

Abstract
From 1 January 2009 to 31 May 2013, 15 287 respiratory specimens submitted to the Clinical Virology Laboratory at the Children’s Hospital Colorado were tested for human coronavirus RNA by reverse transcription-PCR. Human coronaviruses HKU1, OC43, 229E and NL63 co-circulated during each of the respiratory seasons but with significant year-to-year variability, and cumulatively accounted for 7.4–15.6 % of all samples tested during the months of peak activity. A total of 79 (0.5 % prevalence) specimens were positive for human betacoronavirus HKU1 RNA. Genotypes HKU1 A and B were both isolated from clinical specimens and propagated on primary human tracheal–bronchial epithelial cells cultured at the air–liquid interface and were neutralized in vitro by human intravenous immunoglobulin and by polyclonal rabbit antibodies to the spike glycoprotein of HKU1. Phylogenetic analysis of the deduced amino acid sequences of seven full-length genomes of Colorado HKU1 viruses and the spike glycoproteins from four additional HKU1 viruses from Colorado and three from Brazil demonstrated remarkable conservation of these sequences with genotypes circulating in Hong Kong and France. Within genotype A, all but one of the Colorado HKU1 sequences formed a unique subclade defined by three amino acid substitutions (W197F, F613Y and S752F) in the spike glycoprotein and exhibited a unique signature in the acidic tandem repeat in the N-terminal region of the nsp3 subdomain. Elucidating the function of and mechanisms responsible for the formation of these varying tandem repeats will increase our understanding of the replication process and pathogenicity of HKU1 and potentially of other coronaviruses.
Introduction
There are currently six known human coronavirus (hCoV) species: alphacoronaviruses hCoV-229E and hCoV-NL63, and betacoronaviruses group a hCoV-OC43 and hCoV-HKU1, betacoronavirus group b severe acute respiratory syndrome (SARS)-CoV, and the Middle East Respiratory Syndrome virus (MERS-CoV) in betacoronavirus group c. The recent discovery of MERS-CoV in fatal cases of respiratory disease (Assiri et al., 2013; Zaki et al., 2012) and the 2003 SARS pandemic (Drosten et al., 2003; Ksiazek et al., 2003) demonstrate the importance of emerging CoVs in severe human respiratory diseases and the potential for the emergence of new, virulent hCoVs from wildlife reservoirs. The prototype HKU1 strain was discovered in 2005 by reverse transcription (RT)-PCR using conserved CoV primers to a highly conserved region of the CoV 1b gene by screening respiratory samples from adult pneumonia patients in Hong Kong who were negative for SARS-CoV (Woo et al., 2005b). HKU1 has been challenging to study because HKU1 viruses could not be isolated from clinical specimens or propagated in continuous cell lines. Isolation of HKU1 viruses from clinical specimens was accomplished recently by using primary, differentiated human tracheal bronchial epithelial (HTBE) cells cultured at an air–liquid interface (Dijkman et al., 2013; Pyrc et al., 2010). This highly differentiated, primary human respiratory cell-culture system will facilitate studies of the replication, pathogenesis and phylogeny of HKU1 betacoronaviruses and other fastidious human respiratory viruses.
All of the 24 full-length genome sequences of HKU1 that are available in GenBank to date are from primary clinical specimens collected in Hong Kong or France (Pyrc et al., 2010; Woo et al., 2005a, b, 2006). Analysis of these genome sequences identified two distinct genotypes in the HKU1 lineage (A and B), and one genotype (C) that was a recombinant between the A and B genotypes (Woo et al., 2006). To date, there are no full-length HKU1 genome sequences from the western hemisphere. Here, we report the epidemiology, disease association and phylogeny of HKU1 viruses during a 4.5-year period (2009–2013) at the Children’s Hospital Colorado. We used primary HTBE cells to isolate and propagate numerous isolates of HKU1, including both genotypes A and B, at physiologically relevant temperatures (34 and 37 °C), and demonstrated that HKU1 infection of these cells could be neutralized by pooled purified human IgG from healthy donors (intravenous immunoglobulin, IVIG) and by polyclonal rabbit antibody to the HKU1 spike glycoprotein. Sequencing of HKU1 RNA from 15 of our clinical specimens yielded seven full-length genomes. Analysis of these HKU1 genomes and the sequences of seven additional HKU1 spike genes from Colorado and Brazil revealed remarkable conservation of sequences of circulating HKU1 viruses in both the eastern and western hemispheres and identified a subclade within genotype A that has been detected so far only in the USA. Analysis of the first HKU1 genomes from the western hemisphere broadens our understanding of HKU1 CoVs circulating globally.
Results
Epidemiology of hCoVs
To determine and compare the prevalence and seasonal variation of endemic hCoV-HKU1, -OC43, -NL63 and -229E infections in our paediatric population, RNA from 15 287 clinical respiratory specimens collected over a 4.5-year period was analysed by RT-PCR (Fig. 1a). The most common CoV infections were with NL63 and OC43 (1.7 and 1.5 % of all samples submitted, respectively), followed by HKU1 (0.5 %) and finally 299E infections (0.5 %). As noted previously (Monto & Rhodes, 1977), when the prevalence of a particular CoV was high in one year, its prevalence was reduced in one or more of the subsequent years, suggesting the possibility of immune pressure within the community. All four hCoVs, HKU1, OC43, 229E and NL63, co-circulated during each of the respiratory seasons with peak circulation over a 2-month period between the months of December and March (Fig. 1a). In total, 4.3 % of all submitted samples were positive for a CoV. During the peak 2-month period, CoVs accounted for 7.4–15.6 % of all samples tested.
Fig. 1.

Epidemiology of all endemic human CoVs (a) and HKU1 (b). (a) Percentage of respiratory samples (n = 15 287) submitted to the Children’s Hospital Colorado that were positive for the four endemic hCoVs, HKU1 (red), OC43 (orange), 229E (green) and NL63 (blue), and the combined total hCoVs (dashed purple) from 2009 to 2013. (b) Number and percentage of respiratory samples positive for HKU1 RNA from 2009 to 2013.
Epidemiology, clinical characteristics and disease associations of HKU1
Because there are no full-length genomes of HKU1 from the Americas in GenBank, we focused our studies on HKU1. During the 4.5-year course of this study, 79 of the 15 287 respiratory specimens submitted for viral testing were positive for HKU1 viral RNA by RT-PCR, an overall prevalence of 0.5 %. There was considerable variation in the prevalence of HKU1 infection in the paediatric population during the course of the study (Fig. 1b). Notably, during the 2009–2010 winter season, there were more HKU1-positive specimens than in subsequent years. From December 2009 to January 2010, 25 specimens were positive for HKU1, which accounted for 32 % of all of the positive HKU1 specimens during the 53-month study period. The epidemiology and disease associations of the HKU1 viruses in paediatric patients in Brazil have been reported previously (Góes et al., 2011).
Fifty-nine per cent of the Colorado respiratory specimens positive for HKU1 viral RNA during this study period were also positive for one or more additional viruses. The most common co-infections were with human rhinoviruses (24 %), respiratory syncytial virus (20 %), adenoviruses (11 %) and hCoV-NL63 (7 %). The clinical characteristics of the patients from whom the sequenced HKU1 isolates (Table 1) were obtained are shown in Table 2. From the total cohort of children (n = 79) positive for HKU1, 39 % were under 12 months of age and 58 % were under 2 years of age. Sixty-six per cent of the HKU1-positive patients were admitted to the hospital, and 54 % had an underlying medical condition. To characterize the clinical syndromes associated with HKU1, we analysed the primary discharge diagnoses of the patients with HKU1-positive respiratory specimens. The most common discharge diagnoses were upper respiratory tract infection (42 %), bronchiolitis (16 %), pneumonia (12 %), fever and neutropenia (8 %) and seizures (4 %). The most common symptoms in the HKU1-positive patients were fever (50 %), cough (47 %), hypoxia (22 %), seizures (5 %) and diarrhoea (5 %).
Table 1. Sequencing technology, sample names and status of the HKU1 viruses in this study.
| Human betacoronavirus lineage a sample name and source of specimen* | Abbreviated sample name | GenBank accession no. | Sequencing technology | Sequencing status |
| HKU1/USA/1/2005† | HKU1-1 | KF686338 | Illumina and Sanger | Standard draft |
| HKU1/USA/3/2009† | HKU1-3 | KF686339 | Illumina and Sanger | Standard draft |
| HKU1/USA/4/2005† | HKU1-4 | KF430196 | Illumina and Sanger | Standard draft |
| HKU1/USA/5/2009† | HKU1-5 | KF686340 | Sanger | Complete genome |
| HKU1/USA/7/2010† | HKU1-7 | KF430202 | Illumina and Sanger | Standard draft |
| HKU1/USA/10/2010† | HKU1-10 | KF686341 | Sanger | Complete genome |
| HKU1/USA/11/2009† | HKU1-11 | KF686342 | Sanger | Complete genome |
| HKU1/USA/12/2010† | HKU1-12 | KF686346 | Illumina and Sanger | Complete genome |
| HKU1/USA/13/2010† | HKU1-13 | KF686343 | Sanger | Complete genome |
| HKU1/USA/14/2009† | HKU1-14 | KF430199 | Illumina and Sanger | Standard draft |
| HKU1/USA/15/2009† | HKU1-15 | KF686344 | Sanger | Complete genome |
| HKU1/USA/16/2010† | HKU1-16 | KF430200 | Illumina and Sanger | Standard draft |
| HKU1/USA/18/2010† | HKU1-18 | KF430201 | Illumina and Sanger | Complete genome |
| HKU1/USA/20/2010† | HKU1-20 | KF686345 | Illumina and Sanger | Standard draft |
| HKU1/BRA/21/2006‡ | HKU1-21 | KF430198 | Illumina | HE, spike and ORF4 |
| HKU1/BRA/22/2007‡ | HKU1-22 | KF430197 | Illumina | HE, spike and ORF5 |
| HKU1/BRA/23/2006‡ | HKU1-23 | KF430203 | Illumina | HE, spike and ORF6 |
USA indicates Denver, CO, USA, and BRA indicates São Paulo, Brazil.
Sequenced directly from human clinical respiratory specimen.
Sequenced after passage of clinical specimen once on HTBE cells.
Table 2. Clinical and demographic characteristics of patients from the sequenced and cultured Colorado (USA) and Brazilian (BRA) HKU1 viruses.
| Sample | Sample date | Age | Sex | Underlying medical conditions | Clinical presentation | Discharge diagnosis | HKU1 Genotype | Full genome sequence | Spike genome sequence | Cultured on HTBEC |
| HKU1-1/USA | 02/18/2005 | 7 years | F | Congenital myopathy | Fever, cough, tachypnea | Pneumonia | B | Yes | ||
| HKU1-23/BRA | 07/20/2006 | 4 years | M | Unknown | Unknown | Pneumonia | A | Yes | Yes | |
| HKU1-21/BRA | 09/04/2006 | 3 years | M | Unknown | Unknown | Unknown | A | Yes | Yes | |
| HKU1-22/BRA | 11/22/2007 | 20 days | F | Unknown | Fever | Unknown | B | Yes | Yes | |
| HKU1-5/USA | 11/28/2009 | 22 months | F | ASD/VSD repair | Cough, hypoxia, tachypnea | Croup | A | Yes | Yes | |
| HKU1-11/USA | 12/13/2009 | 5 months | M | None | Cough, fever, diarrhoea, haematemesis | Viral gastroenteritis | A | Yes | Yes | |
| HKU1-14/USA | 12/28/2009 | 1 month | M | None | Congestion, fever | Viral URTI | A | Yes | ||
| HKU1-15/USA | 12/28/2009 | 12 years | M | CP, seizure disorder, CLD | Fever, hypoxia, tachypnea | Pneumonia | A | Yes | Yes | |
| HKU1-7/USA | 01/03/2010 | Unknown | Unknown | Unknown | Unknown | Unknown | A | Yes | Yes | |
| HKU1-10/USA | 01/06/2010 | 21 years | F | Heart transplant | Fever, chest pain, cough, tachypnea | DVT+viral URTI | A | Yes | Yes | Yes |
| HKU1-13/USA | 01/08/2010 | 2 years | M | CHD | Increased O2 requirement | Hypoxaemia | A | Yes | Yes | |
| HKU1-16/USA | 01/08/2010 | 18 days | F | None | Cough, congestion | Viral URTI | A | Yes | ||
| HKU1-12/USA | 01/09/2010 | Unknown | Unknown | None | Unknown | Unknown | A | Yes | Yes | Yes |
| HKU1-21/USA | 01/19/2010 | 6 years | M | ALL/BMT | Unknown | Unknown | Unknown | Yes | ||
| HKU1-18/USA | 01/22/2010 | 2 months | F | None | Fever, rhinorrhea | Viral URTI | A | Yes | Yes | Yes |
ASD, atrial septal defect; VSD, ventricular septal defect; URTI, upper respiratory tract infection; DVT, deep venous thrombus; CHD, congenital heart disease; CP, cerebral palsy; CLD, chronic lung disease; ALL, acute lymphoid leukaemia; BMT, bone marrow transplantation.
Genome sequences, phylogenetic analysis and genotypes
Nearly full-length genome sequences were obtained for seven of the 15 Colorado clinical samples submitted for sequencing. All of the samples selected were from the 2009–2010 respiratory season, which corresponded to a mini-outbreak of HKU1 in our community. Poor template quality precluded full-length analysis of the other samples. The sequences ranged from 29 695 to 29 983 nt, lacking only short sequences at the 3′ and 5′ termini (the initial HKU1 isolate was 29 926 nt; Woo et al., 2005a).
All seven full-length HKU1 genomes had the same organization as the 22 full-length HKU1 sequences from Hong Kong and the single sequence from France. As described for the Hong Kong HKU1 specimens (Woo et al., 2006), in our seven full-length genomes, the putative transcription regulatory sequence 5′-AAUCUAAC-3′ was found at the 3′ end of the leader sequence and preceding each of the translated ORFs.
A phylogenetic tree comparing the nucleotide sequences of the seven full-length genomes of the Colorado HKU1 strains combined with the 22 Asian and one French full-length HKU1 genomes is shown in Fig. 2. These 30 HKU1 strains comprised three genotypes, designated genotypes A (21 strains), B (three strains) and C (six strains). All seven of the full-length Colorado genomes were in HKU1 genotype A, and these had remarkable amino acid identity to each other and to the other sequenced genotype A HKU1viruses from Asia and France. Bootscan analysis of the Colorado specimens compared with other genotype A HKU1 specimens did not reveal any potential recombination sites among our samples (data not shown). VISTA analysis using the prototypical HKU1 isolate from Hong Kong in 2005 as a reference showed that the greatest areas of difference between the genotypes were between 3.0 and 3.5 kb (nsp3 in ORF1ab) and between 22.0 and 27.0 kb [which includes the haemagglutinin–esterase (HE) and spike genes] (Fig. S1, available in the online Supplementary Material).
Fig. 2.

Phylogenetic analysis of all known full genomes of HKU1 viruses isolated in different years from Denver, CO, USA, Hong Kong (N) and France (Caen1). One hundred bootstrap replicates were used for reconstruction of the phylogenetic trees, and nodes supported by a bootstrap value greater than 70 % are indicated. The viral sequences fell into three genotypes: A (blue lines), B (green lines) and C (purple lines). A subset of the Colorado HKU1 viruses formed a subclade within genotype A (indicated by the red box). Bar, nucleotide substitutions per site.
A key genetic signature for different strains of HKU1 is a variable number of a 10 aa (NDDEDVVTGD) acidic tandem repeat (ATR) in the N-terminal region of the nsp3 subdomain of gene 1a upstream of PL1pro (Woo et al., 2006). In six of the seven Colorado HKU1 viruses sequenced, the numbers of the first variable tandem repeat ranged from 8 to 17 and a truncated unique variant, NDDED, was found after the conserved variable number of tandem repeats. This signature was unique among the Colorado specimens (Table 3). To determine whether the tandem repeat sequences in the nsp3 protein had any influence on the designation of clades or genotypes based on the full-length genomes, we reconstructed a second phylogenetic tree based on the full genome sequences in which the variable number of tandem repeats region of nsp3 was deleted. No difference in topology between the trees that included and excluded this region was noted (data not shown).
Table 3. Comparison of amino acid sequences of ATRs in the N-terminal region of the nsp3 gene of the Hong Kong and Colorado HKU1 specimens.
Identical repeat sequences are colour coded. The subscript number following a sequence is the number of repeats of that sequence in the indicated viral genome.

As the HKU1 spike gene was one of the most variable regions of the genome and it determined viral clades, we analysed the spike genes from our Colorado HKU1 virus, four additional Colorado clinical specimens and three clinical specimens from Brazilian paediatric patients (Fig. 3). Three of the four additional Colorado viruses and two of the three Brazilian viruses were genotype A, and the other two viruses were both genotype B. The one genotype B specimen from Colorado (HKU1/USA/1/2005) was collected in 2005, whereas the other 10 genotype A sequences were collected during the 2009–2010 respiratory season. The two genotype A sequences from Brazil were collected in 2006 and the genotype B sequence was collected in 2007 (Table 2).
Fig. 3.

Phylogenetic analysis of the amino acid sequences of HKU1 spike genes. One hundred bootstrap replicates were used for reconstruction of the phylogenetic trees, and nodes supported by a bootstrap value greater than 70 % are indicated. Genotypes are defined by different coloured lines, as in Fig. 2. A subset of the Colorado HKU1 viruses formed a subclade within genotype A (indicated by the red box). Bar, amino acid substitutions per site.
Within each HKU1 genotype, the spike sequences (nucleotide and protein) were remarkably conserved. Within genotype A, all of the Colorado HKU1 sequences, except for HKU1/USA/15/2009, formed a subclade defined (Fig. 3) by the same three amino acid substitutions of W197F (in the putative N-terminal domain of S1), F613Y (in the putative C-terminal domain of S1) and S752F, which directly precedes the predicted serine protease cleavage site (RRKRR at residues 756–760) between the S1 and S2 domains of the spike protein (Fig. S2).
Replication kinetics and propagation of HKU1
We propagated primary, differentiated HTBE cells at an air–liquid interface and inoculated them with clinical respiratory specimens positive for HKU1 RNA by RT-PCR at 34 and 37 °C. Infected cells were first detected by immunofluorescence with a polyclonal rabbit antibody against the purified recombinant HKU1 spike protein, between 8 and 12 h post-inoculation (p.i.), and approximately 5–20 % of cells were infected by 48 h p.i. at 34 °C (Fig. 4). As reported previously for growth at 32 and 33 °C in HTBE cells (Dijkman et al., 2013; Pyrc et al., 2010), the yield of HKU1 virus genomes released from the apical surface of the cells increased 1000-fold and plateaued from 48 to 72 h p.i. (Fig. 4). We obtained similar results with seven clinical specimens of HKU1 genotype A and one HKU1 genotype B (Table 2), and observed no differences in the yield of virus released between specimens cultured at 34 versus 37 °C. Serial passage of HKU1 viruses was performed by inoculating HTBE cells at 34 °C with the apical washes collected at 96 or 120 h from cells inoculated with the previous virus passage. HKU1 was serially passaged under these conditions five times without significant loss of titre as evidenced by quantitative real-time PCR (data not shown).
Fig. 4.
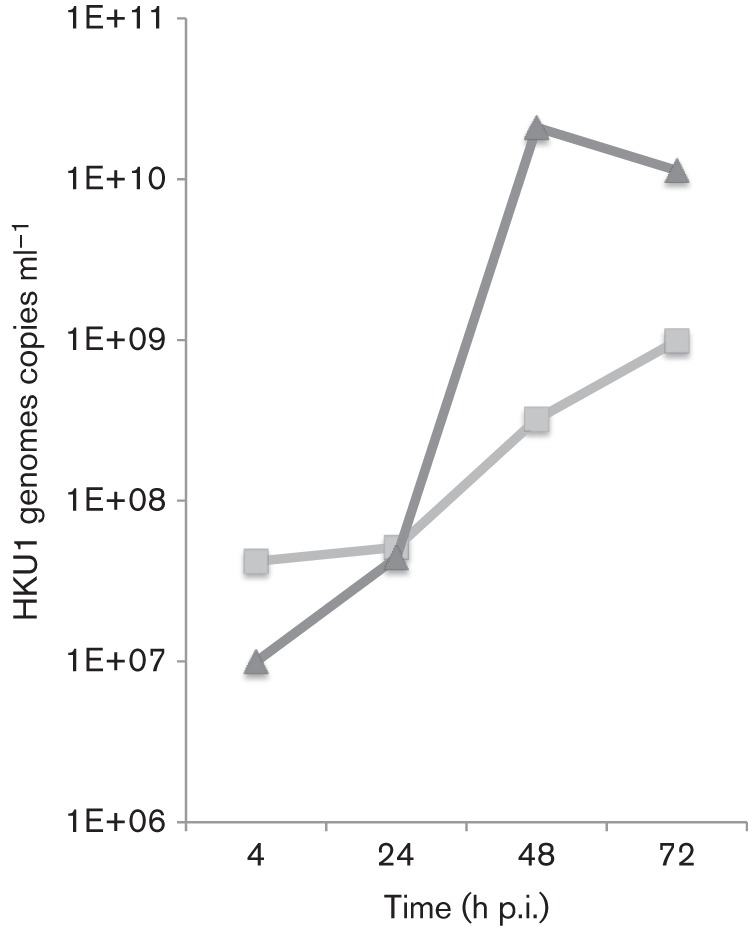
Replication kinetics of HKU1 betacoronavirus in HTBE cells. HTBE cell cultures maintained at 34 °C at an air–liquid interface were inoculated with a clinical specimen of HKU1. Data represent real-time PCR results from two experiments of apical washes from HKU1-inoculated cells harvested at the indicated time points p.i.
In vitro neutralization of HKU1 viruses
HTBE cells were inoculated with HKU1 in the presence or absence of FBS to determine whether serum inhibited infection due either to the potential presence of cross-reactive antibodies to HKU1 or to potential interference with the HE protein. Normal bovine and mouse sera are known to contain mannose-binding lectins that inhibit haemagglutination and neutralize influenza A virus infectivity by binding to carbohydrates at the tip of the influenza HE protein, blocking access of cell-surface receptors to the receptor-binding site on influenza HE (Anders et al., 1990). The percentage of infected cells inoculated with or without serum was identical (data not shown).
To explore the possibility that antibodies could be used as specific antiviral therapy for HKU1 infections, we tested pooled purified human IgG from healthy US donors (IVIG) for its ability to neutralize the infectivity of HKU1 viruses. Both genotypes A and B were incubated with 10-fold serial dilutions of IVIG and inoculated into HTBE cells. IVIG inhibited HKU1 infection at concentration of ≥10 mg ml−1. Similarly, polyclonal rabbit antibodies against the purified HKU1 spike glycoprotein inhibited infection of HTBE cells with HKU1 (Fig. 5).
Fig. 5.
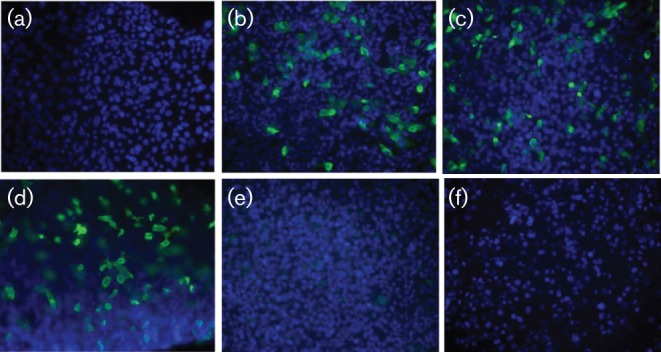
HKU1 neutralization by purified human IVIG and rabbit polyclonal antibodies to the HKU1 spike protein. HKU1 virus was incubated with the antibodies and then inoculated onto HTBE cells at 34 °C for 4 h. Following removal of the inoculum, the cells were maintained at the air–liquid interface, fixed at 24 h p.i. and immunolabelled with a mouse mAb to purified HKU1 spike protein (green fluorescence). Nuclei were stained with DAPI (blue). (a) Mock-infected cells. (b) HKU1-infected HTBE cells incubated with no antibody. (c) HKU1-infected cells incubated with pre-immune rabbit serum. (d) HKU1-infected cells incubated with a control mAb to an irrelevant antigen. (e) HKU1-infected cells incubated with polyclonal rabbit antibody to the HKU1 spike protein. (f) HKU1-infected cells incubated with human IVIG.
Discussion
This is the first study to report full-length genome sequences from clinical isolates of HKU1 from the western hemisphere. HKU1 viruses of genotypes A and B were detected, isolated and propagated from our paediatric specimens. In agreement with previous studies that demonstrated a HKU1 prevalence ranging from 0 to 4.4 % (Dare et al., 2007; Esper et al., 2006; Gaunt et al., 2010; Gerna et al., 2007; Góes et al., 2011; Huo et al., 2012; Jevšnik et al., 2012; Jin et al., 2010; Kuypers et al., 2007; Lau et al., 2006; Lu et al., 2012; Mackay et al., 2012; Prill et al., 2012; Sloots et al., 2006; Talbot et al., 2009; Vabret et al., 2008; Woo et al., 2009), only a small percentage (0.5 %) of all of respiratory samples submitted to the Clinical Virology Laboratory for virus testing were positive for HKU1 RNA during our 4.5-year study period. The cumulative burden of respiratory disease due to all four endemic hCoVs, however, was substantial, accounting for 15 % of all respiratory samples during the month of peak activity.
The prevalence of HKU1 infection and the other hCoVs varied markedly from year to year. For HKU1, there was a mini-outbreak during the 2009–2010 winter respiratory virus infection season when 4 % of respiratory specimens submitted during December were positive for HKU1 RNA. These data are in agreement with other multi-year studies of CoV prevalence, and demonstrate that the prevalence of HKU1, like other hCoVs, varies markedly from year to year (Dare et al., 2007; Gaunt et al., 2010; Gerna et al., 2007; Lau et al., 2006; Talbot et al., 2009; Vabret et al., 2008). This yearly variation may reflect the development of immunity within a community with subsequent development of new susceptible populations (young children) and/or antigenic drift of the virus. The peak in HKU1 activity was immediately preceded by a month with a peak in the activity of OC43, the other human betacoronavirus in group A, where 36 specimens (12 % of all samples submitted) during November 2009 were positive for OC43. This demonstrates that several different CoVs in the same phylogenetic group can co-circulate in the paediatric population during the same season. Indeed, all four hCoVs were present during each of the respiratory virus infection seasons.
HKU1 infection has been detected worldwide. Recent seroepidemiological surveys specific for HKU1 suggest that exposure to HKU1 increases with age but that the overall seroprevalence to HKU1 is lower than for other hCoVs. Approximately 22 % of adults in Hong Kong aged 31–40 years had antibodies specific for HKU1 (Chan et al., 2009), whilst in the USA, about 60 % of adults had antibodies to HKU1, significantly lower than the seropositivity rates for 229E, OC43 and NL63, which were greater than 90 % in adults (Severance et al., 2008). Our data suggest that, in paediatric patients who present for diagnostic testing, the prevalence of NL63 and OC43 significantly exceeded that of HKU1 and 229E.
A recent study of hospitalized children with acute respiratory illness found the same prevalence of HKU1 and other CoVs in hospitalized children and in asymptomatic, outpatient controls. Patients infected with only CoVs had milder illnesses than those infected with other respiratory viruses (Prill et al., 2012). Other studies, however, have found that HKU1 is associated with both upper and lower respiratory tract disease in hospitalized children (Esper et al., 2006; Gerna et al., 2007; Jevšnik et al., 2012; Jin et al., 2010; Kuypers et al., 2007; Regamey et al., 2008; Vabret et al., 2006, 2008) and with adults hospitalized with pneumonia (Woo et al., 2005a, b, 2009). In China, HKU1 was the most commonly found virus in respiratory specimens in patients who presented with influenza-like illness but tested negative for influenza (Huo et al., 2012). In our study, the majority of the HKU1-positive paediatric patients had upper respiratory tract infections, but a subset (28 %) of them presented with bronchiolitis and pneumonia. HKU1 has also been associated with febrile seizures in children (Vabret et al., 2006; Woo et al., 2012), and several HKU1-positive patients in our study also presented with seizures, adding strength to the association of HKU1 with febrile seizures in children. This is an interesting observation, as other betacoronaviruses in group A, including OC43, bovine CoV and murine hepatitis virus (MHV), are neurotrophic (Arbour et al., 1999; Jacomy et al., 2006; Lavi et al., 1987; Phillips & Weiss, 2011).
As others have reported (Dijkman et al., 2013; Pyrc et al., 2010), primary HTBE cells are an excellent in vitro culture system that closely mimics the in vivo environment of the human respiratory tract. These studies demonstrated that HKU1 primarily infects ciliated respiratory epithelial cells. Clinical isolates of our HKU1 specimens were isolated and propagated in HTBE cells at the physiological temperatures of 34 and 37 °C, which mimic the temperatures in the upper and lower respiratory tracts, respectively. Human IVIG and rabbit antibodies against the HKU1 spike glycoprotein inhibited HKU1 infection of HTBE cells. The ability of IVIG to neutralize infection suggests that a significant proportion of individuals in the USA have been exposed to and have mounted a neutralizing antibody response to HKU1. Furthermore, it argues that the target(s) of neutralizing antibody within the HKU1 spike protein might be conserved over time, with minimal antigenic drift or shift. This hypothesis is also supported by the remarkable degree of conservation found across HKU1 spike sequences.
One of the unique features of the HKU1 genome is the presence of the ATRs in the nsp3 protein located in the acidic domain upstream of the PL1pro active site. Interestingly, all of the Colorado HKU1 spike protein sequences from 2009 to 2010 that formed a subclade within genotype A exhibited a similar pattern in the ATR region that was different from that of the Asian genotype A viruses, suggesting that this region might be useful for molecular typing. Despite this similarity in the pattern in the ATRs, the number of repeats varied in the Colorado HKU1 viruses that were circulating in our community over a 2-month period, despite the rest of the genome remaining stable. Although the exact origins and functions of this ATR domain are not known, NSP3 is an essential and important part of the replication complex. Further studies are needed to explore the mechanism surrounding the formation of these repeats and their function in the replication process.
In contrast to NL63, which has regions of marked amino acid diversity, particularly in the N-terminal domain of the spike protein and the 1b gene (Dominguez et al., 2012), HKU1 strains circulating in the western hemisphere had amino acid sequences that were remarkably well conserved and differed little from HKU1 strains circulating in Asia and Europe. The reason for this lack of diversity in HKU1 is unclear. One possible explanation for this observation could be that the proofreading ability of the HKU1 exonuclease might be superior to that of NL63. Alternatively, the HKU1 spike interaction with its unknown receptor might be strictly constrained such that mutant spikes display decreased affinity, leading to impaired viral entry and fitness.
All but one of our Colorado specimens formed a subclade within genotype A determined by three single amino acid substitutions in the spike protein. Based on comparison with MHV, one of the substitutions was located within the N-terminal domain (W197F) and another within the C-terminal domain (F613Y) (Peng et al., 2011). These substitutions could confer antigenic differences or impact on binding of HKU1 viruses to cell surfaces. The third, and most notable, amino acid substitution (S752F), was adjacent to the predicted S1/S2 cleavage site. Although the functional significance of this change is unknown, it could potentially affect the protease cleavage and activation of the membrane fusion activity of the HKU1 spike. In MHV-A59, a mouse betacoronavirus, a single H716D substitution downstream of the S1/S2 cleavage site in the MHV-A59 spike protein causes resistance to cleavage by trypsin (Zelus et al., 2003).
In summary, we report the first full-length genome sequences from HKU1 betacoronavirus specimens in the western hemisphere and comparison with the genomes of other HKU1 viruses circulating in other parts of the world. HKU1 circulated in our community during all five respiratory virus seasons studied, but with considerable differences in yearly prevalence. We demonstrated that the HKU1 genotypes A and B can be neutralized in vitro by human IVIG and by polyclonal antibodies to the spike glycoprotein of HKU1. Remarkably, HKU1 specimens from around the world have highly conserved sequences within individual genotypes. Within genotype A, all but one of the Colorado HKU1 sequences formed a subclade based on their spike gene sequences, which was correlated with a unique ATR sequence in the ATR domain of nsp3 but with varying numbers of repeats. Elucidating the function of and mechanisms responsible for the formation of the varying tandem repeats in the ATR of nsp3 of HKU1 while maintaining a highly conserved sequence will increase our understanding of the replication process and pathogenicity of HKU1 and potentially other CoVs.
Methods
Clinical specimens.
In January 2009, the Children’s Hospital Colorado’s Clinical Virology Laboratory began to use a multiplex PCR assay (xTag Respiratory Virus Panel; Luminex Molecular Diagnostics) that detects 12 respiratory viruses (including the four hCoVs 229E, OC43, HKU1 and NL63). Nucleic acids from respiratory specimens submitted for Respiratory Virus Panel testing were extracted using Virus Minikits v2.0 on BioRobot EZ1 extractors (Qiagen) following the manufacturer’s instructions. Specimens positive for any hCoV were archived at −70 °C in M4 viral transport medium (Remel) for further analysis. Use of the banked specimens and clinical data for this study was approved by the Colorado Multiple Institutional Review Board and the Ethics Committee on Research Involving Human Subjects of the Institute of Biomedical Sciences, University of São Paulo, Brazil.
Generation of HKU1 antibodies.
Codon-optimized genotype A HKU1 spike protein ectodomain (Secto) was constructed with a C-terminal truncation yielding a soluble HKU1 ectodomain (aa 1–1283) that substitutes a C-terminal FLAG tag for the transmembrane domain and tail, and cloned into pcDNA3.1(+) between the BamHI and NotI sites for expression. Proteins were purified by affinity chromatography using anti-FLAG M2 magnetic beads (Sigma).
Polyclonal antisera were generated by immunizing rabbits with HKU1 Secto protein and Freund’s complete adjuvant (Open Biosystems) and mAbs were generated by immunizing mice with HKU1 Secto protein with 100 µl TiterMax gold adjuvant (Sigma-Aldrich) following standard protocols.
Isolation and propagation of HKU1 from clinical specimens.
HBTE cells were obtained from Lifeline Cell Technology. Primary cells were expanded on plastic in BronchiaLife Complete Medium (Lifeline Cell Technology) and then plated at a density of 1×105 cells per well on 12-well Corning Transwell-COL collagen-coated permeable (0.4 µm pore size) membrane inserts (Sigma-Aldrich). Cultures were maintained in growth medium until confluent monolayers were formed on the inserts and then switched to differentiation medium [1 : 1 ratio of BronchiaLife medium and Dulbecco’s modified Eagle’s medium (DMEM) high-glucose medium (Invitrogen) with the addition of 1.1 mM CaCl2 and 25 nM retinoic acid]. Cultures were then grown for 3–4 weeks in differentiation medium at an air–liquid interface at 37 °C to generate well-differentiated cultures that resembled in vivo ciliated respiratory epithelium.
Differentiated HBTE cells were inoculated on the apical surface with 100–150 µl per insert of each clinical sample (primary isolate) diluted 1 : 10 in either DMEM containing 1 % BSA fraction V or FBS, or with a 1 : 10 or 1 : 100 dilution of passage 1 virus stock generated from apical washes of primary cultures from HBTE cells harvested at 72 or 96 h p.i.. Following a 4 h incubation at 34 or 37 °C, the initial inoculum was removed and the HBTE cells were maintained at an air–liquid interface for the remainder of the experiment. The yields of HKU1 were determined at specific time points p.i. by fixing the cells and detecting HKU1-infected cells by immunofluorescence and/or by determination of viral titres in washes of the apical surface of cells on inserts (three consecutive aliquots with 100 µl 1 % BSA in high-glucose DMEM pooled). RNA from the washes was extracted using Virus Minikits v2.0 on a BioRobot EZ1 extractor (Qiagen) following the manufacturer’s instructions. Titres of virus were assessed by real-time PCR targeting the replicase gene using a previously described protocol (Dominguez et al., 2009).
Immunofluorescence.
To detect HKU1 antigens in infected cells, the apical surfaces of HBTE cultures were washed twice with DMEM or PBS and then fixed at 4 °C in 100 % methanol for at least 20 min at −20 °C. HKU1 was detected in infected cells using the rabbit polyclonal antibody directed against the HKU1 spike glycoprotein and visualized using a goat anti-rabbit antibody conjugated to Alexa Fluor 488 (Invitrogen). Immunolabelled cells were imaged using a Zeiss Axioplan 2 or Nikon Eclipse TE2000 U fluorescent microscope.
Antibody neutralization assay.
To assess their ability to neutralize HKU1 infection, polyclonal anti-HKU1 antibodies or human IVIG (CSL Behring) were incubated at 56 °C for 30 min to inactivate complement followed by filter sterilization through a 0.22 µm membrane. Pre-immune serum as well as a mAb directed against an irrelevant antigen (cholera toxin, a kind gift from Randall Holmes, University of Colorado School of Medicine, CO, USA) were used as controls. Antibodies were serially diluted in DMEM+1 % BSA and incubated with passage 1 virus at a 1 : 100 dilution for 1 h at 37 °C. The virus/antibody mixtures were incubated on HTBE cells for 4 h at 34 °C and then removed. Cells were fixed at 24 h p.i. and assayed for infection by immunofluorescence using a mouse mAb directed against the HKU1 spike glycoprotein.
Genome sequencing.
Subsets of HKU1-positive clinical specimens collected from 2009 to 2011, as well as samples collected in earlier years from our previously reported studies in Denver and Brazil (Dominguez et al., 2009; Góes et al., 2011) were selected for genome sequencing. Samples were sequenced using Sanger technology or using a mix of Sanger and Illumina technology (Table 1).
For samples that went through the Sanger sequencing technology, a 96-well plate of degenerate HKU1-related specific primers was designed from a consensus sequence of the 23 related virus reference genomes downloaded from GenBank (accession nos: NC006577, AY884001, DQ339101, DQ415896–DQ415914 and HM034837) using a PCR primer design pipeline developed at J. Craig Venter Institute (JCVI) (Li et al., 2012). This produced tiled amplicons with an optimal length of 550 bp, with a 100 bp overlap and at least twofold amplicon coverage at every base. Additional primer sets specific for the A, B and C viral subtypes were then designed. Subsets of these primers covering regions of significant A/B/C variation were then judiciously combined with the initial primer set to produce a two-plate primer set that could successfully amplify A-, B- or C-type genomes without prior knowledge of the viral subtype. An M13 sequence tag was added to the 5′ end of each primer and was used for sequencing.
RNA extracted from the clinical samples was subjected to RT-PCR using a Qiagen One-step kit to produce cDNA amplicons from each of the primer pairs in the 96-well plate. For each clinical specimen, two independent sets of reactions were performed to produce sequence coverage consistent with JCVI standards for finished Sanger sequences. PCR products were treated with a mixture of exonuclease and shrimp alkaline phosphatase to remove primers and dNTPs, and then subjected to Sanger sequencing using the common M13 primers at the 5′ end of each primer. For HE–spike amplicon sequencing, primer pairs spanning the region were used to generate an approximately 5.6 kb amplicon. For the Denver-originating samples, a hemi-nested strategy was used. The primers were: reverse transcription primer: 5′-GTTCACTATAAGACTTATACAAC-3′, PCR forward primer: 5′-GAAATAGATGGCAATGTTATGC-3′, and PCR reverse primer: 5′-GGTACAATACARTAACCAAC-3′. For the samples originating in Brazil, the PCR primers used were: forward primer: 5′-GAAATAGATGGCAATGTTATGC-3′, and reverse primer: 5′-GTTCACTATAAGACTTATACAAC-3′.
Extracted RNA was reverse transcribed using SuperScript III (Life Technologies) and the appropriate reverse primer, and dsDNA was produced by PCR using Phusion enzyme (New England BioLabs). Bar-coded, Illumina-ready libraries of each amplicon were generated using the Nextera protocol (originally Epicentre; now part of Illumina). A pool of libraries from these and other viral samples was pooled and sequenced in a single lane on the HiSeq platform.
Assembly.
Sanger sequencing reads were assembled de novo using CLC Bio’s clc_novo_assemble (http://www.clcbio.com/files/whitepapers/whitepaper-denovo-assembly-4.pdf). The consensus sequence of the de novo assembly was used to identify the best full-length HKU1 genome downloaded from GenBank to use as a mapping reference sequence. Reads were mapped to the selected reference genome using clc_ref_assemble_long (http://www.clcbio.com/files/whitepapers/CLC_legacy_read_mapper_white_paper.pdf).
For the Illumina sequencing technology, the Illumina reads were mapped to the selected reference genome using the clc_ref_assemble_long (CLCbio – Assembly Cell) program resulting in the final next-gen assembled genome.
Upon review of next-gen assemblies, there were circumstances that required additional sequencing, due to either no or low coverage. For these regions, PCR primer pairs were designed, the DNA was amplified and sequencing was performed as described previously (Ghedin et al., 2005; Li et al., 2012). Sanger reads were assembled together with the Illumina reads using the clc_ref_assemble_long program to map against the selected reference genome.
Annotation.
Viral Genome ORF Reader, vigor (Wang et al., 2010), was used to check segment lengths, perform alignments, ensure the fidelity of ORFs, correlate nucleotide polymorphisms with amino acid changes and detect any potential sequencing errors. The complete genomes were annotated with vigor before submission to GenBank.
Sequence analysis.
The HKU1 individual gene and protein sequences and the complete genomes were multiple aligned using clustal w v.2.0.11 (Thompson et al., 1994). This alignment was used to generate a maximum-likelihood phylogenetic tree with phyml v3.0 (Guindon & Gascuel, 2003) using the HKY85 nucleotide substitution model and the estimated transition/transversion ratio. NL63 was selected as an outgroup for the complete genome phylogenetic tree, and trees were viewed using FigTree v.1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/).
Nucleotide sequence accession numbers.
The clinical specimens positive for HKU1 viruses were named according to the following nomenclature: Virus/<ISO three-digit location >/<specimen number >/<year of isolation in YYYY format > (for example HKU1/USA/18/2010).
Acknowledgements
This work was supported in part by an NIH grant to S. R. D. (1-K08AI073525), in part from federal funds from the National Institute of Allergy and Infectious Diseases, NIH, Department of Health and Human Services (contract no. HHSN272200900007C), in part by a Brazilian National Council of Scientific and Technological Development (CNPq) fellowship grant to L. G. B. G. (201435/2011-0), and in part by the São Paulo Research Foundation (FAPESP) (2010/00397-0). We thank Anna Castano, Christina Osborne, the staff of the Children’s Hospital Colorado Clinical Virology Laboratory, the University of Colorado Protein Production, Monoclonal Antibody, and Tissue Culture Core, and members of the JCVI viral sequencing and informatics groups for their assistance with these studies.
Footnotes
Two supplementary figures are available with the online version of this paper.
References
- Anders E. M., Hartley C. A., Jackson D. C. (1990). Bovine and mouse serum beta inhibitors of influenza A viruses are mannose-binding lectins. Proc Natl Acad Sci U S A 87, 4485–4489 10.1073/pnas.87.12.4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour N., Côté G., Lachance C., Tardieu M., Cashman N. R., Talbot P. J. (1999). Acute and persistent infection of human neural cell lines by human coronavirus OC43. J Virol 73, 3338–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assiri A., McGeer A., Perl T. M., Price C. S., Al Rabeeah A. A., Cummings D. A., Alabdullatif Z. N., Assad M., Almulhim A. & other authors (2013). Hospital outbreak of Middle East respiratory syndrome coronavirus. N Engl J Med 369, 407–416 10.1056/NEJMoa1306742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. M., Tse H., Wong S. S., Woo P. C., Lau S. K., Chen L., Zheng B. J., Huang J. D., Yuen K. Y. (2009). Examination of seroprevalence of coronavirus HKU1 infection with S protein-based ELISA and neutralization assay against viral spike pseudotyped virus. J Clin Virol 45, 54–60 10.1016/j.jcv.2009.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dare R. K., Fry A. M., Chittaganpitch M., Sawanpanyalert P., Olsen S. J., Erdman D. D. (2007). Human coronavirus infections in rural Thailand: a comprehensive study using real-time reverse-transcription polymerase chain reaction assays. J Infect Dis 196, 1321–1328 10.1086/521308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkman R., Jebbink M. F., Koekkoek S. M., Deijs M., Jónsdóttir H. R., Molenkamp R., Ieven M., Goossens H., Thiel V., van der Hoek L. (2013). Isolation and characterization of current human coronavirus strains in primary human epithelial cell cultures reveal differences in target cell tropism. J Virol 87, 6081–6090 10.1128/JVI.03368-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez S. R., Robinson C. C., Holmes K. V. (2009). Detection of four human coronaviruses in respiratory infections in children: a one-year study in Colorado. J Med Virol 81, 1597–1604 10.1002/jmv.21541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez S. R., Sims G. E., Wentworth D. E., Halpin R. A., Robinson C. C., Town C. D., Holmes K. V. (2012). Genomic analysis of 16 Colorado human NL63 coronaviruses identifies a new genotype, high sequence diversity in the N-terminal domain of the spike gene and evidence of recombination. J Gen Virol 93, 2387–2398 10.1099/vir.0.044628-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Günther S., Preiser W., van der Werf S., Brodt H. R., Becker S., Rabenau H., Panning M., Kolesnikova L. & other authors (2003). Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348, 1967–1976 10.1056/NEJMoa030747 [DOI] [PubMed] [Google Scholar]
- Esper F., Weibel C., Ferguson D., Landry M. L., Kahn J. S. (2006). Coronavirus HKU1 infection in the United States. Emerg Infect Dis 12, 775–779 10.3201/eid1205.051316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaunt E. R., Hardie A., Claas E. C., Simmonds P., Templeton K. E. (2010). Epidemiology and clinical presentations of the four human coronaviruses 229E, HKU1, NL63, and OC43 detected over 3 years using a novel multiplex real-time PCR method. J Clin Microbiol 48, 2940–2947 10.1128/JCM.00636-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerna G., Percivalle E., Sarasini A., Campanini G., Piralla A., Rovida F., Genini E., Marchi A., Baldanti F. (2007). Human respiratory coronavirus HKU1 versus other coronavirus infections in Italian hospitalised patients. J Clin Virol 38, 244–250 10.1016/j.jcv.2006.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E., Sengamalay N. A., Shumway M., Zaborsky J., Feldblyum T., Subbu V., Spiro D. J., Sitz J., Koo H. & other authors (2005). Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature 437, 1162–1166 10.1038/nature04239 [DOI] [PubMed] [Google Scholar]
- Góes L. G., Durigon E. L., Campos A. A., Hein N., Passos S. D., Jerez J. A. (2011). Coronavirus HKU1 in children, Brazil, 1995. Emerg Infect Dis 17, 1147–1148 10.3201/eid1706.101381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Gascuel O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52, 696–704 10.1080/10635150390235520 [DOI] [PubMed] [Google Scholar]
- Huo X., Qin Y., Qi X., Zu R., Tang F., Li L., Hu Z., Zhu F. (2012). Surveillance of 16 respiratory viruses in patients with influenza-like illness in Nanjing, China. J Med Virol 84, 1980–1984 10.1002/jmv.23401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacomy H., Fragoso G., Almazan G., Mushynski W. E., Talbot P. J. (2006). Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology 349, 335–346 10.1016/j.virol.2006.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevšnik M., Uršič T., Zigon N., Lusa L., Krivec U., Petrovec M. (2012). Coronavirus infections in hospitalized pediatric patients with acute respiratory tract disease. BMC Infect Dis 12, 365 10.1186/1471-2334-12-365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Song J. R., Xie Z. P., Gao H. C., Yuan X. H., Xu Z. Q., Yan K. L., Zhao Y., Xiao N. G. & other authors (2010). Prevalence and clinical characteristics of human CoV-HKU1 in children with acute respiratory tract infections in China. J Clin Virol 49, 126–130 10.1016/j.jcv.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek T. G., Erdman D., Goldsmith C. S., Zaki S. R., Peret T., Emery S., Tong S., Urbani C., Comer J. A. & other authors (2003). A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348, 1953–1966 10.1056/NEJMoa030781 [DOI] [PubMed] [Google Scholar]
- Kuypers J., Martin E. T., Heugel J., Wright N., Morrow R., Englund J. A. (2007). Clinical disease in children associated with newly described coronavirus subtypes. Pediatrics 119, e70–e76 10.1542/peds.2006-1406 [DOI] [PubMed] [Google Scholar]
- Lau S. K., Woo P. C., Yip C. C., Tse H., Tsoi H. W., Cheng V. C., Lee P., Tang B. S., Cheung C. H. & other authors (2006). Coronavirus HKU1 and other coronavirus infections in Hong Kong. J Clin Microbiol 44, 2063–2071 10.1128/JCM.02614-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi E., Suzumura A., Hirayama M., Highkin M. K., Dambach D. M., Silberberg D. H., Weiss S. R. (1987). Coronavirus mouse hepatitis virus (MHV)-A59 causes a persistent, productive infection in primary glial cell cultures. Microb Pathog 3, 79–86 10.1016/0882-4010(87)90066-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K., Shrivastava S., Brownley A., Katzel D., Bera J., Nguyen A. T., Thovarai V., Halpin R., Stockwell T. B. (2012). Automated degenerate PCR primer design for high-throughput sequencing improves efficiency of viral sequencing. Virol J 9, 261 10.1186/1743-422X-9-261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Yu X., Wang W., Duan X., Zhang L., Zhou W., Xu J., Xu L., Hu Q. & other authors (2012). Characterization of human coronavirus etiology in Chinese adults with acute upper respiratory tract infection by real-time RT-PCR assays. PLoS ONE 7, e38638 10.1371/journal.pone.0038638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay I. M., Arden K. E., Speicher D. J., O’Neil N. T., McErlean P. K., Greer R. M., Nissen M. D., Sloots T. P. (2012). Co-circulation of four human coronaviruses (HCoVs) in Queensland children with acute respiratory tract illnesses in 2004. Viruses 4, 637–653 10.3390/v4040637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monto A. S., Rhodes L. M. (1977). Detection of coronavirus infection of man by immunofluorescence. Proc Soc Exp Biol Med 155, 143–148 10.3181/00379727-155-39761 [DOI] [PubMed] [Google Scholar]
- Peng G., Sun D., Rajashankar K. R., Qian Z., Holmes K. V., Li F. (2011). Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc Natl Acad Sci U S A 108, 10696–10701 10.1073/pnas.1104306108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. M., Weiss S. R. (2011). Pathogenesis of neurotropic murine coronavirus is multifactorial. Trends Pharmacol Sci 32, 2–7 10.1016/j.tips.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prill M. M., Iwane M. K., Edwards K. M., Williams J. V., Weinberg G. A., Staat M. A., Willby M. J., Talbot H. K., Hall C. B. & other authors (2012). Human coronavirus in young children hospitalized for acute respiratory illness and asymptomatic controls. Pediatr Infect Dis J 31, 235–240 10.1097/INF.0b013e31823e07fe [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrc K., Sims A. C., Dijkman R., Jebbink M., Long C., Deming D., Donaldson E., Vabret A., Baric R. & other authors (2010). Culturing the unculturable: human coronavirus HKU1 infects, replicates, and produces progeny virions in human ciliated airway epithelial cell cultures. J Virol 84, 11255–11263 10.1128/JVI.00947-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regamey N., Kaiser L., Roiha H. L., Deffernez C., Kuehni C. E., Latzin P., Aebi C., Frey U., Swiss Paediatric Respiratory Research Group (2008). Viral etiology of acute respiratory infections with cough in infancy: a community-based birth cohort study. Pediatr Infect Dis J 27, 100–105 [DOI] [PubMed] [Google Scholar]
- Severance E. G., Bossis I., Dickerson F. B., Stallings C. R., Origoni A. E., Sullens A., Yolken R. H., Viscidi R. P. (2008). Development of a nucleocapsid-based human coronavirus immunoassay and estimates of individuals exposed to coronavirus in a U.S. metropolitan population. Clin Vaccine Immunol 15, 1805–1810 10.1128/CVI.00124-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloots T. P., McErlean P., Speicher D. J., Arden K. E., Nissen M. D., Mackay I. M. (2006). Evidence of human coronavirus HKU1 and human bocavirus in Australian children. J Clin Virol 35, 99–102 10.1016/j.jcv.2005.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot H. K., Crowe J. E., Jr, Edwards K. M., Griffin M. R., Zhu Y., Weinberg G. A., Szilagyi P. G., Hall C. B., Podsiad A. B. & other authors (2009). Coronavirus infection and hospitalizations for acute respiratory illness in young children. J Med Virol 81, 853–856 10.1002/jmv.21443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Higgins D. G., Gibson T. J. (1994). clustal w: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22, 4673–4680 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret A., Dina J., Gouarin S., Petitjean J., Corbet S., Freymuth F. (2006). Detection of the new human coronavirus HKU1: a report of 6 cases. Clin Infect Dis 42, 634–639 10.1086/500136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret A., Dina J., Gouarin S., Petitjean J., Tripey V., Brouard J., Freymuth F. (2008). Human (non-severe acute respiratory syndrome) coronavirus infections in hospitalised children in France. J Paediatr Child Health 44, 176–181 10.1111/j.1440-1754.2007.01246.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Sundaram J. P., Spiro D. (2010). vigor, an annotation program for small viral genomes. BMC Bioinformatics 11, 451 10.1186/1471-2105-11-451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Chu C. M., Chan K. H., Tsoi H. W., Huang Y., Wong B. H., Poon R. W., Cai J. J. & other authors (2005a). Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79, 884–895 10.1128/JVI.79.2.884-895.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Tsoi H. W., Huang Y., Poon R. W., Chu C. M., Lee R. A., Luk W. K., Wong G. K. & other authors (2005b). Clinical and molecular epidemiological features of coronavirus HKU1-associated community-acquired pneumonia. J Infect Dis 192, 1898–1907 10.1086/497151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Yip C. C., Huang Y., Tsoi H. W., Chan K. H., Yuen K. Y. (2006). Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol 80, 7136–7145 10.1128/JVI.00509-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C., Lau S. K., Yuen K. Y. (2009). Clinical features and molecular epidemiology of coronavirus-HKU1-associated community-acquired pneumonia. Hong Kong Med J 15 (Suppl 9), 46–47 [PubMed] [Google Scholar]
- Woo P. C., Yuen K. Y., Lau S. K. (2012). Epidemiology of coronavirus-associated respiratory tract infections and the role of rapid diagnostic tests: a prospective study. Hong Kong Med J 18 (Suppl 2), 22–24 [PubMed] [Google Scholar]
- Zaki A. M., van Boheemen S., Bestebroer T. M., Osterhaus A. D., Fouchier R. A. (2012). Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367, 1814–1820 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- Zelus B. D., Schickli J. H., Blau D. M., Weiss S. R., Holmes K. V. (2003). Conformational changes in the spike glycoprotein of murine coronavirus are induced at 37°C either by soluble murine CEACAM1 receptors or by pH 8. J Virol 77, 830–840 10.1128/JVI.77.2.830-840.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]