

Abstract
The emergence of drug-resistant pathogens poses a major threat to public health. Although influenced by multiple factors, high-level resistance is often associated with mutations in target-encoding or related genes. The fitness cost of these mutations is, in turn, a key determinant of the spread of drug-resistant strains. Rifampicin (RIF) is a frontline anti-tuberculosis agent that targets the rpoB-encoded β subunit of the DNA-dependent RNA polymerase (RNAP). In Mycobacterium tuberculosis (Mtb), RIF resistance (RIFR) maps to mutations in rpoB that are likely to impact RNAP function and, therefore, the ability of the organism to cause disease. However, while numerous studies have assessed the impact of RIFR on key Mtb fitness indicators in vitro, the consequences of rpoB mutations for pathogenesis remain poorly understood. Here, we examine evidence from diverse bacterial systems indicating very specific effects of rpoB polymorphisms on cellular physiology, and consider these observations in the context of Mtb. In addition, we discuss the implications of these findings for the propagation of clinically relevant RIFR mutations. While our focus is on RIF, we also highlight results which suggest that drug-independent effects might apply to a broad range of resistance-associated mutations, especially in an obligate pathogen increasingly linked with multidrug resistance.
Keywords: epistasis, fitness cost, RNA polymerase, rpoB, TB
INTRODUCTION
The discovery of antibiotics in the 1940s revolutionized the treatment of infectious diseases and, at the same time, suggested the possibility of eradicating bacterial pathogens as a major cause of morbidity and mortality.1 The intervening 70 years have, however, seen the emergence of organisms which are resistant to almost every antibiotic that has been introduced into mainstream use.1,2 Tuberculosis (TB) is no exception: in 2012, there were an estimated 450 000 cases of multidrug-resistant (MDR) TB, which is defined as disease caused by strains of Mycobacterium tuberculosis (Mtb) that are resistant to the frontline anti-TB drugs, isoniazid (INH) and rifampicin (RIF). Almost 10% of these MDR cases were classified as extensively drug-resistant (XDR) TB,3 in which there is additional resistance to any of the fluoroquinolone (FQ) antibiotics and at least one of the second-line injectable aminoglycosides—amikacin, capreomycin or kanamycin. Although accounting for a small number of cases relative to drug-susceptible disease, drug-resistant TB imposes a disproportionate burden on public health systems—especially in endemic regions4—and, for this reason, MDR and XDR Mtb strains are considered emerging pathogens in their own right.5 However, while there has been intensive analysis of the risk factors—primarily social and programmatic—driving the emergence of resistance,6,7,8 the global burden of drug-resistant TB continues to increase. Effective TB control will require a deeper understanding of the impact of drug resistance on the host–pathogen interaction and of the biological factors underlying the relative success of drug-resistant strains.9
Comparative genomic analyses have established that high-level drug resistance in Mtb arises almost exclusively through chromosomal mutations in genes required for antibiotic action,10,11,12,13,14,15 that is, genes encoding the protein targets of the applied drugs, or the enzymes required for prodrug activation. Since antibiotics target essential cellular functions, it might be expected that resistance mutations in target-encoding genes will impact pathogenesis—a concept loosely captured in the term ‘fitness cost'.16 In turn, this raises fundamental questions regarding the ability of Mtb to harbour multiple drug resistance mutations while retaining the ability to infect, persist, and cause disease in its obligate human host. We are interested in RIF resistance (RIFR), which results primarily from single-nucleotide substitution mutations in a small region of rpoB, the gene encoding the β-subunit of the DNA-dependent RNA polymerase (RNAP) (Figure 1). Given the essentiality of RNAP for transcription, it appears likely that mutations in rpoB will have multiple effects on Mtb physiology in addition to RIFR. In this review, we summarize insights obtained from other bacterial systems into the structural and physiological consequences of rpoB mutations, and consider these observations in the context of the available evidence from Mtb. In addition, we assess the potential impact of RIFR on Mtb physiology and pathogenesis and discuss the possible consequences for the continued emergence of drug resistance in a pathogen that is uniquely adapted to human infection.19
Figure 1.

Schematic representation of RNAP structural elements including the RIF resistance determining region (RRDR). The cartoon showing the RNAP holoenzyme is adapted from Borukhuv and Nudler.17 Structural annotations have been simplified, and the promoter sequence has been excluded. The rpoB-encoded β subunit is highlighted in green. A yellow star represents the RNAP active site and a red circle denotes the RIF molecule which approaches within 12 Å of the active site,18 inhibiting transcription. Double-stranded DNA is represented by pink lines and, once unwound, only template DNA is shown, with the growing RNA chain colored in blue. The inset shows a simplified depiction of the RIF binding pocket.18 Amino acids that form hydrogen bonds with RIF are highlighted in blue and those that form van der Waals interactions are colored yellow; amino-acid numbering corresponds to that used for E. coli. Mutations identified in 11 of the 12 residues that surround the RIF binding pocket have been associated with RIF resistance, albeit at different frequencies18 (the sole amino acid, E565, which has not been associated with RIFR mutations is colored in grey). A schematic representation of the rpoB gene which encodes the β subunit of RNA polymerase is shown below the RNAP cartoon (adapted from Campbell et al.18). Amino-acid numbering is shown as dashed demarcations. The RRDR is highlighted in blue and the amino-acid sequence of the RRDR is magnified below. The alignment contains the amino-acid sequences of E. coli, T. aquaticus and M. tuberculosis. Amino acids that interact directly with RIF are indicated by circles and the colors correspond to the inset diagram. Circles highlighted in red indicate residues that are most frequently observed in RIFR isolates.18
WHY IS RIF IMPORTANT FOR TB?
Together with INH, RIF is a major frontline anti-TB agent and has been included in standard chemotherapy since the 1980s.20 RIF is also used for the treatment of asymptomatic Neisseria meningitides carriers and, like other rifamycins, has been prescribed for Streptococcus pneumoniae, Legionella pneumophila and opportunistic Rhodococcus equi infections.21 Moreover, the emergence of methicillin resistance and, more recently, vancomycin resistance (VANR) has resulted in the increasing application of RIF for Staphylococcus aureus infections.21 Unlike most current antibiotics which require active growth and metabolism to exert their anti-bacterial effects,22 RIF is included in a select category of agents (other examples include moxifloxacin and bedaquiline), which retain activity against slow-growing, and even non-replicating, Mtb bacilli.23,24,25 This property is especially important for TB, where low metabolic activity and/or non-replication are considered key factors in persistent Mtb infection.26,27,28 In fact, the role of RIF in sterilizing slowly metabolizing bacillary populations20 is a major factor in the continued reliance of public health programmes on RIF as a frontline anti-TB drug, despite the emergence and spread of RIFR Mtb strains.29
How does RIF kill bacteria?
Most bacteria possess a single DNA-dependent RNAP enzyme comprising a multisubunit αββ′ω core that forms a ‘crab claw-like' structure.17,30,31 The β and β′-subunits constitute the main components of each ‘pincer' of the claw, forming a groove that accommodates the template DNA and provides a catalytic site for phosphodiester bond formation, a secondary channel for incoming nucleotides, and a separate exit for the growing RNA transcript.17,30,31 In a tight complementary fit, RIF binds to the rpoB-encoded β-subunit, thereby inhibiting transcription (Figure 1). Structural analysis of the Thermus aquaticus RNAP has shown that the RIF binding site is located within the DNA/RNA channel, but not at the active site.18 Moreover, RIF-bound RNAP retains the ability to catalyse formation of the first phosphodiester bond in a nascent RNA transcript, suggesting that RIF does not inhibit catalysis. Instead, it seems that the drug obstructs the path of a growing RNA chain of two to three nucleotides in length: once transcriptional elongation is in full progress, RNAP is no longer vulnerable to RIF-mediated inhibition. For this reason, RIF activity is restricted to a very specific stage of transcription.18 However, the precise mechanism by which RIF-mediated transcriptional interference leads to cell death is not well understood.
Mutations in rpoB confer RIFR
Early studies in Escherichia coli mapped RIFR mutations to three distinct clusters (I, II and III) within the rpoB gene.18 It has subsequently been shown that, across all bacterial species, the majority of RIFR mutations occur in an 81 bp region of cluster I—the so-called RIF resistance determining region (RRDR)32,33,34—though some mutations have been identified in other regions of rpoB as well.35,36 As the RRDR was originally defined in E. coli, it is standard practice to use that organism's numbering system even when describing specific features of RpoB from other bacteria;37 that tradition is observed for the remainder of this article.
Of the 12 amino acids that surround the RIF binding pocket, 11 have been associated with RIFR mutations (Figure 1).18 From analyses of clinical Mtb isolates from various geographical regions, it is evident that specific RIFR mutations are identified more frequently than others.12 However, across all bacterial species, RIFR is generally associated with mutations in the binding pocket that substitute an amino acid with a compact side chain with one that is larger (e.g., Ser→Leu), thereby preventing access of the relatively inflexible RIF molecule to RNAP.18 In E. coli and Bacillus subtilis, rpoB mutations have been shown to impact promoter binding38,39 and transcriptional elongation and termination,40,41,42,43,44,45,46 as well as elements of transcription-coupled repair.47,48,49 So profound are these effects that rpoB mutations have been exploited to examine mechanisms of transcription since well before there was an interest in the clinical implications of RIFR.33
There have also been reports of RIF-dependent E. coli strains.50 Similarly, enhanced growth of some Mtb mutants has been observed in vitro in the presence of RIF51 and was recently described in a clinical Mtb strain obtained from a patient whose TB symptoms deteriorated while undergoing treatment with a regimen containing rifamycins.52 The infecting Mtb isolate—carrying the common rpoB S531L mutation together with a less frequently observed F584S polymorphism—exhibited improved growth in RIF-containing versus antibiotic-free growth medium. There is some analogy between this observation and the identification of streptomycin (STR)-dependent clinical isolates such as Mtb 18b, a strain whose inability to grow in the absence of STR has recently been exploited as a useful model for TB drug discovery.25 However, whereas the STR dependence of 18b is thought to result from an insertion mutation in the 16S rRNA sequence that is stabilized by the STR molecule,53 the mechanism(s) underling RIF-dependent growth remains unclear. The presence of an F584S mutation in addition to S531L in the RIF-dependent mutant implies that secondary mutations might contribute to the phenotype of strains that grow better in the presence of RIF; however, this requires formal demonstration. Moreover, it is not known just how often the phenomenon of improved growth in the presence of RIF occurs clinically, notwithstanding its potential implications for TB diagnosis and treatment.
In Mtb, c→t transitions underlie two of the most commonly observed rpoB mutations: S531L (tcg→ttg) and H526Y (cac→tac).12,54 Spontaneous deamination of cytosine to uracil occurs readily in all living cells55 and the high G+C-content of Mtb genomic DNA probably renders the organism especially susceptible to these events.56 Moreover, it is possible that this effect is exacerbated by host immune defences, which include the production of reactive oxygen and nitrogen intermediates57 and, for rpoB in particular, is further amplified by the selective advantage of the S531L mutation.58 There is, however, important recent evidence that, for Mtb, the number of potential RIFR-conferring mutations in rpoB—the effective ‘target size' for resistance—can differ according to strain lineage, at least in vitro.59 This effect is most clearly observed at lower RIF concentrations and, in combination with separate studies identifying putative ‘low-level' resistance and/or compensatory mutations in clinical isolates,10,11 suggests that further work is necessary to determine the contribution of ‘rare' rpoB alleles—and other infrequent resistance-associated mutations—to the emergence of drug resistance. For example, mutations have been identified outside the RRDR in clinical RIFR Mtb strains.60 However, given the focus of genotypic assays on the RRDR for detecting RIFR,61 it is inevitable that any non-RRDR mutations will be underrepresented in molecular epidemiological surveys of resistance.
The conditions favouring the emergence of specific mutations are also poorly understood: for example, it has been shown that the diversity of rpoB mutations in E. coli differs depending on the rate at which susceptible organisms are exposed to increasing RIF concentrations.62 Similarly, adjusting the pH of a chemostat culture of Mtb results in a different spectrum of rpoB mutations from that which is commonly observed during growth under standard conditions in vitro,63 perhaps suggesting the role of a specific selective pressure(s) in ensuring fixation (and even propagation) of the lowest cost mutations.
THE FITNESS COST OF RIFR MUTATIONS
In general, resistance mutations in essential genes have a negative impact on key physiological functions. That is, drug resistance incurs a fitness cost: in the absence of drug pressure, strains without resistance-associated mutations are generally better able to complete the bacterial life cycle in a variety of growth conditions.2,64,65,66,67 The cost of resistance has been identified as a critical determinant of the spread of drug-resistant Mtb strains.68 Given the clinical importance of RIF, it is not surprising that the fitness cost of rpoB mutations has been the subject of intense investigation, both in Mtb and other organisms.65,69,70 Overall, these studies have reported that the fitness of specific rpoB mutants relative to the corresponding drug-susceptible parental strain depends primarily on the strain background and the nature of the assay employed to assess fitness.64,66,67,71,72,73 However, in most cases, strain ‘fitness' is conflated with growth rate under a limited set of in vitro conditions, probably because of the relative ease of performing—and interpreting—bacterial growth assays. In their natural environments, most bacteria seldom encounter conditions which allow for uninterrupted exponential growth.74 Therefore, alternative fitness models might be required to recapitulate the selective pressures that impact competitive fitness, such as adaptation to stationary-phase in vitro75 (discussed below) or virulence in experimental models of infection.76,77 This issue is explored further below (see ‘IN VITRO FITNESS AS A SURROGATE FOR PATHOGEN SUCCESS?' section).
Strain fitness versus epidemiological success
It is likely, too, that for obligate pathogens such as Mtb, growth rate is just one of many factors that might contribute to the ‘success' of the organism. That is, multiple fitness attributes will influence the short-term competitiveness of specific Mtb mutants (or lineages) and, in turn, their longer-term evolution within different hosts and host populations. Therefore, for the purposes of this review, the term ‘fitness' might be defined as the inherent capacity of an organism to drive its lifecycle, the composite phenotype that represents the sum of all genetic and physiological features and abilities. In contrast, ‘success' might be considered the outcome of multiple contributing ‘fitness' attributes, and should be measured by the longevity of a pathogen within a specific environment or host population. For an obligate pathogen, the fitness attributes which contribute to the success of specific lineages (drug-resistant or not) might be usefully considered in terms of discrete steps of the infection lifecycle: that is, the ability to establish an infection, and to replicate and persist within a host, as well as the capacity for transmission.
In turn, this suggests that any resistance cost should be considered in the context of longer-term pathogen success, which is itself a function of multiple biological processes that might not directly influence growth rate, but could impact other core physiological functions. The recent observation that multiple small-effect mutations might contribute to the stepwise emergence of drug resistance or to reducing the cost of acquired resistance,10,11,78 appears to reinforce this notion; such polymorphisms could be critical to the ability of the organism to survive very specific selective pressures that do not correlate with relative growth rate in vitro.
The impact of compensatory mutations
Compensatory mutations in target or related genes can reduce the fitness costs of drug resistance.9,67,79 In a landmark study, Gagneux et al.67 measured the growth rates of RIFR Mtb mutants relative to drug-susceptible parental strains. Their experiments utilized spontaneous RIFR mutants selected in vitro, as well as clinical isolates obtained from TB patients who had developed drug resistance. The strains covered two of the seven main Mtb lineages80 and the results revealed that competitive fitness was dependent not only on the nature of the rpoB mutation (e.g. S531L versus H526Y), but also strain genotype: fitness differed considerably between lineages even where the rpoB mutation was identical.67 As suggested by the authors, these data implied that the presence (or absence) of compensatory mutations might contribute to the inferred fitness cost.67 To address this possibility, the same group recently examined a large database of RIFS and RIFR clinical isolates for compensatory mutations.79 Their analyses revealed that a significant proportion of RIFR strains carried mutations in rpoA or rpoC—encoding the RNAP α and β′ subunits, respectively—whereas the same mutations were not found in susceptible isolates. This observation, together with the location of the polymorphisms in subunits that interact closely with rpoB in the RNAP holoenzyme (Figure 1), identified the observed mutations as compensatory.79 Consistent with this notion, an elevated frequency of rpoC mutations was recently described in closely related strains from South Africa, suggesting a potential association between the propagation of RIFR strains and the presence of rpoC mutations.81 Similar panels of rpoC and rpoB mutations have subsequently been identified among RIFR clinical Mtb isolates in multiple separate studies, reinforcing the inferred role of multiple RNAP mutations in conferring RIFR while maintaining strain fitness.10,11,13
The selective pressure for the acquisition of compensatory mutations to restore competitive fitness is further supported by similar observations in Salmonella enterica, where the acquisition of rpoA and rpoC mutations has been shown to improve the growth rate of slow-growing rpoB mutants during serial passage in media containing RIF.70,82 In that case, though, the authors confirmed the necessity and sufficiency of the identified mutations for reversal of the growth defect by introducing the same mutations into the chromosome of the original rpoB mutant, thereby providing genetic validation of the inferred compensatory effect. Interestingly, the rpoA and rpoC mutations did not alter the susceptibility of the rpoB mutant to RIF, yet the same mutations were associated with small but significant decreases in RIF susceptibility when introduced into the fully susceptible wild-type strain. The combination of Mtb and S. enterica studies strongly suggests that rpoA and rpoC mutations are compensatory;82 however, key questions remain. For example, given that the rpoB mutation identified in the S. enterica study (R529C) is not frequently detected in clinical Mtb isolates,12 what is the role of compensatory mutations in strains carrying more frequently observed rpoB alleles? Also, what are the structural and/or molecular mechanisms underlying the inferred compensatory effects? Comparing the impact of compensatory mutations on diverse physiological and pathogenic features in isogenic strains will be critical to understand the selective advantage conferred by the identified RNAP polymorphisms.
A role for epistasis?
Independent resistance mutations can interact to influence the fitness of drug-resistant bacteria. This concept is captured in the term ‘epistasis', which might be defined broadly as an interaction between genes,83 but is often used to refer to the masking of phenotypic effects of one allele by another allele. To avoid confusion, the term epistasis is used throughout this review to denote the dependence of the phenotypic expression of one allele on another, distinct allele.84 For example, strong positive epistasis has been described for genes associated with resistance to STR, RIF and nalidaxic acid in E. coli.84 Similarly, a very recent in vitro study of engineered FQ-resistant (FQR) Salmonella mutants has demonstrated synergistic epistasis between specific gyrA and parC alleles.85 Critically, these results provide direct evidence of the potential for drug resistance-associated mutations to confer a strong fitness advantage, even in the absence of antibiotic selection.
The interaction of drug resistance determinants need not necessarily be linked to mutation: exposure of RIFR Pseudomonas aeruginosa mutants to sublethal concentrations of the translational inhibitors, STR and chloramphenicol, has been shown to decrease the fitness cost of a RIFR-conferring mutation (as determined by relative growth rate), whereas drugs targeting cellular processes other than translation did not.86 As the authors of this study argued, it is possible that the interdependence of transcription and translation ensures that a decreased demand for RNAP activity in the presence of non-lethal translational inhibition reduces the fitness defect of RIFR mutations. If so, this implies that STRR mutations in rpsL which reduce ribosome functionality could interact similarly with impaired RpoB activity.86 It will be interesting, therefore, to establish whether this relationship is reciprocal; that is, can defective RNAP function mitigate fitness costs that may be associated with STRR-conferring mutations?
The propagation of MDR and XDR Mtb strains suggests that epistatic interactions might contribute to the phenotypes of strains which, by definition, harbour multiple mutations in essential genes. For MDR-TB, it is generally thought that exposure to both INH and RIF results in the fixation of successive, but distinct, mutations that confer resistance to each drug. However, in vitro evidence suggests that the nature of the pre-existing INHR allele can influence the spectrum of subsequent rpoB mutations.87 Consistent with this idea, analyses of drug resistant clinical Mtb isolates have revealed that specific INHR alleles are more frequently associated with resistances to other drugs.88,89 While this might reflect the different fitness costs associated with particular INHR mutations,88 it is plausible that these data indicate epistatic interactions between INHR and RIFR mutations, a possibility which holds significant implications for the success of MDR-TB strains.
In the most advanced work to date, positive epistasis has been demonstrated between FQR mutations in gyrA and common RIFR-associated rpoB alleles.90 Using the non-pathogenic M. smegmatis as surrogate for Mtb, mutants harbouring specific gyrA and rpoB allele combinations were fitter than corresponding strains carrying only a single resistance mutation during competitive growth under standard conditions in vitro. Notably, the same gyrA/rpoB single-nucleotide polymorphism combinations were identified among a panel of clinical XDR Mtb isolates, suggesting that epistasis between resistance mutations may be a major factor in the fitness of drug-resistant Mtb isolates and, further, might determine the trajectory for the acquisition of multiple resistance mutations.90 The established link between transcription rate and DNA supercoiling 91 suggests a plausible mechanism for epistatic interactions between mutations in gyrA and rpoB, though this requires further investigation. More importantly, however, the inferred epistasis signals a need for caution in considering FQs—currently reserved as second-line anti-TB agents—for use in standard regimens for drug-susceptible disease.
IN VITRO FITNESS AS A SURROGATE FOR PATHOGEN SUCCESS?
Drug-resistant Mtb strains carrying low fitness cost mutations should, in principle, result in more secondary TB cases. In turn, this has prompted the idea that the prevalence of specific resistance alleles can be equated with fitness: that is, the most common mutations detected clinically are those which incur the smallest fitness cost.64,73,92,93 The induction of a fitness hierarchy for specific drug resistance mutations from epidemiological data might not be straightforward, however. In particular, this notion ignores critical uncertainties regarding the nature of the selective forces acting on clinical strains—both during host infection and through the process of strain isolation in vitro. For example, while the S531L mutation is frequently observed in clinical Mtb isolates, the extent to which other rpoB alleles might be subject to strong negative selection in vitro is largely unknown, yet could profoundly prejudice apparent strain (and allele) frequencies detected among clinical samples.94 Standard media used for strain isolation (and propagation) for routine diagnostic procedures share common constituents with growth media used in experimental assays of competitive fitness; for example, Middlebrook 7H9 base and dextrose are the major components of the major mycobacterial detection platforms95 and constitute the preferred culture medium in most mycobacterial research laboratories.67 It seems likely, therefore, that a selection bias might be inadvertently imposed through the use of a single, glucose-based growth medium. Critically, the same bias would extend to fitness assays in vitro, thereby reinforcing inferred relationships between clinical frequency and capacity for competitive growth.
A recent study applied deep sequencing to examine the diversity of drug resistance mutations in clinical samples.15 Instead of sequencing individual colonies, a ‘scrape' of colony forming units was taken from the solid medium on which Mtb bacilli had been isolated from sputum. This novel approach uncovered significant levels of heterogeneity of drug resistance-associated mutations within individual patients.15 However, it was still dependent on in vitro culture of Mtb prior to sequencing, which means that only those strains that were culturable on that medium were represented in the analysis. It is possible, therefore, that ‘culture-free' (or metagenomic) techniques will be required to interrogate the potential genetic diversity of the infecting Mtb population in sputum and/or other biological samples.
It is also necessary to consider the environmental context when evaluating relative strain fitness. A rare variant of the Beijing lineage, which is differentiated by a small change in its IS6110 fingerprint, is thought to be less fit owing to the fact that ‘atypical Beijing' strains are infrequently observed in clinical settings. Nevertheless, a RIFR variant sustained transmission in a community with high levels of human immunodeficiency virus (HIV).96 Similarly, a RIFR Mtb isolate carrying an uncommon rpoB mutation was associated with unusually high virulence and transmission in an HIV positive cohort.97 It is possible, therefore, that host populations with compromised immunity impose very different pressures on the infecting (and transmitting) organisms, and so might be associated with altered requirements for strain success.98
THE IMPACT OF RPOB MUTATIONS ON (MYCO)BACTERIAL PHYSIOLOGY
The potential for drug resistance mutations to alter cellular function seems especially relevant to RpoB as an essential component of the highly conserved bacterial RNAP99,100, an idea that is supported by multiple studies demonstrating drug-independent physiological effects of RIFR mutations. In the section below, we briefly review evidence from various bacterial systems that implicates rpoB mutations in diverse phenotypic and functional alterations (summarized in Table 1), and discuss their potential relevance to Mtb.
Table 1. Impact of RIFR-associated rpoB mutations on bacterial physiology.
| Organism | Observed phenotype | Reference |
|---|---|---|
| B. subtilis | The spectrum of rpoB mutations in spores is distinct from those found in vegetative populations and is similar to those associated with clinical Mtb isolates | 101 |
| Widespread changes in globally regulated processes such as competency, germination and sporulation | 102 | |
| The ability to metabolise substrates previously thought to be non-utilisable for B. subtilis and other important changes to B. subtilis carbon source metabolism | 103 | |
| B. subtilis and S. coelicor | Increased antibiotic production and production of cryptic or previously unobserved secondary metabolites | 39,104,105,106 |
| E. coli | In strains unable to produce (p)ppGpp, rpoB mutations mimic (p)ppGpp regulation and a ‘stringent'-like phenotype is observed. | 38,107 |
| Widespread changes in mechanistic aspects of transcription, including pausing, termination and affinity for nucleotides during elongation | 40,42,44,45 | |
| Temperature sensitivity and phage susceptibility | 43 | |
| Adaptation to minimal medium, predominantly as a result of a mutation in rpoB or rpoC | 108,109,110 | |
| Growth advantage during stationary phase growth | 75 | |
| Significant epistatic interactions between antibiotic resistance-associated mutations in rpoB, rpsL and gyrA | 84 | |
| Evolution of RIFR rpoB mutations in response to thermal stress in the absence of RIF | 111 | |
| Mtb | Spectrum of rpoB mutations changes when the pH of a chemostat culture is lowered | 63 |
| After exposure to RIF, strains containing rpoB mutations have increased ofloxacin minimum inhibitory concentrations | 112 | |
| Alteration important cell wall components such as phthiocerol dimycocerosates and fatty acid precursors | 113,114 | |
| Increased dnaE2 expression | 115 | |
| N. meningitidis | Decrease in cell membrane permeability | 116 |
| Nonomurea and Nocardia | A second rpoB gene confers RIFR and activates expression of dormant genes | 117,118,119 |
| P. aeruginosa | Differential carbon source metabolism | 86 |
| Fitness of rpoB mutants is increased when treated with sub-inhibitory concentrations of protein synthesis inhibitors | 86 | |
| S. aureus | Better biofilm formation on catheters in mice, and a distinct set of mutations observed during murine infection compared to in vitro growth | 120 |
| Mutations in rpoB are observed in both clinical and lab-derived isolates that have increased vancomycin resistance | 121,122 |
RpoB mutations mimic the stringent response
Antibiotic production in Streptomyces sp. (like Mycobacterium sp., a genus of the phylum Actinobacteria) is fundamentally dependent on pathways activated by the bacterial alarmones, guanosine tetraphosphate and guanosine pentaphosphate (commonly designated as (p)ppGpp).104,105,123 Structural evidence from Thermus thermophilus suggests that (p)ppGpp binds near the active site of bacterial RNAPs via an interaction with the β and β′ subunits.124 It is interesting, therefore, that rpoB mutations corresponding to those found in RIFR Mtb isolates are associated with increased antibiotic synthesis by antibiotic-producer strains of S. coelicolor.39,104,105 Moreover, the same mutations have been shown to induce production of novel antibiotic compounds in S. coelicolor strains previously characterized as antibiotic ‘non-producers'.39 RIFR mutations have also been shown to activate secondary metabolism, as well as induce (p)ppGpp-independent antibiotic production, in strains that are defective in their inability to produce the alarmone.105
The genomes of some Nonomuraea and Nocardia species contain two closely related but non-identical paralogues of rpoB. In these organisms, the second rpoB gene confers RIFR and its activation induces the expression of cryptic genes.117,118,119 Similarly, heterologous expression of the alternate Nonomuraea rpoB gene in S. lividans induces antibiotic biosynthesis.117,118 Again, it is notable that the sequence polymorphisms that differentiate the second rpoB gene from the canonical paralogue correspond to rpoB mutations commonly detected in RIFR Mtb isolates. In combination, these observations suggest that specific rpoB mutations might phenocopy the effects of the (p)ppGpp-mediated stringent response. Consistent with this idea, work in E. coli has shown that, in spoT and relA deletion mutants which are unable to produce (p)ppGpp, RIFR mutations can decrease RNAP stability at promoters that are transcribed during rapid growth, thereby releasing RNAP for transcription at stringently regulated promoters.38 This study examined a selection of promoters, so it remains to be established whether other stringent response genes might be regulated in this way. However, these results imply that, while rpoB mutations might result in defective growth in nutrient rich environments, the capacity of the mutant alleles to mimic the stringent response may be especially beneficial for competitive survival under nutrient-limited conditions.
The recent observation that (p)ppGpp induces expression of a cryptic STRR determinant in S. enterica suggests that, as a result of its broad effects on transcription, this signalling molecule could mediate resistance to other drugs.125 Given the functional overlap between certain rpoB mutations and (p)ppGpp-mediated transcriptional regulation (discussed above), this raises the additional possibility that RIFR strains might be less susceptible than the wild-type parental strains to other antibiotic classes, as has been proposed elsewhere.125 In Mtb, abrogation of the stringent response through targeted deletion of relMtb results in impaired survival under starvation conditions in vitro126 and during chronic infection in a mouse model.127 It will be interesting, therefore, to determine whether the starvation phenotype of the relMtb mutant can be complemented by specific rpoB mutations.
Do rpoB mutations alter cell wall metabolism?
The physiological implications of different rpoB alleles have not been well studied in Mtb. There are, however, some recent papers describing the utilization of ‘omics approaches to characterize RIFR strains. By applying liquid chromatography/tandem mass spectrometry to analyse the proteomes of clinical RIFR Mtb isolates, Bisson et al.113 detected differences in the expression profiles of S531L rpoB mutants of the Mtb Haarlem lineage and H526D mutants of the Beijing lineage compared to corresponding drug-susceptible counterparts with matched spoligotype and restriction fragment length polymorphism patterns. Conserved hypothetical proteins of unknown function were significantly represented among the differentially expresses proteins, which complicates the biological interpretation of these data. Nevertheless, there were striking differences within—and between—Haarlem and Beijing Mtb lineages for proteins involved in the biosynthesis and regulation of phthiocerol dimycocerosate, a methyl-branched fatty acid which has previously been implicated in mycobacterial virulence.128 In this case, however, the RIFR strains were associated with increased levels of phthiocerol dimycocerosate precursors but not the full-length lipid, an observation that requires further analysis. In a separate study, the fatty acid contents of two in vitro-selected RIFR Mtb mutants with distinct rpoB mutations (S531L and S522L) were analyzed by gas chromatography–mass spectrometry.114 Applying this technique, the authors were able to distinguish the respective rpoB mutants from one other, as well as their parental strain based on fatty acid composition. Although these studies are united in identifying alterations in fatty acid metabolism as a feature of RIFR strains, the precise implications for Mtb pathogenesis are unclear.113 Moreover, no whole-genome sequence data were reported for the strains analysed in these studies, so the possibility that other mutations might contribute to the observed phenotypes cannot be excluded.
It seems, therefore, that establishing an unequivocal relationship between rpoB genotype and RIFR phenotype will require the site-directed transfer of selected RIFR-conferring mutations into sequenced Mtb strains. Moreover, if done in the absence of RIF selection, the impact of a specific rpoB mutation on lipid metabolism—or any other bacillary function—could be established without the complication of potentially confounding genetic (or even epigenetic) effects. This approach was applied to a clinical isolate of methicillin-resistant Staphylococcus aureus that was associated with decreased daptomycin susceptibility following passage in VAN.121 Whole-genome resequencing revealed five-point mutations that distinguished the mutant from the parental isolate, including a single mutation in rpoB. Sequential transfer of the identified mutations into the parental strain established that the rpoB mutation was responsible for the decrease in susceptibility to both VAN and daptomycin and, furthermore, that the mechanism of resistance related to thickening of the cell wall.121 The rpoB mutation examined in this study was located outside of the RRDR; however, in intriguing follow-up work, it has been reported that the introduction of a RIFR-conferring H526Y mutation (located inside the RRDR) into a VAN-sensitive strain also results in increased cell wall thickness and decreased VAN susceptibility.122,129
RIFR mutants and the host–pathogen interaction
The interaction between Mtb and its obligate human host is complex and dynamic. As a critical determinant of the clinical outcome of infection, it is understandable that significant resources have been invested in elucidating the mycobacterial130 and host immunological 131,132 pathways and mechanisms that influence the progression of Mtb through the infection cycle. In contrast, the impact of drug resistance on the host–pathogen interaction remains largely unexplored and so represents a key area for future study given the emergence of drug-resistant Mtb as a major global health concern.
There are several studies which have examined the response to Mtb antigens of peripheral blood mononuclear cells (PBMCs) isolated from MDR-TB patients. In one example, PBMCs from MDR-TB patients were shown to produce lower levels of the pro-inflammatory cytokine, interferon (IFN)-γ, in comparison to healthy controls following exposure to purified protein derivative.133 However, since the antigens in purified protein derivative are not specific to Mtb,134 it is possible that the observed response to purified protein derivative stimulation was complicated by prior vaccination with BCG and/or exposure to other mycobacterial species.135 PBMCs from MDR-TB patients have also been associated with decreased production of IFN-γ and tumor-necrosis factor-α following stimulation with the Mtb-specific antigen, ESAT6.136 Similarly, exposure of PBMCs derived from MDR-TB-infected individuals to a wider set of Mtb-specific antigens—specifically, ESAT6, MPT-51 and GlcB—revealed differences in the levels of cytokines produced, as well as the nature of the producer cell populations, whereas CD4+ and CD8+ T cells from drug-susceptible TB patients produced both IFN-γ and IL-10 in response to all antigens, in cell populations isolated from MDR-TB patients, only CD8+ cells produced IFN-γ in response to ESAT6, and IL-10 in response to GlcB and ESAT6.137 These results suggest that CD4+ T-cell responses are diminished in MDR-TB patients and further, that the outcome of the assay is influenced by the selection of stimulatory antigen.
MDR-TB patients are often infected for longer periods than those with drug-susceptible TB. Therefore, it is possible that immune regulation to dampen the inflammatory response partially explains the decreased immune response observed in MDR-TB patients. Consistent with this idea, several studies of individuals with prolonged MDR-TB disease have revealed increased levels of transforming growth factor β, IL-10 and regulatory T cells, all of which are involved in suppressing the inflammatory response.138,139 In turn, this raises a conundrum regarding causation: does an impaired inflammatory response allow the development of drug-resistant Mtb strains in specific hosts, or do resistance-associated mutations alter Mtb physiology to the extent that the host–pathogen interaction is affected? Resolving this question is complicated by the fact that, by definition, MDR strains harbour more than a single resistance-conferring mutation and, potentially, carry multiple additional single-nucleotide polymorphisms.10,11 It seems critical, therefore, to examine the impact of individual drug resistance mutations—including rpoB alleles—on immunological function.
Do rpoB mutations alter competitive growth under stress conditions?
The relative frequency of RIFR mutants in slow- or non-growing bacterial populations has been widely used as a quantitative measure of the capacity of some organisms to increase mutation rates under stress.140,141,142,143 However, pioneering work by Wrande et al.75 demonstrated that clonal expansion of certain rpoB alleles during competitive growth underlies the (erroneously) inferred generation of RIFR mutants through stress-induced mutagenesis. That is, rpoB mutants propagate owing to a selective advantage in stress conditions,75 a phenomenon recently extended to nalidixic acid-resistant gyrA mutations.144 While the mechanisms remain to be determined, the identification of ‘cheater' rpoB mutants reinforces the idea that RIFR alleles might be selected in the absence of drug pressure. In turn, this raises the possibility that RIFR mutants of obligate pathogens such as Mtb might be associated with different epidemiological prevalence owing to altered disease dynamics, something that requires further investigation.
Further evidence of the potential role of rpoB mutations in modulating growth phenotypes is provided by work in Bacillus subtilis, which has shown that the spectrum of RIFR-conferring mutations differs significantly according to growth state.101 Moreover, single amino acid changes in rpoB have been associated with major metabolic changes in the same organism, enabling the utilization of diverse substrates that had not previously been identified as suitable for supporting growth.103 For example, a S531L mutant was better able to use a variety of β-glucosides, compounds that are likely to be prevalent in the soil owing to breakdown of plant material. As the authors note, this suggests that specific rpoB alleles might be beneficial in environments where β-glucosides represent the primary carbon source and, therefore, could drive the competitive expansion of RIFR clones under nutrient-limited conditions.103
Multiple studies have implicated rpoB mutations in the adaptation of E. coli to in vitro stress.110,111,145 For example, whole-genome resequencing identified rpoB and rpoC among a handful of mutated genes following extended growth of E. coli in glycerol minimal medium.110 Critically, transfer of the mutant alleles into a wild-type E. coli background confirmed that the rpoB and rpoC mutations conferred the greatest increase in growth rate in the same medium.110 These observations were validated in follow-up work, which showed that mutants containing at least one rpoB or rpoC polymorphism were associated with better fitness under nutrient limiting conditions, reinforcing the role of RNAP mutations in the adaptation of E. coli to environmental stress.108 More recently, the same group has demonstrated that rpoC mutations are associated with alterations in transcriptional elongation and pausing—and therefore, gene expression—which suggests this as a plausible molecular mechanism for the observed metabolic changes and growth adaptation.109 In related work, E. coli populations were subjected to different stressors in an investigation of short-term evolution.145 Again, this study identified rpoB mutations in strains that were better adapted to carbon limitation, n-butanol, osmotic and acid stress.145 Similarly, a separate study identified rpoB mutants among those strains that had adapted to elevated temperatures.111 In combination, these observations establish the contribution of rpoB (and rpoC) mutations to altered metabolic capacity and further, reinforce the idea that RIFR-associated mutations might emerge in the absence of drug selection.
FUTURE PROSPECTS
Mutations in rpoB have been associated with altered physiology and metabolic function in a variety of bacterial systems. It seems likely, therefore, that equivalent RIFR-associated mutations in rpoB might be under dual selection in Mtb: that is, the combined benefits of RIFR and the physiological advantage(s) of the causal rpoB allele might fix rpoB mutants in the infecting Mtb population (Figure 2). In turn, this raises key questions for future research. For example, do rpoB alleles impact disease pathology? Can RIFR-associated mutations alter transmissibility and/or the ability of MDR strains to establish an infection? Do mutations in other drug targets (e.g., gyrA) influence physiology, as demonstrated recently in Salmonella?85,146 If so, how do different combinations of resistance mutations influence pathogen success? Addressing these and related questions will be critical in determining the full impact of RIFR—and other drug-resistance alleles—on Mtb, a pathogen whose persistence in the human population suggests a unique ability to adapt to dynamic and often hostile host environments.
Figure 2.
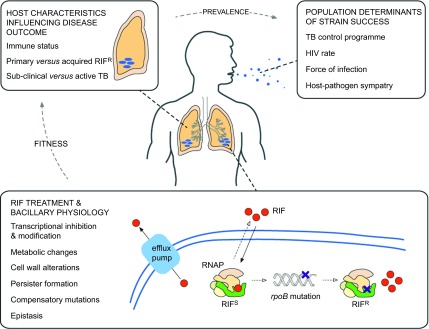
Factors influencing the success of RIFR Mtb strains. Although the focus of this review is on RIF resistance in Mtb, many of the themes are relevant to other drugs and other infectious organisms. RIF treatment and bacillary physiology: within a single bacterial cell, many factors contribute to the development and maintenance of drug resistance. The drug (in this case, RIF) enters the cell by passive diffusion and, once in the cytoplasm, must translocate and bind to its target (here, RNAP). Some organisms encode enzymes that inactivate RIF,147 while recent work suggests that RIF is actively extruded by efflux pumps in Mtb.112,148 The concentration of RIF that is available to bind to RNAP (that is, the effective intracellular concentration) is a major determinant of whether resistance mutations develop or not, and is heavily influenced by the mechanisms described above;149 therefore, the ability to measure this parameter accurately150 will be critical to future efforts to understand the development of drug resistance. As described in the main text, mutations in rpoB might alter the physiology of the RIFR bacterium. Host characteristics influencing disease outcome: any physiological alteration has the potential to influence the interaction of the bacillus with its obligate human host. Similarly, multiple host factors such as age, nutritional status and copathologies will determine infection outcomes, including transmission to other susceptible individuals. Population determinants of strain success: there is an additional layer of complexity when considering the spread of organisms within and between host populations. Factors such as HIV prevalence, force of infection and socioeconomic status will influence the ability of the organism to transmit between hosts.151 While these elements are grouped separately in the figure, it is likely that multiple factors will overlap to influence the success of different Mtb strains.
Acknowledgments
This work was funded by grants from the South African Medical Research Council, the National Research Foundation, the Howard Hughes Medical Institute (Senior International Research Scholar's grant to Valerie Mizrahi) and the South African Tuberculosis and AIDS Training program (NIH/FIC 1U2RTW007370/3) (to Anastasia Koch). We thank Herman de Klerk, freelance designer (http://www.hermandeklerk.com), for technical assistance with the preparation of the figures.
References
- Cars O, Hogberg LD, Murray M, et al. Meeting the challenge of antibiotic resistance. BMJ. 2008;337:a1438. doi: 10.1136/bmj.a1438. [DOI] [PubMed] [Google Scholar]
- Martinez JL, Fajardo A, Garmendia L, et al. A global view of antibiotic resistance. FEMS Microbiol Rev. 2009;33:44–65. doi: 10.1111/j.1574-6976.2008.00142.x. [DOI] [PubMed] [Google Scholar]
- World Health Organization Global tuberculosis report 2013. Geneva: WHO; 2013. Available at http://www.who.int/tb/publications/global_report/en/index.html (accessed 12 December 2013). [Google Scholar]
- Pooran A, Pieterson E, Davids M, Theron G, Dheda K. What is the cost of diagnosis and management of drug resistant tuberculosis in South Africa. PLoS ONE. 2013;8:e54587. doi: 10.1371/journal.pone.0054587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi NR, Nunn P, Dheda K, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet. 2010;375:1830–1843. doi: 10.1016/S0140-6736(10)60410-2. [DOI] [PubMed] [Google Scholar]
- Colijn C, Cohen T, Ganesh A, Murray M. Spontaneous emergence of multiple drug resistance in tuberculosis before and during therapy. PLoS ONE. 2011;6:e18327. doi: 10.1371/journal.pone.0018327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower SM, Chou T. Modeling the emergence of the ‘hot zones': tuberculosis and the amplification dynamics of drug resistance. Nat Med. 2004;10:1111–1116. doi: 10.1038/nm1102. [DOI] [PubMed] [Google Scholar]
- Muller B, Borrell S, Rose G, Gagneux S. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 2012;29:160–169. doi: 10.1016/j.tig.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhat MR, Shapiro BJ, Kieser K, et al. Genomic analysis identifies targets of convergent positive selection in drug resistant Mycobacterium tuberculosis. Nat Genet. 2013;45:1183–1189. doi: 10.1038/ng.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li D, Zhao L, et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat Genet. 2013;45:1255–1260. doi: 10.1038/ng.2735. [DOI] [PubMed] [Google Scholar]
- Sandgren A, Strong M, Muthukrishnan P, Weiner BK, Church GM, Murray MB. Tuberculosis drug resistance mutation database. PLoS Med. 2009;6:e2. doi: 10.1371/journal.pmed.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casali N, Nikolayevskyy V, Balabanova Y, et al. Microevolution of extensively drug-resistant tuberculosis in Russia. Genome Res. 2012;22:735–745. doi: 10.1101/gr.128678.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger TR, Feng Y, Chen X, et al. The non-clonality of drug resistance in Beijing-genotype isolates of Mycobacterium tuberculosis from the Western Cape of South Africa. BMC Genomics. 2010;11:670. doi: 10.1186/1471-2164-11-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Luo T, Yang C, et al. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis. 2012;206:1724–1733. doi: 10.1093/infdis/jis601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance. Nat Rev Microbiol. 2010;8:260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- Borukhov S, Nudler E. RNA polymerase: the vehicle of transcription. Trends Microbiol. 2008;16:126–134. doi: 10.1016/j.tim.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Campbell EA, Korzheva N, Mustaev A, et al. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001;104:901–912. doi: 10.1016/s0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Comas I, Coscolla M, Luo T, et al. Out-of-Africa migration and Neolithic co-expansion of tuberculosis with modern humans. Nat Genet. 2013;45:1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison DA. Role of individual drugs in the chemotherapy of tuberculosis. Int J Tuberc Lung Dis. 2000;4:796–806. [PubMed] [Google Scholar]
- Forrest GN, Tamura K. Rifampin combination therapy for nonmycobacterial infections. Clin Microbiol Rev. 2010;23:14–34. doi: 10.1128/CMR.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsitch M, Levin BR. The population dynamics of antimicrobial chemotherapy. Antimicrob Agents Chemother. 1997;41:363–373. doi: 10.1128/aac.41.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Siddiqi N, Rubin EJ. Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2005;49:4778–4780. doi: 10.1128/AAC.49.11.4778-4780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccaro G, Giannoni F, Filippini P, Mustazzolu A, Fattorini L. Activities of drug combinations against Mycobacterium tuberculosis grown in aerobic and hypoxic acidic conditions. Antimicrob Agents Chemother. 2013;57:1428–1433. doi: 10.1128/AAC.02154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Dhar N, Hartkoorn RC, et al. Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. . Antimicrob Agents Chemother. 2010;54:4150–4158. doi: 10.1128/AAC.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly LE, Edelstein PH, Ramakrishnan L. Why is long-term therapy required to cure tuberculosis. PLoS Med. 2007;4:e120. doi: 10.1371/journal.pmed.0040120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry CE, 3rd, Boshoff HI, Dartois V, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittershaus ES, Baek SH, Sassetti CM. The normalcy of dormancy: common themes in microbial quiescence. Cell Host Microbe. 2013;13:643–651. doi: 10.1016/j.chom.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald PR, van Helden PD. The global burden of tuberculosis—combating drug resistance in difficult times. N Engl J Med. 2009;360:2393–2395. doi: 10.1056/NEJMp0903806. [DOI] [PubMed] [Google Scholar]
- Zhang G, Campbell EA, Minakhin L, Richter C, Severinov K, Darst SA. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 A resolution. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
- Borukhov S, Nudler E. RNA polymerase holoenzyme: structure, function and biological implications. Curr Opin Microbiol. 2003;6:93–100. doi: 10.1016/s1369-5274(03)00036-5. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Musser JM. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuber Lung Dis. 1998;79:3–29. doi: 10.1054/tuld.1998.0002. [DOI] [PubMed] [Google Scholar]
- Jin DJ, Gross CA. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol. 1988;202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Telenti A. Genetics of drug resistance in Mycobacterium tuberculosis. Molecular genetics of mycobacteria. Washington, DC: ASM Press; 2000. [Google Scholar]
- Heep M, Brandstatter B, Rieger U, et al. Frequency of rpoB mutations inside and outside the cluster I region in rifampin-resistant clinical Mycobacterium tuberculosis isolates. J Clin Microbiol. 2001;39:107–110. doi: 10.1128/JCM.39.1.107-110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heep M, Odenbreit S, Beck D, et al. Mutations at four distinct regions of the rpoB gene can reduce the susceptibility of Helicobacter pylori to rifamycins. Antimicrob Agents Chemother. 2000;44:1713–1715. doi: 10.1128/aac.44.6.1713-1715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenti A, Imboden P, Marchesi F, et al. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet. 1993;341:647–650. doi: 10.1016/0140-6736(93)90417-f. [DOI] [PubMed] [Google Scholar]
- Zhou YN, Jin DJ. The rpoB mutants destabilizing initiation complexes at stringently controlled promoters behave like “stringent” RNA polymerases in Escherichia coli. Proc Natl Acad Sci USA. 1998;95:2908–2913. doi: 10.1073/pnas.95.6.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka T, Ohnishi-Kameyama M, Muramatsu H, et al. Antibacterial discovery in actinomycetes strains with mutations in RNA polymerase or ribosomal protein S12. Nat Biotechnol. 2009;27:462–464. doi: 10.1038/nbt.1538. [DOI] [PubMed] [Google Scholar]
- Singer M, Jin DJ, Walter WA, Gross CA. Genetic evidence for the interaction between cluster I and cluster III rifampicin resistant mutations. J Mol Biol. 1993;231:1–5. doi: 10.1006/jmbi.1993.1251. [DOI] [PubMed] [Google Scholar]
- Jin DJ, Gross CA. Characterization of the pleiotropic phenotypes of rifampin-resistant rpoB mutants of Escherichia coli. J Bacteriol. 1989;171:5229–5231. doi: 10.1128/jb.171.9.5229-5231.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin DJ, Gross CA. RpoB8, a rifampicin-resistant termination-proficient RNA polymerase, has an increased Km for purine nucleotides during transcription elongation. J Biol Chem. 1991;266:14478–14485. [PubMed] [Google Scholar]
- Jin DJ, Walter WA, Gross CA. Characterization of the termination phenotypes of rifampicin-resistant mutants. J Mol Biol. 1988;202:245–253. doi: 10.1016/0022-2836(88)90455-x. [DOI] [PubMed] [Google Scholar]
- Landick R, Stewart J, Lee DN. Amino acid changes in conserved regions of the beta-subunit of Escherichia coli RNA polymerase alter transcription pausing and termination. Genes Dev. 1990;4:1623–1636. doi: 10.1101/gad.4.9.1623. [DOI] [PubMed] [Google Scholar]
- Yanofsky C, Horn V. Rifampin resistance mutations that alter the efficiency of transcription termination at the tryptophan operon attenuator. J Bacteriol. 1981;145:1334–1341. doi: 10.1128/jb.145.3.1334-1341.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham CJ, Furneaux PA. Mutations in the ss subunit of the Bacillus subtilis RNA polymerase that confer both rifampicin resistance and hypersensitivity to NusG. Microbiology. 2000;146:3041–3049. doi: 10.1099/00221287-146-12-3041. [DOI] [PubMed] [Google Scholar]
- Trautinger BW, Lloyd RG. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J. 2002;21:6944–6953. doi: 10.1093/emboj/cdf654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan AK, Smith AJ, Savery NJ, Zamos P, Hanawalt PC. Transcription coupled nucleotide excision repair in Escherichia coli can be affected by changing the arginine at position 529 of the beta subunit of RNA polymerase. DNA Repair (Amst) 2007;6:1434–1440. doi: 10.1016/j.dnarep.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharoglu Z, Lestini R, Duigou S, Michel B. RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Mol Microbiol. 2010;77:324–336. doi: 10.1111/j.1365-2958.2010.07208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabbs ER. New tool for studying interactions of components of ribonucleic acid polymerase: rifampin-dependent mutants. J Bacteriol. 1979;139:1072–1074. doi: 10.1128/jb.139.3.1072-1074.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Wang Y, Sun C, Yin H, Wang A, Lou L.[Growth of rifampin-dependent Mycobacterium tuberculosis in conditions without rifampin] Zhonghua Jie He He Hu Xi Za Zhi 200225588–590.Chinese. [PubMed] [Google Scholar]
- Zhong M, Zhang X, Wang Y, et al. An interesting case of rifampicin-dependent/-enhanced multidrug-resistant tuberculosis. Int J Tuberc Lung Dis. 2010;14:40–44. [PubMed] [Google Scholar]
- Honore N, Marchal G, Cole ST. Novel mutation in 16S rRNA associated with streptomycin dependence in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1995;39:769–770. doi: 10.1128/AAC.39.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokrousov I. Multiple rpoB mutants of Mycobacterium tuberculosis and second-order selection. Emerg Infect Dis. 2004;10:1337–1338. doi: 10.3201/eid1007.030598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Warner DF. The role of DNA repair in M. tuberculosis pathogenesis. Drug Discov Today. 2010;7:e5–e11. [Google Scholar]
- Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 2009;11:1170–1178. doi: 10.1111/j.1462-5822.2009.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan DM, McHugh TD, Gillespie SH. Analysis of rpoB and pncA mutations in the published literature: an insight into the role of oxidative stress in Mycobacterium tuberculosis evolution. J Antimicrob Chemother. 2005;55:674–679. doi: 10.1093/jac/dki069. [DOI] [PubMed] [Google Scholar]
- Ford CB, Shah RR, Maeda MK, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013;45:784–790. doi: 10.1038/ng.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu GK, Zhang Y, Lau TC, et al. Mutations outside the rifampicin resistance-determining region associated with rifampicin resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2011;66:730–733. doi: 10.1093/jac/dkq519. [DOI] [PubMed] [Google Scholar]
- Van Deun A, Martin A, Palomino JC. Diagnosis of drug-resistant tuberculosis: reliability and rapidity of detection. Int J Tuberc Lung Dis. 2010;14:131–140. [PubMed] [Google Scholar]
- Lindsey HA, Gallie J, Taylor S, Kerr B. Evolutionary rescue from extinction is contingent on a lower rate of environmental change. Nature. 2013;494:463–467. doi: 10.1038/nature11879. [DOI] [PubMed] [Google Scholar]
- Jenkins C, Bacon J, Allnutt J, et al. Enhanced heterogeneity of rpoB in Mycobacterium tuberculosis found at low pH. J Antimicrob Chemother. 2009;63:1118–1120. doi: 10.1093/jac/dkp125. [DOI] [PubMed] [Google Scholar]
- Mariam DH, Mengistu Y, Hoffner SE, Anderson DI. Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. . Antimicrob Agents Chemother. 2004;48:1289–1294. doi: 10.1128/AAC.48.4.1289-1294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson DI. The biological cost of mutational antibiotic resistance: any practical conclusions. Curr Opin Microbiol. 2006;9:461–465. doi: 10.1016/j.mib.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Gagneux S, Burgos MV, DeRiemer K, et al. Impact of bacterial genetics on the transmission of isoniazid-resistant Mycobacterium tuberculosis. PLos Pathog. 2006;2:e61. doi: 10.1371/journal.ppat.0020061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312:1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- Cohen T, Murray M. Modeling epidemics of multidrug-resistant M. tuberculosis of heterogenous fitness. Nat Med. 2004;10:1117–1121. doi: 10.1038/nm1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell S, Gagneux S. Infectiousness, reproductive fitness and evolution of drug-resistant Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 2009;13:1456–1466. [PubMed] [Google Scholar]
- Brandis G, Hughes D. Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates. J Antimicrob Chemother. 2013;68:2493–2497. doi: 10.1093/jac/dkt224. [DOI] [PubMed] [Google Scholar]
- Billington OJ, McHugh TD, Gillespie SH. Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1999;43:1866–1869. doi: 10.1128/aac.43.8.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AP, Billington OJ, Bannister BA, Weir WR, McHugh TD, Gillespie SH. Comparison of fitness of two isolates of Mycobacterium tuberculosis, one of which had developed multi-drug resistance during the course of treatment. J Infect. 2000;41:184–187. doi: 10.1053/jinf.2000.0711. [DOI] [PubMed] [Google Scholar]
- Gillespie SH, Billington OJ, Breathnach A, McHugh TD. Multiple drug-resistant Mycobacterium tuberculosis: evidence for changing fitness following passage through human hosts. Microb Drug Resist. 2002;8:273–279. doi: 10.1089/10766290260469534. [DOI] [PubMed] [Google Scholar]
- Kolter R, Siegele DA, Tormo A. The stationary phase of the bacterial life cycle. Annu Rev Microbiol. 1993;47:855–874. doi: 10.1146/annurev.mi.47.100193.004231. [DOI] [PubMed] [Google Scholar]
- Wrande M, Roth , Hughes D. Accumulation of mutants in “aging” bacterial colonies is due to growth under selection, not stress-induced mutagenesis. Proc Natl Acad Sci USA. 2008;105:11863–11868. doi: 10.1073/pnas.0804739105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barczak AK, Domenech P, Boshoff HI, et al. In vivo phenotypic dominance in mouse mixed infections with Mycobacterium tuberculosis clinical isolates. J Infect Dis. 2005;192:600–606. doi: 10.1086/432006. [DOI] [PubMed] [Google Scholar]
- Subbian S, Bandyopadhyay N, Tsenova L, et al. Early innate immunity determines outcome of Mycobacterium tuberculosis pulmonary infection in rabbits. Cell Commun Signal. 2013;11:60. doi: 10.1186/1478-811X-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safi H, Lingaraju S, Amin A, et al. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-β-𝒹-arabinose biosynthetic and utilization pathway genes. Nat Genet. 2013;45:1190–1197. doi: 10.1038/ng.2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Borrell S, Roetzer A, et al. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. 2012;44:106–110. doi: 10.1038/ng.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firdessa R, Berg S, Hailu E, et al. Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis, Ethiopia. Emerg Infect Dis. 2013;19:460–463. doi: 10.3201/eid1903.120256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos M, Muller B, Borrell S, et al. Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob Agents Chemother. 2013;57:827–832. doi: 10.1128/AAC.01541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandis G, Wrande M, Liljas L, Hughes D. Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol Microbiol. 2012;85:142–151. doi: 10.1111/j.1365-2958.2012.08099.x. [DOI] [PubMed] [Google Scholar]
- Cordell HJ. Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11:2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- Trindade S, Sousa A, Xavier KB, Dionisio F, Ferreira MG, Gordo I. Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet. 2009;5:e1000578. doi: 10.1371/journal.pgen.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker S, Duy PT, Nga TV, et al. Fitness benefits in fluoroquinolone-resistant Salmonella Typhi in the absence of antimicrobial pressure. Elife. 2013;2:e01229. doi: 10.7554/eLife.01229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AR, James IC, Maclean C. The fitness cost of rifampicin resistance in Pseudomonas aeruginosa depends on demand for RNA polymerase. Genetics. 2011;187:817–822. doi: 10.1534/genetics.110.124628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergval I, Kwok B, Schuitema A, et al. Pre-existing isoniazid resistance, but not the genotype of Mycobacterium tuberculosis drives rifampicin resistance codon preference in vitro. PLoS ONE. 2012;7:e29108. doi: 10.1371/journal.pone.0029108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazbon MH, Brimacombe M, Bobadilla del Valle M, et al. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2006;50:2640–2649. doi: 10.1128/AAC.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Soolingen D, de Haas PE, van Doorn HR, Kuijper E, Rinder H, Borgdorff MW. Mutations at amino acid position 315 of the katG gene are associated with high-level resistance to isoniazid, other drug resistance, and successful transmission of Mycobacterium tuberculosis in the Netherlands. J Infect Dis. 2000;182:1788–1790. doi: 10.1086/317598. [DOI] [PubMed] [Google Scholar]
- Borrell S, Teo Y, Giardina F, et al. Epistasis between antibiotic resistance mutations drives the evolution of extensively drug-resistant tuberculosis. EMPH. 2013;2013:65–74. doi: 10.1093/emph/eot003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovinskiy N, Agbleke AA, Chesnokova O, Pang Z, Higgins NP. Rates of gyrase supercoiling and transcription elongation control supercoil density in a bacterial chromosome. PLoS Genet. 2012;8:e1002845. doi: 10.1371/journal.pgen.1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagneux S, DeRiemer K, Van T, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:2869–2873. doi: 10.1073/pnas.0511240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakov D, Akbergenov R, Matt T, Sander P, Andersson DI, Bottger EC. Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in-vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol Microbiol. 2010;77:830–840. doi: 10.1111/j.1365-2958.2010.07218.x. [DOI] [PubMed] [Google Scholar]
- Saunders NJ, Trivedi UH, Thomson ML, Doig C, Laurenson IF, Blaxter ML. Deep resequencing of serial sputum isolates of Mycobacterium tuberculosis during therapeutic failure due to poor compliance reveals stepwise mutation of key resistance genes on an otherwise stable genetic background. J Infect. 2011;62:212–217. doi: 10.1016/j.jinf.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Drobniewski FA, Caws M, Gibson A, Young D. Modern laboratory diagnosis of tuberculosis. Lancet Infect Dis. 2003;3:141–147. doi: 10.1016/s1473-3099(03)00544-9. [DOI] [PubMed] [Google Scholar]
- Strauss O, Warren R, Jordaan A, et al. Spread of a low-fitness drug-resistant Mycobacterium tuberculosis strain in a setting of high human immunodeficiency virus prevalence. J Clin Microbiol. 2008;46:1514–1516. doi: 10.1128/JCM.01938-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S, Cook VJ, Wolfe JN, Tang P, Elwood K, Sharma MK. A mutation in Mycobacterium tuberculosis rpoB gene confers rifampin resistance in three HIV-TB cases. Tuberculosis (Edinb) 2010;90:152–157. doi: 10.1016/j.tube.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Motiwala AS, Dai Y, Jones-Lopez EC, et al. Mutations in extensively drug-resistant Mycobacterium tuberculosis that do not code for known drug-resistance mechanisms. J Infect Dis. 2010;201:881–888. doi: 10.1086/650999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane WJ, Darst SA. Molecular evolution of multisubunit RNA polymerases: sequence analysis. J Mol Biol. 2010;395:671–685. doi: 10.1016/j.jmb.2009.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane WJ, Darst SA. Molecular evolution of multisubunit RNA polymerases: structural analysis. J Mol Biol. 2010;395:686–704. doi: 10.1016/j.jmb.2009.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson WL, Maughan H. The spectrum of spontaneous rifampin resistance mutations in the rpoB gene of Bacillus subtilis 168 spores differs from that of vegetative cells and resembles that of Mycobacterium tuberculosis. J Bacteriol. 2002;184:4936–4940. doi: 10.1128/JB.184.17.4936-4940.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maughan H, Galeano B, Nicholson WL. Novel rpoB mutations conferring rifampin resistance on Bacillus subtilis: global effects on growth, competence, sporulation, and germination. J Bacteriol. 2004;186:2481–2486. doi: 10.1128/JB.186.8.2481-2486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins AE, Nicholson WL. Uncovering new metabolic capabilities of Bacillus subtilis using phenotype profiling of rifampin-resistant rpoB mutants. J Bacteriol. 2008;190:807–814. doi: 10.1128/JB.00901-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Tozawa Y, Lai C, Hayashi H, Ochi K. A rifampicin resistance mutation in the rpoB gene confers ppGpp-independent antibiotic production in Streptomyces coelicolor A3(2) Mol Genet Genomics. 2002;268:179–189. doi: 10.1007/s00438-002-0730-1. [DOI] [PubMed] [Google Scholar]
- Hu H, Zhang Q, Ochi K. Activation of antibiotic biosynthesis by specified mutations in the rpoB gene (encoding the RNA polymerase beta subunit) of Streptomyces lividans. J Bacteriol. 2002;184:3984–3991. doi: 10.1128/JB.184.14.3984-3991.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaoka T, Takahashi K, Yada H, Yoshida M, Ochi K. RNA polymerase mutation activates the production of a dormant antibiotic 3,3′-neotrehalosadiamine via an autoinduction mechanism in Bacillus subtilis. J Biol Chem. 2004;279:3885–3892. doi: 10.1074/jbc.M309925200. [DOI] [PubMed] [Google Scholar]
- Murphy H, Cashel M. Isolation of RNA polymerase suppressors of a (p) ppGpp deficiency. Methods Enzymol. 2003;371:596–601. doi: 10.1016/S0076-6879(03)71044-1. [DOI] [PubMed] [Google Scholar]
- Applebee MK, Herrgard MJ, Palsson BO. Impact of individual mutations on increased fitness in adaptively evolved strains of Escherichia coli. J Bacteriol. 2008;190:5087–5094. doi: 10.1128/JB.01976-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad TM, Frazier M, Joyce AR, et al. RNA polymerase mutants found through adaptive evolution reprogram Escherichia coli for optimal growth in minimal media. Proc Natl Acad Sci USA. 2010;107:20500–20505. doi: 10.1073/pnas.0911253107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring CD, Raghunathan A, Honisch C, et al. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet. 2006;38:1406–1412. doi: 10.1038/ng1906. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Verdugo A, Gaut BS, Tenaillon O. Evolution of Escherichia coli rifampicin resistance in an antibiotic-free environment during thermal stress. BMC Evol Biol. 2013;13:50. doi: 10.1186/1471-2148-13-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louw GE, Warren RM, Gey van Pittius NC, et al. Rifampicin reduces susceptibility to ofloxacin in rifampicin-resistant Mycobacterium tuberculosis through efflux. Am J Respir Crit Care Med. 2011;184:269–276. doi: 10.1164/rccm.201011-1924OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson GP, Mehaffy C, Broeckling C, et al. Upregulation of the phthiocerol dimycocerosate biosynthetic pathway by rifampin-resistant, rpoB mutant Mycobacterium tuberculosis. J Bacteriol. 2012;194:6441–6452. doi: 10.1128/JB.01013-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Preez I, Loots D. Altered fatty acid metabolism due to rifampicin-resistance conferring mutations in the rpoB gene of Mycobacterium tuberculosis: mapping the potential of pharmaco-metabolomics for global health and personalized medicine. Omics. 2012;16:596–603. doi: 10.1089/omi.2012.0028. [DOI] [PubMed] [Google Scholar]
- Bergval IL, Klatser PR, Schuitema AR, Oskam L, Anthony RM. Specific mutations in the Mycobacterium tuberculosis rpoB gene are associated with increased dnaE2 expression. FEMS Microbiol Lett. 2007;275:338–343. doi: 10.1111/j.1574-6968.2007.00905.x. [DOI] [PubMed] [Google Scholar]
- Abadi FJ, Carter PE, Cash P, Pennington TH. Rifampin resistance in Neisseria meningitidis due to alterations in membrane permeability. Antimicrob Agents Chemother. 1996;40:646–651. doi: 10.1128/aac.40.3.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tala A, Wang G, Zemanova M, Okamoto S, Ochi K, Alifano P. Activation of dormant bacterial genes by Nonomuraea sp. strain ATCC 39727 mutant-type RNA polymerase. J Bacteriol. 2009;191:805–814. doi: 10.1128/JB.01311-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigliotta G, Tredici SM, Damiano F, et al. Natural merodiploidy involving duplicated rpoB alleles affects secondary metabolism in a producer actinomycete. Mol Microbiol. 2005;55:396–412. doi: 10.1111/j.1365-2958.2004.04406.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa J, Chiba K, Kurita H, Satoh H. Contribution of rpoB2 RNA polymerase beta subunit gene to rifampin resistance in Nocardia species. Antimicrob Agents Chemother. 2006;50:1342–1346. doi: 10.1128/AAC.50.4.1342-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Wu J, Francis KP, Purchio TF, Kadurugamuwa JL. Monitoring in vivo fitness of rifampicin-resistant Staphylococcus aureus mutants in a mouse biofilm infection model. J Antimicrob Chemother. 2005;55:528–534. doi: 10.1093/jac/dki053. [DOI] [PubMed] [Google Scholar]
- Cui L, Isii T, Fukuda M, et al. An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob Agents Chemother. 2010;54:5222–5233. doi: 10.1128/AAC.00437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Cui L, Katayama Y, Kozue K, Hiramatsu K. Impact of rpoB mutations on reduced vancomycin susceptibility in Staphylococcus aureus. J Clin Microbiol. 2011;49:2680–2684. doi: 10.1128/JCM.02144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi K. Metabolic initiation of differentiation and secondary metabolism by Streptomyces griseus: significance of the stringent response (ppGpp) and GTP content in relation to A factor. J Bacteriol. 1987;169:3608–3616. doi: 10.1128/jb.169.8.3608-3616.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artsimovitch I, Patlan V, Sekine S, et al. Structural basis for transcription regulation by alarmone ppGpp. Cell. 2004;117:299–310. doi: 10.1016/s0092-8674(04)00401-5. [DOI] [PubMed] [Google Scholar]
- Koskiniemi S, Pranting M, Gullberg E, Nasvall J, Andersson DI. Activation of cryptic aminoglycoside resistance in Salmonella enterica. Mol Microbiol. 2011;80:1464–1478. doi: 10.1111/j.1365-2958.2011.07657.x. [DOI] [PubMed] [Google Scholar]
- Primm TP, Andersen SJ, Mizrahi V, Avarbock D, Rubin H, Barry CE., 3rd The stringent response of Mycobacterium tuberculosis is required for long-term survival. J Bacteriol. 2000;182:4889–4898. doi: 10.1128/jb.182.17.4889-4898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JL, Kraus CN, Boshoff HI, et al. The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc Natl Acad Sci USA. 2003;100:10026–10031. doi: 10.1073/pnas.1631248100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M, Stadthagen G, Gicquel B. Long-chain multiple methyl-branched fatty acid-containing lipids of Mycobacterium tuberculosis: biosynthesis, transport, regulation and biological activities. Tuberculosis (Edinb) 2007;87:78–86. doi: 10.1016/j.tube.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Matsuo M, Hishinuma T, Katayama Y, Cui L, Kapi M, Hiramatsu K. Mutation of RNA polymerase β subunit (rpoB) promotes hVISA-to-VISA phenotypic conversion of strain Mu3. Antimicrob Agents Chemother. 2011;55:4188–4195. doi: 10.1128/AAC.00398-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S, Rhee K. Mycobacterium tuberculosis metabolism and host interaction: mysteries and paradoxes. Curr Top Microbiol Immunol. 2013;374:163–188. doi: 10.1007/82_2012_299. [DOI] [PubMed] [Google Scholar]
- Russell DG. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev. 2011;240:252–268. doi: 10.1111/j.1600-065X.2010.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. 2012;12:581–591. doi: 10.1038/nri3259. [DOI] [PubMed] [Google Scholar]
- Lee J, Song C, Kim C, et al. Profiles of IFN-γ and its regulatory cytokines (IL-12, IL-18 and IL-10) in peripheral blood mononuclear cells from patients with multidrug-resistant tuberculosis. Clin Exp Immunol. 2002;128:516–524. doi: 10.1046/j.1365-2249.2002.01858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad TS, Verma R, Kumar S, et al. Proteomic analysis of purified protein derivative of Mycobacterium tuberculosis. Clin Proteomics. 2013;10:8. doi: 10.1186/1559-0275-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Pinxteren LA, Ravn P, Agger EM, Pollock J, Andersen P. Diagnosis of tuberculosis based on the two specific antigens ESAT-6 and CFP10. Clin Diagn Lab Immunol. 2000;7:155–160. doi: 10.1128/cdli.7.2.155-160.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes A, Pereira K, Antas PR, et al. Detection of in vitro interferon-gamma and serum tumour necrosis factor-alpha in multidrug-resistant tuberculosis patients. Clin Exp Immunol. 2005;141:541–548. doi: 10.1111/j.1365-2249.2005.02872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo-Filho JA, Vasconcelos AC, Jr, Martins de Sousa E, Kipnis A, Ribeiro E, Junqueira-Kipnis AP. Cellular responses to MPT-51, GlcB and ESAT-6 among MDR-TB and active tuberculosis patients in Brazil. Tuberculosis (Edinb) 2008;88:474–481. doi: 10.1016/j.tube.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Castro AZ, Diaz-Bardalez BM, Oliveira EC, et al. Abnormal production of transforming growth factor β and interferon γ by peripheral blood cells of patients with multidrug-resistant pulmonary tuberculosis in Brazil. J Infect. 2005;51:318–324. doi: 10.1016/j.jinf.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Wu YE, Peng WG, Cai YM, et al. Decrease in CD4+ CD25+ FoxP3+ Treg cells after pulmonary resection in the treatment of cavity multidrug-resistant tuberculosis. Int J Infect Dis. 2010;14:e815. doi: 10.1016/j.ijid.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Karunakaran P, Davies J. Genetic antagonism and hypermutability in Mycobacterium smegmatis. J Bacteriol. 2000;182:3331–3335. doi: 10.1128/jb.182.12.3331-3335.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjedov I, Tenaillon O, Gerard B, et al. Stress-induced mutagenesis in bacteria. Science. 2003;300:1404–1409. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- Cairns J, Overbaugh J, Miller S. The origin of mutants. Nature. 1988;335:142–145. doi: 10.1038/335142a0. [DOI] [PubMed] [Google Scholar]
- Rosenberg SM, Hastings PJ. Adaptive point mutation and adaptive amplification pathways in the Escherichia coli Lac system: stress responses producing genetic change. J Bacteriol. 2004;186:4838–4843. doi: 10.1128/JB.186.15.4838-4843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz S, Hershberg R. Elevated mutagenesis does not explain the increased frequency of antibiotic resistant mutants in starved aging colonies. PLOS Genet. 2013;9:e1003968. doi: 10.1371/journal.pgen.1003968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragosits M, Mozhayskiy V, Quinones-Soto S, Park J, Tagkopoulos I. Evolutionary potential, cross-stress behavior and the genetic basis of acquired stress resistance in Escherichia coli. Mol Syst Biol. 2013;9:643. doi: 10.1038/msb.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber MA, Ricci V, Whitehead R, et al. Clinically relevant mutant DNA gyrase alters supercoiling, changes the transcriptome, and confers multidrug resistance. MBio. 2013;4:e00273-13. doi: 10.1128/mBio.00273-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan S, Venter H, Dabbs ER. Ribosylative inactivation of rifampin by Mycobacterium smegmatis is a principal contributor to its low susceptibility to this antibiotic. Antimicrob Agents Chemother. 1997;41:2456–2460. doi: 10.1128/aac.41.11.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams KN, Takaki K, Connolly LE, et al. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. 2011;145:39–53. doi: 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fange D, Nilsson K, Tenson T, Ehrenberg M. Drug efflux pump deficiency and drug target resistance masking in growing bacteria. Proc Natl Acad Sci USA. 2009;106:8215–8220. doi: 10.1073/pnas.0811514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat J, Narayan A, Venkatraman J, Chatterji M. LC-MS based assay to measure intracellular compound levels in Mycobacterium smegmatis: Linking compound levels to cellular potency. J Microbiol Methods. 2013;94:152–158. doi: 10.1016/j.mimet.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Wood R, Lawn SD, Johnstone-Robertson S, Bekker LG. Tuberculosis control has failed in South Africa—time to reappraise strategy. S Afr Med J. 2011;101:111–114. doi: 10.7196/samj.4587. [DOI] [PubMed] [Google Scholar]