. Why does Escherichia coli have two primary pathways for synthesis of glutamate? J. Bacteriol. 176:4664–4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Spanaki C, Plaitakis A. 2012. The role of glutamate dehydrogenase in mammalian ammonia metabolism. Neurotox. Res. 21:117–127. 10.1007/s12640-011-9285-4 [DOI] [PubMed] [Google Scholar]
- 604.Boogerd FC, Bruggeman FJ, Hofmeyr J-HS, Westerhoff HV. 2007. Towards philosophical foundations of systems biology: introduction, p 3–19 In Boogerd FC, Bruggeman FJ, Hofmeyr J-HS, Westerhoff HV. (ed), Systems biology (philosophical foundations), 1st ed. Elsevier, Amsterdam, the Netherlands [Google Scholar]
- 605.Penfound T, Foster JW. 1996. Biosynthesis and recycling of NAD, p 721–730 In Neidhardt FC, Curtiss R, III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC [Google Scholar]
- 606.Andersen KB, Von Meyenburg K. 1977. Charges of nicotinamide adenine nucleotides and adenylate energy charge as regulatory parameters of the metabolism in Escherichia coli. J. Biol. Chem. 252:4151–4156 [PubMed] [Google Scholar]
- 607.Marcus M, Halpern YS. 1969. The metabolic pathway of glutamate in Escherichia coli K-12. Biochim. Biophys. Acta 177:314–320. 10.1016/0304-4165(69)90141-X [DOI] [PubMed] [Google Scholar]
- 608.Tao H, Bausch C, Richmond C, Blattner FR, Conway T. 1999. Functional genomics: expression analysis of Escherichia coli growing on minimal and rich media. J. Bacteriol. 181:6425–6440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Roe AJ, McLaggan D, Davidson I, O'Byrne C, Booth IR. 1998. Perturbation of anion balance during inhibition of growth of Escherichia coli by weak acids. J. Bacteriol. 180:767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Leirmo S, Harrison C, Cayley DS, Burgess RR, Record MT. 1987. Replacement of potassium chloride by potassium glutamate dramatically enhances protein-DNA interactions in vitro. Biochemistry 26:2095–2101. 10.1021/bi00382a006 [DOI] [PubMed] [Google Scholar]
- 611.Gralla JD, Vargas DR. 2006. Potassium glutamate as a transcriptional inhibitor during bacterial osmoregulation. EMBO J. 25:1515–1521. 10.1038/sj.emboj.7601041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Janes BK, Pomposiello PJ, Perez-Matos A, Najarian DJ, Goss TJ, Bender RA. 2001. Growth inhibition caused by overexpression of the structural gene for glutamate dehydrogenase (gdhA) from Klebsiella aerogenes. J. Bacteriol. 183:2709–2714. 10.1128/JB.183.8.2709-2714.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Struhl K, Magasanik B. 1976. Ammonia-sensitive mutant of Klebsiella aerogenes. J. Bacteriol. 126:739–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Fischer EH, Krebs EG. 1955. Conversion of phosphorylase b to phosphorylase a in muscle extracts. J. Biol. Chem. 216:121–132 [PubMed] [Google Scholar]
- 615.Krebs EG, Beavo JA. 1979. Phosphorylation-dephosphorylation of enzymes. Annu. Rev. Biochem. 48:923–959. 10.1146/annurev.bi.48.070179.004423 [DOI] [PubMed] [Google Scholar]
- 616.LaPorte DC, Koshland DE. 1982. A protein with kinase and phosphatase activities involved in regulation of tricarboxylic acid cycle. Nature 300:458–460. 10.1038/300458a0 [DOI] [PubMed] [Google Scholar]
- 617.LaPorte DC, Koshland DE., Jr 1983. Phosphorylation of isocitrate dehydrogenase as a demonstration of enhanced sensitivity in covalent regulation. Nature 305:286–290. 10.1038/305286a0 [DOI] [PubMed] [Google Scholar]
- 618.Zheng J, Jia Z. 2010. Structure of the bifunctional isocitrate dehydrogenase kinase/phosphatase. Nature 465:961–965. 10.1038/nature09088 [DOI] [PubMed] [Google Scholar]
- 619.Huang CY, Ferrell JEJ. 1996. Ultrasensitivity in the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U. S. A. 93:10078–10083. 10.1073/pnas.93.19.10078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Koshland DEJ, Goldbeter A, Stock JB. 1982. Amplification and adaptation in regulatory and sensory systems. Science 217:220–225. 10.1126/science.7089556 [DOI] [PubMed] [Google Scholar]
- 621.Stadtman ER, Chock PB. 1978. Interconvertible enzyme cascades in metabolic regulation. Curr. Top. Cell. Regul. 13:53–95 [DOI] [PubMed] [Google Scholar]
- 622.Szedlacsek SE, Cárdenas ML, Cornish-Bowden A. 1992. Response coefficients of interconvertible enzyme cascades towards effectors that act on one or both modifier enzymes. Eur. J. Biochem. 204:807–813. 10.1111/j.1432-1033.1992.tb16699.x [DOI] [PubMed] [Google Scholar]
- 623.Brown GC, Hoek JB, Kholodenko BN. 1997. Why do protein kinase cascades have more than one level? Trends Biochem. Sci. 22:288. 10.1016/S0968-0004(97)82216-5 [DOI] [PubMed] [Google Scholar]
- 624.Kahn D, Westerhoff HV. 1991. Control theory of regulatory cascades. J. Theor. Biol. 153:255–285. 10.1016/S0022-5193(05)80426-6 [DOI] [PubMed] [Google Scholar]
- 625.Kholodenko BN, Hoek JB, Westerhoff HV, Brown GC. 1997. Quantification of information transfer via cellular signal transduction pathways. FEBS Lett. 414:430–434. 10.1016/S0014-5793(97)01018-1 [DOI] [PubMed] [Google Scholar]
- 626.Goldbeter A, Koshland DE. 1982. Sensitivity amplification in biochemical systems. Q. Rev. Biophys. 15:555–591. 10.1017/S0033583500003449 [DOI] [PubMed] [Google Scholar]
- 627.Small JR, Fell DA. 1990. Covalent modification and metabolic control analysis. Modification to the theorems and their application to metabolic systems containing covalently modifiable enzymes. Eur. J. Biochem. 191:405–411 [DOI] [PubMed] [Google Scholar]
- 628.Westerhoff HV, Aon MA, van Dam K, Cortassa S, Kahn D, van Workum M. 1990. Dynamical and hierarchical coupling. Biochim. Biophys. Acta 1018:142–146. 10.1016/0005-2728(90)90235-V [DOI] [Google Scholar]
- 629.Cárdenas ML, Cornish-Bowden A. 1989. Characteristics necessary for an interconvertible enzyme cascade to generate a highly sensitive response to an effector. Biochem. J. 257:339–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.De Vos D, Bruggeman FJ, Westerhoff HV, Bakker BM. 2011. How molecular competition influences fluxes in gene expression networks. PLoS One 6:e28494. 10.1371/journal.pone.0028494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Cornish-Bowden A, Szedlacsek SE. 1990. Very large response coefficients in interconvertible enzyme cascades. Biomed. Biochim. Acta 49:829–837 [PubMed] [Google Scholar]
- 632.Goldbeter A, Koshland DEJ. 1981. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. U. S. A. 78:6840–6844. 10.1073/pnas.78.11.6840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Goldbeter A, Koshland DEJ. 1984. Ultrasensitivity in biochemical systems controlled by covalent modification. Interplay between zero-order and multistep effects. J. Biol. Chem. 259:14441–14447 [PubMed] [Google Scholar]
- 634.Cornish-Bowden A. 1995. Metabolic control analysis in theory and practice. Adv. Mol. Cell. Biol. 11:21–64. 10.1016/S1569-2558(08)60247-7 [DOI] [Google Scholar]
- 635.Blüthgen N, Bruggeman FJ, Legewie S, Herzel H, Westerhoff HV, Kholodenko BN. 2006. Effects of sequestration on signal transduction cascades. FEBS J. 273:895–906. 10.1111/j.1742-4658.2006.05105.x [DOI] [PubMed] [Google Scholar]
- 636.Ferrell JEJ. 1996. Tripping the switch fantastic: how a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem. Sci. 21:460–466. 10.1016/S0968-0004(96)20026-X [DOI] [PubMed] [Google Scholar]
- 637.LaPorte DC, Walsh K, Koshland DEJ. 1984. The branch point effect. Ultrasensitivity and subsensitivity to metabolic control. J. Biol. Chem. 259:14068–14075 [PubMed] [Google Scholar]
- 638.Mutalik VK, Shah P, Venkatesh KV. 2003. Allosteric interactions and bifunctionality make the response of glutamine synthetase cascade system of Escherichia coli robust and ultrasensitive. J. Biol. Chem. 278:26327–26332. 10.1074/jbc.M300129200 [DOI] [PubMed] [Google Scholar]
- 639.Berg OG, Paulsson J, Ehrenberg M. 2000. Fluctuations and quality of control in biological cells: zero-order ultrasensitivity reinvestigated. Biophys. J. 79:1228–1236. 10.1016/S0006-3495(00)76377-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 640.Goyal S, Yuan J, Chen T, Rabinowitz JD, Wingreen NS. 2010. Achieving optimal growth through product feedback inhibition in metabolism. PLoS Comput. Biol. 6:e1000802. 10.1371/journal.pcbi.1000802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Hornberg JJ, Binder B, Bruggeman FJ, Schoeberl B, Heinrich R, Westerhoff HV. 2005. Control of MAPK signalling: from complexity to what really matters. Oncogene 24:5533–5542. 10.1038/sj.onc.1208817 [DOI] [PubMed] [Google Scholar]
- 642.Westerhoff HV. 2008. Signalling control strength. J. Theor. Biol. 252:555–567. 10.1016/j.jtbi.2007.11.035 [DOI] [PubMed] [Google Scholar]
- 643.Rhee SG, Park R, Chock PB, Stadtman ER. 1978. Allosteric regulation of monocyclic interconvertible enzyme cascade systems: use of Escherichia coli glutamine synthetase as an experimental model. Proc. Natl. Acad. Sci. U. S. A. 75:3138–3142. 10.1073/pnas.75.7.3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Jiang P, Ninfa AJ. 2011. A source of ultrasensitivity in the glutamine response of the bicyclic cascade system controlling glutamine synthetase adenylylation state and activity in Escherichia coli. Biochemistry 50:10929–10940. 10.1021/bi201410x [DOI] [PubMed] [Google Scholar]
- 645.Ventura AC, Jiang P, Van Wassenhove L, Del Vecchio D, Merajver SD, Ninfa AJ. 2010. Signaling properties of a covalent modification cycle are altered by a downstream target. Proc. Natl. Acad. Sci. U. S. A. 107:10032–10037. 10.1073/pnas.0913815107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Jiang P, Ventura AC, Sontag ED, Merajver SD, Ninfa AJ, Del Vecchio D. 2011. Load-induced modulation of signal transduction networks. Sci. Signal. 4:ra67. 10.1126/scisignal.2002152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Straube R. 2012. Comment on “Load-induced modulation of signal transduction networks”: reconciling ultrasensitivity with bifunctionality? Sci. Signal. 5:lc1. 10.1126/scisignal.2002699 [DOI] [PubMed] [Google Scholar]
- 648.Jiang P, Ventura AC, Sontag ED, Merajver SAD, Ninfa AJ, Del Vecchio D. 2012. Reply to “Comment on ‘Load-induced modulation of signal transduction networks': reconciling ultrasensitivity with bifunctionality?” Sci. Signal. 5:1c2. (Reply.) 10.1126/scisignal.2002699 [DOI] [PubMed] [Google Scholar]
- 649.Molenaar D, Kholodenko BN, van Heeswijk WC, Westerhoff HV. 1995. Dynamic aspects of cascade-type metabolic regulation. J. Biol. Syst. 3:187–196. 10.1142/S0218339095000186 [DOI] [Google Scholar]
- 650.Goldbeter A, Koshland DEJ. 1987. Energy expenditure in the control of biochemical systems by covalent modification. J. Biol. Chem. 262:4460–4471 [PubMed] [Google Scholar]
- 651.Tempest DW, Neijssel OM. 1987. Growth yield and energy distribution, p 797–806 In Neidhardt FC, Ingraham JL, Low KB, Magasanik B, Schaechter M, Umbarger HE. (ed), Escherichia coli and Salmonella typhimurium: cellular and molecular biology. ASM Press, Washington, DC [Google Scholar]
- 652.Kim M, Zhang Z, Okano H, Yan D, Groisman A, Hwa T. 2012. Need-based activation of ammonium uptake in Escherichia coli. Mol. Syst. Biol. 8:616. 10.1038/msb.2012.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Buurman ET, Teixeira de Mattos MJ, Neijssel OM. 1991. Futile cycling of ammonium ions via the high affinity potassium uptake system (Kdp) of Escherichia coli. Arch. Microbiol. 155:391–395 [DOI] [PubMed] [Google Scholar]
- 654.Buurman ET, Pennock J, Tempest DW, Teixeira de Mattos MJ, Neijssel OM. 1989. Replacement of potassium ions by ammonium ions in different micro-organisms grown in potassium-limited chemostat culture. Arch. Microbiol. 152:58–63. 10.1007/BF00447012 [DOI] [PubMed] [Google Scholar]
- 655.Boogerd FC, Bruggeman FJ, Richardson RC. April 2013. Mechanistic explanations and models in molecular systems biology. Found. Sci. 10.1007/s10699-012-9302-y [DOI] [Google Scholar]
- 656.Hellingwerf KJ, Postma PW, Tommassen J, Westerhoff HV. 1995. Signal transduction in bacteria: phospho-neural network(s) in Escherichia coli? FEMS Microbiol. Rev. 16:309–321. 10.1111/j.1574-6976.1995.tb00178.x [DOI] [PubMed] [Google Scholar]
- 657.Bruggeman FJ, van Heeswijk WC, Boogerd FC, Westerhoff HV. 2000. Macromolecular intelligence in microorganisms. Biol. Chem. 381:965–972 [DOI] [PubMed] [Google Scholar]
- 658.Hoffer SM, Westerhoff HV, Hellingwerf KJ, Postma PW, Tommassen J. 2001. Autoamplification of a two-component regulatory system results in “learning” behavior. J. Bacteriol. 183:4914–4917. 10.1128/JB.183.16.4914-4917.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Westerhoff HV, van Heeswijk WC, Kholodenko BN, Snoep JL. 1996. BioComplexity. The curly line from dead molecules to live cells, p 323–331 In Kholodenko BN, Snoep JL, Sluse F, Wijker JE. (ed), Biothermokinetics of the living cell. BioThermoKinetics Press, Amsterdam, the Netherlands [Google Scholar]
- 660.Reijenga KA, Bakker BM, van der Weijden CC, Westerhoff HV. 2005. Training of yeast cell dynamics. FEBS J. 272:1616–1624. 10.1111/j.1742-4658.2005.04582.x [DOI] [PubMed] [Google Scholar]
- 661.Bergter F, Roth M. 1977. Bistability of pyruvate production by E. coli ML30 in continuous culture. Z. Allg. Mikrobiol. 17:3–6. 10.1002/jobm.3630170102 [DOI] [PubMed] [Google Scholar]
- 662.Bergter F, Schumann H, Koburger M. 1977. Dependence of the specific growth rate of Escherichia coli ML30 on the ammonia concentration. Z. Allg. Mikrobiol. 17:183–189. 10.1002/jobm.3630170303 [DOI] [PubMed] [Google Scholar]
- 663.Müller PJ, Bergter F. 1977. Transient behavior of the ammonium-limited chemostatic cultures of Escherichia coli ML 30. Z. Allg. Mikrobiol. 17:131–137. 10.1002/jobm.3630170206 [DOI] [PubMed] [Google Scholar]
- 664.Müller PJ, Ivanova II, Bergter F. 1977. Bistability in the activity of glutamine synthetase in ammonium-limiting chemostat cultures of Escherichia coli ML 30. Z. Allg. Mikrobiol. 17:221–225. 10.1002/jobm.3630170307 [DOI] [PubMed] [Google Scholar]
- 665.Westerhoff HV, Verma M, Nardelli M, Adamczyk M, van Eunen K, Simeonidis E, Bakker BM. 2010. Systems biochemistry in practice: experimenting with modelling and understanding, with regulation and control. Biochem. Soc. Trans. 38:1189–1196. 10.1042/BST0381189 [DOI] [PubMed] [Google Scholar]
- 666.Kolodkin AN, Boogerd FC, Bruggeman FJ, Westerhoff HV. 2011. Modeling approaches in systems biology, including silicon cell models, 1st ed. Wiley-Blackwell, Hoboken, NJ [Google Scholar]
- 667.Koleva S, Haidt J. 2012. Let's use Einstein's safety razor, not Occam's Swiss army knife or Occam's chainsaw. Psychol. Inq. 23:175–178. 10.1080/1047840X.2012.667678 [DOI] [Google Scholar]
- 668.Wimsatt WC. 2006. Reductionism and its heuristics: making methodological reductionism honest. Synthese 151:445–475. 10.1007/s11229-006-9017-0 [DOI] [Google Scholar]
- 669.Boogerd FC, Bruggeman F, Richardson R, Achim S, Westerhoff HV. 2005. Emergence and its place in nature: a case study of biochemical networks. Synthese 145:131–164. 10.1007/s11229-004-4421-9 [DOI] [Google Scholar]
- 670.Bruggeman FJ, Westerhoff HV, Boogerd FC. 2002. BioComplexity: a pluralist research strategy is necessary for a mechanistic explanation of the “live” state. Philos. Psychol. 15:411–440. 10.1080/0951508021000041996 [DOI] [Google Scholar]
- 671.Boogerd FC, Bruggeman F, Jonker C, de Jong HL, Tamminga A, Treur J, Westerhoff H, Wijngaards W. 2002. Inter-level relations in computer science, biology, and psychology. Philos. Psychol. 15:463–471. 10.1080/0951508021000042012 [DOI] [Google Scholar]
- 672.Boogerd FC, Bruggeman FJ, Hofmeyr J-HS, Westerhoff HV. 2007. Afterthoughts as foundations for systems biology, p 321–337 In Boogerd FC, Bruggeman FJ, Hofmeyr J-HS, Westerhoff HV. (ed), Systems biology (philosophical foundations), 1st ed. Elsevier, Amsterdam, the Netherlands [Google Scholar]
- 673.Westerhoff HV, Kell DB. 2007. The methodologies of systems biology, p 23–70 In Boogerd FC, Bruggeman FJ, Hofmeyr J-HS, Westerhoff HV. (ed), Systems biology (philosophical foundations), 1st ed. Elsevier, Amsterdam, the Netherlands [Google Scholar]
- 674.Cornish-Bowden A, Cárdenas ML. 2005. Systems biology may work when we learn to understand the parts in terms of the whole. Biochem. Soc. Trans. 33:516–519. 10.1042/BST0330516 [DOI] [PubMed] [Google Scholar]
- 675.Cornish-Bowden A. 2011. Systems biology: how far has it come? Biochemist 2011:16–18 [Google Scholar]
- 676.Inwood WB, Hall JA, Kim KS, Demirkhanyan L, Wemmer D, Zgurskaya H, Kustu S. 2009. Epistatic effects of the protease/chaperone HflB on some damaged forms of the Escherichia coli ammonium channel AmtB. Genetics 183:1327–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Woolfolk CA, Shapiro B, Stadtman ER. 1966. Regulation of glutamine synthetase: I. Purification and properties of glutamine synthetase from Escherichia coli. Arch. Biochem. Biophys. 116:177–192 [DOI] [PubMed] [Google Scholar]