

Background: The glucocorticoid receptor cross-talks with other receptors to integrate cell signaling.
Results: Glucocorticoids and GnRH synergistically up-regulate select mRNA levels via protein kinase C and flotillin-1 to repress cell proliferation.
Conclusion: Cross-talk between membrane-associated receptors modulates protein kinase C-dependent synergistic and gene-specific transcription by intracellular glucocorticoid receptor.
Significance: Glucocorticoid and GnRH receptor cross-talk integrates adrenal and gonadal signaling to fine-tune cell proliferation.
Keywords: Cell Proliferation, Gene Regulation, Glucocorticoid Receptor, Glucocorticoids, Protein Kinase C (PKC), GnRH Receptor, SGK-1, Flotillin-1, Lipid Rafts, Synergism
Abstract
Cross-talk between the glucocorticoid receptor (GR) and other receptors is emerging as a mechanism for fine-tuning cellular responses. We have previously shown that gonadotropin-releasing hormone (GnRH) ligand-independently activates the GR and synergistically modulates glucocorticoid-induced transcription of an endogenous gene in LβT2 pituitary gonadotrope precursor cells. Here, we investigated GR and GnRH receptor (GnRHR) cross-talk that involves co-localization with lipid rafts in LβT2 cells. We report that the GnRHR and a small population of the GR co-localize with the lipid raft protein flotillin-1 (Flot-1) at the plasma membrane and that the GR is present in a complex with Flot-1, independent of the presence of ligands. We found that the SGK-1 gene is up-regulated by Dex and GnRH alone, whereas a combination of both ligands resulted in a synergistic increase in SGK-1 mRNA levels. Using siRNA-mediated knockdown and antagonist strategies, we show that the gene-specific synergistic transcriptional response requires the GR, GnRHR, and Flot-1 as well as the protein kinase C pathway. Interestingly, although several GR cofactors are differentially recruited to the SGK-1 promoter in the presence of Dex and GnRH, GR levels remain unchanged compared with Dex treatment alone, suggesting that lipid raft association of the GR has a role in enhancing its transcriptional output in the nucleus. Finally, we show that Dex plus GnRH synergistically inhibit cell proliferation in a manner dependent on SGK-1 and Flot-1. Collectively the results support a mechanism whereby GR and GnRHR cross-talk within Flot-1-containing lipid rafts modulates cell proliferation via PKC activation and SGK-1 up-regulation.
Introduction
Glucocorticoids (GCs)2 are essential for life and regulate a wide array of physiological functions, including homeostasis, tissue remodeling and repair, immune function, cell cycle, and apoptosis (1–3). GCs mediate their transcriptional genomic effects by binding to the ubiquitous classical cytoplasmic glucocorticoid receptor (GR) followed by nuclear translocation and modulation of transcription of target genes by direct DNA binding of the GR or its tethering to other transcription factors (1–3). Recent evidence suggests, however, that the cellular responses mediated by the classical GR are much more complex and involve multiple parallel mechanisms integrating simultaneous signals from several different hormones involving cross-talk with intracellular signaling proteins. These include rapid, non-genomic, cytoplasmic GC-dependent effects (4–9). The GR has also been shown to cross-talk with both G-protein-coupled receptors, the T-cell receptor, cytokine, and receptor-tyrosine kinase pathways (10–19). Furthermore, several reports show that the GR can be activated by hormones, cytokines as well as by cellular stress and the cell cycle, in the absence of GCs, which modulates the activity of the unliganded GR (10, 11, 19–22). Several lines of evidence suggest that rapid glucocorticoid-dependent and glucocorticoid-independent signaling via classical GRs may occur in specialized membrane lipid raft or caveolae microdomains that are proposed to function as platforms for the assembly of multiprotein signaling complexes (23). Steroid receptors, including the GR, have been localized to the plasma membrane in both the absence and presence of steroid ligands (24–27). These may represent only a small population of the total pool of steroid receptor present in a cell, as suggested by the finding that a subset of the GR resides in the plasma membrane of human leukemic cells (25, 28), mouse monocytes, and B-cells (29). The GR has been shown to localize in caveolae in a human liver cell line and to be involved in GC-induced transactivation (30). Another study showed that the unliganded GR localizes with caveolae to facilitate the rapid Dex-induced phosphorylation of Akt and caveolin-1 (Cav-1) in A549 cells but with no effect on glucocorticoid-response element (GRE) transactivation (27). Caveolae-associated GR has been implicated in a role for Dex-mediated intracellular communication and cell proliferation in mouse neural progenitor cells (31). Collectively these findings suggest that the localization of the GR in lipid rafts or caveolae may regulate several GR-dependent responses in a cell- and/or promoter-specific manner. The association of the GR with the membrane could provide a potential mechanism allowing the reciprocal modulation of downstream signaling pathways between the GR and other membrane-associated receptors. Interestingly, synergistic responses involving GCs and other ligands such as gonadotropin-releasing hormone (GnRH), interleukin-2 (IL-2), IL-13, activin, and tumor necrosis factor α (TNF-α) have been reported (10, 19, 32–34). However, the mechanisms of these synergistic responses and whether they involve membrane-associated GR in complex with other receptors has not been previously reported.
Our previous results in LβT2 pituitary gonadotrope precursor cells, which express the endogenous GnRH receptor (GnRHR) (35), suggest that this is an ideal model to investigate the role of membrane-associated GR in multiple parallel classical and non-classical actions. We reported for the first time that the endogenous GR can be ligand-independently activated by GnRH to result in site-specific phosphorylation and transactivation of an endogenous gene in these cells (10). GnRH, in addition to ligand-independently activating the GR, also induces a synergistic transcriptional response on a GRE reporter gene and an endogenous gene in the presence of GCs (10). The GnRHR is a seven-transmembrane receptor found on the cell surface that has previously been shown to localize exclusively to endogenous flotillin-1 (Flot-1)-enriched lipid rafts in the αT3-1 gonadotrope cell line (36, 37). In the present study we identify Flot-1-associated GR in LβT2 cells and investigate its role in mediating genomic transactivation, ligand-independent GR activation by GnRH, and synergy with the GnRHR signaling pathway.
EXPERIMENTAL PROCEDURES
Cell Culture
P. L. Mellon at the University of California kindly provided the immortalized mouse LβT2 pituitary gonadotrope cell line (38). The COS-7 monkey kidney fibroblast cells were a generous gift from S. Prince at the University of Cape Town, South Africa. Both cell lines were grown in high glucose DMEM supplemented with 10% fetal calf serum (Sigma), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). The cells were maintained in 75-cm2 culture flasks (Greiner Bio-one International) at 37 °C in an environment of 5% CO2 and 90% humidity. Cells were subcultured with 0.25% trypsin, 0.1% EDTA in calcium- and magnesium-free PBS. Cells were routinely tested for mycoplasma infection by Hoechst staining, and only mycoplasma-negative cells were used in experiments.
Antibodies and Materials
GnRH, Dex, phorbol 12-myristate 13-acetate (PMA), Antide, RU486, 8-bromo cyclic-AMP, bisindolylmaleimide (BIM) and the nonspecific rabbit IgG antibody (R1131) were purchased from Sigma. Antibodies for GR (sc-8992), SRC-1 (sc-8995), SRC-3 (sc-25742), CBP (sc-369), p300 (sc-32244), anti-mouse HRP (sc-2005), and anti-rabbit HRP (sc-2313) secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz). Antibodies to caveolin-1 (610406) and flotillin-1 (610822) were purchased from BD Transduction Laboratories. Bovine serum albumin (BSA), Complete protease inhibitor tablets, leupeptin, aprotinin, and FuGENE 6 were purchased from Roche Diagnostics. The anti-histone-H3 antibody (ab1791) was obtained from Abcam. The GRIP-1 antibody (G8970-10) was obtained from United States Biological. The rabbit anti-GnRHR antibody, raised against amino acids 193–212 in the extracellular loop of the ovine receptor, was a generous gift from D. C. Skinner (University of Wyoming, Department of Zoology and Physiology and Neurobiology Program) and has been described before (37). The anti-rabbit AlexaFluor488 (A21206) was purchased from Invitrogen, whereas the donkey anti-mouse Cy3 antibody (715-166-150) was obtained from Jackson ImmunoResearch. The pRK7-flotillin-1 plasmid that encodes a FLAG-tagged mouse Flot-1 protein was a generous gift from A. R. Saltiel (University of Michigan Medical School).
Transient Transfection of Cells
To generate a positive control of the Flot-1 protein, COS-7 cells were seeded into 12-well plates at a density of 1 × 105 cells per well in DMEM with 10% FCS and antibiotics as described above. Twenty-four hours after plating the medium was replaced, and the cells were transfected with 250 ng of pRK7-flotillin-1 plasmid using FuGENE 6 according to the manufacturer's instructions. Forty-eight hours after transfection, cells were washed twice with ice-cold PBS and harvested in 50 μl of SDS sample buffer (5 × SDS sample buffer: 100 mm Tris-Cl (pH 6.8), 5% (w/v) SDS, 20% (v/v) glycerol, 2% β-mercaptoethanol, and 0.1% (w/v) bromphenol blue). The samples were boiled for 10 min at 100 °C before equal amounts of cell lysates were analyzed by Western blotting.
SDS-PAGE and Western Blotting
LβT2 cell lysates were separated on 8–10% SDS-polyacrylamide gels at 120 V in 1× SDS running buffer (25 mm Tris-Cl (pH 8.4), 250 mm glycine, and 0.1% SDS) using a Bio-Rad Mini Protean II electrophoresis cell chamber. Proteins were transferred onto a HyBond ECL nitrocellulose membrane (Amersham Biosciences) for 1 h at 180 mA in a Tris/glycine buffer (25 mm Tris, 250 mm glycine, and 20% (v/v) methanol) using a Mini Protean II blotting system (Bio-Rad). The membranes were blocked for 1 h at room temperature in 4% ECL blocking solution (4% (w/v)) ECL advance blocking powder, (Amersham Biosciences) and Tris-buffered saline (TBS: 50 mm Tris-Cl (pH 7.5) and 150 mm NaCl) containing 0.1% Tween 20 (TBST). After blocking, membranes were incubated with primary antibodies in 4% ECL blocking solution diluted in TBST at 4 °C overnight. The following day the membranes were washed with TBST for 3 × 5 min at room temperature before incubation with secondary HRP-conjugated antibodies for 1 h at room temperature in 5% nonfat milk powder (w/v) in TBST. Membranes were washed for 3 × 5 min with TBST followed by a 1 × 5-min wash at room temperature with TBS and visualized by autoradiography. The membranes were stripped for 30 min at 60 °C in stripping buffer (100 mm β-mercaptoethanol, 2% (w/v) SDS, and 62.5 mm Tris-Cl (pH 6.8)), washed twice for 10 min with TBST, blocked for 1 h at room temperature in 4% ECL blocking solution before incubating with antibody again as described above. The proteins were visualized with ECL Western blotting detection reagents (Amersham Biosciences) and Hyperfilm MP high performance autoradiography film (Amersham Biosciences) according to the manufacturer's instructions. Bands on the autoradiography film were scanned, and quantification was performed with AlphaEaseFC FluorChem 5500 (Alpha Innotech).
Immunofluorescence Staining and Confocal Microscopy
Coverslips were sterilized by flaming and placed in 6-well plates followed by seeding LβT2 cells at a density of 3 × 105 cells per well in DMEM with 10% FCS and antibiotics as described above. After 48 h, cells were washed twice with ice-cold PBS and live cell-stained for 1 h at 4 °C with rabbit anti-GnRHR (1:400) followed by 2 washes with ice-cold PBS. Subsequently, the cells were fixed and permeabilized with methanol at −20 °C for 10 min and washed with PBS for 3 × 5 min. Cells were blocked with 5% BSA in PBS for 1 h at room temperature followed by staining with mouse anti-Flot-1 (1:50) antibody in PBS with 5% BSA for 1 h at room temperature. For the GR and Flot-1 immunofluorescence the cells were stained after fixation with rabbit anti-GR (1:250) and Flot-1 as mentioned above. Subsequently, the cells were washed with 1% BSA in PBS for 3 × 5 min before incubating with anti-rabbit-labeled Alexa488 (1:500) (to detect GnRHR and GR) and anti-mouse-labeled Cy3 (1:1000) (to detect Flot-1) antibodies in PBS with 5% BSA for 1 h at room temperature in the dark. The cells were washed with 1% BSA in PBS for 3 × 5 min followed by incubation with Hoechst (100 μg/ml) in PBS for 5 min. Slides were mounted in Mowiol (475904, Calbiochem) containing n-propyl gallate (Sigma) as the anti-fading agent and allowed to set overnight at room temperature in the dark followed by storage at 4 °C in the dark until visualization. Confocal microscopy was performed with a Zeiss Axiovert 200M LSM 510 Meta NLO Confocal Microscope using the 40× water immersion objective and the 63× oil-immersion objective. A multi-track scanning configuration using the 488-nm (HeNe gas laser), 561-nm (solid state laser), 633-nm (argon laser), and 800-nm (titanium:sapphire femtosecond infrared laser) excitation lines was employed to minimize bleed-through between the fluorophores. The photomultiplier gain and offset were adjusted to exclude any background fluorescence emitted by the cells and fluorophores. At least three different fields of view from three independent experiments were collected. The images were analyzed for co-localization with the Carl Zeiss ZEN software (Version 2009) Manders correlation and overlap coefficients (39) for the two fluorophores.
Lipid Raft Isolation
Plasma membrane lipid rafts were prepared using the Triton X-100 procedure as described by Lafont and Simons with some modifications (40). LβT2 cells were seeded in 150-mm2 dishes at a density of 8 × 106 cells per dish in DMEM with 10% FCS containing antibiotics as described above. The cells were washed twice with PBS and stimulated with 100 nm Dex, 100 nm GnRH, or a combination of both for 30 min in serum-free medium before being washed twice with ice-cold PBS. The cells were scraped on ice in 1 ml of PBS containing 1 mm PMSF, 5 μg/ml leupeptin, and 2 μg/ml aprotinin per dish. Thereafter the cells were centrifuged at 500 × g for 5 min, and each cell pellet was resuspended in 1 ml of solubilization buffer (SB) (25 mm Tris-Cl (pH 7.5), 150 mm NaCl, 5 mm EDTA, 1 mm DTT, 1 mm PMSF, 5 μg/ml leupeptin, and 2 μg/ml aprotinin) containing 0.05% Triton X-100 and incubated on ice water for 45 min. The lysates were adjusted to 60% sucrose in SB and layered at the bottom of SW40 Ultraclear centrifuge tubes (Beckman). A discontinuous sucrose gradient was prepared consisting of 2 ml of extraction lysis buffer (ELB), 10 mm Hepes (pH 7.9), 10 mm NaCl, 3 mm MgCl2, 1 mm DTT, 1 mm PMSF, 5 μg/ml leupeptin, and 2 μg/ml aprotinin), 4 ml of 13% sucrose in ELB, 4 ml of 43% sucrose in ELB, and 4 ml of 60% sucrose containing the sample. Thereafter, the samples were subjected to equilibrium flotation in a SW40Ti rotor (38 000 rpm for 18 h at 4 °C). Flocculent material could be seen at the interfaces, and fractions (1.5 ml) were collected as follows: 1) top of the gradient, 2) ELB/13% interface, 3) 13%/43% interface, 4) remaining 13%/43% interface, 5) middle of 43% sucrose, 6) 43%/60% interface, 7) middle of 60% sucrose (loading fraction), and 8) the pellet. All fractions were sonicated for 30-s pulses in a water bath at room temperature until a homogenous solution was obtained. Fractions were aliquoted and stored at −80 °C. For analysis, equal amounts of fractions were analyzed by Western blotting as described elsewhere. The membranes were probed with specific antibodies against the GR, GnRHR, Flot-1, and histone H3. The results were quantified by scanning the Western blots and determining the intensity of the protein bands with AlphaEaseFC, whereby the GR protein levels were normalized against Flot-1 protein levels for each experiment and expressed relative to vehicle (control).
Co-immunoprecipitation Assays
LβT2 cells were seeded in 100-mm2 dishes at a density of 3 × 106 cells per dish in DMEM with 10% FCS and antibiotics as described above. Seventy-two hours after plating, cells were washed twice with PBS and incubated for 2 h in serum-free DMEM before being stimulated with 100 nm Dex, 100 nm GnRH, or a combination of both for 30 min as indicated in the figure legends. The cells were washed twice with ice-cold PBS and scraped on ice in 1 ml of radioimmuno precipitation assay lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% (v/v) Nonidet P-40, 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate, 1 mm EDTA, 1 mm PMSF, 2 μg/ml aprotinin, 5 μg/ml leupeptin, and 2.5% (w/v) casein). The lysates were briefly vortexed before incubating on ice for 10 min and centrifugation at 5000 × g for 10 min at 4 °C after removing 50 μl aliquots to represent inputs. The supernatants were collected and incubated with 1 μg of rabbit anti-GR, 1 μg of rabbit anti-Flot-1, or nonspecific rabbit IgG antibodies and rotation at 4 °C overnight. The following day the antibodies-protein complexes were incubated with 20 μl of the protein A/G-agarose bead (Santa Cruz) slurry and rotated for 1 h at 4 °C. The protein-immune complexes were collected by centrifugation at 1000 × g for 5 min at 4 °C followed by 2 washes with 1 ml of radioimmuno precipitation assay buffer and 1 ml of PBS. The proteins were eluted from the beads by the addition of 24 μl of 2 × SDS sample buffer and incubated at 100 °C for 5 min. The samples were centrifuged at 20,000 × g at room temperature, and the supernatants were collected and resolved on an 8% SDS-PAGE followed by Western blotting as described elsewhere, probing with antibodies as indicated in the figure legends.
Quantitative Real-time PCR
LβT2 cells were seeded in 12-well plates at a density of 4.5 × 105 cells per well in DMEM with 10% FCS and antibiotics as described elsewhere. Forty-eight hours after plating cells were washed with PBS before being stimulated for 8 h in serum-free medium as indicated in the figure legend. After stimulation, total RNA was isolated with the TRIzol reagent (Sigma) according to the manufacturer's instructions. A total of 0.5 μg of RNA was reverse-transcribed using the Transcriptor first-strand cDNA synthesis kit (Roche Applied Science) according to the manufacturer's instructions. Quantitative real-time PCR was performed with the SensiMix dT kit using the primers serum/glucocorticoid-regulated kinase 1 (SGK-1) forward and SGK-1 reverse primers (5′-ATCTCCAGAGGGAGCGC-3′, 5′-TCAGTGAGGACGATGTGC-3′, respectively), mitogen-activated protein kinase phosphatase-1 (MKP-1) forward and MKP-1 reverse primers (5′-AGTACCCCTCTCTACGATCAGG-3′, 5′-TGATGGAGTCTATGAAGTCAATATG-3′, respectively), follicle-stimulating hormone β (FSHβ) forward and FSHβ reverse primers (5′-GGTGTGCTGCCATATCAGATTCGG-3′, 5′-GCATCAAGTGCTGCTACTCACCTGTG-3′ respectively), glucocorticoid-induced leucine zipper (GILZ) forward and GILZ reverse primers (5′-CCCTAGACAACAAGATTGAGC-3′, 5′-CAGAGCCACTTACACCGC-3′ respectively), FK506-binding protein 5 (FKBP5) forward and FKBP5 reverse primers (5′- GGGCACCAGTAACAATGGAG-3′, 5′- GGCAAATGGCTTCTTTCTGT-3′ respectively), and GAPDH forward and reverse primers (5′-TTCACCACCATGGAGAAGGC-3′ and 5′-GGCATGGACTGTGGTCATCA-3′, respectively) under the following conditions: 95 °C for 10 min followed by 40 cycles of 95 °C for 10 s, 58 °C for 10 s, and 72 °C for 10 s. Melt-curve analysis and gel-electrophoresis were performed to confirm that there was a single product amplified in the PCR reactions. Relative SGK-1, MKP-1, FSHβ, GILZ, and FKBP5 transcript levels were calculated with the Fit Points method (41) and were normalized to relative GAPDH transcript levels.
RNA Interference
LβT2 cells were seeded in 12-well plates at a density of 3.5 × 105 cells per well in 1 ml of DMEM with 10% FCS and antibiotics as described elsewhere. Twenty-four hours after plating, medium was replaced with fresh medium, and the cells were transfected with siRNA using HiPerfect transfection reagent (Qiagen) according to the manufacturer's instructions. Briefly, either a combination of mouse Flot-1 siRNA Mm_FLot1_1, Mm_FLot1_2, Mm_FLot1_3, and Mm_FLot1_4 (FlexiTube siRNA (1027415), Qiagen) or GR siRNA (Mn_Nr3c_3, Mn_Nr3c_4, Mn_Nr3c_5, and Mn_Nr3c_6, FlexiTube siRNA (1027416) or SGK-1 siRNA (01416555) (Qiagen) or just non-silencing scrambled (NSC) siRNA (Negative control siRNA (1027310), Qiagen) was diluted in 50 μl of Opti-MEM+GlutaMAX-I (Invitrogen) with 3.5 μl of HiPerfect. The mixture was incubated for 10 min at room temperature and added dropwise to the cells to obtain a final concentration of 40 nm siRNA per well. Three days after transfection, the cells were washed once with PBS followed by stimulation for 8 h with 100 nm Dex, 100 nm GnRH, and a combination of both in serum-free DMEM for gene expression assays.
ChIP Assay
LβT2 cells were seeded in 150-mm2 dishes at a density of 8 × 106 cells per dish in DMEM with 10% charcoal-stripped FCS and antibiotics as described above. Seventy-two hours after plating the medium was replaced before the cells were stimulated with 100 nm Dex, 100 nm GnRH, or a combination of both for 1 h. Thereafter, proteins were cross-linked with 1% formaldehyde for 40 min before the reaction was quenched with 125 mm glycine for 10 min. The cells were washed and scraped in PBS containing Complete Mini protease inhibitor mixture (Roche Applied Science). Cells were centrifuged and resuspended in 0.5 ml of nuclear lysis buffer (1% SDS, 50 mm Tris-HCl (pH 8.0), 10 mm EDTA, and protease inhibitor mixture). Cross-linked DNA was sheared by sonication. For the input a 30-μg aliquot of chromatin was used, whereas 100 μg of chromatin was diluted in immunoprecipitation dilution buffer (0.01% SDS, 20 mm Tris-HCl (pH 8.0), 1.1% Triton X-100, 167 mm NaCl, 1.2 mm EDTA, and protease mixture inhibitors) before being precleared for 1 h with Protein A/G PLUS beads rotating at 4 °C. The precleared samples were then incubated with 5 μg of anti-GR antibody or 2 μg of anti-SRC-1, anti-GRIP-1 or anti-SRC-3, anti-p300, anti-CBP antibodies rotating at 4 °C overnight. The next day, 40 μl of Protein A/G PLUS-agarose beads were added to the mixture for 6 h at 4 °C. The beads were collected by centrifugation and washed sequentially with 1 ml of each wash buffer I (0.1% (w/v) SDS, 1% (v/v) Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.0, and 150 mm NaCl), II (0.1% (w/v) SDS, 1% (v/v) Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.0, and 500 mm NaCl), and III (1% (v/v) NP-40, 1% (w/v) sodium deoxycholate, 500 mm LiCl, 1 mm EDTA, and 10 mm Tris, pH 8.0) followed by three washes with 1 ml of Tris-EDTA. Complexes were eluted from the beads with 300 μl of immunoprecipitation elution buffer (1% (w/v) SDS and 100 mm NaHCO3). Cross-links were reversed by the addition of 300 mm sodium chloride and incubation at 65 °C overnight. The following day each sample was adjusted to 150 mm EDTA, 125 mm Tris-HCl pH 6.5, and 20 μg of proteinase K followed by incubation for 1 h at 45 °C. The DNA was purified with the QIAquick PCR purification kit according to the manufacturer's instructions. Quantitative real-time PCR were performed using specific primers SGK-1 forward and reverse (5′-CTAACTCGCCACCTCCTCAC-3′, 5′-TCCCAGAACTTGGAAGAGGA-3′, respectively), which span the GRE in the promoter of the SGK-1 gene. Quantitative real-time PCR were performed under the following conditions: 95 °C for 10 min followed by 40 cycles of 95 °C for 10 s, 56 °C for 10 s, and 72 °C for 10 s.
Cell Proliferation Assay
LβT2 cells were seeded in 12-well plates at a density of 3.5 × 105 cells per well in 1 ml of DMEM with 10% FCS and antibiotics as described elsewhere. Twenty-four hours after plating medium was replaced with fresh medium, and the cells were transfected with 40 nm Flot-1 siRNA, 40 nm SGK-1 siRNA, or 40 nm NSC using HiPerfect transfection reagent (Qiagen) as described elsewhere. After 72 h the cells were seeded in 48-well plates at 7.5 × 104 cells per well in 200 μl of DMEM with 10% charcoal-stripped serum and antibiotics as described elsewhere. Twenty-four hours after plating medium was replaced with fresh medium, and the cells (∼70% confluency) were stimulated for 48 h with 100 nm Dex, 100 nm GnRH, or a combination of both for 48 h. Thereafter the cell proliferation was determined with a Cell proliferation BrdU colorimetric kit (Roche Applied Science, 11647229001) according to the manufacturer's instructions.
Statistical Analysis
Statistical analysis were performed with GraphPad Prism software (Version 5) using the one-way ANOVA analysis of variance with either a Dunnett (when comparing all values to a single control) or Tukey (when comparing all values to each other) post-test. Statistical significance is denoted as *, **, or *** to indicate p < 0.05, p < 0.01, or p < 0.001, respectively. The statistical tests performed for each experiment are indicated in the respective figure legends.
RESULTS
GnRHR and GR Co-localize with Flot-1-containing Lipid Rafts Independent of Ligands in LβT2 Cells
To investigate whether LβT2 cells express the lipid raft marker proteins Flot-1 and Cav-1, Western blot analysis was performed with whole cell LβT2 lysates using specific Flot-1 and Cav-1 antibodies. As shown in Fig. 1A, Flot-1, but not Cav-1 protein expression, was detected in LβT2 cells, whereas endogenous Cav-1 was detected in COS-7 cells as previously reported (42). To investigate whether the GnRHR co-localizes with Flot-1 in LβT2 cells, live cells were stained for GnRHR followed by cell fixation, permeabilization, and staining for Flot-1. The cells were visualized with a confocal microscope, and it was found that a substantial portion of Flot-1 was localized at or in the vicinity of the plasma membrane with some distribution in vesicle-like intracellular compartments (Fig. 1B). The GnRHR was also found to mainly localize to the plasma membrane, but a small percentage was also detected in the cytoplasm, possibly due to some internalization (Fig. 1B). The panel on the right shows the co-localized pixels between the green (0.94 at marked region of interest) and red (0.84 at marked region of interest) channels. The data were analyzed using Manders co-localization coefficients, and the values indicate a substantial amount of GnRHR co-localization with Flot-1 at the plasma membrane and intracellular regions of LβT2 cells (Fig. 1B). Furthermore, the co-localization map and overlap coefficients of the GnRHR and Flot-1 signals also indicate a high degree of co-localization of both channels across the entire cell, indicative of potential interactions of both proteins in cellular compartments other than the cell membrane. Having established the co-localization of the GnRHR and Flot-1 at the plasma membrane, we next investigated whether the GR is associated with Flot-1 at the membrane and whether this potential association is affected by the presence of GR agonist. Cells were stimulated for 30 min with saturating concentrations of Dex or GnRH (100 nm each) or a combination of both. Subsequently, cells were fixed, permeabilized, and stained with anti-GR- and anti-Flot-1-specific antibodies. The results presented in Fig. 2, panels A–D, show that a population of Flot-1 is localized to the plasma membrane, whereas some Flot-1 was also detected in intracellular compartments under all treatment conditions. In untreated cells, the GR appears evenly distributed throughout the cell, with a small percentage co-localizing with Flot-1 at the plasma membrane (Fig. 2, panel A) (0.84 and 0.98, at region of interest). Treatment with Dex resulted in distinct nuclear translocation of the GR, but a small fraction remained scattered in the cytoplasm. Interestingly, we found that a small percentage of the GR co-localized with Flot-1 at the membrane even in the presence of the GR agonist (Fig. 2, compare panel A with B) (0.75 and 0.83, at marked region of interest). By contrast, we could not detect GR nuclear localization after GnRH treatment (Fig. 2, compare panels A and C). Co-stimulation with GnRH had no detectable effect on the amount of Dex-induced nuclear import of the GR (Fig. 2, compare panel A with B and D). The percentage nuclear translocation of the GR was quantified, and no difference between the amount of GR that translocated into the nucleus in response to Dex and Dex + GnRH was detected (data not shown). Co-localization of GR and Flot-1 was still detectable after stimulation with GnRH and Dex + GnRH (Fig. 2, panel C and D) (0.82 and 0.74 (C) and 0.97 and 0.84 (D), respectively, at marked region of interest). However, significant differences between co-localization for different conditions could not be established.
FIGURE 1.
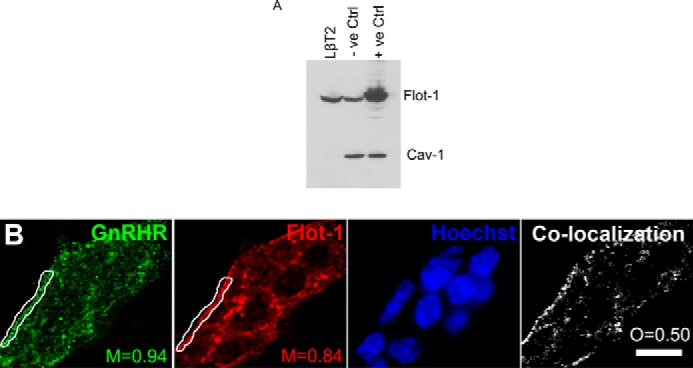
Immunofluorescence shows the GnRHR co-localizes with Flot-1 in LβT2 cells. A, whole LβT2, untransfected COS-7 (−ve Ctrl) and Flot-1 transfected COS-7 (+ve Ctrl) cell lysates were loaded on a 10% SDS-PAGE gel followed by transfer onto nitrocellulose membrane and probing with specific antibodies to Flot-1 and Cav-1. B, LβT2 cells were grown on glass coverslips and live cell-stained with rabbit anti-GnRHR followed by fixation and staining with a mouse anti-Flot-1 antibody. Thereafter, the coverslips were incubated with anti-rabbit Alexa488 (green) and anti-mouse Cy3 (red) antibodies. Nuclei were stained with Hoechst (blue) before mounting in Mowiol. Staining was visualized with a Zeiss LSM510 Meta confocal microscope using the 63× objective, and a representative image is shown for a group of cells, as indicated by the multiple nuclei visible in the Hoechst stain. Manders co-localization coefficients (M) are shown for the red and green channels in regions of interest in the vicinity of the cell membrane. The panel on the right marked Co-localization displays a map of co-localized pixels for the green and red channels across the entire group of cells. The degree of overlap between the two patterns is indicated by the overlap coefficient (O). The scale bar represents 10 μm. The results shown are representative of three independent experiments.
FIGURE 2.
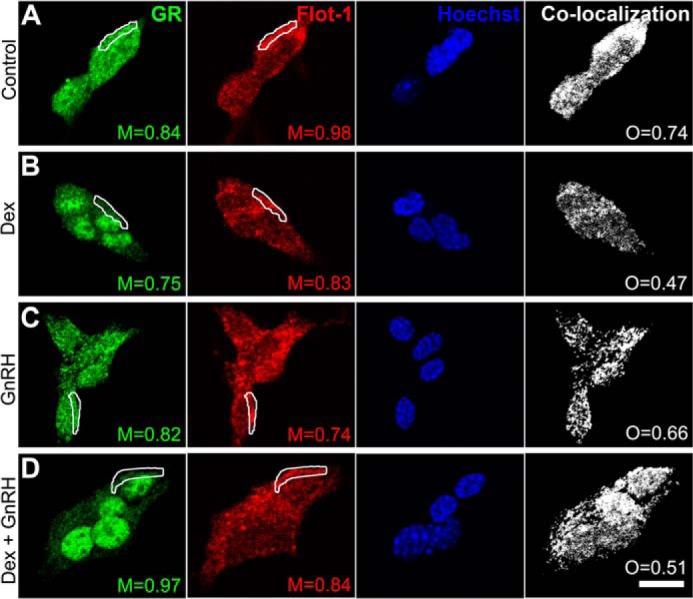
Immunofluorescence shows that the GR co-localizes with Flot-1 in LβT2 cells ligand independently. LβT2 cells were grown on glass coverslips and incubated for 30 min in medium containing charcoal-stripped serum and vehicle control (A), or including 100 nm Dex (B), 100 nm GnRH (C), or a combination of both (D). Cells were stained with rabbit anti-GR and mouse anti-Flot-1 antibodies followed by incubation with anti-rabbit Alexa488 (green) and anti-mouse Cy3 (red) antibodies. Staining and visualization were performed as for Fig. 1B. Manders co-localization coefficients (M) are shown for the red and green channels in regions of interest in the vicinity of the cell membrane. The panels on the right marked Co-localization display maps of co-localized pixels for the green and red channels across the entire group of cells. The degree of overlap between the two patterns is indicated by the overlap coefficient (O). The scale bar represents 10 μm. The results shown are representative of three independent experiments.
Having shown by immunofluorescence that both the GnRHR and a small population of the GR co-localize with Flot-1 at the plasma membrane, a biochemical strategy was pursued to provide additional evidence that these receptors localize to lipid rafts in LβT2 cells. Cells were incubated with 0.05% Triton X-100 before fractionation on a discontinuous sucrose density gradient. The results from Fig. 3A show that histone H3 localized only to fractions 6, 7, and 8, which are the most-dense fractions where the starting material was applied. Importantly, no histone H3 could be detected in the lipid raft fraction (Fig. 3A), indicating the absence of chromatin in the lipid raft fraction. The results from Fig. 3B show that the majority of Flot-1 is located in fraction 4 (lipid rafts) under basal and all stimulated conditions. The results show that the GnRHR localizes exclusively to the lipid raft fraction (fraction 4) in untreated cells and under all stimulated conditions (Fig. 3B, panels i–iv). This together with results presented in Fig. 1B shows that the GnRHR co-localizes with Flot-1 in lipid rafts at the plasma membrane of LβT2 cells. A small percentage of GR was detected in the lipid raft fraction in untreated cells (Fig. 3B, panel i). Despite the resulting nuclear import of the GR with Dex, a small amount of GR was still detected in the lipid raft fraction after 30 min of treatment with Dex (Fig. 3B, panel ii), which is in agreement with results shown in Fig. 2. Similarly, a small amount of GR was detected in the lipid raft fraction after 30 min of stimulation with either GnRH or co-stimulation with Dex and GnRH (Fig. 3B, panels iii and iv). Almost no GR was detected in fraction 5, indicating specific localization of the GR to lipid rafts rather than incomplete separation of the density-gradients (Fig. 3B, panels i–iv). The remaining GR was present in fractions 6, 7, and 8 in unstimulated cells and under all stimulated conditions (Fig. 3B, panels i-iv). These fractions contain all cellular material excluding the low density membrane fractions. To quantify the relative amount of GR associated with lipid rafts in the absence and presence of ligands, the lipid raft GR protein levels were expressed relative to lipid raft Flot-1 protein levels (Fig. 3C). The results show that the extent of GR localized to lipid rafts is independent of short exposures to Dex, GnRH, or a combination of both. This is in agreement with the result from Fig. 2 showing a small percentage of GR co-localizing with Flot-1 in LβT2 cells independent of hormone treatment (Fig. 2, panels A–D). Taken together, the results from Fig. 3B show that GR co-localizes with GnRHR in Flot-1-containing lipid rafts. Furthermore, the localization appears to be independent of short exposures to 100 nm Dex, 100 nm GnRH, or a combination thereof.
FIGURE 3.
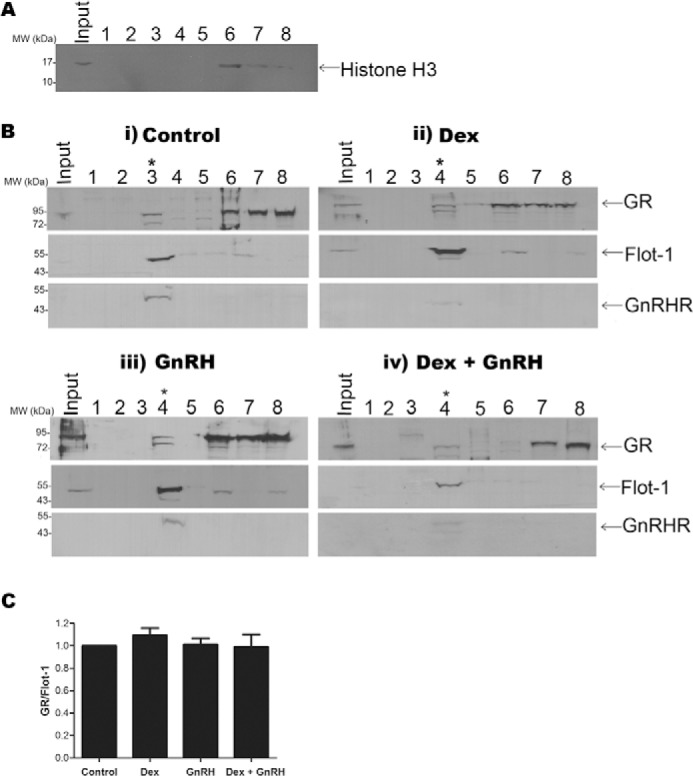
Fractionation shows the GR and GnRHR localize to Flot-1-containing lipid rafts ligand independently. A, lipid rafts were isolated by ultracentrifugation employing the detergent-resistant flotation strategy in a discontinuous sucrose density gradient consisting of 2 ml of ELB, 4 ml of 13% sucrose in ELB, 4 ml of 43% sucrose in ELB, and 4 ml of 60% (containing the sample). Eight fractions were collected: 1, top of the gradient; 2, ELB/13% interface; 3, 13%/43% interface; 4, remaining 13%/43% interface; 5, middle of 43% sucrose; 6, 43%/60% interface; 7, middle of 60% sucrose (loading fraction); 8, the pellet. 15 μl of fraction samples were loaded on an 8% SDS-PAGE gel, transferred onto nitrocellulose membrane, and probed with an antibody against histone H3. The Western blot shown is representative of three independent experiments. B, i-iv, LβT2 cells were serum-starved overnight and incubated for 30 min in serum-free medium (panel i), 100 nm Dex (panel ii), 100 nm GnRH (panel iii), or a combination of both (panel iv). Lipid rafts were isolated as described for A. 15 μl of fraction samples were loaded on an 8% SDS-PAGE gel, transferred onto nitrocellulose membrane, and probed successively with antibodies against GR, Flot-1, and GnRHR. Results shown in panels i–iv are single Western blots that are representative of three independent experiments. Fractions 3 and 4 contain the lipid raft material, indicated with a star. The detectable band in fraction 3 of panel iv in the GR Western blot has a larger size than the GR and is most likely a nonspecific band. The shift of the lipid raft material from fraction 3 (i) to fraction 4 (ii–iv) is most likely due to technical variation between experiments arising from manual collection of the fractions. The variable detection of a small % of total Flot-1 in fractions 6–8, containing unsuspended cells and unbroken nuclei, most likely reflects variations in sensitivity of the blotting procedure for these independent Western blots. C, to determine the ratio of GR lipid raft protein compared with Flot-1 lipid raft protein, only the lipid raft fractions of each condition were re-analyzed on one Western blot, and results were quantified by αEaseFC software. The lipid raft GR protein levels were normalized to lipid raft Flot-1 protein levels for each experiment, and pooled results are expressed relative to vehicle (Control). The graph shows the combined results of three independent experiments.
GR and Flot-1 Are Present Together in a Complex, Independent of Ligand in LβT2 Cells
To investigate whether the GR and Flot-1 are present together in a complex in LβT2 cells, co-immunoprecipitation assays using a GR-specific antibody were performed with whole LβT2 cell lysates. Remarkably, the results show that Flot-1 co-immunoprecipitated with the unliganded GR (Fig. 4A). Furthermore, treating the cells with 100 nm Dex for 30 min did not affect the interaction of Flot-1 with the GR (Fig. 4A). Importantly, no GR or Flot-1 co-immunoprecipitated with the nonspecific IgG under basal or Dex-treated conditions (Fig. 4A), suggesting that the interaction of Flot-1 with the GR is specific. Stimulation with GnRH or co-stimulation with Dex and GnRH did not result in a differential interaction of Flot-1 with the GR in three independent experiments (Fig. 4B). When Flot-1 was precipitated from whole cell lysates, co-immunoprecipitation of the GR occurred independent of Dex treatment (Fig. 4B). There was no statistically significant differential interaction of Flot-1 with the GR in the presence of Dex for 30 min as compared with unstimulated cells in pooled results of three independent experiments (Fig. 4B). These data taken together with results presented in Figs. 2 and 3 strongly suggest that the co-localization of the GR with Flot-1 in lipid rafts in LβT2 cells involves a physical direct or indirect interaction that is independent of ligand.
FIGURE 4.
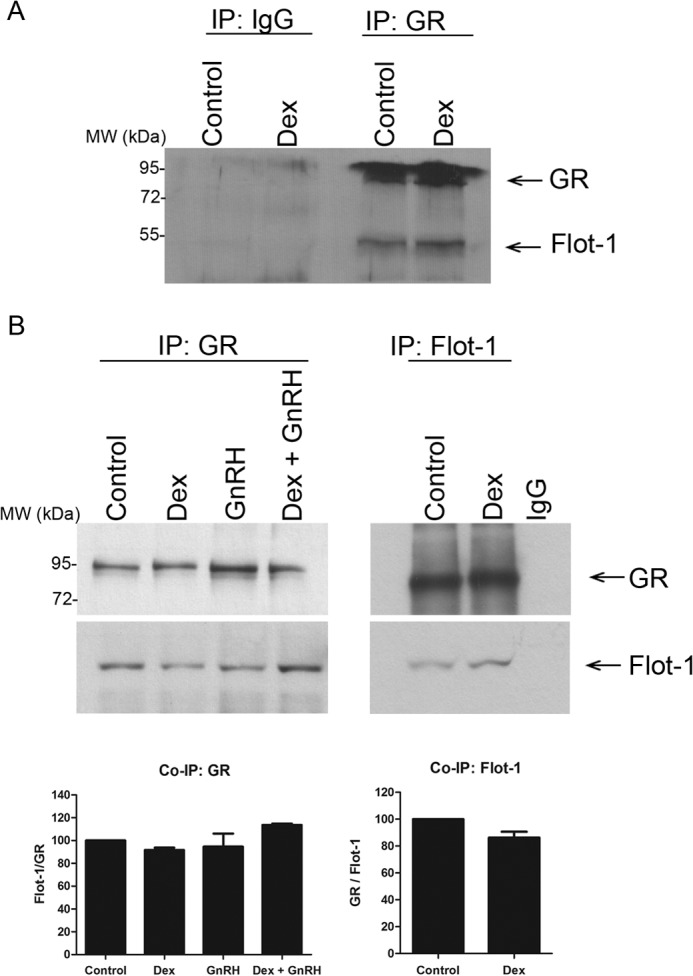
Co-immunoprecipitation (IP) shows that GR and Flot-1 interact ligand independently. A, LβT2 cells were incubated in serum-free medium for 2 h before the addition of 100 nm Dex for 30 min. 400 μl of cell lysates were incubated with GR antibody followed by precipitation with Protein A/G beads. The samples were loaded on an 8% SDS-PAGE gel, transferred onto a nitrocellulose membrane, and probed separately with anti-GR- and anti-Flot-1-specific antibodies. B, as in A, except that cells were also stimulated with 100 nm GnRH and a combination of both Dex plus GnRH, and in the right panel equal amounts of cell lysates were incubated with a rabbit anti-Flot-1 or nonspecific IgG antibody. The top panel shows a single representative Western blot, and the graph shows the combined results of three independent experiments where vehicle (Control) was set to 100%.
Flot-1 Appears Not to Be Required for Site-specific GR Phosphorylation
It has previously been shown that intact lipid rafts are required for Dex-induced GR phosphorylation at Ser-211 in A549 cells (27). Additionally, we have previously shown that Dex induces rapid phosphorylation of Ser-220 and Ser-234 of the mouse GR (mGR), whereas GnRH induces rapid Ser-234 but not Ser-220 mGR phosphorylation in LβT2 cells (10). The human GR residues 203, 211, and 226 are equivalent to the mGR residues 212, 220, and 234, respectively. Therefore, we investigated if the presence of Flot-1, and thus lipid rafts, is required for the GR phosphorylation in LβT2 cells. Flot-1 protein levels were decreased with siRNA-mediated knockdown followed by treatment of these Flot-1-depleted cells with 100 nm Dex, 100 nm GnRH, or both together for 30 min. The results from Fig. 5 show that Flot-1 levels were reduced by ∼60% using this strategy when NSC control is compared with Flot-1 siRNA Ctrl (Fig. 5G). The stimulation also had no effect on the decrease of Flot-1 protein obtained with the siRNA-mediated knockdown strategy. Further reduction in Flot-1 levels was accompanied by cell death (data not shown). The results from Fig. 5, A and B, show that neither Dex, GnRH, or the combination of both resulted in a significant increase in Ser-212 phosphorylation of the mGR in the NSC or Flot-1 siRNA conditions. However, a trend was apparent whereby Dex, GnRH, and Dex plus GnRH all appeared to slightly increase Ser-212 phosphorylation in the NSC, and this increase appeared to be lost when Flot-1 protein levels were reduced, suggesting that phosphorylation of the GR at Ser-212 may require membrane association. However, given the small responses, it was not possible to establish significance above technical error and/or biological variation. In the absence of Flot-1 siRNA, treatment with Dex resulted in a significant increase in GR phosphorylation at Ser-220, whereas GnRH did not increase phosphorylation at that residue, and the combination of Dex plus GnRH increased Ser-220 phosphorylation to the same extent as Dex alone (Fig. 5, C and D). Decreasing Flot-1 protein levels by 60% had no detectable significant effect on Ser-220 phosphorylation of the mGR (Fig. 5, C and D) in the absence or presence of ligands. In the absence of Flot-1 siRNA, treatment with Dex resulted in a significant increase in GR phosphorylation at Ser-234 (Fig. 5, E and F). Although the extent of Ser-234 phosphorylation for Dex plus GnRH appeared to be slightly greater than for Dex alone, statistical significance could not be established (Fig. 5, E and F). As previously reported, GnRH resulted in a similar level of Ser-234 phosphorylation of the mGR as Dex (Fig. 5, E and F) (10). Decreasing Flot-1 protein levels had no detectable significant effect on Ser-234 phosphorylation of the mGR (Fig. 5, E and F) in the absence or presence of ligands. Taken together, decreasing the Flot-1 protein levels by 60% had no detectable significant effect on Ser-212, Ser-220, or Ser-234 phosphorylation of the mGR (basal and ligand-induced), suggesting that lipid raft association is not required for phosphorylation of the GR. However we cannot discount the possibility that a greater percentage knockdown of Flot-1 may have significantly affected ligand-induced mGR phosphorylation.
FIGURE 5.

Flot-1 appears not to be required for site-specific GR phosphorylation. LβT2 cells were transfected with NSC or specific mouse Flot-1 siRNA at a final concentration of 40 nm and incubated for 72 h. The cells were incubated for 30 min in serum-free medium with 100 nm Dex or 100 nm GnRH or a combination of both, and the proteins were harvested. The samples were loaded on an 8% SDS-PAGE gel, transferred onto nitrocellulose membrane, and probed with anti-phospho-Ser-212 (P-S212) GR-, anti-phospho-Ser-220 (P-S220) GR-, and anti-phospho Ser-234 (P-S234)-specific antibodies. The membranes were stripped and probed with specific antibodies to GR, Flot-1, and GAPDH. The panels on the left (A, C, and E) show a single representative Western blot, and the graphs (B, D, and F) show the combined results of four independent experiments where vehicle (Ctrl, control) was set to 1. Panel G shows the average percentage decrease of Flot-1 protein for the control samples in the presence of Flot-1 siRNA. One-way ANOVA with Dunnett's post test was used for statistical analysis and is denoted as *, **, or *** to indicate p < 0.05, p < 0.01, or p < 0.001 respectively.
Co-treatment with Dex plus GnRH Synergistically and Selectively Enhances SGK-1 mRNA Levels
We previously showed that cross-talk between the GR and GnRHR signaling pathways modulates expression of an endogenous AP-1-containing gene, most likely via a GR tethering mechanism involving GR phosphorylation. To investigate whether Dex plus GnRH synergism occurs on endogenous genes containing GREs in their promoters, LβT2 cells were incubated for 8 h with 100 nm Dex, 100 nm GnRH, and both together followed by RNA extraction and cDNA synthesis and real-time PCR. The results from Fig. 6, A–E, show that Dex increased mRNA levels of the GILZ, MKP-1, FKBP5, FSHβ, and SGK-1 genes, consistent with previous reports in the literature (43–47). Treatment with GnRH alone had no effect on mRNA levels of the GILZ, MKP-1, and FKBP5 genes but increased expression of the FSHβ gene (Fig. 6D), as previously reported (43). Interestingly, we found that GnRH alone also significantly increased mRNA levels of the SGK-1 gene (Fig. 6E). Although Dex plus GnRH treatment did not result in a significant change in mRNA expression for the GILZ, MKP-1, FKBP5, and FSHβ genes compared with Dex alone, a statistically significant synergistic response was apparent for the SGK-1 gene. Although Dex and GnRH alone increased SGK-1 mRNA by 8- and 3.5-fold, respectively, the combination increased SGK-1 mRNA levels by 17-fold (Fig. 6E). These results collectively suggest that cross-talk between the GR and GnRHR signaling pathways resulting in synergistic effects on gene expression is selective for only some GRE-containing genes.
FIGURE 6.

Co-treatment of cells with Dex plus GnRH synergistically enhances SGK-1 mRNA levels. LβT2 cells were incubated for 8 h in serum-free medium with 100 nm Dex, 100 nm GnRH, or a combination of both. Total RNA was isolated and reverse-transcribed, and relative mRNA levels of several endogenous GRE-containing genes including GILZ (A), MKP-1 (B), FKBP5 (C), FSHβ (D), and SGK-1 (E) were determined by quantitative real-time PCR. Fold changes in the gene of interest mRNA levels were normalized to GAPDH transcripts and were calculated relative to vehicle-treated (control (Ctrl)) samples. The graph is representative of three independent experiments. One-way ANOVA with Dunnett's (stars above bars) and Tukey (stars above lines joining bars) post tests were used for statistical analysis and denoted as *, **, or *** to indicate p < 0.05, p < 0.01, or p < 0.001, respectively.
Flot-1, GR, and the GnRHR Are Required for the Synergistic Increase in SGK-1 Gene Expression Levels
To investigate whether Flot-1 is required for the observed synergistic increase in SGK-1 mRNA in response to Dex plus GnRH, Flot-1 protein levels were decreased using an siRNA-mediated protein knockdown approach, and the cells were stimulated for 8 h with 100 nm Dex, 100 nm GnRH, or both together. The results from Fig. 7A show that decreased Flot-1 protein levels did not affect the extent of Dex or GnRH-induced increase of SGK-1 mRNA. Fig. 7B shows that Flot-1 levels were reduced by ∼60% using this strategy. It was found that in the NSC, the GnRH-induced increase in SGK-1 expression was not statistical significant. Interestingly, reducing the Flot-1 protein levels by ∼60% decreased the synergistic Dex plus GnRH transcriptional response by ∼50% (Fig. 7A), to a level similar to that of Dex alone. These results suggest that the association of the GR with Flot-1 is not required for the SGK-1 response to Dex alone but is required for the synergistic response to Dex plus GnRH. To obtain more evidence that this synergistic response observed for the SGK-1 gene also requires the GR, the GR protein levels were decreased by ∼55% with siRNA (Fig. 7D). The results from Fig. 7C show that, as expected, the Dex-induced increase in SGK-1 mRNA levels requires the presence of the GR, as GR knockdown completely ablates the response. Similarly, the results are consistent with a requirement for the GR in the Dex plus GnRH response (Fig. 7C), although in these experiments the synergism was not as pronounced. Further support for a requirement for the GR for the synergistic response was obtained using the GR antagonist RU486, which attenuated the transcriptional increase seen when cells where treated with Dex alone and the synergistic response in the presence of both Dex and GnRH (Fig. 7E). To obtain evidence that the synergism is also GnRHR-dependent, cells were incubated with ligands in the absence or presence of the GnRHR antagonist Antide. The results from Fig. 7E show that the GnRHR antagonist had no effect on the Dex-induced increase in SGK-1 mRNA levels. The GnRHR antagonist attenuated the small GnRH-induced increase in SGK-1 mRNA levels, and it abolished the synergistic response induced by co-stimulation with Dex plus GnRH (Fig. 7E). Taken together, these results indicate that the synergistic transcriptional up-regulation of SGK-1 requires membrane-associated Flot-1, GR and the GnRHR.
FIGURE 7.

Flot-1, GR and the GnRHR are required for the synergistic transcriptional response on the SGK-1 gene. A, LβT2 cells were transfected with NSC or specific mouse Flot-1 siRNA at a final concentration of 40 nm and incubated for 72 h. The cells were incubated for 8 h in serum-free medium with 100 nm Dex or 100 nm GnRH or a combination of both. Total RNA was isolated and reverse-transcribed, and relative levels of SGK-1 transcripts were determined by quantitative real-time PCR. Fold changes in SGK-1 mRNA levels were normalized to GAPDH transcripts and calculated relative to vehicle-treated (control (Ctrl)) samples. The graph is representative of three independent experiments. The panels (B and D) on the right show Western blots that are representative images showing the extent of Flot-1 and GR protein knockdown, respectively. The histograms in B and D show the quantitative analysis of the percentage decrease in Flot-1 and GR protein levels, respectively. C, as in A except that the cells were transfected with 20 nm GR siRNA for 96 h. E, LβT2 cells were incubated for 8 h in serum-free medium with 100 nm Dex or 100 nm GnRH or a combination of both in the presence and absence of 100 nm Antide. Total RNA was isolated and reverse-transcribed, and relative levels of SGK-1 transcripts were determined by quantitative real-time PCR. Fold changes in SGK-1 mRNA levels were normalized to GAPDH transcripts and were calculated relative to vehicle-treated (control) samples. The graph is representative of three independent experiments. One-way ANOVA with Dunnett's (stars above bars) and Tukey (stars above lines joining bars) post tests were used for statistical analysis and denoted as *, **, or *** to indicate p < 0.05, p < 0.01, or p < 0.001, respectively.
Synergism Involves Differential Recruitment of SRC Cofactors to the SGK-1 Promoter Compared with Dex and GnRH Alone, Whereas GR Recruitment Remains Unchanged
As we could not detect increased GR phosphorylation in the presence of Dex and GnRH compared with single-hormone treatment, we next investigated whether increased GR recruitment to the GRE present in SGK-1 could explain the augmented transcriptional response. Cells were incubated with 100 nm Dex, GnRH, and both together for 1 h followed by immunoprecipitation using an anti-GR antibody. The results in Fig. 8A show that Dex treatment resulted in a significant 2.5-fold recruitment of the GR to the SGK-1 promoter (Fig. 8A), which is consistent with the GR knockdown results from Fig. 7B. Interestingly, GnRH treatment resulted in a >1.5-fold recruitment of the GR to the SGK-1 promoter compared with control cells, consistent with a previous study reporting Dex-independent promoter recruitment of the GR. However, treatment of cells with both Dex and GnRH together resulted in similar amounts of GR being recruited to the SGK-1 promoter as compared with Dex alone (Fig. 8A). These results suggest that the synergistic transcriptional response induced by Dex plus GnRH on the SGK-1 gene is not a due to increased GR promoter occupancy. On the basis of these results, we next determined whether known GR cofactors are differentially recruited to the promoter. As shown in Fig. 8B, treatment with Dex or GnRH alone resulted in a significant recruitment of SRC-1 to the SGK-1 promoter (2- and 2.25-fold, respectively) (Fig. 8B). Interestingly, co-treatment with Dex and GnRH resulted in significantly less SRC-1 recruitment to the SGK-1 promoter compared with both ligands alone (Fig. 8B). GRIP-1 seemed to only be recruited to the SGK-1 promoter in response to Dex (Fig. 8B), whereas SRC-3 (Fig. 8D), p300, CBP, and p65 (data not shown) were not detected at the gene promoter under any treatment conditions. In summary, the results suggest that increased SGK-1 transcription by Dex, GnRH, or a combination of both is mediated by differential recruitment of cofactors, suggesting that synergistic effects initiated in membrane lipid rafts exert downstream differences in nuclear promoter occupancy compared with the classical response to Dex alone.
FIGURE 8.

Synergism involves differential recruitment of SRC cofactors compared with Dex and GnRH alone without an increase in GR recruitment to the SGK-1 promoter. ChIP assays were performed in LβT2 cells that were treated for 1 h with 100 nm Dex, 100 nm GnRH, and both together using anti-GR (A), anti-SRC-1 (B), anti-GRIP-1 (C), and anti-SRC-3 (D) antibodies. Precipitated complexes that were bound to the SGK-1 promoter were detected with quantitative real-time PCR using primers that span the GRE region. Results were normalized against the input samples and are represented relative to control. The graphs are representative of three independent experiments. One-way ANOVA with Dunnett's post-test was used for statistical analysis and is denoted as *, **, or *** to indicate p < 0.05, p < 0.01, or p < 0.001, respectively.
GnRH Acting via the PKC but Not the PKA Pathway Augments the Dex-mediated Increase in SGK-1 mRNA Levels
Because the GnRHR has been shown to activate both the PKA and PKC signaling pathways in LβT2 cells (48) and our previous results implicated PKC in mediating Dex plus GnRH synergy (10), we investigated the possible involvement of the PKA and PKC signaling pathways in the SGK-1 response. 8-Bromo-cAMP, which is an inducer of the PKA signaling pathway, was used to stimulate the cells in the absence and presence of Dex. We found that 8-bromo-cAMP alone had no effect on the mRNA expression levels of the SGK-1 gene (Fig. 9A). In addition, stimulating cells with Dex and cAMP did not result in a synergistic response, as seen for co-treatment with Dex and GnRH. This result indicates that the PKA signaling pathway is not involved in regulating the expression of the SGK-1 gene in LβT2 cells. To investigate a potential role for the PKC signaling pathway, we incubated cells with PMA, a diacylglycerol analog known to activate PKC. The results show that PMA resulted in a significant 4-fold induction of the SGK-1 mRNA expression (Fig. 9B). Interestingly, stimulating cells with both Dex and PMA resulted in a transcriptional response with a magnitude comparable with treatment with both Dex plus GnRH (Fig. 9B). This similar gene induction strongly suggests that the PKC pathway is involved in mediating the synergistic up-regulation of SGK-1 mRNA levels in response to Dex plus GnRH (Fig. 9B). In support of this notion, the PKC inhibitor BIM not only decreased PMA-induced SGK-1 gene expression but also the synergistic Dex plus GnRH response as well as the synergistic response induced by Dex plus PMA (Fig. 9C).
FIGURE 9.

GnRH acting via the PKC but not the PKA pathway synergistically increases the Dex-mediated up-regulation of the SGK-1 gene. A, LβT2 cells were incubated for 8 h in serum-free medium with 100 nm Dex, 100 nm GnRH, a combination of Dex plus GnRH, 2 mm 8-bromo-cAMP, and a combination of Dex plus 8-bromo-cAMP. Total RNA was isolated and reverse-transcribed, and relative levels of SGK-1 transcripts were determined by quantitative real-time PCR. Fold changes in SGK-1 mRNA levels were normalized to GAPDH transcripts and were calculated relative to vehicle-treated (control (Ctrl)) samples. The graph is representative of three independent experiments. B, as in A, except that 8-bromo-cAMP was replaced with 20 ng/ml PMA. C, as in B, except the cells were stimulated in the presence and absence of 100 nm PKC inhibitor. One-way ANOVA with Dunnett's post-test was used for statistical analysis and is denoted as *, **, or *** to indicate p < 0.05, p < 0.01, or p < 0.001, respectively.
SGK-1 and Flot-1 Are Required for the Synergistic Dex Plus GnRH Decrease in LβT2 Cell Proliferation
Because lipid raft-associated GR was previously implicated in playing a role in cell proliferation of neural mouse progenitor cells (31) and we showed a requirement for Flot-1 in synergistic up-regulation of SGK-1 mRNA, we investigated the role of Dex, GnRH, and a combination of both as well as Flot-1 and SGK-1 in cell proliferation in LβT2 cells. Flot-1 or SGK-1 expression was decreased by siRNA-mediated knockdown, and the cells were replated and stimulated for 48 h with 100 nm Dex, 100 nm GnRH, or both together. Two different SGK-1 antibodies were used to detect the protein, but unfortunately both resulted in many nonspecific bands (data not shown). To establish that SGK-1 gene expression was decreased with the siRNA, SGK-1 mRNA levels were determined with real-time PCR. The results from Fig. 10A show that SGK-1 mRNA was decreased by ∼60%. A cell proliferation assay was performed, and the results from Fig. 10B show that Dex had no effect on cell proliferation in the absence or presence of Flot-1 or SGK-1 knockdown. Interestingly, in the absence of knockdown, GnRH decreased proliferation by ∼20%, whereas treatment with Dex and GnRH together showed a synergistic attenuation in cell proliferation (Fig. 10B). Decreasing the Flot-1 protein levels by ∼60% did not significantly change the effects of Dex or GnRH alone on proliferation of LβT2 cells (Fig. 10B). Decreased Flot-1 protein levels, however, significantly attenuated the repressive effect of Dex plus GnRH to a small extent (Fig. 10B). Interestingly, siRNA-mediated knockdown of SGK-1 lifted the synergistic repression of cell proliferation in response to co-stimulation with Dex and GnRH. Taken together the results suggest that synergistic up-regulation of SGK-1 mRNA levels by Dex plus GnRH as well as the presence of Flot-1 is required for a synergistic repression of LβT2 cell proliferation.
FIGURE 10.

Dex plus GnRH synergistically decreases cell proliferation by up-regulating SGK-1. A, Flot-1 and SGK-1 expression levels were decreased with 40 nm siRNA-mediated knockdown for 72 h, after which the cells were harvested for RNA extraction. Total RNA was isolated and reverse-transcribed, and relative levels of SGK-1 transcripts were determined by quantitative real-time PCR. Fold changes in SGK-1 mRNA levels were normalized to GAPDH transcripts and were calculated relative to vehicle-treated (Ctrl, control) samples. The graph is representative of three independent experiments. B, as in A except that the cells were reseeded after knockdown and stimulated for 48 h with 100 nm Dex, 100 nm GnRH, or a combination of both. During the last hour of stimulation, the thymidine analog, BrdU, was added to the cells, and newly synthesized DNA was quantified with a colorimetric immunoassay. One-way ANOVA with Dunnett's (A) or both Dunnett's and Tukey (B) post-tests were used for statistical analysis and denoted as *, **, or *** to indicate p < 0.05, p < 0.01, or p < 0.001, respectively.
DISCUSSION
Cross-talk between the GR and other receptors is emerging as a potential mechanism for fine-tuning cellular responses via several different genomic and non-genomic mechanisms. We have previously shown that in LβT2 cells the GR is ligand-independently activated by GnRH and that a combination of Dex plus GnRH synergistically increases transcription of a GRE reporter gene in a manner dependent on the presence of the GR and the GnRHR. In the present study we investigated the hypothesis that the localization of the GR to lipid rafts or caveolae is required for some of these genomic or non-genomic GR-mediated effects in LβT2 cells. We show here for the first time that both the GnRHR and a small population of the GR co-localize with the lipid raft protein Flot-1 at the plasma membrane, independent of the presence of ligands. Furthermore, the GR is present in a complex with Flot-1, independent of the presence of ligands. These results are consistent with previous studies showing that localization of the GnRHR to lipid rafts in αT3-1 cells (36) and the mineralocorticoid receptor to lipid rafts in HEK-293 cells (49) is independent of their respective ligands. Our findings suggest that the lipid raft-associated GR-GnRHR complex acts as a signal transduction platform that modulates downstream genomic signaling by the cytoplasmic GR. By a combination of protein knockdown, pharmacological inhibitor/activator and ChIP strategies, and incubation of cells with 1) Dex or 2) GnRH alone or 3) in combination, we have obtained insights into the role of lipid raft-associated GR on gene expression for 1) classical Dex-mediated transactivation, 2) GnRH induced ligand-independent GR responses, and 3) responses to the combination of Dex plus GnRH, for an endogenous GRE-containing gene in LβT2 cells.
Results were first assessed to determine whether Flot-1-associated GR is involved in regulating classical genomic GR agonist-dependent transcription of GRE-containing genes. Because we did not previously investigate endogenous GRE-containing genes (10), we wished to identify such genes in the present study for further mechanistic investigation. We show that Dex increased mRNA levels, as expected, of several GRE-containing genes, namely, GILZ, MKP-1, FKBP5, FSHβ, and SGK-1 genes, consistent with previous reports in the literature (43–46, 50). We chose to further investigate detailed mechanisms only for the SGK-1 gene, as a synergistic up-regulation was observed only on that gene with Dex plus GnRH. Our results show that lipid raft-associated GR does not have a role in mediating the classical Dex-dependent GR transactivation response on the SGK-1 gene and that the PKA and PKC pathways are not required for this response. Most of the GR translocated to the nucleus in the presence of Dex, whereas the small subpopulation of GR associated with lipid rafts did not appear to change location. Moreover, the GR was recruited to the GRE region of the SGK-1 promoter. These data are consistent with a classical mechanism of GR activation as well as with a previous report showing that GR recruitment to the SGK-1 GRE in 10T1/2 cells (51). In contrast, others have shown a functional role for caveolae-associated GR in Dex-mediated GRE reporter gene transactivation (30). The estrogen receptor has also been found in the membrane and was shown to be required for the early membrane estrogenic effects to regulate the slower genomic actions in a nerve cell line (52). Our results suggest that Flot-1-associated GR may act differently to caveolae-associated GR or that the dependence of genomic transactivation on membrane-associated GR responses is cell-specific. Our ChIP results showing increased recruitment of GRIP-1 to the GRE in response to Dex alone are in agreement with reports for the SGK-1 gene in 1470.2 cells (53) as well as published data for other GRE-containing genes (54). Interestingly our results show that SRC-1 is also recruited by this classical GR transactivation mechanism in response to Dex on the SGK-1 promoter in LβT2 cells. Although it has been reported that the GR preferentially recruits GRIP-1 rather than SRC-1 to a GRE reporter (55), others have shown that SRC-1 can act as a GR co-activator (54), suggesting that the recruitment of SRC-1, GRIP-1, or both may be promoter-specific.
Secondly, we assessed whether Flot-1-associated GR is involved in mediating GnRH-induced ligand-independent GR-mediated transcription of the SGK-1 gene. Our results showing that the GR is significantly recruited to the SGK-1 promoter in response to GnRH are novel and suggest that ligand-independently activated GR contributes to the GnRH-induced up-regulation of the endogenous GRE-containing gene. Furthermore, the absence of a response to GnRH for the GRE-containing GILZ, MKP1, and FKBP5 genes suggests that GnRH-activated GR exerts promoter-specific downstream effects. We could not, however, establish conclusively whether Flot-1 is required for the GnRH-induced response on the SGK-1 gene. This was due to the relatively small but statistically significant response on the SGK-1 gene with GnRH, which was even further reduced under NSC knockdown conditions. Note that due to the small response we were also unable to establish whether PKC activation is required for the GnRH response on the SGK gene, although the apparent slight reduction with a PKC inhibitor and mimicking of the response with PMA, but not the cAMP analog, suggest that PKC but not PKA is involved regulating GnRH-induced SGK-1 expression, consistent with our previously published data (10). Besides recruitment of the GR to the SGK-1 promoter, albeit less than with Dex alone, treatment with GnRH also resulted in the recruitment of GRIP-1 but not SRC-1 or SRC-3 to the promoter. By contrast, in Dex-treated cells we found that both GRIP-1 and SRC-1, but not SRC-3, are associated with the promoter. Interestingly, this result for GnRH is different to that obtained for the GnRHR promoter (10), where neither GRIP-1 nor SRC-1 is recruited. These data highlight gene-specific effects of GnRH-activated GR signaling at responsive promoters. It is likely that promoter occupancy of GR cofactors is determined by both the promoter gene architecture as well as the ligands activating the GR, which may result in distinct conformational changes in the GR itself, thus allowing differential interactions with other factors.
Thirdly, we assessed whether Flot-1-associated GR is involved in mediating the combined effects of Dex plus GnRH on transcription of the SGK-1 gene. Our findings provide several novel insights into the mechanism of synergy between the GR and GnRHR signaling pathways. We present evidence for the first time that the GR and GnRHR synergistically up-regulate the SGK-1 gene and, importantly, that this effect, in contrast to the effects elicited by the hormones alone, requires Flot-1-containing lipid rafts. Furthermore, our results show that this synergism only occurs on a subset of GRE-containing endogenous genes. This gene-specific effect supports a model whereby both receptors act in concert to selectively activate only a distinct set of genes to fine-tune the response of a cell to changes in the hormonal environment. We established that the synergistic effect on SGK-1 gene expression is not due to detectable changes in localization of the GR to the membrane or increased GR recruitment to the promoter. Our findings led us to hypothesize that the underlying mechanism of synergistic up-regulation of the SGK-1 gene by Dex plus GnRH involves the activation of a cytoplasmic signaling pathway by the lipid raft-associated GnRHR-GR complex. Consistent with this, our results show a requirement for the PKC pathway in the combined effect of Dex and GnRH and not for the response observed for Dex treatment alone. The observed PMA-dependent and cAMP-independent increase in SGK-1 mRNA levels is in contrast to a previous report showing that PKA and not PKC activates the SGK-1 gene in rat neonatal cardiomyocytes (56), suggesting cell-specific effects. However, the same study showed that PMA enhanced the cortisol-induced mRNA expression of the SGK-1 gene, similar to the effects observed with PMA plus Dex in the current study, suggesting that the PKC pathway augments GC responses in other cells. The data obtained here suggest that in LβT2 cells, cross-talk between the GnRHR and GR involves the activation of the PKC pathway and converges on the SGK-1 gene. As we found a requirement for Flot-1-containing lipid rafts in the synergistic transcriptional response and a direct interaction of Flot-1 and the GR, it is tempting to speculate that these membrane domains provide a platform where GnRHR and GR constitutively assemble and cross-talk to modulate downstream signaling from the membrane.
The finding that Dex plus GnRH did not alter the membrane localization of either receptor together with the result that GR levels at the SGK-1 promoter were similar to single-hormone treatment suggests that other factors mediate the combined transcriptional effect. Similarly, others have found that co-treatment of cells with cortisol plus FGF-2 did not alter the cortisol-induced GR recruitment to the SGK-1 promoter (51). We show that the relative levels of members of the SRC family of co-activators are different for Dex plus GnRH compared with Dex alone, which suggests that another cofactor or an entirely different complex might be recruited to the SGK-1 promoter. Although we investigated a possible role for other co-activators in mediating the synergistic response on the SGK-1 gene, we were unable to identify such a positively acting cofactor. However, in the future it will be important to determine whether other cofactors assemble on the promoter and enhance SGK-1 transactivation compared with Dex and GnRH alone. The genomic synergistic effect is likely to involve non-genomic-driven changes in the phosphorylation status and nuclear translocation of cofactors and/or other transcription factors. Thus it is possible that other factors are not involved but, rather, the same factors that are differentially phosphorylated. On the basis of our results we hypothesize that such phosphorylation and translocation events could be mediated by the PKC pathway.
Previous results from our group suggested a role for PKC in the GnRH-induced but not the Dex-induced transactivation of a GRE reporter and ligand-induced Ser-234 phosphorylation of the GR (10). In the present study we show that Dex and Dex plus GnRH induce phosphorylation of the GR at Ser-234 and Ser-220 of the mGR but that GnRH only induces phosphorylation of the mGR at Ser-234, similar to our previous report (10). Our current novel results suggest that lipid raft association of mouse GR is not required for GR phosphorylation at Ser-212, Ser-220, and Ser-234 in response to GnRH, Dex, or the combination thereof. Furthermore, the SGK-1 response to Dex was unaffected, whereas the response to GnRH was slightly but not significantly reduced, and the synergistic response was significantly inhibited in the presence of the PKC inhibitor. Our results suggest that ligand-induced phosphorylation of the GR does not require lipid raft association. Arguably, a greater reduction in Flot-1 protein levels >60% may have led to detectable changes in GR phosphorylation levels. However, the findings that a 60% reduction of Flot-1 levels results in a significant decrease in the synergistic up-regulation of the SGK-1 gene and a significant reduction of the proliferative effect strongly support the notion that the Flot-1-dependent synergistic response does not require GR phosphorylation at Ser-234 or Ser-220. Furthermore, when taken together with our previous results, we cannot as yet discount the possibility that membrane-associated GR is phosphorylated directly or indirectly by PKC or SGK-1 via a GnRHR-induced pathway.
Signaling by both the GR and the GnRHR has been previously shown to affect cell proliferation in several different cell types (27, 57–63). Our finding that GnRH alone decreases cell proliferation in LβT2 cells is consistent with a previous report (64). The insensitivity of this effect to both Flot-1 and SGK-1 knockdown taken together with the gene expression and ChIP results suggests that the mechanism of repression of cell proliferation by GnRH does not involve lipid raft-associated GR or SGK-1 expression. Because the GnRHR activates several downstream pathways in LβT2 cells (65), which could potentially target several mediators of cell proliferation effects, factors other than the GR and SGK-1 expression are likely to predominate in mediating the anti-proliferative response to GnRH in these cells. Our finding that synergism between the GR and the GnRHR regulates cell proliferation in LβT2 via up-regulating the SGK-1 gene is novel and suggests a mechanism for cross-talk between the hypothalamic pituitary adrenal and hypothalamic pituitary gonadal axes to regulate proliferation of gonadotrope cells in the pituitary. The cell proliferation effects were significantly ablated by reduction of Flot-1 protein levels, consistent with a role for Flot-1 in the synergistic mechanism. Membrane-associated GR has previously been linked to antiproliferative affects in A549 cells (27). However unlike these reports showing anti-proliferative effects with Dex alone, we show that in LβT2 cells, Dex alone does not affect cell proliferation but only does so in combination with GnRH, consistent with cell-specific effects. It is tempting to speculate that lipid raft-associated GR modulates GnRHR signaling to enhance the GnRH-induced anti-proliferative response by direct effects on the GnRHR. However, we did not detect any changes in tyrosine phosphorylation of the GnRHR in the presence of Dex in LβT2 cells (data not shown). Our finding for a role for SGK-1 in cell proliferation is reminiscent of a previous report showing that SGK-1 activity is required for cortisol-induced reduction of cell proliferation in a hippocampal progenitor cell line (66). Because increases in pituitary cell proliferation have been linked to pathologies such as pituitary tumors, Cushings disease, pituitary hyperplasia, and pituitary developmental dysregulation (67–69), synergy between Dex and GnRH may be a mechanism to prevent such negative physiological consequences. Synergy between membrane-associated GR and GnRHR represents an attractive mechanism for fine-tuning responses and interplay between signaling pathways and may be representative of a more widespread mechanism that awaits further discovery.
Acknowledgments
We thank P. Mellon, D. C. Skinner, and A. R. Saltiel for kindly providing the LβT2 cells, the anti-GnRHR antibody, and the flotillin-1 expression vector, respectively. We also thank members of the Hapgood laboratory, especially C. Avenant and C. Kemp, for helpful discussions and methodological support, M. Tomasicchio for help with preparation of the figures, and C. Avenant for critical reading of the manuscript.
This work was supported by the National Research Foundation, South Africa (to J. H.).
- GC
- glucocorticoid
- GR
- glucocorticoid receptor
- mGR
- mouse GR
- GnRH
- gonadotropin-releasing hormone
- GnRHR
- GnRH receptor
- Flot-1
- flotillin-1
- Dex
- dexamethasone
- Cav-1
- caveolin-1
- PMA
- phorbol 12-myristate 13-acetate
- SGK-1
- serum/glucocorticoid regulated kinase 1
- TBST
- TBS with Tween 20
- ELB
- extraction lysis buffer
- NSC
- non-silencing scrambled
- GRE
- glucocorticoid-response element
- GILZ
- glucocorticoid-induced leucine zipper
- MKP-1
- mitogen-activated protein kinase phosphatase-1
- FKBP5
- FK506-binding protein 5
- FSHβ
- follicle-stimulating hormone β
- ANOVA
- analysis of variance
- CBP
- cAMP-response element-binding protein (CREB)-binding protein.
REFERENCES
- 1. Stahn C., Buttgereit F. (2008) Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pract. Rheumatol. 4, 525–533 [DOI] [PubMed] [Google Scholar]
- 2. Newton R., Leigh R., Giembycz M. A. (2010) Pharmacological strategies for improving the efficacy and therapeutic ratio of glucocorticoids in inflammatory lung diseases. Pharmacol. Ther. 125, 286–327 [DOI] [PubMed] [Google Scholar]
- 3. Revollo J. R., Cidlowski J. A. (2009) Mechanisms generating diversity in glucocorticoid receptor signaling. Ann. N.Y. Acad. Sci. 1179, 167–178 [DOI] [PubMed] [Google Scholar]
- 4. Lösel R., Wehling M. (2003) Nongenomic actions of steroid hormones. Nat. Rev. Mol. Cell Biol. 4, 46–56 [DOI] [PubMed] [Google Scholar]
- 5. Croxtall J. D., Choudhury Q., Flower R. J. (2000) Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br. J. Pharmacol. 130, 289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sasson R., Shinder V., Dantes A., Land A., Amsterdam A. (2003) Activation of multiple signal transduction pathways by glucocorticoids, protection of ovarian follicular cells against apoptosis. Biochem. Biophys. Res. Commun. 311, 1047–1056 [DOI] [PubMed] [Google Scholar]
- 7. Bruna A., Nicolàs M., Muñoz A., Kyriakis J. M., Caelles C. (2003) Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J. 22, 6035–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caelles C., González-Sancho J. M., Muñoz A. (1997) Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. 11, 3351–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tasker J. G., Di S., Malcher-Lopes R. (2006) Minireview, rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology 147, 5549–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kotitschke A., Sadie-Van Gijsen H., Avenant C., Fernandes S., Hapgood J. P. (2009) Genomic and nongenomic cross talk between the gonadotropin-releasing hormone receptor and glucocorticoid receptor signaling pathways. Mol. Endocrinol. 23, 1726–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verhoog N. J., Du Toit A., Avenant C., Hapgood J. P. (2011) Glucocorticoid-independent repression of tumor necrosis factor (TNF) α-stimulated interleukin (IL)-6 expression by the glucocorticoid receptor, a potential mechanism for protection against an excessive inflammatory response. J. Biol. Chem. 286, 19297–19310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Löwenberg M., Verhaar A. P., Bilderbeek J., Marle J. v., Buttgereit F., Peppelenbosch M. P., van Deventer S. J., Hommes D. W. (2006) Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN. EMBO Rep. 7, 1023–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Löwenberg M., Tuynman J., Bilderbeek J., Gaber T., Buttgereit F., van Deventer S., Peppelenbosch M., Hommes D. (2005) Rapid immunosuppressive effects of glucocorticoids mediated through Lck and Fyn. Blood 106, 1703–1710 [DOI] [PubMed] [Google Scholar]
- 14. Bartis D., Boldizsár F., Szabó M., Pálinkás L., Németh P., Berki T. (2006) Dexamethasone induces rapid tyrosine-phosphorylation of ZAP-70 in Jurkat cells. J. Steroid Biochem. Mol. Biol. 98, 147–154 [DOI] [PubMed] [Google Scholar]
- 15. Bartis D., Boldizsár F., Kvell K., Szabó M., Pálinkás L., Németh P., Monostori E., Berki T. (2007) Intermolecular relations between the glucocorticoid receptor, ZAP-70 kinase, and Hsp-90. Biochem. Biophys. Res. Commun. 354, 253–258 [DOI] [PubMed] [Google Scholar]
- 16. Löwenberg M., Tuynman J., Scheffer M., Verhaar A., Vermeulen L., van Deventer S., Hommes D., Peppelenbosch M. (2006) Kinome analysis reveals nongenomic glucocorticoid receptor-dependent inhibition of insulin signaling. Endocrinology 147, 3555–3562 [DOI] [PubMed] [Google Scholar]
- 17. Cheung L. W., Leung K. W., Wong C. K., Wong R. N., Wong A. S. (2011) Ginsenoside-Rg1 induces angiogenesis via non-genomic crosstalk of glucocorticoid receptor and fibroblast growth factor receptor-1. Cardiovasc. Res. 89, 419–425 [DOI] [PubMed] [Google Scholar]
- 18. Schmidt P., Holsboer F., Spengler D. (2001) β2-adrenergic receptors potentiate glucocorticoid receptor transactivation via G protein βγ-subunits and the phosphoinositide 3-kinase pathway. Mol. Endocrinol. 15, 553–564 [DOI] [PubMed] [Google Scholar]
- 19. Hu A., Josephson M. B., Diener B. L., Nino G., Xu S., Paranjape C., Orange J. S., Grunstein M. M. (2013) Pro-asthmatic cytokines regulate unliganded and ligand-dependent glucocorticoid receptor signaling in airway smooth muscle. PLoS ONE 8, e60452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eickelberg O., Roth M., Lörx R., Bruce V., Rüdiger J., Johnson M., Block L. H. (1999) Ligand-independent activation of the glucocorticoid receptor by β2-adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J. Biol. Chem. 274, 1005–1010 [DOI] [PubMed] [Google Scholar]
- 21. Mathews W. C., Agmas W., Cachay E. (2011) Comparative accuracy of anal and cervical cytology in screening for moderate to severe dysplasia by magnification guided punch biopsy, a meta-analysis. PLoS ONE 6, e24946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Galliher-Beckley A. J., Williams J. G., Cidlowski J. A. (2011) Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol. Cell. Biol. 31, 4663–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brown D. A., London E. (1998) Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14, 111–136 [DOI] [PubMed] [Google Scholar]
- 24. Liposits Z., Bohn M. C. (1993) Association of glucocorticoid receptor immunoreactivity with cell membrane and transport vesicles in hippocampal and hypothalamic neurons of the rat. J. Neurosci. Res. 35, 14–19 [DOI] [PubMed] [Google Scholar]
- 25. Gametchu B., Watson C. S., Wu S. (1993) Use of receptor antibodies to demonstrate membrane glucocorticoid receptor in cells from human leukemic patients. FASEB J. 7, 1283–1292 [DOI] [PubMed] [Google Scholar]
- 26. Razandi M., Oh P., Pedram A., Schnitzer J., Levin E. R. (2002) ERs associate with and regulate the production of caveolin, implications for signaling and cellular actions. Mol. Endocrinol. 16, 100–115 [DOI] [PubMed] [Google Scholar]
- 27. Matthews L., Berry A., Ohanian V., Ohanian J., Garside H., Ray D. (2008) Caveolin mediates rapid glucocorticoid effects and couples glucocorticoid action to the antiproliferative program. Mol. Endocrinol. 22, 1320–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gametchu B., Chen F., Sackey F., Powell C., Watson C. S. (1999) Plasma membrane-resident glucocorticoid receptors in rodent lymphoma and human leukemia models. Steroids 64, 107–119 [DOI] [PubMed] [Google Scholar]
- 29. Bartholome B., Spies C. M., Gaber T., Schuchmann S., Berki T., Kunkel D., Bienert M., Radbruch A., Burmester G. R., Lauster R., Scheffold A., Buttgereit F. (2004) Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J. 18, 70–80 [DOI] [PubMed] [Google Scholar]
- 30. Jain S., Li Y., Kumar A., Sehgal P. B. (2005) Transcriptional signaling from membrane raft-associated glucocorticoid receptor. Biochem. Biophys. Res. Commun. 336, 3–8 [DOI] [PubMed] [Google Scholar]
- 31. Samarasinghe R. A., Di Maio R., Volonte D., Galbiati F., Lewis M., Romero G., DeFranco D. B. (2011) Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 108, 16657–16662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McGillivray S. M., Thackray V. G., Coss D., Mellon P. L. (2007) Activin and glucocorticoids synergistically activate follicle-stimulating hormone β-subunit gene expression in the immortalized LβT2 gonadotrope cell line. Endocrinology 148, 762–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lannan E. A., Galliher-Beckley A. J., Scoltock A. B., Cidlowski J. A. (2012) Proinflammatory actions of glucocorticoids. Glucocorticoids and TNFα coregulate gene expression in vitro and in vivo. Endocrinology 153, 3701–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fernandes P. A., Bowen R. A., Sawyer H. R., Nett T. M., Gorell T. A. (1989) Concentration of receptors for estradiol and progesterone in canine endometrium during estrus and diestrus. Am. J. Vet. Res. 50, 64–67 [PubMed] [Google Scholar]
- 35. Mellon P. L., Windle J. J, Goldsmith P. C., Padula C. A., Roberts J. L., Weiner R. I. (1990) Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5, 1–10 [DOI] [PubMed] [Google Scholar]
- 36. Navratil A. M., Bliss S. P, Berghorn K. A., Haughian J. M., Farmerie T. A., Graham J. K., Clay C. M., Roberson M. S. (2003) Constitutive localization of the gonadotropin-releasing hormone (GnRH) receptor to low density membrane microdomains is necessary for GnRH signaling to ERK. J. Biol. Chem. 278, 31593–31602 [DOI] [PubMed] [Google Scholar]
- 37. Bliss S. P., Navratil A. M., Breed M., Skinner D. C., Clay C. M., Roberson M. S. (2007) Signaling complexes associated with the type I gonadotropin-releasing hormone (GnRH) receptor. Colocalization of extracellularly regulated kinase 2 and GnRH receptor within membrane rafts. Mol. Endocrinol. 21, 538–549 [DOI] [PubMed] [Google Scholar]
- 38. Turgeon J. L., Kimura Y., Waring D. W., Mellon P. L. (1996) Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol. Endocrinol. 10, 439–450 [DOI] [PubMed] [Google Scholar]
- 39. Manders E., Tyberghein J. (1993) The effects of ventilation tube placement on hearing, speech, language, cognition and behaviour. Acta Otorhinolaryngol. Belg. 47, 27–32 [PubMed] [Google Scholar]
- 40. Lafont F., Simons K. (2001) Raft-partitioning of the ubiquitin ligases Cbl and Nedd4 upon IgE-triggered cell signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 3180–3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nystrom F. H., Chen H., Cong L. N., Li Y., Quon M. J. (1999) Caveolin-1 interacts with the insulin receptor and can differentially modulate insulin signalling in transfected COS-7 cells and rat adipose tissue. Mol. Endocrinol. 13, 2013–2024 [DOI] [PubMed] [Google Scholar]
- 43. Wang J., Roy S. K. (2004) Growth differentiation factor-9 and stem cell factor promote primordial follicle formation in the hamster. Modulation by follicle-stimulating hormone. Biol. Reprod 70, 577–585 [DOI] [PubMed] [Google Scholar]
- 44. Arteaga M. F., Coric T., Straub C., Canessa C. M. (2008) A brain-specific SGK1 splice isoform regulates expression of ASIC1 in neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 4459–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. U. M., Shen L., Oshida T., Miyauchi J., Yamada M., Miyashita T. (2004) Identification of novel direct transcriptional targets of glucocorticoid receptor. Leukemia 18, 1850–1856 [DOI] [PubMed] [Google Scholar]
- 46. Ayroldi E., Riccardi C. (2009) Glucocorticoid-induced leucine zipper (GILZ). A new important mediator of glucocorticoid action. FASEB J. 23, 3649–3658 [DOI] [PubMed] [Google Scholar]
- 47. Klengel T., Mehta D., Anacker C., Rex-Haffner M., Pruessner J. C., Pariante C. M., Pace T. W., Mercer K. B., Mayberg H. S., Bradley B., Nemeroff C. B., Holsboer F., Heim C. M., Ressler K. J., Rein T., Binder E. B. (2013) Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 16, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grafer C. M., Thomas R., Lambrakos L., Montoya I., White S., Halvorson L. M. (2009) GnRH stimulates expression of PACAP in the pituitary gonadotropes via both the PKA and PKC signaling systems. Mol. Endocrinol. 23, 1022–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grossmann C., Husse B., Mildenberger S., Schreier B., Schuman K., Gekle M. (2010) Colocalization of mineralocorticoid and EGF receptor at the plasma membrane. Biochim. Biophys. Acta 1803, 584–590 [DOI] [PubMed] [Google Scholar]
- 50. Asselin-Labat M. L., David M., Biola-Vidamment A., Lecoeuche D., Zennaro M. C., Bertoglio J., Pallardy M. (2004) GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal-induced apoptosis. Blood 104, 215–223 [DOI] [PubMed] [Google Scholar]
- 51. Wu J., Bresnick E. H. (2007) Glucocorticoid and growth factor synergism requirement for Notch4 chromatin domain activation. Mol. Cell. Biol. 27, 2411–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vasudevan N., Koibuchi N., Chin W. W., Pfaff D. W. (2001) Differential crosstalk between estrogen receptor α (ERα) and ERβ and the thyroid hormone receptor isoforms results in flexible regulation of the consensus ERE. Brain Res. Mol. Brain Res. 95, 9–17 [DOI] [PubMed] [Google Scholar]
- 53. Barr F. D., Krohmer L. J., Hamilton J. W., Sheldon L. A. (2009) Disruption of histone modification and CARM1 recruitment by arsenic represses transcription at glucocorticoid receptor-regulated promoters. PLoS ONE 4, e6766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ronacher K., Hadley K., Avenant C., Stubsrud E., Simons S. S, Jr., Louw A., Hapgood J. P. (2009) Ligand-selective transactivation and transrepression via the glucocorticoid receptor. Role of cofactor interaction. Mol. Cell. Endocrinol. 299, 219–231 [DOI] [PubMed] [Google Scholar]
- 55. Li L. B., Leung D. Y., Martin R. J., Goleva E. (2010) Inhibition of histone deacetylase 2 expression by elevated glucocorticoid receptor β in steroid-resistant asthma. Am. J. Respir. Crit. Care Med. 182, 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lister K., Autelitano D. J., Jenkins A., Hannan R. D., Sheppard K. E. (2006) Cross talk between corticosteroids and α-adrenergic signalling augments cardiomyocyte hypertrophy. A possible role for SGK1. Cardiovasc. Res. 70, 555–565 [DOI] [PubMed] [Google Scholar]
- 57. Rogatsky I., Trowbridge J. M., Garabedian M. J. (1997) Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol. Cell. Biol. 17, 3181–3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matthews L., Johnson J., Berry A., Trebble P., Cookson A., Spiller D., Rivers C., Norman M., White M., Ray D. (2011) Cell cycle phase regulates glucocorticoid receptor function. PLoS ONE 6, e22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen W., Dang T., Blind R. D., Wang Z., Cavasotto C. N., Hittelman A. B., Rogatsky I., Logan S. K., Garabedian M. J. (2008) Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol. Endocrinol. 22, 1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang Z., Chen W., Kono E., Dang T., Garabedian M. J. (2007) Modulation of glucocorticoid receptor phosphorylation and transcriptional activity by a C-terminal-associated protein phosphatase. Mol. Endocrinol. 21, 625–634 [DOI] [PubMed] [Google Scholar]
- 61. Hsu S. C., DeFranco D. B. (1995) Selectivity of cell cycle regulation of glucocorticoid receptor function. J. Biol. Chem. 270, 3359–3364 [DOI] [PubMed] [Google Scholar]
- 62. Hsu S. C., Qi M., DeFranco D. B. (1992) Cell cycle regulation of glucocorticoid receptor function. EMBO J. 11, 3457–3468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Abel G. A., Wochnik G. M., Rüegg J., Rouyer A., Holsboer F., Rein T. (2002) Activity of the GR in G2 and mitosis. Mol. Endocrinol. 16, 1352–1366 [DOI] [PubMed] [Google Scholar]
- 64. Feng J., Lawson M. A., Melamed P. (2008) A proteomic comparison of immature and mature mouse gonadotrophs reveals novel differentially expressed nuclear proteins that regulate gonadotropin gene transcription and RNA splicing. Biol. Reprod. 79, 546–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Larivière S., Garrel G., Simon V., Soh J. W., Laverrière J. N., Counis R., Cohen-Tannoudji J. (2007) Gonadotropin-releasing hormone couples to 3′,5′-cyclic adenosine-5′-monophosphate pathway through novel protein kinase Cδ and -ϵ in LβT2 gonadotrope cells. Endocrinology 148, 1099–1107 [DOI] [PubMed] [Google Scholar]
- 66. Anacker C., Cattaneo A., Luoni A., Musaelyan K., Zunszain P. A., Milanesi E., Rybka J., Berry A., Cirulli F., Thuret S., Price J., Riva M. A., Gennarelli M., Pariante C. M. (2013) Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology 38, 872–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rees D. A., Lewis M. D., Lewis B. M., Smith P. J., Scanlon M. F., Ham J. (2002) Adenosine-regulated cell proliferation in pituitary folliculostellate and endocrine cells. Differential roles for the A(1) and A(2B) adenosine receptors. Endocrinology 143, 2427–2436 [DOI] [PubMed] [Google Scholar]
- 68. Langlais D., Couture C., Kmita M., Drouin J. (2013) Adult pituitary cell maintenance. Lineage-specific contribution of self-duplication. Mol. Endocrinol. 27, 1103–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jayakody S. A., Andoniadou C. L., Gaston-Massuet C., Signore M., Cariboni A., Bouloux P. M., Le Tissier P., Pevny L. H., Dattani M. T., Martinez-Barbera J. P. (2012) SOX2 regulates the hypothalamic-pituitary axis at multiple levels. J. Clin. Invest. 122, 3635–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]