

Abstract
The myelodysplastic syndrome (MDS) is a clonal hematologic disorder that frequently evolves to acute myeloid leukemia (AML). Its pathogenesis remains unclear, but mutations in epigenetic modifiers are common and the disease often responds to DNA methylation inhibitors. We analyzed DNA methylation in the bone marrow and spleen in two mouse models of MDS/AML, the NUP98-HOXD13 (NHD13) mouse and the RUNX1 mutant mouse model. Methylation array analysis showed an average of 512/3445 (14.9%) genes hypermethylated in NHD13 MDS, and 331 (9.6%) genes hypermethylated in RUNX1 MDS. Thirty-two percent of genes in common between the two models (2/3 NHD13 mice and 2/3 RUNX1 mice) were also hypermethylated in at least two of 19 human MDS samples. Detailed analysis of 41 genes in mice showed progressive drift in DNA methylation from young to old normal bone marrow and spleen; to MDS, where we detected accelerated age-related methylation; and finally to AML, which markedly extends DNA methylation abnormalities. Most of these genes showed similar patterns in human MDS and AML. Repeat element hypomethylation was rare in MDS but marked the transition to AML in some cases. Our data show consistency in patterns of aberrant DNA methylation in human and mouse MDS and suggest that epigenetically, MDS displays an accelerated aging phenotype.
The myelodysplastic syndrome (MDS) is a heterogeneous group of clonal stem-cell disorders characterized by ineffective hematopoiesis and susceptibility to leukemic transformation to acute myeloid leukemia (AML). A number of chromosomal abnormalities, including deletions, amplifications, inversions, and translocations, have been identified in the malignant cells of patients with MDS, the most common of which are deletions of chromosomes 5q, 7q, and 20q (Haase et al. 2007). Mutations have been detected in genes that encode signal transduction proteins (BRAF, CBL, GNAS, JAK2, KRAS, NRAS, and PTPN11), transcription factors and cofactors (ETV6, NPM1, RUNX1, and TP53), cell cycle regulators (CDKN2A and PTEN) (Bejar et al. 2011), components of the RNA splicing machinery (PRPF40B, SF1, SF3A1, SF3B1, SRSF2, U2AF1, U2AF2, and ZRSR2) (Yoshida et al. 2011), and, most notably, epigenetic modifiers (ASXL1, DNMT3A, EZH2, IDH1/2, and TET2) (Shih et al. 2012). TET2 is the most frequent gene abnormality in MDS (Kosmider et al. 2009; Langemeijer et al. 2009), and DNMT3A mutation is a very early genetic event in MDS (Walter et al. 2011). These mutations in epigenetic modifiers indicate that epigenetic changes contribute to MDS pathogenesis (Graubert and Walter 2011; Nikoloski et al. 2012).
DNA methylation is a well-established epigenetic mechanism that regulates gene transcription through modification of cytosines (usually but not always in the context of CpG dinucleotides) (Xie et al. 2012; Varley et al. 2013) and is frequently aberrant in human cancers. Hypermethylation at promoter CpG sites is common in human MDS, involving genes such as CDH1, CDH13, CDKN2B, PDLIM4, and PGR, and is a poor prognostic factor (Aggerholm et al. 2006; Issa 2010; Shen et al. 2010). Epigenetic therapy using hypomethylating agents such as azacitidine and decitabine has demonstrated clinical effectiveness in this disease. Both drugs are approved by the US Food and Drug Administration for the treatment of MDS (Kantarjian et al. 2006; Kantarjian et al. 2007; Bryan et al. 2010).
Recently, two mouse models for MDS and MDS/AML were established. In the first model, an NUP98-HOXD13 (NHD13) fusion transgene was inserted in the germline of C57BL/6 and FVB/N mice, using vav regulatory elements to direct expression specifically in hematopoietic tissues. NHD13 transgenic mice faithfully recapitulate all the key features of MDS, including peripheral blood cytopenias, bone marrow dysplasia and increased apoptosis, and transformation to acute leukemia between 4 and 14 mo of age (Lin et al. 2005; Slape et al. 2008). A second model uses bone marrow transplantation (BMT) of cells transduced with RUNX1 mutants frequently found in patients with MDS and MDS/AML. A C-terminal truncation mutation of RUNX1 caused by a frame-shift (S291fsX300; hereafter S291fs) induced pancytopenia associated with dysplasia in the erythroid lineage. A RUNX1 point mutation in the Runt homology domain (RHD) (D171N) frequently induced hepatosplenomegaly and leukocytosis associated with marked myeloid dysplasia (Watanabe-Okochi et al. 2008). Cooperating genetic or epigenetic changes in these mouse models are incompletely understood.
Both models mimic aspects of human MDS/AML, including an initial dyspoietic phase and progression to AML. The clinical course of the disease in these mice suggests that they provide a useful model for the human disease. We therefore studied DNA methylation of thousands of CpG sites using CpG island profiling in mouse spleen and bone marrow samples from normal and MDS model mice. We find a high rate of epigenetic deregulation in each model, with a significant number of shared events between the models and with human MDS. Epigenetically, MDS appears to be part of a continuum of changes that start in aging and culminate in AML.
Results
DNA methylation analysis in mouse models of MDS
To study the epigenetic evolution of normal cells to a malignant state in hematopoiesis, we used a total of 122 mouse samples. We summarize sample information such as the genotypes and tissue types analyzed in Supplemental Table S1. We first examined three promoter CpG islands (Cdh13, Esr1, and Pgr) that had previously been reported to be hypermethylated in human MDS (Shen et al. 2010). In all three, we found marked increases in methylation from normal to MDS to AML by using pyrosequencing analysis (Supplemental Fig. S1; Supplemental Table S2). Based on these candidate gene analyses, these MDS mouse models mimic aspects of human MDS methylation patterns, and were therefore studied in more detail.
We performed methylated CpG island amplification microarrays (MCAM) to study DNA methylation of thousands of genes (Estecio et al. 2007b; Yamamoto et al. 2012). Using normal spleen or normal bone marrow (transfected with empty vector) as age-matched control samples, we selected significantly altered autosomal genes based on a signal fold change greater than two (Fig. 1A). We then performed unsupervised hierarchical cluster analysis of the data derived from six mice using variable probes that were changed at least in one of six samples. The samples clustered into two major groups based on the models, as shown in Figure 1B. We studied three distinct NHD13 mice at the MDS stage. Out of 3445 detectable genes, we found that 497, 490, and 548 were hypermethylated (average, 512; 14.9%), and 190, 204, and 236 were hypomethylated (average, 210; 6.1%) (Supplemental Table S3). Figure 1C shows (area-proportional) Venn diagrams indicating that 494 of 3445 detectable genes were hypermethylated in at least 2/3 NHD13 MDS samples, while 199 genes were hypomethylated.
Figure 1.
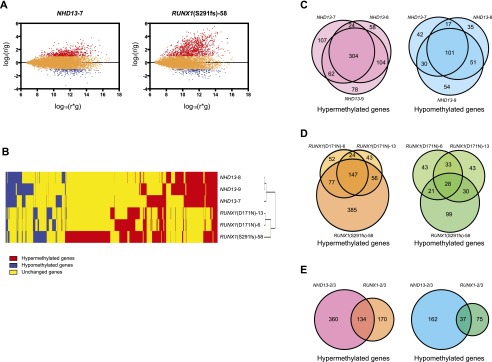
MCAM analysis in mouse MDS models. (A) R-I plot of the probes with FDR at 5% and a fold change greater than two for MCAM. An R-I plot displays the log2(R/G) ratio for each element on the array as a function of the log10(R × G) product intensities and can reveal systematic intensity-dependent effects in the measured log2 (ratio) values. The red and blue spots indicate probes hypermethylated and hypomethylated in diseased samples, respectively. (B) Hierarchical clustering analysis of mouse MCAM. Heat-map analysis showing the MCAM results in six model mice. Red, yellow, and blue correspond to hypermethylated, nonchanged, and hypomethylated loci, respectively. For clarity we excluded probes that were unchanged in all samples. (C) Venn diagram of individual differences of hyper- and hypomethylated genes in NHD13 mice analyzed by MCAM. Each circle represents one individual. (D) Venn diagram of mutational differences of hyper- and hypomethylated genes in RUNX1 mice analyzed by MCAM. Each circle represents one individual. (E) Venn diagram of hyper- and hypomethylated genes overlapped between NHD13 and RUNX1 mice analyzed by MCAM. Each circle represents a genotype.
Next, we studied the RUNX1 model. A total of 300, 270, and 665 genes were hypermethylated, and 125, 134, and 178 genes were hypomethylated in D171N (two individual mice) and S291fs, respectively (Supplemental Table S3). Figure 1D shows the overlapped genes among three individual mice in each model. A total of 147 hypermethylated genes and 28 hypomethylated genes overlapped in the RUNX1 models. Finally, we examined gene overlap between the models. A total of 134 genes were hypermethylated and 37 genes hypomethylated in at least 2/3 NHD13 and 2/3 RUNX1 mice (Fig. 1E). These included several cancer relevant genes such as Gata5, Pax6, Sfrp1, and Sox9 (Toyota et al. 1999; Suzuki et al. 2002; Akiyama et al. 2003; The Cancer Genome Atlas Network 2012). The names of the genes are shown in Supplemental Table S4.
Functional pathways affected
We examined the potential function of genes that showed common hypermethylation and hypomethylation in these MDS models. Transcriptional network and pathway analysis was performed on pairwise comparisons between hypermethylated genes and detectable genes, and between hypomethylated genes and detectable genes, respectively. Supplemental Table S5 shows the top five enriched categories for commonly hypermethylated genes in the mouse models. Hypermethylated genes showed significant enrichment for various developmental processes. The enriched molecular and cellular functions included cell development, signaling, growth, and death. Enriched canonical pathways included Wnt/beta-catenin signaling. The list of hypomethylated genes was too short to analyze.
Age-related methylation drift in normal and malignant hematopoietic cells
To validate and quantitatively extend the array results, pyrosequencing analysis was used to study the methylation of 42 genes. Of these, 15 genes (Asic2, Barhl2, Cdx2, Colec12, Crb3, Gng11, Hoxa2, Hoxc12, Hs3st2, Kcnma1, Ky, Lrig3, Syt10, Tlx3, and Tmem229a) were selected based solely on frequent hypermethylation detected by MCAM. Seven genes (Bmi1, Elovl2, Fbn1, Hand2, Klf14, Pou4f2, and Prdm5) were selected based on prior publications showing age-related methylation (Maegawa et al. 2010; Hannum et al. 2013). Three genes (Cdh13, Esr1, and Pgr) were selected from hypermethylated genes in human MDS, as mentioned earlier (Shen et al. 2010). In addition, we selected three genes (Espnl, Lims2, and Olfr368) based on MCAM data showing hypomethylation in MDS. Finally, 14 genes (Cdh4, Cdkn1c, En2, Gata5, Gpr37, Grm7, Lrrtm1, Nptx2, Pax3, Pcdh10, Sox11, Tmeff2, Twist2, and Wt1) were selected based on MCAM data and also prior information on hypermethylation in mouse aging and/or human leukemias (Kay et al. 1997; Toyota et al. 2001; Shen et al. 2003; Rush et al. 2004; Raval et al. 2005; Ying et al. 2007; Kroeger et al. 2008; Bennett et al. 2009; Tong et al. 2010; Cosialls et al. 2012; Thathia et al. 2012). Of all the selected genes, one (Cdx2) was a false positive (data not shown) and excluded from further analysis.
Quantitative analysis confirmed minimal (4.0%–15.2%) methylation in normal bone marrow and spleen in 33 of 38 genes (Fig. 2A; Supplemental Figs. S1, S2). Five genes (Esr1, Cdkn1c, Klf14, Hoxa2, and Elovl2) showed intermediate methylation percentages in normal tissues. Esr1 corresponds to an exon 2 CpG island, while the Cdkn1c and Klf14 assays are in imprinted CpG islands (Bhogal et al. 2004; Parker-Katiraee et al. 2007; Maegawa et al. 2010). Hoxa2 is ∼50% methylated in normal tissues and could be an imprinted gene (Luedi et al. 2007). The Elovl2 assay is located outside of the CpG island (394 bp upstream of the TSS). We first studied age as a parameter that could influence DNA methylation in normal tissues and focused on the 38 genes hypermethylated in MDS/AML. We found linear hypermethylation with age (Fig. 2B; Supplemental Fig. S3; Supplemental Table S6) for 31/38 genes in normal bone marrow (R > 0.4 and P < 0.05), 32/38 genes in normal spleen (R > 0.4 and P < 0.05) and 36/38 genes in either bone marrow (four), spleen (five), or both (27). Both tissues had a lower degree of methylation change than previously seen in mouse intestine (Maegawa et al. 2010). For bone marrow, there were only minor differences between the C57BL/6 strain (n = 24) and the FVB/N strain (n = 10). For spleen, we only studied C57BL/6 mice (n = 34). Of note, all 15 genes selected solely because of frequent hypermethylation detected by MCAM showed age-related methylation drift. Thus, most genes frequently hypermethylated in mouse MDS also showed age-related methylation in normal hematopoietic tissues.
Figure 2.

DNA methylation profiles by bisulfite pyrosequencing analysis. (A) DNA methylation analysis in MDS mice. The percentages of methylated cytosines in the samples as obtained from pyrosequencing analysis. Each dot corresponds to one animal. We show here data on Grm7, Hand2, Hoxc12, Klf14, and Sox11. The other 33 hypermethylated genes are shown in Supplemental Figures S1 and S2. The averaged data were derived from the methylation data of 38 genes prone to hypermethylation. The bar in the graphs represents the median. (B) Methylation profiles of hypermethylated genes with age in mouse bone marrow and spleen. Association of the averaged percentages of methylated cytosines in the samples as obtained from pyrosequencing (y-axis) with age (x-axis) for significant genes.
Next, we studied these 38 genes in MDS samples from spleen and bone marrow and confirmed that all had higher methylation compared with normal samples (Fig. 2A; Supplemental Figs. S1, S2; Supplemental Table S2). When we averaged all 38 genes, the methylation percentages were 30.4% and 28.8% in RUNX1-spleen and NHD13-bone marrow, respectively, compared to 16.8% and 11.8% in normal spleen and bone marrow (P < 0.001 for both). There was also a progressive increase in hypermethylation in NHD13-AML samples transformed from MDS for all genes in spleen compared with the methylation percentage of normal and NHD13-MDS samples. We found a 20.6% higher-average methylation for the 38 genes in AML-spleen (51.0%) compared to MDS-spleen (30.4%). Most of the genes followed an increasing methylation pattern in a phased manner from normal young (<1 y.o.), normal middle age (>1 y.o., <2 y.o.), and normal old (>2 y.o.), to MDS and finally to AML samples (Fig. 2A; Supplemental Figs. S1, S2; Supplemental Table S2). Hence, we found accelerated age-related hypermethylation in MDS progression, which is similar to what was previously reported in human colorectal cancer tumorigenesis (Toyota and Issa 1999; Issa et al. 2001).
In addition to hypermethylation, we analyzed hypomethylated genes by pyrosequencing analysis. We selected three genes (Espnl, Lims2, and Olfr368) that showed hypomethylation in MDS based on MCAM analysis in mice (Lims2 and Olfr368) or humans. Espnl was not detectable in mouse MCAM and was derived from human MCAM data (see below). All three were confirmed as hypomethylated in mouse MDS/AML (Supplemental Fig. S2; Supplemental Table S2), and all three were hypomethylated in aging spleen but not in aging bone marrow (Supplemental Fig. S3; Supplemental Table S6).
As a measure of CpG methylation specificity provided by the mutations of RUNX1 and truncation of NHD13, we performed a two-dimensional hierarchical cluster analysis using the methylation percentage of the 38 genes studied by pyrosequencing. This analysis of DNA methylation divided all tissue samples into four major clusters (1 to 4) (Supplemental Fig. S4). Cluster 1 contained 50/68 (74%) normal samples and 6/28 (21%) MDS samples. Clusters 2 and 3 contained mostly MDS, while cluster 4 contained mostly AML. When we divided samples into each tissue type, hierarchical clustering also led to an almost perfect segregation of normal tissues depending on age (Supplemental Fig. S5) and tumors based on their diagnostic criteria (Fig. 3). In spleen, we found three major clusters: Cluster 1 includes all normal samples, cluster 2 includes 53% of MDS, and cluster 3 includes 83% of AML (Fig. 3B). The ordering of samples by clustering analysis using the 38 genes closely reflected the number of methylated genes detected by MCAM (Fig. 3), thus validating the array results.
Figure 3.

Hierarchical clustering analysis of methylation markers in mouse. (A) Hierarchical clustering analysis in bone marrow. Green to red cells indicate the range of methylation percentage from 0 to 95.3. The color codes for mouse strain, tissues, age, and diagnosis are listed on the left side. Orange arrowheads indicate samples for MCAM with number of methylated genes by MCAM. (B) Hierarchical clustering analysis in spleen.
Age-related drift and human MDS methylation
The candidate gene analysis suggested conservation between hypermethylated genes in human and mouse MDS. To study this more formally, we used MCAM analysis to study 19 human MDS patients (Supplemental Table S7). Out of 9645 detectable genes, we identified hypermethylated genes ranging from 205 to 1677 genes in all 19 patients (Supplemental Table S7). Unsupervised hierarchical clustering analysis identified two main clusters: one with high levels of DNA hypermethylation (designated CIMP-positive), and one with low levels (designated CIMP-negative) (Fig. 4A). The number of aberrantly hypermethylated genes was significantly higher in CIMP-positive cases (average, 958 genes, 9.9%, compared to 403 genes, 4.2%; P < 0.0001). CIMP-positive cases also had a higher ratio of the number of hypermethylated genes to the number of hypomethylated genes (Supplemental Fig. S6A; Supplemental Table S7). We compared this CIMP classification to methylation levels of nine genes (10 genomic regions), including CDH1, CDH13, CDKN2B, ESR1, NPM2, OLIG2, OSCP1, PDLIM4, and PGR, previously studied by pyrosequencing analysis (Shen et al. 2010) in these cases. Most of the genes showed higher methylation in CIMP-positive cases (Supplemental Fig. S6B). The averaged value of all 10 genomic regions was significantly higher in CIMP-positive patients compared with CIMP-negative patients (8.8 ± 2.5% vs. 21.7 ± 4.6%; P < 0.0001) (Supplemental Fig. S6B). We then looked for common hypermethylation events and found that 973 of 9645 genes (10.1%) were hypermethylated in at least three of seven CIMP-positive patients compared with controls (Supplemental Table S8); these included genes involved in cell signaling, differentiation, and cancer development (Supplemental Table S9). Two hundred ninety-four of 9645 (3.0%) of genes showed MDS specific hypomethylation. In hypomethylated genes, enriched biological functions include cancer and developmental disorders (Supplemental Table S10).
Figure 4.

DNA methylation analyses in humans. (A) Hierarchical clustering analysis of human MCAM. Heat-map analysis showing the MCAM results in 19 MDS patients. Red, yellow, and blue correspond to hypermethylated, nonchanged, and hypomethylated loci, respectively. All probes that were unchanged in all samples were excluded from the analysis. The bar graph on the left shows the methylation percentage of LINE-1 by pyrosequencing analysis. Red-colored boxes on the right side represent the CIMP-positive case defined by MCAM assay. (B) DNA methylation profiles by DREAM analysis in human cord blood, adult blood, MDS, and AML patients. The percentages of methylated cytosines in the samples as obtained from DREAM analysis. Each dot corresponds to one individual. We show here data on HOXC12, SOX11, and ESPNL. The averaged data were derived from the methylation data of 26 hypermethylated genes listed in Supplemental Table S14. The bar in the graphs represents the median.
We next cross-referenced methylated genes in human MDS and age-related genes reported in previous studies using whole blood. Out of 6294 detectable genes in our MCAM array, age-related hypermethylation was found in 573 genes (9.1%) by Horvath et al. (2012), 248 genes (3.9%) by Bell et al. (2012), and 30 (0.35%) by Hannum et al. (2013). This wide range likely reflects different methods and criteria for selecting genes. Nevertheless, age-related methylated genes were significantly enriched among MDS methylated genes (2.1-fold, P < 0.0001 for Horvath et al. [2012]; 2.3-fold, P < 0.0001 for Bell et al. [2012]; 2.1-fold, P = 0.047 for Hannum et al. [2013]). There were relatively few hypomethylated genes in all these data sets precluding a similar analysis. Thus, our data show a significant link between age-related methylation drift and MDS hypermethylation in humans.
Human–mouse MDS comparison
Because of a generally poor sequence conservation between mice and humans at promoter CpG islands (Antequera and Bird 1993; Matsuo et al. 1993), only 2344 genes could be compared directly (detectable on both human and mouse arrays). The accuracy of the comparison is also weakened by the fact that MCAM has limited precision (70%–80% for random loci). We therefore focused on common methylation events. Of 346 genes hypermethylated in NHD13 (2/3 mice) and detectable in the human arrays, 78 (23%) were also hypermethylated in human MDS (at least 2/19). When compared to the RUNX1 models data, 77 of 221 (35%) genes methylated in 2/3 RUNX1 mice were also methylated in human MDS. Among 96 genes methylated in 2/3-NHD13 and 2/3-RUNX1 mice and detectable in human arrays, 31 (32%) were also methylated in human MDS (2/19) (Supplemental Table S11). In a search for driver events, we cross-referenced this list to the Cancer Gene Census (http://www.sanger.ac.uk/genetics/CGP/Census/) in COSMIC databases of cancer genes (Futreal et al. 2004) and identified several hits, including CDH11, PAX3, TLX3, and WT1. Thus there is partial conservation in MDS specific methylation between mice and humans.
Acceleration of age-related drift in human MDS/AML
To increase precision of the mouse–human comparison and to confirm human aging drift and acceleration of methylation from MDS to AML, we analyzed an unpublished data set of Digital Restriction Enzyme Analysis of Methylation (DREAM) in human cord blood (n = 5), adult blood (n = 13), MDS (n = 20), and AML patients (n = 94). DREAM has a high degree of quantitative accuracy thanks to deep sequencing (Challen et al. 2012; Jelinek et al. 2012). We focused on the 41 genes validated in the mouse model. Of these, 27/41 human homologous genes were detectable by DREAM at 118 SmaI sites (Supplemental Tables S12, S13). We averaged the methylation status of SmaI sites (one to 11 sites as listed in Supplemental Table S14) located in the promoter regions of each gene. Of the 27, 14 genes (51.9%) showed aging drift as hyper- or hypomethylation with age (cord blood to adult blood), and 21 genes (77.8%) showed methylation acceleration from MDS to AML as shown in Figure 4B, Supplemental Figure S7, and Supplemental Table S14. Eleven genes showed both features. Thus, the majority of genes that show validated epigenetic drift in mouse models of MDS/AML also show this in human MDS/AML.
Repetitive element methylation
In cancer, DNA hypomethylation typically occurs at repetitive sequences residing in satellite or pericentromeric regions and can result in the reactivation of retrotransposons, leading to disruption of normal gene structure and function (Jones and Baylin 2002; Issa 2004; Estecio et al. 2007a). We studied global DNA methylation using pyrosequencing analysis of multiple DNA repetitive elements, such as long interspersed nuclear elements (LINE-1), major satellite repeats, and short interspersed nuclear elements (SINE B1) in aging normal samples and MDS model samples. In spleen, we detected no significant change with age in LINE-1 or SINE B1 methylation, but major satellite methylation decreased from 74.3% to 68.4% in old mice (P = 0.0006). This hypomethylation was not seen in bone marrow (Fig. 5A).
Figure 5.

Methylation analysis of repetitive elements in mouse. (A) Methylation profiles of LINE-1, SINE B1, and major satellite DNA by pyrosequencing analysis. (B) Association of the methylation percentages between LINE-1 (y-axis), SINE B1 (x-axis), and major satellite DNA (x-axis) in all samples. Sample codes are on the right. The Spearman test was used to determine correlations, with significance set at P < 0.05. R represents a measure of the linear relationship between two variables, and varies from −1 to +1.
In MDS mouse models, repetitive element methylation was generally unchanged or increased, compared with old mice. In contrast, the NHD13-AML samples showed significant hypomethylation in LINE-1 (35.5%–31.7%, P = 0.012) and SINE B1 (38.5%–30.6%, P < 0.0001), compared with normal spleen samples (Fig. 5A). The methylation level of LINE-1 and SINE B1 correlated well (P < 0.0001, R = 0.63). We also found a significant but weaker association between the methylation level of LINE-1 and major satellite status (P = 0.006, R = 0.25) (Fig. 5B).
Relation to polycomb targets and retrotransposon density
DNA hypermethylation in aging and cancer is more frequent among genes targeted by polycomb group (PcG) proteins in embryonic stem (ES) cells (Boyer et al. 2006; Lee et al. 2006; Ohm et al. 2007; Rauch et al. 2007; Schlesinger et al. 2007; Widschwendter et al. 2007; Maegawa et al. 2010; Teschendorff et al. 2010) and among genes with few retrotransposons around their promoter (Estecio et al. 2010). To study this for MDS, we first evaluated whether PcG targets in ES cells are enriched in the group of genes showing MDS-specific methylation in mouse and human, by using databases of PcG occupancy in mouse ES cells (Boyer et al. 2006; Lee et al. 2006). There was 1.5- and 4.7-fold enrichment in SUZ12 targets on average in NHD13 and RUNX1 MDS model-specific hypermethylation compared with nontargets among detectable genes (P < 0.0001, χ2 test). Similarly, EED, RNF2, and PHC1 targets are enriched among hypermethylated genes in each MDS model (Supplemental Table S15). There was no PcG target enrichment among hypomethylated genes (Supplemental Table S15). Similarly, PcG targets in human ES cells are also prone to MDS-specific hypermethylation compared with all detectable genes. SUZ12 and EED targets are associated with a 4.2- and 4.3-fold increase, respectively (P < 0.0001, χ2 test) (Supplemental Table S15).
We next looked at the presence of SINE B1 elements around gene promoters (Estecio et al. 2010). We found that genomic regions hypermethylated in NHD13 and RUNX1 models were less frequently associated with the presence of SINE B1 elements than a set of control genes that have promoter CpG islands (Supplemental Fig. S8). Similar to other human cancers previously reported (Estecio et al. 2010), we also found the depleted presence of SINE B1 around gene promoters in human MDS (Supplemental Fig. S8).
Discussion
Here, we report on epigenetic analysis of mouse models of MDS and show extensive DNA methylation abnormalities that overlap with those seen in human MDS. We further demonstrate that methylation changes arise as a function of age in normal hematopoiesis and are accelerated in MDS and at the transition from MDS to AML.
We analyzed two different MDS mouse models mimicking human MDS and found similar patterns in both, with appreciable overlap with the aberrant DNA methylation phenotype in human MDS. Thus, these oncogene-driven models parallel the human disease both genetically and epigenetically and are worth studying further from a therapeutic perspective, for example, to optimize established treatments and discover new drugs (e.g., epigenetic drugs).
We focused on DNA methylation status because aberrant DNA methylation is believed to be a hallmark of preneoplastic and neoplastic conditions (Jones and Baylin 2002, 2007). Hypermethylation of promoters rich in CpG sites is associated with gene silencing that can be epigenetically inherited across cell divisions (Jones and Baylin 2002, 2007), and methylation provides a memory of the silent gene expression state (Raynal et al. 2012). Hypermethylation-induced silencing of genes was previously described in human MDS for individual genes and limited genome-wide studies (Boultwood and Wainscoat 2007; Issa 2010). Here, using methylation arrays, we show abnormal promoter methylation in about one-tenth of the genes analyzed in human MDS as well as in mouse MDS models compared with control cells. Our results are qualitatively similar to a previous array-based DNA methylation profiling study of 807 genes chosen on the basis of their importance to cancer development and progression (Jiang et al. 2009). One limitation of these studies is the use of unfractionated bone marrow–derived or blood-derived mononuclear cells. Contamination with nonaffected cells can reduce the sensitivity of the analyses, and it is theoretically possible that different methylation patterns can be seen in different MDS cellular fractions. However, we have previously shown that for aberrant promoter hypermethylation, similar results can be seen in human MDS derived from marrow or blood (Shen et al. 2010), and we have also shown that promoter DNA methylation is identical in AML progenitor and differentiated cells (Yamazaki et al. 2013). These previous results and the clonal nature of DNA methylation propagation suggest that the changes observed here are occurring in the MDS stem cell. In addition to promoter hypermethylation and hypomethylation, we also found hypomethylation as measured by repetitive element methylation, but this appeared to be a progression event in these models, as it is in other human malignancies (Chalitchagorn et al. 2004; Estecio et al. 2007a).
In this study, we did not sort undifferentiated leukemic cells. It therefore remains possible that genes we identified could be linked with cell differentiation. Previously, Bock et al. (2012) had reported that methylation changes during differentiation occur mostly in nonpromoter regions. In that study, 162, 160, and 157 genes increased methylation from multipotent progenitor 1 (Flt3 negative) to T helper cell (CD4 positive), cytotoxic T-cell (CD8 positive), and B-cell, respectively. Out of these, none were detected as specifically hypermethylated in mouse MDS among 19, 22, and 24 detectable genes in the MCAM assay. In the same study, 247 genes showed increase methylation during hematopoietic differentiation from hematopoietic stem cells into progenitor cells, and the average levels of increases were 10%–17% of methylation. Among these, 24 were detectable by MCAM, and only two (Rarb and Rtkn) were hypermethylated in mouse MDS. Thus, most of the genes (132/134 genes) detected as hypermethylated in mouse MDS were unrelated to cell differentiation.
Quantitative analysis of methylation using pyrosequencing allowed us to unequivocally show that most of the leukemia-associated changes actually arise in aging hematopoietic cells, be it spleen or bone marrow derived (in mice). The data confirm that epigenetic drift (so-called because both hyper- and hypomethylation can be seen with age depending on the gene studied) is a major cause of neoplasm-associated changes in DNA methylation in both solid (Toyota and Issa 1999; Ahuja and Issa 2000) and liquid tumors and are consistent with a model whereby age-related drift creates epigenetic variation. In turn, we speculate that variation provides the necessary ingredient for natural selection to promote the growth of preneoplastic (and eventually neoplastic) lesions. In spontaneous leukemogenesis, epigenetic drift likely precedes critical genetic changes, but the mouse models show that the order can be reversed, with selection for epigenetic changes being part of the secondary changes that lead to disease evolution in NHD13 and RUNX1 models. Interestingly, MDS has intermediate levels of methylation drift compared with AML and, epigenetically, cell studies at the MDS stage appear to have an accelerated aging phenotype, as previously seen in chronic inflammatory conditions (Hsieh et al. 1998; Issa et al. 2001; Sato et al. 2002; Ushijima and Okochi-Takada 2005; Hahn et al. 2008; Chiba et al. 2012). The factors that lead to epigenetic drift in the first place remain unclear, though we show here that both polycomb occupancy in ES cells and depletion of retrotransposons around transcription sites are accelerating factors, as previously seen in other tissues (Widschwendter et al. 2007; Estecio et al. 2010).
The methylation data in the 19 human MDS patients studied by MCAM was heterogeneous, with a subset of cases having very high levels of promoter-CpG island methylation, suggestive of CIMP. Some of these same patients were previously studied for a panel of nine genes (10 genomic regions), and the results were concordant with the current study, thus confirming the extensive nature of CIMP in MDS. The causes of this phenotype are unknown. It was previously suggested that TET2 or IDH1/2 mutations can cause CIMP in AML (Figueroa et al. 2010), but we have not found an association between TET2 mutations and CpG island methylation in CMML (Yamazaki et al. 2012), and this was not seen in TCGA data sets either (The Cancer Genome Atlas Research Network 2013). It has been previously suggested that methylation heterogeneity might be related to heterogeneity in genetic backgrounds and/or differences in exposures to carcinogens or diet. However, even in the MDS mouse models with a uniform genetic background and exposures, we also found methylation-prone and methylation-resistant MDS by pyrosequencing analysis. Thus, the mechanisms responsible for producing methylation variation must involve stochastic events that, however, remain unknown.
The methylated genes we identified here have roles in multiple cellular functions, including cell-signaling, regulation of development, differentiation, and tumorigenesis. This observation agrees with other reports that many fundamental pathways related to tumor pathogenesis are inactivated by methylation (Baylin and Ohm 2006). Some of them overlap with the genes listed as cancer genes related to leukemias and lymphomas (Futreal et al. 2004). However, it is important to keep in mind that only a minority of the genes aberrantly methylated in cancer are driver events, and identification of these remains a difficult proposition. For genetic events, frequency points to drivers, but for DNA methylation, frequency may point to intrinsic gene susceptibility rather than function/selection (Estecio et al. 2010). Indeed, many of the known tumor-suppressors hypermethylated in cancer and leukemias have methylation frequencies of 10%–30% only in target malignancies (e.g., CDKN2B in AML, CDKN2A in colon cancer and melanoma, VHL in renal cancer, etc.) (Herman et al. 1994; Gonzalgo et al. 1997; Baylin and Herman 2000; Rashid et al. 2001; Raj et al. 2007; Jonsson et al. 2010). It is possible that shared lesions in mouse and human leukemogenesis may point to driver genes, and we identified some potential examples such as PAX3, TLX3, ALK, and GATA2. The functional significance of hypermethylation of these genes needs to be determined in future studies.
Methods
Tissue samples
We studied a total of 122 mouse samples including normal tissues, NHD13 mice, and RUNX1 mice (Supplemental Table S1). We also studied 19 samples of human MDS whose characteristics were previously reported (Supplemental Table S7; Shen et al. 2010).
MCAM
MCAM was performed as previously described (Maegawa et al. 2010). We used genomic DNA form normal whole bone marrow to test the malignant bone marrow tissues derived from model mice as a control. Previously, we found minimal differences in DNA methylation of CpG islands between CD34+ bone marrow cells and mononuclear cells and between CD34+ bone marrow cells and unsorted peripheral white blood cells in human MCAM experiments, and we found no differences between stem cells and progenitor cells (Yamazaki et al. 2013), suggesting that the chosen control was appropriate for MCAM analysis. Mouse and human promoter arrays were purchased from Agilent Technologies. After washing, arrays were scanned on an Agilent scanner and analyzed using Agilent feature extraction software at the MD Anderson Microarray Core Facility. We analyzed array data using a database that includes probe sequences, length of SmaI fragments, chromosomal regions, and localization of repetitive elements. The ratios of hybridization intensities were adjusted by using Lowess normalization.
Pathway analyses
Functional class scoring analysis was performed on hyper- or hypomethylated genes with each genotype by using the Ingenuity pathway analysis software. We analyzed biological processes, molecular functions, and cellular components that were relatively enriched by the gene lists of interest using the detectable genes as a reference set.
Bisulfite pyrosequencing for promoter and global DNA methylation analysis
Bisulfite treatment using the EpiTect bisulfite kit was performed according to the manufacturer’s instructions (Qiagen). We used a quantitative bisulfite pyrosequencing method for all DNA methylation analyses as previously reported (Maegawa et al. 2010). Primer sequences and PCR conditions for bisulfite pyrosequencing assays are listed in Supplemental Table S16. Primer sequences and conditions used for human promoter methylation determination are described in a previous publication (Shen et al. 2010). For each assay, we used fully methylated DNA prepared by treating genomic DNA with SssI methylase (New England Biolabs) as a positive control.
Statistics
All correlation was calculated using Spearman’s correlation analysis. Calculations were done using GraphPad Prism 4.0 (GraphPad Software). All P-values are two-sided. Hierarchical clustering for the MCAM result was performed with Partek software. Euclidean distance dissimilarities and Ward’s method were used to cluster both genes and samples. For clustering analysis in pyrosequencing results, we used ArrayTrack Software available at http://edkb.fda.gov/webstart/arraytrack/.
Data access
The microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE46067.
Acknowledgments
This work was supported by National Institutes of Health grants CA100632, CA158112, and CA108631 and by a grant from the Ellison Medical Foundation. J-P.J.I. is an American Cancer Society Clinical Research professor supported by a generous gift from the F.M. Kirby Foundation.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.157529.113.
References
- Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P 2006. Promoter hypermethylation of p15INK4B, HIC1, CDH1, and ER is frequent in myelodysplastic syndrome and predicts poor prognosis in early-stage patients. Eur J Haematol 76: 23–32 [DOI] [PubMed] [Google Scholar]
- Ahuja N, Issa JP 2000. Aging, methylation and cancer. Histol Histopathol 15: 835–842 [DOI] [PubMed] [Google Scholar]
- Akiyama Y, Watkins N, Suzuki H, Jair KW, van Engeland M, Esteller M, Sakai H, Ren CY, Yuasa Y, Herman JG, et al. 2003. GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric cancer. Mol Cell Biol 23: 8429–8439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antequera F, Bird A 1993. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci 90: 11995–11999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Herman JG 2000. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends in Genet 16: 168–174 [DOI] [PubMed] [Google Scholar]
- Baylin SB, Ohm JE 2006. Epigenetic gene silencing in cancer: A mechanism for early oncogenic pathway addiction? Nat Rev Cancer 6: 107–116 [DOI] [PubMed] [Google Scholar]
- Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, et al. 2011. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364: 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, et al. 2012. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet 8: e1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett LB, Schnabel JL, Kelchen JM, Taylor KH, Guo J, Arthur GL, Papageorgio CN, Shi H, Caldwell CW 2009. DNA hypermethylation accompanied by transcriptional repression in follicular lymphoma. Genes Chromosomes Cancer 48: 828–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogal B, Arnaudo A, Dymkowski A, Best A, Davis TL 2004. Methylation at mouse Cdkn1c is acquired during postimplantation development and functions to maintain imprinted expression. Genomics 84: 961–970 [DOI] [PubMed] [Google Scholar]
- Bock C, Beerman I, Lien WH, Smith ZD, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi DJ, Meissner A 2012. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol Cell 47: 633–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boultwood J, Wainscoat JS 2007. Gene silencing by DNA methylation in haematological malignancies. Br J Haematol 138: 3–11 [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353 [DOI] [PubMed] [Google Scholar]
- Bryan J, Jabbour E, Prescott H, Garcia-Manero G, Issa JP, Kantarjian H 2010. Current and future management options for myelodysplastic syndromes. Drugs 70: 1381–1394 [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network 2013. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalitchagorn K, Shuangshoti S, Hourpai N, Kongruttanachok N, Tangkijvanich P, Thong-ngam D, Voravud N, Sriuranpong V, Mutirangura A 2004. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene 23: 8841–8846 [DOI] [PubMed] [Google Scholar]
- Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, Bock C, Vasanthakumar A, Gu H, Xi Y, et al. 2012. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44: 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T, Marusawa H, Ushijima T 2012. Inflammation-associated cancer development in digestive organs: Mechanisms and roles for genetic and epigenetic modulation. Gastroenterology 143: 550–563 [DOI] [PubMed] [Google Scholar]
- Cosialls AM, Santidrian AF, Coll-Mulet L, Iglesias-Serret D, Gonzalez-Girones DM, Perez-Perarnau A, Rubio-Patino C, Gonzalez-Barca E, Alonso E, Pons G, et al. 2012. Epigenetic profile in chronic lymphocytic leukemia using methylation-specific multiplex ligation-dependent probe amplification. Epigenomics 4: 491–501 [DOI] [PubMed] [Google Scholar]
- Estecio MR, Gharibyan V, Shen L, Ibrahim AE, Doshi K, He R, Jelinek J, Yang AS, Yan PS, Huang TH, et al. 2007a. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS ONE 2: e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estecio MR, Yan PS, Ibrahim AE, Tellez CS, Shen L, Huang TH, Issa JP 2007b. High-throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res 17: 1529–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estecio MR, Gallegos J, Vallot C, Castoro RJ, Chung W, Maegawa S, Oki Y, Kondo Y, Jelinek J, Shen L, et al. 2010. Genome architecture marked by retrotransposons modulates predisposition to DNA methylation in cancer. Genome Res 20: 1369–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. 2010. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18: 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR 2004. A census of human cancer genes. Nat Rev Cancer 4: 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalgo ML, Bender CM, You EH, Glendening JM, Flores JF, Walker GJ, Hayward NK, Jones PA, Fountain JW 1997. Low frequency of p16/CDKN2A methylation in sporadic melanoma: Comparative approaches for methylation analysis of primary tumors. Cancer Res 57: 5336–5347 [PubMed] [Google Scholar]
- Graubert T, Walter MJ 2011. Genetics of myelodysplastic syndromes: New insights. Hematology Am Soc Hematol Educ Program 2011: 543–549 [DOI] [PubMed] [Google Scholar]
- Haase D, Germing U, Schanz J, Pfeilstocker M, Nosslinger T, Hildebrandt B, Kundgen A, Lubbert M, Kunzmann R, Giagounidis AA, et al. 2007. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: Evidence from a core dataset of 2124 patients. Blood 110: 4385–4395 [DOI] [PubMed] [Google Scholar]
- Hahn MA, Hahn T, Lee DH, Esworthy RS, Kim BW, Riggs AD, Chu FF, Pfeifer GP 2008. Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res 68: 10280–10289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. 2013. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 49: 359–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, et al. 1994. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci 91: 9700–9704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, van den Berg LH, Ophoff RA 2012. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol 13: R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R 1998. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Res 58: 3942–3945 [PubMed] [Google Scholar]
- Issa JP 2004. CpG island methylator phenotype in cancer. Nat Rev Cancer 4: 988–993 [DOI] [PubMed] [Google Scholar]
- Issa JP 2010. Epigenetic changes in the myelodysplastic syndrome. Hematol Oncol Clin North Am 24: 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA 2001. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res 61: 3573–3577 [PubMed] [Google Scholar]
- Jelinek J, Liang S, Lu Y, He R, Ramagli LS, Shpall EJ, Estecio MR, Issa JP 2012. Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique. Epigenetics 7: 1368–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe C, Sekeres M, Saunthararajah Y, Maciejewski JP 2009. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 113: 1315–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Baylin SB 2002. The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415–428 [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB 2007. The epigenomics of cancer. Cell 128: 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson A, Tuominen R, Grafstrom E, Hansson J, Egyhazi S 2010. High frequency of p16INK4A promoter methylation in NRAS-mutated cutaneous melanoma. J Invest Dermatol 130: 2809–2817 [DOI] [PubMed] [Google Scholar]
- Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, et al. 2006. Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 106: 1794–1803 [DOI] [PubMed] [Google Scholar]
- Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O’Brien S, Cortes J, Faderl S, Bueso-Ramos C, Ravandi F, Estrov Z, et al. 2007. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 109: 52–57 [DOI] [PubMed] [Google Scholar]
- Kay PH, Spagnolo DV, Taylor J, Ziman M 1997. DNA methylation and developmental genes in lymphomagenesis: More questions than answers? Leuk Lymphoma 24: 211–220 [DOI] [PubMed] [Google Scholar]
- Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, Viguie F, Quesnel B, Beyne-Rauzy O, Solary E, et al. 2009. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs). Blood 114: 3285–3291 [DOI] [PubMed] [Google Scholar]
- Kroeger H, Jelinek J, Estecio MR, He R, Kondo K, Chung W, Zhang L, Shen L, Kantarjian HM, Bueso-Ramos CE, et al. 2008. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood 112: 1366–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, et al. 2009. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 41: 838–842 [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. 2006. Control of developmental regulators by polycomb in human embryonic stem cells. Cell 125: 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YW, Slape C, Zhang Z, Aplan PD 2005. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood 106: 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ 2007. Computational and experimental identification of novel human imprinted genes. Genome Res 17: 1723–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP 2010. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 20: 332–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K, Clay O, Takahashi T, Silke J, Schaffner W 1993. Evidence for erosion of mouse CpG islands during mammalian evolution. Somat Cell Mol Genet 19: 543–555 [DOI] [PubMed] [Google Scholar]
- Nikoloski G, van der Reijden BA, Jansen JH 2012. Mutations in epigenetic regulators in myelodysplastic syndromes. Int J Hematol 95: 8–16 [DOI] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. 2007. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39: 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker-Katiraee L, Carson AR, Yamada T, Arnaud P, Feil R, Abu-Amero SN, Moore GE, Kaneda M, Perry GH, Stone AC, et al. 2007. Identification of the imprinted KLF14 transcription factor undergoing human-specific accelerated evolution. PLoS Genet 3: e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj K, John A, Ho A, Chronis C, Khan S, Samuel J, Pomplun S, Thomas NS, Mufti GJ 2007. CDKN2B methylation status and isolated chromosome 7 abnormalities predict responses to treatment with 5-azacytidine. Leukemia 21: 1937–1944 [DOI] [PubMed] [Google Scholar]
- Rashid A, Shen L, Morris JS, Issa JP, Hamilton SR 2001. CpG island methylation in colorectal adenomas. Am J Pathol 159: 1129–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch T, Wang Z, Zhang X, Zhong X, Wu X, Lau SK, Kernstine KH, Riggs AD, Pfeifer GP 2007. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci 104: 5527–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval A, Lucas DM, Matkovic JJ, Bennett KL, Liyanarachchi S, Young DC, Rassenti L, Kipps TJ, Grever MR, Byrd JC et al. 2005. TWIST2 demonstrates differential methylation in immunoglobulin variable heavy chain mutated and unmutated chronic lymphocytic leukemia. J Clin Oncol 23: 3877–3885 [DOI] [PubMed] [Google Scholar]
- Raynal NJ, Si J, Taby RF, Gharibyan V, Ahmed S, Jelinek J, Estecio MR, Issa JP 2012. DNA methylation does not stably lock gene expression but instead serves as a molecular mark for gene silencing memory. Cancer Res 72: 1170–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush LJ, Raval A, Funchain P, Johnson AJ, Smith L, Lucas DM, Bembea M, Liu TH, Heerema NA, Rassenti L, et al. 2004. Epigenetic profiling in chronic lymphocytic leukemia reveals novel methylation targets. Cancer Res 64: 2424–2433 [DOI] [PubMed] [Google Scholar]
- Sato F, Harpaz N, Shibata D, Xu Y, Yin J, Mori Y, Zou TT, Wang S, Desai K, Leytin A, et al. 2002. Hypermethylation of the p14ARF gene in ulcerative colitis-associated colorectal carcinogenesis. Cancer Res 62: 1148–1151 [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. 2007. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 39: 232–236 [DOI] [PubMed] [Google Scholar]
- Shen L, Toyota M, Kondo Y, Obata T, Daniel S, Pierce S, Imai K, Kantarjian HM, Issa JP, Garcia-Manero G 2003. Aberrant DNA methylation of p57KIP2 identifies a cell-cycle regulatory pathway with prognostic impact in adult acute lymphocytic leukemia. Blood 101: 4131–4136 [DOI] [PubMed] [Google Scholar]
- Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X, Berry D, Ahmed S, Zhu W, Pierce S et al. 2010. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol 28: 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Abdel-Wahab O, Patel JP, Levine RL 2012. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 12: 599–612 [DOI] [PubMed] [Google Scholar]
- Slape C, Liu LY, Beachy S, Aplan PD 2008. Leukemic transformation in mice expressing a NUP98-HOXD13 transgene is accompanied by spontaneous mutations in Nras, Kras, and Cbl. Blood 112: 2017–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, Herman JG, Baylin SB 2002. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 31: 141–149 [DOI] [PubMed] [Google Scholar]
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al. 2010. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res 20: 440–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thathia SH, Ferguson S, Gautrey HE, van Otterdijk SD, Hili M, Rand V, Moorman AV, Meyer S, Brown R, Strathdee G 2012. Epigenetic inactivation of TWIST2 in acute lymphoblastic leukemia modulates proliferation, cell survival and chemosensitivity. Haematologica 97: 371–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong WG, Wierda WG, Lin E, Kuang SQ, Bekele BN, Estrov Z, Wei Y, Yang H, Keating MJ, Garcia-Manero G 2010. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics 5: 499–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M, Issa JP 1999. CpG island methylator phenotypes in aging and cancer. Semin Cancer Biol 9: 349–357 [DOI] [PubMed] [Google Scholar]
- Toyota M, Ho C, Ahuja N, Jair KW, Li Q, Ohe-Toyota M, Baylin SB, Issa JP 1999. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res 59: 2307–2312 [PubMed] [Google Scholar]
- Toyota M, Kopecky KJ, Toyota MO, Jair KW, Willman CL, Issa JP 2001. Methylation profiling in acute myeloid leukemia. Blood 97: 2823–2829 [DOI] [PubMed] [Google Scholar]
- Ushijima T, Okochi-Takada E 2005. Aberrant methylations in cancer cells: Where do they come from? Cancer Sci 96: 206–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al. 2013. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 23: 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, Fulton R, Schmidt H, Kalicki-Veizer J, O’Laughlin M, et al. 2011. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25: 1153–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe-Okochi N, Kitaura J, Ono R, Harada H, Harada Y, Komeno Y, Nakajima H, Nosaka T, Inaba T, Kitamura T 2008. AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood 111: 4297–4308 [DOI] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al. 2007. Epigenetic stem cell signature in cancer. Nat Genet 39: 157–158 [DOI] [PubMed] [Google Scholar]
- Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, Ren B 2012. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148: 816–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto E, Suzuki H, Yamano HO, Maruyama R, Nojima M, Kamimae S, Sawada T, Ashida M, Yoshikawa K, Kimura T, et al. 2012. Molecular dissection of premalignant colorectal lesions reveals early onset of the CpG island methylator phenotype. Am J Pathol 181: 1847–1861 [DOI] [PubMed] [Google Scholar]
- Yamazaki J, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Shen L, Kantarjian HM, Estecio MR, Jelinek J, Godley LA et al. 2012. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 7: 201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Estecio MR, Lu Y, Long H, Malouf GG, Graber D, Huo Y, Ramagli L, Liang S, Kornblau SM et al. 2013. The epigenome of AML stem and progenitor cells. Epigenetics 8: 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying J, Gao Z, Li H, Srivastava G, Murray PG, Goh HK, Lim CY, Wang Y, Marafioti T, Mason DY, et al. 2007. Frequent epigenetic silencing of protocadherin 10 by methylation in multiple haematologic malignancies. Br J Haematol 136: 829–832 [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69 [DOI] [PubMed] [Google Scholar]