

Abstract
BACKGROUND AND PURPOSE
Cardiolipin plays an important role in mitochondrial respiration and cardiolipin peroxidation is associated with age-related diseases. Hydrophobic interactions between cytochrome c and cardiolipin converts cytochrome c from an electron carrier to a peroxidase. In addition to cardiolipin peroxidation, this impedes electron flux and inhibits mitochondrial ATP synthesis. SS-31 (D-Arg-dimethylTyr-Lys-Phe-NH2) selectively binds to cardiolipin and inhibits cytochrome c peroxidase activity. Here, we examined whether SS-31 also protected the electron carrier function of cytochrome c.
EXPERIMENTAL APPROACH
Interactions of SS-31 with cardiolipin were studied using liposomes and bicelles containing phosphatidylcholine alone or with cardiolipin. Structural interactions were assessed by fluorescence spectroscopy, turbidity and nuclear magnetic resonance. Effects of cardiolipin on electron transfer kinetics of cytochrome c were determined by cytochrome c reduction in vitro and oxygen consumption using mitoplasts, frozen and fresh mitochondria.
KEY RESULTS
SS-31 interacted only with liposomes and bicelles containing cardiolipin in about 1:1 ratio. NMR studies demonstrated that the aromatic residues of SS-31 penetrated deep into cardiolipin-containing bilayers. SS-31 restored cytochrome c reduction and mitochondrial oxygen consumption in the presence of added cardiolipin. In fresh mitochondria, SS-31 increased state 3 respiration and efficiency of ATP synthesis.
CONCLUSIONS AND IMPLICATIONS
SS-31 selectively targeted cardiolipin and modulated its interaction with cytochrome c. SS-31 inhibited the cytochrome c/cardiolipin complex peroxidase activity while protecting its ability to serve as an electron carrier, thus optimizing mitochondrial electron transport and ATP synthesis. This novel class of cardiolipin therapeutics has the potential to restore mitochondrial bioenergetics for treatment of numerous age-related diseases.
LINKED ARTICLES
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: cardiolipin, cyt c, cardiolipin peroxidation, mitochondria, electron transport, ATP synthesis, mitochondria-targeted peptides, Szeto-Schiller peptides, SS peptides, SS-31, Bendavia®
Introduction
Living mammalian cells require a continuous input of energy, in the form of ATP, to support complex biological functions that are essential for both survival and interaction with the environment. The mitochondrial electron transport chain (ETC), coupled to ATP synthesis, is responsible for conversion of the chemical energy of sugars, amino acids and fatty acids into ATP, and decline in this mitochondrial function is thought to be the basis for aging and many complex diseases (Wallace, 2013a,2013b). In the conditions of bioenergetic deficiency, such as ischaemia and aging, protection of the ETC and ATP synthesis in affected cells is essential for normalizing structural and functional recovery.
Mitochondrial cardiolipin (CL) is known to be required for normal mitochondrial bioenergetics and its peroxidation and depletion are thought to contribute to age-related decline in mitochondrial function. The major functions of CL are (i) to support spatial organization of mitochondrial cristae, (ii) to create the proton trap necessary for sustaining the proton gradient and ATP synthesis by the F0F1 ATP synthase and (iii) to act as a scaffold for assembly of respiratory complexes and super-complexes to facilitate optimal electron transfer among the redox partners (Schlame and Ren, 2009; Sorice et al., 2009; Paradies et al., 2010). Many of the respiratory complexes require CL for optimal function (Böttinger et al., 2012; Gonzalvez et al., 2013). CL also plays an important role in anchoring cytochrome c (cyt c) to the inner mitochondrial membrane and facilitates electron transfer from Complex III to Complex IV (Rytömaa and Kinnunen, 1994; 1995).
Cyt c is the only non-integral protein of the ETC on the inner mitochondrial membrane. While coenzyme Q within the inner mitochondrial membrane facilitates electron transfer from complex I to complex III, cyt c is a soluble protein in the inner membrane space, which transfers electrons from complex III to complex IV. Cyt c mediates electron transfer via its hexa-coordinated haem iron, which switches between the reduced ferrous (Fe2+) and oxidized ferric (Fe3+) state. Met80 and His18 are the axial ligands of the haem iron in the native protein, and they are essential for stabilizing both its native conformation and its electron transfer function (Fisher et al., 1973; Hamada et al., 1993). To assist in electron transfer from complex III to complex IV, cyt c must be within close proximity of these complexes. CL provides an anionic platform for electrostatic interaction with the highly cationic cyt c (9 + net charge) so that it is loosely attached to the ETC. The electrostatic interaction of cyt c with CL is supported by high levels of ATP under normal physiological conditions (Sinibaldi et al., 2011; Snider et al., 2013). However, when ATP concentration decreases in pathological conditions such as ischaemia, cyt c becomes tightly associated with CL via hydrophobic interactions. This cyt c/CL complex results in unfolding of cyt c and disrupts the iron-Met80 coordination, thus interfering with π-π* interaction within the haem environment, and converts cyt c from an electron carrier into a peroxidase/oxygenase (Santucci and Ascoli, 1997; Kagan et al., 2004; Sinibaldi et al., 2008; Hanske et al., 2012), which cannot participate in electron transfer (Basova et al., 2007). As a peroxidase, cyt c serves to catalyse the oxidation of CL, leading to its degradation and depletion from the inner mitochondrial membrane. Thus cyt c is Janus-faced with two contrasting functions, and the predominant activity is determined by its interaction with CL.
Compounds that can inhibit peroxidase activity and preserve electron carrier function in the cyt c/CL complex may potentially be beneficial for the ischaemia and other age-related diseases. This has led to limited attempts in designing molecules that can inhibit the peroxidase activity of cyt c. Most efforts rely on reducing mitochondrial reactive oxygen species to inhibit CL peroxidation (Bayir et al., 2007). This approach is not as effective as directly inhibiting the catalytic peroxidase activity of cyt c. Recently, alternative sacrificial substrates for cyt c peroxidase, such as alkyl-hydroxylamine and imidazole derivatives of fatty acids were designed to block the catalytic site of cyt c (Kagan et al., 2009). It is not clear if disruption of the haem iron might interfere with the electron transfer function of cyt c. Furthermore, the triphenylphosphonium ion (TPP+) was used to deliver these inhibitors to mitochondria, and TPP+ has recently been shown to inhibit mitochondrial bioenergetics (Fink et al., 2012; Reily et al., 2013). Yet, another approach used a poorly peroxidizable TPP+-conjugated octadecanoic acid to remodel the endogenous pool of CL, but it is not known how this modified CL may affect the rest of the ETC (Tyurina et al., 2012).
We recently reported the discovery of a compound that selectively targets the cyt c/CL complex and inhibits its peroxidase activity (Birk et al., 2013). The Szeto-Schiller peptides (SS peptides) represent the only known class of cell-permeable compounds that concentrate specifically in the inner mitochondrial membrane (Zhao et al., 2004). SS-31 (D-Arg-dimethylTyr-Lys-Phe-NH2) has an alternating aromatic-cationic motif and a 3 + net charge (Figure 1A). SS-31 selectively binds to CL via electrostatic interaction and penetrates into the cyt c/CL complex to protect the Met80-haem coordination, thus inhibiting the peroxidase activity of cyt c (Birk et al., 2013).
Figure 1.

Structures of SS-31, [ald]SS-31, and various phospholipids. (A) SS-31. (B) [ald]SS-31. (C) TOCL. (D) TLCL. (E) POPC. (F) DHPC.
In this study, we provide further structural evidence for the interaction of SS-31 with CL in liposomes, bicelles and mitoplasts. Besides inhibiting cyt c peroxidase activity, we now show that SS-31 can also improve electron transfer through the cyt c/CL complex and promote mitochondrial ATP synthesis. These new findings help us understand the remarkable efficacy that SS-31 has shown in diverse animal disease models associated with bioenergetic failure, including ischaemia-reperfusion injury, heart failure, skeletal muscle atrophy and neurodegenerative diseases (Yang et al., 2009; Min et al., 2011; Szeto and Schiller, 2014; Kloner et al., 2012; Sloan et al., 2012; Dai et al., 2013; Talbert et al., 2013).
Methods
Preparation of liposomes and bicelles
Lipids in chloroform were combined in 12 × 75 mm glass tubes in the ratios for forming liposomes and bicelles. To control for the hygroscopic nature of some lipids we mostly purchased them directly in chloroform. When purchased in powder form, we weighed the sealed container before and after removing the entire lipid content to determine the actual mass of lipid used for bicelles formulation. Therefore, our lipid concentrations could have an error of about 5%, which is unlikely to affect the Q value of 0.15–0.3 for our bicelles. We observed no difference in results from different bicelle preparations. Liposomes contained either 150 μM TOCL : 150 μM POPC or 300 μM POPC. Bicelles contained either 150 μM TOCL : 1500 μM POPC : 4500 μM DHPC or 1500 μM POPC : 4500 μM DHPC. The solvent was evaporated slowly under N2 gas for 30 min (Techair, Naugatuck, CT, USA), and the resulting lipid film was rehydrated in an aqueous solution of 20 mM HEPES pH 7.4 or deuterated water (D2O; for NMR studies). The resulting multilamellar vesicles were vortexed lightly, and sized into small unilamellar vesicles by 25 min of heated bath sonication (Solid State Ultrasonic FS-9, 40 kHz, Fisher Scientific, Waltham, MA, USA). All sonicated liposomes were cooled to ambient temperature before use. Bicelles were not sonicated. All bicelles used for NMR studies, which were hydrated in D2O, were buffered with 5 mM H2PO4 and titrated with HCL to a final pH of 6.5. No peak due to residual chloroform was observed.
Preparation of rat kidney mitochondria
All animal care and experimental protocols were in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and received prior approval by the Cornell University Institutional Animal Care and Use Committee. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 10 animals were used in the experiments described here. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Mitochondria were isolated from the kidneys of 250–300 g Sprague-Dawley rats (Charles River Laboratories International, Inc., Wilmington, MA, USA). Rats were housed in a light-controlled room under a 12 h light : dark cycle and given free access to water and standard rat chow. Excised kidneys were cut and incubated for 10 min in an ice-cold wash buffer consisting of 200 mM mannitol, 10 mM sucrose, 5 mM HEPES, 1 g·L−1 fatty-acid-free BSA and KOH balanced to pH 7.4. Samples were washed twice in isolation buffer (wash buffer with 1 mM EGTA), homogenized for 3 min, and then centrifuged in 20 mL of isolation buffer at 900× g for 10 min. The white fatty acid layer was removed and the pellet was discarded. The supernatant was centrifuged at 11 000× g for 10 min, and the resulting pellet was resuspended in 800 μL of wash buffer. Protein content of the mitochondrial preparation was measured by the BCA assay (Thermo Scientific, Rockfold, IL, USA). Mitochondria were frozen immediately at −80°C until use. The integrity of mitochondria was demonstrated by observing no effect of exogenous cyt c on mitochondrial respiration. As expected, mitochondria that been frozen had uncoupled oxygen consumption and ADP did not increase succinate-induced oxygen consumption (data not shown). Also, addition of exogenous cyt c promoted respiration of un-frozen mitochondria, without affecting fresh mitochondria, suggesting that the outer membrane of frozen mitochondria is ruptured.
Preparation of cyt c-deficient mitoplasts
The outer membranes of fresh or once-frozen mitochondria were removed by 45 min exposure to 3.3 mg·mL−1 digitonin on ice. To remove electrostatically bound cyt c, 300 mM KCl was added and the mixture was centrifuged for 30 min at 14 000× g. The supernatant was discarded, and the pellet was washed with 300 mM KCl and centrifuged twice more. The final pellet was re-dissolved in wash buffer and stored on ice until use. After removal of cyt c, mitoplasts were subjected to respiration experiments with succinate, and only those preparations that showed an increased mitochondrial respiration by fivefold to sixfold upon addition of exogenous cyt c were used. Protein content of the mitoplast preparation was measured by the BCA assay (Thermo Scientific, Rockfold, IL, USA).
Measurement of fluorescence of [ald]SS-31 in the presence of CL liposomes, bicelles and mitoplasts
The interaction of [ald]SS-31 with CL in liposomes, bicelles or mitoplasts was observed through the intrinsic fluorescence of [ald] at λex/λem = 360 nm/535 nm (SPECTRAmax GeminiXPS, Molecular Devices, Sunnyvale, CA, USA). Liposome or bicelle solutions were added to 1 μM [ald]SS-31 in 20 mM HEPES pH 7.4 such that the CL concentration in the mixtures was kept at 30 μM. Mitoplasts were prepared as described above and used at a final concentration of ∼40 μg·mL−1.
Measurement of turbidity of SS-31 and CL complex
Turbidity was measured in liposomes and bicelles via right-angle scattering at λ = 350 nm (Hitachi F-4500 Fluorescent Spectrophotometer, Tokyo, Japan). The baseline scattering of POPC-DHPC bicelles was significantly higher than that of TOCL-POPC-DHPC bicelles, suggesting that TOCL itself may decrease the overall size of the bicelles (data not shown).
NMR analysis of SS-31 and CL interaction
NMR data were collected on a Bruker Avance III 500 MHz NMR spectrometer (Bruker Corporation, Billerica, MA, USA) equipped with a 5 mm broadband equipped with z-gradient pulse field gradients. The variable temperature unit was calibrated using a thermocouple placed inside a filled NMR tube within the probe. The chemical shifts were referenced with 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) as an internal reference. Samples of SS-31 examined at 75 μM to 10 mM concentrations were collected in 90% H2O 10% D2O or 100% D2O. 2D COSY, TOCSY, 13C-HSQC and 13C-HMBC experiments were acquired to verify chemical shift assignments in peptide and lipid resonances under the conditions used for these studies. SS-31 lipid-binding experiments were carried out using anisotropic bicelles based on the ratio of long-and short-chain phospholipids (Whiles et al., 2002). Bicelles contained either 150 μM TOCL : 1500 μM POPC : 4500 μM DHPC or 1500 μM POPC : 4500 μM DHPC in 5 mM sodium phosphate adjusted to pH 6.5 (uncorrected for isotope shifts) using 2HCl or NaO2H. Bicelles are small lipid bilayers that form disc-like particles in solution. They are composed of long-chain lipids, which form the planar surface surrounded by short-chain lipids coating the edges (Figure 2B). The small size of these bicelles allows proteins/peptides to be analysed by solution NMR, providing good spectral characteristics (∼21 kDa for POPC/DHPC q = 0.15 to 0.3; Prosser et al., 2006). Importantly, even small unilamellar liposomes are unsuitable for solution NMR due to their large diameter (20–100 nm). The POPC/DHPC bicelles have been demonstrated to best approximate biological lipids in membrane protein crystallography studies and NMR studies (Prosser et al., 2006). NMR data were processed and analysed using Topspin 2.1 (Bruker Corporation) and MestReNova (Mestrelab Research S.L., Santiago de Compostela, Spain).
Figure 2.
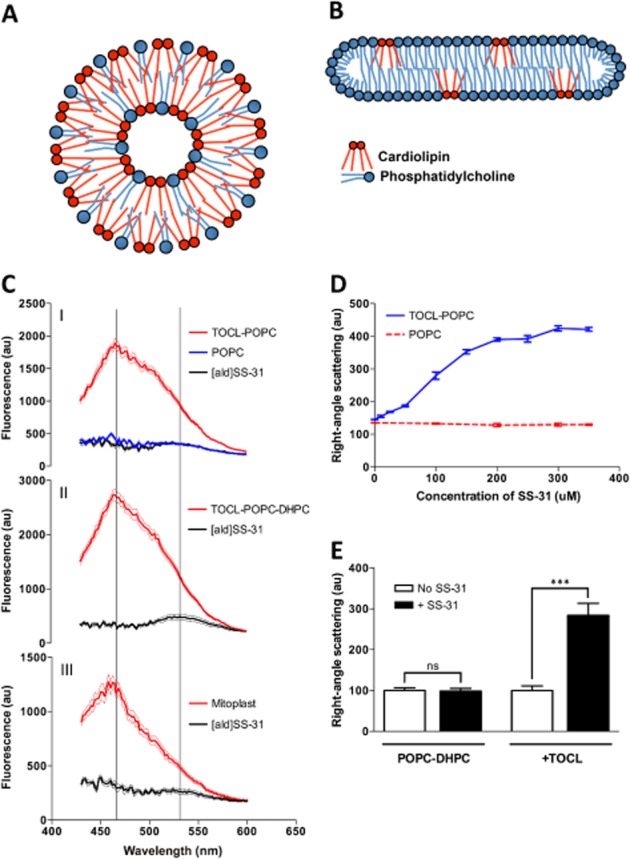
Selective interaction of SS-31 and [ald]SS-31 with CL-containing bilayer membranes and the mitochondrial inner membrane. (A) Diagram of the cross-section through a TOCL-POPC liposome. (B) Diagram of the cross-section through a CL-POPC-DHPC bicelle. (C) The fluorescence emission spectra (in arbitrary units; au) of [ald]SS-31 alone and in the presence of POPC liposomes (upper panel, I), TOCL-POPC liposomes (upper panel, I), TOCL-POPC-DHPC bicelles (middle panel, II), or cyt c-depleted mitoplasts (bottom panel, III). Two lines correspond to 535 and 465 nm peaks for [ald]SS-31 alone and in the presence of CL-containing bilayers respectively. (D) SS-31 dose dependently increases turbidity of TOCL-POPC liposomes, but not POPC liposomes. (E) SS-31 increases the turbidity of CL-containing bicelles, but not those containing only PC. All data are shown as means ± SEM, n = 4–6. ***P< 0.001, significant effects of SS-31; Student's t-test.
Reduction of cyt c in the presence of SS-31 and CL
Reduction of 20 μM horse heart cyt c by 50 μM ascorbate or 500 μM glutathione was measured by observing the changes in cyt c absorbance at 550 and 570 nm (SPECTRAmax 190, Molecular Devices). For CL-containing reactions, cyt c was pre-incubated with TOCL in the presence or absence of SS-31 for 1 min. The concentration of exogenous CL used was based on the concentration required to inhibit O2 consumption by 90% (IC90). Afterwards, ascorbate or glutathione was added and absorbance was read. The rate of reduction was calculated based on the slope of the change in absorbance intensity at 550 nm over 570 nm.
Oxygen consumption in mitochondria and mitoplasts
Oxygen consumption was measured as described previously (Birk et al., 2013). Briefly, mitochondria (40 μg protein) were incubated in 1 mL of respiration buffer (composition 3.25 mM potassium dihydrogen phosphate, 320 mM sucrose, 7.5 mM HEPES and 0.5 mM EGTA, balanced to pH 7.4 with KOH and HCl). The mitochondria were equilibrated with ADP (400 μM, all final concentrations) representing state 2, at 30°C for 1 min, after which glutamate (500 μM)/malate (500 μM) or succinate (500 μM) was added to initiate state 3 respiration. A TMPD (N,N,N′,N′-tetramethyl-p-phenylenediamine) (3 μM)/ascorbate (500 μM) system was used to directly reduce cyt c in intact mitochondria. Mitochondrial respiration was allowed to proceed into state 4.
Oxygen consumption in mitoplasts was measured with 40 μg of mitoplast protein in 1 mL of 20 mM HEPES buffer, pH 7.4, after initiating respiration with TMPD (250 μM)/ascorbate (5 mM) in the presence of antimycin (2 μM) to block complex III. SS-31 was added to the mitoplasts before initiating respiration. After initial low respiration in mitoplasts, exogenous horse heart cyt c (400 nM) was added to promote respiration. The concentration of exogenous CL used was based on the concentration required to inhibit O2 consumption by 90% (IC90). The concentration of SS-31 was based on our previously published in vivo pharmacokinetic data, in which the maximal plasma concentration after sc injection of 2.5 mg·kg−1 is ∼10–15 μM (Szeto and Schiller, 2014). SS-31 is distributed to all tissues with the highest concentration in kidneys, where the concentration is 3.5 times higher than plasma concentration (Birk et al., 2013). As SS-31 concentrates in mitochondria ∼5000-fold (Zhao et al., 2005), it is certainly feasible to achieve the levels of SS-31 (10–100 μM) used in these studies in mitochondria.
Measurement of ATP synthesis during respiration of isolated intact mitochondria
In accordance with previously published methodology regarding determination of ATP synthesis in isolated mitochondria (Lee et al., 1996), after 1 min of mitochondrial respiration in state 3 with succinate (500 μM), the reaction was stopped and centrifuged at 18 000× g at 4°C for 10 min and supernatants were neutralized with K2CO3. Protein content was determined using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA). ATP was measured by HPLC according to published methods (Vives-Bauza et al., 2007). The equipment used included a Perkin Elmer (Norwalk, CT, USA) M-250 binary LC pump, a Waters (Milford, MA, USA) 717 plus autosampler, a Waters 490 programmable multiwavelength UV detector, and an ESA (Chelmsford, MA, USA) 501 chromatography data process system. External standards were prepared in 0.4 M perchloric acid, neutralized and treated in exactly the same way as the mitochondrial samples. 50 μL of prepared sample or standard mixture were auto-injected and monitored by UV at 260 nm from 15 to 45 min. Peaks for phosphorylated nucleotides were identified by their retention times. AMP formation was not detected.
Data analysis
All results are expressed as mean ± SEM. Statistical analyses were carried out using one-way anova analysis with multiple comparisons or Student's t-test (GraphPad Software, Inc., San Diego, CA, USA).
Materials
SS-31 was provided by Stealth Peptides Inc. (Newton Centre, MA, USA). Aladan [ald] was synthesized from commercially available 6-methoxy-2-acetonaphthone, as described by Birk et al., 2013). D-Arg-2′6'dimethyl-Tyr-Lys-Ald-NH2 ([ald]SS-31) was synthesized by Dalton Pharma Services (Toronto, Ontario, Canada). 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC) and 1′,3′-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol (TOCL) were obtained from Avanti Polar Lipids Inc. (Alabaster, AL, USA). CL from bovine heart, containing more than 80% of tetra-linoleoyl CL (TLCL) and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Results
[ald]SS-31 interacts selectively with CL in liposomes, bicelles and mitoplasts
Previously, we demonstrated that the SS-31 analogue, [ald]SS-31 (Figure 1B), interacts selectively with CL-inverted micelles (Birk et al., 2013). To investigate the interaction of SS-31 and [ald]SS-31 with CL in biologically relevant bilayer membranes, we used CL-containing liposomes (50 mole % TOCL; Figure 2A) and bicelles (2.5 mole % TOCL) (Figure 2B). Importantly, low abundance of CL in bicelles increases the stringency of SS-31 and CL interaction.
[ald] undergoes a blue shift and a strong increase in fluorescence intensity when it moves from aqueous solution to a more hydrophobic environment (Cohen et al., 2002). [ald] alone was demonstrated to have no interaction with CL (Birk et al., 2013). [ald]SS-31 showed a blue shift from 535 to 465 nm and increased intensity in the presence of liposomes containing TOCL and POPC (1:1), but not POPC alone (Figure 2C, I), suggesting that [ald]SS-31 interacts with the hydrophobic environment unique to CL-containing membranes. A similar blue shift was seen in TOCL-containing bicelles (Figure 2C, II). Interestingly, the same fluorescence pattern was also observed in cyt c-deficient mitoplasts isolated from rat kidney mitochondria, which consist of only mitochondrial inner-membrane encapsulating matrix (Figure 2C, III). Therefore, these data suggest that [ald]SS-31 can interact with the endogenous CL of the mitochondrial inner membrane. Given that these results mimic those of SS-31 and inverted micelles of CL (Birk et al., 2013), it is most likely that SS-31 interacts with the hydrophobic moieties of CL within those model membranes.
SS-31 changes turbidity in the presence of CL-containing membranes vesicles
With strong evidence of [ald]SS-31 interacting with CL-containing membranes, it next became important to validate the interaction of SS-31 itself with CL. We found that SS-31 selectively increased the turbidity of CL-containing liposome solutions, both by visual observation and right-angle scattering. Right-angle scattering, which indicates an increase in turbidity, can be correlated with increased vesicular volume in homogenous solutions (Matsuzaki et al., 2000). In our context, an increase in turbidity clearly indicates interaction of SS-31 with the liposome or bicelle containing CL. No significant turbidity changes were observed from any liposomes or bicelles alone, or from SS-31 alone (Figure 2D and E). Turbidity increased dose dependently when SS-31 was added to TOCL-POPC liposomes with an EC50 of ≈109 ± 9 μM, which is about a 1:1.5 ratio of SS-31 to CL (Figure 2D). Similarly, turbidity of TOCL-POPC-DHPC bicelles increased greatly upon addition of SS-31 (Figure 2E).
NMR identification of specific peptide interactions with CL-containing bicelles
Since the aromatic [ald] of [ald]SS-31 interacts with the hydrophobic environment of CL-containing bicelles, it can reasonably be predicted that the dimethyl-tyrosine and phenylalanine rings of SS-31 might also interact with the hydrophobic environment of CL. To further test the specificity of SS-31 binding and identify specific side chains in contact with CL, 1D 1H NMR spectra were collected for SS-31 alone and in TOCL-POPC-DHPC bicelles or POPC-DHPC bicelles. The assignments for free SS-31 were determined using a combination of 1D-and 2D-NMR (Figure 3). Mixed with POPC-DHPC bicelles, SS-31 displays sharp resonances that match those observed in solution (Figure 3) indicating an absence of lipid interaction. However, in the presence of a 1:1 ratio of peptide : CL in TOCL-POPC-DHPC bicelles, the side chain resonances of dimethyl-tyrosine (6.55 ppm) and phenylalanine (7.2–7.5 ppm) are substantially altered. The dimethyl-tyrosine peak at 2.20 ppm was also changed in the presence of TOCL (data not shown). In the presence of CL, the phenylalanine aromatic signals coalesce into a broad hump, while the dimethyl-tyrosine aromatic ring and methyl protons (6.55 and 2.20 ppm, respectively) change in both chemical shift and increased linewidths (Figure 3), indicating significant environmental changes. Titration with increased concentrations of peptide begins to resolve the sharper phenylalanine resonances and narrow the SS-31 linewidths (data not shown), indicating the presence of rapid chemical exchange averaging. These results are in basic agreement with the fluorescence shift of [ald]SS-31, suggesting a model in which the aromatic residues of SS-31 dip into the hydrophobic interior of the membrane, specifically at CL-enriched sites, as depicted in the diagram in Figure 6A.
Figure 3.
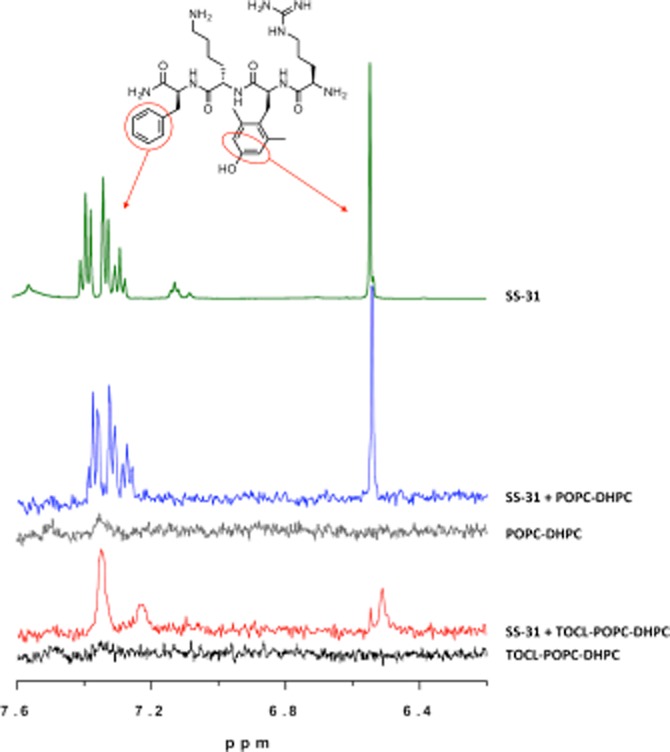
Aromatic amino acids of SS-31 interact with CL. Representative 500 MHz 1H-NMR of the aromatic region (7.6-6.2 ppm). From top, as indicated: (i) SS-31 aromatic protons of phenylalanine (7.3 ppm) and dimethyl-tyrosine (6.5 ppm). (ii) POPC-DHPC bicelles do not substantially affect aromatic proton peaks; (iii) POPC does not contribute to the spectrum in the aromatic region. (iv) TOCL-POPC-DHPC bicelles remodel and broaden the aromatic protons of SS-31. (v) TOCL-POPC-DHPC bicelles themselves contribute no peaks in this range.
Figure 6.
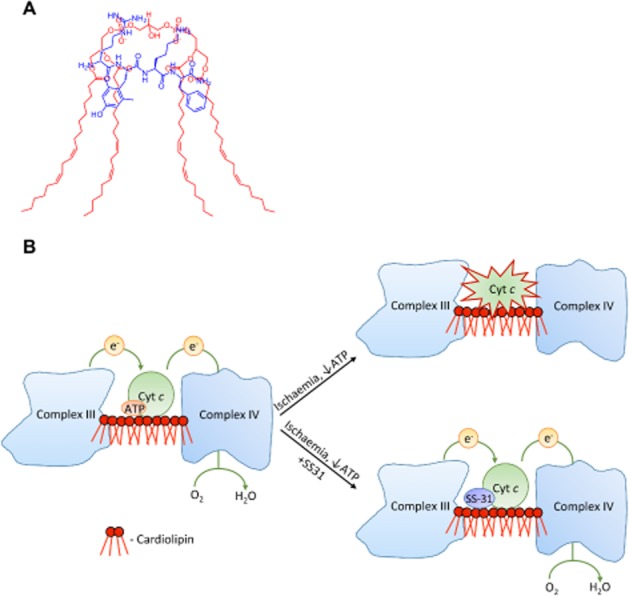
(A) Proposed model of the 1:1 interaction of SS-31 with CL shows favourable overlap of opposite electrostatic charges and hydrophobic regions. (B) Schematic diagram of mitochondrial dysfunction during early post-ischaemia. Unfolding of cyt c due to hydrophobic interaction with CL on the outer leaflet of the inner membrane prevents electron transport through complex III and IV. SS-31 localizes to CL, stabilizes cyt c in its folded conformation and restores electron transport.
SS-31 preserves cytochrome c reduction and electron transport in the presence of CL
SS-31 alone had no effect on the reduction of ferric cyt c by either glutathione or ascorbate. As previously reported, CL inhibited cyt c reduction by glutathione and ascorbate (P < 0.0001; Sinibaldi et al., 2011; Snider et al., 2013). Addition of SS-31 dose dependently abolished inhibitory effects of CL (P < 0.0001; Figure 4A).
Figure 4.

SS-31 promotes electron transfer through the cyt c-CL complex. (A) SS-31 promotes reduction of cyt c in the presence of CL. Dotted line represents reduction (%) of cyt c with either GSH or ascorbate. 100 μM SS-31 does not promote reduction of cyt c in its native conformation, without CL. In the presence of CL, SS-31 dose dependently rescues reduction of cyt c. **P<0.01. ***P<0.001, significantly different from ‘no CL’ values; one-way ANOVA with Dunnett's multiple comparison test. (B) SS-31 promotes oxygen consumption in mitoplasts where cyt c is inhibited by excess CL. SS-31 did not affect TMPD/ascorbate-induced oxygen consumption in cyt c-depleted mitoplasts, in the presence or absence of exogenous cyt c. When exogenous cyt c-activated oxygen consumption was inhibited by CL, SS-31 completely restored respiration. All data are shown as means ± SEM, n = 4–8. ***P<0.001, significant effects of SS-31 on respiration in mitoplasts; Student's t-test.
To investigate if SS-31 promotes electron transfer through the cyt c-CL complex to complex IV, we measured TMPD-induced respiration in cyt c-depleted mitoplasts in the presence of exogenous free cyt c and or exogenously formed cyt c-CL complex. In this system, electron flow from complex III to cyt c is prevented by antimycin, allowing us to focus on the role of electron transfer through exogenous cyt c or cyt c-CL complex to complex IV, which is measured by the oxygen consumption at complex IV. TMPD, which is selective to cyt c, can have minor stimulatory effects on the cytochromes at complex IV, which is detected as a small increase in the rate of oxygen consumption (Figure 4B). Addition of 400 nM exogenous horse heart cyt c increased TMPD-induced oxygen consumption by threefold to fourfold, and this was inhibited by addition of CL (Figure 4B). The results show that SS-31 had no effect on respiration of cyt c-depleted mitoplasts alone or in the presence of exogenous cyt c, which is consistent with cyt c reduction data (Figure 4A). Blocking exogenous cyt c-dependent oxygen consumption of mitoplasts with CL was fully prevented by treatment with SS-31 (P < 0.001), demonstrating that SS-31 promotes electron transfer though the cyt c-CL complex.
SS-31 increases oxygen consumption in once-frozen and fresh mitochondria, and promotes ATP production in fresh mitochondria
To study the effect of SS-31 on electron transport in mitochondria without influences of membrane potential, we used oxygen consumption in once-frozen mitochondria, where the outer membrane is ruptured, making it impossible to maintain a proton gradient across the inner membrane (Petrenko et al., 1982; Schober et al., 2007). SS-31 increased mitochondrial oxygen consumption in a dose-dependent manner (Figure 5A). We next asked whether the inhibitory function of the endogenous cyt c/CL complex plays a role in coupled respiration and ATP production in freshly isolated mitochondria, and whether SS-31 can overcome this inhibition. Indeed, addition of SS-31 to isolated mitochondria significantly and dose-dependently increased mitochondrial state 3 (exogenous substrate and ADP) oxygen consumption regardless of whether substrates for complex I (glutamate-malate; Figure 5B) or complex II (succinate; Figure 5C) were used. Oxygen consumption was also increased when cyt c was directly reduced by TMPD/ascorbate, supporting the idea that SS-31 regulates the ETC at the level of endogenous cyt c (Figure 5D). It should be mentioned that SS-31 had no effect on state 4 respiration, suggesting that SS-31 does not promote electron transfer by uncoupling mitochondria. Furthermore, the increase in state 3 respiration was actually coupled to enhanced ATP production (Figure 5E). Importantly, the calculated P/O ratio demonstrates that SS-31 promotes efficiency of ATP synthesis, in isolated mitochondria (Figure 5F).
Figure 5.
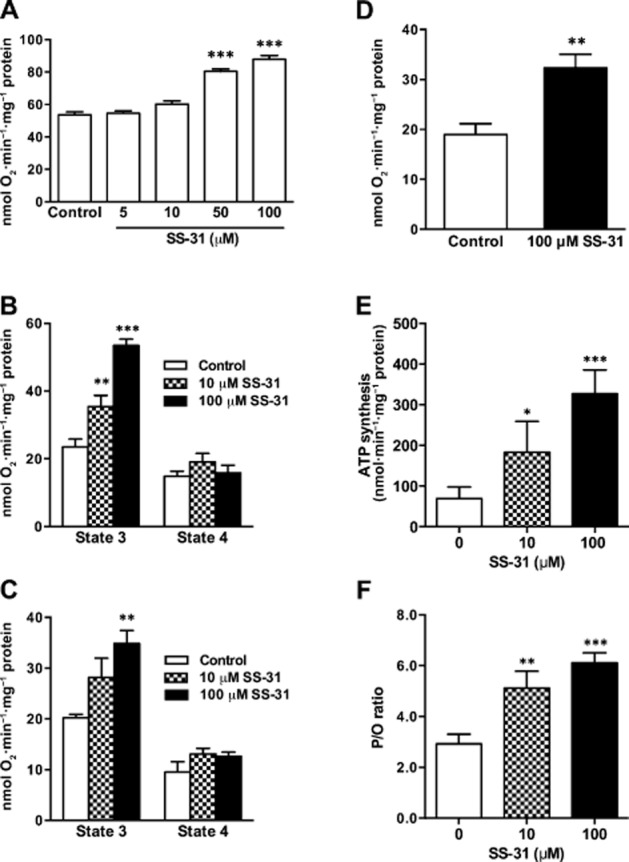
SS peptides promote mitochondrial oxygen consumption and ATP synthesis. (A) Electron flux was measured as oxygen consumption in frozen mitochondria, where SS-31 dose dependently increased oxygen consumption. (B) SS-31 dose dependently increases O2 consumption during state 3 respiration in the presence of complex I substrates (glutamate/malate). (C) SS-31 dose dependently increases oxygen consumption during state 3 respiration in the presence of complex II substrate (succinate). (D) SS-31 promotes oxygen consumption after direct reduction of cyt c by TMPD/ascorbate. (E) SS-31 dose dependently promotes ATP synthesis during state 3 respiration in the presence of succinate. (F) SS-31 dose dependently increases P/O ratio during state 3 respiration in the presence of complex II substrate (succinate). Data are shown as means ± SEM, n = 5–10. * P< 0.05, ** P< 0.01, *** P<0,001, significantly different from control; one-way anova with Dunnett's multiple comparison test (A, B, C, E) or Student's t-test (D).
Discussion
Earlier studies have shown that the SS peptides selectively partition to the inner mitochondrial membrane and they can interact with CL micelles (Zhao et al., 2004; Birk et al., 2013). Here, we provide further evidence that SS-31 can selectively interact with lipid membranes containing various amounts of CL. Studies with TOCL/POPC/DHPC bicelles, where CL is present only in a 1:40 ratio, revealed that [ald]SS-31 can still cause a threefold to fivefold increase in fluorescence intensity and a large blue shift from 535 to 465 nm. Importantly, the same blue shift to 465 nm and increased fluorescence intensity were observed when mitoplasts were added to [ald]SS-31, suggesting that the peptide recognizes endogenous mitochondrial CL. Our results provide the first evidence that [ald]SS-31 interacts selectively with CL in the inner mitochondrial membrane and inserts its aromatic residues into the hydrophobic environment of CL. Turbidity studies supported our hypothesis that SS-31 interacted selectively with CL in both liposomes and bicelles, at the 1:1.5 ratio of SS-31 to CL, which suggests that SS-31 behaved similarly to [ald]SS-31 (Birk et al., 2013).
We further used NMR to investigate the structural interaction of SS-31 with CL. NMR showed chemical shifts and smoothing of both the dimethyl-tyrosine and phenylalanine peaks in SS-31 only in bicelles containing CL. Importantly, SS-31, added in a 1:1 ratio with CL, had to be very selective to recognize this phospholipid since CL constitutes only 2.5% of the overall phospholipids in the bicelles. Our data suggest that the two aromatic residues of SS-31 are embedded deep into CL, allowing 1:1 interaction, as illustrated in Figure 6A.
As SS-31 interacts with CL on the inner mitochondrial membrane, it was important to ascertain whether this interaction might affect cyt c/CL interaction. In our previous publication, we demonstrated that SS-31 can specifically target the cyt c/CL complex and penetrate deep into the haem environment. Addition of cyt c to [ald]SS-31 and CL resulted in an immediate and dramatic quenching of the fluorescent signal of [ald]SS-31, suggesting that the [ald]SS-31 is residing within angstroms of the haem, which is a large resonance acceptor (Birk et al., 2013). Interestingly, addition of cyt c to [ald]SS-31 alone did not result in quenching, suggesting that SS-31 does not interfere with cyt c structure or cause unfolding. Structural studies further showed that SS-31 preserves the stability of the iron-Met80 coordination in cyt c/CL complex and, as a result, inhibits cyt c peroxidase activity in vitro and in mitochondrial preparations (Birk et al., 2013).
In addition to preserving the iron-Met80 bond, SS-31 provides additional aromatic groups that may also help promote π-π* interaction and prevent the effect of CL on both oxidation and reduction of the haem iron. Indeed, our results show that SS-31 can promote reduction of cyt c in complex with CL by either GSH or ascorbate. Importantly, SS-31 does not increase reduction of cyt c in its native conformation, suggesting that the peptide recognizes only unfolded cyt c in its complex with CL. Using once-frozen mitochondria, where electron flow is uncoupled from ATP synthesis, we obtained evidence that SS-31 can increase electron flux in the respiratory chain. Furthermore, SS-31 increased state 3 respiration in freshly isolated mitochondria with either complex I or complex II substrates, or by direct reduction of cyt c with TMPD, suggesting that SS-31 can optimize electron flux in the respiratory chain by improving electron transfer through the cyt c/CL complex. The ability of SS-31 to promote electron flux and reduce electron leak is supported by previous findings that SS-31 can reduce mitochondrial superoxide flashes (Ma et al., 2011) and spontaneous mitochondrial H2O2 emission (Zhao et al., 2004; Szeto, 2008b).
In early post-ischaemia, the hydrophobic interaction between CL and cyt c is enhanced by low ATP concentration (Sinibaldi et al., 2011; Snider et al., 2013) and this would further inhibit mitochondrial respiration at a time when ATP synthesis is necessary (Figure 6B). Both once-frozen mitochondria and isolated mitochondria systems have low levels of ATP (5–20 μM), and SS-31 was able to increase oxygen consumption in a dose-dependent manner in both systems. Because the cyt c/CL complex is likely to be present already in frozen and fresh mitochondria, our experiments also show that SS-31 can reverse the compromised function of CL-modified cyt c (Figure 6B). This is supported by our structural studies using circular dichroism (Birk et al., 2013).
In early post-ischaemic conditions, efficient coupling of oxygen consumption to ATP synthesis is necessary to minimize cell death and promote organ recovery. Our results showed that SS-31-mediated increase in electron flux was accompanied by an increase in ATP production. Furthermore, SS-31 increased P/O ratio in isolated kidney mitochondria from ∼3 (Bode et al., 1992) to 5–6, suggesting that SS-31 was capable of increasing coupled respiration. Our results are consistent with previous reports in which SS-31 significantly increased P/O ratio and rate of ATP synthesis, while decreasing ROS production, in aged animals or in pathological conditions(Min et al., 2011; Dai et al., 2013; Siegel et al., 2013; Talbert et al., 2013). Importantly, SS-31 had no effect on mitochondrial morphology, mitochondrial proteome, respiration, ATP synthesis or ROS production in young healthy animals. This has significant implications in post-ischaemic conditions, and may explain the remarkable ability of SS-31 to preserve mitochondria structure and allow for rapid recovery of ATP synthesis upon reperfusion (Szeto et al., 2011; Birk et al., 2013). The prompt restoration of ATP helped maintain cellular structure and function, and prevented cell death and organ dysfunction (Szeto et al., 2011; Birk et al., 2013). SS-31 has demonstrated remarkable efficacy in ameliorating injury from myocardial, renal and cerebral ischaemia (Cho et al., 2007a,2007b; Szeto et al., 2011; Kloner et al., 2012; Sloan et al., 2012; Birk et al., 2013; Frasier et al., 2013; Szeto, 2014).
In summary, the hydrophobic interaction between CL and cyt c is advantageous for keeping cyt c close to the respiratory complexes and preventing its loss from the inter-membrane space. However, this hydrophobic interaction can also elicit peroxidase activity and inhibit electron transfer from the exposed haem iron, thus creating the rate-limiting step in the ETC. SS-31 can minimize this rate-limiting step by preserving the tight association between CL and cyt c without inducing peroxidase activity while also restoring π−π* interaction within cyt c to facilitate electron transfer. Targeting and optimizing CL and the cyt c/CL complex represents a novel and innovative approach for the treatment of mitochondrial dysfunction and bioenergetic failure. Other than ischaemia-reperfusion injury, SS-31 is effective for many other diseases that are associated with bioenergetic failure (Szeto, 2008a,2008b; Calkins et al., 2011; Min et al., 2011; Dai et al., 2013) and the idea of restoring bioenergetics with SS-31 (Bendavia) as a common approach to prevent and treat diverse age-associated diseases is currently being evaluated in several clinical trials.
Acknowledgments
This research was supported, by the National Institute of Health (PO1-AG001751), and the Research Program in Mitochondrial Therapeutics at Weill Cornell Medical College that is supported by a gift from Stealth Peptides Inc. We thank Dr. Lichuan Yang for helping us with the ATP analysis.
Glossary
- [ald]
Aladan
- CL
cardiolipin
- cyt c
cytochrome c
- DHPC
1,2-dihexanoyl-sn-glycero-3-phosphocholine
- ETC
electron transport chain
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- SS-31
(D-Arg-dimethylTyr-Lys-Phe-NH2)
- [ald]SS-31
(D-Arg-dimethylTyr-Lys-Ald-NH2; where Ald is aladan)
- TMPD
N,N,N′,N′-tetramethyl-p-phenylenediamine
- TOCL
1′,3′-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol
- TPP+
triphenylphosphonium ion
Conflict of interest
The SS peptides described in this article are licensed for commercial research and development to Stealth Peptides Inc., a clinical stage biopharmaceutical company, in which H. H. S. and A. V. B., and the Cornell Research Foundation have financial interests
References
- Basova LV, Kurnikov IV, Wang L, Ritov VB, Belikova NA, Vlasova II, et al. Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry. 2007;46:3423–3434. doi: 10.1021/bi061854k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, et al. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol. 2007;62:154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol. 2013;24:1250–1261. doi: 10.1681/ASN.2012121216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AM, Miller LA, Faber J, Saari JT. Mitochondrial respiration in heart, liver, and kidney of copper-deficient rats. J Nutr Biochem. 1992;3:668–672. [Google Scholar]
- Böttinger L, Horvath SE, Kleinschroth T, Hunte C, Daum G, Pfanner N, et al. Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J Mol Biol. 2012;423:677–686. doi: 10.1016/j.jmb.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, et al. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007a;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS-31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007b;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- Cohen BE, McAnaney TB, Park ES, Jan YN, Boxer SG, Jan LY. Probing protein electrostatics with a synthetic fluorescent amino acid. Science. 2002;296:1700–1703. doi: 10.1126/science.1069346. [DOI] [PubMed] [Google Scholar]
- Dai DF, Hsieh EJ, Chen T, Menendez LG, Basisty NB, Tsai L, et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ Heart Fail. 2013;6:1067–1076. doi: 10.1161/CIRCHEARTFAILURE.113.000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink BD, Herlein JA, Yorek MA, Fenner AM, Kerns RJ, Sivitz WI. Bioenergetic effects of mitochondrial-targeted coenzyme Q analogs in endothelial cells. J Pharmacol Exp Ther. 2012;342:709–719. doi: 10.1124/jpet.112.195586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher WR, Taniuchi H, Anfinsen CB. On the role of heme in the formation of the structure of cytochrome c. J Biol Chem. 1973;248:3188–3195. [PubMed] [Google Scholar]
- Frasier CR, Moukdar F, Patel HD, Sloan RC, Stewart LM, Alleman RJ, et al. Redox-dependent increases in glutathione reductase and exercise preconditioning: role of NADPH oxidase and mitochondria. Cardiovasc Res. 2013;98:47–55. doi: 10.1093/cvr/cvt009. [DOI] [PubMed] [Google Scholar]
- Gonzalvez F, D'Aurelio M, Boutant M, Moustapha A, Puech JP, Landes T, et al. Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim Biophys Acta. 2013;1832:1194–1206. doi: 10.1016/j.bbadis.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Hamada D, Hoshino M, Kataoka M, Fink AL, Goto Y. Intermediate conformational states of apocytochrome c. Biochemistry. 1993;32:10351–10358. doi: 10.1021/bi00090a010. [DOI] [PubMed] [Google Scholar]
- Hanske J, Toffey JR, Morenz AM, Bonilla AJ, Schiavoni KH, Pletneva EV. Conformational properties of cardiolipin-bound cytochrome c. Proc Natl Acad Sci U S A. 2012;109:125–130. doi: 10.1073/pnas.1112312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang J, Potapovich AI, et al. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med. 2004;37:1963–1985. doi: 10.1016/j.freeradbiomed.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Kagan VE, Bayir A, Bayir H, Stoyanovsky D, Borisenko GG, Tyurina YY, et al. Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: a new strategy in anti-apoptotic drug discovery. Mol Nutr Food Res. 2009;53:104–114. doi: 10.1002/mnfr.200700402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ, et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective peptide. J Am Heart Assoc. 2012;1:e001644. doi: 10.1161/JAHA.112.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CP, Gu Q, Xiong Y, Mitchell RA, Ernster L. P/O ratios reassessed: mitochondrial P/O ratios consistently exceed 1.5 with succinate and 2.5 with NAD-linked substrates. FASEB J. 1996;10:345–350. doi: 10.1096/fasebj.10.2.8641569. [DOI] [PubMed] [Google Scholar]
- Ma Q, Fang H, Shang W, Liu L, Xu Z, Ye T, et al. Superoxide flashes: early mitochondrial signals for oxidative stress-induced apoptosis. J Biol Chem. 2011;286:27573–27581. doi: 10.1074/jbc.M111.241794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K, Murase O, Sugishita K, Yoneyama S, Akada K, Ueha M, et al. Optical characterization of liposomes by right angle light scattering and turbidity measurement. Biochim Biophys Acta. 2000;1467:219–226. doi: 10.1016/s0005-2736(00)00223-6. [DOI] [PubMed] [Google Scholar]
- Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol. 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic Biol Med. 2010;48:1286–1295. doi: 10.1016/j.freeradbiomed.2010.02.020. [DOI] [PubMed] [Google Scholar]
- Petrenko A, Belous AM, Lemeshko VV. [Effect of freezing-thawing rates on the functional state and ionic permeability of rat liver mitochondria] Biokhimiia. 1982;47:626–632. [PubMed] [Google Scholar]
- Prosser RS, Evanics F, Kitevski JL, Al-Abdul-Wahid MS. Current applications of bicelles in NMR studies of membrane-associated amphiphiles and proteins. Biochemistry. 2006;45:8453–8465. doi: 10.1021/bi060615u. [DOI] [PubMed] [Google Scholar]
- Reily C, Mitchell T, Chacko BK, Benavides G, Murphy MP, Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1:86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytömaa M, Kinnunen PK. Evidence for two distinct acidic phospholipid-binding sites in cytochrome c. J Biol Chem. 1994;269:1770–1774. [PubMed] [Google Scholar]
- Rytömaa M, Kinnunen PK. Reversibility of the binding of cytochrome c to liposomes. Implications for lipid-protein interactions. J Biol Chem. 1995;270:3197–3202. doi: 10.1074/jbc.270.7.3197. [DOI] [PubMed] [Google Scholar]
- Santucci R, Ascoli F. The Soret circular dichroism spectrum as a probe for the heme Fe(III)-Met(80) axial bond in horse cytochrome c. J Inorg Biochem. 1997;68:211–214. doi: 10.1016/s0162-0134(97)00100-1. [DOI] [PubMed] [Google Scholar]
- Schlame M, Ren M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim Biophys Acta. 2009;1788:2080–2083. doi: 10.1016/j.bbamem.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober D, Aurich C, Nohl H, Gille L. Influence of cryopreservation on mitochondrial functions in equine spermatozoa. Theriogenology. 2007;68:745–754. doi: 10.1016/j.theriogenology.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, et al. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell. 2013;12:763–771. doi: 10.1111/acel.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinibaldi F, Fiorucci L, Patriarca A, Lauceri R, Ferri T, Coletta M, et al. Insights into cytochrome c-cardiolipin interaction. Role played by ionic strength. Biochemistry. 2008;47:6928–6935. doi: 10.1021/bi800048v. [DOI] [PubMed] [Google Scholar]
- Sinibaldi F, Droghetti E, Polticelli F, Piro MC, Di Pierro D, Ferri T, et al. The effects of ATP and sodium chloride on the cytochrome c-cardiolipin interaction: the contrasting behavior of the horse heart and yeast proteins. J Inorg Biochem. 2011;105:1365–1372. doi: 10.1016/j.jinorgbio.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, et al. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J Mol Cell Cardiol. 2012;52:1009–1018. doi: 10.1016/j.yjmcc.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Snider EJ, Muenzner J, Toffey JR, Hong Y, Pletneva EV. Multifaceted effects of ATP on cardiolipin-bound cytochrome c. Biochemistry. 2013;52:993–995. doi: 10.1021/bi301682c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorice M, Manganelli V, Matarrese P, Tinari A, Misasi R, Malorni W, et al. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 2009;583:2447–2450. doi: 10.1016/j.febslet.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Szeto HH. Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. Ann N Y Acad Sci. 2008a;1147:112–121. doi: 10.1196/annals.1427.013. [DOI] [PubMed] [Google Scholar]
- Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008b;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 171:2029–2050. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto HH, Schiller PW. Novel therapies targeting inner mitochondrial membrane – from discovery to clinical development. Pharm Res. 2011;28:2669–2679. doi: 10.1007/s11095-011-0476-8. [DOI] [PubMed] [Google Scholar]
- Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert EE, Smuder AJ, Min K, Kwon OS, Szeto HH, Powers SK. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. J Appl Physiol. 2013;115:529–538. doi: 10.1152/japplphysiol.00471.2013. [DOI] [PubMed] [Google Scholar]
- Tyurina YY, Tungekar MA, Jung MY, Tyurin VA, Greenberger JS, Stoyanovsky DA, et al. Mitochondria targeting of non-peroxidizable triphenylphosphonium conjugated oleic acid protects mouse embryonic cells against apoptosis: role of cardiolipin remodeling. FEBS Lett. 2012;586:235–241. doi: 10.1016/j.febslet.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Yang L, Manfredi G. Assay of mitochondrial ATP synthesis in animal cells and tissues. Methods Cell Biol. 2007;80:155–171. doi: 10.1016/S0091-679X(06)80007-5. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial bioenergetic etiology of disease. J Clin Invest. 2013a;123:1405–1412. doi: 10.1172/JCI61398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Bioenergetics in human evolution and disease: implications for the origins of biological complexity and the missing genetic variation of common diseases. Philos Trans R Soc Lond B Biol Sci. 2013b;368:20120267. doi: 10.1098/rstb.2012.0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiles JA, Deems R, Vold RR, Dennis EA. Bicelles in structure-function studies of membrane-associated proteins. Bioorg Chem. 2002;30:431–442. doi: 10.1016/s0045-2068(02)00527-8. [DOI] [PubMed] [Google Scholar]
- Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid Redox Signal. 2009;11:2095–2104. doi: 10.1089/ars.2009.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Zhao GM, Wu D, Soong Y, Birk AV, Schiller PW, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- Zhao K, Luo G, Giannelli S, Szeto HH. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem Pharmacol. 2005;70:1796–1806. doi: 10.1016/j.bcp.2005.08.022. [DOI] [PubMed] [Google Scholar]