

Abstract
Mesorhizobium huakuii 7653R occurs either in nitrogen-fixing symbiosis with its host plant, Astragalus sinicus, or free-living in the soil. The M. huakuii 7653R genome has recently been sequenced. To better understand the complex biochemical and developmental changes that occur in 7653R during bacteroid development, RNA-Seq and Microarrays were used to investigate the differential transcriptomes of 7653R bacteroids and free-living cells. The two approaches identified several thousand differentially expressed genes. The most prominent up-regulation occurred in the symbiosis plasmids, meanwhile gene expression is concentrated to a set of genes (clusters) in bacteroids to fulfill corresponding functional requirements. The results suggested that the main energy metabolism is active while fatty acid metabolism is inactive in bacteroid and that most of genes relevant to cell cycle are down-regulated accordingly. For a global analysis, we reconstructed a protein-protein interaction (PPI) network for 7653R and integrated gene expression data into the network using Cytoscape. A highly inter-connected subnetwork, with function enrichment for nitrogen fixation, was found, and a set of hubs and previously uncharacterized genes participating in nitrogen fixation were identified. The results described here provide a broader biological landscape and novel insights that elucidate rhizobial bacteroid differentiation, nitrogen fixation and related novel gene functions.
Introduction
Rhizobia are specific soil bacteria, which can establish an intricate symbiotic relationship with Legumes. This interaction results in the formation of specialized root structures called nodules [1]. In the indeterminate nodules of the Inverted Repeat Lacking Clade (IRLC) legumes [2] (e.g. Medicago, Pisum, and Astragalus sinicus), bacteria undergo dramatic changes in size, shape, and DNA content to become terminally differentiated bacteroids [3]. The host plant provides rhizobia with dicarboxylic acids as a source of carbon, energy, and reductant [4] and ammonium (via N2 reduction that occurs in these bacteroids) is secreted back to the plant. This biological process serves as a natural form of fertilization and has significant potential for use in sustainable agricultural programs, especially if optimized. Thus, Rhizobium-legume symbiosis has been the subject of much study for decades.
Mesorhizobium huakuii 7653R is a α-proteobacterium that occurs either in a nitrogen-fixing symbiosis with its host plant, A. sinicus, or free-living in the soil. M. huakuii 7653R is a narrow-host-range rhizobium that induces indeterminate-type nitrogen-fixing nodules on the roots of A. sinicus, which is an economically important forage and green manure growing in winter throughout Eastern Asia. The whole-genome sequence of M. huakuii 7653R has been recently completed (http://www.ncbi.nlm.nih.gov/genome/11322). Transcriptome analysis has always played a central role for unraveling the complexity of gene regulation in the field of functional genomics. Transcriptome profiling is traditionally done using either real time quantitative PCR to examine a few genes [5], or Microarrays to examine genome-wide transcriptional activity [6]. As the genome sequences of several rhizobia are readily available [7]–[10], it is possible to use Microarrays to study complex host-microbe interaction. Formation of determinate nodules in the Bradyrhizobium japonicum-Glycine max symbiosis [11], [12] and the Mesorhizobium loti-Lotus japonicus symbiosis [13] have been investigated with Microarrays. Indeterminate-type nodule formation, as occurs in Sinorhizobium meliloti-Medicago sativa symbiosis [14] or Rhizobium/pea-vetch symbiosis [15] have been studied with Microarrays as well. These studies have provided significant insight into bacteroid function and development. Most of the publications focus on the analysis of individual metabolic pathways, a global perspective of the interaction mechanism is still limited. Therefore, it is difficult to fully understand the complex metabolic changes and gene networks that occur in bacteroids relative to free-living cells.
Recently, as a result of the decreased cost of next generation sequencing technologies [16], RNA sequencing (RNA-Seq) is becoming the method of choice in transcriptome studies[17]. However, study of the bacterial transcriptomes has not progressed as quickly as the study of eukaryotic transcriptomes [18], [19] due to the lack of mRNA polyA tails in bacteria, which prevents specific targeting of the mRNA versus the much larger rRNA pool. Therefore, the application of RNA-Seq in prokaryotes requires additional steps in the RNA preparation procedure to increase the relative abundance of mRNA and new methods to replace the poly (T)-primed approach in cDNA synthesis. Yoder-Himes et al. (2009) successfully resolved these limitations and subsequently used RNA-Seq to investigate the bacterial transcriptome in Burkholderia cenocepacia [20]. However, few have investigated the Rhizobium transcriptome using this method, until recently in Sinorhizobium sp. NGR234 bacteroids in determinate nodules of Vigna unguiculata and indeterminate nodules of Leucaena leucocephala have been studied with RNA-Seq [21]. To date, Microarrays remains a useful and accurate tool for measuring expression levels, and RNA-Seq complements and extends data obtained from Microarrays. Studies have demonstrated that RNA-Seq and Microarrays do in fact complement each other in transcriptome profiling. There is an obvious advantage to applying multiple transcriptome profiling methods, providing a comprehensive picture of a transcriptome, rather than relying solely on one method [22]. To better understand the complex biochemical and developmental changes that occur in M. huakuii 7653R during bacteroid development, we performed genome-wide transcriptome profiling by using a combination RNA-Seq and Microarrays technology. As a result, we have provided a broader biological landscape and novel insight that advances our understanding of rhizobium response to alterations in the symbiotic environment, and what physiological changes occur during bacteroid differentiation.
Materials and Methods
Bacterial strains and growth conditions
M. huakuii 7653R was grown at 28°C in TY medium (5 g/liter tryptone, 3 g/liter yeast extract, and 1.3 g/liter CaCl2·6H2O[pH 7.0])[23]. To isolate RNA from free-living cells, M. huakuii 7653R was grown in 100 mL of medium up to an optical density at 600 nm of 1 to 2. Cultured cells were harvested by centrifugation at 12 000 g at 4°C for 1 min. The pellets were frozen in liquid nitrogen and stored at −80°C until RNA was isolated.
Plant growth, inoculation, and nodules harvest
A. sinicus seeds were sterilized, germinated, planted, and inoculated with M. huakuii 7653R strains as previously described[24]. At 32 days post-inoculation, the nodules of plants were harvested, immediately frozen in liquid nitrogen, and stored at −80°C until RNA was isolated.
RNA extraction and bacteroids RNA isolation
Total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction. To remove contaminating genomic DNA, approximately 1 μg of RNA was treated with 1 U of DNaseI (Fermentas, Burlington, VT, Canada) at 37 °C for 20−30 min. These reactions were terminated by the addition of 1 μL of 25 mM EDTA (Invitrogen) and incubation at 65°C for 10 min. To isolate bacteroid RNA, total nodules RNA passed though MICROBEnrich kit (Ambion) according to the manufacturer's instruction. This step was intended to remove plant mRNAs and also 18S and 28S rRNAs. However, as it was necessary to reduce variation resulting from sample preparations, RNA substrates were extracted from the same phase (OD600 of 1 to 2 free-living cells and 32 days post-inoculation nodules) and were carried out using independently isolated RNA preparations from independent cultures and set of plants.
RNA-Seq experimental design, libraries construction and sequencing
RNA-Seq libraries were prepared for each sample following standard protocols (Illumina, San Diego, CA). Briefly, mRNA was isolated from total bacteroids RNA using MICROBExpress kit (Ambion). Isolated mRNA was fragmented using divalent cations under increasing temperatures followed by ethanol precipitation. The fragmented mRNA was reverse-transcribed into cDNA using Superscript III and random primers. The cDNA was end-repaired to create blunt-ended fragments, and an ‘A’ base was ligated to the 3′ ends of the fragments to create an ‘A’ base overhang. Adapters with a ‘T’ base overhang were ligated to each end of the cDNA fragments. The ligated fragments were run on an agarose gel, and a thin slice of DNA fragments corresponding to approximately 200 bp was excised and purified from the gel. The purified size-selected fragments were enriched by PCR to generate the final library fragments. Sequencing reactions were carried out on the Illumina HiSeq 2000. The read length was 90 bp, paired end sequencing method was used. RNA-Seq experiments produced 1.07 Gb and 1.13 Gb sequences for the free-living and bacteroids, respectively. Total mapped reads to genome is 94.93% and 17.31% for the free-living and bacteroids, respectively. Around 200 Mb sequences from the bacteroids' sample were uniquely mapped to M. huakuii 7653R genome (6.6 Mb).
The experimental design and gene expression analysis of Microarrays
Microarrays oligonucleotides were designed according to the 7,453 annotated open reading frames (ORFs) of M. huakuii 7653R. For each gene, three probes were designed and each duplicated three times. Microarrays were prepared for each sample following the User's Guide (Roche NimbleGen, Madison, WI). Briefly, total RNA was added a poly(A) tail using Poly(A) Polymerase. Poly(A) RNA was reverse transcribed with a T7 promoter site Oligo dT primer to synthesize a first-strand cDNA using SuperScript II. T7 promoter site was completed by a second strand cDNA synthesis. The RNA Polymerase driven in vitro transcription converts the double-stranded cDNA with T7 RNA Polymerase promoter site into the final product, amplified-antisense RNA (aRNA). 5 μg cRNA was reverse transcribed with random primer by using CbcScript, then it was purified by using NucleoSpin Extract II Kit (MN). Double-stranded cDNA was primed with random primer Cy3 or Cy5-dCTP (GE Healthcare) using Klenow enzyme. At the completion of the procedure, the quality of the purified Cy3 or Cy5 labeled cDNA was assessed by using a spectrophotometer. Pairs of samples designed for hybridization to the same array were labeled in parallel. Bacteroids samples were labeled with Cy3 and free-living cells samples were labeled with Cy5. Cy-labeled cDNA was hybridized with high-density, 70-mer oligonucleotide Microarrays slide mixer in a Roche NimbleGen Hybridization System 12 for 14-16 h at 42°C. The hybridized slides were washed and scanned by using Roche-NimbleGen MS200. A total of three independent biological replicates were designed for each condition, which included a dye-swap for each replicate, resulting in a total of 6 slides for the experimental condition.
Statistical analysis of RNA-Seq and microarrays data
Microarrays images and intensity data were uploaded to NimbleScan (version:2.6.0.0) to analyze data extracted from the image. Briefly,a tif image was uploaded into NimbleScan along with NimbleGen design files, which describe both probe identities and locations, to generate a tab-delimited report with probe identities and feature signal intensities. At the completion of the procedure, data were present in an appropriate format for normalization and comparative analysis. Microarrays images with uniformity hybridization signal and the area of scratches, bubbles and other dust no more than 5% were analyzed. The Alignment Oligo is a mixture of Cy3- and Cy5-labeled 48 mer oligonucleotides that hybridize to alignment features on NimbleGen arrays. Twelve Sample Tracking Controls (STCs) were used. Each STC is a Cy3-labeled 48 mer oligonucleotide. When a unique STC was added to each sample before hybridization to a multiplex array, the STC could be used to confirm that the correct sample was hybridized to each array. This control hybridized to probes on the Microarrays and enabled to confirm the sample identity on each array and the integrity of the experiment. STC probes were placed as repeating sets of 20 along the perimeter of each array and as two 4 × 5 blocks in the upper left corner and in the center of the array. The data files were normalized for correcting the signal value by RMA (Robust Multichip Analysis) and then analyzed by SAM (Significance Analysis of Microarrays) [25].
RNA-Seq images generated by sequencers were converted by base calling into nucleotide sequences, which were called raw data or raw reads. Reads with adaptors, unknown nucleotides larger than 10% and low quality (more than half of the bases' qualities are less than 5) were removed, and the clean reads were then obtained. Clean reads were mapped to M. huakuii 7653R genome and gene sequences respectively using SOAP2[26]. Mismatches of no more than 5 bases were allowed in the alignment. To eliminate the influence of different gene length and sequencing discrepancy on the calculation of gene expression, the RPKM (reads per kilobase per million mapped reads) method was used to calculate gene expression level [27]. Genes with the ratio of RPKMs of the two samples above 2, Benjamini FDR (False Discovery Rate) ≤0.001 were defined as the differentially expressed genes between two samples.
Differentially expressed genes of statistical significance from both RNA-Seq and Microarrays data were determined by applying a threshold of p/q value ≤ 0.05 (5%) and absolute fold-change ≥ 2. In order to identify those genes with a relevant role in bacteroid metabolism and, in turn, form a set of genes that serve as a benchmark for computational assessment, we took into account both sources of data following an integrative rather than a selective strategy.
PPI network reconstruction and analyses
Information found in PPIs databases supports the construction of interaction networks. Here, we elucidated the homologous protein-protein interactions network through the concepts of orthologous conservation (or interologs) [27]. In this work, M. huakuii bv loti MAFF303099[28] and Escherichia coli [29] PPI network databases were used as references, and pairwise genome comparison for bidirectional best hits (BBH) [30] was adopted to acquire high-quality PPI network of M. huakuii 7653R. Briefly, in pairwise genome comparison for BBH, we set the e-value < 1e-5. Identity≥0.3, coverage≥0.7. The workflow is illustrated in Figure S1. In total, 10 306 pairs of non-redundant PPIs were isolated (Table S1). Cytoscape[31] was used for network visualization and analysis. jActiveModules[32] plugin was used to search subnetworks of differentially expressed genes and BiNGO[33] plugin was used to find the up-regulated GO terms and to display the functional enrichment of the GO hierarchy where the up-regulated terms were from as a graph in Cytoscape. The CtrA subnetwork was dissected from its first two neighbors. Briefly, first found the first neighbors of selected Nodes (CtrA) that we can get several genes, second found first neighbors of these genes.
RNA-Seq and microarrays data accession number
RNA-Seq and Microarrays data were submitted to the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under GEO accession no. GSE48105. The accession numbers of RNA-Seq and Microarrays data are GSE48103 and GSE48090, respectively.
Results and Discussion
In this study, we analyzed the global gene expression profiles that are present during bacteroid differentiation. Therefore, it was critical to prepare sufficient and purified RNA samples. As bacteroids are enclosed by a peribacteroid membrane of plant origin [34], it is difficult to extract bacteroids RNA from nodules, particularly, from the nodules of A. sinicus induced by M. huakuii 7653R because of their smaller size.
With traditional protocols [15], [35], the bacteroids are usually isolated from nodules via physical separation, prior to bacteroid RNA extraction. It is inherently difficult to preserve bacteroid structure and thus prevent contamination by plant materials. This isolation process also takes longer to accomplish, thereby making it difficult to investigate real-time bacteroid gene expression. Traditional isolation procedures are somewhat complicated and also require large quantities of nodules; even still the resulting RNA is often of unsatisfactory quality. Since RNA-Seq requires large quantities of high-quality total RNA, we isolated the total RNA directly from the freshly harvested nodules immediately using a TRIzol protocol. We then used a MICROBEnrich kit to enrich bacterial RNA from the mixtures of plant and bacterial RNA samples. Our subsequent use of a MICROBExpress kit resulted in obtaining large quantities of high-quality targeted bacteroid mRNA for use in transcriptome analysis.
1 Transcriptome overall analysis based on RNA-Seq and Microarrays
1.1 Global gene expression profile
In total 6967 and 6662 expressed genes were detected in M. huakuii 7653R during symbiosis through RNA-Seq and Microarray, respectively. Among them, 2788 (RNA-Seq) and 2897 (Microarrays) genes were differentially expressed (|log2| ≥ 1, p/q value <0.05) in bacteroids compared with free-living cells. For the differentially expressed genes, 1267 genes were up-regulated and 1521 genes were down-regulated (as determined by RNA-Seq), while 1462 genes were up-regulated and 1435 genes were down-regulated (as determined by Microarrays). Any overlap of differentially expressed genes as identified by these methods is presented in Figure 1A. For the overlapping 1865 genes, the proportions of up- and down-regulation in plasmid and chromosome were calculated (Table 1). An overall trend showed that the plasmid genes were up-regulated, while chromosomal genes were down-regulated under symbiosis. These data suggest that a significant number of plasmid genes are required for nitrogen fixation, whereas housekeeping genes, those essential for active growth are located primarily on the chromosome.
Figure 1. Schematic view of data from RNA-Seq and Microarrays.

A: Venn diagram summarizing the overlap between differentially expressed genes from RNA-Seq (left circle) and Microarrays (right circle). The number of genes called by both methods is indicated by the overlap between the two circles. It also contains the numbers of up/down-regulated genes for each set in the Venn-Diagram. B: Comparison and correlations analyses of differentially expressed genes in RNA-Seq and Microarrays. The plot shows log2 ratios (bacteroids/free-living cells) of expressed genes in RNA-Seq (x axis) and Microarrays (y axis) (7043 genes in total). Pearson's correlation coefficient (r) is indicated for each comparison. C: Distribution of differentially expressed genes detected by RNA-Seq and Microarrays. Genes detected by RNA-Seq are shown with blue bars. Genes detected by Microarrays are shown with orange bars.
Table 1. The proportions of differentially expressed genes in plasmid and chromosome of M. huakuii 7653R during symbiosis.
| Category | Number of genes | Number of up-regulated genes | Percentagea | Number of down-regulated genes | Percentagea |
| pMHa | 257 | 59 | 22.96% | 18 | 7.00% |
| pMHb | 447 | 105 | 23.49% | 26 | 5.82% |
| Chromosome | 6562 | 702 | 10.70% | 955 | 14.56% |
| Total | 7266 | 866 | 11.92% | 999 | 13.75% |
The percentage represents the proportion of the up- or down-regulated genes to the total number of genes in the plasmids and chromosome, respectively.
Total expressions of differentially expressed genes were calculated using the expression levels of 2784 and 2750 genes (RNA-Seq) in free-living cells and bacteroids, respectively. There had several genes only been tested in one condition, accordingly the numbers (2784 and 2750) were less than the total differentially expressed genes (2889) in RNA-Seq. This analysis indicated that 80% of the total expression amount is attributed to 506 and 365 genes, respectively. Following the assignment of these genes to functional categories and calculation the distribution of genes among functional categories (Figure S2), from the percentage of each category, we noticed that most types of functional categories were high in free-living cells, and only the energy production and conversion, replication, recombination and repair and transcription were high in bacteroids.
As such most housekeeping genes are switched off in bacteroids, and those involved in reduction of N2 to NH3 are up-regulated. These data reflect the fundamental biological difference in that free-living cells have a primary role in maintaining basal metabolism, whereas bacteroids have a primary role in nitrogen fixation. More importantly, the expression amount of the top 10 most highly-expressed genes, which located in plasmids in exception of MCHK_1372, in bacteroids (Table S2) accounted for up to 32% of the total expression amount. Meanwhile, these top 10 genes in free-living cells accounted for up to 0.18% of the total expression amount. These suggest that gene expression is concentrated to a set of genes (gene clusters) in bacteroids to fulfill corresponding functional requirements.
1.2 Gene expression comparison and correlation
The expression levels for all the listed (6291) genes as measured by RNA-Seq and Microarrays were examined. Correlation between the two methods was high (Pearson's correlation coefficient R = 0.83), in spite of a compression effect resulting in smaller ratios in Microarrays (Figure 1B). This has been reported previously and is due in part to the limited dynamic range of array experiments [36], [37]. The comparison and distribution of the differentially expressed genes detected by RNA-Seq and Microarrays are presented in Figure 1C. It is apparent that RNA-Seq and Microarrays are consistent with each other in transcriptome profiling ability. Although RNA-Seq had greater accuracy and sensitivity relative to Microarrays, which had a relatively limited dynamic range for the detection of transcript levels due to background, saturation, and spot density and quality [36], [37]. A number of differentially expressed genes with exceptionally high change were detected by RNA-Seq. We listed the top 25 up- (Table S3) and down-regulated (Table S4) genes as called by both RNA-Seq and Microarrays. Most of the top 25 up-regulated genes in RNA-Seq and Microarrays were in parallel (16 genes were identical)and related to nitrogen fixation. In addition, we displayed the global gene expressions by plot visualization (Figure 2). This plot shows the overall location of differentially expressed genes in the chromosome and two plasmids. It was observed that the most prominent up-regulation occurred in the symbiosis plasmids (assigned as region III) and the most prominent down-regulation occurred in chromosome, assigned as regions I and II. The up-regulation regions in the symbiosis plasmids have a remarkable lower GC content. The accumulation of those genes in the symbiotic plasmids was correlated with nitrogen fixation. The two down-regulation regions in the chromosome are correlated with maintaining basal metabolism. The region I has several fab genes participating in fatty acid biosynthesis, while the region II encodes many of the flagellar assembly genes. Most genes in these two regions were down-regulated, which reflected the cellular and biological functional changes in the bacteroids to fulfill the symbiotic nitrogen fixation. It is speculated that analyses of the genes within these regions may reveal some novel insights into molecular mechanism of bacteroid differentiation and nitrogen fixation.
Figure 2. Visualization of global transcriptional profiles as determined by RNA-Seq and Microarrays.
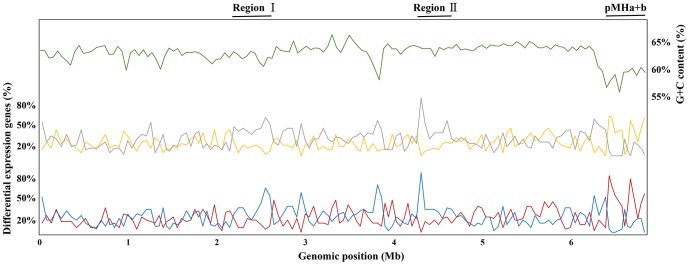
The plot shows a summary of RNA-Seq and Microarrays expression data at all regions in bacteroids. Across the entire genome, the percentages of up-regulated genes of RNA-Seq and Microarrays are shown in yellow and red, while those of the down-regulated shown in gray and blue, respectively. The G+C content is in green. The percentage of differentially expressed genes and the GC content were computed with a sliding window of every 50 genes. Note the most prominent up-regulation in the symbiosis plasmids and the most prominent down-regulation in regions I and II in the chromosome.
1.3 Genetic pathways altered in the bacteroid in response to nitrogen fixation
Even though a variety of sophisticated regulatory mechanisms occur at diverse levels of biological organization [38], we assumed that bacteroid genes significantly up-regulated were closely connected with the functional mechanisms of nitrogen fixation.
The assignment of genes to functional categories, as is commonly applied to transcriptome data [39], assists with the identification of differentially regulated pathways, and reduces our dependence on assumptions derived from individuals of differentially expressed genes within large-scale data sets. To survey the role that these genes play in supporting symbiotic nitrogen fixation, we classified them in accordance to the COG functional categories. Approximately 20% of up-regulated genes and 14% of down-regulated genes were classified as uncategorized genes in COG (data not shown). In the functional categories (Figure 3) which were identified as either up- or down-regulated (p/q<0.05), a large proportion of differentially regulated genes were categorized into either: 1) energy production and conversion, 2) amino acid transport and metabolism, 3) carbohydrate transport and metabolism, or 4) transcription. However, most of the down-regulated genes of M. huakuii 7653R were associated with cellular processes and signaling. Similar observations have been reported in S. meliloti [40] and R. etli [41] bacteroids. S. meliloti induces indeterminate-type nodules, while R. etli forms determinate-type nodules. Hence, the expression profile of bacteroids resembles that of non-growing cells, regardless of whether they are terminally differentiated or not. It is worthy of note that two functional groups, “general function prediction only” and “function unknown” genes, encompassed a large number of differentially expressed genes. This provided novel genetic material, and suggests a need for further investigation.
Figure 3. COG functional categories of differentially expressed genes identified by both RNA-Seq and Microarrays.

All differentially expressed genes during bacteroids were assigned to the 26 COG functional categories using RPS-BLAST (uncategorized genes not shown). Bars represent the number of up-regulated (orange) and down-regulated (blue) genes in bacteroids compared with free-living cells. The number in each bar represents its up/down-regulated percentage (%).
As there are many known genes important to bacteroid nitrogen fixation, we also identified genes participating in the regulatory mechanisms of nitrogen fixation and nodulation, flagellar assembly, urea cycle, poly-β-hydroxybutyrate (PHB) biosynthesis, protein secretion systems, two-component systems and polysaccharide synthesis (Figure S3). These genes reflect general genetic pathways adapted for use in nitrogen fixation. Some pathways are activated, whereas others are inactivated in bacteroids. For example, the nif and fix genes in 7653R bacteroids are all up-regulated, while the nod genes are down-regulated or at least retain the same expression level.
1.4 Gene set enrichment analysis
Gene set enrichment analysis (GSEA) [42] was performed to compare bacteroids and free-living cells in this work. GSEA of pathways and genes was performed by using the Category package in Bioconductor ver. 2.6.0 [43]. In this analysis, the gene sets of fewer than 10 genes were excluded. Using a permutation test 10 times, the cutoff of the significance level p values was chosen as 0.01 for the most significant pathways related to WD. Accordingly, the significant pathways were then identified when comparing bacteroids and free-living cells. The dataset has 6646 native features, and after collapsing features into gene symbols, there are 6646 genes. Gene set size filters (min = 15, max = 500) resulted in filtering out 123/197 gene sets, and the remaining 74 gene sets were used in the analysis. In the 74 gene sets, there were 35 and 39 gene sets are up-regulated in free-living cells and bacteroids, respectively. For these gene sets, 14 gene sets are significant at FDR < 25% and significantly enriched at nominal p value < 1% in free-living cells, while 30 gene sets are significant at FDR < 25% and 23 gene sets are significantly enriched at nominal p value < 1% in bacteroids.
In the list of enrichment in free-living cells (Table S5), there were gene sets related to membrane transport and cell motility, growth and death. While in the list of enrichment in bacteroids (Table S6), there were gene sets related to nitrogen metabolism and TCA cycle. GSEA showed that in our transcriptome dataset, free-living cells have a primary role in maintaining basal metabolism, whereas bacteroids have a primary role in nitrogen fixation.
2. Central biological metabolic pathways in bacteroids
At the molecular level, symbiotic nitrogen fixation arises as a consequence of the coordinated actions of a variety of genes and metabolites that in turn activate signal transduction cascades within the bacteroid. To survey the role that these differential expression genes played in supporting symbiotic nitrogen fixation, we tested for enriched KEGG pathways. In the list of enriched KEGG pathways (Table S7, Table S8), in the most 25 prominent KEGG pathways in free-living cells and bacteroids, we can see some pathways were different. In free-living cells, there were fatty acid metabolism, cell cycle, flagellar assembly and bacterial chemotaxis, while in bacteroids, there were glycolysis/gluconeogenesis, carbon fixation pathways, pentose phosphate pathway, citrate cycle (TCA cycle) and nitrogen metabolism. These results also support the point that free-living cells have a primary role in maintaining basal metabolism, whereas bacteroids have a primary role in nitrogen fixation.
To analyze the central metabolism in bacteroids and to visually identify these metabolic reactions, we connected a set of differentially expressed genes (identified via high-throughput analyses) and the enzymes from each metabolic pathway as defined in the KEGG database. A comparative analysis between bacteroid and free-living cell in each pathway allowed us to visualize and highlight any differences. We selected several important KEGG metabolic pathways and illustrated them in a cellular framework (Figure 4) to clearly demonstrate and identify the metabolic pathways activated or suppressed. Overall, C4-dicarboxylic acids provided by the host plant were transported into bacteroids and the main energy metabolism pathway remained active, as were ammonium synthesis pathways which supply ammonium to the host plant as a return.
Figure 4. Schematic overview of important KEGG metabolic pathways in bacteroids.

Arrows indicate the direction of the metabolism reaction. The enzymes participating in a specific step of reaction were shown in red for up-regulation, green for down-regulation and black for unchanged expression level. Several important differentially expressed genes of two-component systems and transporters (responsible for transportation of glutamine, asparagine, sulfate, molybdenum and C4-dicarboxylates) were displayed with the same patterns for direction and color.
2.1. Glycolysis, gluconeogenesis, Entner-Doudoroff and pentose phosphate pathways
As shown in Figure 4, most of the glycolytic and gluconeogenic enzymes identified were up-regulated. It accords with the report that a common metabolic trait for some rhizobia is the intense activity of gluconeogenesis pathway [44]. This indicated that the gluconeogenesis pathway actively participates in nitrogen fixation. There is evidence that the pentose phosphate pathway also actively participates in nitrogen fixation in R. etli [45]. While the pentose phosphate and Entner-Doudoroff pathways coordinately serve as probable major routes for the metabolism of sugars in fast-growing rhizobia [46]. In our results, most of the enzymes in pentose phosphate and Entner-Doudoroff pathways were up-regulated, which is consistent with the needs of the highly energy-dependent symbiotic nitrogen fixation reactions.
2.2. Citric acid cycle
Plants provide rhizobia with C4-dicarboxylic acids such as malate, fumarate, and succinate, which are imported into bacteroids via the dicarboxylate transport system (Dct) [47]. The Dct system of rhizobia consists of a dicarboxylate carrier protein DctA and a two-component regulatory system of DctB and DctD that serve as sensor kinases and transcriptional activators, respectively [48]. In this study, dctA, dctB and dctD genes were up-regulated. As C4-dicarboxylic acids are the sole carbon source to support bacteroid nitrogen fixation and are metabolized exclusively by the tricarboxylic acid (TCA) cycle, it was not surprising to find that dct genes were active in bacteroids. Interestingly, a part of the decarboxylating arm of the TCA cycle (involved in citrate synthase, aconitase, isocitrate dehydrogenase, and 2-ketoglutarate dehydrogenase) was transcriptionally up-regulated in bacteroids. Meanwhile the metabolism of malate and succinate was highly up-regulated since α-ketoglutarate is the source of glutamate. Glutamate is transformed into glutamine, which bacteroids subsequently provide to the host plant. Only partial sections of TCA cycle that function in nitrogen fixation program were specifically up-regulated in M. huakuii 7653R.
However, not all reports in the literature are in agreement. For example, up-regulation of TCA-cycle enzymes was recently reported in 28-day-old bacteroids of R. leguminosarum [15]. But in R. etli, the transcription data suggest that the TCA cycle is largely inactive [41]. A similar down-regulation was observed in S. meliloti [49]. Although α-ketoglutarate dehydrogenase mutants of B. japonicum have lower rates of nitrogen fixation per plant, they fix N2 at the same rate as the wild-type on a per-bacteroid basis [50]. These data suggest that, although α-ketoglutarate dehydrogenase mutants of B. japonicum are impaired for normal growth and development, once a bacteroid has formed it does not need a fully functioning TCA cycle. In this present study, we found that all essential steps were tailored to being up-regulated in bacteroids adapt to nitrogen fixation. These results counter that presented in the classical model of N2 reduction and suggest that either a bypass of the TCA cycle must operate in bacteroids. For example a α-ketoglutarate decarboxylase step and C4-dicarboxylic acids dehydrogenase step, or that the TCA cycle is non-cyclic, with intermediates leaving at a key branch point.
2.3. Polyhydroxybutyrate (PHB) biosynthesis
In this study, expression of PHB synthesis genes was down-regulated, and those of PHB degradation genes were unchanged. The bacterial carbon storage compound PHB accumulates in rhizobia cells within infection threads [51], [52]. It is possible that intracellular PHB stores may fuel cell division and growth during root infection and invasion as PHB can be used as a carbon and energy source for bacteroid formation. Most species of rhizobia synthesize PHB, but not all accumulate it during symbiosis with legumes. During the formation of bacteroids in indeterminate-type nodules, the PHB granules are broken down. Biochemically, PHB synthesis directly competes with N2 fixation for reductant [53]. As acetyl-CoA can either enter the TCA cycle via condensation with oxaloacetate, or form PHB, N2-fixing rhizobium bacteroids face a tradeoff between nitrogen fixation and PHB accumulation. The situation in determinate-type nodules is even more complex. Mature bacteroids continue to accumulate large amounts of PHB. One of the reasons might be that N2 was fixed as ureides rather than glutamine or asparagine. The other reason might be attributed to the fact that in the determinate-type nodules bacteroids are reversibly differentiated and have the reproductive capacity [3]. Therefore, PHB granules may fuel rhizobial cell division in the determinate-type nodules, but PHB is not essential for the terminally differentiated bacteroids in the indeterminate-type nodules. Trainer and Trevor (2007) demonstrated that PhaP proteins are required for PHB granule accumulation in S. meliloti [54]. But phaP is absent in the M. huakuii 7653R genome, which might be another reason for the lack of PHB accumulation in 7653R indeterminate-type nodules.
2.4. Branched-chain amino acids transport
Rhizobia are capable of utilizing a wide range of compounds from soil and rhizosphere as evident by the large number of transport systems they possess [7], [55]. For example, ATP binding cassette (ABC) transporters are abundant in rhizobia. Amino acid transport in Rhizobia occurs predominantly via two ABC transport systems, L-amino acid permease (Aap) and branched chain amino acid transport system (Bra). Both systems have broad-range specificity for amino acids and are together required for effective symbiotic nitrogen fixation [51]. The Aap, encoded by the aapJQMP operon, belongs to the polar amino acid transporter (PAAT) subclass of ABC transport systems [56]. The Bra, encoded by braC-DEFG, belongs to the hydrophobic amino acid transporter (HAAT) subclass of ABC transporters [57]. Bacteroids require branched-chain amino acids (LIV) transport, and this cannot be achieved via endogenous glutamate synthesis alone. Thus, although free-living cultures are prototrophic for LIV, bacteroids do become auxotroph [58]. ABC uptake systems are important in symbiosis, but they may only be active prior to bacteroid maturation [59]. Strains of R. leguminosarum (with mutations in both aap and bra) within nodulated peas only fixed N2 at around 30% of wild-type rates [51]. In this work, Aap and Bra systems in 7653R, which were homologous to R. leguminosarum Aap (AapJQMP) and Bra (BraDEFGC) respectively, were either down-regulated or remained unchanged. It is important to note, that several other bra gene clusters organized in one operon in 7653R genome exists, and all are up-regulated (Table 2). This suggests the presence of an alternative transport system for branched-chain amino acids in M. huakuii 7653R bacteroids, for which the specific functional mechanism remains to be determined.
Table 2. Genes associated with branched-chain amino acid transport in M. huakuii 7653R.
| GeneID | Gene | Function | Up or Down-Regulation |
| MCHK_1484 | aapJ | lysine-arginine-ornithine-binding periplasmic family protein | Unchanged |
| MCHK_1485 | aapQ | binding-dependent transport system inner membrane component family protein | Down |
| MCHK_1486 | aapM | inner membrane amino-acid ABC transporter permease protein yhdY | Down |
| MCHK_1487 | aapP | ABC transporter family protein | Down |
| MCHK_5281 | braC | ABC transporter, substrate binding protein | Unchanged |
| MCHK_5283 | braG | ABC transporter family protein | Unchanged |
| MCHK_5284 | braF | ABC transporter family protein | Unchanged |
| MCHK_5285 | braE | branched-chain amino acid transport system/permease component family protein | Unchanged |
| MCHK_5286 | braD | branched-chain amino acid transport system/permease component family protein | Unchanged |
| MCHK_5954 | braG | ABC transporter family protein | Up |
| MCHK_5955 | braF | ABC transporter family protein | Up |
| MCHK_5956 | braE | branched-chain amino acid transport system/permease component family protein | Up |
| MCHK_5957 | braD | branched-chain amino acid transport system/permease component family protein | Up |
| MCHK_6267 | braF | ABC transporter family protein | Up |
| MCHK_6268 | braD | branched-chain amino acid transport system/permease component family protein | Up |
| MCHK_6269 | braE | branched-chain amino acid transport system/permease component family protein | Up |
| MCHK_6270 | braC | branched-chain amino acid transport system substrate-binding protein | Up |
| MCHK_6271 | braG | ABC transporter family protein | Up |
| MCHK_0689 | braE | branched-chain amino acid transport system/permease component family protein | Up |
| MCHK_0690 | braD | branched-chain amino acid transport system/permease component family protein | Up |
| MCHK_0691 | braG | ABC transporter family protein | Up |
| MCHK_0692 | braF | ABC transporter family protein | Up |
2.5. Fatty acid metabolism
Analysis of transcriptome data indicated metabolism of fatty acids was not occurring in the 7653R bacteroids. RNA-Seq and Microarrays detected a variety of fab genes participating in fatty acid biosynthesis, yet their expression remained unchanged (Table S9). Fatty acid metabolism plays an important role in bacteroid metabolism as it supplies a variety of precursors, (e.g. components of rhizobial membranes, or lipopolysaccharides and coenzymes required in signal transduction). In the 18 dpi bacteroids, metabolism of fatty acid in R. etli is active [45]. In contrast, in the 28 dpi bacteroids, a drastic reduction in lipid biosynthesis was observed in B. japonicum [60]. As the host plant does not supply fatty acids; the requirement for lipids is reduced during the later stages of bacteroid nitrogen fixation, due in part to its non-dividing nature. More importantly, fatty acid synthesis requires NADPH as a reducing agent and acetyl-CoA as a precursor. The down-regulation of fatty acid metabolism provides NADPH and acetyl-CoA for use in bacteroid nitrogen fixation during this stage.
3. Bacteroid differentiation
3.1. Cell cycle and c-di-GMP
Indeterminate nodules are occupied by a single bacteroid, which has enlarged cells, increased genome amplification, and a loss of membrane integrity and reproductive capacity. In contrast, bacteroids in determinate nodules have no significant morphological differentiation and do maintain their capacity for cell division [3]. The bacteroid development in indeterminate nodules therefore represents a symbiosis-specific bacterial cell cycle event, which is closely connected with either intracellular persistence, or efficient nitrogen fixation. Thus, we sought to better understand how the bacterial cell cycle might be regulated for bacteroid differentiation and nitrogen fixation in indeterminate nodules.
Rhizobia cell cycle regulation has not been studied in great detail, with the exception of that in S. meliloti [61]; however, this has been examined in detail in Caulobacter crescentus [62]. It is apparent that rhizobia under free-living and symbiotic conditions undergo asymmetrical division as C. crescentus; the cell cycle of C. crescentus is providing insight for our analysis of cell cycle relevant gene expression in M. huakuii 7653R.
The CtrA protein was first characterized for C. crescentus [3], in which it is essential for viability and acts as a master regulator of the cell cycle [63]. CtrA controls at least 25% of the genes involved in cell cycle progression, including those required for DNA replication, DNA methylation, cell division, and biogenesis of flagella and pili [64]. The ctrA genes of S. meliloti [65], Brucella abortus [61], Rhodobacter capsulatus [66], and Ruegeria sp. strain TM1040 [67] have also been studied.
Based on analysis of RNA-Seq and Microarrays data in this study, in accordance with the center protein CtrA, a significant number of housekeeping genes were down-regulated at the nitrogen fixation stage except dnaA, thus we presumed that these genes are associated with the cell cycle (Table S10). The biological functions of these genes were associated with DNA replication, DNA methylation, cell division, flagellar assembly, flagellar ejection, CtrA degradation, polar morphogenesis and pili biogenesis (Figure 5). As DnaA protein activates the initiation of DNA replication in prokaryotes, dnaA was up-regulated in bacteroids, demonstrating its agreement with their genome endoreduplication at this stage. The gene expression profiles were congruent with cellular events during bacteroid differentiation, when the cells underwent successive rounds of genome multiplication, without accompanying cell division.
Figure 5. The putative CtrA regulatory network in M. huakuii 7653R according to the regulation model of cell cycle in C. crescentus.

Blue arrows indicate positive effects and blunt blue lines indicate negative effects. Genes up-regulated were shown in red and down-regulated shown in green in M. huakuii 7653R bacteroids. Phosphorylated CtrA autoregulated its own transcription and CtrA regulated genes associated with DNA replication, DNA methylation, cell division, flagellar assembly, CtrA degradation, cell division, polar morphogenesis and pili biogenesis.
The c-di-GMP relevant genes were down-regulated as well in the bacteroids. The intracellular concentration of c-di-GMP is controlled by diguanylate cyclase (DGC) which contains a GGDEF [68] or GGEEF [69] domain, and hydrolytic enzymes, phosphodiesterases (PDE) containing either a EAL [70] or HD-GYP [71] domain. In the RNA-Seq and Microarrays data of M. huakuii, we list genes containing the above conserved protein domains, and their expression (Table S11); most were down-regulated. Interestingly, two genes mhl3535 and mhl8229 were up-regulated. This suggests that the function of these two genes might differ from others. Actually, not all EAL or GGDEF domain-containing proteins are enzymatically active as are DGC or PDE. c-di-GMP may be involved in the regulation of bacteria and plant symbiosis [72]. But their function has not been investigated using genetic, or biochemical approaches. It remains to be determined when c-di-GMP plays and important role during nodulation and nitrogen fixation. Thus, the intracellular level and dynamic changes of c-di-GMP in bacteroids should be further studied.
3.2. BacA and TypA
Bacteroids in indeterminate nodules of Inverted Repeat Lacking Clade (IRLC) legumes undergo terminal differentiation caused by Nodule-specific Cysteine-Rich peptides (NCRs) [73], [74]. The BacA protein from S. meliloti was the first protein shown to be essential for bacteroid development [75]. It has also been shown to be essential for the symbiosis between M. huakuii 7653R and A. sinicus [76]. BacA provides a critical protective role for bacteria entering infected plant cells. The essential function of BacA in symbiosis seems limited to indeterminate-nodules-formation host plant, which produce NCRs. However, it remains to be determined whether this occurs via a direct interaction of BacA with the NCR peptides, or whether the peptide transport function of BacA could be involved in maintaining an oxidizing environment within the periplasm.
TypA, also named BipA, belongs to a superfamily of ribosome-binding GTPases within the TRAFAC class (translation factors) of GTPases [77], [78]. TypA has been suggested to be involved in the regulation of virulence and stress responses in pathogenic E. coli [79], Salmonella enterica Serovar Typhimurium [80] and Pseudomonas aeruginosa [81], and stress responses in non-pathogenic S. meliloti [82] and Bacillus subtilis.[83].
The establishment of the Rhizobium-legume symbiosis is similar to pathogenesis in several respects. In both cases, bacteria have to penetrate the plant tissues, avoid the host defenses, and adapt to the physiological conditions found in the new environment [84], [85]. During nodule invasion and bacteroid differentiation, rhizobia are exposed to new physiological conditions (pH, osmotic concentration, oxidative stress, defense responses and NCR peptides). TypA was required for survival of S. meliloti under certain stress conditions, such as growth at low temperature or low pH and in the presence of sodium dodecyl sulfate (SDS). It is noteworthy that TypA is involved in bacteroid development and is indispensable for symbiosis depending on the host plant type (indeterminate-nodules-formation host plant) [82]. This demonstrates that TypA and BacA have highly similarity in symbiotic phenotypes. It is promoted us to speculate the possible functional associations between TypA and BacA.
In a parallel study, we found that M. huakuii 7653R TypA protein interacts with BacA in a bacterial two-hybrid system. In addition, a typA knockout mutant exhibited abnormal development of bacteroids in 7653R (unshown) with highly similarity to the symbiotic phenotypes of the bacA mutant. Consistent with these data, the gene expressions of bacA and typA are both up-regulated in bacteroids. These results suggest that TypA, as a regulatory factor, might regulate the activity of BacA and therefore jointly mediate bacteroid differentiation.
4. Mapping gene expression dataset into 7653R PPI network
The M. huakuii 7653R genome has been recently sequenced (Accession Nos. CP006581-CP006583, http://www.ncbi.nlm.nih.gov/genome/11322). It encodes around 7000 proteins; nearly 40% of them are of unknown function, although some are defined “belonging to a superfamily”. High-throughput experimental data can provide valuable information for linking unknown genes with specific biological roles; and is thus widely used to predict gene function. For example, much protein-protein interaction (PPI) data has been acquired for several microorganisms [28], [86]–[88]. Global viewing of PPI is also a powerful tool for assigning biological role to large numbers of proteins predicted by whole-genome sequencing. Identification of interactions between characterized and uncharacterized proteins infers functional relatedness, thus integrative analysis of datasets may reveal function of uncharacterized genes. Accordingly, we constructed PPI networks for M. huakuii 7653R based on the two published PPI reference networks of M. huakuii bv loti MAFF303099 [28] and E. coli [29] published. The former MAFF303099 strain belongs to the same genus and its genome is highly similar to M. huakuii 7653R. The inferred PPI network of M. huakuii 7653R has 2607 nodes and 10307 edges (Table S1). We integrated gene expression data and protein interaction to identify inter-connected, differentially-expressed components, which may represent important pathways or protein complexes that are altered in bacteroids of 7653R.
4.1. Nitrogen fixation
By selecting those genes with a 5-fold change in expression and mapping the data into the PPI network of 7653R, an interesting subnetwork was identified (Figure 6, Table 3, see Methods). The subnetwork had 113 nodes and 214 edges, including 78 genes with 5-fold change in expression. Enriched GO term analysis found that the most significant GO term was that over-represented were those involved with nitrogen fixation (Figure S4). These results indicate that the constructed PPI network of 7653R and the transcriptomes data identified in this study were reliable.
Figure 6. An inter-connected, differentially-expressed PPI subnetwork in bacteroids altered for nitrogen fixation.

The genes with differential expression fold change greater than 5 were shown in red; those greater than 20 were shown in dark red; those down-regulated were shown in green and others were shown in yellow.
Table 3. The set of genes in the M. huakuii 7653R subnetwork of nitrogen fixation.
| ID | Name | Function | Fold change |
| MCHK_8175 | nifD | nitrogenase molybdenum-iron protein alpha chain | 228.1625 |
| MCHK_8172 | nifE | nitrogenase MoFe cofactor biosynthesis protein NifE | 272.4621 |
| MCHK_8188 | nifQ | nifQ family protein | 36.087 |
| MCHK_1835 | nifS | aminotransferase class-V family protein | 11.1585 |
| MCHK_8164 | nifT | putative nitrogen fixation protein FixU | 2.7712 |
| MCHK_8219 | fixC | FAD dependent oxidoreductase family protein | 153.2337 |
| MCHK_2178 | ntrX | response regulator | 0.9891 |
| MCHK_5476 | glnZ | PII-like protein Pz | 43.3614 |
| MCHK_2333 | gltA | citrate (Si)-synthase | 6.0617 |
| MCHK_8190 | rpoN | sigma-54 factor, core binding domain protein | 28.7755 |
| MCHK_7137 | rpoN | RNA polymerase sigma-54 factor | 40.125 |
| MCHK_5199 | rpoH | RNA polymerase sigma-32 factor | 5.7607 |
| MCHK_2096 | rpsN | 30S ribosomal protein S14 | 0.5787 |
| MCHK_1837 | sufC | feS assembly ATPase SufC | 6.589 |
| MCHK_1838 | sufD | feS assembly protein SufD | 7.4377 |
| MCHK_1111 | sufE | fe-S metabolism associated domain protein | 0.5652 |
| MCHK_1839 | sufS | cysteine desulfurase, SufS subfamily protein | 9.8869 |
| MCHK_2452 | urtB | urea ABC transporter, permease protein UrtB | 16.8354 |
| MCHK_2450 | urtD | urea ABC transporter, ATP-binding protein UrtD | 9.3897 |
| MCHK_2449 | urtE | urea ABC transporter, ATP-binding protein UrtE | 5.1336 |
| MCHK_8359 | hybC | nickel-dependent hydrogenase family protein | 52.028 |
| MCHK_8347 | hypD | hydrogenase expression/formation protein HypD | 5.6356 |
| MCHK_8345 | hypE | hydrogenase expression/formation protein HypE | 5.6193 |
| MCHK_5539 | acnA | aconitate hydratase 1 | 9.0088 |
| MCHK_0267 | aldH | aldehyde dehydrogenase family protein | 0 |
| MCHK_1080 | betC | choline-sulfatase | 5.9333 |
| MCHK_1499 | betI | transcriptional repressor BetI | 5.379 |
| MCHK_1081 | betI | transcriptional repressor BetI | 7.0643 |
| MCHK_5793 | cysK | cysteine synthase A | 0 |
| MCHK_6087 | degPch1 | protease Do family protein | 12.494 |
| MCHK_1148 | degPch2 | protease Do family protein | 22.5974 |
| MCHK_5827 | dnaJ | chaperone protein DnaJ | 0.6215 |
| MCHK_5828 | dnaK | chaperone protein DnaK | 1.7466 |
| MCHK_1003 | dsbA | DSBA-like thioredoxin domain protein | 0.6718 |
| MCHK_0964 | dxs | 1-deoxy-D-xylulose-5-phosphate synthase | 5.6548 |
| MCHK_1205 | era | GTP-binding protein Era | 6.0056 |
| MCHK_3376 | exsG | putative two-component sensor histidine kinase protein | 6.334 |
| MCHK_8419 | folD1 | tetrahydrofolate dehydrogenase/cyclohydrolase, catalytic domain protein | 8.8978 |
| MCHK_1496 | fumC | fumarate hydratase, class II | 7.3542 |
| MCHK_2080 | fusA | translation elongation factor G | 0.9551 |
| MCHK_0391 | galE5 | short chain dehydrogenase family protein | 0.9348 |
| MCHK_4109 | gatB | yqey-like family protein | 14.0403 |
| MCHK_7093 | glmS | glutamine-fructose-6-phosphate transaminase | 8.8581 |
| MCHK_4716 | grpE | grpE family protein | 1.1287 |
| MCHK_5101 | grx | redoxin family protein | 34.8731 |
| MCHK_1705 | hemB | delta-aminolevulinic acid dehydratase | 1.0837 |
| MCHK_6079 | hisB | imidazoleglycerol-phosphate dehydratase family protein | 6.858 |
| MCHK_5153 | hisP | ABC transporter family protein | 5.6978 |
| MCHK_3552 | htpG | histidine kinase-, DNA gyrase B-, and HSP90-like ATPase family protein | 0.3735 |
| MCHK_3523 | kamA | kamA family protein | 1.2954 |
| MCHK_6499 | kdsB | 3-deoxy-D-manno-octulosonate cytidylyltransferase | 0.8333 |
| MCHK_1589 | kdtA | 3-Deoxy-D-manno-octulosonic-acid transferase family protein | 5.6107 |
| MCHK_5594 | leuD | 3-isopropylmalate dehydratase, small subunit | 5.2354 |
| MCHK_3575 | llpU | short chain dehydrogenase family protein | 5.5047 |
| MCHK_2788 | maiA | maleylacetoacetate isomerase | 1.0738 |
| MCHK_4398 | mdtA | efflux transporter, RND family, MFP subunit | 5.1515 |
| MCHK_6536 | metK | methionine adenosyltransferase | 0.9219 |
| MCHK_5860 | metQ | conserved hypothetical protein | 5.4162 |
| MCHK_0867 | mexF | RND transporter, hydrophobe/amphiphile efflux-1 family protein | 58.6485 |
| MCHK_2510 | mfd | transcription-repair coupling factor | 1.5562 |
| MCHK_4118 | mntH | metal ion transporter, metal ion family protein | 14.1307 |
| MCHK_5902 | msrB | methionine-R-sulfoxide reductase | 10.4404 |
| MCHK_1856 | murI | glutamate racemase | 7.7059 |
| MCHK_1985 | panB | 3-methyl-2-oxobutanoate hydroxymethyltransferase | 6.6055 |
| MCHK_1984 | panC | pantoate—beta-alanine ligase | 6.8577 |
| MCHK_6052 | phnN | phosphonate metabolism protein/1,5-bisphosphokinase (PRPP-forming) PhnN | 2.4099 |
| MCHK_6535 | phrR | helix-turn-helix family protein | 10.2277 |
| MCHK_1763 | potA | ABC transporter family protein | 23.1647 |
| MCHK_0211 | potF | bacterial extracellular solute-binding family protein | 0.3783 |
| MCHK_5082 | pstB | phosphate ABC transporter, ATP-binding protein | 2.405 |
| MCHK_5085 | pstS | phosphate ABC transporter, phosphate-binding protein PstS | 6.2484 |
| MCHK_2812 | putA | delta-1-pyrroline-5-carboxylate dehydrogenase | 0.2091 |
| MCHK_2418 | queA | S-adenosylmethionine:tRNA ribosyltransferase-isomerase | 0 |
| MCHK_6121 | rplT | ribosomal protein L20 | 5.046 |
| MCHK_5489 | sdhA | succinate dehydrogenase, flavoprotein subunit | 5.6783 |
| MCHK_6358 | sdhB | universal stress family protein | 1.4018 |
| MCHK_2704 | tig | trigger factor | 1.6301 |
| MCHK_5374 | typA | GTP-binding protein TypA/BipA | 3.7535 |
| MCHK_3307 | ugpC | ABC transporter family protein | 6.1079 |
| MCHK_0451 | uxuA | mannonate dehydratase | 16.059 |
| MCHK_3900 | xfp | D-xylulose 5-phosphate/D-fructose 6-phosphate phosphoketolase family protein | 0.2442 |
| MCHK_8196 | xylB | aryl-alcohol dehydrogenase | 5.2102 |
| MCHK_8197 | xylC | benzaldehyde dehydrogenase NAD+ | 19.882 |
| MCHK_2864 | yfbK | von Willebrand factor type A domain protein | 13.9193 |
| MCHK_0866 | mhl0866 | efflux transporter, RND family, MFP subunit | 40.6938 |
| MCHK_2865 | mhl2865 | RNA polymerase sigma factor, sigma-70 family protein | 15.6472 |
| MCHK_4854 | mhl4854 | luciferase-like monooxygenase family protein | 7.7335 |
| MCHK_4912 | mhl4912 | bacterial regulatory helix-turn-helix, lysR family protein | 12.7467 |
| MCHK_5957 | mhl5957 | branched-chain amino acid transport system/permease component family protein | 6.4752 |
| MCHK_8229 | mhl8229 | sensory box protein | 8.4814 |
| MCHK_0275 | mhr0275 | tetratricopeptide repeat family protein | 0.4469 |
| MCHK_0381 | mhr0381 | chain length determinant family protein | 1.1847 |
| MCHK_0693 | mhr0693 | bacterial regulatory s, gntR family protein | 10.9707 |
| MCHK_1555 | mhr1555 | marR family protein | 0.8471 |
| MCHK_5071 | mhr5071 | HWE histidine kinase family protein | 5.2004 |
| MCHK_5151 | mhr5151 | amino ABC transporter, permease, 3-TM region, His/Glu/Gln/Arg/opine family domain protein | 7.8098 |
| MCHK_4160a | gmhB | mll2559 protein | 1.3228 |
| MCHK_0168a | mhl0168 | mll6478 protein | 5.9853 |
| MCHK_6013a | mhl6013 | mll4959 protein | 6.0062 |
| MCHK_0893a | mhr0893 | putative uncharacterized protein | 1.5161 |
| MCHK_2058a | mhl2058 | conserved hypothetical protein | 0.4313 |
| MCHK_2321a | mhl2321 | putative esterase/lipase/thioesterase | 0.8216 |
| MCHK_5691a | mhl5691 | putative uncharacterized domain protein | 8.5342 |
| MCHK_3151a | mhl3151 | ykud domain protein | 8.1921 |
| MCHK_1970a | mhr1970 | bacterial transferase hexapeptide family protein | 8.2936 |
| MCHK_3571a | mhl3571 | ABC transporter family protein | 12.0058 |
| MCHK_4853a | mhl4853 | flavin reductase like domain protein | 5.2647 |
| MCHK_5785a | mhl5785 | FMN-binding domain protein | 6.9947 |
| MCHK_6017a | mhl6017 | bacterial regulatory s, lacI family protein | 5.7227 |
| MCHK_0963a | mhr0963 | pirin C-terminal cupin domain protein | 5.4469 |
| MCHK_3741a | mhr3741 | periplasmic binding s and sugar binding domain of the LacI family protein | 6.9341 |
| MCHK_5181a | mhr5181 | bacterial regulatory s, tetR family protein | 7.5806 |
| MCHK_7135a | mhr7135 | putative peroxiredoxin sll1621 | 14.553 |
: The unknown function genes.
An overview analysis on the biological function of gene set (Table 3), indicated that genes in the subnetwork reflected the main features of symbiotic nitrogen fixation. (e.g. highly expressed genes such as nif, fix, suf, sigma factor σ54, ammonium assimilation associated, amino acid transport, etc.). Nif and Fix are important components of the symbiotic nitrogen fixation machinery. The nif gene encodes dinitrogenase, nitrogenase reductase, and iron-molybdenum cofactor synthesis proteins, whereas the fix gene encodes electron transfer relevant ferredoxins or flavoproteins. The Suf gene is involved in iron-sulfur (Fe-S) cluster assembly. SufS is cysteine desulfurase, similar to NifS, and supplies sulfur for Fe-S centers. The SufBCD complex activates the cysteine desulfurase activity of SufS in conjunction with the SufE that is a sulfur acceptor protein. The high expression of these suf genes suggests that bacteroids require a large amount of Fe-S clusters and that Fe-S clusters provided by SufS proteins may be used to sustain nitrogen fixation as well as the NifS protein. Lastly, although it has been previously reported that expression levels of genes encoding the PII-like protein of R. etli [89] and S. meliloti [90] are lower in bacteroids than in free-living cells, our results suggest otherwise, as MCHK_5476 (PII-like protein Pz) was highly expressed in 7653R bacteroids. This suggests that transcriptional regulation of nitrogen sensing and assimilation in M. huakuii 7653R bacteroids is different from that of R. etli and S. meliloti. In addition, several amino acid transport system associated proteins were also members of the identified subnetwork, reflecting the importance of amino acids, particularly branched-chain amino acids that are exchanged between bacteroids and host cells (refer to the section 3.2.4). It is interesting to note the importance of rpoH (RNA polymerase sigma-32 factor) in the nitrogen fixation subnetwork. The transcriptional regulation of nitrogen fixation associated genes is generally mediated through RpoN, a RNA polymerase sigma-54 factor. In S. meliloti, RpoH1 is required for nitrogen-fixing symbiosis with alfalfa, the rpoH1 mutant exhibits a nitrogen fixation defect (Fix−) and the rpoH1 is expressed within a symbiotic nodule [91]. Similarly in 7653R, MCHK_5199 (encoding RNA polymerase sigma-32 factor) is up-regulated, which supports RpoH as having an essential role in the PPI complex of nitrogen fixation. Some hub genes which can be simultaneously targeted by a comparatively large number of genes are related to the resistance/nodulation/cell division (RND) family, such as MCHK_0866 and MCHK_4398. Transport systems of the RND family are the major cause of antibiotic resistance in clinically relevant Gram-negative bacteria [92]. Lindemann, et al reported that a mutant lacking this predicted RND transport system (designated BdeAB), confers antibiotic resistance and is required for an efficient symbiosis; specifically within soybean [93]. This suggests that the RND family might be involved in nitrogen fixation. Earlier studies have demonstrated that two genes encoding a putative multidrug efflux pump of the RND/MFP family are co-transcribed with an rpoH gene in B. japonicum [94]. Our results were similar in that RpoH and RND family proteins were in the subnetwork of nitrogen fixation. Lastly, nearly 15% of the genes in the subnetwork were of “unknown function”; are ascribed to novel candidates essential to nitrogen fixation; and therefore certainly worthy of further examination.
4.2. CtrA-centered cell cycle
In order to identify the subnetwork mediated by CtrA connected with the cell cycle and bacteroid differentiation, we dissected the 7653R PPI network by using CtrA as a specific target. Then we analyzed this CtrA subnetwork which consists of 30 genes (Table 4, Figure 7). Based on the analysis of RNA-Seq and Microarrays data we found that expression levels for most of the genes in the subnetwork were down-regulated or unchanged. It was found that four genes directly interacting with CtrA, which regulated cell cycle progression [95]–[98]. In particular, six genes (MCHK_0391, MCHK_3151, MCHK_5082, MCHK_5476, MCHK_5691 and MCHK_6536) present in the CtrA-centered subnetwork (Table 4), existed in the “nitrogen fixation” subnetwork (Table 3) as well. Among these genes, the PII-like protein Pz was a nitrogen sensing protein [99]; function of the remaining genes is unknown. Therefore, the CtrA subnetwork might be related to nitrogen fixation through a yet to be determined “cross-talk” mechanism. Several phosphate ABC transporters, responsible for phosphate uptake, were also found. This suggests the involvement of some level of coordination to meet the higher phosphate demand that occurs during bacteroid nitrogen fixation. There were also several glucose-methanol-choline (GMC) oxidoreductase family proteins in the CtrA-centered subnetwork. This is perhaps not surprising, as oxidation-reduction reactions are a fundamental reaction occurring in all organisms. The family of glucose–GMC oxidoreductases is represented by a wide variety of enzymes present in both prokaryotic and eukaryotic organisms. The previously unidentified GMC oxidoreductase gene was present in the subnetwork of 7653R; it is plausible that this gene is involved in the cell cycle. This subnetwork provided novel insights into the CtrA-centralized network in M. huakuii 7653R relevant to cell cycle, cell differentiation, and nitrogen sensing in bacteroids.
Table 4. The subnetwork of CtrA with interacting proteins in M. huakuii 7653R.
| ID | Name | Function | Up or Down-Regulation |
| MCHK_1078 | betA | choline dehydrogenase | Up |
| MCHK_5082a | pstB | phosphate ABC transporter, ATP-binding protein | Up |
| MCHK_5476a | glnZ | PII-like protein Pz | Up |
| MCHK_5691a | mhl5691 | putative uncharacterized domain protein | Up |
| MCHK_3151a | mhl3151 | ykud domain protein | Up |
| MCHK_4201 | betA | GMC oxidoreductase family protein | Unchanged |
| MCHK_0414 | betA | GMC oxidoreductase family protein | Unchanged |
| MCHK_0614 | mhr0614 | GMC oxidoreductase family protein | Unchanged |
| MCHK_1015 | wbnF | short chain dehydrogenase family protein | Unchanged |
| MCHK_0391a | galE5 | short chain dehydrogenase family protein | Unchanged |
| MCHK_1025 | wbiB | NAD dependent epimerase/dehydratase family protein | Unchanged |
| MCHK_1847 | recA | protein RecA | Unchanged |
| MCHK_2439 | clpP1 | clp protease family protein | Unchanged |
| MCHK_4371 | ctrA | response regulator | Unchanged |
| MCHK_6536a | metK | methionine adenosyltransferase | Unchanged |
| MCHK_1820 | mhl1820 | putative oxidoreductase | Unchanged |
| MCHK_5541 | mhr5541 | putative uncharacterized domain protein | Unchanged |
| MCHK_5341b | ftsK1 | ftsK/SpoIIIE family protein | Down |
| MCHK_5471b | ftsK1 | ftsK/SpoIIIE family protein | Down |
| MCHK_4567 | mhr4567 | GMC oxidoreductase family protein | Down |
| MCHK_4720 | alkJ | GMC oxidoreductase family protein | Down |
| MCHK_1789 | clpP | ATP-dependent Clp protease, proteolytic subunit ClpP | Down |
| MCHK_1790b | clpX | ATP-dependent Clp protease, ATP-binding subunit ClpX | Down |
| MCHK_6106 | fadB | UDP-glucose/GDP-mannose dehydrogenase family, NAD binding domain protein | Down |
| MCHK_1018 | rfbB | dTDP-glucose 4,6-dehydratase | Down |
| MCHK_4463 | flgK | flagellar hook-associated protein FlgK | Down |
| MCHK_4772 | phnG | phosphonate C-P lyase system protein PhnG | Down |
| MCHK_4842 | clpB | ATP-dependent chaperone ClpB | Down |
| MCHK_5239b | rcdA | regulator of CtrA degradation | Down |
| MCHK_1035 | mhr1035 | short chain dehydrogenase family protein | Down |
The protein also in the nitrogen fixation subnetwork.
The protein interacts directly with CtrA.
Figure 7. The subnetwork of CtrA and its interacting proteins in M. huakuii 7653R.
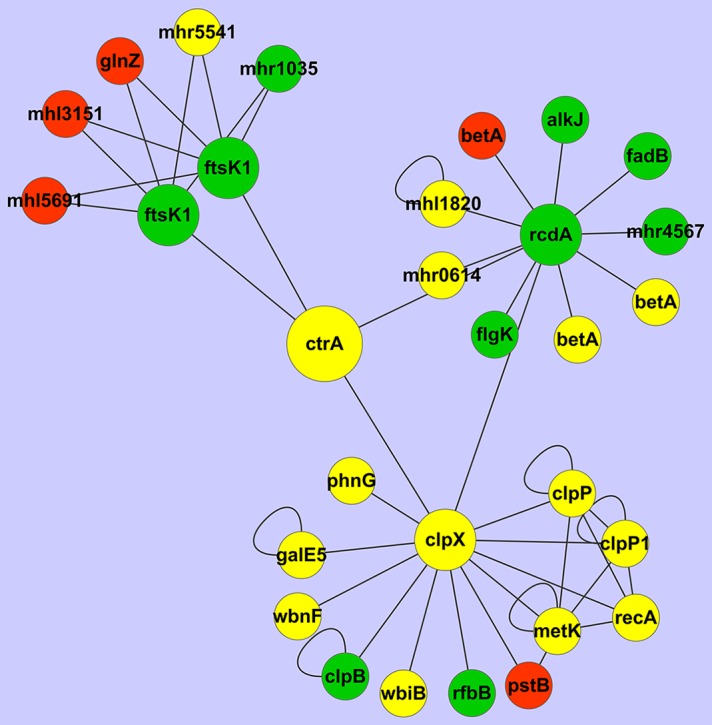
The genes up-regulated were shown in red, those down-regulated were shown in green and those of no change were shown in yellow.
Conclusion
In the past decade, several genome-wide transcriptome studies have described differential gene expression in bacteroids. These studies identified many genes involved in nitrogen-fixing symbiosis. However, the RNA-Seq method used in this study (in combination with Microarrays) examines a larger range of differential gene expression levels. In addition, we developed an improved method for the isolation and purification of bacteroid RNA for use in subsequent transcriptome analysis of this, and other Rhizobium-legume symbiosis systems. To examine the regulatory mechanisms involved in bacteroid differentiation, we focused our analysis on the putative PPI subnetwork, and the molecules participating in bacteroid differentiation. We found that CtrA/c-di-GMP may be involved in regulation of the cell cycle, bacteroid development, and nitrogen fixation. These data also suggest that bacA/typA might coordinately mediate bacteroid differentiation, and adaption to intracellular environments of infected host cells. In order to further understand how the genes determine the progression to bacteroid differentiation and nitrogen fixation, we expanded our analyses to include interaction of genes, rather than a single, or at best several gene functions. We integrated gene expression data and protein interaction data of 7653R to examine interaction between differentially expressed genes. Functional enrichment analysis revealed that biological processes are altered in nodule bacteroids compared with free-living cells. A few hubs include critical proteins or transcription factors, which alter expression level and therefore have a significant role in bacteroid differentiation and nitrogen fixation. Hence, the results described in this study provide a global biological landscape and novel insight that advances our understanding of bacteroid differentiation and nitrogen fixation in indeterminate-type root nodules. It also provides a valuable dataset for further functional confirmation, and use as a reference for investigation of other Rhizobium-legume symbiosis systems.
Supporting Information
The workflow of constructing PPI networks for M. huakuii 7653R. A: the schematic of the homologous protein-protein interactions network through the concepts of orthologous conservation (or interologs); B: the schematic illustration of the search for M. huakuii 7653R protein–protein interactions.
(TIFF)
The distribution of differentially expressed genes (bacteroids vs. free-living cells) which account for 80% of total expression amounts in COG functional categories. All genes were assigned to 21 COG functional categories using RPS-BLAST (uncategorized genes not shown). Bars represent the numbers of corresponding genes in free-living cells (green) and bacteroids (blue). The number in each bar represents its percentage (%).
(TIFF)
Schematic view of expressions of known genes associated with pathways of important biological function. An overview of the differentially expressed genes in bacteroids compared with free-living cells. The up-regulated genes are shown in red; the down-regulated genes are shown in green; the unchanged genes are shown in black. Genes were grouped according to their biological function or process involved; membrane-associated proteins were positioned over the inner and outer membranes.
(TIFF)
Up-regulated GO terms associated with bacteroids identified by BiNGO plugin. The enriched GO term of the subnetwork was nitrogen fixation.
(TIFF)
The PPI network of M. huakuii 7653R inferred from the two published PPI reference networks of M. huakuii bv loti MAFF303099 and E. coli.
(XLSX)
The top 10 of highly-expressed genes in M. huakuii 7653R bacteroids revealed by RNA-Seq. a The percentage represents the proportion of the gene transcriptional amounts to the total transcriptional amounts.
(DOC)
Top 25 up-regulated genes in M. huakuii 7653R bacteroids revealed by RNA-Seq and Microarrays.
(DOC)
Top 25 down-regulated genes in M. huakuii 7653R bacteroids revealed by RNA-Seq and Microarrays.
(DOC)
The significantly up-regulated pathways based on GSEA in M. huakuii 7653R free-living cells.
(XLSX)
The significantly up-regulated pathways based on GSEA in M. huakuii 7653R.
(XLSX)
Enriched KEGG pathways of differential expression genes in M. huakuii 7653R free-living cells.
(XLSX)
Enriched KEGG pathways of differential expression genes in M. huakuii 7653R.
(XLSX)
fab genes participating in fatty acid biosynthesis in M. huakuii 7653R.
(XLSX)
Genes associated with cell cycle and putatively regulated by CtrA in M. huakuii 7653R.
(XLSX)
Genes associated with c-di-GMP synthesis and degradation in M. huakuii 7653R.
(XLSX)
Funding Statement
This work was supported by funds from the National Basic Research Program of China (973 program grant no. 2010CB126502), Research Fund for the Doctoral Program of Higher Education of China (20110146110012), the National Natural Science Foundation of China (grant no. 31371549 and 31100602), and the Fundamental Research Funds for the Central Universities (2009PY020, 2010QC016 and 2011QC066). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Oldroyd GE, Downie JA (2008) Coordinating nodule morphogenesis with rhizobial infection in legumes. Annu Rev Plant Biol 59: 519–546. [DOI] [PubMed] [Google Scholar]
- 2. Wojciechowski MF, Lavin M, Sanderson MJ (2004) A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am J Bot 91: 1846–1862. [DOI] [PubMed] [Google Scholar]
- 3. Mergaert P, Uchiumi T, Alunni B, Evanno G, Cheron A, et al. (2006) Eukaryotic control on bacterial cell cycle and differentiation in the Rhizobium-legume symbiosis. Proc Natl Acad Sci U S A 103: 5230–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prell J, Poole P (2006) Metabolic changes of rhizobia in legume nodules. Trends Microbiol 14: 161–168. [DOI] [PubMed] [Google Scholar]
- 5. Becker-Andre M, Hahlbrock K (1989) Absolute mRNA quantification using the polymerase chain reaction (PCR). A novel approach by a PCR aided transcript titration assay (PATTY). Nucleic Acids Res 17: 9437–9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270: 467–470. [DOI] [PubMed] [Google Scholar]
- 7. Galibert F, Finan TM, Long SR, Puhler A, Abola P, et al. (2001) The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293: 668–672. [DOI] [PubMed] [Google Scholar]
- 8. Gonzalez V, Santamaria RI, Bustos P, Hernandez-Gonzalez I, Medrano-Soto A, et al. (2006) The partitioned Rhizobium etli genome: genetic and metabolic redundancy in seven interacting replicons. Proc Natl Acad Sci U S A 103: 3834–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goodner B, Hinkle G, Gattung S, Miller N, Blanchard M, et al. (2001) Genome sequence of the plant pathogen and biotechnology agent Agrobacterium tumefaciens C58. Science 294: 2323–2328. [DOI] [PubMed] [Google Scholar]
- 10. Kaneko T, Nakamura Y, Sato S, Asamizu E, Kato T, et al. (2000) Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti (supplement). DNA Res 7: 381–406. [DOI] [PubMed] [Google Scholar]
- 11. Brechenmacher L, Kim MY, Benitez M, Li M, Joshi T, et al. (2008) Transcription profiling of soybean nodulation by Bradyrhizobium japonicum. Mol Plant Microbe Interact 21: 631–645. [DOI] [PubMed] [Google Scholar]
- 12. Pessi G, Ahrens CH, Rehrauer H, Lindemann A, Hauser F, et al. (2007) Genome-wide transcript analysis of Bradyrhizobium japonicum bacteroids in soybean root nodules. Mol Plant Microbe Interact 20: 1353–1363. [DOI] [PubMed] [Google Scholar]
- 13. Uchiumi T, Ohwada T, Itakura M, Mitsui H, Nukui N, et al. (2004) Expression islands clustered on the symbiosis island of the Mesorhizobium loti genome. J Bacteriol 186: 2439–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Becker A, Berges H, Krol E, Bruand C, Ruberg S, et al. (2004) Global changes in gene expression in Sinorhizobium meliloti 1021 under microoxic and symbiotic conditions. Mol Plant Microbe Interact 17: 292–303. [DOI] [PubMed] [Google Scholar]
- 15. Karunakaran R, Ramachandran VK, Seaman JC, East AK, Mouhsine B, et al. (2009) Transcriptomic analysis of Rhizobium leguminosarum biovar viciae in symbiosis with host plants Pisum sativum and Vicia cracca. J Bacteriol 191: 4002–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26: 1135–1145. [DOI] [PubMed] [Google Scholar]
- 17. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gonzalez-Ballester D, Casero D, Cokus S, Pellegrini M, Merchant SS, et al. (2010) RNA-seq analysis of sulfur-deprived Chlamydomonas cells reveals aspects of acclimation critical for cell survival. Plant Cell 22: 2058–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, et al. (2008) A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321: 956–960. [DOI] [PubMed] [Google Scholar]
- 20. Yoder-Himes DR, Chain PS, Zhu Y, Wurtzel O, Rubin EM, et al. (2009) Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc Natl Acad Sci U S A 106: 3976–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y, Tian CF, Chen WF, Wang L, Sui XH, et al. (2013) High-resolution transcriptomic analyses of Sinorhizobium sp. NGR234 bacteroids in determinate nodules of Vigna unguiculata and indeterminate nodules of Leucaena leucocephala. PLoS One 8: e70531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kogenaru S, Qing Y, Guo Y, Wang N (2012) RNA-seq and microarray complement each other in transcriptome profiling. BMC Genomics 13: 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beringer JE (1974) R factor transfer in Rhizobium leguminosarum. J Gen Microbiol 84: 188–198. [DOI] [PubMed] [Google Scholar]
- 24. Chen DS, Li YG, Zhou JC (2007) The symbiosis phenotype and expression patterns of five nodule-specific genes of Astragalus sinicus under ammonium and salt stress conditions. Plant Cell Rep 26: 1421–1430. [DOI] [PubMed] [Google Scholar]
- 25. Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98: 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
- 27. Matthews LR, Vaglio P, Reboul J, Ge H, Davis BP, et al. (2001) Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or "interologs". Genome Res 11: 2120–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shimoda Y, Shinpo S, Kohara M, Nakamura Y, Tabata S, et al. (2008) A large scale analysis of protein-protein interactions in the nitrogen-fixing bacterium Mesorhizobium loti. DNA Res 15: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xenarios I, Rice DW, Salwinski L, Baron MK, Marcotte EM, et al. (2000) DIP: the database of interacting proteins. Nucleic Acids Res 28: 289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mushegian AR, Koonin EV (1996) A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci U S A 93: 10268–10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ideker T, Ozier O, Schwikowski B, Siegel AF (2002) Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 18 Suppl 1S233–240. [DOI] [PubMed] [Google Scholar]
- 33. Maere S, Heymans K, Kuiper M (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21: 3448–3449. [DOI] [PubMed] [Google Scholar]
- 34. Ivanov S, Fedorova E, Bisseling T (2010) Intracellular plant microbe associations: secretory pathways and the formation of perimicrobial compartments. Curr Opin Plant Biol 13: 372–377. [DOI] [PubMed] [Google Scholar]
- 35. Streeter JG, Gomez ML (2006) Three enzymes for trehalose synthesis in Bradyrhizobium cultured bacteria and in bacteroids from soybean nodules. Appl Environ Microbiol 72: 4250–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patterson TA, Lobenhofer EK, Fulmer-Smentek SB, Collins PJ, Chu TM, et al. (2006) Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat Biotechnol 24: 1140–1150. [DOI] [PubMed] [Google Scholar]
- 37. Barnes M, Freudenberg J, Thompson S, Aronow B, Pavlidis P (2005) Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic Acids Res 33: 5914–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dixon R, Kahn D (2004) Genetic regulation of biological nitrogen fixation. Nat Rev Microbiol 2: 621–631. [DOI] [PubMed] [Google Scholar]
- 39. Khatri P, Draghici S (2005) Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics 21: 3587–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Capela D, Filipe C, Bobik C, Batut J, Bruand C (2006) Sinorhizobium meliloti differentiation during symbiosis with alfalfa: a transcriptomic dissection. Mol Plant Microbe Interact 19: 363–372. [DOI] [PubMed] [Google Scholar]
- 41. Vercruysse M, Fauvart M, Beullens S, Braeken K, Cloots L, et al. (2011) A comparative transcriptome analysis of Rhizobium etli bacteroids: specific gene expression during symbiotic nongrowth. Mol Plant Microbe Interact 24: 1553–1561. [DOI] [PubMed] [Google Scholar]
- 42. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chiaretti S, Li X, Gentleman R, Vitale A, Vignetti M, et al. (2004) Gene expression profile of adult T-cell acute lymphocytic leukemia identifies distinct subsets of patients with different response to therapy and survival. Blood 103: 2771–2778. [DOI] [PubMed] [Google Scholar]
- 44. Lodwig E, Poole P (2003) Metabolism of Rhizobium bacteroids. Critical Reviews in Plant Sciences 22: 37–78. [Google Scholar]
- 45. Resendis-Antonio O, Hernandez M, Salazar E, Contreras S, Batallar GM, et al. (2011) Systems biology of bacterial nitrogen fixation: high-throughput technology and its integrative description with constraint-based modeling. BMC Syst Biol 5: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Romanov VI, Hernandez-Lucas I, Martinez-Romero E (1994) Carbon Metabolism Enzymes of Rhizobium tropici Cultures and Bacteroids. Appl Environ Microbiol 60: 2339–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ronson CW, Astwood PM, Downie JA (1984) Molecular cloning and genetic organization of C4-dicarboxylate transport genes from Rhizobium leguminosarum. J Bacteriol 160: 903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yurgel SN, Kahn ML (2004) Dicarboxylate transport by rhizobia. FEMS Microbiol Rev 28: 489–501. [DOI] [PubMed] [Google Scholar]
- 49. Barnett MJ, Toman CJ, Fisher RF, Long SR (2004) A dual-genome Symbiosis Chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc Natl Acad Sci U S A 101: 16636–16641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Green LS, Li Y, Emerich DW, Bergersen FJ, Day DA (2000) Catabolism of alpha-ketoglutarate by a sucA mutant of Bradyrhizobium japonicum: evidence for an alternative tricarboxylic acid cycle. J Bacteriol 182: 2838–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lodwig EM, Hosie AH, Bourdes A, Findlay K, Allaway D, et al. (2003) Amino-acid cycling drives nitrogen fixation in the legume-Rhizobium symbiosis. Nature 422: 722–726. [DOI] [PubMed] [Google Scholar]
- 52. Lodwig EM, Leonard M, Marroqui S, Wheeler TR, Findlay K, et al. (2005) Role of polyhydroxybutyrate and glycogen as carbon storage compounds in pea and bean bacteroids. Mol Plant Microbe Interact 18: 67–74. [DOI] [PubMed] [Google Scholar]
- 53. Anderson AJ, Dawes EA (1990) Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol Rev 54: 450–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang C, Sheng X, Equi RC, Trainer MA, Charles TC, et al. (2007) Influence of the poly-3-hydroxybutyrate (PHB) granule-associated proteins (PhaP1 and PhaP2) on PHB accumulation and symbiotic nitrogen fixation in Sinorhizobium meliloti Rm1021. J Bacteriol 189: 9050–9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Young JP, Crossman LC, Johnston AW, Thomson NR, Ghazoui ZF, et al. (2006) The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol 7: R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Walshaw DL, Poole PS (1996) The general L-amino acid permease of Rhizobium leguminosarum is an ABC uptake system that also influences efflux of solutes. Mol Microbiol 21: 1239–1252. [DOI] [PubMed] [Google Scholar]
- 57. Hosie AH, Allaway D, Galloway CS, Dunsby HA, Poole PS (2002) Rhizobium leguminosarum has a second general amino acid permease with unusually broad substrate specificity and high similarity to branched-chain amino acid transporters (Bra/LIV) of the ABC family. J Bacteriol 184: 4071–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Prell J, White JP, Bourdes A, Bunnewell S, Bongaerts RJ, et al. (2009) Legumes regulate Rhizobium bacteroid development and persistence by the supply of branched-chain amino acids. Proc Natl Acad Sci U S A 106: 12477–12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Terpolilli JJ, Hood GA, Poole PS (2012) What determines the efficiency of N(2)-fixing Rhizobium-legume symbioses? Adv Microb Physiol 60: 325–389. [DOI] [PubMed] [Google Scholar]
- 60. Sarma AD, Emerich DW (2006) A comparative proteomic evaluation of culture grown vs nodule isolated Bradyrhizobium japonicum. Proteomics 6: 3008–3028. [DOI] [PubMed] [Google Scholar]
- 61. Barnett MJ, Hung DY, Reisenauer A, Shapiro L, Long SR (2001) A homolog of the CtrA cell cycle regulator is present and essential in Sinorhizobium meliloti. J Bacteriol 183: 3204–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Laub MT, McAdams HH, Feldblyum T, Fraser CM, Shapiro L (2000) Global analysis of the genetic network controlling a bacterial cell cycle. Science 290: 2144–2148. [DOI] [PubMed] [Google Scholar]
- 63. Quon KC, Marczynski GT, Shapiro L (1996) Cell cycle control by an essential bacterial two-component signal transduction protein. Cell 84: 83–93. [DOI] [PubMed] [Google Scholar]
- 64. Skerker JM, Laub MT (2004) Cell-cycle progression and the generation of asymmetry in Caulobacter crescentus. Nat Rev Microbiol 2: 325–337. [DOI] [PubMed] [Google Scholar]
- 65. Laub MT, Chen SL, Shapiro L, McAdams HH (2002) Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc Natl Acad Sci U S A 99: 4632–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mercer RG, Callister SJ, Lipton MS, Pasa–Tolic L, Strnad H, et al. (2010) Loss of the response regulator CtrA causes pleiotropic effects on gene expression but does not affect growth phase regulation in Rhodobacter capsulatus. J Bacteriol 192: 2701–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miller TR, Belas R (2006) Motility is involved in Silicibacter sp. TM1040 interaction with dinoflagellates. Environ Microbiol 8: 1648–1659. [DOI] [PubMed] [Google Scholar]
- 68. Hengge R (2009) Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol 7: 263–273. [DOI] [PubMed] [Google Scholar]
- 69. Tal R, Wong HC, Calhoon R, Gelfand D, Fear AL, et al. (1998) Three cdg operons control cellular turnover of cyclic di-GMP in Acetobacter xylinum: genetic organization and occurrence of conserved domains in isoenzymes. J Bacteriol 180: 4416–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schmidt AJ, Ryjenkov DA, Gomelsky M (2005) The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J Bacteriol 187: 4774–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ryan RP, McCarthy Y, Andrade M, Farah CS, Armitage JP, et al. (2010) Cell-cell signal-dependent dynamic interactions between HD-GYP and GGDEF domain proteins mediate virulence in Xanthomonas campestris. Proc Natl Acad Sci U S A 107: 5989–5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang Y, Xu J, Chen A, Wang Y, Zhu J, et al. (2010) GGDEF and EAL proteins play different roles in the control of Sinorhizobium meliloti growth, motility, exopolysaccharide production, and competitive nodulation on host alfalfa. Acta Biochim Biophys Sin (Shanghai) 42: 410–417. [DOI] [PubMed] [Google Scholar]
- 73. Mergaert P, Nikovics K, Kelemen Z, Maunoury N, Vaubert D, et al. (2003) A novel family in Medicago truncatula consisting of more than 300 nodule-specific genes coding for small, secreted polypeptides with conserved cysteine motifs. Plant Physiol 132: 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Van de Velde W, Zehirov G, Szatmari A, Debreczeny M, Ishihara H, et al. (2010) Plant peptides govern terminal differentiation of bacteria in symbiosis. Science 327: 1122–1126. [DOI] [PubMed] [Google Scholar]
- 75. Glazebrook J, Ichige A, Walker GC (1993) A Rhizobium meliloti homolog of the Escherichia coli peptide-antibiotic transport protein SbmA is essential for bacteroid development. Genes Dev 7: 1485–1497. [DOI] [PubMed] [Google Scholar]
- 76. Tan XJ, Cheng Y, Li YX, Li YG, Zhou JC (2009) BacA is indispensable for successful Mesorhizobium-Astragalus symbiosis. Appl Microbiol Biotechnol 84: 519–526. [DOI] [PubMed] [Google Scholar]
- 77. Scott K, Diggle MA, Clarke SC (2003) TypA is a virulence regulator and is present in many pathogenic bacteria. Br J Biomed Sci 60: 168–170. [DOI] [PubMed] [Google Scholar]
- 78. Farris M, Grant A, Richardson TB, O'Connor CD (1998) BipA: a tyrosine-phosphorylated GTPase that mediates interactions between enteropathogenic Escherichia coli (EPEC) and epithelial cells. Mol Microbiol 28: 265–279. [DOI] [PubMed] [Google Scholar]
- 79. Grant AJ, Farris M, Alefounder P, Williams PH, Woodward MJ, et al. (2003) Co-ordination of pathogenicity island expression by the BipA GTPase in enteropathogenic Escherichia coli (EPEC). Mol Microbiol 48: 507–521. [DOI] [PubMed] [Google Scholar]
- 80. deLivron MA, Robinson VL (2008) Salmonella enterica serovar Typhimurium BipA exhibits two distinct ribosome binding modes. J Bacteriol 190: 5944–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Neidig A, Yeung AT, Rosay T, Tettmann B, Strempel N, et al. (2013) TypA is involved in virulence, antimicrobial resistance and biofilm formation in Pseudomonas aeruginosa. BMC Microbiol 13: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kiss E, Huguet T, Poinsot V, Batut J (2004) The typA gene is required for stress adaptation as well as for symbiosis of Sinorhizobium meliloti 1021 with certain Medicago truncatula lines. Mol Plant Microbe Interact 17: 235–244. [DOI] [PubMed] [Google Scholar]
- 83. Beckering CL, Steil L, Weber MH, Volker U, Marahiel MA (2002) Genomewide transcriptional analysis of the cold shock response in Bacillus subtilis. J Bacteriol 184: 6395–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tsolis RM (2002) Comparative genome analysis of the alpha -proteobacteria: relationships between plant and animal pathogens and host specificity. Proc Natl Acad Sci U S A 99: 12503–12505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hentschel U, Steinert M, Hacker J (2000) Common molecular mechanisms of symbiosis and pathogenesis. Trends Microbiol 8: 226–231. [DOI] [PubMed] [Google Scholar]
- 86. Arifuzzaman M, Maeda M, Itoh A, Nishikata K, Takita C, et al. (2006) Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res 16: 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643. [DOI] [PubMed] [Google Scholar]
- 88. Rodriguez-Llorente I, Caviedes MA, Dary M, Palomares AJ, Canovas FM, et al. (2009) The Symbiosis Interactome: a computational approach reveals novel components, functional interactions and modules in Sinorhizobium meliloti. BMC Syst Biol 3: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tate R, Riccio A, Merrick M, Patriarca EJ (1998) The Rhizobium etli amtB gene coding for an NH4+ transporter is down-regulated early during bacteroid differentiation. Mol Plant Microbe Interact 11: 188–198. [DOI] [PubMed] [Google Scholar]
- 90. Ampe F, Kiss E, Sabourdy F, Batut J (2003) Transcriptome analysis of Sinorhizobium meliloti during symbiosis. Genome Biol 4: R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mitsui H, Sato T, Sato Y, Ito N, Minamisawa K (2004) Sinorhizobium meliloti RpoH1 is required for effective nitrogen-fixing symbiosis with alfalfa. Mol Genet Genomics 271: 416–425. [DOI] [PubMed] [Google Scholar]
- 92. Piddock LJ (2006) Multidrug-resistance efflux pumps - not just for resistance. Nat Rev Microbiol 4: 629–636. [DOI] [PubMed] [Google Scholar]
- 93. Lindemann A, Koch M, Pessi G, Muller AJ, Balsiger S, et al. (2010) Host-specific symbiotic requirement of BdeAB, a RegR-controlled RND-type efflux system in Bradyrhizobium japonicum. FEMS Microbiol Lett 312: 184–191. [DOI] [PubMed] [Google Scholar]
- 94. Krummenacher P, Narberhaus F (2000) Two genes encoding a putative multidrug efflux pump of the RND/MFP family are cotranscribed with an rpoH gene in Bradyrhizobium japonicum. Gene 241: 247–254. [DOI] [PubMed] [Google Scholar]
- 95. Van der Henst C, Beaufay F, Mignolet J, Didembourg C, Colinet J, et al. (2012) The histidine kinase PdhS controls cell cycle progression of the pathogenic alphaproteobacterium Brucella abortus. J Bacteriol 194: 5305–5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McGrath PT, Iniesta AA, Ryan KR, Shapiro L, McAdams HH (2006) A dynamically localized protease complex and a polar specificity factor control a cell cycle master regulator. Cell 124: 535–547. [DOI] [PubMed] [Google Scholar]
- 97. Wang SC, West L, Shapiro L (2006) The bifunctional FtsK protein mediates chromosome partitioning and cell division in Caulobacter. J Bacteriol 188: 1497–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jenal U, Fuchs T (1998) An essential protease involved in bacterial cell-cycle control. EMBO J 17: 5658–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Arcondeguy T, Jack R, Merrick M (2001) P(II) signal transduction proteins, pivotal players in microbial nitrogen control. Microbiol Mol Biol Rev 65: 80–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The workflow of constructing PPI networks for M. huakuii 7653R. A: the schematic of the homologous protein-protein interactions network through the concepts of orthologous conservation (or interologs); B: the schematic illustration of the search for M. huakuii 7653R protein–protein interactions.
(TIFF)
The distribution of differentially expressed genes (bacteroids vs. free-living cells) which account for 80% of total expression amounts in COG functional categories. All genes were assigned to 21 COG functional categories using RPS-BLAST (uncategorized genes not shown). Bars represent the numbers of corresponding genes in free-living cells (green) and bacteroids (blue). The number in each bar represents its percentage (%).
(TIFF)
Schematic view of expressions of known genes associated with pathways of important biological function. An overview of the differentially expressed genes in bacteroids compared with free-living cells. The up-regulated genes are shown in red; the down-regulated genes are shown in green; the unchanged genes are shown in black. Genes were grouped according to their biological function or process involved; membrane-associated proteins were positioned over the inner and outer membranes.
(TIFF)
Up-regulated GO terms associated with bacteroids identified by BiNGO plugin. The enriched GO term of the subnetwork was nitrogen fixation.
(TIFF)
The PPI network of M. huakuii 7653R inferred from the two published PPI reference networks of M. huakuii bv loti MAFF303099 and E. coli.
(XLSX)
The top 10 of highly-expressed genes in M. huakuii 7653R bacteroids revealed by RNA-Seq. a The percentage represents the proportion of the gene transcriptional amounts to the total transcriptional amounts.
(DOC)
Top 25 up-regulated genes in M. huakuii 7653R bacteroids revealed by RNA-Seq and Microarrays.
(DOC)
Top 25 down-regulated genes in M. huakuii 7653R bacteroids revealed by RNA-Seq and Microarrays.
(DOC)
The significantly up-regulated pathways based on GSEA in M. huakuii 7653R free-living cells.
(XLSX)
The significantly up-regulated pathways based on GSEA in M. huakuii 7653R.
(XLSX)
Enriched KEGG pathways of differential expression genes in M. huakuii 7653R free-living cells.
(XLSX)
Enriched KEGG pathways of differential expression genes in M. huakuii 7653R.
(XLSX)
fab genes participating in fatty acid biosynthesis in M. huakuii 7653R.
(XLSX)
Genes associated with cell cycle and putatively regulated by CtrA in M. huakuii 7653R.
(XLSX)
Genes associated with c-di-GMP synthesis and degradation in M. huakuii 7653R.
(XLSX)