

Abstract
Signal transducer and activator of transcription 3 (Stat3) transmits signals from growth factors and interleukin-6 family cytokines by binding to their receptors via its Src homology 2 (SH2) domain. This results in phosphorylation of Tyr705, dimerization, translocation to the nucleus, and regulation of transcription of downstream genes. Stat3 is constitutively activated in several human cancers and is a target for anti-cancer drug design. We have shown previously phosphorylation of Tyr705 in intact cancer cells can be inhibited with prodrugs of phosphopeptide mimics targeting the SH2 domain. In a series of prodrugs consisting of bis-pivaloyloxymethyl esters of 4′-phosphonodifluoromethyl cinnamoyl-Haic-Gln-NHBn, appending methyl group to the β-position of the cinnamate increased potency ca. twofold, which paralleled the increase in affinity of the corresponding phosphopeptide models. However, dramatic increases in potency were observed when the C-terminal C(O)NHBn of Gln-NHBn was replaced with a simple methyl group. In this communication we continue to explore the effects of structural modifications of prodrugs on their ability to inhibit Tyr705 phosphorylation. A set of 4-substituted prolines incorporated into β-methyl-4-phosphocinnamoyl-leucinyl-Xaa-4-aminopentamide model peptides exhibited affinities of 88–317 nM by fluorescence polarization (Pro IC50 = 156 nM). In corresponding prodrugs, Pro inhibited constitutive Stat3 phosphorylation at 10 μM in MDA-MB-468 breast tumor cells. However, 4,4-difluoroproline and 4,4-dimethylproline resulted in complete inhibition at 0.5 μM. These results suggest that the prodrug with native proline undergoes metabolism that those with substituted prolines do not. In conclusion, changes in structure with minimal impact on intrinsic affinity can nevertheless have profound effects on the cellular potency of prodrug inhibitors of Stat3.
Keywords: Signal transducer and activator of transcription 3, Stat3, Src homology domain 2, SH2 domain, Peptidomimetic, Phosphopeptide, Prodrug
Introduction
Signal transducer and activator of transcription 3 (Stat3) was first found as an immediate transducer of interleukin-6 (IL-6) signaling in immune responses to disease causing organisms and was originally named the acute phase response factor (Akira et al. 1994). On IL-6 binding to its receptor, Janus kinases (JAKs) are recruited which phosphorylate tyrosine residues on the cytoplasmic domain of the co-receptor, gp130. Via its Src homology 2 (SH2) domain, Stat3 is recruited to the pTyr residues on gp130 where Stat3 becomes phosphorylated at Tyr705 (pStat3) by the JAKs. pStat3 dimerizes via reciprocal pTyr-SH2 domain interactions, is translocated to the nucleus, and participates in the transcription of acute phase response genes (Levy and Darnell 2002). Stat3 was found to be activated (phosphorylated on Tyr705) in numerous human cancer types and cell lines and was hypothesized to transcribe genes involving cell cycling (cyclin D1), invasion and metastasis (MMP9), angiogenesis (vascular endothelial growth factor, VEGF) and resistance to apoptosis (Bcl-2-family proteins) (Yu and Jove 2004). In addition to IL-6 and IL-6-family cytokines, Stat3 was found to transmit signals from growth factors implicated in cancer such as epidermal growth factor, platelet derived growth factor, and VEGF. Inhibition of Stat3 activity by antisense, dominant negative, and siRNA oligonucleotides led to reduced expression of VEGF and MMP9, as well as apoptosis in cultured tumor cells. Taken together, these observations led to the hypothesis that blocking recruitment of Stat3 to receptors by targeting the SH2 domain with phosphopeptide mimics would lead to inhibition of Tyr705 phosphosphorylation, dimerization, translocation to the nucleus, and transcription of cancer associated genes, resulting in anti-tumor activity (Darnell 2002; Yu and Jove 2004; Fletcher et al. 2008).
We and others (Turkson et al. 2004; Fletcher et al. 2009; Gunning et al. 2007, 2008, Shahani et al. 2011a, b, Siddiquee et al. 2007; Zhang et al. 2010; Chen et al. 2007, 2010) have been targeting the SH2 domain of Stat3 with phosphopeptide mimics. Our laboratory found that the hexapeptide Ac-pTyr-Leu-Pro-Gln-Thr-Val-NH2 inhibited phosphoStat3 binding to DNA with an IC50 of 150 nM using the electrophoretic mobility shift assay (Ren et al. 2003) and 290 nM when using fluorescence polarization (Coleman et al. 2005). This peptide possesses the Tyr-Xaa-Yaa-Gln recognition determinant for Stat3 (Stahl et al. 1995). Extensive structure affinity studies (Coleman et al. 2005, 2008; Mandal et al. 2007, 2009b, c) showed substitution of Ac-pTyr with conformationally constrained 4-phosphorylcinnamate (pCinn) enhanced affinity and the C-terminal Thr-Val could be replaced with a simple benzyl group. The pY + 1 could be either Nle or Leu, the pY + 2 residue could be Pro or mPro (cis-3,4-methanoproline), or the pY + 1-pY + 2 dipeptide could be replaced with the tricyclic heterocycle, Haic ((2S,5S)-5-amino-1,2,4,5,6, 7-hexahydro-4-oxo-azepino[3,2,1-hi]indole-2-carboxylic acid) (1a, 2a, 3a, and 4a, Fig. 1). Analysis of the crystal structure of Stat3 (Becker et al. 1998) and models generated in our laboratory (Mandal et al. 2009b; McMurray 2008) suggested that a methyl group appended to the β-position of pTyr or pCinn would enhance affinity due to increased hydrophobic interaction with the side chain CH2 groups of Glu638. Methyl groups attached to the β carbon of pCinn (βMpCinn) resulted in a 2–3-fold increase in affinity in a set of modified phosphopeptides (Mandal et al. 2011) (1b, 2b, 3b, and 4b, Fig. 1).
Fig. 1.

High affinity inhibitors of the SH2 domain of Stat3. Addition of a methyl group to the β position of 4′-phosphorylcinnamate enhances affinity (from Mandal et al. 2011)
Having discovered high affinity ligands to the SH2 domain, the next step was to modify the structure to inhibit the target within intact live cells. To prevent cleavage of the phosphate by phosphatases, we employed the difluoromethylphosphonate group, first used for pTyr by Burke et al. (1994). The dual negative charge of a phosphate or difluoromethyl phosphonate prevents passive diffusion across cell membranes. Bioreversible esters have been employed to deliver phosphonates and phosphates of a variety of drugs into cells (Hecker and Erion 2008; Krise and Stella 1996; Schultz 2003). We employed carboxyesterase-labile pivaloyloxymethyl (POM) esters, that were pioneered by Farquhar and colleagues for the delivery of antiviral and antitumor nucleotides (Farquhar et al. 1994). Our first prodrug, BP-PM6 (5, Fig. 2), completely inhibited constitutive phosphorylation of Stat3 in MDA-MB-468 breast cancer cells after 2 h treatment at 10 μM. This suggested that the prodrug entered the cell, the POM groups were cleaved, and the free phosphonate bound to the SH2 domain of Stat3, preventing recruitment to receptors and subsequent phosphorylation (Mandal et al. 2009a). It also showed that Stat3 exists in a dynamic equilibrium between phosphorylation and dephosphorylation. Herein, we report our observations on the effects of structure on the potency of prodrugs of phosphopeptide mimics targeting the SH2 domain of Stat3 in intact cancer cells. We summarize previously published results on the effects of alterations to the cinnamate and the C-terminus and we present new data showing that peptides with proteinogenic proline are substantially less potent than analogues with proline substituted on the ring.
Fig. 2.

The effect of β-methyl cinnamate and C-terminal modification on the potency of prodrugs for the inhibition of constitutive Stat3 phosphorylation in MDA-MB-468 breast cancer cells (from Mandal et al. 2011). Compound names in parentheses are given for reference to future manuscripts on further biological evaluation. The procedure for evaluation is given in “Materials and Methods” section
Materials and Methods
Nα-Protected amino acids were purchased from NovaBiochem, ChemImpex, or Anaspec. 1-Hydroxybenzotriazole (HOBt) was from ChemImpex. Anhydrous DMF for amino acid solutions was from Aldrich. Other solvents were reagent grade and were used without further purification. NMR spectra were obtained on either a Bruker DPX 300 MHz spectrometer or a Bruker DRX 500 MHz spectrometer. Pentachlorophenyl 4′-phosphoryloxy-β-methylcinnamate was synthesized as described (Mandal et al. 2011). (R)-4-(9-Fluorenylmethoxycarbonlyamino)-pentanoic acid was prepared as described (Mandal et al. 2009c). Racemic Fmoc-cis-3,4-methanoproline was purchased from EMD Biosciences (Novabiochem). Peptides were assayed for affinity to Stat3 using fluorescence polarization as described (Coleman et al. 2005). For the synthesis of phosphopeptides, Rink resin with a loading of 0.6 mmol/g was employed. For the synthesis of prodrugs, Rink resin with a loading of 1.2 mmol/g was used. Resins were obtained from Advanced Chemtech, Inc. Phosphopeptides and prodrug intermediates were dried in vacuo over P2O5 at 37 °C for 24 h prior to use (Coleman et al. 2005). All compounds were >95 % pure (HPLC) before evaluation. Purities, yields, and mass spectral characteristics of phosphopeptides and prodrugs are provided in subsequent tables.
General Procedure for the Synthesis of Phosphopeptides
Solid phase syntheses were carried out manually using commercially available Rink resin. Resin, 0.2 g, was placed in a manual reactor and swollen and washed with 5 × 10 mL of DMF/CH2Cl2. Fmoc groups were removed with 3 × 6 mL of 20 % piperidine/DMF for 5 min each. For coupling, threefold excesses of Fmoc-amino acids, diisopropylcarbodiimide (DIC), and HOBt were used in 8–10 mL of DMF/CH2Cl2 and were allowed to proceed until resin samples tested negative with ninhydrin tests. For the phosphocinnamate derivatives, Leu-Xaa-Apa-Resins were capped with pentachlorophenyl 4′-phosphoryl-β-methylcinnamate in the presence of HOBt and DIPEA (2.0 equivalent each) in DMF/CH2Cl2 overnight or until ninhydrin tests were negative. After coupling and deprotection steps, resins were washed with 5 × 10 mL of DMF/CH2Cl2. On completion of the peptide chain, resins were washed with CH2Cl2 (3 × 10 mL) and were treated with trifluoroacetic acid (TFA):triisopropylsilane(TIS):H2O (95:2.5:2.5) (Pearson et al. 1989) (3 × 5 mL) for 15 min each. The combined filtrates sat at rt for 1–2 h and the volumes were reduced in vacuo. Peptides were precipitated in ice cold Et2O, collected by centrifugation, and washed twice more with the same solvent and centrifuged. After drying, peptides were purified by reverse phase HPLC on a Rainin Rabbit HPLC or a Varian Dynamax HPLC using a Phenomenex Luna C18(2) 10 μM 2.1 × 25 cm column. Gradients of MeCN in H2O at 10–20 mL/min containing 0.1 % TFA were employed. Peptides were tested for purity by reverse phase HPLC on an Agilent 1090 HPLC or an Agilent 1100 HPLC using a Phenomenex Luna C18(2) 5 μM 4.6 × 250 mm column. A gradient of 0–40 % MeCN/30 min at 1.5 mL/min was used.
General Procedure for the Synthesis of Prodrugs. Synthesis of 27
Nle-4,4-Me2Pro-Apa was synthesized on Rink resin, cleaved and purified by HPLC as described for the phosphopeptides above. To a stirred solution of TFA·H-Nle-4,4-Me2Pro-Apa (0.050 g, 0.106 mmol), N-methylmorpholine (0.046 mL, 0.42 mmol) and DMAP (0.005 g, 0.042 mmol) in 2 mL of dry NMP, was added a solution of pentachlorophenyl 4′-(bispivaloyloxymethyl)phosphinyldifluoromethyl-β-methylcinnamate (Mandal et al. 2011) (0.107 g, 0.14 mmol) in 2 mL of dry MeCN under inert atmosphere. Reaction progress and purity were assessed on the same HPLC systems as the phosphopeptides except the gradient was 10–80 % MeCN/30 min with no TFA in the mobile phase. After completion, about 2 h, the reaction mixture was concentrated under vacuum, and 50 mL of 40 % ether–hexane was added and mixed. The resultant emulsion was centrifuged and the crude residue then purified by reverse phase HPLC using gradients of MeCN in H2O with no TFA. Collected pure fractions were then lyophilized to give 34 mg of 27 as a white fluffy material (37 %). HRMS (M + H) calc: 857.4277; found: 857.4200. 1H NMR in CDCl3 revealed approximately 10 % cis conformation about the Nle-4,4-Me2Pro peptide bond. The resonances are tabulated and the spectra are included in the supporting information file.
Evaluation of Phosphopeptide Binding to Stat3 by Fluorescence Polarization
The procedure of Coleman et al. (2005) was utilized. Briefly, a Packard 204DT liquid handling robot was used to dispense 50 μL aliquots of a solution of 0.4 μg of full length Stat3 (160 nM) and 20 nM of FAM-Ala-pTyr-Leu-Pro-Gln-Thr-Val-NH2 (FAM = 5 carboxyfluorescein) in 50 mM NaCl, 10 mM Hepes, 1 mM Na4EDTA, 2 mM DTT, and 1 % NP-40 into wells of a black, opaque, flat bottom 96 well microtiter plate (Corning 3650). Serial dilutions of phosphopeptides in the same buffer were made in a separate 96-well plate with the robot and 50 μL of were added to the Stat3-FAM-peptide solution in corresponding wells in the black plate. Fluorescence polarization was then read in a Tecan Polarian plate reader. mP values were plotted against the log of the peptide concentration and IC50 values were obtained from linear regression analysis in the one site competition mode using Prizm Version 5 from GraphPad Software, Inc. IC50 values are reported as the mean of three independent IC50 determinations ± the standard deviation. Full length Stat3 was provided by Dr. Xiaomin Chen.
Inhibition of Stat3 Tyrosine 705 Phosphorylation in Tumor Cells
Procedures were carried out as described (Mandal et al. 2011). Briefly, 4 × 105 MDA-MB-468 breast tumor cells were added to 6-well culture dishes in DMEM media containing 10 % FCS and were allowed to grow overnight. The media was discarded. Prodrugs, 10 mM stock solutions in DMSO, were formulated immediately before use and aliquots were added to media to give the desired concentrations. After 2 h the cells were washed with ice cold phosphate buffered saline and were treated with lysis buffer (50 mM Hepes, pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM sodium pyrophosphate, 10 % glycerol, 1 % Triton X-100, 1 mM PMSF, 1 mM Na3VO4, 10 μg/mL leupeptin and 10 μg/mL aprotinin). Cell-free detergent extracts were centrifuged (15,000 rpm) for 30 min at 4 °C and protein concentrations of the supernatants were determined. Aliquots containing 12 μg of protein were separated on 8 % SDS-PAGE gels and were transferred to PVDF membranes. The filters were blocked with 5 % bovine serum albumin and were probed with pStat3Y705 antibody (Cell Signaling) followed by secondary antibody (Fisher Scientific), whose signal was detected with an enhanced chemiluminescence kit (ECL, Amersham, Chicago, IL). Filters were stripped with stripping buffer (62.5 mM tris, pH 6.8, 2 % SDS, and 0.1 M 2-mercaptoethanol) at 50 °C for 30 min. Filters were then probed with total Stat3 antibody (Cell Signaling) and visualized with chemiluminescence as above.
Results and Discussion
Effect of β-Methyl Cinnamate and C-Terminal Modifications on Cellular Potency (Mandal et al. 2011)
To evaluate the effect of a methyl group on the β-position of the cinnamate, PM-70G (6) was synthesized and the ability to inhibit Stat3 phosphorylation in MDA-MB-468 breast cancer cells was compared to BP-PM6. This modification increased cellular inhibitory activity about twofold, which is in keeping with the 2–3-fold increase in affinity of the corresponding phospho derivatives toward the isolated protein (Fig. 2). However, dramatic increases in potency occurred when Gln-NHBn was replaced with (R)-4-aminopentamide (Apa), i.e., the C-terminal C(O)NHBn was substituted with a simple methyl group (5 vs. 7 and 6 vs. 8). Compounds 7 and 8 are very potent prodrugs and complete inhibition of pStat3 was observed at 0.5 μM, tenfold lower than 5 and 6 (Mandal et al. 2011). This contrasts with the corresponding phosphopeptides, in which replacing the C-terminal Gln-NHBn with Apa resulted in a twofold loss of affinity (Ki values were 57 and 105 nM, respectively) (Mandal et al. 2011).
Effect of Substituted Proline on Affinity for Stat3
Two of our most potent prodrugs, PM-72G-1 (9) and PM-274G-1 (10) (Fig. 3), possess mPro at the pY + 2 position (Mandal et al. 2011). mPro increases affinity of phosphopeptides for the SH2 domain of Stat3 1.5–3-fold relative to native proline (Coleman et al. 2005; Mandal et al. 2009c, 2011) (Table 1). This unnatural amino acid analogue is no longer commercially available and methodology for its synthesis is laborious (Witkop et al. 1971; Sagnard et al. 1995). Therefore we sought more readily available alternatives with substituents on the pyrrolidine ring that might contact Stat3 effectively.
Fig. 3.
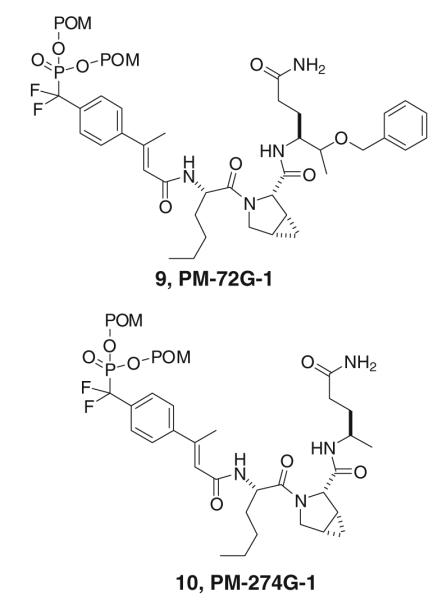
Potent prodrug inhibitors of Stat3 with Nle-mPro as the central dipeptide (from Mandal et al. 2011)
Table 1.
cis-3,4-Methanoproline enhances affinity of phosphopeptides and mimics for Stat3
| # | Substrate | IC50 (nm) | Reference |
|---|---|---|---|
| 11 | Ac-pTyr-Leu-Pro-Gln-Val- NH2 |
739 ± 31 | Coleman et al. 2005 |
| 12 | Ac-pTyr-Leu-mPro-Gln-Val- NH2 |
265 ± 14a | Coleman et al. 2005 |
| 13 | pCinn-Leu-Pro-Gln-NHBn | 138 ± 8 | Mandal et al. 2009b |
| 14 | pCinn-Leu-mPro-Gln-NHBn | 68 ± 8 | Mandal et al. 2009b |
| 15 |
βMpCinn-Leu-Pro-Gln- NHBn |
70 ± 15 | Mandal et al. 2011 |
| 16 |
βMpCinn-Leu-mPro-Gln- NHBn |
46 ± 5 | Mandal et al. 2011 |
| 17 | βMpCinn-Leu-Pro-Apa | 154 ± 10 | Mandal et al. 2011 |
| 18 | βMpCinn-Leu-mPro-Apa | 88 ± 11 | Mandal et al. 2011 |
pCinn 4-phosphoryloxycinnamoyl, βMpCinn 4-phosphoryloxy-β-methylcinnamoyl, Apa (R)-4-aminopentamide
Commercially available mPro is a racemic mixture of enantiomers. Only the more active diastereomer was reported
A set of 4-substituted proline derivatives was incorporated into the high affinity scaffold, βMpCinn-Leu-Xaa-Apa (17 and 18, Table 1) and each was assayed for affinity using our fluorescence polarization assay (Coleman et al. 2005). This series contains the second generation phosphotyrosine mimic, β-methylcinnamate (βMpCinn) (Mandal et al. 2011) and the simplified glutamine surrogate, Apa (Mandal et al.2009c) that increased potency in intact cells.
Fmoc-4,4-dimethyl-l-proline, used for inhibitor 20, was synthesized from benzyl N-Boc-l-pyroglutamate following the published procedures (Ezquerra et al. 1994). Fmoc-4,4-difluoro-l-proline and Fmoc-cis-4-fluoro-l-proline, used for the synthesis of inhibitors 21 and 22, were prepared as reported (Demange et al. 1998). Commercially available Fmoc-trans-4-tert-butoxy-l-proline and Fmoc-cis-4-tert-butoxy-l-proline were used for inhibitors 23 and 24, respectively. For inhibitor 25, Fmoc-trans-4-methoxy-l-proline was used, which was synthesized from Boc-trans-4-methoxy-l-proline (Chiba et al. 2006). The Fmoc group was introduced following one pot Boc group deprotection by treating with TFA and subsequent capping with Fmoc-OSu to give identical compound as described (Chiang et al. 2009).
Phosphopeptides were synthesized using Fmoc-based solid phase peptide synthesis starting with the attachment of N-Fmoc-Apa to Rink amide resin (30) (Mandal et al. 2009c) (Scheme 1). Couplings were mediated with DIC/HOBt or PyBOP/HOBt/DIEA. After the synthesis of Leu-Xaa-Apa-Resin (32), peptides were capped with pentachlorophenyl 4′-phosphoryloxy-β-methylcinnamate, prepared as described (Mandal et al. 2011). Peptides were cleaved with TFA:TIS:H2O (95:2.5:2.5) (Pearson et al. 1989), purified by reverse phase HPLC to >95 % purity as judged by analytical HPLC with the correct mass spectrum, and were dried at 37 °C under vacuum over P2O5 to remove residual water from lyophilization (Coleman et al. 2005) (Table 2).
Scheme 1.

Synthesis of phosphopeptides and prodrugs containing substituted proline analogues
Table 2.
Characterization of phosphopeptides
| Yield (mg) | HPLC tR (min)a | Mass (calc) | Mass (found) | |
|---|---|---|---|---|
| 17 b | ||||
| 18 b | ||||
| 19 | 42 | 24.80 | 541.23 | 541.25 |
| 20 | 22 | 22.21 | 595.30 | 595.30 |
| 21 | 18 | 20.92 | 603.20 | 603.20 |
| 22 | 22 | 23.62 | 585.30 | 585.30 |
| 23 | 19 | 21.10 | 583.30 | 583.30 |
| 24 | 29 | 16.95 | 583.25 | 583.25 |
| 25 | 28 | 10.97 | 597.30 | 597.30 |
HPLC gradient: 0–40 % MeCN/30 min, 0.1 % TFA, 1.5 mL/min
from Mandal et al. (2011)
Evaluation of the effect of the substituted prolines on affinity for Stat3 is summarized in Table 3. Replacement of proline by its linear analogue, sarcosine, peptide 19, resulted tenfold loss of avidity suggesting that conformational restraint contributes to affinity. Though 4,4-dimethyl proline in peptide 20 retained the same affinity as parent compound 17, 4,4-difluoroproline (21) produced a slight increase. cis 4-monofluoroproline (22) resulted in almost twofold reduced affinity compared to proline. The configuration of 4-hydroxyproline (4-HOPro) was important. Peptide 23, incorporating trans 4-hydroxyproline exhibited higher affinity than 24, possessing cis 4-hydroxyproline. However, the methylated trans 4-methoxyproline containing peptide (25) exhibited a twofold loss in affinity compared to the 4-hydroxy analogue.
Table 3.
Effect of 4-substituted prolines on the affinity of peptidic inhibitor βMpCinn-Leu-Xaa-Apa
| # | Xaa | IC50 (nM)a | |
|---|---|---|---|
| 17 | Proline | Pro | 154 ± 10 |
| 19 | Sarcosine | Sar | 1540 ± 34 |
| 18 | cis-3,4-Methanoproline | mPro | 88 ± 11 |
| 20 | 4,4-Dimethylproline | 4,4-Me2Pro | 155 ± 31 |
| 21 | 4,4-Difluoroproline | 4,4-F2Pro | 121 ± 16 |
| 22 | cis-4-Fluoroproline | cis-4-FPro | 246 ± 62 |
| 23 | trans-4-Hydroxyproline | trans-4-HOPro | 166 ± 10 |
| 24 | cis-4-Hydroxyproline | cis-4-HOPro | 220 ± 27 |
| 25 | trans-4-Methoxyproline | trans-4-MeOPro | 317 ± 14 |
Affinity was determined by fluorescence polarization (Coleman et al. 2005)
Effect of Substituted Proline on Cellular Potency
To measure the effect of 4-substituted prolines on the ability to inhibit Stat3 phosphorylation in intact cells, we converted phosphopeptides 17, 20, and 21 to their corresponding prodrugs 26, 27, and 28, respectively. Compound 10 (Mandal et al. 2011) is included for comparison. In these prodrugs, leucine was replaced by norleucine to reduce the “natural” peptide character. Nle exhibited identical affinity as Leu for isolated Stat3 by fluorescence polarization (Coleman et al. 2005). Prodrugs were synthesized using the convergent strategy of synthesizing Nle-Xaa-Apa on solid supports and capping it in solution with pentachlorophenyl F2Pm(POM2)-β-methylcinnamate (Mandal et al. 2011) (Scheme 2; Table 4).
Scheme 2.

Synthesis of prodrugs containing substituted proline analogues
Table 4.
Characterization of prodrugs
| Yield (mg) | HPLC tR (min)a | Mass (calc) | Mass (found) | |
|---|---|---|---|---|
| 26 | 67 | 28.76 | 829.3964 | 829.3925 |
| 27 | 34 | 30.92 | 857.4277 | 857.4200 |
| 28 | 29 | 26.90 | 865.3776 | 865.3731 |
HPLC gradient: 10–80 % MeCN/30 min, no TFA, 0.4 mL/min
All four prodrugs inhibited the constitutive phosphorylation of Tyr705 Stat3 in MDA-MB-468 cells (Fig. 4). Interestingly, compared to the substituted analogues, proline (26) exhibited reduced potency for the target. Approximately 10 μM of inhibitor was required for complete inhibition. The prodrugs with substituted prolines had significant inhibition as low as 10 nM with complete inhibition at or below 500 nM. Given that the phosphopeptides possessing Pro, 4,4-dimethylproline (4,4-Me2Pro) and 4,4-difluoroproline (4,4-F2Pro) were essentially equally avid for the SH2 domain of Stat3, the reduced potency of 26 suggests that the unsubstituted derivative may undergo metabolic processing that the others do not. We found that in tumor cells grown in 2D cultures, inhibition of Stat3 phosphorylation with 8, 9, or 10 is neither cytotoxic nor does it result in the reduction in expression of canonical Stat3 downstream genes at 5 μM, tenfold higher concentration than that which completely inhibited Stat3 phosphorylation (Mandal et al. 2011). This phenomena was also observed with JAK kinase inhibitors (Hedvat et al. 2009; Kreis et al. 2007; Looyenga et al. 2012), calling into question the hypothesis that phosphorylated Stat3 is necessary for growth and survival of tumor cells of epithelial origin. Furthermore, cytotoxic concentrations of prodrug 8 (25 μM) resulted in off-target effects (Mandal et al. 2011). Therefore these parameters were not evaluated for prodrugs 26–28.
Fig. 4.

Effect of proline substitution on the inhibition of constitutive phosphorylation of Tyr705 of Stat3 in MDA-MB-468 breast cancer cells. Compound names in parentheses are given for reference to future manuscripts on further biological evaluation. The procedure for evaluation is given in “Materials and Methods” section
In conclusion, we report here that 4-substituted proline at position pY + 2 has minimal effect on intrinsic affinity of a set of phosphopeptides for the SH2 domain of Stat3. However, substitution dramatically enhances the ability of the prodrugs to inhibit the constitutive phosphorylation of Stat3 in intact breast tumor cells. In phosphopeptides in which the central dipeptide unit was Haic, replacing the C-terminal Gln-NHBn with Apa resulted in a twofold loss of affinity. However this substitution led to a striking increase in potency of the corresponding pro-drugs as the concentration for complete inhibition of pStat3 decreased 20-fold from 10 to 0.5 μM. Thus, changes in the structures of these SH2 domain-targeted ligands that have minimal effect on affinity for the isolated protein (≤2-fold) can have major impacts on potency in intact cells.
Supplementary Material
Acknowledgments
We are grateful to the National Cancer Institute (CA096652) for support of this work. Partial support by a Grant from the Center for Targeted Therapy of The University of Texas M. D. Anderson Cancer Center and the Texas Institute of Drug and Diagnostic Development at The University of Texas at Austin is acknowledged. We also acknowledge the NCI Cancer Center Support Grant CA016672 for the support of both our NMR facility and the Translation Chemistry Core Facility which performed the mass spectrometry. Funding as an Odyssey Fellow (Z.R.) was supported by the Odyssey Program and the Cockrell Foundation Award for Scientific Achievement at UTMDACC.
Abbreviations
- 4,4-Me2Pro
4,4-Dimethylproline
- 4,4-F2Pro
4,4-Difluoroproline
- 4-FPro
4-Fluoroproline
- 4-HOPro
4-Hydroxyproline
- 4-MeOPro
4-Methoxyoxyproline
- Apa
(R)-4-Aminopentamide
- DIEA
Diisopropylethylamine
- DIC
Diisopropylcarbodiimide
- Haic
5-(Amino)-1,2,4,5,6,7-hexahydro-4-oxo-(2S,5S)-azepino[3,2,1-hi]indole-2-carboxylic acid
- HOBt
1-Hydroxybenzotriazole
- IL-6
Interleukin-6
- JAK
Janus kinase
- mPro
cis-3,4-Methanoproline
- pCinn
4-Phosphoryloxycinnamide
- βMpCinn
β-Methyl pCinn or [2E] 3-(4-phosphoryloxyphenyl)-2-butenamide
- POM
Pivaloyloxymethyl
- pStat3
Tyr705 phosphorylated Stat3
- PyBOP
1H-Benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate
- SH2 domain
Src homology 2 domain
- Stat3
Signal transducer and activator of transcription 3
- TFA
Trifluoroacetic acid
- TIS
Triisopropylsilane
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10989-012-9313-0) contains supplementary material, which is available to authorized users.
Supporting Information NMR chemical shifts for the two conformations of 27 characterization prodrugs 27–29 by HPLC, purity, mass spectra and yield.
Conflict of interest The authors have no association with commercial entities associated with this work.
Contributor Information
Pijus K. Mandal, Department of Experimental Therapeutics, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030, USA
Zhiyong Ren, Department of Biochemistry and Molecular Biology, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030, USA.
Xiaomin Chen, Department of Biochemistry and Molecular Biology, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030, USA.
Kumar Kaluarachchi, Department of Experimental Therapeutics, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030, USA.
Warren S.-L. Liao, Department of Experimental Therapeutics, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030, USA
John S. McMurray, Department of Experimental Therapeutics, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030, USA
References
- Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- Burke TR, Jr, Smyth MS, Otaka A, Nomizu M, Roller PP, Wolf G, Case R, Shoelson SE. Nonhydrolyzable phosphotyrosyl mimetics for the preparation of phosphatase-resistant SH2 domain inhibitors. Biochemistry. 1994;33:6490–6494. doi: 10.1021/bi00187a015. [DOI] [PubMed] [Google Scholar]
- Chen J, Nikolovska-Coleska Z, Yang CY, Gomez C, Gao W, Krajewski K, Jiang S, Roller P, Wang S. Design and synthesis of a new, conformationally constrained, macrocyclic small-molecule inhibitor of STAT3 via ‘click chemistry’. Bioorg Med Chem Lett. 2007;17:3939–3942. doi: 10.1016/j.bmcl.2007.04.096. [DOI] [PubMed] [Google Scholar]
- Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, Yi H, Wang S. Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors. ACS Med Chem Lett. 2010;1:85–89. doi: 10.1021/ml100010j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang YC, Lin YJ, Horng JC. Stereoelectronic effects on the transition barrier of polyproline conformational interconversion. Protein Sci. 2009;18:1967–1977. doi: 10.1002/pro.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba J, Takayama G, Takashi T, Yokoyama M, Nakayama A, Baldwin JJ, McDonald E, Moriarty KJ, Sarko CR, Saionz KW, Swanson R, Hussain Z, Wong A, Machinaga N. Synthesis, biological evaluation, and pharmacokinetic study of prolyl-1-piperazinylacetic acid and prolyl-4-piperidinylacetic acid derivatives as VLA-4 antagonists. Bioorg Med Chem. 2006;14:2725–2746. doi: 10.1016/j.bmc.2005.11.058. [DOI] [PubMed] [Google Scholar]
- Coleman DR, IV, Ren Z, Mandal PK, Cameron AG, Dyer GA, Muranjan S, Campbell M, Chen X, McMurray JS. Investigation of the binding determinants of phosphopeptides targeted to the SRC homology 2 domain of the signal transducer and activator of transcription 3. Development of a high-affinity peptide inhibitor. J Med Chem. 2005;48:6661–6670. doi: 10.1021/jm050513m. [DOI] [PubMed] [Google Scholar]
- Coleman DR, IV, Kaluarachchi K, Ren Z, Chen X, McMurray JS. Solid phase synthesis of phosphopeptides incorporating 2,2-dimethyloxazolidine pseudoproline analogs: evidence for trans Leu-Pro peptide bonds in Stat3 inhibitors. Int J Pept Res Ther. 2008;14:1–9. [Google Scholar]
- Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- Demange L, Menez A, Dugave C. Practical synthesis of Boc and Fmoc protected 4-fluoro and 4-difluoroprolines from trans-4-hydroxyproline. Tetrahedron Lett. 1998;39:1169–1172. [Google Scholar]
- Ezquerra J, Pedregal C, Rubio A, Vaquero JJ, Matia MP, Martin J, Diaz A, Navio JLG, Deeter JB. Stereoselective double alkylation of ethyl N-boc-pyroglutamate. J Org Chem. 1994;59:4327–4331. [Google Scholar]
- Farquhar D, Khan S, Srivastva DN, Saunders PP. Synthesis and antitumor evaluation of bis[(pivaloyloxy)methyl] 2′-deoxy-5-fluorouridine 5′-monophosphate (FdUMP): a strategy to introduce nucleotides into cells. J Med Chem. 1994;37:3902–3909. doi: 10.1021/jm00049a009. [DOI] [PubMed] [Google Scholar]
- Fletcher S, Turkson J, Gunning PT. Molecular approaches towards the inhibition of the signal transducer and activator of transcription 3 (Stat3) protein. ChemMedChem. 2008;3:1159–1168. doi: 10.1002/cmdc.200800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S, Singh J, Zhang X, Yue P, Page BD, Sharmeen S, Shahani VM, Zhao W, Schimmer AD, Turkson J, Gunning PT. Disruption of transcriptionally active Stat3 dimers with non-phosphorylated, salicylic acid-based small molecules: potent in vitro and tumor cell activities. ChemBioChem. 2009;10:1959–1964. doi: 10.1002/cbic.200900172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning PT, Katt WP, Glenn M, Siddiquee K, Kim JS, Jove R, Sebti SM, Turkson J, Hamilton AD. Isoform selective inhibition of STAT1 or STAT3 homo-dimerization via peptidomimetic probes: structural recognition of STAT SH2 domains. Bioorg Med Chem Lett. 2007;17:1875–1878. doi: 10.1016/j.bmcl.2007.01.077. [DOI] [PubMed] [Google Scholar]
- Gunning PT, Glenn MP, Siddiquee KA, Katt WP, Masson E, Sebti SM, Turkson J, Hamilton AD. Targeting protein–protein interactions: suppression of Stat3 dimerization with rationally designed small-molecule, nonpeptidic SH2 domain binders. ChemBioChem. 2008;9:2800–2803. doi: 10.1002/cbic.200800291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker SJ, Erion MD. Prodrugs of phosphates and phosphonates. J Med Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, Wang L, Ye M, McEachern K, Chen H, Morosini D, Bell K, Alimzhanov M, Ioannidis S, McCoon P, Cao ZA, Yu H, Jove R, Zinda M. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis S, Munz GA, Haan S, Heinrich PC, Behrmann I. Cell density dependent increase of constitutive signal transducers and activators of transcription 3 activity in melanoma cells is mediated by Janus kinases. Mol Cancer Res. 2007;5:1331–1341. doi: 10.1158/1541-7786.MCR-07-0317. [DOI] [PubMed] [Google Scholar]
- Krise JP, Stella VJ. Prodrugs of phosphates, phosphonates, and phosphinates. Adv Drug Deliv Rev. 1996;19:287–310. [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Looyenga BD, Hutchings D, Cherni I, Kingsley C, Weiss GJ, Mackeigan JP. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS One. 2012;7:e30820. doi: 10.1371/journal.pone.0030820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Heard PA, Ren Z, Chen X, McMurray JS. Solid-phase synthesis of Stat3 inhibitors incorporating O-carbamoylserine and O-carbamoylthreonine as glutamine mimics. Bioorg Med Chem Lett. 2007;17:654–656. doi: 10.1016/j.bmcl.2006.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Liao WS, McMurray JS. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org Lett. 2009a;11:3394–3397. doi: 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Limbrick D, Coleman DR, Dyer GA, Ren Z, Birtwistle JS, Xiong C, Chen X, Briggs JM, McMurray JS. Conformationally constrained peptidomimetic inhibitors of signal transducer and activator of transcription 3: evaluation and molecular modeling. J Med Chem. 2009b;52:2429–2442. doi: 10.1021/jm801491w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Ren Z, Chen X, Xiong C, McMurray JS. Structure-affinity relationships of glutamine mimics incorporated into phosphopeptides targeted to the SH2 domain of signal transducer and activator of transcription 3. J Med Chem. 2009c;52:6126–6141. doi: 10.1021/jm901105k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen X, Bast RC, Liao WS, McMurray JS. Potent and Selective phosphopeptide mimetic prodrugs targeted to the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3. J Med Chem. 2011;54:3549–5463. doi: 10.1021/jm2000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray JS. Structural basis for the binding of high affinity phosphopeptides to Stat3. Biopolymers. 2008;90:69–79. doi: 10.1002/bip.20901. [DOI] [PubMed] [Google Scholar]
- Pearson DA, Blanchette M, Baker ML, Guindon CA. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett. 1989;30:2739–2742. [Google Scholar]
- Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg Med Chem Lett. 2003;13:633–636. doi: 10.1016/s0960-894x(02)01050-8. [DOI] [PubMed] [Google Scholar]
- Sagnard I, Sasaki NA, Chiaroni A, Riche C, Potier P. Enantioselective synthesis of cyclopropane a-amino acids: synthesis of N-Boc-cis. (2S,3R,4S)-3,4-methanoproline and N-Boc-(2S,3R,4S)-3,4-methanoglutamic acid. Tetrahedron Lett. 1995;36:3149–3152. [Google Scholar]
- Schultz C. Prodrugs of biologically active phosphate esters. Bioorg Med Chem. 2003;11:885–898. doi: 10.1016/s0968-0896(02)00552-7. [DOI] [PubMed] [Google Scholar]
- Shahani VM, Yue P, Fletcher S, Sharmeen S, Sukhai MA, Luu DP, Zhang X, Sun H, Zhao W, Schimmer AD, Turkson J, Gunning PT. Design, synthesis, and in vitro characterization of novel hybrid peptidomimetic inhibitors of STAT3 protein. Bioorg Med Chem. 2011a;19:1823–1838. doi: 10.1016/j.bmc.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahani VM, Yue P, Haftchenary S, Zhao W, Lukkarila JL, Zhang X, Ball D, Nona C, Gunning PT, Turkson J. Identification of purine-scaffold small-molecule inhibitors of Stat3 activation by QSAR studies. ACS Med Chem Lett. 2011b;2:79–84. doi: 10.1021/ml100224d. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schrock C, Sebti SM, Jove R, Hamilton AD, Turkson J. An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects. ACS Chem Biol. 2007;2:787–798. doi: 10.1021/cb7001973. [DOI] [PubMed] [Google Scholar]
- Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267:1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, Haura E, Sebti S, Hamilton AD, Jove R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3:261–269. [PubMed] [Google Scholar]
- Witkop B, Fujimoto Y, Irreverre F, Karle JM, Karle IL. Synthesis and x-ray analysis of cis-3,4-methylene-l-proline, the new natural amino acid from horse chestnuts, and of its trans isomer. J Am Chem Soc. 1971;93:3471–3477. doi: 10.1021/ja00743a030. [DOI] [PubMed] [Google Scholar]
- Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochem Pharmacol. 2010;79:1398–1409. doi: 10.1016/j.bcp.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.