

Abstract
Tumor suppressors function in a coordinated regulatory network, and their inactivation is a key step in carcinogenesis. The tumor suppressor Par-4 is a novel integral player in the PTEN network. Thus, Par-4 is absent in a high percentage of human prostate carcinomas, and its loss is concomitantly associated with PTEN loss. Genetic ablation of Par-4 induces fully invasive prostate carcinomas in PTEN-heterozygous mice. In contrast, Par-4 deficiency alone, like PTEN heterozygosis, results in lesions that are unable to progress beyond the benign neoplastic stage known as PIN. At this PIN transition, the mutual induction of Par-4 and PTEN is an additional regulatory step in preventing cancer progression. Par-4 deficiency cooperates with PTEN haploinsufficiency in prostate cancer initiation and progression and their simultaneous inactivation, in addition to enhancing Akt activation, sets in motion a unique mechanism involving the synergistic activation of NFκB. These results suggest that the concurrent interruption of complementary signaling pathways targeting PI3K/Akt and NFκB activation could provide new and effective strategies for cancer therapy.
Keywords: Par-4, PTEN, aPKC, PKCζ, Akt, NFκB, prostate cancer, tumor suppressors
Introduction
Prostate cancer (PCa) is one of the most common malignancies in men. The prevalence of PCa is on the increase in western societies. It is among the leading causes of male cancer-related morbidity and death, second only to lung cancer, representing approximately 10% of all cancer deaths among men in the United States. Indeed, one in six men in the United States will be diagnosed with PCa during their lifetime. PCa is a complex disease in its development and response to therapy.
PCa proceeds through a series of defined steps, including prostatic intraepithelial neoplasia (PIN), invasive cancer, and hormone-dependent or hormone-independent metastasis. All these different stages have been well defined histologically, although the molecular mechanisms contributing to the initiation and progression of PCa are not fully understood. Diagnosis is based mainly on histology and Gleason scoring and, while effective for disease identification and determining general prognosis, these tools have limited usefulness in deciding the best course of treatment for patients with intermediate grade tumors.1 Treatment is further complicated by the fact that prostate cancer initially responds well to androgen-ablation or anti-androgen therapy, but eventually enters an androgen-independent stage with no effective therapy.2 Therefore, the development of new therapies and better diagnostic techniques will depend on increasing our understanding of the molecular basis of this disease.
Genetic loss or mutation of tumor suppressor genes is a frequent event initiating and/or promoting tumorigenesis.3 The tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome 10) is an important player in human prostate carcinogenesis and, with p53, represents one of the most frequently mutated genes in PCa. PTEN maps to 10q23, a locus that is highly susceptible to mutation in primary human cancers4,5 and is commonly lost in metastatic prostate cancer.6 Functionally, PTEN is a plasma-membrane lipid phosphatase that antagonizes the PI-3K/Akt pathway7,8 by hydrolyzing phosphatidylinositol 3,4,5-trisphosphate (PIP3) to generate phosphatidylinositol 4,5-trisphosphate PIP2. Upon PTEN loss, PIP3 accumulates and promotes the recruitment to the membrane of pleckstrin homology domain-containing proteins, including AKT, and their subsequent activation. Activation of Akt through deregulated PI3K signaling, resulting from genetic inactivation of PTEN or an activating mutation in PI3K, is a frequent molecular event in human cancer and one of the major signaling pathways implicated in advanced PCa.9–12 Consistent with this, genetic inactivation of Akt1 suppresses tumor development in PTEN+/− mice. However, transgenic expression of activated Akt in the murine prostate is not sufficient to promote the development of invasive PCa, suggesting that Akt-independent pathways or the cooperation of complementary networks of tumor suppressors might be required for tumor progression.13 In keeping with this, PTEN haploinsufficiency has been shown to promote cell proliferation and the development of PIN, but these lesions do not progress to invasive disease14 unless there is a concomitant loss of other tumor suppressors. This opens an important avenue of study that involves the identification of novel tumor suppressors and the elucidation of the means by which they coordinate with each other to form an intricate regulatory network to protect against tumorigenesis.
The Tumor Suppressor Par-4 at the Crossroad of NFκB and Akt Signaling
Par-4 is a tumor suppressor originally identified in an in vitro differential screen of prostate cancer cells undergoing apoptosis following androgen withdrawal.15 The Par-4 gene maps to chromosome 12q21, a region frequently deleted in certain malignancies.16 Par-4’s role as a tumor suppressor matches its tissue distribution in that it appears to be active in tissues in which is highly expressed such as prostate, endometrium and lung.17,18 Consistent with this, in vivo studies of Par-4 KO mice show reduced lifespan, enhanced benign tumor formation, and low-frequency carcinogenesis. Par-4-deficient mice develop increased benign neoplasia in hormone-dependent tissues and cooperate with Ras to induce lung carcinoma in vivo. In addition, it has been shown that Par-4 is downregulated in approximately 40% of human endometrial carcinomas, human prostate carcinomas, and human lung adenocarcinomas.17,19,20
The Par-4 gene encodes a protein that harbors a leucine-zipper domain in the carboxy-terminal region, which interacts with several proteins including the atypical PKCs (aPKCs), the Wilms’ tumor 1 (WT1) protein, and the kinase MUK/DLK/ZPK.21,22 The interaction with WT1 and MUK/DLK/ZPK points to a nuclear role for Par-4 as a transcriptional repressor; however, in vivo genetic evidence supporting this function is sorely lacking. The available data based on genetic evidence support a model according to which the direct binding of Par-4 to the zinc-finger domain of both aPKC isoforms, PKCζ and PKCζ/ι, results in inhibition of their enzymatic activity.23 This leads to the subsequent impairment of NFκB activation, as both aPKCs are relevant pro-inflammatory molecules for the regulation of the NFκB pathway.24,25 In fact, multiple studies independently demonstrated that overexpression of Par-4 leads to inhibition of NFκB, thus potentiating TNFα-induced cell death.26–28 In this regard, the loss of Par-4 in embryo fibroblasts leads to the hyperactivation of PKCζ and of NFκB transcriptional activity.29 Consistent with this, the NFκB-dependent anti-apoptotic protein XIAP is expressed at significantly elevated levels in Par-4-null cells, which correlates with reduced caspase-3 activation and apoptosis.29 In addition, Par-4 deficiency is associated with increased NFκB activation in both lung and prostate cells. Moreover, this hyperactivation is reversed upon loss of PKCζ in Par-4/PKCζ DKO mice, suggesting that PKCζ is a bona fide target of Par-4 in vivo. Interestingly, Par-4-deficient mice also have higher levels of activated Akt in lung and prostate epithelial cells, and, as is the case for NFκB, this activation is mediated by PKCζ.17 Akt is a direct substrate of PKCζ at the phosphorylation site Ser124, which helps to control basal Akt activity by allowing the efficient phosphorylation of Akt at two other critical residues, Thr308 and Ser473, which are required for full activation.17 This places Par-4 as a common step in the regulation of the Akt and NFκB pathways. A question that deserves further investigation is whether or not Akt is involved in the regulation of NFκB by the Par-4/PKCζ cassette.
Par-4, a Novel Tumor Suppressor in the PTEN Network
There are two primary ways that tumor suppressors could coordinate their activities in a regulatory network: they could impinge on a single signaling pathway to increase a required molecular threshold, or they could activate different complementary and downstream pathways that interact to create a synergistic effect. In this regard, a common mechanism of action underlying the cooperation of PTEN with other tumor suppressors is through the modulation of Akt activation. For example, the tumor suppressor PML cooperates with PTEN inside the nucleus to inhibit Akt through its recruitment and inhibition by the phosphatase PP2a in the PML nuclear bodies.30 Another tumor suppressor, NEP, cooperates with PTEN through synergistic inhibition of the PI3K/Akt pathway by direct interaction of and stabilization of PTEN.31 In addition, PTEN synergizes with other tumor suppressors, such as NKx3.1, p18 and Tsc2, through cooperation in Akt activation.32–35 However, in addition to Akt activation, there could be other complementary mechanisms set in motion by the cooperative loss of PTEN and other tumor suppressors that have an important impact on the progression to invasive carcinoma. In fact, PTEN cooperates with Rb and p18 in a complementary collaboration through their role in controlling cell cycle progression.33 PTEN haploinsufficiency cooperates with the overexpression of Rheb, an upstream activator of mTOR complex 1 and with the overexpression of FGF8b, a commonly occurring genetic aberration of human PCa, impinging on different signaling pathways.36,37 Also, two recent studies show that the common recurrent gene fusion between TMPRSS2 and ERG promotes PCa when PTEN is concurrently lost.36,38 ERG can act together with PTEN by inducing the transcription of downstream checkpoint genes that would usually be blocked by Akt and have a crucial role on cell migration and invasion. Such collaboration could provide a selective advantage at the cellular level to allow benign lesions to progress to cancer.
Par-4 is a newly identified player in the network of tumor suppressors that cooperate with PTEN (Fig. 1). Recent studies from our laboratory demonstrate that the loss of Par-4 in the context of PTEN haploinsufficiency leads to invasive PCa in mice.20 Concomitant deficiency of both tumor suppressors has an impact not only on the progression of PIN lesions to invasive carcinoma, but also on tumor initiation, with a higher incidence of PIN lesions upon Par-4 loss. Interestingly, the combined mutation of Par-4 and PTEN regulates both proliferation and survival of prostatic epithelial cells, in contrast to the cooperation between PTEN and other tumor suppressors, which only affect proliferation. This is a unique feature of Par-4 and PTEN interplay that could be explained by the synergy of the two mutations on activation of the NFκB cascade, an important pathway in cell survival. Of note, the inactivation of both tumor suppressors results in the synergistic stimulation of NFκB, not only in PIN lesions but also in preneoplastic prostates.20 This suggests that the activation of NFκB in the preneoplastic glands of the compound mutants could be a causative mechanism to promote invasive PCa. Par-4 deficiency also leads to an increase in Akt activation, and this effect is enhanced in the context of PTEN heterozygosity. Thus, the concomitant loss of PTEN and Par-4, in addition to modulating the Akt pathway, impinges on the NFκB cascade, which could unleash signals complementary to those elicited by Akt (Fig. 1). In this regard, two important inflammatory targets of NFκB, the cytokine IL-6 and the chemokine IL-8, are increased in the Par-4/PTEN compound-mutant prostates.20 This might mediate the recruitment of inflammatory cells and facilitate an angiogenic response that could collaborate with proliferative and survival signals in the progression to an invasive phenotype.
Figure 1.

Cooperation of Par-4 deficiency and PTEN haploinsufficiency in prostate cancer progression. Par-4 loss cooperates with PTEN heterozygosity to promote invasive prostate carcinoma. The simultaneous inactivation of Par-4 and PTEN enhances Akt and leads to a synergistic stimulation of the NFκB pathway. This sets in motion complementary signals regulating cell growth, cell survival, inflammation and angiogenesis that collaborate in prostate cancer progression. It is not known whether Akt is able to directly impinge on the NFκB pathway in this system.
Interestingly, the synergy between the inactivation of Par-4 and PTEN observed in the double-mutant mouse model is consistent with a significant association of Par-4 and PTEN expression levels in human PCa. That is, Par-4 loss is associated with PTEN loss and correlates with high Gleason scores in human PCa. Par-4 inactivation is mostly associated with aberrant de novo methylation of the Par-4 promoter.20 Of note, there is also an inverse correlation between Par-4 and PTEN levels and activation of the NFκB pathway, measured as p65 nuclear translocation and IL-6 levels, indicating that, in fact, activation of this pathway might account for the collaboration between these two tumor suppressors.
Par-4 and PTEN Interplay as a Safeguard Checkpoint in Tumor Progression
The combined loss of tumor suppressors is a hallmark of advanced human PCa and suggests a “one-by-one” hit model for tumor development in which there is a sequential loss of tumor suppressor genes. Research suggests that there is a line of defense against tumorigenesis composed of a number of tumor suppressors, each with the ability to control one or more cellular process through the specific pathways on which they act. This suggests that a network exists through which the different signaling and molecular events are integrated and coordinated to fine-tune cancer progression. To add to the complexity, tumor suppressor genes are also subject to countless regulatory mechanisms that ultimately control their activity, protein levels and function.
The fact that Par-4 KO prostates display a hyper-plastic phenotype and do not progress to later stages in PCa, except in the context of PTEN haploinsufficiency, suggests that PTEN could act as a safeguard mechanism in the absence of Par-4 to prevent cancer progression. Thus, it is possible that PTEN levels could increase as a consequence of Par-4 deficiency, dampening the tumorigenic signaling cascades unleashed by the loss of Par-4. Figure 2 shows that this is actually the case, in that Par-4 KO prostates have increased PTEN protein and mRNA levels. These observations are consistent with the notion that PTEN acts as a checkpoint that limits hyperplastic proliferation and malignant transformation. That is, upon the loss of one tumor suppressor, the cell sets in motion compensatory mechanisms that induce other tumor suppressors to restrain tumorigenesis. Interestingly, such interplay between Par-4 and PTEN is also evident in PTEN heterozygous animals. That is, as shown in Figure 3, Par-4 levels are increased in PTEN+/− prostates, both at the protein and mRNA levels. These results suggest that the mutual regulatory interplay between Par-4 and PTEN expression may represent an additional safeguard mechanism in the transition from preneoplastic lesions to invasive cancer. This could also explain the cooperation of the two tumor suppressors, as the dual loss would result in the inactivation of this molecular brake. Invasive cancer, where checkpoint loss may have already occurred, would then be associated with reduced levels of both Par-4 and PTEN, a prediction that was confirmed by analysis of human prostate tumors.20
Figure 2.
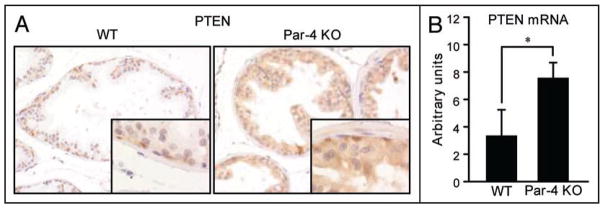
Increased levels of PTEN in Par-4 KO prostates. (A) Immunostaining showing increased PTEN levels in prostate epithelial cells in Par-4 KO mice. (B) PTEN mRNA levels are also induced upon Par-4 deficiency, as measured by QRT-PCR. n = 4 mice per genotype. *p < 0.02.
Figure 3.
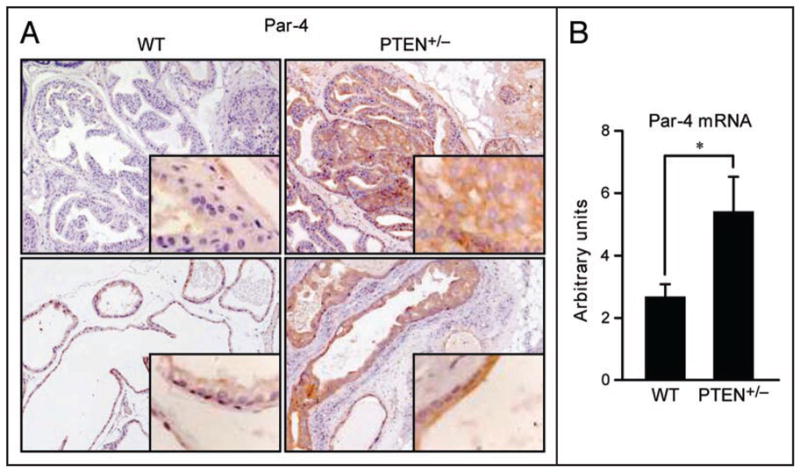
Induction of Par-4 in PTEN+/− prostates. (A) Immunostaining showing increased Par-4 levels in prostate epithelial cells of the anterior (upper) and dorsal (lower) prostate lobes in PTEN+/− mice. (B) Induction of Par-4 mRNA levels in dorsal prostates of PTEN+/− mice, as measured by QRT-PCR. n = 4 mice per genotype. *p < 0.01.
A similar scenario has previously been described in which PTEN is linked with other tumor suppressors, such as p53, in a complex relationship. PTEN has been reported to be a downstream target of p53 in mediating apoptosis,39 and also to act upstream of p53 to regulate its expression levels and activity.40,41 The physical binding of p53 and PTEN gives further support to their functional crosstalk.42,43 Furthermore, total deletion of PTEN results in enhanced expression of p53 as well as p21, a direct transcription target of p53.44 Consistent with this, combined inactivation of p53 and PTEN cooperates and accelerates tumor development.44 Importantly, acute PTEN inactivation triggers p53-dependent cellular senescence as a checkpoint to restrain tumorigenesis.44 Cellular senescence is commonly seen in early or precursor stages of cancer.45,46 Interestingly, p27-mediated senescence has been also described in AKT1 transgenic and PTEN homozygous mice, where it is associated with the PIN phenotype. Consistent with this, loss of p27 in the context of AKT1 transgenic mice leads to increased proliferation, loss of senescence, and progression of PIN lesions to invasive PCa.47 This crosstalk between PTEN and p53 or p27 resembles that of PTEN and Par-4, although whether or not the PTEN-Par-4 interplay triggers senescence is not known. The connections between Par-4 and other players in the PTEN tumor suppressor network and the mutual regulatory mechanisms controlling their levels, location and activity is still an open question that deserves further investigation.
Concluding Remarks
Par-4 is a novel tumor suppressor that is well positioned to be an integrator of the currently growing PTEN network. New studies are unveiling the cooperation of tumor suppressors to increase the threshold of a shared signaling event, or to set in place new molecular mechanisms. In this regard, Par-4 deficiency cooperates with PTEN haploinsufficiency to promote invasive PCa, and their simultaneous inactivation (in addition to enhancing Akt activation) sets in motion a unique mechanism involving the synergistic stimulation of NFκB. These observations provide further support for the increasingly recognized role of NFκB in cancer. Akt and NFκB pathways are both deregulated during prostate tumorigenesis, and their activation could offer complementary advantage to cells to further progress towards an invasive phenotype. These findings also suggest that the concurrent interruption of complementary signaling pathways could provide a new and effective avenue for cancer therapy. Specifically, combinatorial therapies targeting PI3K/Akt and NFκB signaling pathways may be an effective treatment for PCa.
In summary, recent evidences are unveiling that tumor suppressors exist in a finely tune and regulated network that integrates signals to coordinate molecular events that protect cells against tumorigenesis. Understanding the cross talks among each component of these cascades, and unraveling the intricate molecular mechanisms that govern their connections will undoubtedly contribute to the development of new therapeutic strategies sorely needed for the treatment of aggressive forms of cancer.
Acknowledgments
We thank Maryellen Daston for editing this manuscript and Glenn Doerman for preparing the figures. This work was funded in part by the University of Cincinnati-CSIC Collaborative Agreement, the NIH grant 1R01CA134530, and the Barrett/UC Cancer Center Pilot Grant.
Abbreviations
- Akt (PKB)
protein kinase B
- aPKC
atypical PKC
- DLK
dual leucine zipper kinase
- ERG
ETS related gene
- FGF8b
fibroblast growth factor 8, isoform b
- IL-6
interleukin-6
- IL-8
interleukin-8
- mTOR
mammalian target of rapamycin
- MUK, NFκB
nuclear factor κB
- Par-4
prostate apoptosis response-4
- PCa
prostate cancer
- PI 3-kinase
phosphoinositide 3-kinase
- PIN
prostatic intraepithelial neoplasia
- PIP
phosphatidylinositol
- PKC
protein kinase C
- PML
promyelocytic leukemia
- PTEN
phosphatase and tensin homolog gene
- Rb
retinoblastoma
- Rheb
Ras homolog-enriched in brain
- TNFα
tumor necrosis factor α
- Tsc2
tuberous sclerosis complex 2
- WT1
Wilms’ tumor 1
- XIAP
X-linked inhibitor of apoptosis protein
- ZPK
zipper protein kinase
References
- 1.Isaacs W, De Marzo A, Nelson WG. Focus on prostate cancer. Cancer Cell. 2002;2:113–6. doi: 10.1016/s1535-6108(02)00103-4. [DOI] [PubMed] [Google Scholar]
- 2.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 5.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23. 3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 6.Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57:4997–5000. [PubMed] [Google Scholar]
- 7.Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA. 1997;94:9052–7. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–5. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malik SN, Brattain M, Ghosh PM, Troyer DA, Prihoda T, Bedolla R, et al. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clin Cancer Res. 2002;8:1168–71. [PubMed] [Google Scholar]
- 11.McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999;59:4291–6. [PubMed] [Google Scholar]
- 12.Kremer CL, Klein RR, Mendelson J, Browne W, Samadzedeh LK, Vanpatten K, et al. Expression of mTOR signaling pathway markers in prostate cancer progression. Prostate. 2006;66:1203–12. doi: 10.1002/pros.20410. [DOI] [PubMed] [Google Scholar]
- 13.Majumder PK, Yeh JJ, George DJ, Febbo PG, Kum J, Xue Q, et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proc Natl Acad Sci USA. 2003;100:7841–6. doi: 10.1073/pnas.1232229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 15.Sells SF, Wood DP, Jr, Joshi-Barve SS, Muthukumar S, Jacob RJ, Crist SA, et al. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth Differ. 1994;5:457–66. [PubMed] [Google Scholar]
- 16.Johnstone RW, Tommerup N, Hansen C, Vissing H, Shi Y. Mapping of the human PAWR (par-4) gene to chromosome 12q21. Genomics. 1998;53:241–3. doi: 10.1006/geno.1998.5494. [DOI] [PubMed] [Google Scholar]
- 17.Joshi J, Fernandez-Marcos PJ, Galvez A, Amanchy R, Linares JF, Duran A, et al. Par-4 inhibits Akt and suppresses Ras-induced lung tumorigenesis. EMBO J. 2008;27:2181–93. doi: 10.1038/emboj.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia-Cao I, Duran A, Collado M, Carrascosa MJ, Martin-Caballero J, Flores JM, et al. Tumour-suppression activity of the proapoptotic regulator Par4. EMBO Rep. 2005;6:577–83. doi: 10.1038/sj.embor.7400421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moreno-Bueno G, Fernandez-Marcos PJ, Collado M, Tendero MJ, Rodriguez-Pinilla SM, Garcia-Cao I, et al. Inactivation of the candidate tumor suppressor par-4 in endometrial cancer. Cancer Res. 2007;67:1927–34. doi: 10.1158/0008-5472.CAN-06-2687. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Marcos PJ, Abu-Baker S, Joshi J, Galvez A, Castilla EA, Canamero M, et al. Simultaneous inactivation of Par-4 and PTEN in vivo leads to synergistic NF{kappa}B activation and invasive prostate carcinoma. Proc Natl Acad Sci USA. 2009 doi: 10.1073/pnas.0813055106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnstone RW, See RH, Sells SF, Wang J, Muthukkumar S, Englert C, et al. A novel repressor, par-4, modulates transcription and growth suppression functions of the Wilms’ tumor suppressor WT1. Mol Cell Biol. 1996;16:6945–56. doi: 10.1128/mcb.16.12.6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Page G, Kogel D, Rangnekar V, Scheidtmann KH. Interaction partners of Dlk/ZIP kinase: co-expression of Dlk/ZIP kinase and Par-4 results in cytoplasmic retention and apoptosis. Oncogene. 1999;18:7265–73. doi: 10.1038/sj.onc.1203170. [DOI] [PubMed] [Google Scholar]
- 23.Diaz-Meco MT, Municio MM, Frutos S, Sanchez P, Lozano J, Sanz L, et al. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell. 1996;86:777–86. doi: 10.1016/s0092-8674(00)80152-x. [DOI] [PubMed] [Google Scholar]
- 24.Moscat J, Diaz-Meco MT. The atypical protein kinase Cs. Functional specificity mediated by specific protein adapters. EMBO Rep. 2000;1:399–403. doi: 10.1093/embo-reports/kvd098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moscat J, Diaz-Meco MT, Rennert P. NFkappaB activation by protein kinase C isoforms and B-cell function. EMBO Rep. 2003;4:31–6. doi: 10.1038/sj.embor.embor704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barradas M, Monjas A, Diaz-Meco MT, Serrano M, Moscat J. The downregulation of the pro-apoptotic protein Par-4 is critical for Ras-induced survival and tumor progression. EMBO J. 1999;18:6362–9. doi: 10.1093/emboj/18.22.6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diaz-Meco MT, Lallena MJ, Monjas A, Frutos S, Moscat J. Inactivation of the inhibitory kappaB protein kinase/nuclear factor kappaB pathway by Par-4 expression potentiates tumor necrosis factor alpha-induced apoptosis. J Biol Chem. 1999;274:19606–12. doi: 10.1074/jbc.274.28.19606. [DOI] [PubMed] [Google Scholar]
- 28.Nalca A, Qiu SG, El-Guendy N, Krishnan S, Rangnekar VM. Oncogenic Ras sensitizes cells to apoptosis by Par-4. J Biol Chem. 1999;274:29976–83. doi: 10.1074/jbc.274.42.29976. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Cao I, Lafuente M, Criado L, Diaz-Meco M, Serrano M, Moscat J. Genetic inactivation of Par4 results in hyperactivation of NFκB and impairment of JNK and p38. EMBO Rep. 2003;4:307–12. doi: 10.1038/sj.embor.embor769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–7. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sumitomo M, Iwase A, Zheng R, Navarro D, Kaminetzky D, Shen R, et al. Synergy in tumor suppression by direct interaction of neutral endopeptidase with PTEN. Cancer Cell. 2004;5:67–78. doi: 10.1016/s1535-6108(03)00331-3. [DOI] [PubMed] [Google Scholar]
- 32.Ma L, Teruya-Feldstein J, Behrendt N, Chen Z, Noda T, Hino O, et al. Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev. 2005;19:1779–86. doi: 10.1101/gad.1314405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai F, Pei XH, Pandolfi PP, Xiong Y. p18 Ink4c and Pten constrain a positive regulatory loop between cell growth and cell cycle control. Mol Cell Biol. 2006;26:4564–76. doi: 10.1128/MCB.00266-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim MJ, Cardiff RD, Desai N, Banach-Petrosky WA, Parsons R, Shen MM, et al. Cooperativity of Nkx3. 1 and Pten loss of function in a mouse model of prostate carcinogenesis. PNAS. 2002;99:2884–9. doi: 10.1073/pnas.042688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei Q, Jiao J, Xin L, Chang CJ, Wang S, Gao J, et al. NKX3. 1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer Cell. 2006;9:367–78. doi: 10.1016/j.ccr.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 36.Nardella C, Chen Z, Salmena L, Carracedo A, Alimonti A, Egia A, et al. Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev. 2008;22:2172–7. doi: 10.1101/gad.1699608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong C, Saribekyan G, Liao CP, Cohen MB, Roy-Burman P. Cooperation between FGF8b overexpression and PTEN deficiency in prostate tumorigenesis. Cancer Res. 2006;66:2188–94. doi: 10.1158/0008-5472.CAN-05-3440. [DOI] [PubMed] [Google Scholar]
- 38.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41:524–6. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- 40.Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002;27:462–7. doi: 10.1016/s0968-0004(02)02166-7. [DOI] [PubMed] [Google Scholar]
- 41.Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–30. doi: 10.1016/s1535-6108(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 42.Tang Y, Eng C. PTEN autoregulates its expression by stabilization of p53 in a phosphatase-independent manner. Cancer Res. 2006;66:736–42. doi: 10.1158/0008-5472.CAN-05-1557. [DOI] [PubMed] [Google Scholar]
- 43.Flores-Delgado G, Liu CW, Sposto R, Berndt N. A limited screen for protein interactions reveals new roles for protein phosphatase 1 in cell cycle control and apoptosis. J Proteome Res. 2007;6:1165–75. doi: 10.1021/pr060504h. [DOI] [PubMed] [Google Scholar]
- 44.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 46.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 47.Majumder PK, Grisanzio C, O’Connell F, Barry M, Brito JM, Xu Q, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14:146–55. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]