

This paper presents a culture system that can be used as an in vitro surrogate for xenotransplantation and that has the potential to dramatically increase the throughput of the investigation of leukemia-initiating cells, providing the means to identify and target the functionality of the different signaling pathways involved in the maintenance and resistance of leukemia-initiating cells to improve acute myeloid leukemia treatments.
Keywords: Acute myeloid leukemia, Leukemia initiating cells, Leukemic long-term culture initiating cell, Xenograft
Abstract
Acute myeloid leukemia-initiating cells (LICs) are responsible for the emergence of leukemia and relapse after chemotherapy. Despite their identification more than 15 years ago, our understanding of the mechanisms responsible for their self-renewal activity and their chemoresistance remains poor. The slow progress in this area is partly due to the difficulty of studying these cells ex vivo. Indeed, current studies are reliant on xenotransplantation assays in immunodeficient mice. In this paper, we report that by modeling key elements of the bone marrow niche using different stromal feeder layers and hypoxic culture conditions, we can maintain LICs over at least 3 weeks and support their self-renewal properties demonstrated through primary and secondary successful xenograft. We provide a proof of principle that this niche-like culture system can be used to study LIC chemoresistance following in vitro cytarabine treatment similarly to the xenograft chemotherapy model. We found that although LICs are believed to be more chemoresistant than non-LICs, functionally defined LICs are not enriched after cytarabine treatment, and heterogeneity in their resistance to treatment can be seen between patients and even within the same patient. We present a culture system that can be used as an in vitro surrogate for xenotransplantation and that has the potential to dramatically increase the throughput of the investigation of LICs. This would further provide the means by which to identify and target the functionality of the different signaling pathways involved in the maintenance and resistance of LICs to improve acute myeloid leukemia treatments.
Introduction
Acute myeloid leukemia (AML) is characterized by the infiltration of leukemic myeloid blasts in the bone marrow that have been arrested at various maturation steps. Although 50%–75% of patients respond to induction chemotherapy and enter remission, the majority relapse, and only 20%–30% of patients can achieve long-term survival [1]. One established paradigm of leukemogenesis is that leukemia arises from the malignant transformation of a single cell and is maintained by a small population of leukemia-initiating cells (LICs) [2, 3]. It is commonly admitted that LICs are typically chemoresistant due to intrinsic and extrinsic properties [4]. Efficient DNA repair pathways, high levels of drug efflux pumps, and quiescence account for intrinsic drug resistance, whereas environment-mediated drug resistance arises from a complex interplay between soluble factors and cell adhesion-mediated drug-resistance mechanisms [5, 6]. Chemoresistant LICs are responsible for AML relapse and represent the targets for future innovative therapies [7].
Techniques currently available for culturing primary AML samples have been adapted from standardized protocols for culturing normal hematopoietic cells. Although in vitro short-term liquid culture and colony assays allow quantification of leukemic precursors, the identification of LICs is still dependent on in vivo xenograft assays in which LICs can self-renew and have the ability to recapitulate the disease. Although new immunodeficient mice strains and xenograft chemotherapy protocols are being defined [8], the AML xenograft assay still requires a time frame of 8–20 weeks before analysis and remains prospectively blind with little ability to monitor progress. These limitations make its use difficult for routine drug screening and investigation of the role of specific genes and pathways involved in LIC maintenance. The recently established LIC phenotype heterogeneity [9–12] and the lack of a reliable and easy in vitro bioassay for monitoring LICs is slowing down the development of innovative therapeutic strategies. Different coculture systems achieving some degree of support of normal or leukemic hematopoiesis over a few weeks have been described [13–17]; however, only a few of these studies truly demonstrated the maintenance of normal hematopoietic stem cells (HSCs) validated in parallel through xenotransplantation assessment. In the context of leukemia, leukemic cobblestone area-forming cells and/or leukemic long-term culture initiating cells (L-LTC-ICs) can be maintained for a few weeks in coculture [18, 19]; however, so far, there have been no reports demonstrating the ex vivo maintenance of LICs through subsequent xenograft assessment. Consequently, in vitro studies investigating environment-mediated drug resistance have never been applied to human primary LICs.
A primary goal of establishing long-term cultures (LTCs) for HSCs or LICs has been to mimic the bone marrow microenvironment ex vivo. Cytokines and extracellular matrix proteins have been shown to play an essential role in providing a supportive environment, but HSCs or LICs are also influenced by cues provided through cell-cell contact (the “stem cell niche synapses”) between different cell types in the bone marrow microenvironment such as preosteoblasts, osteoblasts, endothelial cells, and mesenchymal cells [20–24]. In the leukemic context, survival and proliferative benefits have been reported for primary AML samples when cocultured with the osteosarcoma SaOS-2 or human umbilical vein endothelial cell (HUVEC) lines [25–28]. A mesenchymal MS-5 coculture supplemented with interleukin 3 (IL3), granulocyte colony-stimulating factor (G-CSF), and thrombopoietin (TPO) can also sustain primary AML samples over 24 weeks with successful generation of leukemic cobblestone area-forming cells and primary L-LTC-ICs [18]; however, it is not known whether all of these coculture systems can actually sustain LICs. Hypoxia is also known to favor normal HSC quiescence and maintenance, whereas normoxia induces proliferation and differentiation, followed by exhaustion [29–31]. In AML, a recent study has demonstrated a requirement for the hypoxia-inducible factor 1α pathway for LIC maintenance in vivo [32]. The impact that hypoxia has on AML survival in steady-state condition or under drug treatment is less clear, depending on the population of cells and the drug being studied [33–35]. Furthermore, the chemoprotective role of hypoxia in LICs in vitro remains to be explored.
Our work represents the first direct comparison between different coculture systems and the assessment of the impact of low oxygen on functionally defined human LICs. This was achieved by secondary leukemia-long-term culture and in vivo repopulating assays in limiting dilution. We report a reliable, easy, and reproducible niche-based culture system suitable for the maintenance of human primary LICs. In addition, we show that this system could be used to study the chemoresistance of LICs and to screen the activity of new therapeutic agents that will specifically target and eliminate LICs.
Materials and Methods
Cells
AML cells were obtained after informed consent at St. Bartholomew’s Hospital (London, U.K.). The protocol was approved by the East London Ethical Committee and in accordance with the Declaration of Helsinki. Samples were collected at diagnosis and first screened for their ability to engraft in immunodeficient mice. Details of the patient samples are listed in supplemental online Table 1. The stromal cell line MS-5 was obtained from the DSMZ cell bank (Braunschweig, Germany, http://www.dsmz.de) and maintained in Iscove’s modified Dulbecco’s medium (IMDM) plus 10% fetal calf serum plus 2 mM l-glutamine.
Short-Term Culture, LTC, and Secondary Plating LTC
Cocultures were performed as bulk culture or using a limiting dilution analysis on confluent monolayer of MS-5, supplemented as indicated with recombinant human IL3, G-CSF, and TPO (MS-5+3GT) (20 ng/ml each; Peprotech, London, U.K., http://www.peprotech.com) in MyeloCult H5100 (StemCell Technologies, Vancouver, BC, Canada, http://www.stemcell.com). Cells were cultured at 37°C in 5% CO2 -humidified incubators at 20% or 3% O2 conditions. Low-oxygen cultures were performed in a two-gas HERAcell incubator (Thermo Fisher Scientific, Waltham, MA, http://www.thermoscientific.com/en/home.html). For cytosine β-d-arabinofuranoside (Ara-C; Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com) treatment, AML cells were preincubated in coculture 72 hours prior to the addition once of Ara-C at 3 μM followed by an additional 7 days of culture. In some experiments, cocultures were washed four times with phosphate-buffered saline after Ara-C treatment, and then fresh MyeloCult H5100 medium was added, and cocultures were maintained for two additional weeks. For primary LTC for limiting dilution analysis (LDA; 1° LDA) and secondary LTC for LDA (2° LDA), cells were plated in 20 replicates in 96-well microplates containing confluent MS-5 monolayer. After 5 weeks, LTC medium was replaced by methylcellulose H4435 (StemCell Technologies). After an additional 2 weeks, each well was scored as negative if no colonies were present. All 2° LDA and colony assays were performed in normoxia. To determine the frequency of L-LTC-ICs, LDA was done using LCalc software (StemCell Technologies) according to the Poisson statistics and method of maximum likelihood.
Flow Cytometry Analysis
Fluorescence-activated cell sorting (FACS) analysis was performed with a BD LSRII flow cytometer, and cell sorting was performed with a BD FACSAria (BD Biosciences, San Diego, CA, http://www.bdbiosciences.com). After 1–5 weeks of coculture, nonadherent and adherent cells were harvested through trypsinization. Recovered cells were resuspended and stained in Annexin-binding buffer (BD Biosciences). Sca-1 was identified as a specific marker for 100% of MS-5. Human hematopoietic cells were stained with anti-CD45-APC-Cy7, anti-CD34-Percp, anti-CD38-PE-Cy7 antibodies, and Lin-FITC as well as with Alexa Fluor 647-conjugated Annexin V (Invitrogen, Carlsbad, CA, http://www.invitrogen.com). At the end, cells were resuspended in phosphate-buffered saline containing 4′,6-diamidino-2-phenylindole (DAPI). All antibodies were from BD Biosciences. Only viable (both DAPI and Annexin V-negative fraction) human hematopoietic cells (CD45-APC-Cy7 positive and Sca-1-PE negative) were assessed and/or sorted for all analyses. For some AML samples, LIC and non-LIC phenotypes were used according to pre-established xenograft experiments with sorted subpopulations (previously published in [10] and data not shown). For the precise cell counts, CountBright absolute counting beads (Molecular Probes, Paisley, U.K., http://probes.invitrogen.com) were used to assess the total number of cells, following manufacturer’s recommendations.
Adoptive Transfer of Human Hematopoietic Cells in Immunodeficient Mice
All animal experiments were performed in compliance with Home Office and Cancer Research UK guidelines. NOD/SCID/β2-microglobulin null (β2m−/−)mice or NOD/SCID IL-2Rγ common chain null (NSG) were originally obtained from Dr. Leonard Schultz (Jackson Laboratory, Bar Harbor, ME, http://www.jax.org) and bred at the animal facility of the London Research Institute. Mice were irradiated at 375 cGy (137Cs source) 24 hours before transplantation. The same mouse strain (β2m−/− or NSG) was used to address one experimental parameter per patient. Cells were injected via intratibia injection because of previous report of an HSC homing defect described after in vitro manipulation [36]. MS-5 cells were eliminated by cell sorting prior to cell inoculation. Animals were sacrificed at 12 weeks, and bone marrow cells were collected from the injected bone and contralateral noninjected long bones separately. For LDA experiments, cell doses of 102 to 107 cells from primary AML samples were injected into three to five recipients per dose. A mouse was scored as engrafted if a distinct human CD45+ and murine CD45− population was detectable in the contralateral bone marrow. AML engraftment was defined by the presence of a single CD45+CD33+CD19− population.
Please refer to the supplemental online data for the remaining materials and methods information.
Results
Selection of Feeder Layer and Oxygen Concentration for AML Initiating Cell Maintenance In Vitro Over 3 Weeks
Based on the heterogeneity of AML, we first aimed to compare osteoblasts and endothelial and mesenchymal coculture systems with a large number of AML samples at 20% O2. Therefore, we evaluated the cell viability of 38–57 AML samples (supplemental online Table 1) after coculturing them for 1 week with mesenchymal-related MS-5, osteoblast-derived SaOS-2, and endothelial HUVEC lines without cytokine supplement. We observed a highly variable level of cell viability, ranging from 3.23% to 96.4%, but this variability was seen across the three systems. This was in contrast to normal HSCs and hematopoietic stem/progenitor cells, in which we observed a homogenous cell viability of 84% ± 8% (p < .001, n = 16; data not shown) during coculture. A cross-comparison analysis for each individual AML sample revealed that MS-5 was the most supportive coculture system (Fig. 1A). Furthermore, with the MS-5 condition, we also observed maintenance of the cell number (with a mean fold expansion of 1.22 ± 0.32; 28 samples). Phenotypic analysis also revealed MS-5-based coculture as the most efficient system for keeping the initial CD34 and CD38 expression pattern, as compared with SaOS-2 or HUVEC, with which CD34+ cells were decreased and CD38+ cells were increased (Fig. 1B). Thus, we focused on the MS-5 feeder because it appeared to be the most efficient system for supporting leukemic cells. A previous report demonstrated that MS-5 supplemented with IL3+G-CSF+TPO (MS-5+3GT) allows the expansion of leukemic cells [18]. Thus, the impact of the 3GT cytokine cocktail was next investigated. After 3 days in suspension culture, 3GT alone did not increase the post-thawed viability of AML cells; however, the MS-5+3GT condition increased viable cells by 18.8% ± 5.7% (n = 23, p < .005) (supplemental online Fig. 1A). AML long-term cocultures performed with or without 3GT also demonstrated a requirement of cytokine supplement for the maintenance of L-LTC-ICs over a 5-week period (supplemental online Fig. 1B). Preliminary results obtained with few patients among the ones presented in this study were also cultured for 5 weeks. In contrast to normal hematopoietic stem/progenitor cells, there was no clear benefit in terms of leukemic pool expansion going from 3 weeks to 5 weeks (data not shown); therefore, all subsequent experiments were shortened to 3 weeks.
Figure 1.

One-week coculture on mesenchymal-related MS-5 cells promoted better AML cell viability and better retained the initial phenotype compared with osteoblast-derived SaOS-2 and endothelial-like HUVEC. (A): Viability comparison, MS-5 and sample 12 (57 samples), MS-5 and HUVEC (38 samples), and SaOS-2 and HUVEC (43 samples). Putative equivalent cell viability is represented by the bold black line. The thin black line shows experimentally derived simple linear regression trend line with the 95% confidence band (dashed lines). A paired t test was applied for each comparison. (B): Fluorescence-activated cell sorting plot of CD34 and CD38 expression profiles of a CD34+ representative AML sample (sample 6) just after thawing (left panel) or after 1-week coculture with MS-5, SOS-2, or HUVECs. A 4′,6-diamidino-2-phenylindole-negative/Annexin V-negative population was used for analysis. Numbers represent the percentages of cells within each region. Abbreviations: AML, acute myeloid leukemia; HUVEC, human umbilical vein endothelial cell.
Of note, all the AML samples used for the rest of this study were pre-evaluated for their capacity to give rise to an exclusive leukemic engraftment (verified by a unique high-scatter CD45+CD33+CD19− myeloid blast population by flow cytometry analyses). We further confirmed the leukemic origin of the cells after ex vivo culture. For patients with nucleophosmin mutations, for example, we show by polymerase chain reaction that nucleophosmin mutation can be detected in the cells after culture (supplemental online Fig. 1C). Furthermore, in the read-out colony-forming unit assay, we observed only blast-type colony formation, thus excluding the possibility of having expanded normal hematopoietic cells.
We were then interested in the impact of the 3% O2 condition on AML cells in vitro. Primary AML cells showed a significant decrease in growth when cultured at 3% O2 compared with 20% O2 (p < .05) (Fig. 2A). We quantified the replating potential of three AML samples maintained for 3 weeks either in 3% O2 or 20% O2 compared with day 0 (Fig. 2B). We observed 2.7-, 4.5-, and 1.96-fold increases in L-LTC-IC frequencies in 2° LDA for sample 6 (p < .005), sample 2 (p < .0001), and sample 3 (p < .05), respectively, when cultured at 3% O2 compared with 20% O2 (Fig. 2C). When comparing the initial L-LTC-IC frequency (1° LDA, day 0) with the frequency after the 3 weeks of culture (2° LDA) and taking into account the fold expansion, we were able to determine the difference between the total L-LTC-IC input and output numbers. In 20% O2 coculture, L-LTC-ICs capable of secondary plating were found to be either decreased (sample 6), maintained (sample 2), or increased (sample 3) compared with their respective input numbers (Fig. 2D). A low-oxygen coculture system was found beneficial for samples 2 and 3 in terms of total L-LTC-IC number, whereas a decrease was observed for sample 6 compared with their respective input numbers. Altogether, low-oxygen coculture appears to be superior to the 20% O2 coculture condition.
Figure 2.

Low oxygen concentration favors long-term in vitro maintenance of leukemia-initiating cells (LICs) over 3 weeks. (A): 3% O2 reduced AML expansion. AML samples were cocultured for 3 weeks with MS-5+3GT at 20% O2 or 3% O2 in parallel. Data shown represent fold expansion of five AML samples (samples 2, 7, 10, 3, and 6) determined in 20% O2 or 3% O2. ∗, p < .05 in paired t test. (B): Flowchart illustrates the experiment design for (C) and (D). (C): Replating potential at week 3. Cells from AML samples 6, 2, and 3 were cocultured at 20% O2 or 3% O2 for 3 weeks and then sorted for CD45+ cells and replated in 2° LDA for an additional 5 weeks at 20% O2. (D): Total L-LTC-IC numbers calculated from 1° LDA and 2° LDA after 3 weeks of coculture at 20% O2 or 3% O2 for samples 6, 2, and 3. ∗, p < .05; ∗∗∗, p < .001; NS, p > .05 (90% confidence interval is displayed). (E): Flowchart illustrates the experiment design for (F) and (G). (F): In vivo LDA of cells from AML samples 6, 2, and 3 recovered from each coculture and injected at different doses into NOD/SCID interleukin-2Rγ common chain null mice (for sample 6, n = 24, and sample 3, n = 42) or NOD/SCID/β2-microglobulin null mice (for sample 2, n = 52) to calculate the frequency of LICs. (G): Total LIC counts (90% confidence interval) determined for cells from samples 6, 2, and 3 for uncultured (T0) or cocultured cells for 3 weeks at 20% O2 or 3% O2. ∗, p < .05; NS, p > .05. For (D) and (G), data presented take into account the fold expansion in culture. (H): Primary and secondary leukemic engraftment in NOD/SCID/β2-microglobulin null mice of AML samples cocultured for 3 weeks. The data show the percentage of human leukemic chimerism in primary recipient mice (first) for six samples (samples 1, 2, 3, 4, 5, and 6) and secondary recipients (second) for samples 1, 2, and 3. Plain lines represent mean levels of leukemic cell engraftment in each group. Each symbol represents a single transplanted mouse. Abbreviations: 1° LDA, LDA from post-thawed cells; 2° LDA, LDA from cultured cells; AML, acute myeloid leukemia; CFC, colony forming cells; LDA, limiting dilution analysis; L-LTC-IC, leukemic long-term culture initiating cell; NS, not significant; w, weeks.
Xenograft Potential Maintenance After Ex Vivo Culture
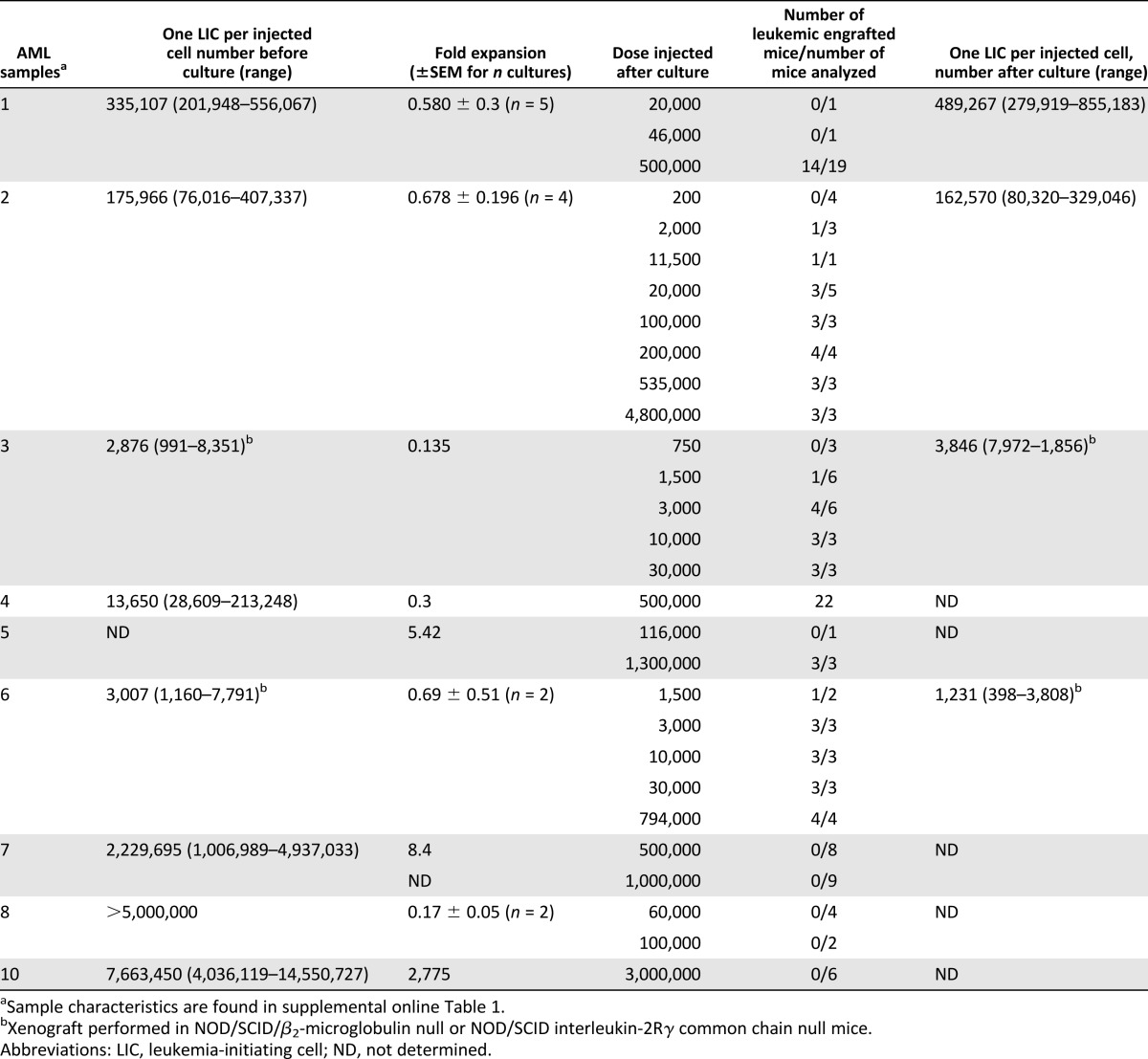
We then wondered whether the in vitro L-LTC-IC replating potential observed was indicative of LIC activity. In vivo LDA was performed for these three samples after coculturing either at 20% O2 or 3% O2 for 3 weeks (Fig. 2E). Leukemic (myeloid-restricted) engraftment was observed in all cases (supplemental online Fig. 3). There were 138.8-, 6.5-, and 4.7-fold increases in LIC frequency at 3% O2 compared with 20% O2 for samples 6, 2, and 3, respectively (Fig. 2F) (p < .0001). We also determined for samples 6, 2, and 3 the total LIC input and output numbers. For samples 6 and 2, the 3% O2 coculture condition allowed for maintenance of LICs compared with their initial input number, whereas significant 57.3- and 6.1-fold decreases were obtained, respectively, in the normoxic culture system (p < .05) (Fig. 2G). For sample 3, we observed a reduction in total LICs after culture compared with day 0, and this change appears to be independent of the culture conditions used (3% O2 vs. 20% O2) (Fig. 2G). To further validate the beneficial impact of the low-oxygen coculture system, we repeated the experiments (3 weeks at 3% O2) with an additional six primary AML samples prior to transplantation into NOD/SCID/β2m−/− mice. In six of nine samples tested (including samples 6, 2, and 3, already presented), leukemic myeloid-restricted engraftment could be detected after culture at 3% O2 (Table 1). In three of these six samples, self-renewal capacity was also investigated by performing serial transplants, and leukemic engraftment was observed in all secondary recipients (Fig. 2H; supplemental online Fig. 3). To further investigate the engraftment failure observed in three patients after culture (samples 7, 8, and 10), we evaluated the LIC frequency before in vitro culture (Table 1). We found that these samples had the lowest LIC frequency of the patients analyzed and that the cells injected after culture fell below the initial LIC frequency observed in noncultured cells. Altogether, these results support the notion that functionally defined LICs can be maintained in vitro over a 3-week period in MS-5+3GT at 3% O2 but not at 20% O2.
Table 1.
LIC frequency is retained over 3 weeks of in vitro coculture with MS-5+3GT at 3% O2
Chemoresistant LICs Can Be Studied In Vitro
We next investigated whether these culture conditions might be suitable for studying chemoresistance of LICs to cytarabine (Ara-C), a commonly known drug for AML treatment. We decided to use Ara-C at a concentration of 3 μM, which is >25 times higher than the IC50 dose obtained on three AML cell lines (supplemental online Fig. 2A) but 3.8 times lower than the IC50 dose for MS-5 feeder cells. We selected this dose because it was safe for the MS-5 stroma cells used, and we wished to evaluate the chemoresistance of LICs. After 7 days with Ara-C treatment in normoxic conditions, we observed more than 94% eradication of primary leukemic cells in both suspension and MS-5-based culture systems (supplemental online Fig. 2B1). However, coculture on MS-5 improved the percentage of viable cells fourfold in the residual population for leukemic samples compared with the suspension system (p < .05) (supplemental online Fig. 2B2). We observed that culturing the cells at 3% O2 improved the recoverable fraction of live leukemic cells after Ara-C treatment (Fig. 3A, 3B). When replating live cells after Ara-C-treated groups cultured under 3% O2 or 20% O2 (Fig. 3C), we observed that cells that were able to reinitiate leukemic LTC were enriched in the 3% O2 group (Fig. 3D) by 34-fold for sample 6 (p < .0001) and 3-fold for sample 2 (p < .01). More important, a chemoprotective effect of low oxygen was also observed when looking at the total yield of L-LTC-ICs in secondary plating for these samples: 32 times and 3 times more secondary L-LTC-ICs were recovered in 3% O2 cultures (Fig. 3E) for sample 6 (p < .01) and sample 2 (p < .05), respectively. We further demonstrated that residual cells were still capable of engrafting immunodeficient mice. However, cultures at 3% O2 without any feeder layer did not support the engraftment of cells that remained after Ara-C treatment (no MS-5), demonstrating that the maintenance of LICs required stroma support for their chemoresistance under 3% O2 (Fig. 3F).
Figure 3.

Low oxygen concentration enhances in vitro chemoresistance of AML cells in coculture and allows the preservation of leukemia-initiating cell activity. (A): Flowchart shows the experiment design for (B). (B): Data represent mean percentage (±SEM) of four AML samples (samples 2, 3, 6, and 10) retrieved after 1 week of ARA-C at 3 μM compared with untreated control cells counts when cocultured with MS-5+3GT at 20% O2 or 3% O2. (C): Flowchart shows the experiment design for (D) and (E). (D): Replating potential after ARA-C treatment. Cells from AML samples 2 and 6 were treated during the first week with 3 μM ARA-C in cocultures at 20% O2 or 3% O2. Cocultures were washed and maintained for an additional 2 weeks. Viable leukemic cells were sorted in week 3 and then replated in 2° LDA for an additional 5 weeks. (E): Total leukemic long-term culture initiating cell numbers were calculated from 1° LDA of uncultured cells and 2° LDA of 3-week cultured and ARA-C-treated cells at 20% O2 or 3% O2 for samples 6 and 2. ARA-C treatment occurred during the first week only. Data presented take into account the first culture fold expansion. ∗, p < .05; ∗∗, p < .01; ∗∗∗, p < .001. (F): Cells from AML sample 1 were cocultured with MS-5+3GT or cultured in suspension only with 3GT (no MS-5) in the presence of 3 μM ARA-C at 3% O2. Recovered live cells were sorted for human CD45+ cells at 3 weeks and then injected into NOD/SCID interleukin-2Rγ common chain null mice (n = 8). We injected 5 × 104 cells, which represented 17% of the input cell number for the MS-5 condition and 23% for the no MS-5 condition. Data shown represent the percentage of human leukemic engraftment in recipient bone marrow determined by fluorescence-activated cell sorting analysis at 12 weeks after injection. Solid lines represent mean levels of leukemic cell engraftment in each group. Dashed lines represent the threshold of positivity. Each symbol represents a single transplanted mouse. ∗, p < .005. Abbreviations: 1° LDA, limiting dilution analysis from post-thawed cells; 2° LDA, limiting dilution analysis from cultured cells; AML, acute myeloid leukemia; ARA-C, cytosine β-d-arabinofuranoside; MS-5, MS-5+3GT; NO MS-5; no MS-5+3GT; w, weeks.
In Vitro Ara-C Treatment Does Not Enrich for LICs
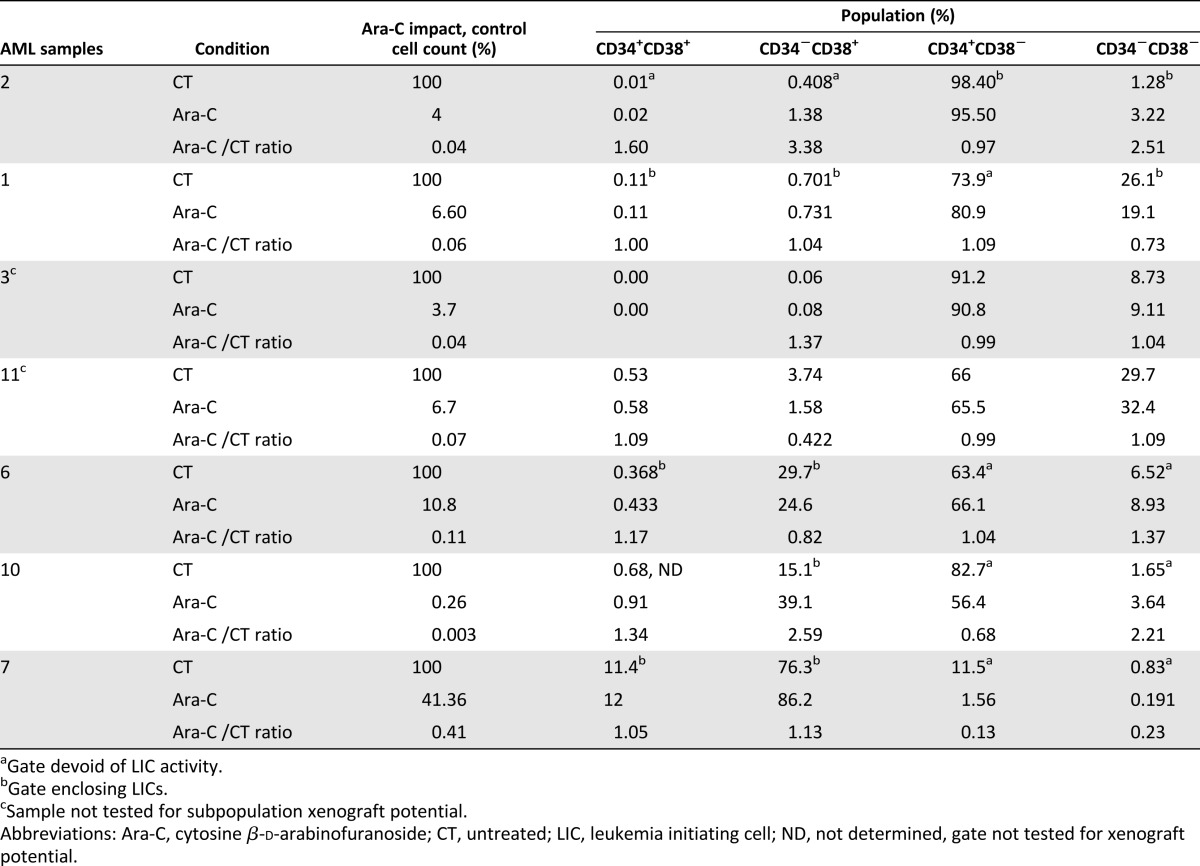
In the dose tested, Ara-C treatment was really effective at killing leukemic cells in vitro (Fig. 4A; supplemental online Fig. 2B1). Because LICs were reported to be quiescent [37], we wondered if the in vitro Ara-C treatment could preferentially kill non-LIC blasts, hence enriching for LICs. For seven AML samples, the CD34/CD38 phenotype appeared globally unchanged after Ara-C treatment, despite the drastic impact of Ara-C on reducing the cell number (Table 2; supplemental online Fig. 4). For five AML samples, we functionally determined the phenotype of the LIC compartment before treatment in vivo (Table 2). As recently published by our group and others [9–12], LIC phenotype was not restricted to the CD34+CD38− population. When comparing the proportion of defined LIC and non-LIC fractions before and after treatment, we did not observe any significant enrichment of LICs after treatment (Fig. 4B).
Figure 4.

Chemoresistant LIC maintenance can be assessed through in vitro replating potential. (A): Histograms represent the percentage of viable CD45+ cells counts at 1 week (±SEM ; n = 3) for four AML samples (samples 3, 2, 1, and 6) cocultured with MS-5+3GT at 3% O2 with 3 μM ARA-C compared with untreated (100%) control. (B): The percentage of LICs versus non-LICs was determined by fluorescence-activated cell sorting for five AML samples (samples 1, 2, 6, 7, and 10) before and after ARA-C treatment. Data represent the frequency of LICs and non-LICs in the ARA-C condition compared with control untreated. Values shown were normalized to the respective untreated condition, and error bars are ±SEM. Refer to Table 2 and to the Materials and Methods for LIC and non-LIC population definitions. NS, p > .05. (C): Flowchart illustrating the experimental design for (D–G). (D): L-LTC-IC frequencies in secondary plating relative to control were calculated during LDA with cultured cells. Untreated or in vitro ARA-C-treated samples 3, 2, and 6 are shown. (E): Xenograft potential of AML cells after ex vivo chemotherapy. A single cell dose (20,000 cells; samples 3, 2, and 6) of sorted leukemic cells were injected into NOD/SCID interleukin-2Rγ common chain null (NSG) mice. Scatter plot represents leukemic engraftment in recipient bone marrow at 12 weeks after injection with untreated cells or in vitro ARA-C-treated cells. Each symbol represents a single transplanted mouse. ∗∗∗, p < .001; NS, p > .05. (F): In vivo LDA for sample 2. Untreated or in vitro ARA-C-treated cells were injected at LDA into NSG mice, and LIC frequency was calculated (22 mice). (G): Total LIC numbers calculated for sample 2 were derived from transplantation data using NSG mice. Culture fold expansion was taken into account. ∗∗∗, p < .001; NS, p > .05. (H): Flowchart illustrating the in vivo ARA-C treatment. (I): At week 12, after establishing leukemia in NSG mice with samples 3, 2, and 6, recipients were treated with ARA-C (10 mg/kg per day) for 7 days. Data show the mean absolute number of leukemic cells on day 7 in the first recipient mice treated or not with ARA-C (∗∗∗, p < .001; ∗, p < .05). (J): LDA in secondary NSG mice after in vivo ARA-C treatment in primary recipients for samples 3 and 2. Cells from the first recipients were sorted and injected into secondary recipients at LDA to calculate the frequency of LICs. (K): Data shown represent leukemic engraftment in NSG mouse bone marrow after secondary xenotransplantation of indicated cell doses from sample 6 cells recovered from primary recipient. NS, p > .05. Abbreviations: AML, acute myeloid leukemia; ARA-C, cytosine β-d-arabinofuranoside; CT, untreated; L-LTC-IC, leukemic long-term culture initiating cells; LDA, limiting dilution analysis; LIC, leukemia-initiating cell; NS, not significant; w, weeks.
Table 2.
Impact of 1 week of Ara-C treatment on CD34 and CD38 subpopulations
Nevertheless, to further confirm the persistence of L-LTC-ICs in the residual Ara-C-treated population, we performed 2° LDA to determine the frequency of L-LTC-ICs capable of secondary plating (Fig. 4C, 4D). We saw a 3.5- and 8.7-fold decrease of L-LTC-IC frequency in the Ara-C condition for samples 3 and 2, respectively, whereas for sample 6, the frequency was not altered. To evaluate the correlation between L-LTC-ICs capable of secondary plating and LICs, we first injected the same cell dose (control and Ara-C group) in NSG mice and observed a reduction in the level of engraftment with Ara-C treatment compared with control for samples 3 and 2, whereas for sample 6, similar engraftment was observed with Ara-C treatment and control (Fig. 4E). These results mimic the in vitro L-LTC-IC results. We further performed an in vivo LDA for sample 2, and that allowed us to calculate the frequency and the total number of LICs after culture with or without Ara-C. We found a 16-fold reduction in the LIC frequency after treatment (p < .01) (Fig. 4F) and a 460-fold reduction of the total number of LICs (p < .01) (Fig. 4G). Interestingly, comparable 8-fold and 313-fold reductions in secondary L-LTC-IC frequency and total number, respectively, could also be observed through 2° LDA (both p < .0001) (Figs. 2E, 4D). Our data show that simply determining in vitro killing or performing phenotype analysis were not informative parameters for assessing drug-targeting efficiency toward LICs versus non-LICs, whereas the 2° LDA was as efficient as the xenograft assay to interrogate LIC persistence.
Comparative Effect of Ara-C on LICs Treated Ex Vivo and In Vivo
To determine whether the in vitro Ara-C treatment in our culture condition would reproduce an in vivo chemotherapy treatment protocol, we compared the impact of the two treatments on three AML samples (samples 3, 2, and 6). Twelve weeks after establishing leukemia in NSG mice, one group of animals was treated with Ara-C (at 10 mg/kg for 1 week) and sacrificed at the end of the treatment (Fig. 4H). We observed a mean leukemic burden that was 4 times lower (sample 3), 17 times lower (sample 2), and 2.53 times lower (sample 6) in the treated group (Fig. 4I). To look at the impact on LICs, we performed an LDA in a secondary transplant setting. We could determine the LIC frequency. and we found 4.7- and 6.9-fold reductions in the LIC frequency for sample 3 (p < .05) and sample 2 (p < .01), respectively, after treatment (Fig. 4J). In contrast, in all three doses tested for sample 6, we did not observe any difference between the control and Ara-C-treated groups, indicating that, at least for this patient, the LIC compartment was not significantly affected during the in vivo Ara-C treatment (Fig. 4K). These results show a good correlation between in vitro and in vivo treatment of cells with Ara-C. We propose that L-LTC-ICs in secondary cultures (2° LDA) could serve as a good surrogate assay for the xenotransplantation model, allowing screening of LICs affecting drugs in vitro.
Discussion
In this study, we have defined the MS-5+3GT 3% O2 culture condition as an efficient niche-like culture system to maintain human LICs in vitro.
A recent study demonstrated a requirement for the hypoxia signaling pathway for LIC maintenance in vivo [32]. Our data support these results and further demonstrate that hypoxic conditions are also essential for LIC maintenance ex vivo. Culturing AML on MS-5+3GT at 3% O2 for 3 weeks preserved the LIC frequency and maintained the total number at the initial level. By using serial transplantation, we were also able to demonstrate that LIC self-renewal potential was preserved during the 3-week coculture period. Myeloid-restricted mouse engraftment was observed in all cases, indicating that the residual normal HSCs that could have been present within leukemic samples did not outcompete the AML cells during ex vivo culture, contrary to what has been described previously in other culture systems [19].
More important, we demonstrated that this niche-like culture system is also capable of supporting functionally defined human chemoresistant LICs as efficiently as the in vivo assay, with the latter being a more costly and time-consuming experimental approach. In a rare case where AML cells could be cultured in absence of stroma for 3 weeks, we were able to demonstrate that 3% O2 improved LIC chemoresistance in a feeder-dependent fashion. Our study also shows that phenotypic analysis of treated AML cells is an inefficient method for determining the LIC targeting efficacy. Recent studies from our laboratory and others demonstrate the LIC surface antigen phenotype is more heterogeneous than initially believed [9–12]. In this study, our effort to identify the actual LIC phenotype per sample by coupling heavy sorting strategies with the xenotransplantation assay proved to be a useless approach in the context of cytarabine treatment because the CD34 and CD38 expression profile was not perturbed. Alternatively, we repeatedly observed that quantifying the frequency of L-LTC-ICs in secondary cultures (2° LDA) is a reliable functional read-out for monitoring the LICs. The 2° LDA or xenograft LDA also proved that LICs are not systematically enriched after chemotherapy, whereas the total leukemic pool is effectively decreased after Ara-C, at least for the samples tested. Although further samples should be investigated to cover AML heterogeneity, our observations suggest—contrary to conventional belief—that LICs do not always preferentially survive antineoplastic treatment compared with non-LICs. Because our 1-week Ara-C treatment mimics the clinical treatment duration, we believe that our experimental setting represents a good model for dissecting the sequence of molecular mechanisms by which LICs escape cell death. Although this ex vivo system is faster and requires less tissue material than the xenograft assay, our data also demonstrate that its use might be limited to the most aggressive samples, in which LICs are relatively frequent.
Conclusion
The 2° LDA read-out for L-LTC-ICs described in this paper could represent the first system in which sensitivity to drugs, small compounds, or short hairpin RNA library screening could be tested on a relatively large scale with LICs. This appears even more crucial in the context of preclinical and clinical studies, which have demonstrated that assessment of bulk tumor shrinkage is not faithfully related to patient survival [38–40]. Moreover, this tool should enable both pharmaceutical and academic research laboratories to design innovative therapeutic approaches to target and monitor the roots of AML.
Supplementary Material
Acknowledgments
This work was funded by Cancer Research UK and by a European grant (contract no. 037632) to D.B. We are indebted to patients who gave samples. We thank Finlay McDougall for providing diagnostic information, Dr. Jude Fitzgibbon for helping us obtain patients' materials, and Dr. Arnaud Gandillet for performing some of the experiments. We also gratefully acknowledge Erin Currie and Yasmin Reyal for critical reading of the manuscript. We thank the Animal Care and Flow Cytometry core facilities staff at the London Research Institute for valuable technical help.
Author Contributions
E.G.: design and performance of experiments, data analysis and interpretation, manuscript writing; F.A.-A.: design and performance of experiments, data analysis and interpretation; I.P., K.R.-P., and J.V.: research, data analysis; D.T. and J.G..: provision of vital patients, materials, and data; F.L.: assistance in conception and design; D.B.: design of experiments, data analysis, manuscript writing.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Löwenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 3.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 5.Damiano JS, Cress AE, Hazlehurst LA, et al. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 6.Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–2526. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 7.Zhou BB, Zhang H, Damelin M, et al. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8:806–823. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 8.Wunderlich M, Mizukawa B, Chou FS, et al. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121:e90–e97. doi: 10.1182/blood-2012-10-464677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taussig DC, Miraki-Moud F, Anjos-Afonso F, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- 10.Taussig DC, Vargaftig J, Miraki-Moud F, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood. 2010;115:1976–1984. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarry JE, Murphy K, Perry R, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest. 2011;121:384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 13.Rafii S, Shapiro F, Pettengell R, et al. Human bone marrow microvascular endothelial cells support long-term proliferation and differentiation of myeloid and megakaryocytic progenitors. Blood. 1995;86:3353–3363. [PubMed] [Google Scholar]
- 14.Lu LS, Wang SJ, Auerbach R. In vitro and in vivo differentiation into B cells, T cells, and myeloid cells of primitive yolk sac hematopoietic precursor cells expanded > 100-fold by coculture with a clonal yolk sac endothelial cell line. Proc Natl Acad Sci USA. 1996;93:14782–14787. doi: 10.1073/pnas.93.25.14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodan SB, Imai Y, Thiede MA, et al. Characterization of a human osteosarcoma cell line (Saos-2) with osteoblastic properties. Cancer Res. 1987;47:4961–4966. [PubMed] [Google Scholar]
- 16.Taichman RS, Reilly MJ, Emerson SG. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood. 1996;87:518–524. [PubMed] [Google Scholar]
- 17.Gillette JM, Larochelle A, Dunbar CE, et al. Intercellular transfer to signalling endosomes regulates an ex vivo bone marrow niche. Nat Cell Biol. 2009;11:303–311. doi: 10.1038/ncb1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Gosliga D, Schepers H, Rizo A, et al. Establishing long-term cultures with self-renewing acute myeloid leukemia stem/progenitor cells. Exp Hematol. 2007;35:1538–1549. doi: 10.1016/j.exphem.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 19.Ailles LE, Gerhard B, Hogge DE. Detection and characterization of primitive malignant and normal progenitors in patients with acute myelogenous leukemia using long-term coculture with supportive feeder layers and cytokines. Blood. 1997;90:2555–2564. [PubMed] [Google Scholar]
- 20.Méndez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiel MJ, Yilmaz OH, Iwashita T, et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 22.Hooper AT, Butler JM, Nolan DJ, et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009;4:263–274. doi: 10.1016/j.stem.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 24.Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 25.Bruserud O, Ryningen A, Wergeland L, et al. Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica. 2004;89:391–402. [PubMed] [Google Scholar]
- 26.Dias S, Choy M, Alitalo K, et al. Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood. 2002;99:2179–2184. doi: 10.1182/blood.v99.6.2179. [DOI] [PubMed] [Google Scholar]
- 27.Glenjen NI, Hatfield K, Bruserud O. Coculture of native human acute myelogenous leukemia blasts with fibroblasts and osteoblasts results in an increase of vascular endothelial growth factor levels. Eur J Haematol. 2005;74:24–34. doi: 10.1111/j.1600-0609.2004.00333.x. [DOI] [PubMed] [Google Scholar]
- 28.Liesveld JL, Rosell KE, Lu C, et al. Acute myelogenous leukemia—microenvironment interactions: Role of endothelial cells and proteasome inhibition. Hematology. 2005;10:483–494. doi: 10.1080/10245330500233452. [DOI] [PubMed] [Google Scholar]
- 29.Danet GH, Pan Y, Luongo JL, et al. Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest. 2003;112:126–135. doi: 10.1172/JCI17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammoud M, Vlaski M, Duchez P, et al. Combination of low O(2) concentration and mesenchymal stromal cells during culture of cord blood CD34(+) cells improves the maintenance and proliferative capacity of hematopoietic stem cells. J Cell Physiol. 2012;227:2750–2758. doi: 10.1002/jcp.23019. [DOI] [PubMed] [Google Scholar]
- 31.Hermitte F, Brunet de la Grange P, Belloc F, et al. Very low O2 concentration (0.1%) favors G0 return of dividing CD34+ cells. Stem Cells. 2006;24:65–73. doi: 10.1634/stemcells.2004-0351. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Liu Y, Malek SN, et al. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011;8:399–411. doi: 10.1016/j.stem.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan N, Koritzinsky M, Zhao H, et al. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 2008;68:605–614. doi: 10.1158/0008-5472.CAN-07-5472. [DOI] [PubMed] [Google Scholar]
- 34.Comerford KM, Wallace TJ, Karhausen J, et al. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387–3394. [PubMed] [Google Scholar]
- 35.Erler JT, Cawthorne CJ, Williams KJ, et al. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol. 2004;24:2875–2889. doi: 10.1128/MCB.24.7.2875-2889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szilvassy SJ, Meyerrose TE, Ragland PL, et al. Homing and engraftment defects in ex vivo expanded murine hematopoietic cells are associated with downregulation of beta1 integrin. Exp Hematol. 2001;29:1494–1502. doi: 10.1016/s0301-472x(01)00751-2. [DOI] [PubMed] [Google Scholar]
- 37.Guan Y, Gerhard B, Hogge DE. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML) Blood. 2003;101:3142–3149. doi: 10.1182/blood-2002-10-3062. [DOI] [PubMed] [Google Scholar]
- 38.Durie BG, Jacobson J, Barlogie B, et al. Magnitude of response with myeloma frontline therapy does not predict outcome: Importance of time to progression in southwest oncology group chemotherapy trials. J Clin Oncol. 2004;22:1857–1863. doi: 10.1200/JCO.2004.05.111. [DOI] [PubMed] [Google Scholar]
- 39.Ardeshna KM, Smith P, Norton A, et al. Long-term effect of a watch and wait policy versus immediate systemic treatment for asymptomatic advanced-stage non-Hodgkin lymphoma: A randomised controlled trial. Lancet. 2003;362:516–522. doi: 10.1016/s0140-6736(03)14110-4. [DOI] [PubMed] [Google Scholar]
- 40.Huff CA, Matsui W, Smith BD, et al. The paradox of response and survival in cancer therapeutics. Blood. 2006;107:431–434. doi: 10.1182/blood-2005-06-2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.