

Abstract
The oxidative phosphorylation (OXPHOS) system in mitochondria is responsible for the generation of the majority of cellular energy in the form of ATP. Patients with genetic OXPHOS disorders form the largest group of inborn errors of metabolism. Unfortunately, there is still a lack of efficient therapies for these disorders other than management of symptoms. Developing therapies has been complicated because, although the total group of OXPHOS patients is relatively large, there is enormous clinical and genetic heterogeneity within this patient population. Thus there has been a lot of interest in generating relevant mouse models for the different kinds of OXPHOS disorders. The most common treatment strategies tested in these mouse models have aimed to up-regulate mitochondrial biogenesis, in order to increase the residual OXPHOS activity present in affected animals and thereby to ameliorate the energy deficiency. Drugs such as bezafibrate, resveratrol and AICAR target the master regulator of mitochondrial biogenesis PGC-1α either directly or indirectly to manipulate mitochondrial metabolism. This review will summarize the outcome of preclinical treatment trials with these drugs in mouse models of OXPHOS disorders and discuss similar treatments in a number of mouse models of common diseases in which pathology is closely linked to mitochondrial dysfunction. In the majority of these studies the pharmacological activation of the PGC-1α axis shows true potential as therapy; however, other effects besides mitochondrial biogenesis may be contributing to this as well.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: mitochondria, oxidative phosphorylation, PGC-1α, mitochondrial biogenesis, bezafibrate, resveratrol, AICAR, mouse models, mitochondrial disease
Mitochondria as the powerhouse and mitochondrial disease
Mitochondria are involved in numerous processes but they are best known for their role as the powerhouse of the cell. The oxidative phosphorylation (OXPHOS) system located in the mitochondrial inner membrane integrates the production of cellular energy in the form of ATP, with all the major catabolic pathways including fatty acid oxidation (FAO), the tricarboxylic acid cycle and amino acid oxidation. The OXPHOS system consists of five multimeric protein complexes, complex I–V (CI–V). Although critical OXPHOS subunits are encoded by the circular DNA molecules residing inside mitochondria (mtDNA), the majority of the subunits, together with all the proteins required for mtDNA transcription and translation, are encoded by the nuclear DNA (nDNA). This forms an extra layer of regulatory complexity in order to get the right balance in both the nuclear and mitochondrial subunits together for a functional OXPHOS system.
Historically, the term ‘respiratory chain’ refers only to CI–IV and has its starting point at the oxidation of NADH by CI (NADH-ubiquinone oxidoreductase), or succinate by CII (succinate-ubiquinone oxidoreductase). These metabolites originate from the degradation of sugars, protein and lipids, and their oxidation initiates a series of redox reactions up until the actual respiration at CIV, where oxygen is the final electron acceptor. The chain of redox reactions coincides with proton pumping at CI, CIII and CIV from the matrix into the intermembrane space, giving rise to an electrochemical gradient known as the mitochondrial membrane potential which is used by CV to drive ATP synthesis.
Inherited OXPHOS disorders, also known as mitochondrial diseases, are the most common group of inborn errors of metabolism, affecting approximately 1:5000 people (Skladal et al., 2003). These diseases may affect any organ at any age of onset and most often affect tissues that have a high-energy demand such as heart, brain, skeletal muscle and liver. Because of the complex nature of the genetics of the OXPHOS system, OXPHOS disorders can occur via almost any mode of inheritance. Moreover, a clear genotype–phenotype relationship is only sometimes apparent. Mutations have been found in nuclear and mtDNA genes encoding OXPHOS subunits, mtDNA, tRNA and rRNA genes, and nuclear genes encoding proteins required for the assembly of the OXPHOS complexes or for mtDNA replication, transcription and translation (Ylikallio and Suomalainen, 2012).
In recent years enormous progress has been made in the molecular diagnosis of patients with clinically diagnosed OXPHOS disorders by combining technological advances in the area of bioinformatics and molecular biology (Calvo et al., 2012; Falk et al., 2012; Wong, 2013). This is very important for parents as this shortens their search for diagnosis and may provide them with reproductive options in case of planning for subsequent pregnancies. On the other hand, there is unfortunately still no effective treatment for patients with OXPHOS disorders (Pfeffer et al., 2012). Current practice consists of management of symptoms and may include dietary intervention based on anecdotal success (Wallace et al., 2010).
One of the reasons for the lack of therapeutic options has been a lack of relevant animal models, especially in mammals, for preclinical trials. The generation of viable mouse models with useful phenotypes has been difficult because of the crucial role of the OXPHOS system. Moreover, genetic manipulation of mtDNA has been a real technical challenge. Nevertheless, recently, there have been various mouse models for OXPHOS disorders generated with mutations in nDNA or mtDNA. It is no surprise that one model does not suffice for all OXPHOS disorders when considering the entire range of phenotypes in patients. For an overview of mouse models of OXPHOS disorders. please see the following reviews: Dogan and Trifunovic (2011); Komen and Thorburn (2012); and Torraco et al. (2009).
Mitochondrial biogenesis induction as treatment for mitochondrial disease
A popular treatment strategy for mouse models of OXPHOS disorders has been the stimulation of mitochondrial biogenesis. Boosting mitochondrial numbers would hypothetically increase the residual OXPHOS activity present in all patients. This would enhance the cellular ATP synthesis capacity and hopefully ameliorate the cellular energy deficit. This review will focus on the outcome of studies in mouse models of OXPHOS disorders in which pharmacological induction of mitochondrial biogenesis has been trialled as therapy. In addition, similar treatment studies with mouse models of generic diseases in which mitochondrial dysfunction has been linked to pathogenesis will be discussed with the emphasis on mitochondrial biology. These mouse models include models for Duchenne muscular dystrophy (DMD), Huntington's disease (HD), amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD) and Alzheimer's disease (AD). Effects of the various treatments on these models outside the context of mitochondrial function will mainly be beyond the scope of this review.
Furthermore, mitochondrial dysfunction has been implicated as an integral part in the pathology of non-genetic metabolic disorders, such as obesity and metabolic syndrome (Joseph et al., 2012; Szendroedi et al., 2012). Whole-body energy balance is a key concept in these disorders, with a long-standing interest in the role of brown fat, which is rich in mitochondria and can dissipate energy efficiently through non-shivering thermogenesis due to high levels of the uncoupling protein 1 (Wu et al., 2013). More recently, attention has also focused on the role of so-called beige fat, which can be generated from white adipose tissue by induction of a thermogenic pathway in which mitochondrial biogenesis plays a key role (Schulz et al., 2013). Hence, stimulation of mitochondrial biogenesis has been a major target in the treatment of obesity and metabolic syndrome, as reviewed elsewhere (Joseph et al., 2012; Wu et al., 2013). Pharmacological stimulation of mitochondrial biogenesis simulates the positive effect of caloric restriction and exercise on these generic metabolic disorders. Inevitably, many of these drugs that aim to provide exercise in a pill have now been tested in mouse models of genetic OXPHOS disorders as well and will be reviewed mainly in the context of inherited mitochondrial disease here. On the other hand, outcome of these studies using mouse models in which pathogenesis is caused directly by a genetic mitochondrial defect may be very useful for understanding the outcome of studies in models of more complex diseases in which there is an indirect mitochondrial defect.
Not surprisingly, activation of mitochondrial biogenesis is not the only treatment strategy for OXPHOS disorders and mitochondrial dysfunction in general. For an overview of other treatment strategies, please see the following reviews: Schon et al. (2010); Wallace et al. (2010); Koene and Smeitink (2011); Suomalainen (2011); Yu-Wai-Man et al. (2011); and Andreux et al. (2013).
Activation of mitochondrial biogenesis and the central role of PPAR-γ coactivator 1 (PGC)-1α
It is well known that certain stressors such as caloric restriction, exercise and cold adaptation can induce proliferation of mitochondria, which may be accompanied by changes in size and shape (Scarpulla et al., 2012). Members of the PGC-1 family play a central role in this process of mitochondrial biogenesis. The PGC-1 family consists of three members: PGC-1α, PGC-1β and PGC-1-related coactivator. Although all members have been shown to play a part in the regulatory network surrounding mitochondrial biogenesis, the major research focus has been on PGC-1α, which is considered to be the master regulator of this process (Figure 1; Fernandez-Marcos and Auwerx, 2011; Scarpulla, 2011; Wenz, 2013). This has made PGC-1α of special interest as a target of treatment intervention for various diseases in which mitochondrial dysfunction has been implicated, including the primary OXPHOS disorders. As a coactivator, PGC-1α requires other transcription factors for the trans-activation of mitochondrial biogenesis genes. Among these are the nuclear respiratory factors 1 and 2 (NRF1/2), and the PPAR-and oestrogen-related receptor nuclear receptor families (see Scarpulla, 2011; Scarpulla et al., 2012).
Figure 1.
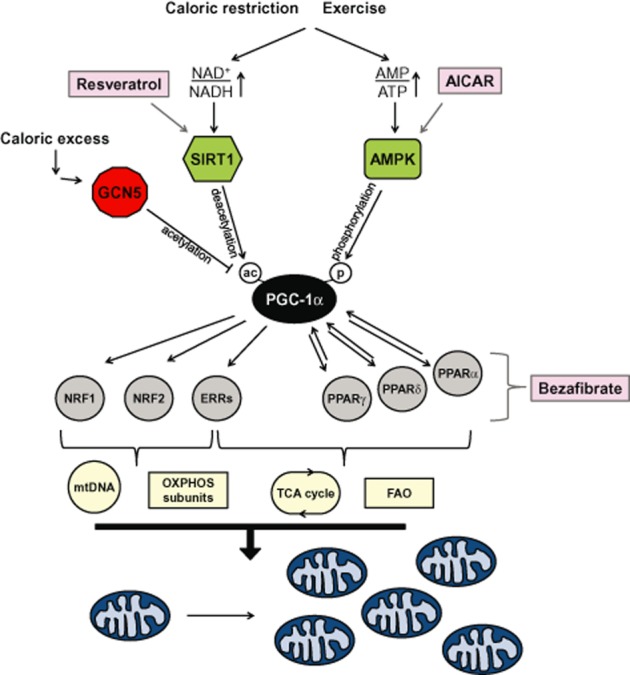
Overview of the induction of PGC1-α-mediated mitochondrial biogenesis by resveratrol, bezafibrate and AICAR. During caloric excess PGC-1α, the master regulator of mitochondrial biogenesis is in an inactive acetylated (ac) state. In times of caloric restriction, or after exercise, energy becomes scarce resulting in increases in the AMP/ATP ratio and NAD+/NADH ratio. The former has a direct impact on the activation of AMPK as binding of AMP (or ADP) facilitates phosphorylation (p) of AMPK by upstream kinases. Activated AMPK phosphorylates PGC-1α directly, which may either activate this coactivator or prime PGC-1α for activation via deacetylation by SIRT1. SIRT1 requires increased NAD+ levels for its activity as this nucleotide is a cosubstrate of the enzyme. Once in its deacetylated activated form PGC-1α is able to coactivate transcription of nuclear mitochondrial genes via transcription factors including NRF1/2, oestrogen-related receptor (ERR)α,β,γ (ERRs) and PPARα,β,γ. Consequently, this activates the mtDNA transcription, translation and replication machinery (represented by mtDNA); production of OXPHOS subunits; tricarboxylic acid (TCA) cycle enzymes and FAO enzymes. This ultimately leads to mitochondrial proliferation and increased mitochondrial mass. Resveratrol is an activator of SIRT1. The exact mechanism of activation is still unresolved but ultimately results in activation of the PGC-1α pathway via increased deacetylation of the coactivator. AICAR is phosphorylated into its ZMP analogue, an AMP mimetic, and can directly activate AMPK by promoting the phosphorylation of AMPK by upstream kinases. Activation of AMPK causes phosphorylation of PGC-1α, and SIRT1 activation via AMPK-induced increases in the NAD+/NADH ratio. Bezafibrate is a PPAR pan-agonist and therefore able to activate PPAR-regulated gene expression, which includes the expression of PGC-1α via the PPAR-responsive element in its promoter region. The increase in PGC-1α levels is believed to be sufficient for activation of the mitochondrial biogenesis pathway. For more details see the text or reviews (Fernandez-Marcos and Auwerx, 2011; Scarpulla et al., 2012; Wenz, 2013).
Activation of PGC-1α can be induced via gene expression and/or by post-translational modification. Transcriptional activation of PGC-1α expression is under the regulation of various transcription factors including cAMP response element-binding protein, thyroid hormones and PPARs (Wenz, 2013). The latter comprise three family members – PPARα, PPARβ/δ (in this review δ) and PPARγ – each responsible for tissue-specific activation of PGC-1α gene expression, albeit with some overlap. Although their endogenous ligands in vivo are still not well defined, each of the PPARs can be pharmacologically activated by specific agonists (Poulsen et al., 2012; Monsalve et al., 2013). PPARs form heterodimers with retinoid X receptors in the nucleus. These heterodimers bind to DNA together with corepressors. Ligand binding releases the corepressor and allows activation of coactivator, and results in changes in gene expression. Pharmacological activation of any of the PPARs is able to activate PGC-1α gene expression via the peroxisome proliferator response element (PPRE) located in the promoter region of the PGC-1α gene (Figure 1; Hondares et al., 2006; 2007; 2011).
In addition to the induction of gene expression, PGC-1α activity is also tightly regulated via various post-translational modifications. These modifications make PGC-1α an inportant integration site for intra-and extracellular metabolic signals. For example, increases in cellular AMP levels, the indicator for cellular energy status, result in the activation of AMP-activated kinase (AMPK). Phosphorylation of PGC-1α by AMPK activates the coactivator and consequently induces PGC-1α gene expression via a feedback loop (Jager et al., 2007). More recent studies have shown that phosphorylation of PGC-1α by AMPK may not directly activate PGC-1α but only prime PGC-1α for activation via deacetylation (Canto et al., 2009).
Silent information regulator two (Sir2) protein 1 (sirtuin 1, SIRT1) is the enzyme responsible for deacetylation of PGC-1α in the nucleus (Rodgers et al., 2005). SIRT1 is part of the sirtuin family of metabolic regulators that in mammals consists of seven proteins (SIRT1–7) and uses cellular NAD+ as cofactor for the deacetylation of proteins. The activation of SIRT1 is closely linked to the availability of its substrate NAD+ and therefore linked to the cellular redox state [the NAD(P)+/NAD(P)H balance] (Fulco et al., 2008; Canto et al., 2009).
This coordinated response of AMPK and SIRT1 is very important as a physiological response to fasting and exercise in skeletal muscle, but the mechanism is not without controversy as recently shown by the study of Higashida et al. (Canto et al., 2010; Higashida et al., 2013). Downstream of PGC-1α activation the major mitochondrial sirtuin, SIRT3, plays an important role in this response although the exact mechanism is still somewhat unclear (Hirschey et al., 2010; 2011,; Qiu et al., 2010; Shimazu et al., 2010; Someya et al., 2010; Hallows et al., 2011; Jing et al., 2011; 2013,; Fernandez-Marcos et al., 2012). SIRT3 modulates activities of mitochondrial enzymes via NAD+-dependent deacetylation and has been shown to have a broad substrate specificity acting on mitochondrial proteins present in all major mitochondrial pathways including FAO, OXPHOS and reactive oxygen species (ROS) metabolism (Rardin et al., 2013; see also Verdin et al., 2010; Giralt and Villarroya, 2012; Pirinen et al., 2012). Overexpression of SIRT3 induced mitochondrial biogenesis in HIB1B brown adipocytes and was anti-proliferative in cancer cells by decreasing ROS and thereby destabilizing HIF-1α (Finley et al., 2011; Bell et al., 2011). Furthermore, this SIRT3-mediated stimulation of the antioxidant response was also able to protect transgenic, SIRT3-overexpressing, mice against cardiac hypertrophy (Sundaresan et al., 2009). No data were provided on activation of mitochondrial biogenesis as a result of the SIRT3 overexpression in the latter studies, so it is unknown whether this played any part in the observed effects.
In times of caloric excess, deactivation of PGC-1α occurs by general control of amino acid synthesis 5 through acetylation (Lerin et al., 2006). Additionally, inhibition of PGC-1α also occurs by phosphorylation of a specific site by Akt/PKB in response to insulin signalling (Li et al., 2007).
PGC-1α knockout and transgenic mouse models
Considering the fact that PGC-1α is the master regulator of mitochondrial biogenesis, the knockout of the PGC-1α gene in mice gives rise to a relatively mild phenotype (Lin et al., 2004; Leone et al., 2005). These mice are hyperactive, have reduced muscle function and exercise capacity, and an abnormal thermogenic response. The most logical explanation for the mild phenotype is the redundant function of the three PGC-1 family members. Accordingly, the PGC-1α/PGC-1β double knockout has a very severe phenotype, and the majority of pups die of cardiac failure within 24 h after birth (Lai et al., 2008).
Besides these studies with knockout mice, studies with transgenic overexpressing PGC-1α mice have been very insightful for determining the function of PGC-1α in various tissues (Table 1). Transgenic overexpression of PGC-1α in mouse heart has been shown to induce proliferation of enlarged mitochondria (Lehman et al., 2000). However, this coincides with the development of cardiomyopathy and oedema, which can be reversible depending on the period of PGC-1α overexpression (Lehman et al., 2000; Russell et al., 2004).
Table 1.
Rescue of mitochondrial dysfunction in mouse models by PGC-1α overexpression
| Mouse model (mode of overexpression) | Effect on phenotype | Evidence for mitochondrial biogenesis | Reference |
|---|---|---|---|
| mdx model of DMD (MCK-PGC-1α) | Amelioration of muscle damage and motor performance | ND | Handschin et al. (2007) |
| mdx model of DMD (recombinant adeno-associated virus mediated in skeletal muscle) | Increased resistance against contraction-induced muscle damage | + | Selsby et al. (2012) |
| cox10 model of mitochondrial myopathy (MCK-PGC-1α) | Increase in lifespan and reduced progression of myopathy | + | Wenz et al. (2008) |
| Surf1−/− model of COX deficiency (MCK-PGC-1α) | No distinct phenotype | + | Viscomi et al. (2011) |
| Polg Mut mouse model of mitochondrial disease/premature ageing (MCK-PGC-1α) | Improvement of motor performance and cardiac function | + | Dillon et al. (2012b) |
| R6/2 model of HD (lentiviral vector via injection in striatum) | Prevention of atrophy of striatal neurons | ND | Cui et al. (2006) |
| SOD1 (G93A) DL model of ALS (Liang et al., 2011, low-level whole-body human PGC-1α) | Slower disease progression and improved motor performance, prevention of neuron loss | ND | Liang et al. (2011) |
| SOD1 (G93A) Gurl model of ALS (hPGC-1α with rat neuron-specific enolase promoter) | Improvement of survival and motor performance, prevention of neuron loss | + | Zhao et al. (2011) |
| SOD1 (G37R) model of ALS (MCK-PGC-1α) | Improved motor function but no effect on motor neuron loss and survival | ND | Da Cruz et al. (2012) |
| MTPT-induced mouse model of PD (PGC-1α behind a neuronal thy-1 promotor) | Neuroprotective effect, amelioration of decreased dopamine levels | + | Mudo et al. (2012) |
ND, not determined.
The overexpression of PGC-1α in skeletal muscle is phenotypically more beneficial and results in fibre-type switching towards the more oxidative type I (slow-twitch) fibres, which rely on mitochondrial OXPHOS for the generation of ATP rather than glycolysis (Lin et al., 2002). Furthermore, it protects the mice against age-induced muscle wasting and metabolic disease, and has been shown to increase lifespan (Wenz et al., 2009b). Interestingly, PGC-1β overexpression results in different fibre-type switching into type IIx fibres (Arany et al., 2007), indicating slight differences in the regulatory network of these two closely related coactivators.
These initial studies led to the use of the PGC-1α transgenic mice in proof of principle experiments to determine the effect of induction of PGC-1α on the pathology in mouse models of various diseases as discussed below and summarized in Table 1.
Rescue of mitochondrial dysfunction by transgenic PGC-1α expression
The phenotype in a mouse model of DMD, mdx mice, benefitted significantly from transgenic PGC-1α expression in skeletal muscle (Lin et al., 2002; Handschin et al., 2007). Mdx mice lack the dystrophin protein and develop a mild muscular dystrophy most pronounced in sedentary mice around 4–6 weeks of age (Handschin et al., 2007). Transgenic PGC-1α overexpression decreased skeletal muscle damage in sedentary young mice and provided protection against exercise-induced muscle damage in older mdx mice. Unfortunately, no details were reported on changes in mitochondrial function due to increased expression of PGC-1α in the muscle of mdx mice. Recently, however, it was shown that PGC-1α overexpression by virally mediated gene transfer in skeletal muscle of mdx mice could result in increased expression of mitochondrial OXPHOS components suggestive of induction of mitochondrial biogenesis (Selsby et al., 2012).
The same approach as used for the mdx mice was deployed using mice with mitochondrial CIV [cytochrome c oxidase (COX)] deficiency in skeletal muscle due to a conditional knockout of the CIV assembly factor gene Cox10 (Diaz et al., 2005; Wenz et al., 2008). It was observed that muscle-specific PGC-1α overexpression improved the survival of ΔCox10 mice in both males and females by slowing the progression of myopathy. Analysis of the muscle showed that the ΔCox10 transgenic mice had more COX activity per muscle volume, whereas COX activity per mitochondrion was not increased and remained reduced compared with wild type (20–25%). The activity of citrate synthase, a marker enzyme for mitochondrial quantity, was also increased four-to fivefold in muscle of transgenic ΔCox10 mice in agreement with a large induction of mitochondrial proliferation. On the other hand, non-transgenic ΔCox10 mice already had a two-to threefold increase in citrate synthase activity compared with wild-type mice as a natural compensation mechanism for the OXPHOS defect. A similar induction has often been observed in humans with mitochondrial myopathy, as well as in the first mouse model of mitochondrial myopathy/cardiomyopathy (Ant1−/− mice) (Graham et al., 1997).
In a different COX-defective mouse model with a mild decrease in COX activity due to ablation of the Surf1 COX assembly factor gene, transgenic overexpression of PGC-1α in muscle induced an increase in mitochondrial mass as shown by increases in mtDNA and citrate synthase activity and increases in specific activities of most OXPHOS enzymes including the defective COX (Viscomi et al., 2011). Because Surf1−/− mice have no overt phenotype it is not possible to look at correction of pathology in this model; however, no negative effects of the expression of the transgene were reported either (Dell'agnello et al., 2007; Viscomi et al., 2011).
Recently the same strategy has been used in another mouse model with mitochondrial dysfunction, the mutator (Mut) mouse (Dillon et al., 2012b). Mut mice have a proofreading mutation in the gene coding for polymerase γ (POLG). POLG is the polymerase responsible for mtDNA replication, and the mutation causes the accumulation of mutations in mtDNA causing progressive mitochondrial dysfunction and symptoms of premature ageing (Trifunovic et al., 2004; Kujoth et al., 2005). Transgenic expression of PGC-1α in muscle of Mut mice was analysed at 10 months of age and shown to result in increases in OXPHOS subunits, respiratory chain enzyme activity, citrate synthase activity and mtDNA per muscle volume. Surprisingly, cardiac function in transgenic PGC-1α Mut mice was restored to wild-type levels at 10 months of age despite the fact that there was no significant increase in PGC-1α expression and mitochondrial biogenesis in this tissue. There was an increased accumulation of low-abundance mtDNA mutations in muscle, which are assumed to be mainly acquired during the lifespan of mice rather than inherited. This was suggested to be caused by the increase in mtDNA replication rate that coincides with mitochondrial proliferation.
In addition to muscle, PGC-1α also plays an important role in the brain as demonstrated by the neurological component in the phenotype of the PGC-1α−/− mice (Lin et al., 2004; Leone et al., 2005). Moreover, it has been shown that part of the role of PGC-1α is as a regulator of ROS metabolism (St-Pierre et al., 2006). This role is very relevant to OXPHOS disorders, many of which manifest with a form of neurodegeneration or brain abnormalities. Furthermore, the OXPHOS system is a major site of ROS generation, which may contribute to pathology when increased in the case of a malfunctioning system. In this respect the lentiviral-mediated delivery of PGC-1α in a mouse model of the fatal neurological disease HD is of note (Cui et al., 2006). PGC-1α is down-regulated in the striata of HD patients and in a mouse model for HD (R6/2), which is suggested to play a role in the energy imbalance in HD striata (Cui et al., 2006; Weydt et al., 2006). Administration of a PGC-1α expression vector via lentivirus to the striata of HD mice prevented neural atrophy. Conversely, breeding a different and milder HD mouse model onto a PGC-1α−/− background aggravated the phenotype emphasizing the importance of PGC-1α in disease progression (Cui et al., 2006).
Recently, studies have tested whether constitutive transgenic overexpression of PGC-1α in neuronal tissues would ameliorate the pathology in mouse models of the neurological diseases ALS (Liang et al., 2011; Zhao et al., 2011) and PD (Mudo et al., 2012) (Table 1). In the ALS mouse models, transgenic overexpression of mutant SOD1 (G93A), a mutation found in familial ALS, causes progressive neurodegeneration. PGC-1α expression improved motor performance, attenuated neurodegeneration and, in the most severely affected model, extended survival (Liang et al., 2011; Zhao et al., 2011). Mitochondrial function was only assessed by in situ histological experiments, which showed a restoration towards wild-type levels for CI and CIV activity in the spinal cords of PGC-1α ALS mice while no change in CII activity observed (Zhao et al., 2011).
A subsequent study using the ALS G37R SOD1 mouse model took a different approach and showed that overexpression of PGC-1α in the skeletal muscle specifically could not slow the disease progression and survival (Da Cruz et al., 2012). Still, the improvement in muscle function in the mice did show that targeting peripheral activation of PGC-1α may improve quality of life in ALS patients (Da Cruz et al., 2012).
In order to study the effect of PGC-1α in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mouse model, a new mouse line with constitutive transgenic overexpression of PGC-1α behind a neuronal thy-1 promotor was generated (Mudo et al., 2012). In the substantia nigra of these PD mice, PGC-1α expression resulted in induction of the mitochondrial antioxidant enzymes SOD2 and TRX2 as well as the OXPHOS protein COX IV (Mudo et al., 2012). The increased amount of mitochondrial proteins is in line with minimal experimental evidence showing a higher respiratory control ratio in isolated brain mitochondria from transgenic animals. These biochemical changes were suggested to contribute to the neuroprotective effect of PGC-1α expression against cell degeneration and decreases in dopamine levels by the PD inducing neurotoxin MPTP. The active component of this toxin, 1-methyl-4-phenylpyridinium (MPP+), is a very potent inhibitor of CI in substantia nigra mitochondria, which is believed to be causing the Parkinson's-like symptoms in this model. Hence, this model is not only a PD model but also a model with a direct OXPHOS defect. Therefore, this study highlights PGC-1α up-regulation as a potential therapeutic method for OXPHOS disorders in the brain.
The studies with PGC-1α transgenic mice clearly reveal the potential of pharmacological activation of this coactivator as part of treatment of various diseases affecting muscle, heart and brain. In the next sections we will discuss the current status on pharmacological treatments in mouse models, which aim to do just this.
Induction of mitochondrial biogenesis via PPAR activation: bezafibrate
Pharmacological activation of mitochondrial biogenesis can be achieved with a large range of compounds (see Andreux et al., 2013). Here the main focus is on the treatments used in mouse models of OXPHOS disorders (Figure 1 and Tables 2,3,4).
Table 2.
Mouse models studies trialling PPAR pan-agonist bezafibrate for the activation of PGC-1α
| Mouse model | Effect on phenotype | Evidence for mitochondrial biogenesis | Reference |
|---|---|---|---|
| Cox10 model of mitochondrial myopathy | Prolonged lifespan and delayed onset of myopathy | + | Wenz et al. (2008) |
| Surf1−/− model of COX deficiency | Loss of body weight, hepatomegaly | − | Viscomi et al. (2011) |
| ACTA-Cox15−/− model of mitochondrial myopathy | Lethality within 48 h | − | Viscomi et al. (2011) |
| Peo1 Deletor mouse model of late-onset mitochondrial myopathy | Loss of body weight, hepatomegaly, reduction in body temperature, reduction in COX-negative fibres and mtDNA deletion load | − | Yatsuga and Suomalainen (2012) |
| Polg Mut mouse model of mitochondrial disease/premature ageing | Delay in hair loss, reduction in skin damage, reduction in spleen size, reduction of body weight, hepatomegaly | − | Dillon et al. (2012a) |
| Forebrain-specific ΔCox10 mice, a model of mitochondrial encephalopathy | Overall amelioration of the phenotype including attenuation of the loss of motor function, protection of brain damage, reduction of loss of bodyweight | + | Noe et al. (2013) |
| R6/2 model of HD | Increase in motor performance, increase in lifespan. Amelioration of phenotype in brain, skeletal muscle and BAT. Slight increase in liver weight and lipid vacuolization | + | Johri et al. (2012) |
| P301S transgenic mouse model of AD | Attenuation of locomotor and anxiety abnormalities, histological improvement in the brain pathology, improvement in lipid vacuoles in BAT, reduced body weight | + (BAT) | Dumont et al. (2012) |
Table 3.
Treatment studies using resveratrol for the activation of PGC-1α in mouse models
| Mouse model | Effect on phenotype | Evidence for mitochondrial biogenesis | Reference |
|---|---|---|---|
| The YG8R transgenic mice containing FXN with GAA repeat expansion in intron 1 as model for Friedreich's ataxia | ND, only increased FXN expression | ND | Li et al. (2013) |
| mdx model of DMD (skeletal muscle) | Amelioration of muscle damage and motor performance, increased mortality rate when dosage at 400 mg·kg−1·day−1 | ND | Hori et al. (2011); Selsby et al. (2012); Gordon et al. (2013) |
| mdx model of DMD (cardiac muscle) | Prevention of cardiomyopathy and cardiac function | ND | Kuno et al. (2013) |
| Tg19959 mouse model of AD | Diminished plaque formation in specific brain areas | ND | Karuppagounder et al. (2009) |
| APP/PS1 mouse model of AD | Diminished plaque formation in specific brain areas | ND | Vingtdeux et al. (2010) |
| N171-82Q HD transgenic mouse model of HD | No effect on motor performance and striatal atrophy | + (BAT) | Ho et al. (2010) |
| MTPT-induced mouse model of PD | Neuroprotective effect, amelioration of dopamine levels | ND | Blanchet et al. (2008); Mudo et al. (2012); Lofrumento et al. (2010) |
| SOD1 (G93A) Gurl model of ALS | No effect when administrated via diet (0.06%). Delay of disease onset and prolonged survival when administered via i.p. injection | ND | Markert et al. (2010); Han et al. (2012) |
ND, not determined.
Table 4.
Treatment studies using AICAR for the activation of mitochondrial biogenesis in mouse models
| Mouse model | Effect on phenotype | Evidence for mitochondrial biogenesis | Reference |
|---|---|---|---|
| Surf1−/− model of COX deficiency | Improved biochemical phenotype, no motor impairment to improve | + | Viscomi et al. (2011) |
| ACTA-Cox15−/− model of mitochondrial myopathy | Improved biochemical phenotype without improved motor impairment | + | Viscomi et al. (2011) |
| Sco2KO/KI model of COX deficiency | Improved biochemical phenotype and improved motor impairment | + | Viscomi et al. (2011) |
| mdx model of DMD (skeletal muscle) | Amelioration of muscle pathology. May have positive effect on muscle function | + | Ljubicic et al. (2011); Jahnke et al. (2012) |
| mdx model of DMD (diaphragm) | Amelioration of muscle pathology. Evidence of increased mitophagy | − | Pauly et al. (2012) |
The pan-PPAR (α, β/δ, γ) agonist bezafibrate is a commonly used drug for the treatment of dyslipidaemia (Tenenbaum et al., 2005). Although historically fibrates are strongly linked to FAO and peroxisome proliferation, studies in recent years have shown that PPARs via PGC-1α are able to regulate other aspects of mitochondrial metabolism as well (Hondares et al., 2006;, 2007;, 2011,). Bezafibrate stimulates heart and skeletal muscle PGC-1α expression in mice in a PPARδ-dependent manner (Hondares et al., 2007). Moreover, in vitro experiments have shown that bezafibrate treatment or overexpression of PGC-1α is able to up-regulate respiratory capacity and OXPHOS function in fibroblasts and myoblasts from OXPHOS patients (Bastin et al., 2008; Srivastava et al., 2009; Casarin et al., 2012).
Bezafibrate was the drug of choice for Wenz et al. (2008) to study whether they could pharmacologically replicate the beneficial effect of transgenic overexpression of PGC-1α in ΔCox10 mice. Dietary bezafibrate administration at a concentration of 0.5% (w/w) in the diet, as used in all other studies described in this review, resulted in an induction of mitochondrial biogenesis in skeletal muscle of ΔCox10 mice. Bezafibrate caused increased expression of Ppar and Pgc-1 genes and induction of mtDNA and mitochondrial proteins in muscle homogenates. Importantly, treatment delayed the onset of myopathy and prolonged survival of ΔCox10 mice.
After this first study demonstrated a beneficial effect of bezafibrate treatment for mitochondrial myopathy, more groups reported on the use of the drug in other mouse models of mitochondrial disease with varying success (Table 2). In the Surf1−/− model of mild COX deficiency, treatment with bezafibrate was not able to replicate the effect of PGC-1α overexpression (Viscomi et al., 2011). Instead, treatment caused an induction in both PPARα and δ and resulted in the up-regulation of FAO in skeletal muscle. Phenotypically, bezafibrate caused a loss in body weight and severe hepatomegaly, clearly indicating toxicity of the drug. Viscomi et al. also evaluated the use of bezafibrate as treatment for a new mouse model with severe muscle-specific COX deficiency (ACTA-Cox15−/− mice) and early onset mitochondrial myopathy. In these mice bezafibrate was lethal within 48 h after the start of the treatment at the same dose used for ΔCox10 and Surf1−/− mice previously (Wenz et al., 2008; Viscomi et al., 2011).
The studies of Viscomi et al. and Wenz et al. showed that the success rate with bezafibrate may be very dependent on the particular model and the severity of disease in the model. Therefore, it was not a surprise that bezafibrate was tolerated in Deletor mice, a mouse model for adult-onset mitochondrial myopathy (Yatsuga and Suomalainen, 2012). However, in this model significant induction of hepatic liver FAO, hepatomegaly and decreases in adipose tissue were also reported as a result of the treatment. The Deletor mice accumulate mtDNA deletions because of the transgenic expression of a mutated Twinkle helicase protein. The mutated Twinkle contains a mutation in Peo1 similar to a mutation observed in the C10orf2 gene in patients with progressive external ophthalmoplegia (Tyynismaa et al., 2005; Yatsuga and Suomalainen, 2012). The effect of bezafibrate in Deletor mice was limited as it delayed the accumulation of COX negative fibres and reduced mtDNA deletion load without a change in phenotype and without induction of muscle mitochondrial biogenesis. The treatment strategy in this study was different from the previous studies in that treatment was started at the onset of symptoms at 1 year of age instead of at 5 weeks of age in the previous studies (Wenz et al., 2008; Viscomi et al., 2011).
Intermediate beneficial effects of bezafibrate were also observed in another mouse model with defects in mtDNA maintenance, Mut mice, which had previously been shown to be substantially rescued by PGC-1α expression, as described above (Dillon et al., 2012a; 2012b,). Again no evidence of mitochondrial biogenesis was observed in mice treated with bezafibrate. However, there was a delay of hair loss, reduction in damaged dorsal skin structure and amelioration of the abnormal spleen size and weight observed in Mut mice receiving bezafibrate. Biochemically there was evidence of increased FAO in skeletal muscle and liver of treated mice, as previously observed by Viscomi et al. (2011) and Yatsuga and Suomalainen (2010) in their studies.
The results described above show considerable variability in the outcome of treatment with bezafibrate in mouse models with muscle disease due to mitochondrial dysfunction. Notwithstanding, bezafibrate has recently been reported to be beneficial in mouse models with neurological disorders, apparently mainly by improvement of mitochondrial function. Forebrain-specific ΔCox10 mice develop progressive neurodegeneration and shrinkage of the cortex, which can be ameliorated by treatment with bezafibrate (Fukui et al., 2007; Noe et al., 2013). Mitochondrial mass was increased in the brain after treatment, including specific activity of COX and citrate synthase. Additionally, Tfam and PGC-1α gene expression were shown to be increased, which may have contributed to mitochondrial proliferation. Antioxidant enzymes SOD2 and catalase were induced in brain of treated mice, and may have protected against oxidative damage that is believed to contribute to the pathology in forebrain-specific ΔCox10 mice. The treatment resulted in an overall amelioration of the phenotype including attenuation of the loss of motor function.
In models with neurological disease not caused by primary mitochondrial dysfunction, bezafibrate was able to ameliorate pathology as well. Johri et al. (2012) demonstrated that the beneficial effect of lentiviral PGC-1α transgene expression in brain of the R6/2 HD mouse model by Cui et al. (2006) could be mimicked via bezafibrate treatment. This included restoring the expression levels of genes involved in mitochondrial biogenesis to the levels in non-treated wild-type mice in brain, skeletal muscle and brown adipose tissue (BAT) (Johri et al., 2012). Phenotypically, motor performance was improved by treatment and survival was increased by 20% in HD mice.
The same group also showed the amelioration of disease progression in a mouse model of AD (P301S transgenic mice) by bezafibrate diminishing locomotor and anxiety-related abnormalities (Dumont et al., 2012). The overall protective mechanism of bezafibrate in this model remains inconclusive despite some biochemical changes including activation of FAO in various tissues. Evidence of mitochondrial proliferation as a result of treatment was minimal in brain with slight increases in mtDNA quantity and Sirt1 and Tfam gene expression, but more apparent in BAT with increased expression of regulators Pgc-1α, Nrf1, Tfam and Sirt1.
In summary, bezafibrate has been able to stimulate mitochondrial biogenesis in some mouse models with mitochondrial dysfunction (Wenz et al., 2008; Johri et al., 2012; Noe et al., 2013) (Table 2). In the majority of the other studies, increases in FAO have been found which may contribute to bezafibrate-induced changes in disease progression and normal development. Toxicity issues were identified in a number of studies, including impairment of growth. Bezafibrate causes a strong anabolic effect in mice including stimulation of FAO, most likely due to activation of PPARα by bezafibrate. Activation of PPARα is the major contributor responsible for the hepatomegaly resulting from bezafibrate treatment in mice and can result in hepatocarcinogenesis (Hays et al., 2005). Small differences between the mouse and human PPARα orthologues prevent proliferation of hepatic tissue in humans. This is demonstrated in the studies of Yang et al. (2008), which showed that the PPARα activator fenofibrate was unable to induce hepatomegaly in the PPARα-humanized mice whereas other metabolic effects of the drug were similar to wild type. Therefore, bezafibrate is relatively safe in humans although rhabdomyolysis and kidney failure have been reported as side effects (Charach et al., 2005; Wu et al., 2009). On a positive note, long-term bezafibrate treatment of patients with the mitochondrial FAO disorder CPT2 deficiency has shown a significant clinical improvement (Bonnefont et al., 2009; 2010,).
Induction of mitochondrial biogenesis via SIRT1 activation: resveratrol
Resveratrol (3,5,4′-trihydroxy-trans-stilbene) is a natural component present in low concentrations in the skin of red grapes and has been extensively studied for its ability to activate sirtuins, including SIRT1 in mammals (Howitz et al., 2003). Resveratrol was originally shown to extend lifespan in various species and provide protection against the damaging effects of metabolic stress in mice (Howitz et al., 2003; Wood et al., 2004; Baur et al., 2006; Lagouge et al., 2006). Although the direct link between activation of SIRT1 and lifespan extension remains inconclusive (Bass et al., 2007; Burnett et al., 2011), the importance of SIRT1 as a metabolic sensor/regulator is well accepted but not without controversy. Resveratrol activates SIRT1 either directly or indirectly, which may be dose-dependent (Borra et al., 2005; Kaeberlein et al., 2005; Pacholec et al., 2010; Price et al., 2012; Hubbard et al., 2013). On the other hand, it is well documented that the activation of SIRT1 by resveratrol involves the activation of AMPK (Baur et al., 2006; Zang et al., 2006; Dasgupta and Milbrandt, 2007; Um et al., 2010). Consequently, AMPK activation results in increases in cellular NAD+ levels, thereby activating SIRT1 through increasing its substrate (Fulco et al., 2008; Canto et al., 2009). The subsequent concerted action of both activated AMPK and SIRT1 on PGC-1α activates this coactivator, resulting in the induction of mitochondrial biogenesis (Canto et al., 2009). Recently, a direct inhibition of cAMP PDEs by resveratrol was demonstrated, which may be an important aspect in its mechanism of action (Park et al., 2012). Then again, the large number of proteins that are able to interact with resveratrol makes it very difficult to interpret the contribution of the effect of direct inhibition of PDEs by this compound, in the overall scheme of interactions (Pacholec et al., 2010).
In healthy mice, resveratrol increased lifespan and was protective under conditions of diet-induced obesity, although it did not extend their lifespan under normal conditions (Baur et al., 2006; Lagouge et al., 2006; Pearson et al., 2008). Induction of mitochondrial biogenesis via PGC-1α activation is a major contributor of the beneficial effects of the drug. Hence it is not surprising that resveratrol has been trialled for the treatment in mouse models of genetic disorders associated with mitochondrial dysfunction. No treatment studies with resveratrol in mouse models of primary OXPHOS disorders have been published so far. In this respect it is interesting to mention the use of resveratrol in a study with a mouse model of Friedreich's ataxia (Al-Mahdawi et al., 2006; Li et al., 2013). This neurological condition is caused by impaired expression of the FXN gene due to a pathological expansion of GAA repeats in intron 1 of this gene encoding mitochondrial frataxin (Li et al., 2013). Frataxin appears to be involved in biosynthesis of iron-sulfur clusters, which form integral parts of OXPHOS CI, CII and CIII. Accordingly, the OXPHOS system has been found to be dysfunctional in Friedreich's ataxia. Subcutaneous resveratrol injections (200 mg·kg−1 for 3 days) increased FXN mRNA and frataxin protein levels in the mice indicative of the potential of resveratrol for the treatment of Friedreich's ataxia (Li et al., 2013).
Some of the other mouse models previously described in the PGC-1α overexpression studies (Table 1) or bezafibrate trials (Table 2) have been treated with resveratrol as well (Table 3). In the mdx model of DMD, long-term resveratrol treatment mimicked the effect observed by PGC-1α overexpression including reduction of muscle tissue loss, changes in fibre-type composition and reduction of oxidative damage in skeletal muscle of mdx mice (Handschin et al., 2007; Hori et al., 2011). Additional studies confirmed the positive results of resveratrol treatment on skeletal muscle in mdx mice (Selsby et al., 2012; Gordon et al., 2013). However, 8 weeks of 400 mg·kg−1·day−1 was reported to result in a high mortality rate (Selsby et al., 2012).
In a subsequent study, the effect of resveratrol on the development of cardiomyopathy in mdx mice was studied (Kuno et al., 2013). Treatment with resveratrol prevented cardiomyopathy and restored cardiac function in mdx mice. This effect was suggested to be caused via attenuation of expression of the p300 coactivator, which is a key contributor to cardiac hypertrophy and fibrosis in the heart. In the context of this review it is unfortunate that the effect of resveratrol on mitochondrial status was not reported in any of the studies using mdx mice despite the important function of mitochondria in muscle and heart during health and disease.
Resveratrol has become a popular compound to test as treatment for a range of neurological disorders (Herskovits and Guarente, 2013; Table 3). In the majority of these disorders, SIRT1 activation by resveratrol is not primarily aiming to ameliorate mitochondrial metabolism but to improve other SIRT1-regulated biological processes (Herskovits and Guarente, 2013). Resveratrol has been used in mouse models of AD, HD, PD and ALS with some success but with minimal evidence of the mode of action.
Importantly, some of these studies have confirmed the stability of resveratrol in mouse food when stored at room temperature, which has been under debate (Karuppagounder et al., 2009; Vingtdeux et al., 2010). Furthermore, Vingtdeux et al. (2010) demonstrated the presence of resveratrol in the brain after oral treatment, indicating resveratrol can have a direct mode of action in neurological disorders. These two studies followed an initial treatment trial with p25 transgenic mice with features of AD in which resveratrol was injected directly into the brain via i.c.v. injections. In these mice neurodegeneration and cognitive decline was decreased (Kim et al., 2007). Both subsequent studies, using two different models of AD (Table 3) showed that dietary administration of this compound [0.2 and 0.35% (w/w)] resulted in amelioration of plaque pathology in specific areas of the brain.
As mentioned previously, lentiviral-mediated overexpression of PGC-1α was beneficial in the R6/2 mouse model of HD (Cui et al., 2006). However, administration of resveratrol (oral gavage, 25 mg/mouse/day) could not replicate this effect in N171-82Q HD transgenic mice where it was found to have an effect on PGC1α gene expression in BAT and not in the striatum, which is likely to explain the lack of improvement of phenotype (Ho et al., 2010).
In terms of PD, several studies have shown the neuroprotective effect of resveratrol in the substantia nigra after MPTP-induced neurotoxicity in mice (Blanchet et al., 2008; Mudo et al., 2012; Lofrumento et al., 2010). The outcomes of these studies were suggested to result from the induction of an antioxidant response and/or an anti-inflammatory effect induced by resveratrol (Mudo et al., 2012; Lofrumento et al., 2010).
In the SOD1G93A ALS mouse model, treatment with dietary resveratrol (0.06%, w/w) was ineffective (Markert et al., 2010) but resveratrol injected i.p. (20 mg·kg−1 twice weekly) delayed disease onset and prolonged survival of mice (Han et al., 2012), suggesting the dose and mode of administration can be critical when using resveratrol. SIRT1 activation by resveratrol resulted in deacetylation of heat shock factor 1 and a subsequent unfolded protein response. The latter effect is likely to be a significant contributor to the mechanism of action of resveratrol in this model in which the pathology is caused by misfolding of the mutated SOD1G93A protein (Westerheide et al., 2009; Han et al., 2012).
The studies described above illustrate the enormous potential for resveratrol in the treatment of a wide variety of diseases. The positive treatment outcomes of resveratrol in these studies have been attributed to induction of an antioxidant response, anti-inflammatory effect or an unfolded protein response. None of these studies have looked at the effect of resveratrol on mitochondrial status, which may well play an additional role in the amelioration of pathology in these disease models.
Induction of mitochondrial biogenesis via AMPK activation: AICAR
The activation of AMPK has been implicated as a key player for the mechanism of action of resveratrol on SIRT1 and consequent mitochondrial biogenesis (Baur et al., 2006; Zang et al., 2006; Dasgupta and Milbrandt, 2007; Um et al., 2010). AMPK activation occurs through phosphorylation of Thr172 by AMPK kinases, including Ca2+/calmodulin-dependent kinase kinase and liver kinase B1 (see Hardie et al., 2011; Oakhill et al., 2012). AMPK phosphorylation is directly promoted by AMP and ADP binding, and therefore linked to the cellular energy status that is reflected by the AMP/ATP and the ADP/ATP ratios. Furthermore, AMP is also able to stimulate the activity of phosphorylated AMPK via allosteric activation of the enzyme, although the significance of this effect in vivo is not known (Oakhill et al., 2012). All these aspects make AMPK an ideal metabolic sensor that monitors and reacts to changes in the cellular energy status.
Moreover, it makes AMPK also an ideal drug target. AMPK can be directly activated via treatment with the drug 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR). AICAR requires phosphorylation inside the cell and it is this phosphorylated derivative known as ZMP, that acts as an AMP mimetic (Sullivan et al., 1994; Corton et al., 1995; Henin et al., 1995;, 1996; Merrill et al., 1997). Unlike other AMPK stimuli, AICAR promotes phosphorylation and activation of AMPK while not disturbing the energy status of the cell, that is, the AMP/ATP ratio remains the same.
AICAR is orally effective in mice and shown to increase endurance performance in sedentary mice by increasing expression of oxidative metabolism genes (Narkar et al., 2008). This is believed to be via the direct phosphorylation/activation of PGC-1α by activated AMPK and subsequent induction of mitochondrial biogenesis (Winder et al., 2000; Jager et al., 2007). Unfortunately AICAR has a very poor ability to penetrate the blood–brain barrier, which limits its use to peripheral tissues (Marangos et al., 1990). Nevertheless peripheral stimulation of AMPK and mitochondrial biogenesis may still have beneficial effects on brain function as demonstrated in healthy mice (Kobilo et al., 2011).
In the case of OXPHOS disorders, in which there are typically defects in ATP generation, partial activation of AMPK may be already occurring in certain tissues. Additional stimulation of this pathway to maximum with AICAR may bring this rescue mechanism to full potential. Accordingly, in a drug screen using skin fibroblasts from a cohort of CI patients AICAR was shown to be the most promising candidate in ameliorating mitochondrial defects (Golubitzky et al., 2011).
In mice, Viscomi et al. (2011) have demonstrated the potential of AICAR for OXPHOS disorders by the successful treatment of their muscle-specific COX-deficient mouse models of mitochondrial myopathy. The drug was injected s.c. from 8 weeks of age for 1 month (0.25 mg·day−1·g−1). AICAR treatment resulted in AMPK phosphorylation and induction of FAO and OXPHOS gene expression in muscle in Surf1−/−, Sco2KO/KI and the ACTA-Cox15−/− mice (Table 4; Dell'agnello et al., 2007; Yang et al., 2010; Viscomi et al., 2011). Moreover, COX enzyme activity in muscle was increased in all models as a result of increased amounts of COX subunits. As well as increasing the deficient COX activity, AICAR also increased other OXPHOS enzyme activities, indicating a general increase in mitochondrial biogenesis. Motor performance impairment present in Sco2KO/KI mice could be rescued by AICAR treatment. No changes were observed in the more severely affected ACTA-Cox15−/− mice, even when doubling the AICAR dose. This may be due to the severity of the muscle damage present in these mice at the start of treatment.
AICAR is also able to improve skeletal muscle pathology in the mdx model of DMD (Ljubicic et al., 2011; Jahnke et al., 2012). This coincided with the induction of mitochondrial biogenesis as shown by increases in COX activity in fast-twitch muscle (extensor digitorum longus) and a switch to more oxidative fibre types. In addition, AICAR evoked the expression of utrophin and PGC-1α plus increased expression of the OXPHOS components cytochrome c and COX IV. However, in the more oxidative diaphragm muscle, AICAR treatment did not result in changes in fibre type or induction of mitochondrial biogenesis (Pauly et al., 2012). Contrariwise, data from this study suggested that AICAR caused increased mitophagy in the diaphragm of mdx mice (Pauly et al., 2012). Mitophagy, the process responsible for degradation of dysfunctional and damaged mitochondria, has been shown to be impaired in mdx muscle (De Palma et al., 2012). AICAR had a positive effect on diaphragm structure and contractile capacity, suggesting improvement of mitophagy may be an alternative pathway to target for the treatment of muscle diseases including DMD and mitochondrial myopathies (Pauly et al., 2012).
Induction of mitochondrial biogenesis: miscellaneous compounds
This review has focussed on the treatment of mitochondrial dysfunction by bezafibrate, resveratrol and AICAR, with the rationale that these compounds have all been used in OXPHOS mouse models to induce mitochondrial biogenesis. Various other drugs have been used in preclinical trials with the other mouse models discussed in this review. Some of these may have potential for the induction of mitochondrial biogenesis. Among these is the drug metformin, which is widely used for the treatment of type 2 diabetes (Viollet et al., 2012). The mode of action of metformin is still not completely resolved, but AMPK is believed to contribute to the effect of the drug. AMPK is not directly activated by metformin but through changes in the cellular energy state as a consequence of the mild inhibitory effect of metformin on CI of the OXPHOS system. Of course this makes metformin unlikely to be a treatment option for OXPHOS disorders where a pathogenic OXPHOS defect is already present.
NO is a gaseous signalling molecule that has been shown to be involved in mitochondrial biogenesis (see Nisoli and Carruba, 2006; Tengan et al., 2012). The role of NO in mitochondrial biogenesis is not exactly clear but the downstream induction of PGC-1α expression is essential for this effect (Nisoli et al., 2003). NO is able to activate PGC-1α through a cyclic GMP-dependent pathway, via AMPK, or in a p53-dependent manner, with a possibility of multiple pathways overlapping as well (Nisoli et al., 2003; Lira et al., 2010; McConell et al., 2010; Aquilano et al., 2013). Not only is NO involved in mitochondrial biogenesis but it is also implicated in myogenic differentiation in which the inhibition of the mitochondrial fission protein Drp1 by NO through activation of guanylate cyclases plays an important role (De Palma et al., 2010). Because of the physiological effects that are under control of NO, its metabolism has been an attractive therapeutic target especially in case of muscle wasting conditions such as DMD and mitochondrial myopathy. NO levels can be influenced by stimulating the endogenous synthesis with arginine, the substrate for the NOS, or via drugs with NO-donating properties. These strategies have been trialled in the mdx mouse model of DMD and beneficial effects have been observed (see De Luca, 2012; De Palma and Clementi, 2012).
Moreover, positive results have been reported from attempts to treat the potential NO deficiency present in patients with the mitochondrial disease MELAS (mitochondrial encephalopathy, lactic acidosis, stroke-like episodes) with arginine or citrulline (Koga et al., 2005; see also El-Hattab et al., 2012; Tengan et al., 2012). However, the rationale in these studies is focused on restoring the NO deficiency observed in some forms of mitochondrial disease and not increasing mitochondrial biogenesis. Contrariwise, mitochondrial proliferation resulting from a compensation mechanism in these mitochondrial disease patients has been hypothesized to be one of the underlying causes of impaired NO synthesis in the first place (El-Hattab et al., 2012).
Apart from its role in mitochondrial biogenesis, NO also has a direct inhibitory effect on the protein complexes of the OXPHOS system. NO competitively inhibits oxygen binding at COX and, via reaction with haem iron, iron sulfur clusters or reduced protein thiols, NO also inhibits NADH-dehydrogenase activity at CI and the transfer of electrons at CIII and CI. All these direct effects on the respiratory chain make manipulation of NO levels in patients with OXPHOS defects a complex strategy (Tengan et al., 2012). On the other hand, this effect of NO is cardioprotective during ischaemia-reperfusion injury. Recently, using a mitochondria-selective S-nitrosating agent, it was shown that reversible mitochondrial S-nitrosation of CI forms a major part of the mechanism underlying the protective effect of NO during ischaemic reperfusion, thereby forming a therapeutic target for the prevention of injury after infarct or stroke (Chouchani et al., 2013).
The PPARδ agonist GW501516 induced the generation of oxidative myofibers and increased endurance, especially in combination with exercise or AMPK activation (Narkar et al., 2008). This was followed by studies in mdx models in which GW501516 improved the DMD phenotype (Miura et al., 2009; Bueno Junior et al., 2012; Jahnke et al., 2012).
In humans, GW501516 has beneficial effects on parameters linked to the metabolic syndrome (Olson et al., 2012). Unfortunately long-term preclinical treatment trials have led to safety concerns, and clinical development for humans has been discontinued. Nevertheless, stimulation of PPARδ still has enormous potential for treatment of multiple conditions once a safer alternative for GW501516 can be identified.
PPARγ activation by drugs of the thiazolidinedione (TZD) class is widely used as treatment for diabetes. Although PPARγ interacts with PGC-1α in many different tissues, TZDs have been extensively tested in models of neurodegeneration because of their anti-inflammatory properties (Carta, 2013; Choi and Bothwell, 2012). The first studies were performed with the TZD pioglitazone in the G93A SOD1 mouse model of ALS (Kiaei et al., 2005; Schutz et al., 2005). Both studies found increases in lifespan, which were suggested to be due to the anti-inflammatory effect of pioglitazone in both models. This was subsequently translated into a clinical trial with ALS patients but, disappointingly, there was no additional effect of pioglitazone on the standard treatment with the drug riluzole (Dupuis et al., 2012).
TZDs have also been tested successfully on multiple models of HD recently (Chiang et al., 2010; Napolitano et al., 2011; Jin et al., 2013). Data from these studies suggest amelioration of both PPARγ and PGC-1α levels contributes to the improved phenotype in treated mice.
Furthermore, PPARγ activation has been used extensively for the treatment of various mouse models of PD (see Carta, 2013; Ridder and Schwaninger, 2012) and AD (see Mandrekar et al., 2011). In all these models PPARγ activation was used to suppress the inflammatory response and not the induction of mitochondrial biogenesis. However, TZDs may still be beneficial for the treatment of OXPHOS disorders, especially when an inflammatory response in the brain is part of the symptoms.
The last treatment option discussed here does not require any pharmacological compounds but consists of endurance exercise, which is a strong endogenous inducer of PGC-1α, especially in skeletal muscle. Indeed, the endurance exercise regime that Wenz et al. (2008; 2009a,) used on the ΔCox10 mice was sufficient for prolonged survival and delayed onset of disease, mimicking transgenic PGC-1α overexpression or treatment with bezafibrate. The study showed clear evidence of mitochondrial proliferation and improved OXPHOS parameters in skeletal muscle. Similarly, a substantial effect of endurance exercise was also observed in the Mut mice (Safdar et al., 2011). Again this mimicked the effect of overexpression of PGC-1α, but without a decrease in muscle weight indicating that exercise may act on additional metabolic pathways as well (Dillon et al., 2012b). Both these studies in mice provide additional evidence for the current use of exercise therapy in the treatment of mitochondrial myopathies (Pfeffer and Chinnery, 2013).
Concluding remarks
The hypothesis that induction of mitochondrial biogenesis could be used as a treatment for OXPHOS disorders has been explored in various studies using bezafibrate, RSV and AICAR, as summarized here. In vitro studies in cell models have initially shown great potential for these compounds, and the recent development of appropriate mouse models for various OXPHOS disorders has resulted in the translation of this in vitro work into preclinical treatment trials.
The studies described in this review show that the translation from the in vitro results to the in vivo pharmacological outcomes is often not straightforward. There has been substantial variation in the success of treatment for each tested compound, even in terms of whether mitochondrial biogenesis is induced. In this review this has been summarized in detail for bezafibrate, but recently one study reported that dietary resveratrol treatment had no effect on mitochondrial biogenesis in skeletal muscle of wild-type mice and rats when using the same dosage shown previously by others to induce mitochondrial proliferation (Lagouge et al., 2006; Higashida et al., 2013). Differences of treatment effect may be partly due to the physical condition, age, genetic background and which tissues are affected in each mouse model. Bioavailability of the drugs could also be influenced by differences in drug quantities and administration routes. This has been demonstrated for both bezafibrate and resveratrol, for which the dose can contribute to differences in the specific mode of action (Nakajima et al., 2009; Price et al., 2012). This is even more important in the context of toxic side effects that have been reported for both compounds in studies with mouse models (Tables 2 and 3). However, it remains unclear why even wild-type mice treated with apparently the same quantity and administration route of bezafibrate show clear up-regulation of skeletal muscle mitochondrial biogenesis in some studies (Wenz et al., 2008) but not others (Viscomi et al., 2011; Yatsuga and Suomalainen, 2012).
Both resveratrol and bezafibrate are relatively safe compounds and widely used by humans. The damaging effects of bezafibrate on the liver in mice are not observed in humans, due to differences in PPARα signalling between species and the much lower doses typically used in humans (Hays et al., 2005; Tenenbaum et al., 2005; Yang et al., 2008; Djouadi and Bastin, 2011). Kidney and muscle wasting have been reported as side effects of the drug and should be taken into account when trialling bezafibrate for disorders in which these tissues are already affected (Charach et al., 2005; Wu et al., 2009). Nevertheless, a clinical trial with bezafibrate in patients with CPT2 deficiency, who have muscle involvement as part of the pathology, has generated a positive outcome (Bonnefont et al., 2009, 2010; Djouadi and Bastin, 2011).
Resveratrol is readily available as part of food and vitamin supplements but, unfortunately,it also has a very poor bioavailability and therefore has been used in trials at high doses in humans. At these high doses, resveratrol can have deleterious side effects, of which gastrointestinal symptoms are the most common (Patel et al., 2011). Despite this, recent trials with the drug in humans have shown the potential of resveratrol for the treatment of metabolic diseases (Brasnyo et al., 2011; Timmers et al., 2011; Bhatt et al., 2012; Yoshino et al., 2012; Poulsen et al., 2013). The low bioavailability has resulted in the development of more potent small activators of sirtuins, also known as sirtuin-activating compounds, which may cause fewer off-target effects (Milne et al., 2007; Feige et al., 2008).
Bezafibrate, resveratrol and AICAR have been used in trials for the treatment of other mouse models with more common neurological and muscular diseases. Mitochondrial dysfunction has been linked to the pathology of all of these more common diseases described in this review (Kuznetsov et al., 1998; Lin and Beal, 2006). Surprisingly, the majority of these treatment studies have had little focus on the effect of the treatment on mitochondrial parameters, despite an expectation that they may well have contributed to the outcomes (see Tables 2,3,4).
Finally the concept that inducing mitochondrial biogenesis may lead to increased numbers of dysfunctional mitochondria should be discussed. On the one hand, all patients with OXPHOS disorders have residual enzyme activity because a complete loss of activity is not compatible with life. Hence, up-regulation of the amount of less active mitochondria may increase ATP synthesis capacity above a certain cellular threshold. Of course, pharmacological induction may not help every cell in every tissue in a multicellular organism but could be helpful in a limited set of tissues. This is particularly relevant to AICAR, which does not cross the blood–brain barrier. Many studies have shown that, especially, skeletal muscle cells with their long multinucleated structure seem good candidates for the induction of mitochondria, resulting in fibre type switching towards a more oxidative type.
On the other hand, the mitochondrial dysfunction may not only lead to an ATP generation defect but also to an increased generation of ROS. ROS may be cellular signalling molecules under normal conditions but also have the capacity to damage protein, DNA and lipids. First of all, mitochondrial ROS production is not increased in all OXPHOS disorders. In those OXPHOS conditions where ROS are increased, it may be better to consider treatment with antioxidants instead of targeting biogenesis and potentially increasing the number of ROS generators in the cell. Second, activation of the PGC-1α pathway leads not only to mitochondrial biogenesis but also to a SIRT3-mediated antioxidant response, which may counteract any increases in ROS production (Bell and Guarente, 2011; Scarpulla, 2012; Scarpulla et al., 2012). Third, recently it has been found that AMPK activation directly activates the autophagy/mitophagy pathway via phosphorylation of mitophagy regulator ULK1 (Egan et al., 2011; Kim et al., 2011). Mitophagy regulates the degradation/recycling of dysfunctional and damaged mitochondria. Simultaneous stimulation of mitophagy and mitochondrial biogenesis is likely to provide a protection mechanism against the proliferation of dysfunctional mitochondria.
Furthermore, the study of Dillon et al. (2012b) showed that induction of the PGC-1α pathway in Mut mice mainly resulted in an increase in newly acquired mtDNA mutations in skeletal muscle compared with control and not in an increase in the heteroplasmic mutant load of inherited mtDNA mutations. The increased introduction of new mtDNA mutations probably occurs during the proliferation of mtDNA and is due to the DNA proofreading defect in POLG in the Mut mice. Stability of the inherited mutations is very important when considering patients with mitochondrial DNA mutations as candidates for treatment with mitochondrial biogenesis inducers.
This review describes a number of mouse models of conditions that appear to benefit from the induction of mitochondria. These studies have identified challenges related to variation with genetic background or bioavailability, side effects, the blood–brain barrier, differing response between tissues and potential risks of increasing mtDNA mutation load or ROS generation. These issues are clearly relevant to human trials. Because of the enormous clinical variability in patients with OXPHOS disorders, it would be unrealistic to expect to develop a one-size-fits-all treatment. Current and future clinical trials in humans will reveal how well these mouse studies translate into actual therapeutic use.
Acknowledgments
This work was supported by grants and a Principal Research Fellowship (D. R. T.) from the Australian National Health and Medical Research Council, and the Victorian Government's Operational Infrastructure Support Program (D. R. T.).
Glossary
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- AMPK
AMP-activated kinase
- BAT
brown adipose tissue
- CI–V
complex I–V
- DMD
Duchenne muscular dystrophy
- FAO
fatty acid oxidation
- HD
Huntington's disease
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- mtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- NRF
nuclear respiratory factor
- OXPHOS
oxidative phosphorylation
- PD
Parkinson's disease
- PGC-1
PPAR-γ coactivator 1
- POLG
polymerase γ
- SIRT1
silent information regulator two (Sir2) protein 1, sirtuin 1
Conflict of interest
The authors have no financial links with manufacturers of any of the materials or devices described in the manuscript.
References
- Al-Mahdawi S, Pinto RM, Varshney D, Lawrence L, Lowrie MB, Hughes S, et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–590. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov. 2013;12:465–483. doi: 10.1038/nrd4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquilano K, Baldelli S, Pagliei B, Cannata SM, Rotilio G, Ciriolo MR. p53 orchestrates the PGC-1alpha-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid Redox Signal. 2013;18:386–399. doi: 10.1089/ars.2012.4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, Lebrasseur N, Morris C, Smith E, Yang W, Ma Y, et al. The transcriptional coactivator PGC-1beta drives the formation of oxidative type IIX fibers in skeletal muscle. Cell Metab. 2007;5:35–46. doi: 10.1016/j.cmet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Bass TM, Weinkove D, Houthoofd K, Gems D, Partridge L. Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech Ageing Dev. 2007;128:546–552. doi: 10.1016/j.mad.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Bastin J, Aubey F, Rötig A, Munnich A, Djouadi F. Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients' cells lacking its components. J Clin Endocrinol Metab. 2008;93:1433–1441. doi: 10.1210/jc.2007-1701. [DOI] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell. 2011;42:561–568. doi: 10.1016/j.molcel.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Emerling BM, Ricoult SJ, Guarente L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011;30:2986–2996. doi: 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt JK, Thomas S, Nanjan MJ. Resveratrol supplementation improves glycemic control in type 2 diabetes mellitus. Nutr Res. 2012;32:537–541. doi: 10.1016/j.nutres.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Blanchet J, Longpre F, Bureau G, Morissette M, DiPaolo T, Bronchti G, et al. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1243–1250. doi: 10.1016/j.pnpbp.2008.03.024. [DOI] [PubMed] [Google Scholar]
- Bonnefont JP, Bastin J, Behin A, Djouadi F. Bezafibrate for an inborn mitochondrial beta-oxidation defect. N Engl J Med. 2009;360:838–840. doi: 10.1056/NEJMc0806334. [DOI] [PubMed] [Google Scholar]
- Bonnefont JP, Bastin J, Laforet P, Aubey F, Mogenet A, Romano S, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther. 2010;88:101–108. doi: 10.1038/clpt.2010.55. [DOI] [PubMed] [Google Scholar]
- Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- Brasnyo P, Molnar GA, Mohas M, Marko L, Laczy B, Cseh J, et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br J Nutr. 2011;106:383–389. doi: 10.1017/S0007114511000316. [DOI] [PubMed] [Google Scholar]
- Bueno Junior CR, Pantaleao LC, Voltarelli VA, Bozi LH, Brum PC, Zatz M. Combined effect of AMPK/PPAR agonists and exercise training in mdx mice functional performance. PLoS One. 2012;7:e45699. doi: 10.1371/journal.pone.0045699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvari M, Piper MD, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011;477:482–485. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Compton AG, Hershman SG, Lim SC, Lieber DS, Tucker EJ, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4:118ra110. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta AR. PPAR-gamma: therapeutic prospects in Parkinson's disease. Curr Drug Targets. 2013;14:743–751. doi: 10.2174/1389450111314070004. [DOI] [PubMed] [Google Scholar]
- Casarin A, Giorgi G, Pertegato V, Siviero R, Cerqua C, Doimo M, et al. Copper and bezafibrate cooperate to rescue cytochrome c oxidase deficiency in cells of patients with SCO2 mutations. Orphanet J Rare Dis. 2012;7:21. doi: 10.1186/1750-1172-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charach G, Grosskopf I, Rotmensch HH, Kitzis I, Weintraub M. Bezafibrates cause moderate, reversible impairment in renal function in patients without prior renal disease. Nephron Clin Pract. 2005;100:c120–c125. doi: 10.1159/000085291. [DOI] [PubMed] [Google Scholar]
- Chiang MC, Chen CM, Lee MR, Chen HW, Chen HM, Wu YS, et al. Modulation of energy deficiency in Huntington's disease via activation of the peroxisome proliferator-activated receptor gamma. Hum Mol Genet. 2010;19:4043–4058. doi: 10.1093/hmg/ddq322. [DOI] [PubMed] [Google Scholar]
- Choi JM, Bothwell AL. The nuclear receptor PPARs as important regulators of T-cell functions and autoimmune diseases. Mol Cells. 2012;33:217–222. doi: 10.1007/s10059-012-2297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Da Cruz S, Parone PA, Lopes VS, Lillo C, McAlonis-Downes M, Lee SK, et al. Elevated PGC-1alpha activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab. 2012;15:778–786. doi: 10.1016/j.cmet.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A. Pre-clinical drug tests in the mdx mouse as a model of dystrophinopathies: an overview. Acta Myol. 2012;31:40–47. [PMC free article] [PubMed] [Google Scholar]
- De Palma C, Clementi E. Nitric oxide in myogenesis and therapeutic muscle repair. Mol Neurobiol. 2012;46:682–692. doi: 10.1007/s12035-012-8311-8. [DOI] [PubMed] [Google Scholar]
- De Palma C, Falcone S, Pisoni S, Cipolat S, Panzeri C, Pambianco S, et al. Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ. 2010;17:1684–1696. doi: 10.1038/cdd.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012;3:e418. doi: 10.1038/cddis.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, et al. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet. 2005;14:2737–2748. doi: 10.1093/hmg/ddi307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon LM, Hida A, Garcia S, Prolla TA, Moraes CT. Long-term bezafibrate treatment improves skin and spleen phenotypes of the mtDNA mutator mouse. PLoS One. 2012a;7:e44335. doi: 10.1371/journal.pone.0044335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon LM, Williams SL, Hida A, Peacock JD, Prolla TA, Lincoln J, et al. Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Hum Mol Genet. 2012b;21:2288–2297. doi: 10.1093/hmg/dds049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouadi F, Bastin J. Species differences in the effects of bezafibrate as a potential treatment of mitochondrial disorders. Cell Metab. 2011;14:715–716. doi: 10.1016/j.cmet.2011.11.003. author reply 717. [DOI] [PubMed] [Google Scholar]
- Dogan SA, Trifunovic A. Modelling mitochondrial dysfunction in mice. Physiol Res. 2011;60(Suppl 1):S61–S70. doi: 10.33549/physiolres.932179. [DOI] [PubMed] [Google Scholar]
- Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova N, et al. Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum Mol Genet. 2012;21:5091–5105. doi: 10.1093/hmg/dds355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, Dengler R, Heneka MT, Meyer T, Zierz S, Kassubek J, et al. A randomized, double blind, placebo-controlled trial of pioglitazone in combination with riluzole in amyotrophic lateral sclerosis. PLoS One. 2012;7:e37885. doi: 10.1371/journal.pone.0037885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Emrick LT, Craigen WJ, Scaglia F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol Genet Metab. 2012;107:247–252. doi: 10.1016/j.ymgme.2012.06.018. [DOI] [PubMed] [Google Scholar]
- Falk MJ, Pierce EA, Consugar M, Xie MH, Guadalupe M, Hardy O, et al. Mitochondrial disease genetic diagnostics: optimized whole-exome analysis for all MitoCarta nuclear genes and the mitochondrial genome. Discov Med. 2012;14:389–399. [PMC free article] [PubMed] [Google Scholar]
- Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, et al. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008;8:347–358. doi: 10.1016/j.cmet.2008.08.017. [DOI] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. 2011;93:884S–8890. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Jeninga EH, Canto C, Harach T, de Boer VC, Andreux P, et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci Rep. 2012;2:425. doi: 10.1038/srep00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell. 2011;19:416–428. doi: 10.1016/j.ccr.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Diaz F, Garcia S, Moraes CT. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2007;104:14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, Villarroya F. SIRT3, a pivotal actor in mitochondrial functions: metabolism, cell death and aging. Biochem J. 2012;444:1–10. doi: 10.1042/BJ20120030. [DOI] [PubMed] [Google Scholar]
- Golubitzky A, Dan P, Weissman S, Link G, Wikstrom JD, Saada A. Screening for active small molecules in mitochondrial complex I deficient patient's fibroblasts, reveals AICAR as the most beneficial compound. PLoS One. 2011;6:e26883. doi: 10.1371/journal.pone.0026883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BS, Delgado Diaz DC, Kostek MC. Resveratrol decreases inflammation and increases utrophin gene expression in the mdx mouse model of Duchenne muscular dystrophy. Clin Nutr. 2013;32:104–111. doi: 10.1016/j.clnu.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- Hallows WC, Yu W, Smith BC, Devries MK, Ellinger JJ, Someya S, et al. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol Cell. 2011;41:139–149. doi: 10.1016/j.molcel.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Choi JR, Soon Shin K, Kang SJ. Resveratrol upregulated heat shock proteins and extended the survival of G93A-SOD1 mice. Brain Res. 2012;1483:112–117. doi: 10.1016/j.brainres.2012.09.022. [DOI] [PubMed] [Google Scholar]
- Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007;21:770–783. doi: 10.1101/gad.1525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci. 2011;36:470–477. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Hays T, Rusyn I, Burns AM, Kennett MJ, Ward JM, Gonzalez FJ, et al. Role of peroxisome proliferator-activated receptor-alpha (PPARalpha) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis. 2005;26:219–227. doi: 10.1093/carcin/bgh285. [DOI] [PubMed] [Google Scholar]
- Henin N, Vincent MF, Gruber HE, Van den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995;9:541–546. doi: 10.1096/fasebj.9.7.7737463. [DOI] [PubMed] [Google Scholar]
- Henin N, Vincent MF, Van den Berghe G. Stimulation of rat liver AMP-activated protein kinase by AMP analogues. Biochim Biophys Acta. 1996;1290:197–203. doi: 10.1016/0304-4165(96)00021-9. [DOI] [PubMed] [Google Scholar]
- Herskovits AZ, Guarente L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 2013;23:746–758. doi: 10.1038/cr.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashida K, Kim SH, Jung SR, Asaka M, Holloszy JO, Han DH. Effects of resveratrol and SIRT1 on PGC-1alpha activity and mitochondrial biogenesis: a reevaluation. PLoS Biol. 2013;11:e1001603. doi: 10.1371/journal.pbio.1001603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DJ, Calingasan NY, Wille E, Dumont M, Beal MF. Resveratrol protects against peripheral deficits in a mouse model of Huntington's disease. Exp Neurol. 2010;225:74–84. doi: 10.1016/j.expneurol.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Hondares E, Mora O, Yubero P, Rodriguez de la Concepcion M, Iglesias R, Giralt M, et al. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1alpha gene transcription: an autoregulatory loop controls PGC-1alpha expression in adipocytes via peroxisome proliferator-activated receptor-gamma coactivation. Endocrinology. 2006;147:2829–2838. doi: 10.1210/en.2006-0070. [DOI] [PubMed] [Google Scholar]
- Hondares E, Pineda-Torra I, Iglesias R, Staels B, Villarroya F, Giralt M. PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem Biophys Res Commun. 2007;354:1021–1027. doi: 10.1016/j.bbrc.2007.01.092. [DOI] [PubMed] [Google Scholar]
- Hondares E, Rosell M, Diaz-Delfin J, Olmos Y, Monsalve M, Iglesias R, et al. Peroxisome proliferator-activated receptor alpha (PPARalpha) induces PPARgamma coactivator 1alpha (PGC-1alpha) gene expression and contributes to thermogenic activation of brown fat: involvement of PRDM16. J Biol Chem. 2011;286:43112–43122. doi: 10.1074/jbc.M111.252775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori YS, Kuno A, Hosoda R, Tanno M, Miura T, Shimamoto K, et al. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J Pharmacol Exp Ther. 2011;338:784–794. doi: 10.1124/jpet.111.183210. [DOI] [PubMed] [Google Scholar]
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science. 2013;339:1216–1219. doi: 10.1126/science.1231097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnke VE, Van Der Meulen JH, Johnston HK, Ghimbovschi S, Partridge T, Hoffman EP, et al. Metabolic remodeling agents show beneficial effects in the dystrophin-deficient mdx mouse model. Skelet Muscle. 2012;2:16. doi: 10.1186/2044-5040-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Albertz J, Guo Z, Peng Q, Rudow G, Troncoso JC, et al. Neuroprotective effects of PPAR-gamma agonist rosiglitazone in N171-82Q mouse model of Huntington's disease. J Neurochem. 2013;125:410–419. doi: 10.1111/jnc.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A. 2011;108:14608–14613. doi: 10.1073/pnas.1111308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E, O'Neill BT, Rardin MJ, Kleinridders A, Ilkeyeva OR, Ussar S, et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes. 2013;62:3404–3417. doi: 10.2337/db12-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, Calingasan NY, Hennessey TM, Sharma A, Yang L, Wille E, et al. Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington's disease. Hum Mol Genet. 2012;21:1124–1137. doi: 10.1093/hmg/ddr541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph AM, Joanisse DR, Baillot RG, Hood DA. Mitochondrial dysregulation in the pathogenesis of diabetes: potential for mitochondrial biogenesis-mediated interventions. Exp Diabetes Res. 2012;2012:642038. doi: 10.1155/2012/642038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, et al. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- Karuppagounder SS, Pinto JT, Xu H, Chen HL, Beal MF, Gibson GE. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer's disease. Neurochem Int. 2009;54:111–118. doi: 10.1016/j.neuint.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;191:331–336. doi: 10.1016/j.expneurol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilo T, Yuan C, van Praag H. Endurance factors improve hippocampal neurogenesis and spatial memory in mice. Learn Mem. 2011;18:103–107. doi: 10.1101/lm.2001611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koene S, Smeitink J. Metabolic manipulators: a well founded strategy to combat mitochondrial dysfunction. J Inherit Metab Dis. 2011;34:315–325. doi: 10.1007/s10545-010-9162-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Tanabe Y, et al. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64:710–712. doi: 10.1212/01.WNL.0000151976.60624.01. [DOI] [PubMed] [Google Scholar]
- Komen JC, Thorburn DR. Modeling mitochondrial dysfunction in neurodegenerative disease. In: Reeve AK, Krishnan KJ, Duchen MR, Turnbull DM, editors. Mitochondrial Dysfunction in Neurodegenerative Disorders. London: Springer; 2012. pp. 193–212. [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Kuno A, Hori YS, Hosoda R, Tanno M, Miura T, Shimamoto K, et al. Resveratrol improves cardiomyopathy in dystrophin-deficient mice through SIRT1 protein-mediated modulation of p300 protein. J Biol Chem. 2013;288:5963–5972. doi: 10.1074/jbc.M112.392050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov AV, Winkler K, Wiedemann FR, von Bossanyi P, Dietzmann K, Kunz WS. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol Cell Biochem. 1998;183:87–96. doi: 10.1023/a:1006868130002. [DOI] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, et al. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Li L, Voullaire L, Sandi C, Pook MA, Ioannou PA, Delatycki MB, et al. Pharmacological screening using an FXN-EGFP cellular genomic reporter assay for the therapy of Friedreich ataxia. PLoS One. 2013;8:e55940. doi: 10.1371/journal.pone.0055940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- Liang H, Ward WF, Jang YC, Bhattacharya A, Bokov AF, Li Y, et al. PGC-1alpha protects neurons and alters disease progression in an amyotrophic lateral sclerosis mouse model. Muscle Nerve. 2011;44:947–956. doi: 10.1002/mus.22217. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KSS, Pierre J, Zhang CY, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lira VA, Brown DL, Lira AK, Kavazis AN, Soltow QA, Zeanah EH, et al. Nitric oxide and AMPK cooperatively regulate PGC-1 in skeletal muscle cells. J Physiol. 2010;588:3551–3566. doi: 10.1113/jphysiol.2010.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubicic V, Miura P, Burt M, Boudreault L, Khogali S, Lunde JA, et al. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum Mol Genet. 2011;20:3478–3493. doi: 10.1093/hmg/ddr265. [DOI] [PubMed] [Google Scholar]
- Lofrumento DD, Nicolardi G, Cianciulli A, De Nuccio F, La Pesa V, Carofiglio V, et al. Neuroprotective effects of resveratrol in an MPTP mouse model of Parkinson's-like disease: possible role of SOCS-1 in reducing pro-inflammatory responses. Innate Immun. doi: 10.1177/1753425913488429. DOI: 10.1177/1753425913488429. [DOI] [PubMed] [Google Scholar]
- McConell GK, Ng GP, Phillips M, Ruan Z, Macaulay SL, Wadley GD. Central role of nitric oxide synthase in AICAR and caffeine-induced mitochondrial biogenesis in L6 myocytes. J Appl Physiol. 2010;108:589–595. doi: 10.1152/japplphysiol.00377.2009. [DOI] [PubMed] [Google Scholar]
- Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. 2011;54:2185–2197. doi: 10.1002/hep.24599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangos PJ, Loftus T, Wiesner J, Lowe T, Rossi E, Browne CE, et al. Adenosinergic modulation of homocysteine-induced seizures in mice. Epilepsia. 1990;31:239–246. doi: 10.1111/j.1528-1157.1990.tb05371.x. [DOI] [PubMed] [Google Scholar]
- Markert CD, Kim E, Gifondorwa DJ, Childers MK, Milligan CE. A single-dose resveratrol treatment in a mouse model of amyotrophic lateral sclerosis. J Med Food. 2010;13:1081–1085. doi: 10.1089/jmf.2009.0243. [DOI] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol Endocrinol Metab. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura P, Chakkalakal JV, Boudreault L, Belanger G, Hebert RL, Renaud JM, et al. Pharmacological activation of PPARbeta/delta stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum Mol Genet. 2009;18:4640–4649. doi: 10.1093/hmg/ddp431. [DOI] [PubMed] [Google Scholar]
- Monsalve FA, Pyarasani RD, Delgado-Lopez F, Moore-Carrasco R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediators Inflamm. 2013;2013:549627. doi: 10.1155/2013/549627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudo G, Makela J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, et al. Transgenic expression and activation of PGC-1alpha protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Sci. 2012;69:1153–1165. doi: 10.1007/s00018-011-0850-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Tanaka N, Kanbe H, Hara A, Kamijo Y, Zhang X, et al. Bezafibrate at clinically relevant doses decreases serum/liver triglycerides via down-regulation of sterol regulatory element-binding protein-1c in mice: a novel peroxisome proliferator-activated receptor alpha-independent mechanism. Mol Pharmacol. 2009;75:782–792. doi: 10.1124/mol.108.052928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano M, Costa L, Palermo R, Giovenco A, Vacca A, Gulino A. Protective effect of pioglitazone, a PPARgamma ligand, in a 3 nitropropionic acid model of Huntington's disease. Brain Res Bull. 2011;85:231–237. doi: 10.1016/j.brainresbull.2011.03.011. [DOI] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Carruba MO. Nitric oxide and mitochondrial biogenesis. J Cell Sci. 2006;119:2855–2862. doi: 10.1242/jcs.03062. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- Noe N, Dillon L, Lellek V, Diaz F, Hida A, Moraes CT, et al. Bezafibrate improves mitochondrial function in the CNS of a mouse model of mitochondrial encephalopathy. Mitochondrion. 2013;13:417–426. doi: 10.1016/j.mito.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakhill JS, Scott JW, Kemp BE. AMPK functions as an adenylate charge-regulated protein kinase. Trends Endocrinol Metab. 2012;23:125–132. doi: 10.1016/j.tem.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Olson EJ, Pearce GL, Jones NP, Sprecher DL. Lipid effects of peroxisome proliferator-activated receptor-delta agonist GW501516 in subjects with low high-density lipoprotein cholesterol: characteristics of metabolic syndrome. Arterioscler Thromb Vasc Biol. 2012;32:2289–2294. doi: 10.1161/ATVBAHA.112.247890. [DOI] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012;148:421–433. doi: 10.1016/j.cell.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KR, Scott E, Brown VA, Gescher AJ, Steward WP, Brown K. Clinical trials of resveratrol. Ann N Y Acad Sci. 2011;1215:161–169. doi: 10.1111/j.1749-6632.2010.05853.x. [DOI] [PubMed] [Google Scholar]
- Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol. 2012;181:583–592. doi: 10.1016/j.ajpath.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Chinnery PF. Diagnosis and treatment of mitochondrial myopathies. Ann Med. 2013;45:4–16. doi: 10.3109/07853890.2011.605389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;(4) doi: 10.1002/14651858.CD004426.pub3. CD004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirinen E, Lo Sasso G, Auwerx J. Mitochondrial sirtuins and metabolic homeostasis. Best Pract Res Clin Endocrinol Metab. 2012;26:759–770. doi: 10.1016/j.beem.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen L, Siersbaek M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol. 2012;23:631–639. doi: 10.1016/j.semcdb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Poulsen MM, Vestergaard PF, Clasen BF, Radko Y, Christensen LP, Stodkilde-Jorgensen H, et al. High-dose resveratrol supplementation in obese men: an investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes. 2013;62:1186–1195. doi: 10.2337/db12-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A. 2013;110:6601–6606. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridder DA, Schwaninger M. In search of the neuroprotective mechanism of thiazolidinediones in Parkinson's disease. Exp Neurol. 2012;238:133–137. doi: 10.1016/j.expneurol.2012.08.012. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, et al. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A. 2011;108:4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. 2011;1813:1269–1278. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC. Nucleus-encoded regulators of mitochondrial function: integration of respiratory chain expression, nutrient sensing and metabolic stress. Biochim Biophys Acta. 2012;1819:1088–1097. doi: 10.1016/j.bbagrm.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459–466. doi: 10.1016/j.tem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, DiMauro S, Hirano M, Gilkerson RW. Therapeutic prospects for mitochondrial disease. Trends Mol Med. 2010;16:268–276. doi: 10.1016/j.molmed.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, et al. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature. 2013;495:379–383. doi: 10.1038/nature11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schurmann B, et al. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25:7805–7812. doi: 10.1523/JNEUROSCI.2038-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsby JT, Morine KJ, Pendrak K, Barton ER, Sweeney HL. Rescue of dystrophic skeletal muscle by PGC-1alpha involves a fast to slow fiber type shift in the mdx mouse. PLoS One. 2012;7:e30063. doi: 10.1371/journal.pone.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010;12:654–661. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Diaz F, Iommarini L, Aure K, Lombes A, Moraes CT. PGC-1α/β induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Human Molecular Genetics. 2009;18:1805–1812. doi: 10.1093/hmg/ddp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353:33–36. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A. Therapy for mitochondrial disorders: little proof, high research activity, some promise. Semin Fetal Neonatal Med. 2011;16:236–240. doi: 10.1016/j.siny.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8:92–103. doi: 10.1038/nrendo.2011.138. [DOI] [PubMed] [Google Scholar]
- Tenenbaum A, Motro M, Fisman EZ. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: the bezafibrate lessons. Cardiovasc Diabetol. 2005;4:14. doi: 10.1186/1475-2840-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengan CH, Rodrigues GS, Godinho RO. Nitric oxide in skeletal muscle: role on mitochondrial biogenesis and function. Int J Mol Sci. 2012;13:17160–17184. doi: 10.3390/ijms131217160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers S, Konings E, Bilet L, Houtkooper RH, van de Weijer T, Goossens GH, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14:612–622. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torraco A, Diaz F, Vempati UD, Moraes CT. Mouse models of oxidative phosphorylation defects: powerful tools to study the pathobiology of mitochondrial diseases. Biochim Biophys Acta. 2009;1793:171–180. doi: 10.1016/j.bbamcr.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JH, Park SJ, Kang H, Yang S, Foretz M, McBurney MW, et al. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes. 2010;59:554–563. doi: 10.2337/db09-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010;35:669–675. doi: 10.1016/j.tibs.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010;285:9100–9113. doi: 10.1074/jbc.M109.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci. 2012;122:253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, et al. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011;14:80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenz T. Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion. 2013;13:134–142. doi: 10.1016/j.mito.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wenz T, Diaz F, Hernandez D, Moraes CT. Endurance exercise is protective for mice with mitochondrial myopathy. J Appl Physiol. 2009a;106:1712–1719. doi: 10.1152/japplphysiol.91571.2008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A. 2009b;106:20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol. 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- Wong LJ. Next generation molecular diagnosis of mitochondrial disorders. Mitochondrion. 2013;13:379–387. doi: 10.1016/j.mito.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- Wu J, Song Y, Li H, Chen J. Rhabdomyolysis associated with fibrate therapy: review of 76 published cases and a new case report. Eur J Clin Pharmacol. 2009;65:1169–1174. doi: 10.1007/s00228-009-0723-7. [DOI] [PubMed] [Google Scholar]
- Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. 2013;27:234–250. doi: 10.1101/gad.211649.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Brosel S, Acin-Perez R, Slavkovich V, Nishino I, Khan R, et al. Analysis of mouse models of cytochrome c oxidase deficiency owing to mutations in Sco2. Hum Mol Genet. 2010;19:170–180. doi: 10.1093/hmg/ddp477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Nagano T, Shah Y, Cheung C, Ito S, Gonzalez FJ. The PPAR alpha-humanized mouse: a model to investigate species differences in liver toxicity mediated by PPAR alpha. Toxicol Sci. 2008;101:132–139. doi: 10.1093/toxsci/kfm206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsuga S, Suomalainen A. Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum Mol Genet. 2012;21:526–535. doi: 10.1093/hmg/ddr482. [DOI] [PubMed] [Google Scholar]
- Ylikallio E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med. 2012;44:41–59. doi: 10.3109/07853890.2011.598547. [DOI] [PubMed] [Google Scholar]
- Yoshino J, Conte C, Fontana L, Mittendorfer B, Imai S, Schechtman KB, et al. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab. 2012;16:658–664. doi: 10.1016/j.cmet.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies – disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–114. doi: 10.1016/j.preteyeres.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang M, Xu S, Maitland-Toolan KA, Zuccollo A, Hou X, Jiang B, et al. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55:2180–2191. doi: 10.2337/db05-1188. [DOI] [PubMed] [Google Scholar]
- Zhao W, Varghese M, Yemul S, Pan Y, Cheng A, Marano P, et al. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1alpha) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener. 2011;6:51. doi: 10.1186/1750-1326-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]