

Abstract
Diabetic nephropathy (DN) is a progressive microvascular complication arising from diabetes. Within the kidney, the glomeruli, tubules, vessels and interstitium are disrupted, ultimately impairing renal function and leading to end-stage renal disease (ESRD). Current pharmacological therapies used in individuals with DN do not prevent the inevitable progression to ESRD; therefore, new targets of therapy are urgently required. Studies from animal models indicate that disturbances in mitochondrial homeostasis are central to the pathogenesis of DN. Since renal proximal tubule cells rely on oxidative phosphorylation to provide adequate ATP for tubular reabsorption, an impairment of mitochondrial bioenergetics can result in renal functional decline. Defects at the level of the electron transport chain have long been established in DN, promoting electron leakage and formation of superoxide radicals, mediating microinflammation and contributing to the renal lesion. More recent studies suggest that mitochondrial-associated proteins may be directly involved in the pathogenesis of tubulointerstitial fibrosis and glomerulosclerosis. An accumulation of fragmented mitochondria are found in the renal cortex in both humans and animals with DN, suggesting that in tandem with a shift in dynamics, mitochondrial clearance mechanisms may be impaired. The process of mitophagy is the selective targeting of damaged or dysfunctional mitochondria to autophagosomes for degradation through the autophagy pathway. The current review explores the concept that an impairment in the mitophagy system leads to the accelerated progression of renal pathology. A better understanding of the cellular and molecular events that govern mitophagy and dynamics in DN may lead to improved therapeutic strategies.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: diabetes, mitochondria, diabetic nephropathy, kidney, mitochondrial dysfunction, mitophagy, autophagy, dynamics
Introduction
Diabetic nephropathy (DN) refers to chronic kidney disease (CKD) initiated by diabetes mellitus (2007). DN, which affects approximately 30% of patients with diabetes (Parving et al., 2006; Thomas et al., 2006; 2007; Nathan et al., 2009), is the most common cause of end-stage renal disease (ESRD) in Western societies (Gilbertson et al., 2005). Patients with DN carry a higher risk for co-morbidities such as cardiovascular disease (Borch-Johnsen and Kreiner, 1987), and indeed, renal dysfunction is a major predictor of all-cause mortality (Matsushita et al., 2010). Not only does diabetes and its complications impose a substantial burden on the economy, but it also inflicts high costs on society, both in terms of reduced quality of life, and pain and suffering of people with diabetes (2013). As the prevalence of both type 1 and type 2 diabetes is increasing (Whiting et al., 2011), these costs are set to escalate, making diabetes and its complications an urgent public health issue.
The natural history of DN is the development of persistent microalbuminuria (presence of albumin in the urine), which in some patients progresses to overt proteinuria followed by a gradual decline in glomerular filtration rate (GFR), eventually leading to renal failure (Parving et al., 1981). However, not all patients fit the classical paradigm, as there is increasing evidence of renal impairment in the absence of albuminuria in some patients with DN (Perkins et al., 2003; Hovind et al., 2004; de Boer et al., 2011). The presence and severity of nephropathy is associated with premature mortality due to cardiovascular causes (Ninomiya et al., 2009; Whitman et al., 2012). Changes in renal function are associated with ultrastructural changes involving all four renal compartments: glomeruli, tubules, interstitium and vessels. Within the glomerular and tubulointerstitial compartments, there is an accumulation of extracellular matrices (ECMs) (Raparia et al., 2013). Thickening of the glomerular basement membrane (Parving et al., 1992) is paralleled by thickening of the tubular basement membrane (Brito et al., 1998). Other renal lesions include afferent and efferent arteriolar hyalinosis (Mauer et al., 1981). Progressive renal functional loss results with advanced disease, due to glomerular collapse and scarring (i.e. sclerosis), by capillary lumen obliteration resulting from mesangial expansion and by tubular atrophy (Steinke, 2009).
The Diabetes Control and Complications Trial, which examined whether long-term complications of diabetes could be prevented or delayed by implementing therapies aimed at achieving blood glucose levels as close as possible to the normal range (HbA1c < 6.05%), revealed that intensive therapy reduced the risks for the development and progression of microalbuminuria (1993). This implies that hyperglycaemia is a fundamental cause of microvascular complications. However, more recent studies have confirmed that intensive treatment does not prevent loss of GFR or progression to ESRD (Foundation, 2012). Hence, other variables are needed to explain why only a subset of patients with DN progress to ESRD.
Inhibition of the renin angiotensin system (RAS) is the major focus of the current clinical therapy. However, agents that target the RAS, such as ACE inhibitors or angiotensin receptor-1 antagonists, have been only partially successful as these compounds just delay, rather than prevent, the progression to ESRD (Brenner et al., 2001; Bilous et al., 2009; Mauer et al., 2009). Thus, there is an urgent need to identify better targets to prevent ESRD.
The pathogenesis of DN is extremely complex as the kidney is not only composed of multiple cell populations, but is involved in several essential regulatory functions including, but not limited to, BP regulation, filtration of toxins, maintenance of acid–base balance, regulation of electrolytes, reabsorption of nutrients and hormone secretion (Brenner and Rector, 2008). Several key pathways of pathology thought to promote renal dysfunction leading to DN include an abnormally active RAS, overproduction of reactive oxygen species (ROS), increased glucose metabolite flux, advanced glycation end-products, endoplasmic reticulum (ER) stress and enhanced pro-inflammatory cytokine signalling (recently reviewed in Forbes and Cooper, 2013; Rask-Madsen and King, 2013). Increasing evidence indicates that the disruption of mitochondrial bioenergetics may be important in the development and progression of DN (Forbes et al., 2008; Sivitz and Yorek, 2010; Sharma et al., 2013). As the kidney relies on oxidative phosphorylation (OXPHOS) to provide the bulk requirements of ATP for tubular reabsorption (Soltoff, 1986), it is not surprising that mitochondrial homeostasis is strictly essential for an optimally functioning kidney. Indeed, in animal models of DN, it has been shown that mitochondrial ATP is depleted in the renal cortex in late diabetes, coinciding with glomerular and tubulointerstitial pathology (Tan et al., 2010).
Mitochondrial dysfunction occurs following inhibition of OXPHOS, which results in decreased ATP production, loss of mitochondrial membrane potential (ΔΨm) and can ultimately lead to changes in cationic gradients (e.g. cytosolic Ca2+ concentrations), increased ROS from various sites of the electron transport chain (ETC) and, ultimately, redistribution of mitochondrial cell death proteins. Whereas much attention has been given to the molecular events that govern mitochondrial homeostasis in cancer (Wallace, 2012) and neurodegeneration, such as Parkinson's disease and Alzheimer's disease (Higgins et al., 2010; Lezi and Swerdlow, 2012), limited investigations have been undertaken on these events in DN. The high energy demands of the brain and malignant tumours depend heavily on functional mitochondria. Similarly, cells in the kidney – particularly the mitochondria-rich proximal tubule epithelial cells (PTECs) – have high ATP requirements to drive tubular reabsorption (Harris et al., 1981; Balaban and Mandel, 1988). Yet, in the context of DN, there has been comparatively less research undertaken on mitochondrial biology.
Autophagy is the process responsible for recycling organelles and long-lived proteins to maintain cellular homeostasis. Autophagy can be stimulated in the face of cellular stress or starvation to meet increased energy demands or to remove toxic protein aggregates and damaged organelles (Yang and Klionsky, 2010a). Autophagy comes in three forms: micro-, macro-and chaperone-mediated autophagy. Macro-autophagy (herein referred to as autophagy) is distinguished from other forms of autophagy, as it is the only process that requires the encapsulation of cellular debris in double membrane vesicles, known as autophagosomes. Autophagosomes transport their contents to lysosomes where fusion occurs, and the contents are transferred to the acidic organelle for degradation. The contents, once degraded, are recycled to generate new proteins and organelles. The removal of dysfunctional or redundant mitochondria from cells can occur by selective or non-selective autophagy. Targeted degradation of mitochondria by autophagic machinery is termed ‘mitophagy’ (Lemasters, 2005). The role of mitophagy in DN is of particular interest given that accumulations of damaged mitochondria have been observed in the kidney, raising the possibility that an impairment in the mitophagy system may be present.
The current review aims to focus on the role of impaired mitochondrial homeostasis in the aetiology of DN and to provide new insights into bioenergetic and morphological regulation of mitochondria in the kidney. In particular, the role of autophagy and mitophagy and the potential contribution to mitochondrial dysfunction in DN will be examined. Knowledge on mitochondria and autophagy gained from other fields of cell biology will be discussed, in addition to putative targets for therapeutic intervention in DN. As there has been increasing emphasis on the importance of mitochondria and autophagy in the development and progression of many other chronic diseases, particularly cancer and neurodegeneration, it is likely that these pathways could also be aberrant in the diabetic kidney.
Cause or consequence: mitochondrial dysfunction during DN
Hyperglycaemia is thought to promote oxidative stress during diabetic complications such as DN (Baynes, 1991; Brownlee, 2001). Oxidative stress can be directly toxic to cells, causing damage to DNA, proteins and lipids. However, the significance of excessive ROS to the pathology of DN remains to be fully determined, as it is not yet known whether oxidative stress has a primary role in the pathogenesis of complications (Baynes and Thorpe, 1999). ROS is produced from several sources in DN, with mitochondria considered one of the major sites (Forbes et al., 2008). Mitochondrial dysfunction in DN may have far wider implications for the pathogenesis of the disease as it can result in a decline in ATP required to maintain cellular homeostasis, in particular to drive the Na+/K+-ATPase in the proximal tubule to preserve the Na+ gradient (Soltoff, 1986). Furthermore, end-stage mitochondrial dysfunction can lead to intrinsic cell death. Ultimately, the question of whether mitochondrial dysfunction is the cause or consequence of DN becomes irrelevant. Without healthy mitochondria, cellular homeostasis within the proximal tubule will degenerate (Figure 1). Thus, therapeutic intervention of DN must incorporate strategies directed towards alleviating mitochondrial dysfunction (discussed later).
Figure 1.
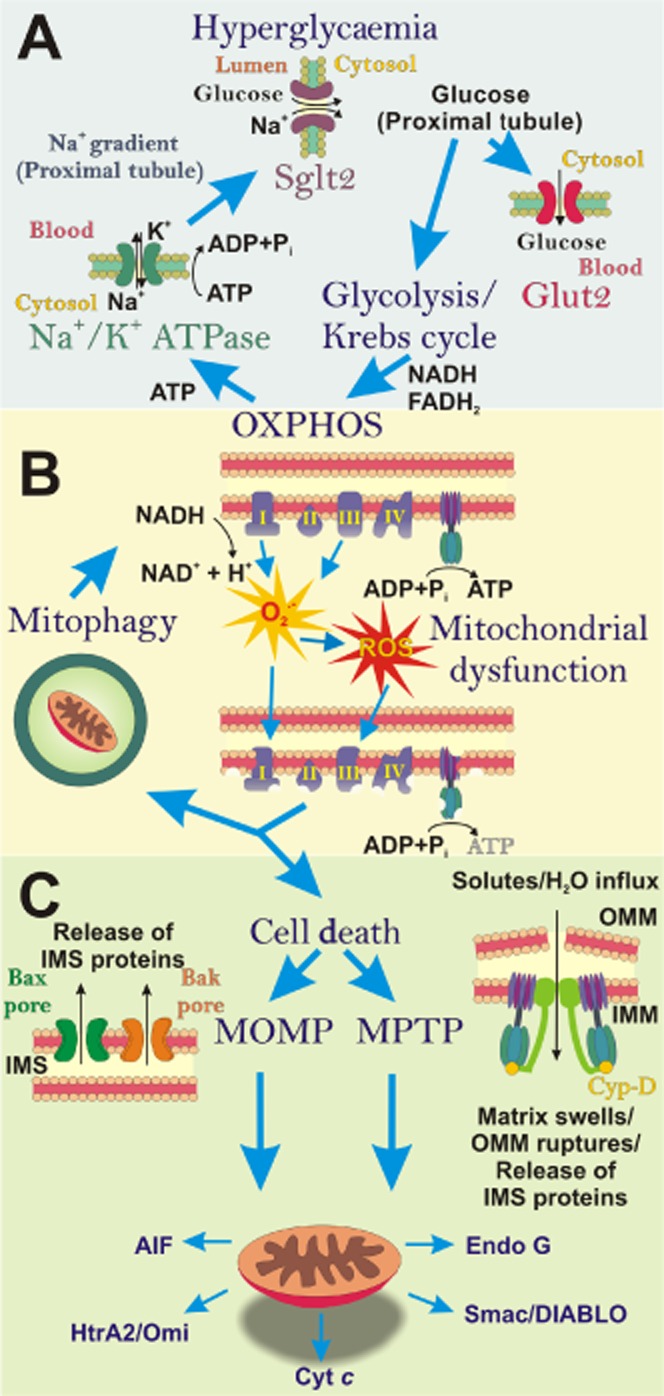
The ‘vicious cycle’ of diabetic nephropathy (DN): impact of hyperglycaemia on mitochondrial function. (A) In the diabetic milieu, excess blood glucose, filtered by the kidneys has to be reabsorbed, predominantly by the S1 segment of the proximal tubule. Glucose is transported through the proximal tubule epithelial cell (PTEC) via Sglt2 (on the apical/luminal surface) and GLUT2 (on the basolateral surface). Within the PTEC, this increased pool of glucose is metabolized and ATP is produced by oxidative phosphorylation (OXPHOS). The resultant ATP is utilized to fuel the Na+/K+-ATPase pump, which drives the Na+ gradient across the proximal tubule, required for co-transport of glucose via Sglt2. (B) During hyperglycaemia, superoxide (O2−) is generated from the electron transport chain (ETC) and can be subsequently converted into other reactive oxygen species (ROS). Excess ROS causes damage to mitochondria and the ETC, resulting in impaired ATP production. These dysfunctional mitochondria can be eliminated by mitophagy. However, when the number of damaged mitochondria overwhelms the mitophagy process, dysfunctional mitochondria can initiate cell death. (C) Apoptosis or programmed necrosis ensues via mitochondria outer membrane permeabilization (MOMP) or mitochondrial permeability transition pore (MPTP). These mechanisms result in the release of inter-membrane space (IMS) proteins from mitochondria into the cytosol, which promote cell death.
During hyperglycaemia, plasma glucose exceeds the filtration rate of the proximal tubule, resulting in glucose excretion in urine (Mather and Pollock, 2011). In the early onset of DN, an increased glucose load is thus placed on the apical sodium glucose transporter 2 (SGLT2), as glucose is transported from the tubular lumen into the PTECs against the glucose gradient (Figure 1A). SGLT2 relies on active basolateral Na+/K+-ATPase pumps to remove Na+ from the PTECs into the peritubular capillary, creating a downhill Na+ gradient between the tubular lumen and the PTECs. The downhill Na+ gradient allows co-transport of Na+ and glucose into the proximal tubule from the tubular lumen (Vallon, 2011). Glucose is, in turn, passed from the PTECs into the peritubular capillary via the glucose uniporter GLUT2. This complex process is reliant on functional mitochondria to produce an ample pool of ATP to drive the Na+/K+-ATPase pump (Figure 1A). Under the diabetic milieu, in order to maintain the glucose gradient, there is an increased demand for ATP, which, unless satisfied, will lead to PTEC impairment. Indeed, a decrease in the ATP pool is the initiating factor in the development of both lethal and sub-lethal renal PTEC injury, and the degree of ATP decline determines the severity of injury (Bonventre and Weinberg, 2003).
Mitochondria are required to increase OXPHOS in order to meet this additional demand for ATP during DN. OXPHOS is the process by which a membrane potential (ΔΨm) is generated across the inner mitochondrial membrane (IMM) via the ETC. The ETC is composed of a series of protein complexes (complex I–IV). This ΔΨm drives the transfer of H+ across the IMM into the inter-membrane space (IMS). The passive diffusion of H+ via ATP synthase back across the IMM into the matrix results in a proton motive force that drives ATP production. The ETC receives electrons from NADH, and FADH2, which are produced via the citric acid cycle. In DN, where there is an abundance of substrate in the form of glucose, the citric acid cycle is readily fuelled to produce NADH and FADH2. However, during the transfer of electrons, leakage can occur from complex I and complex III. These leaked electrons react with oxygen to create ROS in the form of superoxide (extensively reviewed by Murphy, 2009). Hence, over a sustained period of time, the accumulative effects of excessive ROS production may overwhelm the mitochondria's antioxidant defence, resulting in oxidative stress. The essence of this idea is born from Denham Harman's free radical theory of ageing, whereby uncontrolled mitochondrial reactive oxygen species (mROS) production resulted in damage to mitochondria, particularly mitochondrial DNA (mtDNA) (Harman, 1972). The mtDNA codes for subunits of the ETC, thus damage caused by oxidative stress can result in impaired formation of complexes leading to inefficient OXPHOS, further superoxide production and decreased ATP production. Hence, over time, excessive amounts of ROS can lead to a vicious cycle, resulting in mitochondrial dysfunction (Figure 1B).
Studies from animal models indicate that disturbances in mitochondrial homeostasis are central to the pathogenesis of DN. Aberrations in mitochondrial function within the kidney in the context of experimental diabetes have long been observed. Lesions have been seen at the level of the ETC, with a decline in the activity of complex I (Coughlan et al., 2009) and complex III (Rosca et al., 2005), observed within the renal cortex in rats with streptozotocin (STZ)-induced diabetes. Current dogma is that hyperglycaemia provides excess substrate to the respiratory chain, which, in the face of impaired ETC subunits, leaks electrons to form superoxide (Brownlee, 2005). This decline in OXPHOS results in a reduced ATP pool. Indeed, mitochondrial ATP depletion has been shown in the renal cortex in a diabetic setting (Tan et al., 2010). ATP depletion in tubular epithelial cells can lead to cytoskeletal changes as actin polymerization is ATP-dependent (Atkinson et al., 2004). Disruption of the cytoskeleton results in the degradation of the brush border, loss of cell to cell contact, disruption of barrier function and cell detachment (Sharfuddin and Molitoris, 2011).
Increased formation of ROS has been widely observed in the diabetic kidney (Raza et al., 2004; Moreira et al., 2006; Coughlan et al., 2007; 2009,; Fujita et al., 2009; Galloway et al., 2012; Sourris et al., 2012; Wang et al., 2012), and, in tandem with an inherent manganese superoxide dismutase (MnSOD or SOD2) functional decline (Coughlan et al., 2007; de Cavanagh et al., 2008) would serve to induce oxidative stress. Moreover, an increase in oxygen consumption is generally seen (Korner et al., 1994; Katyare and Satav, 2005; Palm et al., 2008) and is thought to be a manifestation of mitochondrial uncoupling (Hansell et al., 2013). In contrast, two studies found no change in mitochondrial respiration in the renal cortices of Ins2-Akita mice, a genetic model of type 1 diabetes (Bugger et al., 2009; Chacko et al., 2010). Although these studies demonstrate somewhat variable results, taken together, they indicate an impairment in mitochondrial bioenergetics in the kidney in the diabetic setting. Since each study differed in methodology, that is, differences in substrate utilization, rodent model, age and diabetes duration, it is not surprising that inconsistencies are apparent between data sets. The overall paradigm that can be drawn from these studies is that the metabolic and bioenergetic changes in experimental diabetes may occur before the onset of structural changes, lending support for a causal role for disturbed mitochondrial homeostasis in the development of DN.
Less is known about mitochondrial function in human DN. This is due to the lack of availability of fresh renal biopsy material, which is essential for tests of bioenergetics. However, indirect measures of mitochondrial homeostasis have been determined in patients with DN. Recently, Sharma et al. performed metabolomics on urine from patients with type 1 or type 2 diabetes and found that metabolites from the citric acid cycle, pyrimidine metabolism, amino acid, propionate, fatty acid and oxalate metabolism, were significantly reduced in DN (Sharma et al., 2013). Because the majority of these metabolites are produced within the mitochondria, the authors suggested that a generalized decrease in several aspects of mitochondrial function is present in patients with DN. Validation of these pathways led to the discovery that protein levels of complex IV of the ETC were decreased in renal biopsies from patients with DN, and mtDNA content of urine exosomes was likewise decreased. Peroxisome proliferator-activated receptor γ co-activator 1α (PGC1α) mRNA was also down-regulated within microdissected cortical tubulointersitial samples from patients with DN compared with non-diabetic individuals, suggesting that mitochondrial biogenesis may also be impaired. Other mitochondrial proteins have been shown to be altered in the kidney in patients with DN. In a gene expression database, renal biopsies of human DN demonstrated higher expression of genes encoding key mitochondrial proteins, including cytochrome c (cyt c) and MnSOD, compared with control biopsies (Hickey et al., 2011). These studies, however, are unable to establish a causal relationship for mitochondrial dysfunction in the pathogenesis of DN.
Mitochondria and renal fibrosis
Tubulointerstitial fibrosis is the terminal pathway leading to ESRD in the majority of all CKDs (Liu, 2006). Fibrosis, the consequence of excessive deposition of ECM, is considered a failed wound-healing process (for a comprehensive review, see Liu, 2011). Fibrosis of the kidney results in glomerulosclerosis, tubulointerstitial fibrosis, tubular atrophy and dilatation, and rarefaction of the glomerular and peritubular capillaries (Boor et al., 2010). This process is highly complex, as most cell types that reside within the kidney, in addition to infiltrating cells such as fibrocytes, macrophages and lymphocytes, participate in fibrogenesis. The main cellular events that occur in the pathogenesis of renal fibrosis include infiltration of inflammatory cells, fibroblast activation and expansion from various sources, production and deposition of ECM, tubular atrophy and microvascular rarefaction (Liu, 2011). All of these events lead to a loss of renal function. So which mechanisms could link mitochondrial dysfunction to fibrosis? During the initial stages of fibrosis, following injury, inflammatory cells infiltrate and generate fibrogenic cytokines and growth factors (Duffield, 2010). It is conceivable that ROS generated from mitochondria in the diabetic milieu initiates the inflammatory response. Indeed, mROS has been shown to activate the NLRP3 inflammasome (Zhou et al., 2011), which is also induced in DN (Fang et al., 2013a). There also is an extensive body of evidence that mROS trigger pro-inflammatory cytokine production in an inflammasome-independent manner (Naik and Dixit, 2011).
During renal fibrosis, profibrotic cytokines primary fibroblasts and other cells including tubular epithelial cells undergo phenotypic activation or transition and generate and secrete ECM. It is thought that fibroblasts, pericytes and perivascular cells acquire a myofibroblast phenotype, which may also lead to cytoskeletal rearrangements, altered cell polarity and synthesis of profibrotic and pro-inflammatory mediators (Boor et al., 2010). TGF-β is a key mediator of fibrosis and is up-regulated in DN (Ziyadeh, 2004). It induces ECM accumulation and proliferation of myoblasts and fibroblasts. The link between TGF-β and mitochondrial bioenergetics has been recently reviewed (Casalena et al., 2012). An increase in TGF-β activity is associated with mitochondrial dysfunction and increased mROS synthesis with apoptosis of podocytes and tubular epithelial cells, driving the progression of glomerulosclerosis and nephron loss (Casalena et al., 2012). A growing body of work involving the mitochondrial-associated protein induced-by-high-glucose-1 (IHG-1) has provided a critical molecular link between mitochondria and renal fibrosis in DN (Murphy et al., 2013). IHG-1 was identified in a screen for genes differentially expressed in renal cells exposed to high glucose (Murphy et al., 1999) and was subsequently found to be increased tenfold in tubule-enriched microdissected renal biopsies from patients with DN (Murphy et al., 2008). IHG-1 expression co-localized with activated Smad3, a downstream mediator of TGF-β1-induced fibrosis. Further investigation determined that IHG-1 amplifies TFG-β1 action in renal tubular HK-2 cells, leading to typical fibrotic responses such as increased expression of connective tissue growth factor and fibronectin. IHG-1 was subsequently found to be localized to the mitochondria and to modulate mitochondrial mass by stabilization of PGC1α (Hickey et al., 2011). Mitochondrial localization of IHG-1 was shown to be pivotal for the amplification of TGF-β1 signalling (Corcoran et al., 2013). As IHG-1 regulates mitochondrial biogenesis, it is likely to play a role in renal cell survival during oxidative stress. In pathological conditions, this would result in dedifferentiation of the renal tubule epithelial cell and fibrosis (Murphy et al., 2013).
ROS also activate a range of other pathways implicated in the pathogenesis in renal fibrosis, including the NFκB-monocyte chemoattractant protein-1 pathway, which promotes monocyte recruitment and profibrotic processes; the plasminogen activator inhibitor pathway, leading to ECM accumulation via suppression of plasmin activity; and finally, the TGF-β1-induced myofibroblast transformation of renal PTECs (epithelial-mesenchymal transition) contributing to tubulointerstitial fibrosis (Ha and Lee, 2005).
It is widely appreciated that activation of glomerular mesangial cells by angiotensin II, a vasoactive peptide and growth factor, leads to ECM accumulation. A recent study demonstrated a link between angiotensin II and the mitochondria, whereby angiotensin II stimulated NADPH oxidase 4 (NOX4) up-regulation within the mitochondria, with mROS generation, loss of NO availability and fibronectin accumulation (Lee et al., 2013). There is an increasing body of evidence for crosstalk between different enzymatic sources of oxidative stress. Mitochondrial ROS has been shown to trigger NOX activation in phagocytes and cardiovascular tissue, leading to endothelial dysfunction (Kroller-Schon et al., 2013).
Mitochondrial dynamics and DN
It is important to note that other aspects of mitochondrial biology can modulate mitochondrial functional capacity, such as mitochondrial morphology or dynamics. Altered mitochondrial morphology was first demonstrated in the proximal tubule cells of patients with type 2 diabetes and albuminuria more than 20 years ago (Takebayashi and Kaneda, 1991). Renal biopsy specimens showed enlarged mitochondria with cellular hypertrophy and thickening of the tubular basement membrane. The same research group then showed that enlarged mitochondria in proximal tubule cells correlated with microalbuminuria in early diabetes in a rat model of STZ-induced diabetes (Kaneda et al., 1992). This has been recently confirmed, both in an STZ-mouse model (Sun et al., 2008; Galloway et al., 2012) and in a mouse model of type 2 diabetes, the db/db mouse (Persson et al., 2012). In the latter study, although an increase in mitochondrial fragmentation was found, only two mice per group were studied. In another study, evidence was obtained of mitochondrial fragmentation in the podocytes of mice with STZ diabetes, in parallel with enhanced mitochondrial superoxide production and up-regulation of glomerular dynamin-related protein 1 (Drp-1) (Wang et al., 2012), a GTPase of the dynamin superfamily that plays an essential role in mitochondrial fission. Taken together, these studies suggest that perturbed mitochondrial dynamics may be important in the pathogenesis of DN. The role of mitochondrial fragmentation in the pathogenesis of renal pathophysiology is a newly emerging area (recently reviewed by Zhan et al., 2013); however, the molecular events that govern mitochondrial dynamics in DN are not yet known, and the mechanisms whereby alterations in dynamics lead to renal fibrosis have not been elucidated. It is clear that further studies in this area are warranted.
Mitochondrial function and cell death
Unabated mitochondrial dysfunction can ultimately result in the activation of the intrinsic cell death pathways and cellular demise (Davis and Williams, 2012; Smith et al., 2012b). In addition to being considered the cellular powerhouse, mitochondria are also known to be the judge and executioner during programmed cell death (Tait and Green, 2010; Kubli and Gustafsson, 2012). Programmed cell death occurs in several forms, including apoptosis, autophagic cell death (ACD) and programmed necrosis [caspase-independent cell death that is highly regulated, yet with features similar to accidental necrosis (Baines, 2010)], which encompasses necroptosis (RIP1-dependent) (Christofferson and Yuan, 2010) and parthanatos (PARP-1-dependent) (Andrabi et al., 2008). Despite these cell death pathways being distinct, Bcl-2 family members are implicated in each, directly or indirectly regulating mitochondrial involvement (Youle and Strasser, 2008; Martinou and Youle, 2011). Bcl-2 family members, such as Bnip3, Nix, Bax, Bak, Bmf and Bcl-XL, have been implicated in programmed necrosis (Vande Velde et al., 2000; Kubasiak et al., 2002; Moubarak et al., 2007; Hitomi et al., 2008; Diwan et al., 2009; Chen et al., 2010; Messner et al., 2012; Whelan et al., 2012; Michels et al., 2013). As will be discussed later, a number of autophagy proteins have been shown to interact with Bcl-2 family members.
In the case of apoptosis and programmed necrosis, the redistribution of IMS cell death proteins such as cyt c, Smac/DIABLO and HtrA2/Omi, which are involved in caspase-dependent cell death and caspase-independent signallers apoptosis-inducing factor (AIF) and endonuclease G are central (Kubli and Gustafsson, 2012) (Figure 1C). However, these IMS cell death signalling proteins depend on two distinct mechanisms for redistribution. Apoptosis involves the pro-apoptotic Bcl-2 family members Bax and Bak to form a pore on the outer mitochondrial membrane (MOMP), resulting in transient hyperpolarization followed by dissipation of ΔΨm, the release of proteins from the IMS to the cytosol (Tait and Green, 2010) (Figure 1C).
In contrast to mitochondrial membrane permeabilization during apoptosis, programmed necrosis appears to involve the initiation of mitochondrial permeability transition pore (MPTP) (Baines, 2010). This pore causes dissipation of ΔΨm, allows solutes (less than 1.5 kDa in size) to enter the mitochondria resulting in mitochondrial matrix swelling and subsequent rupture of the outer mitochondrial membrane (OMM) releasing the contents of the IMS (Zorov et al., 2009). However, unlike MOMP, much conjecture remains as to the composition of the MPTP. It was initially thought that the MPTP was composed of the voltage-dependent anion channel 1 (VDAC1), adenine nucleotide translocator (ANT) and cyclophilin D (Cyp-D), spanning the OMM and IMM (Armstrong, 2006; Zorov et al., 2009). The subsequent demonstration that VDAC1 and ANT are not needed for mitochondrial permeability transition (MPT) to occur has cast doubt over this classical model (Kokoszka et al., 2004; Baines et al., 2007). Nonetheless, Cyp-D was identified as a critical component of the MPTP, with Cyp-D-deficient cells unable to undergo MPT (Baines et al., 2005; Nakagawa et al., 2005). More recently, dimers of ATP synthase have been shown to form the MPTP within the IMM, with Cyp-D binding to the lateral stalk of ATP synthase (Giorgio et al., 2013) (Figure 1C). The implications of this discovery are further detailed in Bernardi (2013), although it is evident that this already remarkable enzyme is at the centre of the crossroads between biogenesis and cell death, which also affects mitophagy.
Non-selective autophagy signalling: indiscriminate removal of mitochondria
Classical autophagy, as it was first described by De Duve, is a non-selective process whereby cytosolic contents (long-lived proteins and organelles) are sequestered into autophagosomes for transport to lysosomes for bulk degradation (De Duve and Wattiaux, 1966). Although selective autophagy (which encompasses mitophagy) may appear to be a more refined and efficient means of removing dysfunctional mitochondria, much remains unknown as to how these two autophagy processes interact. In order to develop effective therapeutics to modulate the accumulation of dysfunctional mitochondria, both process must be given due consideration. Non-selective autophagy activity can be modulated by growth factors, such as negative regulation via insulin and insulin-like growth factor-1 (Troncoso et al., 2012; Naito et al., 2013; Paula-Gomes et al., 2013; Renna et al., 2013) and positively by TGF-β (Ding et al., 2010b; Koesters et al., 2010; Xu et al., 2012), cellular stress (e.g. oxidative stress, hypoxia, advanced glycation end-products) (Miyata and de Strihou, 2010; Hu et al., 2012; Xu et al., 2013) and changes in nutrient levels (e.g. glucose, AMP/ATP ratio) (Moruno et al., 2012; Lempiainen et al., 2013; Vallon et al., 2013). It is likely that these same stimuli, many of which have been linked to the development of functional or structural defects of DN (Forbes and Cooper, 2013), play critical roles in modulating selective autophagy activity in the DN milieu (Figure 2).
Figure 2.
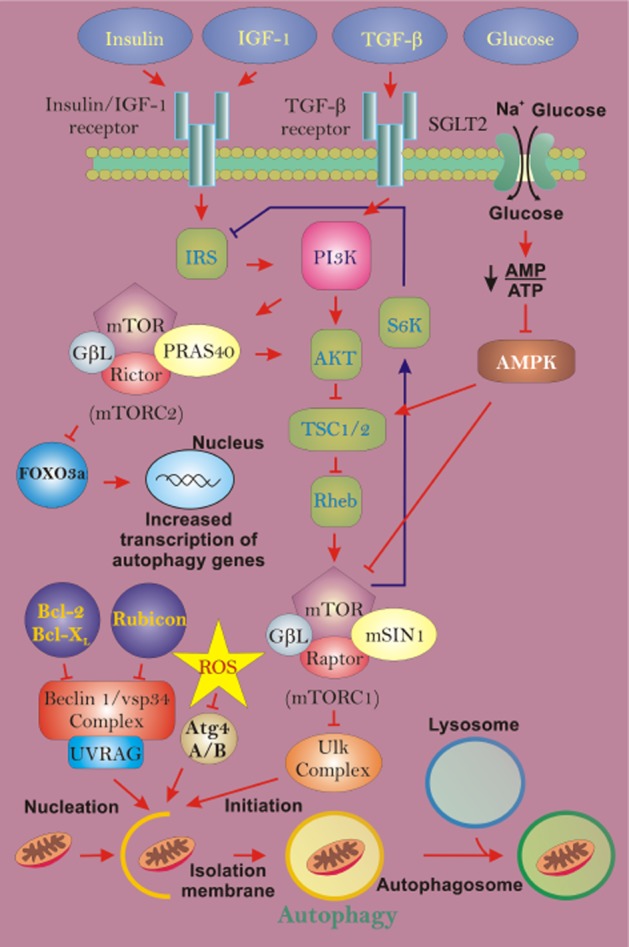
Non-selective autophagy regulation during diabetic nephropathy (DN). During DN, autophagic turnover of mitochondria can be influenced by changes in several contributing factors, including insulin, IGF-1 (insulin-like growth factor-1), TGF-β, glucose and reactive oxygen species (ROS). These influences can act on the mechanistic target of rapamycin (mTOR) signalling pathway, which negatively regulates autophagy activity. Insulin, IGF-1 and TGF-β can modulate the mTOR pathway via the PI3K pathway. Increases in insulin or IGF-1 will suppress autophagy activity, while an increase in TGF-β will stimulate autophagy activity. Increases in glucose, with hyperglycaemia, can result in suppression of autophagy through activation of AMPK, which can activate the mTOR pathway. Finally, increases in ROS production can stimulate autophagy activation via inhibition of Atg4A/B or by directly acting on other components of the mTOR pathway, impairing activation.
In the face of cellular starvation, stress or growth factor withdrawal, cells undergo non-selective autophagy regulated by the mechanistic target of rapamycin (mTOR) signalling pathway to recycle cellular components and maintain energy levels (Foster and Fingar, 2010). Inhibition of mTOR stimulates gross autophagy, which indiscriminately encapsulates damaged and healthy mitochondria alike, along with other organelles and long-lived proteins for bulk degradation. This pathway is a relatively rudimentary means of removing damaged mitochondria. Acute treatments with rapamycin inhibit mechanistic target of rapamycin complex 1 (mTORC1), making it a widely used inducer of autophagy activity in experimental studies (Klionsky et al., 2012). There have been several seminal studies demonstrating that attenuating mTORC1 signalling can cause longevity in eukaryotic organisms such as Caenorhabditis elegans (Vellai et al., 2003), Drosophila (Kapahi et al., 2004) and mice (Harrison et al., 2009).
Mitophagy: selective removal of dysfunctional mitochondria
Selective autophagy is the process by which damaged or surplus proteins and organelles are specifically targeted for degradation, as eloquently reviewed recently (Feng et al., 2013; Shaid et al., 2013). In the last decade, the discovery of selective autophagy has seen the emergence of many sub-types of autophagy. Such terms as mitophagy, reticulophagy and ribophagy have been coined for the select removal of mitochondria, ER and ribosomes respectively (Lemasters, 2005; Cebollero et al., 2012; Jin and Youle, 2012). Although each could potentially play a role in DN, attention will only be given to mitophagy here based on the evidence provided that the accumulation of damaged mitochondria contribute to its pathology. Mitophagy is distinguished by its regulation through the ubiquitin proteasome system (UPS), although the emergence of UPS-independent adaptor proteins suggests that alternative pathways may exist, or that this process has cell-type specific variances. Cellular recognition of dysfunctional mitochondria is determined by loss of ΔΨm. Changes in mitochondrial morphology and microtubule transport of damaged mitochondria have secondary roles in determining whether mitophagy is engaged. This mitophagy activity may, in turn, be regulated by ATP and mROS production, which may determine the rate at which mitochondria are turned over. These aspects of mitophagy regulation will be covered in detail in the following sections.
Recognition of damaged mitochondria via loss of ΔΨm
The key determinant that a mitochondrion should be targeted for degradation is the cessation of OXPHOS. Dysfunctional mitochondria that do not have an ΔΨm are tagged for mitophagy clearance via PTEN-induced putative kinase 1 (PINK1) on the organelle's outer membrane (Matsuda et al., 2010; Narendra et al., 2010b; Vives-Bauza et al., 2010). PINK1, which is synthesized in the cytosol, is imported into mitochondria via the translocase of the outer mitochondrial membrane (TOM) complex into the mitochondrial IMS. In functional mitochondria (with ΔΨm), the mitochondrial targeting sequence (MTS) of PINK1 guides the protein through the IMS into the translocase of the inner membrane (TIM) complex where it spans the inner membrane (Jin et al., 2010). The MTS of PINK1 is cleaved by MPP (mitochondrial processing peptidase), while the protein itself undergoes processing via presenilin-associated rhomboid-like protease (PARL) located within the IMM (Jin et al., 2010; Meissner et al., 2011; Greene et al., 2012). This processed form of PINK1 is rapidly degraded by a further mitochondrial protease. In the case of dysfunctional mitochondria that do not have ΔΨm, PINK1 is unable to pass through the TIM complex in the IMM, leaving it anchored via the TOM complex on the OMM unprocessed (Lazarou et al., 2012). Here, PINK1 is recognized by Parkin, an E3 ubiquitin ligase, which translocates to the mitochondria from the cytosol and is phosphorylated by PINK1 at Ser65 to become active and initiate mitophagy (Narendra et al., 2008; 2010b; Matsuda et al., 2010; Vives-Bauza et al., 2010; Kondapalli et al., 2012; Iguchi et al., 2013). However, whereas the generally accepted view is that PINK1 phosphorylates Parkin at Ser65, this has been shown to be non-essential in some circumstances (Shiba-Fukushima et al., 2012; Sarraf et al., 2013). Upon binding with PINK1, Parkin mediates the ubiquitination of OMM proteins such as VDAC1, the TOM complex, the pro-apoptotic protein Bak and proteins involved in mitochondrial dynamics and transport such as mitofusins 1/2 (Mfn1/2) and mitochondria Rho GTPase (Miro) respectively. The purpose of ubiquitinating these proteins and the possible degradation of these proteins via the UPS will be discussed in the subsequent sections below.
Encapsulation of mitochondria within autophagosomes is dependent on adaptor proteins
One defining characteristic of selective autophagy that differentiates it from non-selective autophagy is the dependence on adaptor (or receptor) proteins to encapsulate its target molecule or organelle. As a form of selective autophagy, mitophagy can engage adaptor proteins such as p62/SQSTM1 (sequestosome 1) (Geisler et al., 2010), Nix (Bnip3L) (Novak et al., 2010), Bnip3 (Hanna et al., 2012), FUN14 domain containing 1 (FUNDC1) (L. Liu et al., 2012) and histone deacetylase 6 (HDAC6) (Lee et al., 2010), which bind to the OMM and aid the docking of the isolation membrane of the pre-autophagosome with damaged mitochondria. Each of the aforementioned adaptor proteins has a unique composition that encompasses binding sites that interact with either light chain 3 (LC3) via an LC3 interacting region (LIR) or ubiquitin via an ubiquitin-binding domain (UBD) during mitophagy.
The adaptor proteins Bnip3, Nix and FUNDC1 possess an LIR, which allows these proteins to dock LC3 to the OMM (Novak et al., 2010; Hanna et al., 2012; L. Liu et al., 2012). The mechanism that allows these adaptor proteins to recruit LC3 to the OMM remains unclear. Yet, it is evident that FUNDC1, Bnip3 and Nix have different roles in mitophagy as shown by their differential binding affinities to different orthologues of LC3. FUNDC1 and BNIP3 have a high affinity for LC3B (L. Liu et al., 2012; Zhu et al., 2013), whereas Nix has a weak affinity for LC3B, but a high affinity for LC3A and GABARAP-L1 (Novak et al., 2010). Under hypoxic conditions, these LIR-only adaptor proteins have been shown to be involved in mitophagy, with Bnip3 and Nix expression up-regulated, whereas FUNDC1 is down-regulated, again suggesting functional differences between these proteins (Bellot et al., 2009; L. Liu et al., 2012). Whereas hypoxia-inducible factor 1α (HIF-1α) has been shown to mediate Bnip3 and Nix-dependent mitophagy (Bellot et al., 2009), it remains to be determined whether FUNDC1 is similarly regulated. The implications of these discoveries are pertinent to DN, where the onset of hypoxia has been demonstrated to occur in early stage renal failure (Palm and Nordquist, 2011). Moreover, Bnip3 has recently been shown experimentally to regulate autophagy and mitophagy in proximal tubular cells during acute kidney injury (AKI) (Ishihara et al., 2013).
Recognition of dysfunctional mitochondria by the PINK1/Parkin pathway appears to be dependent on the involvement of Bnip3 and Nix, despite their lack of a UBD (Ding et al., 2010a; Lee et al., 2011). Both Bnip3 and Nix have been shown to promote the translocation of Parkin to mitochondria. Interestingly, Bnip3 and Nix both have the ability to induce mitochondrial depolarization, which can be a trigger for mitophagy as discussed earlier (Sandoval et al., 2008; Rikka et al., 2011). However, given the involvement of Bnip3 and Nix in cell death signalling, FUNDC1 might be the more dependable adaptor protein in which to develop a therapeutic strategy to stimulate mitophagy.
Unlike Bnip3, Nix and FUNDC1 that act independently of ubiquitin, HDAC6 and p62 can directly attach to polyubiquitin chains via their UBD domains. The recruitment of HDAC6 and p62 during mitophagy is therefore dependent on ubiquitination of OMM proteins mediated by Parkin (Geisler et al., 2010; Lee et al., 2010). HDAC6 only possess a UBD in the form of a ubiquitin-binding zinc finger (Seigneurin-Berny et al., 2001; Hook et al., 2002), whereas p62 is distinct in that it comprises a UBD as well as an LIR, allowing it to interact with both LC3 and ubiquitin respectively (Bjorkoy et al., 2005). Ubiquitin can attach to its substrate as a single ubiquitin monomer (monoubiquitination) or form chains of sequentially conjugated ubiquitin (polyubiquitination). Each ubiquitin monomer has seven lysine (K) residues that can act as a point of polyubiquitination. Polyubiquitin chains are therefore identified based on which lysine of the ubiquitin monomer it is attached to. Ligation of OMM substrates by Parkin can result in either K27-, K48-or K63-linked polyubiquitin chains (Geisler et al., 2010; Narendra et al., 2010a; Rana et al., 2013). K63-linked polyubiquitin chains are considered ‘non-classical’, as substrates with these labels are predominantly targeted for selective autophagy rather than degradation via the UPS (Olzmann et al., 2007; Tan et al., 2008; Geisler et al., 2010; Narendra et al., 2010a). The adaptors p62 and HDAC6 can recognize and attach to K63-polyubiquitinated proteins for transport via dynein motors along microtubules to the perinuclear or juxtanuclear region to form aggresomes, which are clusters of misfolded proteins (Kawaguchi et al., 2003; Seibenhener et al., 2004). The LIR on p62 binds with LC3 to encapsulate and degrade aggresomes via autophagy.
Mitochondria are also capable of aggregating via p62 and HDAC6 in a manner similar to misfolded proteins when ubiquitinated (Lee et al., 2010; Narendra et al., 2010a; Okatsu et al., 2010). However, whereas several groups have reported that transport and aggregation of damaged mitochondria by p62 is critical for mitophagy (Geisler et al., 2010; Lee et al., 2010), p62 has also been reported to be dispensable (Okatsu et al., 2010; Narendra et al., 2010a). HDAC6 transport has so far been shown to be essential for mitophagy (Lee et al., 2010). It is important to note that the role of either adaptor has not been fully investigated in a pathological in vivo model, and whereas mitophagy may still occur in the absence of perinuclear clustering, the degradation efficiency may be compromised.
K48-linked polyubiquitin chains can also form on the OMM (Sun et al., 2009). K48-linked ubiquitin chains are considered ‘classical’ in that proteins tagged with these chains are generally degraded by proteasomes (Shaid et al., 2013). OMM substrates, such as Mfn1 and Mfn2 that are ligated by Parkin have been shown to possess K48-linkages (Chan et al., 2011; Lazarou et al., 2013). Through the use of proteasome inhibitors, the degradation of these proteins was shown to be dependent on the UPS. Of further interest was the demonstration that mitophagy is dependent on proteasomal activity (Chan et al., 2011). Parkin has also been reported to ligate VDAC1 with K27-linked polyubiquitin chains (Geisler et al., 2010), although mitophagy is not dependent on this step (Narendra et al., 2010a). This apparent multi-specificity of Parkin ligation is most likely dependent upon which E2 enzymes are recruited to each substrate (Lim et al., 2006). HDAC6 and p62 have both been reported to influence the activity of the UPS as both are capable of binding to K48-polyubiquitin chains, providing a potential compensatory crosstalk mechanism between the two degradation pathways (Korolchuk et al., 2009; Du and Jiao, 2011; Shaid et al., 2013). This close relationship between the UPS and autophagy is further demonstrated by the regulation of mitophagy by p97/VCP (Tanaka et al., 2010). p97 is a AAA-ATPase that was originally discovered as a chaperone for delivery of substrates to the UPS, but has more recently been determined to have a role in regulating autophagy and lysosomal degradation (Meyer et al., 2012).
Impedance of mitochondrial dynamics and transport facilitates mitophagy
Dysfunctional mitochondria undergo retrograde transport to the perinuclear region where they cluster during mitophagy (Narendra et al., 2010a; Okatsu et al., 2010; Vives-Bauza et al., 2010). The perinuclear region of the cell is the location of the ER, as well as being rich in lysosomes and proteasomes. During mitophagy, encapsulated mitochondria are probably transported along the microtubules via dynein motors to the perinuclear region where degradation via lysosomes can occur. This phenomenon may allow for more efficient recycling of mitochondria, which can be broken down into basic constituents that can be utilized by the ER. Under severe stress, perinuclear clustering may be advantageous to cope with an influx in mitophagy activity. The population of mitochondria within any cell type are dynamic, constantly being transported along microtubules, while undergoing constant fission and fusion between adjoining mitochondria within the network. To initiate mitophagy, disengagement of mitochondria from the mitochondrial network is required. Overall, regulation of mitochondrial dynamics requires PINK1 and Parkin interaction with the OMM proteins, Miro involved in microtubule transport (Wang et al., 2011), Drp1, Mfn 1 and 2 that regulate mitochondrial dynamics and ultimately the adaptors p62 and HDAC6 for perinuclear transport as discussed earlier.
Miro links with another adaptor protein Milton and kinesin-1 heavy chain (KHC) to form the KHC complex. The KHC complex allows anterograde transport of mitochondria via the kinesin motor (Saotome et al., 2008; Wang and Schwarz, 2009). Miro has also been shown to mediate retrograde transport, suggesting that Miro may interact with the dynein motor as well (Wang and Schwarz, 2009). During mitophagy, PINK1 phosphorylates the Ser156 of Miro, which is followed by ubiquitination via Parkin, which labels Miro for proteosomal degradation (Wang et al., 2011). The degradation of Miro results in arrest of mitochondrial motility. The purpose of this action during mitophagy remains unclear, although it may help to quarantine damaged mitochondria or aid the encapsulation of the organelle within autophagosomes by rendering the organelle stationary. Given that Miro is located on the OMM, mitochondrial encapsulation and subsequent retrograde transport during mitophagy does not involve Miro.
Dynamic morphological changes in mitochondria are required to maintain a homogenous population of functional mitochondria to ensure continuous OXPHOS. Mitochondrial fusion allows cross-complementation by sharing mtDNA, lipids and proteins between adjoining organelles, which is thought to preserve mitochondrial integrity (Youle and van der Bliek, 2012). Given the relatively high level of superoxide generated during OXPHOS, this dynamic process may occur to minimize the effects of mtDNA mutations, which code for subunits of the ETC, as well as diluting the pool of damaged proteins and lipids. Mitochondrial dynamics are tightly regulated by two distinct groups of proteins. Mitochondrial fission is regulated by the cytosolic Drp1, which is recruited to the OMM where it interacts with Fis1, Mid49, Mid51 and mitochondrial fission factor to form coils that constrict and separate mitochondria in two (Elgass et al., 2013). Conversely, mitochondrial fusion is mediated by dynamin family members Mfn1 and Mfn2, which fuse the OMM and optic atrophy 1 (OPA1), which mediates fusion of the inner membrane (Youle and van der Bliek, 2012). These opposing mitochondrial dynamics mechanisms are regulated in concert by proteolysis and post-translational modifications. As previously discussed, there is increasing evidence that mitochondrial morphology is altered in the PTECs in DN, suggesting that changes in mitochondrial dynamics may be glucose-driven or modulated by reactive glucose-derived molecules. Indeed, glycation of proteins within the kidney is integral to the pathophysiology of DN (Coughlan et al., 2008).
In order for mitophagy to occur, mitochondria must undergo fission or fragmentation into spheroids that can be encapsulated within autophagic vesicles (Twig et al., 2008). The PINK1/Parkin pathway disrupts the mitochondrial network, which is essential to isolate damaged mitochondria for engulfment by autophagosomes (Poole et al., 2008; Y. Yang et al., 2008; Gegg et al., 2010). Mitochondrial ubiquitin E3 ligases such as MULAN and MARCH5 are thought to facilitate the polyubiquitination of fission and fusion proteins during mitochondrial biogenesis (Karbowski et al., 2007; Li et al., 2008; Park et al., 2010). Similarly, as an E3 ligase, Parkin has been shown to ubiquitinate mitofusins for degradation by the UPS to disengage mitochondrial biogenesis (Poole et al., 2008; Gegg et al., 2010; Tanaka et al., 2010; Ziviani et al., 2010; Rakovic et al., 2011). Although, recently MULAN (also known as Mul1) has also been implicated in mitophagy in skeletal muscle (Lokireddy et al., 2012). The targeting of mitofusins to the UPS prevents damaged mitochondria from fusing with adjoining healthy mitochondria. The transport of mitofusins to the UPS for degradation is mediated by p97 (Tanaka et al., 2010). Furthermore, in cardiomyocytes, phosphorylation of Mfn2 by PINK1 is needed for translocation of Parkin to mitochondria and ubiquitination of Mfn2 and other proteins to ensue (Chen and Dorn, 2013). Ablation of Mfn2 in cardiomyocytes prevented translocation of Parkin and subsequent mitophagy to occur in response to mitochondrial depolarisation. Mfn2 can therefore act as a receptor for Parkin when phosphorylated. However, this observation may be cell-type specific given that Parkin translocation has also been demonstrated in cells lacking Mfn1 and Mfn2 (Narendra et al., 2008; Chan et al., 2011). Furthermore, whereas mitofusins may have an essential role in the occurrence of mitophagy, proteasomal degradation of mitofusins alone does not instigate mitochondrial fission.
The regulation of mitochondrial fragmentation during mitophagy involves the recruitment of Drp1. PINK1 and Parkin have been shown to mediate the recruitment of Drp1 to mitochondria. Loss of PINK1 or Parkin function has been shown to cause an increase in Drp1-dependent mitochondrial fragmentation, which can be reversed by the overexpression of Mfn2, Opa1, a dominant negative of Drp1 or by use of the Drp1 inhibitor mdivi-1 (Lutz et al., 2009; Sandebring et al., 2009; Cui et al., 2010). Similarly, cell culture studies have also revealed that the adaptor protein Bnip3 can induce translocation of Drp1 to mitochondria during mitophagy (Lee et al., 2011). Moreover, Bnip3 has also been reported to disrupt Opa1, leading to mitochondrial fragmentation during apoptosis (Landes et al., 2010). However, it remains to be investigated whether the Bnip3–Opa1 interaction is also involved in mitochondrial fragmentation during mitophagy. Overexpression of Fis1 can also stimulate mitophagy (Gomes and Scorrano, 2008). However, this outcome was shown to be dependent on the ability of Fis1 to cause mitochondrial dysfunction rather than its role in fission. In contrast, Fis1 silencing has been shown to reduce mitophagy activity (Twig et al., 2008). Lastly, the mitochondrial protein p32 has been shown to effect changes in mitochondrial morphology via Parkin, by way of fragmentation when suppressed (Li et al., 2011). Whereas the mechanisms that mediate Parkin translocation to mitochondria remain to be fully defined, the action of p32 may warrant further attention. Taken together, these findings demonstrate that while mitochondrial dysfunction can trigger mitophagy activation, this process is dependent on the regulation of mitochondrial dynamics via the PINK1/Parkin pathway to ensure damaged mitochondria are cleared from the cell.
Regulation of mitophagy activity by ROS
Once mitophagy is activated, this process must be modulated to ensure that the clearance rate is sufficient to prevent the accumulation of damaged mitochondria. Autophagy activity is ROS-dependent and is tightly regulated by intracellular redox signalling (Scherz-Shouval and Elazar, 2011). OXPHOS can be a major source of ROS and, as such, under homeostatic conditions, this is managed by an array of antioxidants. Moreover, mitochondria have their own antioxidant defence network that resides within these organelles to minimize the escape of ROS into the cytosol (Crane et al., 1957; Weisiger and Fridovich, 1973; Ursini et al., 1982). As discussed earlier, in DN, an influx of ROS caused by high glucose has been postulated to contribute to oxidative stress, which contributes to the development of the disease (Forbes et al., 2008). It can be further postulated that this excess ROS may stimulate mitophagy activity in the kidney and regulate the turnover of damaged mitochondria (Chen et al., 2007; Scherz-Shouval et al., 2007; Bensaad et al., 2009). This section will focus on the mitochondria's antioxidant defence and how it may influence redox signalling and the regulation of mitophagy activity.
Transcriptional regulation of autophagy activity can be stimulated by oxidative stress, as well as by mROS produced in response to hypoxia. An influx of ROS or oxidative stress can result in activation of transcription factors HIF-1α, Forkhead box O3a (FOXO3a) or nuclear factor (erythroid-derived 2)-like 2 (Nrf2), which each, in turn, induce the transcription of autophagy genes (Scherz-Shouval and Elazar, 2011). As discussed earlier, HIF-1α and FOXO3a regulate the transcription of Bnip3 and Nix, which are both involved in mitophagy. Nrf2 induces transcription of p62 (Jain et al., 2010). Another mechanism that can regulate autophagy in response to ROS is the p53-target gene TIGAR (TP53-induced glycolysis and apoptosis regulator), which, in this capacity, acts independently of p53 and mTOR (Bensaad et al., 2009). TIGAR promotes activation of the pentose phosphate pathway, which produces NADPH. NADPH is required to reduce oxidized glutathione, which is required to lower ROS levels, which, in turn, decreases autophagy activity. Of potential relevance to mitophagy regulation is the recent discovery that under hypoxic conditions, TIGAR can translocate to the mitochondria in an HIF-1α-dependent manner, where it forms a complex with mitochondrial HK2 (hexokinase 2), which limits mROS levels (Cheung et al., 2012).
The cytosolic protein DJ-1 has been implicated in cellular signalling in response to oxidative stress and linked to mitophagy. DJ-1 has a conserved cysteine residue (Cys106) that is sensitive to ROS. Oxidation of Cys106 promotes the translocation of DJ-1 to mitochondria at times of oxidative stress (Canet-Aviles et al., 2004; Zhang et al., 2005; Junn et al., 2009). DJ-1 has been shown to protect against oxidative stress, but this is more likely via ROS suppression through its role in cellular signalling rather than acting as an antioxidant (Canet-Aviles et al., 2004; Taira et al., 2004; Junn et al., 2009). Loss of DJ-1 results in increased ROS production and MPTP opening in mitochondria of murine embryonic fibroblasts (Giaime et al., 2012). Furthermore, DJ-1 has been shown to be important for ATP production and mitochondrial function in skeletal muscle and can restore PINK1 loss of function, but not Parkin in Drosophila (Hao et al., 2010). Interestingly, DJ-1 has also been shown to associate with Parkin under oxidative stress conditions (Moore et al., 2005; Joselin et al., 2012), and it is suggested to work in parallel with the PINK1/Parkin pathway to regulate mitophagy (Thomas et al., 2011). DJ-1 has also been reported to be involved in proteasomal degradation of Fis1 (Zhang et al., 2012). In the kidney, DJ-1 is expressed in proximal tubule cells where it has been implicated in regulating ROS production via NOX4 during hypertension (Cuevas et al., 2012). Although it is unclear what the exact function of DJ-1 is, it is evident that it may have an important role in regulating mitophagy, particularly under oxidative stress.
Cellular ROS can also promote autophagy via a post-transcriptional mechanism via inactivation of Atg4 (Scherz-Shouval et al., 2007). Atg4 has four paralogues: Atg4A-Atg4D also known as autophagins (Marino et al., 2003). The Atg4 family members are responsible for proteolytic activation and subsequent delipidation (removal from autophagosomes) of Atg8 family members (e.g. LC3 and GABARAP). Atg4A and Atg4B have a redox-sensitive cysteine residue, which can be oxidized by H2O2 to inactivate this protease (Scherz-Shouval et al., 2007). It has been hypothesized that when Atg4A or Atg4B are within close proximity of dysfunctional mitochondria producing ROS, these proteases become inactivated, stimulating LC3 lipidation. However, when the autophagosomes translocate to the perinuclear lysosomes, this region must be low in ROS, allowing active Atg4A and Atg4B to delipidate LC3.
The mitochondrial antioxidant network that may regulate mitophagy consists of a diverse range of proteins that are each capable of detoxifying distinct ROS. Electrons that escape the ETC during OXPHOS react with oxygen to produce superoxide (O2−). This O2− can be detoxified by SOD into hydrogen peroxide (H2O2). H2O2 can, in turn, be reduced to H2O via glutathione peroxidase (Gpx). Mitochondria have their own copies of each enzyme, SOD2 (Weisiger and Fridovich, 1973) and mitochondrial Gpx4 (mGpx4) (Ursini et al., 1999). mGpx4 has dual roles; it can reduce hydroperoxides in lipoproteins and is also thought to be responsible for repairing oxidative damage to biomembranes (Ursini et al., 1999). The role of mGpx4 has not been the focus of DN studies to date, as attention has been given to SOD2, largely due to a polymorphism in its targeting sequence.
In a KK/Ta-Akita mouse model of DN, SOD2 activity was found to be unchanged (Fujita et al., 2009). Furthermore, SOD2 activity and expression were also unchanged in a STZ rat model and in vitro in rat proximal tubule cells exposed to high glucose (25 mM) despite evidence of mitochondrial dysfunction and elevated mitochondrial superoxide (Munusamy and MacMillan-Crow, 2009). In contrast, studies from our laboratory (Coughlan et al., 2007) and others (de Cavanagh et al., 2008) have found that MnSOD (SOD2) activity is suppressed in the renal cortex in rodent models of DN. Moreover, SOD2 has been implicated in human DN through a polymorphism, V16A (Val16Ala) in Danish, Swedish and Finnish type 1 diabetes patients (Mollsten et al., 2007; 2009,) and Korean, Chinese and Japanese type 2 diabetes patients (Nomiyama et al., 2003; Lee et al., 2006; Liu et al., 2009). An increase in the Val-allele has also been reported in Egyptian type 1 patients (el-Masry et al., 2005). Although this Val-allele increase was statistically insignificant, the cohort in this study was smaller than the other studies reported here. Having valine instead of alanine in the 16th amino acid position of the targeting sequence of SOD2 results in less efficient transport of the antioxidant into the mitochondrial matrix (Shimoda-Matsubayashi et al., 1996; Sutton et al., 2003). Studies on SOD2-kidney specific knockout mice (SOD2f/f/Ksp1.3-Cre, knockout in distal tubules, collecting ducts, Loops of Henle, ureteric buds, and developing genitourinary tract) show an influx in autophagy and mitochondrial biogenesis compared with wild-type mice, suggesting that SOD2 deficiencies may stimulate mitophagy (Parajuli and MacMillan-Crow, 2013).
Yet another antioxidant residing in mitochondria is coenzyme Q (CoQ). CoQ has a primary role as an electron carrier between complex I and II and complex II and III in the ETC (Mellors and Tappel, 1966a). Due to CoQ's ability to easily oxidize and reduce in transporting electrons, it is also adapt at behaving as an antioxidant, preventing lipid peroxidation within the IMM (Mellors and Tappel, 1966b). Endogenous CoQ is decreased in the renal cortex in DN (Sourris et al., 2012), and it has been the subject of several nephropathy studies to date through the development of mitoquinol (MitoQ) (Chacko et al., 2010; Mukhopadhyay et al., 2012). MitoQ is a mitochondria-targeted compound based on the CoQ antioxidant conjugated to a triphenylalkylphosphonium cation (TPP+) that is dependent on ΔΨm to be taken up into the mitochondrial matrix (Murphy and Smith, 2007). MitoQ has been shown to be renoprotective in Ins2-Akita mice (reminiscent of type 1 diabetes), improving tubular and glomerular function, decreasing albuminuria, normalizing glomerular basement membrane thickening and tubulointerstitial fibrosis (Chacko et al., 2010). MitoQ also blocked phospho-Smad2/3-mediated signalling in the kidney, directly implicating ROS derived from mitochondria as a crucial mediator of renal fibrosis in DN. MitoQ may therefore prove to be a suitable tool for regulating mitophagy activity.
Mitophagy and programmed cell death: dangerous liaisons
When considering the potential of exploiting mitophagy in therapeutic strategies, one must remember that the pathways that regulate this paradigm are intricately linked to programmed cell death. Much evidence has emerged that apoptosis and autophagy pathways share common proteins (Figure 3). There is also much evidence to suggest that in the absence of apoptosis, overactivation of autophagy can elicit cell death in mammalian systems (Chen et al., 2007; Oppenheim et al., 2008; Elgendy et al., 2011; Higgins et al., 2011). However, it is unresolved as to whether ACD is physiologically relevant in vivo. Yet, whether ACD exists in an intrinsic physiological setting becomes irrelevant when pharmacological intervention is brought into the milieu, given that the manipulation of any molecular pathway no longer renders the situation biologically innate. This section will look at the potential caveats of targeting mitophagy in cells that are already under severe stress.
Figure 3.
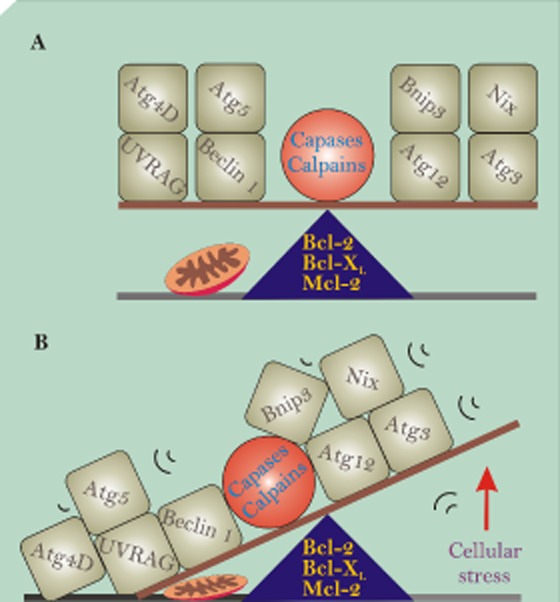
Mitophagy players in life and death. (A) Depicts key autophagy proteins in perfect balance allowing the maintenance of mitochondrial homeostasis. (B) However, with increased cellular stress (e.g. glucose, reactive oxygen species), the balance can be tipped, resulting in some of these proteins being activated by proteases, such as caspases and calpains, whereas others interact with Bcl-2 family members through BH3-only domains.
Many of the key autophagic proteins that have been implicated in programmed cell death are involved in regulating mitochondrial turnover. Atg5 was the first Atg protein to be identified to have a role in cell death (Yousefi et al., 2006). Atg5 was shown to be cleaved by calpain-1 or calpain-2, with cleaved Atg5 translocating to the mitochondria where it associated with Bcl-XL before enacting cyt c redistribution and caspase activation. Similarly, Beclin-1 was also reported to become cleaved by caspases at the C-terminus, converting it from a pro-autophagy protein to a pro-apoptotic protein (Wirawan et al., 2010). Once cleaved, Beclin-1 translocates to the mitochondria where it induces the release of cyt c and HtrA2/Omi from the IMM (Wirawan et al., 2010). In addition, Beclin-1 also possesses a BH3-only domain and is thought to mediate binding with anti-apoptotic proteins Bcl-2 and Bcl-XL (Liang et al., 1998; Maiuri et al., 2007; Oberstein et al., 2007). Binding of Beclin-1 with Bcl-2 and Bcl-XL inhibits autophagy function (Pattingre et al., 2005). UVRAG (ultraviolet irradiation resistance-associated gene), a key regulator of autophagy activity, is also capable of regulating apoptosis via mitochondria through the regulation of Bax (Yin et al., 2011). The interaction between UVRAG and Bax inhibits apoptosis, preventing the induction of the intrinsic apoptotic pathway.
Additionally, several studies have also reported varying roles for the ubiquitin-like modifier Atg12 in mediating mitochondria homeostasis and involvement in cell death (Radoshevich et al., 2010; Rubinstein et al., 2011). Atg12 conjugation with Atg3 has been shown to regulate mitochondrial mass, mitochondrial fusion and cell death involving mitochondria via Bcl-XL (Radoshevich et al., 2010). This differs greatly from its role in autophagy, where Atg12 conjugates with Atg5 to mediate phosphatidylethanolamine lipidation of LC3 (Hanada and Ohsumi, 2005). Atg12 is further implicated in cell death through a BH3-like domain that allows it to bind with Bcl-2 family members (Rubinstein et al., 2011). This action of Atg12 is independent of conjugation with Atg5 or Atg3, with inhibition of Bcl-2 and Mcl-1 triggering Bax activation and cyt c release (Rubinstein et al., 2011). Atg5, Atg12 and Beclin-1 have all been reported to be cleaved during cisplatin-induced AKI in renal proximal tubule cells, which was attenuated by the pan-caspase inhibitor z-VAD-fmk (Herzog et al., 2012). Given that z-VAD-fmk can inhibit other proteases such as calpains and cathepsins, this result in cisplatin-induced injury could be the effect of caspase or calpain activation (Rozman-Pungercar et al., 2003; Bizat et al., 2005). Taken together, these results suggest that crosstalk between autophagy/mitophagy and apoptotic cell death pathways is invoked in acute renal injury and that these interactions should be the focus of future studies on DN.
The mitophagy adaptor proteins Bnip3 and Nix were both initially discovered as pro-apoptotic BH3-only proteins (Chen et al., 1997; 1999,). Unlike other BH3-only proteins, neither Bnip3 nor Nix rely on the BH3-only domain to interact with anti-apoptotic Bcl-2 or Bcl-XL. Rather, it is the C-terminal that is indispensible for binding with these Bcl-2 family members and for mitochondrial translocation (Chen et al., 1999; Ray et al., 2000). A further distinction from other BH3-only proteins lies with their complex roles in cell death, with Nix (Chen et al., 2010) and Bnip3 (Vande Velde et al., 2000; Quinsay et al., 2010; Chavez-Valdez et al., 2012) having dual roles being involved in both apoptosis and programmed necrosis. In cardiomyocytes, Nix is capable of mediating MOMP and apoptosis via a Bax/Bak-dependent pathway, as well as initiating the MPTP for programmed necrosis (Chen et al., 2010). However, Nix evokes programmed necrosis by localizing to the ER where it induces Ca2+ release, which is taken up by mitochondria stimulating the MPT (Diwan et al., 2009; Chen et al., 2010).
Bnip3 has been shown to activate during ischaemia–reperfusion injury or hypoxia in various cell systems (Vande Velde et al., 2000; Kubasiak et al., 2002; Kubli et al., 2008; Chavez-Valdez et al., 2012). Like Nix, Bnip3 localizes at both the mitochondria and the ER to regulate apoptosis and programmed necrosis (Zhang et al., 2009; 2010,; Landes et al., 2010; Qi et al., 2012). However, the mechanism involving Bnip3 in these cell death pathways has not been as succinctly defined as with Nix. Nonetheless, evidence suggests that when Bnip3 translocates to the OMM, it induces cell death by inducing MPT via a mechanism that is independent of Cyp-D, dissipating ΔΨm and inducing necrotic cell death (Quinsay et al., 2010). Interestingly, the induction of MPT by Bnip3 appears to be dependent on Bax and Bak (Kubli et al., 2007). A further role for Bnip3 in cell death includes inducing mitochondrial fragmentation via interaction with OPA1 (Landes et al., 2010). In addition, Bnip3 and AIF have been shown to cooperate to induce apoptosis in embryonic epithelial morphogenesis under hypoxic conditions (Qi et al., 2012).
Of particular interest was the discovery that Atg4D can be cleaved by caspase-3 at its N-terminus to reveal a latent mitochondrial leader sequence (Betin and Lane, 2009; Betin et al., 2012). Mitochondrial recruitment of cleaved Atg4D precedes apoptosis induction and is imported into the matrix where it undergoes further processing (Betin et al., 2012). Atg4D recruitment to mitochondria appears to be highly specific, with H2O2 inducing Atg4D translocation, an effect not observed during starvation or when using the apoptotic inducer staurosporine (Betin and Lane, 2009; Betin et al., 2012). Notwithstanding, caspase cleavage of Atg4D was shown to be non-essential for mitochondrial recruitment and apoptosis can occur independently of Atg4D translocation (Betin and Lane, 2009). Although, overexpression of cleaved Atg4B did sensitize the cells towards apoptosis (Betin et al., 2012). Further confounding its role in cell death is the fact that Atg4D has a BH3-only domain at its C-terminus, which appears to be required for cell death to occur (Betin and Lane, 2009). Compared to the other autophagins, Atg4C is the only other member to possess a DEVD, caspase-3 cleavage site and mitochondrial leader sequence, although it does not possess a BH3-only domain and its function remains undefined (Betin et al., 2012).
Cleaved Atg4D has a specificity for GABARAP-L1 delipidation, restricting GABARAP-L1 autophagosome formation, suggesting it also has a role in autophagy regulation (Betin and Lane, 2009). Evidently, each Atg4 family member possesses a unique specificity to Atg8 family members and has distinct roles in autophagy. In addition, it has been reported that Atg4A can be cleaved at the C-terminus by caspase-3 and centrally by calpain-1, with the later protease also capable of cleaving Atg4B at the C-terminus (Norman et al., 2010). A role for each of these autophagins in cell death in vivo has yet to be established. However, it is apparent that proteases are a key determinant in deciding how Atg4 family members are recruited post-transcription.
The importance of autophagy/mitophagy in renal injury
Autophagy has been implicated in the pathogenesis of a number of diseases such as cancer and neurodegenerative diseases (recently reviewed by Choi et al., 2013). There is a growing body of evidence to indicate that autophagy is linked to the pathogenesis of renal diseases such as DN, AKI and polycystic kidney disease (Huber et al., 2012). Autophagy may have a protective role in renal injury as shown by studies using conditional-knockout mice of the autophagy-related genes Atg5 or Atg7, in the proximal tubule (Kimura et al., 2011; Jiang et al., 2012; S. Liu et al., 2012; Takahashi et al., 2012). Atg5 and Atg7 are both required for the elongation and expansion of the isolation membrane during the formation of the autophagosome (Yang and Klionsky, 2010a,b2010b). Knockdown of Atg5 or Atg7 sensitizes the kidneys to ischaemia–reperfusion injury (Kimura et al., 2011; Jiang et al., 2012; S. Liu et al., 2012) or to nephrotoxic drugs such as cisplatin (Periyasamy-Thandavan et al., 2008; Yang C. et al., 2008; Takahashi et al., 2012) resulting in an increase in tubular damage, blood urea nitrogen, serum creatinine levels and cell death. Moreover, several of these studies also reported that the accumulation of damaged mitochondria was exacerbated when autophagy was impaired (Kimura et al., 2011; S. Liu et al., 2012; Takahashi et al., 2012).
There is a growing body of evidence that autophagy or mitophagy is altered in DN. An early study in diabetic rats found an association between decreased autophagic activity, as demonstrated by a decrease in volume and densities of autophagic vacuoles in PTECs detected by electron microscopy (Barbosa Junior Ade et al., 1992). The caveat to this study was that the rodents were studied only three days after injection with STZ, in which there is likely to be acute effects of STZ that are unrelated to diabetes (Tay et al., 2005). A similar study showed that regulation of blood glucose with insulin treatment led to the normalization of the density of autophagic vesicles, suggesting that this phenomenon was glucose-mediated (Han et al., 1992). However, insulin itself has been shown to promote mTOR signalling, which negatively regulates autophagy activity (Dennis et al., 2011) (Figure 2), although the sensitivity of this effect can be tissue specific (Naito et al., 2013). Other reports have found markers of autophagy to be increased in the kidney in DN. In particular, Beclin-1 was increased 8 weeks after STZ injection in rats (Wu et al., 2011). Recent studies reveal an up-regulation of p62/SQSTM1, a marker of impaired autophagy, in the kidneys of mice with STZ-induced diabetes (Vallon et al., 2013) and Wistar fatty (fa/fa) rats (Kitada et al., 2011), a model of type 2 diabetes. In glomerular lysates of STZ diabetic mice, LC3-II, Beclin-1 and Atg12-Atg5 levels were decreased over the course of DN (Fang et al., 2013b). Importantly, in that paper, decreased immunofluorescence staining for LC3 was shown in glomerular podocytes within human renal biopsies obtained from patients with DN. Furthermore, in vitro studies have demonstrated that high glucose can induce LC3-II and Beclin-1 in podocytes (Ma et al., 2013). Less is known about mitophagy in DN. In a rat model of STZ diabetes, in early diabetes (four weeks after diabetes induction), PINK1 protein was increased in the renal cortex (Smith et al., 2012a). In the same study, up-regulation of PINK1 was not observed in diabetic rats treated with insulin, suggesting that activation of mitophagy was glucose-driven. It is conceivable that early in diabetes, the kidney activates mitophagy to clear dysfunctional mitochondria, but as DN progresses, this process becomes overwhelmed, leading to impairment of mitophagy, accumulation of fragmented mitochondria and cell death. This concept, including the molecular mechanisms that may regulate changes in mitophagy, is discussed in more detail in the subsequent sections.
Several mechanisms have been postulated whereby autophagy could modulate renal fibrosis (Wang and Choi, 2013). In response to injury, glomerular mesangial cells proliferate and produce ECM leading to the development of glomerulosclerosis and renal fibrosis. Collagens are the primary constituents of the ECM. A recent study identified a novel role for autophagy in regulating ECM deposition in mesangial cells by promoting the degradation of intracellular collagen-1 (Kim et al., 2012). Kidneys from mice deficient in the autophagic protein Beclin-1 exhibited a profibrotic phenotype, with increased collagen-I deposition. In mesangial cells treated with bafilomycin A1 (an inhibitor of autolysosomal protein degradation) increased collagen-1 accumulation and co-localization of collagen-1 with LC3, or LAMP-1 (a lysosome marker) was observed. Conversely, treatment with trifluoperazine, an inducer of autophagy, resulted in decreased TGF-β1-induced Collagen accumulation. Previous studies have demonstrated the ability of TGF-β1 itself to induce autophagy in renal mesangial cells. A study by Ding et al. showed that TGF-β1 up-regulated LC3 via the Akt pathway (Ding et al., 2010b). Kim et al. demonstrated that TGF-β1 induces autophagy in mesangial cells via the TAK1-MKK3-p38 signalling pathway and concluded that the dual functions of TGF-β1, as both an inducer of Col-1 synthesis and an inducer of autophagy and Col-1 degradation underscore the pleiotropic nature of TGF-β1 (Kim et al., 2012). These findings suggest that autophagy is a cytoprotective mechanism to negatively regulate and prevent excess collagen accumulation in the glomeruli (Wang and Choi, 2013). In the light of these studies, it is plausible that an impairment in autophagic flux in DN would lead to a failure in the clearance of ECM, contributing to enhanced renal fibrosis.
The loss of podocytes, which form the filtration barrier of glomeruli, is considered a key feature of progressive glomerular disease. Within the podocyte, a post-mitotic cell type, a high level of constitutive autophagy has been detected (Hartleben et al., 2010). Podocyte-specific deletion of Atg5 leads to glomerulopathy in ageing mice that is accompanied by an accumulation of oxidized and ubiquitinated proteins, ER stress, proteinuria, podocyte loss and glomerulosclerosis. Moreover, activation of autophagy has been observed in glomeruli from mice with induced proteinuria and in glomeruli from patients with acquired proteinuric diseases (Hartleben et al., 2010). These findings underscore the significance of autophagy as a key homeostatic mechanism to maintain podocyte integrity (Hartleben et al., 2010). Several further studies using rodent models with podocyte-specific deletion of genes encoding proteins involved in autophagy have supported a critical role for autophagy in the maintenance of renal function (Cina et al., 2012b; Bechtel et al., 2013). Interestingly, angiotensin II promotes autophagy in podocytes in an mROS-dependent manner (Yadav et al., 2010).
As discussed earlier, there is some evidence that autophagy may also mediate tubulointerstital fibrosis. In addition, in tubule-specific TGF-β1 overexpressing mice, tubular cells were dedifferentiated and finally decomposed by autophagy, with evidence of tubulointerstitial fibrosis, but not epithelial-to-mesenchymal transition, nor was there any sign of apoptosis, suggesting that autophagy might mediate tubular cell death (Koesters et al., 2010).
A deficiency in mitophagy induces oxidative stress in kidneys, as mitophagy blockade leads to the accumulation of damaged mitochondria. In the context of a healthy cell, mitophagy is also a critical negative regulator of inflammasome activation. Damaged mitochondria promote mROS with subsequent mtDNA release and activation of the NLRP3 inflammasome promoting microinflammation (Naik and Dixit, 2011).
Pharmacological targeting of autophagy and mitophagy
As autophagy/mitophagy is either activated or impaired in a variety of pathological processes, pharmacological approaches to promote or to inhibit these pathways are receiving considerable interest (Rubinsztein et al., 2012) and highlight the translational potential of this area of research. Previous studies indicate that impairment of autophagy is a feature of DN, although mitophagy/autophagy may be stimulated in the kidney early in diabetes. Indeed, defects in autophagy and/or mitophagy can occur at different stages of a disease process, which will affect any treatment strategies. Various compounds have been identified as modulating the autophagy/mitophagy process including sirolimus (rapamycin), an mTORC1 inhibitor that inhibits autophagy. The benefits of rapamycin in treating DN have been supported by experimental studies that have shown that inhibition of mTOR activation protects against renal hypertrophy (Sakaguchi et al., 2006), glomerulosclerosis and proteinuria (Godel et al., 2011; Inoki et al., 2011), and mesangial expansion (Lloberas et al., 2006; Yang et al., 2007; Mori et al., 2009) in rodent models of diabetes.
However, in the nephrology field, rapamycin is best known as an immunosuppressant commonly used during kidney transplantation and has side effects that are limiting to therapy (Morath et al., 2007). Moreover, kidney transplant patients that are treated with rapamycin are more susceptible to new-onset diabetes (Teutonico et al., 2005; Johnston et al., 2008). The differences between experimental and clinical findings may lie in results from recent studies that suggest that rapamycin, once considered to be exclusively an mTORC1 inhibitor, can in fact inhibit mTORC2 under more chronic treatment regimes. Recently, rapamycin through chronic inhibition of mTORC2 was shown to impair glucose tolerance and insulin activation in in vivo mouse studies akin to type 2 diabetes (Lamming et al., 2012). mTORC2 differs from mTORC1 in that it is regulated by rictor (rapamycin-insensitive companion of mTOR), whereas mTORC1 is regulated by raptor (rapamycin-associated protein of mTOR). The emerging function of mTORC2 appears to be regulating autophagy gene expression through acting on Akt, which phosphorylates FOXO3a (Mammucari et al., 2007; Zhao et al., 2007) (Figure 2). In muscle cells, some of the autophagy genes that FOXO3a regulates include LC3, Bnip3, Nix (Mammucari et al., 2007), Atg4b and Atg12l (Zhao et al., 2007), all of which play key roles in mitophagy. These findings may help to explain the adverse effect of conditional knockout of mTOR in podocytes, which disrupted autophagic flux and led to renal failure 5 weeks after birth (Cina et al., 2012a).
The mTOR signalling pathway evidently contributes to the impairment of autophagy during DN. However, the above studies suggest that rapamycin or similar drugs, such as rapalogs (derivatives of rapamycin) or dual mTOR inhibitors like the chemotherapeutic drug AZD8055 (Huang et al., 2011) may not be suitable for the long-term stimulation of autophagy required for a chronic treatment regime, such as in people with DN. Recently, rilmenidine, a clinically approved anti-hypertensive drug was demonstrated to stimulate autophagy activity via an mTOR-independent pathway (Rose et al., 2010). Rilmenidine is an imidazoline receptor 1 agonist that has been shown to attenuate polyglutamine toxicity in the brain, through increased autophagy in a Huntington's disease mouse model (Rose et al., 2010). Rilmenidine has been shown to bind to imidazoline receptor 1 in the proximal tubule (Lachaud et al., 1989) and to reduce microalbuminuria in patients with type 2 diabetes and hypertension (Reid, 2000). Although activation of autophagy via mTOR signalling is a non-selective means of removing dysfunctional mitochondria, this pathway may be important in modulating mitophagy activity. However, drugs such as rilmenidine (Renna et al., 2010), which bypass mTOR and are not nephrotoxic may be required to promote autophagy activity in DN.
Because mitophagy is dependent on mitochondrial dynamics, modulation of fission and fusion may target the process of mitophagy. It is interesting to note that in AKI, treatment of mice with mdivi-1, a Drp1 inhibitor (Cassidy-Stone et al., 2008), was renoprotective in an ischaemia–reperfusion injury model; however, mdivi-1 only partially suppressed ischaemia-induced mitochondrial fragmentation in proximal tubular cells, from 43 to 32% (Brooks et al., 2009). Treatment with mdivi-1 reduced serum creatinine and blood urea nitrogen, tubular damage and apoptosis, but these studies were performed in an acute setting. Preclinical studies are required to investigate efficacy and possible adverse effects in a chronic setting such as DN.
The relationship between ROS production and removal must be delicately balanced in order to stimulate mitophagy activity (Frank et al., 2012). Too much ROS is detrimental, but too little can result in disruption to redox signalling. This paradox may explain the lack of efficacy of antioxidant therapies to date (Benfeito et al., 2013), with other sources of ROS also needing to be considered when treating DN (Forbes et al., 2008; Singh et al., 2011). To this end, the development of mitochondrial targeted antioxidants may be beneficial for controlling mROS production, while minimizing interference with cellular redox signalling. However, the benefits of using antioxidants of this nature need context. Although mitochondria-targeted antioxidants may limit oxidative damage and be capable of repairing lipid membranes, they may not restore functionality to damaged mitochondria (e.g. repair damaged proteins or mtDNA). The balance of healthy mitochondria within a cell may only be restored by removal of damaged mitochondria through mitophagy. It is plausible that antioxidants may also target the process of mitophagy. A previous study showed that treatment with CoQ decreased both mitochondrial fragmentation and size (Persson et al., 2012), indicating that mitochondrion targeted antioxidant treatment may protect against altered morphology and function in diabetes, and indeed may modulate mitophagic flux.
Distinct from MitoQ, other forms of mitochondria-targeted antioxidants known as SS-peptides that are not dependent on ΔΨm have also been developed (Szeto, 2008). These cationic antioxidants can passively diffuse into the mitochondria and reside in the IMM. Mitochondria-targeted antioxidant SS-31 has been demonstrated to be beneficial in AKI studies involving ischaemic–reperfusion injury and unilateral ureteral obstruction (Mizuguchi et al., 2008; Szeto et al., 2011; Birk et al., 2013). However, it remains to be established whether these antioxidants will be equally beneficial in more chronic nephropathy settings such as DN, particularly in situations where treatments more often than not will need to be administered following the diagnosis of the onset of the disease. Furthermore, in the case of mitophagy regulation, where ΔΨm and ROS production are considered critical signals for the turnover of damaged mitochondria, TPP+ compounds might be more advantageous than SS-peptides. In manipulating mitophagy activity, preferential antioxidant treatment of healthy or partially damaged mitochondria with ΔΨm would minimize damage and oxidative stress production, allowing autophagosomes to target damaged mitochondria (without ΔΨm and producing oxidative stress). In chronic situations where mitophagy machinery may be overwhelmed by the magnitude of dysfunctional mitochondria, enhanced preferential selection of these organelles would be a considerable advantage. Although SS-peptides act independently of ΔΨm, the rate of mitophagy activity may be compromised with blanket suppression of ROS masking redox signalling from damaged mitochondria.
Closing comments
Although there is ample evidence that there is a disturbance of mitochondrial bioenergetics in the kidney in DN, it is clear that there is a lack of studies characterizing the role of mitophagy in this disease process. How the process of mitophagy is changed in DN and if this change is beneficial or detrimental to kidney function still remains to be fully understood. What we know of mitophagy is drawn from studies across many mammalian cellular systems. From these studies, it is apparent that mitochondrial homeostasis is a tightly regulated balance between several different processes that are responsible for maintaining dynamics, motility, redox signalling and, ultimately, the decision between life and death. Mitophagy and the UPS are intricately intertwined with each of these processes to ensure a healthy pool of mitochondria is maintained (Figure 4). Yet, it is the mode and severity of the insult as well as unique cellular characteristics (e.g. presence of SGLT2, GLUT2 and Na+/K+-ATPase transporters to facilitate the glucose gradient and relatively rich mitochondria content in PTECs) that will influence the type of mitophagic response. Thus, underscoring the need to elucidate the mechanisms that regulate mitophagy in the kidney.
Figure 4.
Balancing mitochondrial homeostasis. This schematic illustrates the interaction between mitochondrial biogenesis, dynamics, organelle turnover and cell death, in addition to the major players involved in these processes. Mitochondria biogenesis involves oxidative phosphorylation (OXPHOS) via the electron transport chain (ETC). This process generates energy for the cell in the form of ATP, but in the process also generates reactive oxygen species (ROS). These ROS are involved in redox signalling regulated by antioxidants and transcription factors (orange circles). Mitochondria are dynamic organelles that are in constant motion, being transported around the cytoplasm while undergoing changing morphology (yellow circles). When mitochondria become dysfunctional via oxidative stress, mitochondrial dynamics is arrested and the ubiquitin proteasome system and mitophagy come into play to turnover the damaged organelles (pale blue circles). Central in regulating this process is the interaction between PARL (presenilin-associated rhomboid-like protease), PINK1 and Parkin (dark blue circles), and the involvement of adapter proteins (green circles). When cellular stress (e.g. damage to mitochondria and ER stress) overwhelms cellular homeostasis, cells can undergo diverse forms of death. Depending on the type and severity of the stress, mitochondria can regulate multiple cell death pathways involving several different signalling pathways (pink circles).
Much attention has been given to studying mitochondrial dysfunction in DN, raising the prospects of pharmacological agents that may impede progression of the disease. However, DN diagnosis usually occurs once the onset of the disease is well advanced. Strategies to impede mitochondrial dysfunction may not be sufficient to restore kidney homeostasis and only delay the progression of DN. Despite the lack of studies focused on mitophagy in DN, it stands to reason that this process may be implicated in its pathology. The accumulation of dysfunctional mitochondria in DN is evidence of either an impairment of mitophagy, or an inability of this process to meet the influx of damaged mitochondria. An improved understanding of the mechanisms that regulate mitophagy may lead to the discovery of new targets for therapeutic approaches of DN that can prevent or reverse pathology. Drug screening for agents that interact with mitophagy regulators could provide further therapeutic targets. Therapeutic strategies that promote mitophagy activity offer the prospect of not only eliminating dysfunctional mitochondria, but also the possibility of restoring mitochondrial biogenesis.
Acknowledgments
M. C. is supported by an Australian and New Zealand Society of Nephrology Career Development award. This work was supported, in part, by the National Health and Medical Research Council of Australia and the Diabetes Australia Research Trust.
Glossary
- ΔΨm
mitochondrial membrane potential
- ACD
autophagic cell death
- AIF
apoptosis-inducing factor
- AKI
acute kidney injury
- ANT
adenine nucleotide translocator;
- CKD
chronic kidney disease
- CoQ
coenzyme Q
- Cyp-D
cyclophilin D
- cyt c
cytochrome c
- DN
diabetic nephropathy
- Drp1
dynamin-related protein 1
- ECM
extracellular matrix
- ER
endoplasmic reticulum
- ESRD
end-stage renal disease
- ETC
electron transport chain
- FOXO3a
Forkhead box O3a
- FUNDC1
FUN14 domain containing 1
- GFR
glomerular filtration rate
- Gpx
glutathione peroxidase
- HDAC6
histone deacetylase 6
- HIF-1α
hypoxia-inducible factor 1α
- HK2
hexokinase 2
- IHG-1
induced-in-high-glucose-1
- IMM
inner mitochondrial membrane
- IMS
inter-membrane space
- KHC
kinesin-1 heavy chain
- LC3
light chain 3
- LIR
LC3 interacting region
- Mfn1/2
mitofusin 1/2
- Miro
mitochondria Rho GTPase
- MitoQ
mitoquinol
- MOMP
mitochondrial outer membrane pore
- MPT
mitochondrial permeability transition
- MPTP
mitochondrial permeability transition pore
- mROS
mitochondrial reactive oxygen species
- mtDNA
mitochondrial DNA
- mTOR
mechanistic target of rapamycin
- mTORC1/2
mechanistic target of rapamycin complex 1/2
- MTS
mitochondria targeting sequence
- NOX4
NADPH oxidase 4
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- OMM
outer mitochondrial membrane
- OPA1
optic atrophy 1
- OXPHOS
oxidative phosphorylation
- PGC1α
peroxisome proliferator-activated receptor γ co-activator 1α
- PINK1
PTEN-induced putative kinase 1
- PTEC
proximal tubule epithelial cell
- RAS
renin angiotensin system
- ROS
reactive oxygen species
- SGLT2
sodium glucose transporter 2
- SQSTM1
sequestosome 1
- STZ
streptozotocin
- TIM
translocase of the inner mitochondrial membrane
- TIGAR
TP53-induced glycolysis and apoptosis regulator
- TOM
translocase of the outer mitochondrial membrane
- TPP+
triphenylalkylphosphonium cation
- UBD
ubiquitin-binding domain
- UPS
ubiquitin proteasome system
- UVRAG
ultraviolet resistance-associated gene
- VDAC1
voltage-dependent anion channel 1
Conflict of interest
None.
References
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. Economic costs of diabetes in the U.S. in 2012. Diabetes Care. 2013;36:1033–1046. doi: 10.2337/dc12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci. 2008;1147:233–241. doi: 10.1196/annals.1427.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JS. The role of the mitochondrial permeability transition in cell death. Mitochondrion. 2006;6:225–234. doi: 10.1016/j.mito.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Atkinson SJ, Hosford MA, Molitoris BA. Mechanism of actin polymerization in cellular ATP depletion. J Biol Chem. 2004;279:5194–5199. doi: 10.1074/jbc.M306973200. [DOI] [PubMed] [Google Scholar]
- Baines CP. Role of the mitochondrion in programmed necrosis. Front Physiol. 2010;1:156. doi: 10.3389/fphys.2010.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Mandel LJ. Metabolic substrate utilization by rabbit proximal tubule. An NADH fluorescence study. Am J Physiol. 1988;254(3 Pt 2):F407–F416. doi: 10.1152/ajprenal.1988.254.3.F407. [DOI] [PubMed] [Google Scholar]
- Barbosa Junior Ade A, Zhou H, Hultenschmidt D, Totovic V, Jurilj N, Pfeifer U. Inhibition of cellular autophagy in proximal tubular cells of the kidney in streptozotocin-diabetic and uninephrectomized rats. Virchows Arch B Cell Pathol Incl Mol Pathol. 1992;61:359–366. doi: 10.1007/BF02890439. [DOI] [PubMed] [Google Scholar]
- Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- Bechtel W, Helmstadter M, Balica J, Hartleben B, Kiefer B, Hrnjic F, et al. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J Am Soc Nephrol. 2013;24:727–743. doi: 10.1681/ASN.2012070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfeito S, Oliveira C, Soares P, Fernandes C, Silva T, Teixeira J, et al. Antioxidant therapy: still in search of the ‘magic bullet’. Mitochondrion. 2013;13:427–435. doi: 10.1016/j.mito.2012.12.002. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28:3015–3026. doi: 10.1038/emboj.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol. 2013;4:95. doi: 10.3389/fphys.2013.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betin VM, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci. 2009;122(Pt 14):2554–2566. doi: 10.1242/jcs.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betin VM, MacVicar TD, Parsons SF, Anstee DJ, Lane JD. A cryptic mitochondrial targeting motif in Atg4D links caspase cleavage with mitochondrial import and oxidative stress. Autophagy. 2012;8:664–676. doi: 10.4161/auto.19227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilous R, Chaturvedi N, Sjolie AK, Fuller J, Klein R, Orchard T, et al. Effect of candesartan on microalbuminuria and albumin excretion rate in diabetes: three randomized trials. Ann Intern Med. 2009;151:11–20. doi: 10.7326/0003-4819-151-1-200907070-00120. W13–W14. [DOI] [PubMed] [Google Scholar]
- Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24:1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizat N, Galas MC, Jacquard C, Boyer F, Hermel JM, Schiffmann SN, et al. Neuroprotective effect of zVAD against the neurotoxin 3-nitropropionic acid involves inhibition of calpain. Neuropharmacology. 2005;49:695–702. doi: 10.1016/j.neuropharm.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer IH, Rue TC, Cleary PA, Lachin JM, Molitch ME, Steffes MW, et al. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: an analysis of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications cohort. Arch Intern Med. 2011;171:412–420. doi: 10.1001/archinternmed.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–2210. doi: 10.1097/01.asn.0000079785.13922.f6. [DOI] [PubMed] [Google Scholar]
- Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol. 2010;6:643–656. doi: 10.1038/nrneph.2010.120. [DOI] [PubMed] [Google Scholar]
- Borch-Johnsen K, Kreiner S. Proteinuria: value as predictor of cardiovascular mortality in insulin dependent diabetes mellitus. Br Med J (Clin Res Ed) 1987;294:1651–1654. doi: 10.1136/bmj.294.6588.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner BM, Rector FC. Brenner & Rector's the Kidney. Philadelphia, PA: Elsevier/Saunders; 2008. [Google Scholar]
- Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- Brito PL, Fioretto P, Drummond K, Kim Y, Steffes MW, Basgen JM, et al. Proximal tubular basement membrane width in insulin-dependent diabetes mellitus. Kidney Int. 1998;53:754–761. doi: 10.1046/j.1523-1755.1998.00809.x. [DOI] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119:1275–1285. doi: 10.1172/JCI37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, et al. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes. 2009;58:1986–1997. doi: 10.2337/db09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, et al. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalena G, Daehn I, Bottinger E. Transforming growth factor-beta, bioenergetics, and mitochondria in renal disease. Semin Nephrol. 2012;32:295–303. doi: 10.1016/j.semnephrol.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cavanagh EM, Ferder L, Toblli JE, Piotrkowski B, Stella I, Fraga CG, et al. Renal mitochondrial impairment is attenuated by AT1 blockade in experimental type I diabetes. Am J Physiol Heart Circ Physiol. 2008;294:H456–H465. doi: 10.1152/ajpheart.00926.2007. [DOI] [PubMed] [Google Scholar]
- Cebollero E, Reggiori F, Kraft C. Reticulophagy and ribophagy: regulated degradation of protein production factories. Int J Cell Biol. 2012;2012:182834. doi: 10.1155/2012/182834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, et al. Prevention of diabetic nephropathy in Ins2(+/)(-)(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem J. 2010;432:9–19. doi: 10.1042/BJ20100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Valdez R, Martin LJ, Flock DL, Northington FJ. Necrostatin-1 attenuates mitochondrial dysfunction in neurons and astrocytes following neonatal hypoxia-ischemia. Neuroscience. 2012;219:192–203. doi: 10.1016/j.neuroscience.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ray R, Dubik D, Shi L, Cizeau J, Bleackley RC, et al. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med. 1997;186:1975–1983. doi: 10.1084/jem.186.12.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, et al. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem. 1999;274:7–10. doi: 10.1074/jbc.274.1.7. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci. 2007;120(Pt 23):4155–4166. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW., 2nd Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:9035–9042. doi: 10.1073/pnas.0914013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci U S A. 2012;109:20491–20496. doi: 10.1073/pnas.1206530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cina DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J, et al. Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol. 2012a;23:412–420. doi: 10.1681/ASN.2011070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cina DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J, et al. MTOR regulates autophagic flux in the glomerulus. Autophagy. 2012b;8:696–698. doi: 10.4161/auto.19386. [DOI] [PubMed] [Google Scholar]
- Corcoran JB, McCarthy S, Griffin B, Gaffney A, Bhreathnach U, Borgeson E, et al. IHG-1 must be localised to mitochondria to decrease Smad7 expression and amplify TGF-β1-induced fibrotic responses. Biochim Biophys Acta. 2013;1833:1969–1978. doi: 10.1016/j.bbamcr.2013.03.027. [DOI] [PubMed] [Google Scholar]
- Coughlan MT, Thallas-Bonke V, Pete J, Long DM, Gasser A, Tong DC, et al. Combination therapy with the advanced glycation end product cross-link breaker, alagebrium, and angiotensin converting enzyme inhibitors in diabetes: synergy or redundancy? Endocrinology. 2007;148:886–895. doi: 10.1210/en.2006-1300. [DOI] [PubMed] [Google Scholar]
- Coughlan MT, Mibus AL, Forbes JM. Oxidative stress and advanced glycation in diabetic nephropathy. Ann N Y Acad Sci. 2008;1126:190–193. doi: 10.1196/annals.1433.018. [DOI] [PubMed] [Google Scholar]
- Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol. 2009;20:742–752. doi: 10.1681/ASN.2008050514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane FL, Hatefi Y, Lester RL, Widmer C. Isolation of a quinone from beef heart mitochondria. Biochim Biophys Acta. 1957;25:220–221. doi: 10.1016/0006-3002(57)90457-2. [DOI] [PubMed] [Google Scholar]
- Cuevas S, Zhang Y, Yang Y, Escano C, Asico L, Jones JE, et al. Role of renal DJ-1 in the pathogenesis of hypertension associated with increased reactive oxygen species production. Hypertension. 2012;59:446–452. doi: 10.1161/HYPERTENSIONAHA.111.185744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Tang X, Christian WV, Yoon Y, Tieu K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J Biol Chem. 2010;285:11740–11752. doi: 10.1074/jbc.M109.066662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RE, Williams M. Mitochondrial function and dysfunction: an update. J Pharmacol Exp Ther. 2012;342:598–607. doi: 10.1124/jpet.112.192104. [DOI] [PubMed] [Google Scholar]
- De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- Dennis MD, Baum JI, Kimball SR, Jefferson LS. Mechanisms involved in the coordinate regulation of mTORC1 by insulin and amino acids. J Biol Chem. 2011;286:8287–8296. doi: 10.1074/jbc.M110.209171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010a;285:27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Kim JK, Kim SI, Na HJ, Jun SY, Lee SJ, et al. TGF-{beta}1 protects against mesangial cell apoptosis via induction of autophagy. J Biol Chem. 2010b;285:37909–37919. doi: 10.1074/jbc.M109.093724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, et al. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest. 2009;119:203–212. doi: 10.1172/JCI36445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du G, Jiao R. To prevent neurodegeneration: HDAC6 uses different strategies for different challenges. Commun Integr Biol. 2011;4:139–142. doi: 10.4161/cib.4.2.14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS. Macrophages and immunologic inflammation of the kidney. Semin Nephrol. 2010;30:234–254. doi: 10.1016/j.semnephrol.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgass K, Pakay J, Ryan MT, Palmer CS. Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta. 2013;1833:150–161. doi: 10.1016/j.bbamcr.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol Cell. 2011;42:23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Fang L, Xie D, Wu X, Cao H, Su W, Yang J. Involvement of endoplasmic reticulum stress in albuminuria induced inflammasome activation in renal proximal tubular cells. PLoS ONE. 2013a;8:e72344. doi: 10.1371/journal.pone.0072344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W, et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS ONE. 2013b;8:e60546. doi: 10.1371/journal.pone.0060546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D, Liu L, Zhu Y, Chen Q. Molecular signaling toward mitophagy and its physiological significance. Exp Cell Res. 2013;319:1697–1705. doi: 10.1016/j.yexcr.2013.03.034. [DOI] [PubMed] [Google Scholar]
- Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Kidney Foundation. KDOQI clinical practice guideline for diabetes and CKD: 2012 update. Am J Kidney Dis. 2012;60:850–886. doi: 10.1053/j.ajkd.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, et al. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta. 2012;1823:2297–2310. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Fujita H, Fujishima H, Chida S, Takahashi K, Qi Z, Kanetsuna Y, et al. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J Am Soc Nephrol. 2009;20:1303–1313. doi: 10.1681/ASN.2008080844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway CA, Lee H, Nejjar S, Jhun BS, Yu T, Hsu W, et al. Transgenic control of mitochondrial fission induces mitochondrial uncoupling and relieves diabetic oxidative stress. Diabetes. 2012;61:2093–2104. doi: 10.2337/db11-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Giaime E, Yamaguchi H, Gautier CA, Kitada T, Shen J. Loss of DJ-1 does not affect mitochondrial respiration but increases ROS production and mitochondrial permeability transition pore opening. PLoS ONE. 2012;7:e40501. doi: 10.1371/journal.pone.0040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson DT, Liu J, Xue JL, Louis TA, Solid CA, Ebben JP, et al. Projecting the number of patients with end-stage renal disease in the United States to the year 2015. J Am Soc Nephrol. 2005;16:3736–3741. doi: 10.1681/ASN.2005010112. [DOI] [PubMed] [Google Scholar]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest. 2011;121:2197–2209. doi: 10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta. 2008;1777:860–866. doi: 10.1016/j.bbabio.2008.05.442. [DOI] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi: 10.1038/embor.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha H, Lee HB. Reactive oxygen species amplify glucose signalling in renal cells cultured under high glucose and in diabetic kidney. Nephrology (Carlton) 2005;10(Suppl):S7–10. doi: 10.1111/j.1440-1797.2005.00448.x. [DOI] [PubMed] [Google Scholar]
- Han K, Lehringer-Polzin M, Zhou H, Pfeifer U. Cellular autophagy in proximal tubules of early diabetic rats following insulin treatment and islet transplantation. Virchows Arch B Cell Pathol Incl Mol Pathol. 1992;61:367–373. doi: 10.1007/BF02890440. [DOI] [PubMed] [Google Scholar]
- Hanada T, Ohsumi Y. Structure-function relationship of Atg12, a ubiquitin-like modifier essential for autophagy. Autophagy. 2005;1:110–118. doi: 10.4161/auto.1.2.1858. [DOI] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansell P, Welch WJ, Blantz RC, Palm F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin Exp Pharmacol Physiol. 2013;40:123–137. doi: 10.1111/1440-1681.12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao LY, Giasson BI, Bonini NM. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc Natl Acad Sci U S A. 2010;107:9747–9752. doi: 10.1073/pnas.0911175107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Harris SI, Balaban RS, Barrett L, Mandel LJ. Mitochondrial respiratory capacity and Na+-and K+-dependent adenosine triphosphatase-mediated ion transport in the intact renal cell. J Biol Chem. 1981;256:10319–10328. [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest. 2010;120:1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog C, Yang C, Holmes A, Kaushal GP. zVAD-fmk prevents cisplatin-induced cleavage of autophagy proteins but impairs autophagic flux and worsens renal function. Am J Physiol Renal Physiol. 2012;303:F1239–F1250. doi: 10.1152/ajprenal.00659.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey FB, Corcoran JB, Docherty NG, Griffin B, Bhreathnach U, Furlong F, et al. IHG-1 promotes mitochondrial biogenesis by stabilizing PGC-1α. J Am Soc Nephrol. 2011;22:1475–1485. doi: 10.1681/ASN.2010111154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins GC, Beart PM, Shin YS, Chen MJ, Cheung NS, Nagley P. Oxidative stress: emerging mitochondrial and cellular themes and variations in neuronal injury. J Alzheimers Dis. 2010;20(Suppl. 2):S453–S473. doi: 10.3233/JAD-2010-100321. [DOI] [PubMed] [Google Scholar]
- Higgins GC, Devenish RJ, Beart PM, Nagley P. Autophagic activity in cortical neurons under acute oxidative stress directly contributes to cell death. Cell Mol Life Sci. 2011;68:3725–3740. doi: 10.1007/s00018-011-0667-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook SS, Orian A, Cowley SM, Eisenman RN. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc Natl Acad Sci U S A. 2002;99:13425–13430. doi: 10.1073/pnas.172511699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovind P, Tarnow L, Rossing P, Jensen BR, Graae M, Torp I, et al. Predictors for the development of microalbuminuria and macroalbuminuria in patients with type 1 diabetes: inception cohort study. BMJ. 2004;328:1105. doi: 10.1136/bmj.38070.450891.FE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med. 2012;29:613–618. doi: 10.3892/ijmm.2012.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Yang ZJ, Yu C, Sinicrope FA. Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J Biol Chem. 2011;286:40002–40012. doi: 10.1074/jbc.M111.297432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber TB, Edelstein CL, Hartleben B, Inoki K, Dong Z, Koya D, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8:1009–1031. doi: 10.4161/auto.19821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, et al. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem. 2013;288:22019–22032. doi: 10.1074/jbc.M113.467530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest. 2011;121:2181–2196. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, et al. Sestrin2 and BNIP3 (Bcl-2/adenovirus E1B 19kDa-interacting protein3) regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol. 2013;305:F495–509. doi: 10.1152/ajprenal.00642.2012. [DOI] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–1283. doi: 10.1038/ki.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Youle RJ. PINK1-and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125(Pt 4):795–799. doi: 10.1242/jcs.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston O, Rose CL, Webster AC, Gill JS. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol. 2008;19:1411–1418. doi: 10.1681/ASN.2007111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joselin AP, Hewitt SJ, Callaghan SM, Kim RH, Chung YH, Mak TW, et al. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet. 2012;21:4888–4903. doi: 10.1093/hmg/dds325. [DOI] [PubMed] [Google Scholar]
- Junn E, Jang WH, Zhao X, Jeong BS, Mouradian MM. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J Neurosci Res. 2009;87:123–129. doi: 10.1002/jnr.21831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda K, Iwao J, Sakata N, Takebayashi S. Correlation between mitochondrial enlargement in renal proximal tubules and microalbuminuria in rats with early streptozotocin-induced diabetes. Acta Pathol Jpn. 1992;42:855–860. doi: 10.1111/j.1440-1827.1992.tb01890.x. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katyare SS, Satav JG. Effect of streptozotocin-induced diabetes on oxidative energy metabolism in rat kidney mitochondria. A comparative study of early and late effects. Diabetes Obes Metab. 2005;7:555–562. doi: 10.1111/j.1463-1326.2004.00429.x. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- KDOQI. KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis. 2007;49(2 Suppl. 2):S12–154. doi: 10.1053/j.ajkd.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J Biol Chem. 2012;287:11677–11688. doi: 10.1074/jbc.M111.308460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902–913. doi: 10.1681/ASN.2010070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada M, Takeda A, Nagai T, Ito H, Kanasaki K, Koya D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabetes Res. 2011;2011:908185. doi: 10.1155/2011/908185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R, et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010;177:632–643. doi: 10.2353/ajpath.2010.091012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korner A, Eklof AC, Celsi G, Aperia A. Increased renal metabolism in diabetes. Mechanism and functional implications. Diabetes. 1994;43:629–633. doi: 10.2337/diab.43.5.629. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroller-Schon S, Steven S, Kossmann S, Scholz A, Daub S, Oelze M, et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2012.4953. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci U S A. 2002;99:12825–12830. doi: 10.1073/pnas.202474099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J. 2007;405:407–415. doi: 10.1042/BJ20070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2008;295:H2025–H2031. doi: 10.1152/ajpheart.00552.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaud V, Bidet M, Coupry I, Podevin RA, Poujeol P, Parini A. [Interaction of rilmenidine with renal imidazoline-guanidine sites] Arch Mal Coeur Vaiss. 1989;82(Spec No 5):15–18. [PubMed] [Google Scholar]
- Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnaune-Pelloquin L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010;11:459–465. doi: 10.1038/embor.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol. 2013;200:163–172. doi: 10.1083/jcb.201210111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Wauquier F, Eid AA, Roman LJ, Ghosh-Choudhury G, Khazim K, et al. Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: role of mitochondrial reactive oxygen species. J Biol Chem. 2013;288:28668–28686. doi: 10.1074/jbc.M113.470971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Choi MG, Kim DS, Kim TW. Manganese superoxide dismutase gene polymorphism (V16A) is associated with stages of albuminuria in Korean type 2 diabetic patients. Metabolism. 2006;55:1–7. doi: 10.1016/j.metabol.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;301:H1924–H1931. doi: 10.1152/ajpheart.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- Lempiainen J, Finckenberg P, Mervaala E, Sankari S, Levijoki J, Mervaala E. Caloric restriction ameliorates kidney ischemia reperfusion injury through PGC-1α-eNOS-pathway and enhanced autophagy. Acta Physiol (Oxf) 2013;208:410–421. doi: 10.1111/apha.12120. [DOI] [PubMed] [Google Scholar]
- Lezi E, Swerdlow RH. Mitochondria in neurodegeneration. Adv Exp Med Biol. 2012;942:269–286. doi: 10.1007/978-94-007-2869-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, et al. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS ONE. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wan OW, Xie W, Chung KK. p32 regulates mitochondrial morphology and dynamics through parkin. Neuroscience. 2011;199:346–358. doi: 10.1016/j.neuroscience.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KL, Dawson VL, Dawson TM. Parkin-mediated lysine 63-linked polyubiquitination: a link to protein inclusions formation in Parkinson's and other conformational diseases? Neurobiol Aging. 2006;27:524–529. doi: 10.1016/j.neurobiolaging.2005.07.023. [DOI] [PubMed] [Google Scholar]
- Liu L, Zheng T, Wang N, Wang F, Li M, Jiang J, et al. The manganese superoxide dismutase Val16Ala polymorphism is associated with decreased risk of diabetic nephropathy in Chinese patients with type 2 diabetes. Mol Cell Biochem. 2009;322:87–91. doi: 10.1007/s11010-008-9943-x. [DOI] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8:826–837. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int. 2006;69:213–217. doi: 10.1038/sj.ki.5000054. [DOI] [PubMed] [Google Scholar]
- Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloberas N, Cruzado JM, Franquesa M, Herrero-Fresneda I, Torras J, Alperovich G, et al. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol. 2006;17:1395–1404. doi: 10.1681/ASN.2005050549. [DOI] [PubMed] [Google Scholar]
- Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, et al. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab. 2012;16:613–624. doi: 10.1016/j.cmet.2012.10.005. [DOI] [PubMed] [Google Scholar]
- Lutz AK, Exner N, Fett ME, Schlehe JS, Kloos K, Lammermann K, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem. 2009;284:22938–22951. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Zhu J, Chen X, Zha D, Singhal PC, Ding G. High glucose induces autophagy in podocytes. Exp Cell Res. 2013;319:779–789. doi: 10.1016/j.yexcr.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Marino G, Uria JA, Puente XS, Quesada V, Bordallo J, Lopez-Otin C. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J Biol Chem. 2003;278:3671–3678. doi: 10.1074/jbc.M208247200. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Masry TM, Zahra MA, el-Tawil MM, Khalifa RA. Manganese superoxide dismutase alanine to valine polymorphism and risk of neuropathy and nephropathy in Egyptian type 1 diabetic patients. Rev Diabet Stud. 2005;2:70–74. doi: 10.1900/RDS.2005.2.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather A, Pollock C. Glucose handling by the kidney. Kidney Int Suppl. 2011;120:S1–S6. doi: 10.1038/ki.2010.509. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010;375:2073–2081. doi: 10.1016/S0140-6736(10)60674-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer M, Zinman B, Gardiner R, Suissa S, Sinaiko A, Strand T, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med. 2009;361:40–51. doi: 10.1056/NEJMoa0808400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer SM, Steffes MW, Brown DM. The kidney in diabetes. Am J Med. 1981;70:603–612. doi: 10.1016/0002-9343(81)90582-9. [DOI] [PubMed] [Google Scholar]
- Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem. 2011;117:856–867. doi: 10.1111/j.1471-4159.2011.07253.x. [DOI] [PubMed] [Google Scholar]
- Mellors A, Tappel AL. The inhibition of mitochondrial peroxidation by ubiquinone and ubiquinol. J Biol Chem. 1966a;241:4353–4356. [PubMed] [Google Scholar]
- Mellors A, Tappel AL. Quinones and quinols as inhibitors of lipid peroxidation. Lipids. 1966b;1:282–284. doi: 10.1007/BF02531617. [DOI] [PubMed] [Google Scholar]
- Messner B, Ploner C, Laufer G, Bernhard D. Cadmium activates a programmed, lysosomal membrane permeabilization-dependent necrosis pathway. Toxicol Lett. 2012;212:268–275. doi: 10.1016/j.toxlet.2012.05.026. [DOI] [PubMed] [Google Scholar]
- Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- Michels J, Kepp O, Senovilla L, Lissa D, Castedo M, Kroemer G, et al. Functions of BCL-X L at the interface between cell death and metabolism. Int J Cell Biol. 2013;2013:705294. doi: 10.1155/2013/705294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, de Strihou C. Diabetic nephropathy: a disorder of oxygen metabolism? Nat Rev Nephrol. 2010;6:83–95. doi: 10.1038/nrneph.2009.211. [DOI] [PubMed] [Google Scholar]
- Mizuguchi Y, Chen J, Seshan SV, Poppas DP, Szeto HH, Felsen D. A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2008;295:F1545–F1553. doi: 10.1152/ajprenal.00395.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollsten A, Marklund SL, Wessman M, Svensson M, Forsblom C, Parkkonen M, et al. A functional polymorphism in the manganese superoxide dismutase gene and diabetic nephropathy. Diabetes. 2007;56:265–269. doi: 10.2337/db06-0698. [DOI] [PubMed] [Google Scholar]
- Mollsten A, Jorsal A, Lajer M, Vionnet N, Tarnow L. The V16A polymorphism in SOD2 is associated with increased risk of diabetic nephropathy and cardiovascular disease in type 1 diabetes. Diabetologia. 2009;52:2590–2593. doi: 10.1007/s00125-009-1550-1. [DOI] [PubMed] [Google Scholar]
- Moore DJ, Zhang L, Troncoso J, Lee MK, Hattori N, Mizuno Y, et al. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum Mol Genet. 2005;14:71–84. doi: 10.1093/hmg/ddi007. [DOI] [PubMed] [Google Scholar]
- Morath C, Arns W, Schwenger V, Mehrabi A, Fonouni H, Schmidt J, et al. Sirolimus in renal transplantation. Nephrol Dial Transplant. 2007;22(Suppl. 8):viii61–viii65. doi: 10.1093/ndt/gfm652. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Rolo AP, Sena C, Seica R, Oliveira CR, Santos MS. Insulin attenuates diabetes-related mitochondrial alterations: a comparative study. Med Chem. 2006;2:299–308. doi: 10.2174/157340606776930754. [DOI] [PubMed] [Google Scholar]
- Mori H, Inoki K, Masutani K, Wakabayashi Y, Komai K, Nakagawa R, et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem Biophys Res Commun. 2009;384:471–475. doi: 10.1016/j.bbrc.2009.04.136. [DOI] [PubMed] [Google Scholar]
- Moruno F, Perez-Jimenez E, Knecht E. Regulation of autophagy by glucose in mammalian cells. Cells. 2012;1:372–395. doi: 10.3390/cells1030372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J, et al. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Horvath B, Zsengeller Z, Zielonka J, Tanchian G, Holovac E, et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic Biol Med. 2012;52:497–506. doi: 10.1016/j.freeradbiomed.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munusamy S, MacMillan-Crow LA. Mitochondrial superoxide plays a crucial role in the development of mitochondrial dysfunction during high glucose exposure in rat renal proximal tubular cells. Free Radic Biol Med. 2009;46:1149–1157. doi: 10.1016/j.freeradbiomed.2009.01.022. [DOI] [PubMed] [Google Scholar]
- Murphy M, Godson C, Cannon S, Kato S, Mackenzie HS, Martin F, et al. Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J Biol Chem. 1999;274:5830–5834. doi: 10.1074/jbc.274.9.5830. [DOI] [PubMed] [Google Scholar]
- Murphy M, Docherty NG, Griffin B, Howlin J, McArdle E, McMahon R, et al. IHG-1 amplifies TGF-beta1 signaling and is increased in renal fibrosis. J Am Soc Nephrol. 2008;19:1672–1680. doi: 10.1681/ASN.2007101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Hickey F, Godson C. IHG-1 amplifies TGF-β1 signalling and mitochondrial biogenesis and is increased in diabetic kidney disease. Curr Opin Nephrol Hypertens. 2013;22:77–84. doi: 10.1097/MNH.0b013e32835b54b0. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208:417–420. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito T, Kuma A, Mizushima N. Differential contribution of insulin and amino acids to the mTORC1-autophagy pathway in the liver and muscle. J Biol Chem. 2013;288:21074–21081. doi: 10.1074/jbc.M113.456228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin d-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010a;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010b;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan DM, Zinman B, Cleary PA, Backlund JY, Genuth S, Miller R, et al. Modern-day clinical course of type 1 diabetes mellitus after 30 years' duration: the diabetes control and complications trial/epidemiology of diabetes interventions and complications and Pittsburgh epidemiology of diabetes complications experience (1983–2005) Arch Intern Med. 2009;169:1307–1316. doi: 10.1001/archinternmed.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya T, Perkovic V, de Galan BE, Zoungas S, Pillai A, Jardine M, et al. Albuminuria and kidney function independently predict cardiovascular and renal outcomes in diabetes. J Am Soc Nephrol. 2009;20:1813–1821. doi: 10.1681/ASN.2008121270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomiyama T, Tanaka Y, Piao L, Nagasaka K, Sakai K, Ogihara T, et al. The polymorphism of manganese superoxide dismutase is associated with diabetic nephropathy in Japanese type 2 diabetic patients. J Hum Genet. 2003;48:138–141. doi: 10.1007/s100380300021. [DOI] [PubMed] [Google Scholar]
- Norman JM, Cohen GM, Bampton ET. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy. 2010;6:1042–1056. doi: 10.4161/auto.6.8.13337. [DOI] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282:13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15:887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, et al. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol. 2007;178:1025–1038. doi: 10.1083/jcb.200611128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW, Blomgren K, Ethell DW, Koike M, Komatsu M, Prevette D, et al. Developing postmitotic mammalian neurons in vivo lacking Apaf-1 undergo programmed cell death by a caspase-independent, nonapoptotic pathway involving autophagy. J Neurosci. 2008;28:1490–1497. doi: 10.1523/JNEUROSCI.4575-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm F, Nordquist L. Renal oxidative stress, oxygenation, and hypertension. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1229–R1241. doi: 10.1152/ajpregu.00720.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm F, Friederich M, Carlsson PO, Hansell P, Teerlink T, Liss P. Reduced nitric oxide in diabetic kidneys due to increased hepatic arginine metabolism: implications for renomedullary oxygen availability. Am J Physiol Renal Physiol. 2008;294:F30–F37. doi: 10.1152/ajprenal.00166.2007. [DOI] [PubMed] [Google Scholar]
- Parajuli N, MacMillan-Crow LA. Role of reduced manganese superoxide dismutase in ischemia-reperfusion injury: a possible trigger for autophagy and mitochondrial biogenesis? Am J Physiol Renal Physiol. 2013;304:F257–F267. doi: 10.1152/ajprenal.00435.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci. 2010;123(Pt 4):619–626. doi: 10.1242/jcs.061481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parving HH, Smidt UM, Friisberg B, Bonnevie-Nielsen V, Andersen AR. A prospective study of glomerular filtration rate and arterial blood pressure in insulin-dependent diabetics with diabetic nephropathy. Diabetologia. 1981;20:457–461. doi: 10.1007/BF00253407. [DOI] [PubMed] [Google Scholar]
- Parving HH, Gall MA, Skott P, Jorgensen HE, Lokkegaard H, Jorgensen F, et al. Prevalence and causes of albuminuria in non-insulin-dependent diabetic patients. Kidney Int. 1992;41:758–762. doi: 10.1038/ki.1992.118. [DOI] [PubMed] [Google Scholar]
- Parving HH, Lewis JB, Ravid M, Remuzzi G, Hunsicker LG. Prevalence and risk factors for microalbuminuria in a referred cohort of type II diabetic patients: a global perspective. Kidney Int. 2006;69:2057–2063. doi: 10.1038/sj.ki.5000377. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Paula-Gomes S, Goncalves DA, Baviera AM, Zanon NM, Navegantes LC, Kettelhut IC. Insulin suppresses atrophy-and autophagy-related genes in heart tissue and cardiomyocytes through AKT/FOXO signaling. Horm Metab Res. 2013;45:849–855. doi: 10.1055/s-0033-1347209. [DOI] [PubMed] [Google Scholar]
- Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74:631–640. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS. Regression of microalbuminuria in type 1 diabetes. N Engl J Med. 2003;348:2285–2293. doi: 10.1056/NEJMoa021835. [DOI] [PubMed] [Google Scholar]
- Persson MF, Franzen S, Catrina SB, Dallner G, Hansell P, Brismar K, et al. Coenzyme Q10 prevents GDP-sensitive mitochondrial uncoupling, glomerular hyperfiltration and proteinuria in kidneys from db/db mice as a model of type 2 diabetes. Diabetologia. 2012;55:1535–1543. doi: 10.1007/s00125-012-2469-5. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Tian X, Liu J, Han Y, Graham AM, Simon MC, et al. Bnip3 and AIF cooperate to induce apoptosis and cavitation during epithelial morphogenesis. J Cell Biol. 2012;198:103–114. doi: 10.1083/jcb.201111063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinsay MN, Lee Y, Rikka S, Sayen MR, Molkentin JD, Gottlieb RA, et al. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J Mol Cell Cardiol. 2010;48:1146–1156. doi: 10.1016/j.yjmcc.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. 2010;142:590–600. doi: 10.1016/j.cell.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakovic A, Grunewald A, Kottwitz J, Bruggemann N, Pramstaller PP, Lohmann K, et al. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS ONE. 2011;6:e16746. doi: 10.1371/journal.pone.0016746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Rera M, Walker DW. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci U S A. 2013;110:8638–8643. doi: 10.1073/pnas.1216197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raparia K, Usman I, Kanwar YS. Renal morphologic lesions reminiscent of diabetic nephropathy. Arch Pathol Lab Med. 2013;137:351–359. doi: 10.5858/arpa.2012-0243-RA. [DOI] [PubMed] [Google Scholar]
- Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab. 2013;17:20–33. doi: 10.1016/j.cmet.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray R, Chen G, Vande Velde C, Cizeau J, Park JH, Reed JC, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275:1439–1448. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- Raza H, Prabu SK, Robin MA, Avadhani NG. Elevated mitochondrial cytochrome P450 2E1 and glutathione S-transferase A4-4 in streptozotocin-induced diabetic rats: tissue-specific variations and roles in oxidative stress. Diabetes. 2004;53:185–194. doi: 10.2337/diabetes.53.1.185. [DOI] [PubMed] [Google Scholar]
- Reid JL. Rilmenidine: a clinical overview. Am J Hypertens. 2000;13(6 Pt 2):106S–111S. doi: 10.1016/s0895-7061(00)00226-0. [DOI] [PubMed] [Google Scholar]
- Renna M, Jimenez-Sanchez M, Sarkar S, Rubinsztein DC. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem. 2010;285:11061–11067. doi: 10.1074/jbc.R109.072181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renna M, Bento CF, Fleming A, Menzies FM, Siddiqi FH, Ravikumar B, et al. IGF-1 receptor antagonism inhibits autophagy. Hum Mol Genet. 2013;22:4528–4544. doi: 10.1093/hmg/ddt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18:721–731. doi: 10.1038/cdd.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca MG, Mustata TG, Kinter MT, Ozdemir AM, Kern TS, Szweda LI, et al. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol. 2005;289:F420–F430. doi: 10.1152/ajprenal.00415.2004. [DOI] [PubMed] [Google Scholar]
- Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Hum Mol Genet. 2010;19:2144–2153. doi: 10.1093/hmg/ddq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozman-Pungercar J, Kopitar-Jerala N, Bogyo M, Turk D, Vasiljeva O, Stefe I, et al. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ. 2003;10:881–888. doi: 10.1038/sj.cdd.4401247. [DOI] [PubMed] [Google Scholar]
- Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44:698–709. doi: 10.1016/j.molcel.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M, Isono M, Isshiki K, Sugimoto T, Koya D, Kashiwagi A. Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochem Biophys Res Commun. 2006;340:296–301. doi: 10.1016/j.bbrc.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Sandebring A, Thomas KJ, Beilina A, van der Brug M, Cleland MM, Ahmad R, et al. Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS ONE. 2009;4:e5701. doi: 10.1371/journal.pone.0005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneurin-Berny D, Verdel A, Curtet S, Lemercier C, Garin J, Rousseaux S, et al. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21:8035–8044. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. 2013;20:21–30. doi: 10.1038/cdd.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7:189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- Sharma K, Karl B, Mathew AV, Gangoiti JA, Wassel CL, Saito R, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol. 2013;24:1901–1912. doi: 10.1681/ASN.2013020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda-Matsubayashi S, Matsumine H, Kobayashi T, Nakagawa-Hattori Y, Shimizu Y, Mizuno Y. Structural dimorphism in the mitochondrial targeting sequence in the human manganese superoxide dismutase gene. A predictive evidence for conformational change to influence mitochondrial transport and a study of allelic association in Parkinson's disease. Biochem Biophys Res Commun. 1996;226:561–565. doi: 10.1006/bbrc.1996.1394. [DOI] [PubMed] [Google Scholar]
- Singh DK, Winocour P, Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat Rev Endocrinol. 2011;7:176–184. doi: 10.1038/nrendo.2010.212. [DOI] [PubMed] [Google Scholar]
- Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12:537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Covington MD, Schnellmann RG. Loss of calpain 10 causes mitochondrial dysfunction during chronic hyperglycemia. Arch Biochem Biophys. 2012a;523:161–168. doi: 10.1016/j.abb.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Hartley RC, Cocheme HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012b;33:341–352. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Soltoff SP. ATP and the regulation of renal cell function. Annu Rev Physiol. 1986;48:9–31. doi: 10.1146/annurev.ph.48.030186.000301. [DOI] [PubMed] [Google Scholar]
- Sourris KC, Harcourt BE, Tang PH, Morley AL, Huynh K, Penfold SA, et al. Ubiquinone (coenzyme Q10) prevents renal mitochondrial dysfunction in an experimental model of type 2 diabetes. Free Radic Biol Med. 2012;52:716–723. doi: 10.1016/j.freeradbiomed.2011.11.017. [DOI] [PubMed] [Google Scholar]
- Steinke JM. The natural progression of kidney injury in young type 1 diabetic patients. Curr Diab Rep. 2009;9:473–479. doi: 10.1007/s11892-009-0077-7. [DOI] [PubMed] [Google Scholar]
- Sun F, Kanthasamy A, Anantharam V, Kanthasamy AG. Mitochondrial accumulation of polyubiquitinated proteins and differential regulation of apoptosis by polyubiquitination sites Lys-48 and-63. J Cell Mol Med. 2009;13(8B):1632–1643. doi: 10.1111/j.1582-4934.2009.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Xie P, Wada J, Kashihara N, Liu FY, Zhao Y, et al. Rap1b GTPase ameliorates glucose-induced mitochondrial dysfunction. J Am Soc Nephrol. 2008;19:2293–2301. doi: 10.1681/ASN.2008030336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton A, Khoury H, Prip-Buus C, Cepanec C, Pessayre D, Degoul F. The Ala16Val genetic dimorphism modulates the import of human manganese superoxide dismutase into rat liver mitochondria. Pharmacogenetics. 2003;13:145–157. doi: 10.1097/01.fpc.0000054067.64000.8f. [DOI] [PubMed] [Google Scholar]
- Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol. 2012;180:517–525. doi: 10.1016/j.ajpath.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Takebayashi S, Kaneda K. Mitochondrial derangement: possible initiator of microalbuminuria in NIDDM. J Diabet Complications. 1991;5:104–106. doi: 10.1016/0891-6632(91)90034-m. [DOI] [PubMed] [Google Scholar]
- Tan AL, Sourris KC, Harcourt BE, Thallas-Bonke V, Penfold S, Andrikopoulos S, et al. Disparate effects on renal and oxidative parameters following RAGE deletion, AGE accumulation inhibition, or dietary AGE control in experimental diabetic nephropathy. Am J Physiol Renal Physiol. 2010;298:F763–F770. doi: 10.1152/ajprenal.00591.2009. [DOI] [PubMed] [Google Scholar]
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–439. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay YC, Wang Y, Kairaitis L, Rangan GK, Zhang C, Harris DC. Can murine diabetic nephropathy be separated from superimposed acute renal failure? Kidney Int. 2005;68:391–398. doi: 10.1111/j.1523-1755.2005.00405.x. [DOI] [PubMed] [Google Scholar]
- Teutonico A, Schena PF, Di Paolo S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol. 2005;16:3128–3135. doi: 10.1681/ASN.2005050487. [DOI] [PubMed] [Google Scholar]
- Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, et al. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet. 2011;20:40–50. doi: 10.1093/hmg/ddq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MC, Weekes AJ, Broadley OJ, Cooper ME, Mathew TH. The burden of chronic kidney disease in Australian patients with type 2 diabetes (the NEFRON study) Med J Aust. 2006;185:140–144. doi: 10.5694/j.1326-5377.2006.tb00499.x. [DOI] [PubMed] [Google Scholar]
- Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, et al. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 2012;93:320–329. doi: 10.1093/cvr/cvr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710:197–211. doi: 10.1016/0005-2760(82)90150-3. [DOI] [PubMed] [Google Scholar]
- Ursini F, Heim S, Kiess M, Maiorino M, Roveri A, Wissing J, et al. Dual function of the selenoprotein PHGPx during sperm maturation. Science. 1999;285:1393–1396. doi: 10.1126/science.285.5432.1393. [DOI] [PubMed] [Google Scholar]
- Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1009–R1022. doi: 10.1152/ajpregu.00809.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol. 2013;304:F156–F167. doi: 10.1152/ajprenal.00409.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186–200. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Choi ME. Autophagy in kidney health and disease. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5363. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisiger RA, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J Biol Chem. 1973;248:4793–4796. [PubMed] [Google Scholar]
- Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting DR, Guariguata L, Weil C, Shaw J. IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res Clin Pract. 2011;94:311–321. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- Whitman IR, Feldman HI, Deo R. CKD and sudden cardiac death: epidemiology, mechanisms, and therapeutic approaches. J Am Soc Nephrol. 2012;23:1929–1939. doi: 10.1681/ASN.2012010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010;1:e18. doi: 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WH, Zhang MP, Zhang F, Liu F, Hu ZX, Hu QD, et al. The role of programmed cell death in streptozotocin-induced early diabetic nephropathy. J Endocrinol Invest. 2011;34:e296–e301. doi: 10.3275/7741. [DOI] [PubMed] [Google Scholar]
- Xu Y, Yang S, Huang J, Ruan S, Zheng Z, Lin J. Tgf-β1 induces autophagy and promotes apoptosis in renal tubular epithelial cells. Int J Mol Med. 2012;29:781–790. doi: 10.3892/ijmm.2012.911. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ruan S, Wu X, Chen H, Zheng K, Fu B. Autophagy and apoptosis in tubular cells following unilateral ureteral obstruction are associated with mitochondrial oxidative stress. Int J Mol Med. 2013;31:628–636. doi: 10.3892/ijmm.2013.1232. [DOI] [PubMed] [Google Scholar]
- Yadav A, Vallabu S, Arora S, Tandon P, Slahan D, Teichberg S, et al. ANG II promotes autophagy in podocytes. Am J Physiol Cell Physiol. 2010;299:C488–C496. doi: 10.1152/ajpcell.00424.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol. 2008;294:F777–F787. doi: 10.1152/ajprenal.00590.2007. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wang J, Qin L, Shou Z, Zhao J, Wang H, et al. Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am J Nephrol. 2007;27:495–502. doi: 10.1159/000106782. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010a;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010b;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Cao L, Kang R, Yang M, Wang Z, Peng Y, et al. UV irradiation resistance-associated gene suppresses apoptosis by interfering with BAX activation. EMBO Rep. 2011;12:727–734. doi: 10.1038/embor.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013;83:568–581. doi: 10.1038/ki.2012.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Shimoji M, Thomas B, Moore DJ, Yu SW, Marupudi NI, et al. Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum Mol Genet. 2005;14:2063–2073. doi: 10.1093/hmg/ddi211. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li L, Liu H, Borowitz JL, Isom GE. BNIP3 mediates cell death by different pathways following localization to endoplasmic reticulum and mitochondrion. FASEB J. 2009;23:3405–3414. doi: 10.1096/fj.08-124354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li L, Leavesley HW, Zhang X, Borowitz JL, Isom GE. Cyanide-induced apoptosis of dopaminergic cells is promoted by BNIP3 and Bax modulation of endoplasmic reticulum-mitochondrial Ca2+ levels. J Pharmacol Exp Ther. 2010;332:97–105. doi: 10.1124/jpet.109.159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Wu J, Wu R, Ma J, Du G, Jiao R, et al. DJ-1 promotes the proteasomal degradation of Fis1: implications of DJ-1 in neuronal protection. Biochem J. 2012;447:261–269. doi: 10.1042/BJ20120598. [DOI] [PubMed] [Google Scholar]
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288:1099–1113. doi: 10.1074/jbc.M112.399345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A. 2010;107:5018–5023. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziyadeh FN. Mediators of diabetic renal disease: the case for tgf-Beta as the major mediator. J Am Soc Nephrol. 2004;15(Suppl. 1):S55–S57. doi: 10.1097/01.asn.0000093460.24823.5b. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res. 2009;83:213–225. doi: 10.1093/cvr/cvp151. [DOI] [PMC free article] [PubMed] [Google Scholar]