

Abstract
Crystallographic and solution studies have shown that IgE molecules are acutely bent in their Fc region. Crystal structures reveal the Cε2 domain pair folded back onto the Cε3-Cε4 domains, but is the molecule exclusively bent or can the Cε2 domains adopt extended conformations and even “flip” from one side of the molecule to the other? We report the crystal structure of IgE-Fc captured in a fully extended, symmetrical conformation and show by molecular dynamics, calorimetry, stopped-flow kinetic, SPR and FRET analyses, that the antibody can indeed adopt such extended conformations in solution. This diversity of conformational states available to IgE-Fc offers a new perspective on IgE function in allergen recognition, as part of the B cell receptor and as a therapeutic target in allergic disease.
INTRODUCTION
Immunoglobulin E (IgE) antibodies play a central role in allergic disease1. They recognise allergens in two very different contexts, either in a membrane-bound form as part of the B-cell receptor (BCR), or bound to the receptor FcεRI on effector cells such as mast cells and basophils. FcεRI-bound IgE causes long-term sensitisation of these cells, and cross-linking by allergen leads to cell degranulation, release of inflammatory mediators and an immediate allergic response. Disruption of the IgE-FcεRI interaction is a validated strategy for therapeutic intervention in allergic diseases including asthma: an anti-IgE monoclonal IgG antibody, omalizumab (Xolair™, Novartis Pharmaceuticals Ltd), inhibits IgE binding to FcεRI and is effective in the treatment of severe persistent asthma and other allergic diseases2.
IgE consists of a dimer of two identical heavy and two identical light chains, but unlike IgG in which the antigen-binding Fab region is separated from the receptor-binding Fc region by a flexible hinge, IgE contains an additional disulphide-linked pair of domains, (Cε2)2, forming a (Cε2-Cε3-Cε4)2 dimer1. Fluorescence depolarisation studies to assess segmental flexibility have shown IgE to be less flexible than IgG3-6, and Förster resonance energy transfer (FRET) studies that determined distances both intra-molecular and to the membrane led to a model of a compact, bent structure both for IgE free in solution and when bound to FcεRI6-9. Although an extended model was also proposed10, X-ray and neutron scattering studies in solution confirmed that IgE and IgE-Fc adopt a compact, bent structure11,12. Nevertheless no one anticipated the acutely and asymmetrically bent conformation that was subsequently observed in the crystal structure of IgE-Fc (Fig. 1a)13. In this bent structure, the (Cε2)2 domain pair folds back onto the Cε3-Cε4 domains, forming an extensive intra-molecular interface (1,520Å2). The subsequent structure of the complex of IgE-Fc bound to the extracellular domains of the FcεRI α-chain (sFcεRIα) revealed an even more acute bend upon receptor binding14, consistent with FRET and fluorescence depolarisation studies that indicated reduced segmental flexibility6,15,16. At this point the existence of an extended conformation of IgE-Fc was all but dismissed.
Figure 1. Bent and extended structures adopted by IgE-Fc.

(a) The bent structure of free IgE-Fc, with the (Cε2)2 domain pair making contact with the Cε3-4 domains. IgE-FcA is shown in blue, and IgE-FcB in orange. (b) The structure of IgE-Fc bound symmetrically by two aεFab molecules (shown with heavy chains in dark green, and light chains in light green). (c) The extended conformation of IgE-Fc as seen in the complex (rotated 90° relative to b).
Although the Cε2 domains are not directly involved in binding FcεRIα, they do contribute to the kinetics of the interaction, decreasing both the association and dissociation rate constants14,17. Interest in their structural and functional role intensified following the discovery that the Fab fragment of omalizumab binds to a partially “unbent” conformation of IgE-Fc, as detected in a FRET experiment16. This first indication that IgE-Fc may not always be bent, raises the question of whether the molecule transiently explores more extended conformations, and perhaps even flips between bent structures with the Cε2 domains folded back on opposite sides of the Cε3-Cε4 domains. Trapping of transiently populated conformational states has previously been achieved by antibody binding18, and so to explore the potential conformational diversity of IgE-Fc we generated an IgG antibody Fab fragment that binds to IgE-Fc (anti-ε-chain Fab; aεFab) and discovered that it had captured an extended conformation.
RESULTS
Structure of IgE-Fc bound by two aεFab fragments
The aεFab-IgE-Fc crystal structure was solved at 2.9Å resolution (see Table 1 for data collection and refinement statistics). Remarkably, the IgE-Fc adopts a fully extended conformation, with two aεFab molecules bound, one on each side of the almost perfectly symmetrical IgE-Fc (aεFab1-IgE-Fc-aεFab2, Fig. 1b and c). Compared with the structure of IgE-Fc alone, the molecule has undergone a drastic “unbending” of 120° (Fig. 1a and c), losing completely the extensive intra-molecular interface between the Cε2 and Cε3-Cε4 domains. This unbending appears to derive largely from movements in the Cε2-Cε3 linker region, in particular residues Pro333, Arg334 and Gly335 (Supplementary Video 1). While the Cε2 domains display the greatest structural change, the Cε3 domains also undergo considerable movement. Conformational flexibility has been seen in a number of structures of the Fcε3-4 sub-fragment of IgE-Fc, with the Cε3 domains described as “open” or “closed” (together with a “swinging” of one Cε3 domain relative to the other, Supplementary Fig. 1a)19. In the bent structure of IgE-Fc alone, one Cε3 is “open” (chain B) and one is “closed” (chain A), whereas in the extended conformation of IgE-Fc revealed here, both Cε3 domains adopt an “open” conformation (Supplementary Fig. 1b). Cε3A thus moves more than Cε3B upon aεFab binding, with a change in the Cε3-Cε4 inter-domain angle of 15°. The (Cε4)2 pair are unchanged upon complex formation. Such is the symmetry of the IgE-Fc in the complex that the local two-fold axes of all three domain pairs are virtually coincident.
Table 1. Data collection and refinement statistics.
| aεFab1–IgE-Fc–aεFab2 | |
|---|---|
| Data collection | |
| Space group | P2l2121 |
| Cell dimensions | |
| a, b, c (Α̊) | 84.59, 100.81, 219.68 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution (Α̊) | 2.91 (2.98 – 2.91)* |
| R merge | 0.055 (0.796) |
| I / σI | 18.4 (3.0) |
| Completeness (%) | 99.7 (99.2) |
| Redundancy | 5.7 (6.0) |
| Refinement | |
| Resolution (Α̊) | 67.01 – 2.91 |
| No. unique reflections | 41987 (3078) |
| Rwork / Rfree | 0.236 / 0.284 |
| No. atoms | 11712 |
| Protein | 11541 |
| Ligand/ion | 122 |
| Water | 49 |
| β factors | |
| Protein | 103.8 |
| Ligand/ion | 110.9a |
| Water | 88.4 |
| r.m.s. deviations | |
| Bond lengths (Α̊) | 0.010 |
| Bond angles (°) | 1.566 |
Values in parentheses are for highest-resolution shell.
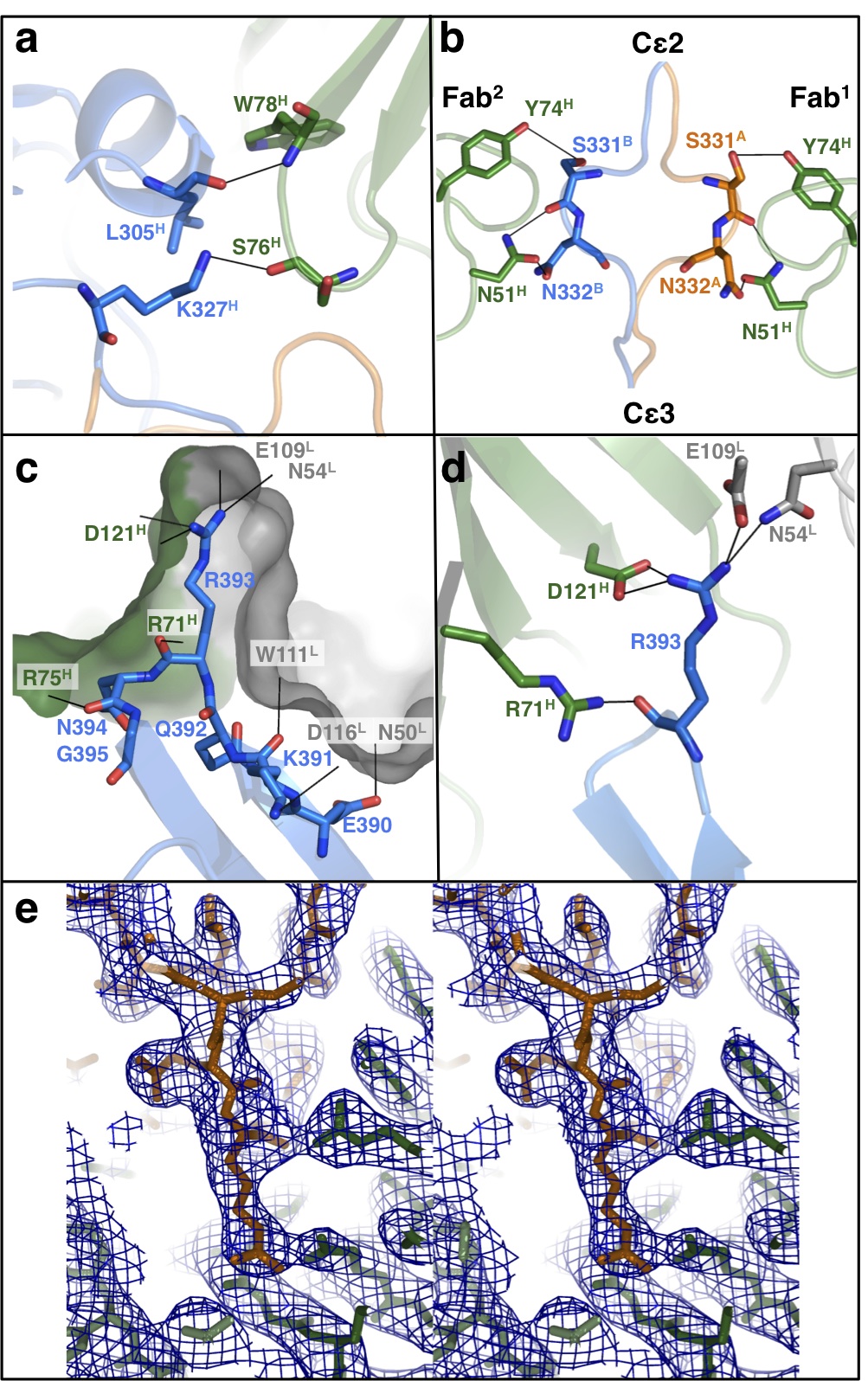
As a result of this symmetry the two aεFab interfaces are structurally equivalent (each ~1,400Å2), mainly involving contact of the aεFab heavy and light chains with Cε3, but with a small interaction with Cε2 (Supplementary Fig. 2a). Each aεFab molecule principally contacts a single IgE-Fc chain (aεFab1 to IgE-FcA and aεFab2 to IgE-FcB), with the exception of a 315Å2 interface with the Cε2-Cε3 linker region (including Ser331 and Asn332) of the other IgE-Fc chain (Supplementary Fig. 2b). The ‘hot spot’ of the aεFab binding surface on IgE-Fc appears to centre on Cε3 residue Arg393, which protrudes into a pocket at the heavy/light chain interface of aεFab (Supplementary Fig. 2c) forming a salt bridge (to Asp121H) and hydrogen bonds (to Glu109L and Asn54L; Supplementary Fig. 2d and e). The adjacent Cε3 residues also contribute extensively (Supplementary Fig. 2c).
Importantly, comparison of the aεFab1–IgE-Fc–aεFab2 complex with free IgE-Fc shows that the latter presents only one aεFab binding site. When superimposed on the Cε3A domains, the aεFab-binding hot spot is both unchanged and accessible in bent IgE-Fc, making it a likely point for aεFab engagement. However, the Cε2 domains in the bent structure completely occlude aεFab binding to the other side of the molecule (Cε3B), and would have to move substantially towards an extended conformation before aεFab access is possible (Fig. 1a and b).
Molecular dynamics simulation of IgE-Fc unbending
The crystal structure of the aεFab1–IgE-Fc–aεFab2 complex reveals that an extended conformation of IgE-Fc is feasible. The key question however is whether this is just a consequence of aεFab binding, or whether it (or other extended conformations) exists in solution as an intrinsic property of IgE. If the latter, then could the Cε2 domain pair make the transition via an extended structure from one side of the Cε3-Cε4 domains to the other and “flip” between two symmetrically-related bent conformations (Supplementary Video 2)? We used metadynamics, an enhanced molecular dynamics (MD) method20-23, to produce a detailed atomistic simulation of IgE-Fc unbending and its extended conformations. This simulation provides a free-energy surface (potential of mean force) calculated and presented in terms of the two principal components of the molecule’s unbending dynamics (Fig. 2a and Supplementary Fig. 3).
Figure 2. Molecular dynamics simulation of IgE-Fc unbending.

(a) Free-energy surface representing the IgE-Fc unbending process generated through metadynamics simulation. Axes show the projection along the two lowest frequency collective motions (extracted from a biased trajectory) for unbending using equations described in Plumed-1.3 documentation46. Contours are drawn every 5 kJ/mol and colored accordingly. The simulation covers the transition across the linear conformational states (at × = 0) but does not encompass the complete “flip”. The conformation seen in the aεFab1–IgE-Fc–aεFab2 complex is indicated (black cross). A possible pathway between energy minima is shown (dotted line). (b) Conformations of IgE-Fc corresponding to the energy minima in a are numbered accordingly and colored as in Fig. 1. (c) Sketch of the smoothed free-energy profile corresponding to the pathway indicated in a; numbers correspond to panels a and b.
The lowest energy state is a well-defined minimum that corresponds to the bent conformation seen in the free IgE-Fc crystal structure13 (Fig. 2, conformation 1). A number of other energy minima may be seen at ~20 kJ/mol above that of the bent state, suggesting possible paths (one of which is indicated, Fig. 2a), leading away from the bent structure towards more extended conformations (Fig. 2a and b, conformations 2 and 3). The dynamic simulation identifies another minimum (Fig. 2a, conformation 4) symmetrically related to conformation 3 and separated by a small energy barrier that corresponds to a linear conformation (indicated in Fig. 2c). As IgE-Fc crosses this low barrier, the Cε2 domain pair passes from one side of the molecule to the other. Having traversed the mid-point (indicated at x=0 in Fig. 2a), the Cε2 domains may fold onto the other side of the Cε3-Cε4 domains, completing a full transition from one bent conformation to the other. (The complete trajectory beyond conformation 4 was not simulated). The extended conformation seen in the aεFab1–IgE-Fc–aεFab2 complex, indicated by a cross in Figure 2a, does not lie directly between conformations 3 and 4. The shift from the proposed pathway corresponds to twisting of (Cε2)2, presumably stabilised by aεFab binding.
While comparison of the bent and extended crystal structures identified Pro333, Arg334 and Gly335 in the Cε2-Cε3 linkers as the residues that undergo the most substantial structural rearrangement accompanying (Cε2)2 domain motions, the simulation revealed additional changes around Gly335A and Asn332B; smaller movements in the Cε3-Cε4 linker around Ser437A are associated with the change in the inter-domain angle. The simulation additionally revealed a twisting of (Cε2)2 relative to the Cε3-Cε4 domains during the unbending process. The mechanism by which domain rearrangements and unbending occur may not simply be rigid body movements facilitated by linkers that undergo changes from one ordered structure to another (“mechanical hinges”), but may involve order-disorder transitions as described in other systems24-26. Local disorder or unfolding/re-folding within linkers, or regions of the domains themselves, can facilitate large-scale conformational changes in proteins, providing entropic compensation to reduce high enthalpic barriers26,27. We can now search for evidence of such order-disorder transitions within the wealth of structural information generated by this simulation, but such detailed mechanistic analyses are beyond the scope of the present manuscript.
Regardless of the mechanism, the highest barrier encountered in a complete flip, and thus the rate-determining step, is clearly that involved in leaving the bent conformation; once this is achieved, the barriers to exploring the extended conformations are relatively low. However, the energy difference between the wells corresponding to the bent and extended conformations is such that the fraction of molecules in the extended state will be very small, consistent with the experimental observation that the bent conformation predominates in solution9,12,16.
aεFab binding to IgE-Fc by isothermal titration calorimetry
One way to establish whether IgE-Fc has an intrinsic capacity to unbend in solution is to determine the number of available aεFab binding sites. If rigidly and exclusively bent, only one site will ever be accessible, but if flexible, then two sites will be accessible. We therefore studied the interaction between aεFab and IgE-Fc by isothermal titration calorimetry (ITC) and compared this with the binding to Fcε3-4 which, lacking the Cε2 domains, always has two accessible sites. The results for both IgE-Fc and Fcε3-4 are similar, showing that both molecules do indeed present two binding sites (Fig. 3a and b). The Kd values for aεFab binding to IgE-Fc and Fcε3-4 were found to be 40 nM and 50 nM respectively (Fig. 3a and b; Table 2), but in order to distinguish between the binding affinities of the two aεFab molecules, the ITC experiment was then conducted by titrating aεFab into either IgE-Fc or Fcε3-4 (Fig. 3c and d). This revealed Kd values for IgE-Fc of 76 nM and 1.5 μM for the first and second binding sites respectively (with similar values for Fcε3-4; Table 2); the second aεFab clearly binds more weakly than the first, and small ΔH values indicate that both binding of aεFab1 and aεFab2 are entropically driven.
Figure 3. ITC and FRET analyses of aεFab binding to IgE-Fc.

Titration of IgE-Fc (a) or Fcε3-4 (b) into aεFab, showing that two aεFab-binding sites are available on IgE-Fc and Fcε3-4 (molar ratio of 0.5:1 for IgE-Fc or Fcε3-4 to aεFab, i.e. 2:1 aεFab:IgE-Fc or Fcε3-4). Titration of aεFab into IgE-Fc (c) or Fcε3-4 (d), showing that aεFab1 and aεFab2 bind with different affinities (see Table 2). Molar ratio 1:1 corresponds to an aεFab1–IgE-Fc complex and 2:1 corresponds to the aεFab1–IgE-Fc–aεFab2 complex. (e) FRET signal (ratio (E520/E485) × 104) from labeled IgE-Fc at 25 nM as a function of titrated aεFab concentration. The binding affinities of aεFab1 and aεFab2 are indicated, showing that minimum FRET only occurs upon binding of aεFab2. ITC and FRET data were fitted to a sequential, two-step binding mechanism, and FRET error bars are s.e.m (n=6).
Table 2. ITC and stopped-flow analysis of the interaction between aεFab and IgE-Fc or Fcε3-4.
| Kdl ITC (μM) | Kd2 ITC (μM) | Kon(M−1s−1) | koff(s−1) | kon2(M−1s−1) | koff2 (s−1) | ΔH1 ITC (Kcal/mol) | ΔH2ITC (Kcal/mol) | |
|---|---|---|---|---|---|---|---|---|
| 1aεFab–1IgE-Fc | 0.040 (±0.02) | – | 6.7 (±0.2) × 105 | n/m | – | – | −3.5 (±0.1) | – |
| 2aεFab–1IgE-Fc | 0.076 (±0.07) | 1.5 (±1.3) | 3.5 (±0.2) × 105 | 3.2 (± 0.1) | 1.2 (±0.2) × 105 | n/m | −3.5 (±0.1) | −1.3 (±0.1) |
| 1aεFab–1Fce3-4 | 0.050 (±0.02) | – | 1.0 (±0.1) × 106 | n/m | – | – | −3.9 (±0.1) | – |
| 2aεFab–1Fce3-4 | 0.034 (±0.03) | 0.98 (±0.45) | 1.0 (±0.1) × 106 | 0.95 (± 0.2) | 3.7 (±0.3) × 105 | n/m | −4.3 (±0.1) | −2.3 (±0.1) |
n/m not measurable, too slow to measure.
Assembly mechanism of aεFab1–IgE-Fc–aεFab2 in solution
The observed 2:1 stoichiometry demonstrates that in solution IgE-Fc can adopt the extended structures needed to expose both aεFab binding sites. The binding of aεFab1 mediates an allosteric change in IgE-Fc, which may occur through induced fit28, conformational selection29 or a combination of both mechanisms30. In order to investigate the assembly mechanism of the aεFab1–IgE-Fc–aεFab2 complex, we monitored the intrinsic tryptophan fluorescence upon binding in stopped-flow kinetic experiments. When IgE-Fc (or Fcε3-4) was in excess over aεFab, thus restricting the stoichiometry of the aεFab-IgE-Fc (or Fcε3-4) complex to 1:1, a single binding event was observed (Supplementary Fig. 4a and b). This has a fast association rate constant and a dissociation rate too slow to measure by this technique (Table 2). The observation of monophasic kinetics implies that no conformational change is coupled to formation of aεFab1-IgE-Fc (although we cannot rule out the possibility of such a change occurring on a timescale faster than that of the experiment).
When repeated with aεFab in excess over IgE-Fc, two-step binding was observed for both IgE-Fc and Fcε3-4 (Supplementary Fig. 4a and b) consistent not only with aεFab1 binding faster than aεFab2, but also the difference in affinities of the two sites determined by ITC (Table 2). Furthermore, a linear concentration dependence for the observed rate constants (kobs) of both aεFabs to either IgE-Fc or Fcε3-4 suggest that aεFab1 does not cause a conformational change (such as unbending of IgE-Fc) that is rate-limiting for aεFab2 binding (Supplementary Fig. 4c-f). In the absence of any evidence for an induced conformational change, our results are most consistent with a conformational selection model, with extended structures selected from a dynamic population of IgE-Fc conformations.
In order to test whether aεFab1 binding alone traps IgE-Fc in an extended conformation or still allows it to flex between extended and bent structures, we employed intra-molecular FRET, with IgE-Fc labelled with donor and acceptor fluorophores in the Cε2 and Cε4 domains respectively. Upon titration into labelled IgE-Fc, a concentration of aεFab sufficient to occupy the first binding site resulted in a partial decrease of FRET signal (Fig. 3e). This implies that under these conditions the IgE-Fc is not exclusively extended, as would be expected if aεFab1 binding caused full linearization; nor does IgE-Fc remain bent. Taken together with the stopped-flow data (linear concentration dependence of the kinetics of aεFab2 binding and similar behaviour of IgE-Fc and Fcε3-4), the FRET data show that IgE-Fc in the aεFab1-IgE-Fc complex remains conformationally dynamic. It is only upon saturation of the second binding site that no further reduction in the FRET signal occurs (Fig. 3e), corresponding to a fully extended IgE-Fc.
These results are summarized in Figure 4, which depicts a possible mechanism for the formation of the aεFab1–IgE-Fc–aεFab2 complex (see also Supplementary Video 3): 1. IgE-Fc is predominantly bent in solution (consistent with X-ray and neutron scattering12 and FRET16), but may transiently adopt extended conformations such as those identified in the MD simulation (Fig. 4a). 2. aεFab1 engages IgE-Fc in the bent or an extended conformation. With aεFab1 bound, IgE-Fc can still flex between bent and extended conformations (Fig. 4b). 3. When IgE-Fc is transiently extended, aεFab2 engages and completes the aεFab1–IgE-Fc–aεFab2 complex (Fig. 4c).
Figure 4. Proposed mechanism of IgE-Fc flexibility and aεFab binding in solution.

(a) IgE-Fc is predominantly bent in solution, but is capable of adopting transiently extended conformations through which the (Cε2)2 domains can flip from one side of the molecule to the other. (b) aεFab1 engages one of the binding sites of IgE-Fc restricting its range of flexibility, but IgE-Fc is neither exclusively bent nor exclusively extended. (c) aεFab2 engages the extended form of IgE-Fc, capturing the molecule in this otherwise transiently occupied conformation. Colored as in Fig. 1.
Effect of aεFab on FcεRI binding
We have demonstrated the existence of a range of conformational states available to IgE-Fc, but how do these different states affect binding of IgE to FcεRIα? Receptor-bound IgE-Fc is even more acutely bent than free IgE-Fc, and interacts with high affinity (Kd ≈ 0.1 nM)1 through two sub-sites, one on each Cε3 domain (Fig. 5a)14,31. In the extended conformation of IgE-Fc that results from aεFab binding, both of the FcεRIα binding sub-sites are disrupted (Fig. 5b). In sub-site 1 on IgE-FcA, Arg334 forms a critically important salt bridge with FcεRIα, but this residue is part of the linker that undergoes structural rearrangement upon extension of (Cε2)2, and is no longer in a position to engage in FcεRIα binding. The second sub-site on IgE-FcB involves a proline (Pro426 of Cε3B) sandwiched between two tryptophan residues (Trp87 and Trp110 of FcεRIα), but this proline moves 6Å away from its receptor-bound conformation in the aεFab complex. Even if receptor engagement with Pro426 of sub-site 2 alone occurs, modelling of sFcεRIα in this position on the extended IgE-Fc results in clashes between both Cε3A and (Cε2)2 of IgE-Fc with the receptor (Fig. 5c).
Figure 5. Structural basis for inhibition of IgE-Fc interaction with FcεRIα by aεFab.

(a) The structure of IgE-Fc bound to sFcεRIα (PDB 2Y7Q14). IgE-Fc is shown in grey cartoon representation (IgE-FcA in lighter shade, IgE-FcB in darker shade); sFcεRIα is shown as purple surface. (b) Detail of the receptor binding region following overlay of the Cε3 and Cε4 domains of IgE-Fc in receptor-bound (grey) and aεFab-bound (IgE-FcA in blue, IgE-FcB in orange) conformations. Key residues involved in receptor binding are shifted, as indicated by the arrows. (c) Location of sFcεRIα (purple, cartoon representation) after superposition of the IgE-Fc–sFcεRIα complex onto the aεFab1–IgE-Fc–aεFab2 complex (using the Cε3B domain and thus maintaining the P426 interaction with receptor) and displaying only aεFab2. The clashes between receptor and both the Cε3A and (Cε2)2 domains are indicated in red. (d) Location of aεFab1 (cartoon representation, heavy chain in dark green, light chain in light green) after superposition of the aεFab1–IgE-Fc–aεFab2 complex onto the IgE-Fc–sFcεRIα complex (using the Cε4 domains), but displaying only aεFab1 from the complex. aεFab1 and sFcεRIα compete for the same region in space, as shown by the overlap region in red.
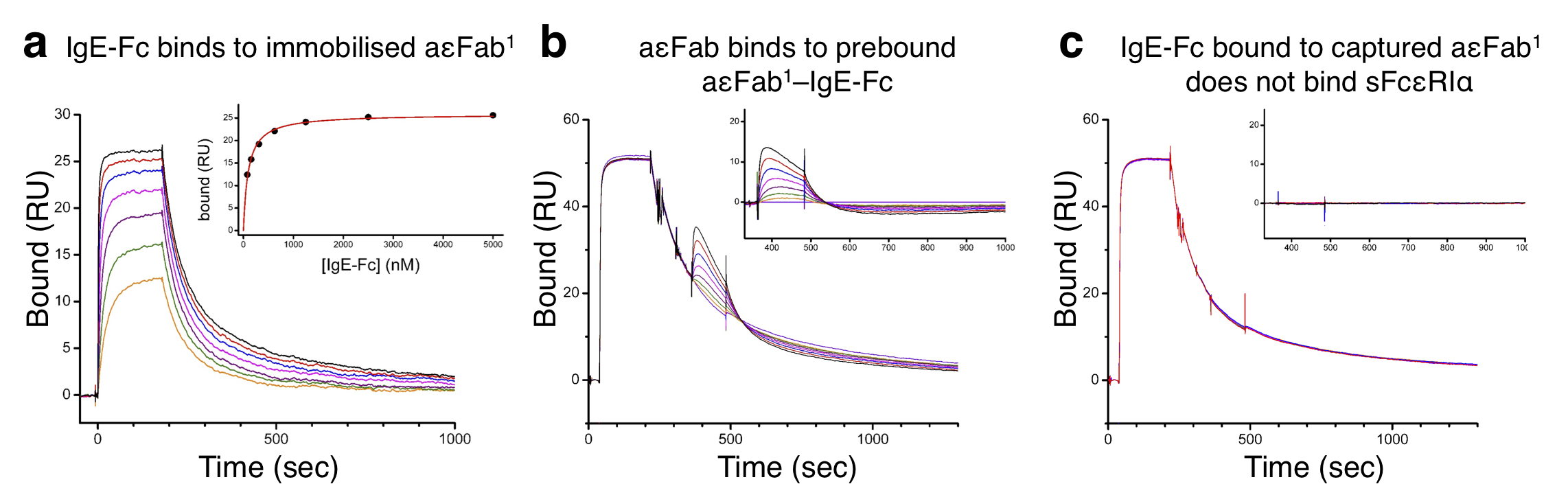
Thus aεFab binding allosterically prevents FcεRIα binding to IgE-Fc by trapping the molecule in an extended state that cannot engage with receptor. But does aεFab1 alone compete with FcεRIα binding to bent IgE-Fc? We compared the aεFab-bound structure with that of IgE-Fc bound to sFcεRIα. Modelling of a single aεFab onto the IgE-Fc–sFcεRIα structure shows that although the residues involved in the two interactions are distinct, the aεFab and receptor molecules clash (Fig. 5d). Therefore, aεFab1 and sFcεRIα compete for binding to the bent form of IgE-Fc. This was confirmed using surface plasmon resonance (SPR) to determine whether aεFab1 alone is capable of inhibiting sFcεRIα binding to IgE-Fc, or whether aεFab2 is required to trap the molecule in an extended conformation before inhibition occurs.
aεFab was first immobilised on an SPR biosensor surface and binding of IgE-Fc was tested over a range of concentrations (Supplementary Fig. 5a). The Kd of aεFab1 binding to IgE-Fc was found to be 95 nM (± 6 nM), in agreement with ITC results (Table 2). Subsequently, binding of aεFab2 to form aεFab1–IgE-Fc–aεFab2 was characterised. After immobilisation of aεFab on the SPR biosensor surface, IgE-Fc was flowed over the chip to form the aεFab1–IgE-Fc complex, followed by aεFab2 to complete the trimolecular complex (Supplementary Fig. 5b). Based on an equilibrium analysis, the Kd of aεFab2 binding was estimated to be 1.4 μM (± 0.1 μM), again in good agreement with ITC results. To determine how binding of aεFab1 to IgE-Fc affects the binding of sFcεRIα, the aεFab1–IgE-Fc complex was first formed as described above, and before this complex could dissociate, sFcεRIα was passed over the surface in increasing concentrations. No binding of sFcεRIα was observed up to a concentration of 1 μM, confirming that binding of a single aεFab is sufficient to inhibit receptor binding to IgE-Fc (Supplementary Fig. 5c). aεFab thus inhibits receptor binding by a steric mechanism when IgE-Fc is bent, and also allosterically when extended.
Modelling membrane-bound IgE
We previously modelled the conformations accessible to the Fab arms of IgE based upon the bent, receptor-bound structure of IgE-Fc16. This revealed little overlap between the spaces explored by the two Fab arms, one of which was directed away from, the other parallel to, the membrane, consistent with both allergen recognition and receptor cross-linking (Figs. 6a & b). However, it is difficult to see how a rigidly bent IgE molecule could function in allergen recognition in the BCR, since the Fab arms would be directed towards the membrane (Figs. 6c). To test this, the ensemble of whole IgE models reported previously16 was orientated in a membrane-bound position, with a spacer to represent the extra membrane proximal domains (EMPD) that separate the C-termini of the Cε4 domains from the membrane (Fig. 6d). Irrespective of spacer length (since the EMPD structure is unknown), the Fab arms only explore space close to the membrane. To determine how an extended conformation of IgE-Fc would present the Fab arms, the same method of model generation was implemented and 2,000 models of IgE were positioned relative to the membrane. Extended IgE-Fc conformations orient the Fab arms away from membrane, optimally for allergen binding (Fig. 6e and f).
Figure 6. Modeled structure of the entire IgE molecule in different biological contexts.

On the right in each panel, the allowed range of locations for each Fab arm is represented by a sphere (blue or yellow for each Fab) placed at the center of the allergen binding site. Cartoon (a) and schematic (b) depictions of acutely and rigidly bent IgE bound to FcεRIα (purple). IgE (chain A in dark and light blue, chain B in dark and light orange), and FcεRIα (purple) are shown. Cartoon (c) and schematic (d) depictions of membrane bent IgE as part of the BCR. The extra membrane-proximal domains of mIgE are indicated by a circle (in c) and a black spacer bar (in d), since their structure is unknown. Igα/β, the BCR accessory proteins, are shown in c (green). Cartoon (e) and schematic (f) depictions of extended IgE conformations as part of the BCR. Igα/β, the BCR accessory proteins, are shown in e (green).
DISCUSSION
Ever since the first model of a bent structure for IgE6-9, the determination of the acutely and asymmetrically bent IgE-Fc by X-ray crystallography13 and demonstration by X-ray solution scattering and FRET that this structure predominates in solution9,11,12,16, the idea that the (Cε2)2 domain pair might flip from one side to the other has been only a tantalising possibility. Fluorescence depolarisation analyses of segmental flexibility in IgE and electron microscopy (EM) of immune complexes had shown that the molecule was less flexible than IgG3-6,15,32, and in the crystal structure of its complex with sFcεRIα14, IgE-Fc was found to be even more acutely bent, consistent with FRET studies in solution16 and when receptor-bound on cells6. Only the binding of omalizumab Fab to IgE-Fc in solution, monitored by FRET, indicated that a partial “unbending” of IgE-Fc was possible16. The observation of a fully extended IgE-Fc in the aεFab complex reported here was therefore a considerable surprise. We have now demonstrated by MD simulation and solution biophysical studies that IgE-Fc can indeed unbend and adopt a range of transiently extended conformations, suggesting a possible pathway for the Cε2 domains to flip from one side of the Cε3-Cε4 to the other.
The degree of flexibility described for IgE-Fc here is unlike any reported for other antibody classes. Flexibility between the V and C domains in Fab fragments (“elbow bending”) and within the hinge separating Fab and Fc regions in IgG antibodies is well documented, but conformational variation within the Fc has to date been limited to minor changes in the orientation of the Cγ2 domains of IgG33 or the Cε3 domains of IgE19. “Open” and “closed” conformations for the latter have been reported in structures of IgE-Fc and Fcε3-4 both free and when bound to either sFcεRIα14,31 or to sCD23, the soluble IgE-binding domain of the ‘low affinity’ receptor on B-cells34. The conformational diversity in IgE-Fc reported here is substantially greater than previously seen in Fc structures. In fact the earlier fluorescence depolarisation data on IgE should be re-interpreted in terms of bending at the Cε2/Cε3 linker regions in addition to bending between Fab and Fc.
IgM, an evolutionary precursor of IgE, also contains a domain pair, (Cμ2)2, rather than a flexible hinge, and is known to undergo substantial conformational changes. Free in solution it adopts planar, star-shaped pentameric or hexameric structures, as determined by small angle X-ray and neutron scattering and EM studies35,36, but when bound by Fab arms of two or more subunits to an antigenic surface, it adopts “table-like” structures with the disc of penta/hexameric Fc regions at 90° to the Fab arms35. However, the location of the bend in the molecule is unknown. Modelling of IgM structures against solution scattering data placed the bend between Cμ2 and Cμ336, as suggested earlier in fluorescence depolarisation studies37, whereas later modelling against cryo-EM data was consistent with bending between Cμ1 and Cμ238. Recent crystallographic analyses of individual IgM-Fc domains leaves the question unresolved39. It is clear however, that at least in the context of the polymeric structures, the monomeric IgM subunit can adopt either linear or bent conformations, and if the flexibility in IgE is evolutionarily related to that of IgM, bending of the latter would be predicted to occur between Cμ2 and Cμ3. Conformational change in IgM is critical for function, since dislocation upon antigen binding reveals sites for complement binding that are hidden in the planar structures. There is therefore a precedent for an extended conformation of an antibody with the “additional”, hinge-replacing domains that has a clear biological function.
That IgE evolved conformational diversity to allow both a compact bent and an ensemble of extended, flexible structures may be understood in terms of its allergen-binding functions in two distinct contexts. Bound to FcεRI on effector cells (mast cells, basophils or antigen-presenting cells), bent IgE presents its Fab arms appropriately for allergen recognition and cross-linking16, which triggers the allergic response (Fig. 6a and b). In contrast, as part of the BCR, in which the role of membrane IgE is to capture allergen and ensure B cell survival and proliferation, only an extended molecule presents the Fab arms optimally for detection of allergen (Fig. 6c-f). One recognised advantage of conformational flexibility is “fly-casting”, i.e. the ability to sample a greater range of conformational space and make ligand (allergen) capture more effective40. Conventional depictions of the BCR for all antibody classes show extended Fab arms, and the extended IgE-Fc structure reported here demonstrates that such a conformation is feasible.
In the extensively studied IgM BCR, which contains a single IgM subunit, it has been assumed in the absence of a crystal structure that IgM adopts an extended conformation. It has also been proposed that conformational changes occur in the IgM-Fc upon antigen binding as part of the signalling function41,42. Our results for IgE-Fc provide support for the notion of an extended IgM-Fc and suggest one way in which a conformational change might occur. Without knowing the structure of the EMPDs that connect the Cε4 domains to the transmembrane regions of the ε-chains, or how the accessory Igα and Igβ chains interact with mIgE or mIgM, it remains a matter for speculation how extended structures might be stabilised. A model for the association between Igα and Cμ4 of IgM has been proposed based upon the crystal structure of Igβ43, but it is hard to see how interaction with Cμ4 alone could stabilise the extended conformation. Structural studies of the EMPD and the interactions between IgE-Fc and Igα/Igβ are clearly required.
We now consider the nature of the free IgE molecule. The MD analysis shows that the bent conformation exists in a relatively deep energy well (Fig. 2c, conformation 1), and that the rate-limiting step to reaching the extended conformations and then flipping to the alternative bent conformation with the Cε2 domains packed against the other side of the Fcε3-4, is the escape from this well. Once this occurs, the energy barriers between various extended conformations, including the barrier as the IgE-Fc passes through the linear conformation, are much lower (Fig. 2b & c, conformations 2–4). Different pathways for unbending may be envisaged, but the most highly populated are expected to have free-energy profiles similar to that sketched in Figure 2c. Although kinetic information may not be directly extracted from the simulations presented here as biasing forces were applied, an estimate for the rate of unbending may be made if we assume that the process of escape from the well that corresponds to the bent conformation can be described by passage over a single free-energy barrier26,44. The resulting rate of 0.15 s−1 (see Methods for equation and parameters used26,44) must however be considered a very approximate calculation. The free-energy difference between the bent and the various extended states (~8 kBT, Figure 2c), implies that only a very small fraction of the molecules occupy an extended conformation at any one time. This is in accord with studies of IgE-Fc in solution9,12,16, which indicate that the bent conformation predominates.
Comparison of the structures of IgE-Fc or Fcε3-4 with sFcεRIα and sCD23 has shown that, although the binding sites are spatially distinct, at either end of the Cε3 domains14,31,34, they are allosterically linked such that IgE cannot bind both receptors simultaneously34. This mutual exclusion is vital for controlling IgE function. For example, cross-linking of FcεRI-bound IgE by oligomeric CD23 would trigger the allergic response in the absence of allergen. Flexibility in IgE-Fc, in particular the relative disposition of the Cε3 domains, is key to determining their receptor binding capacity, therefore understanding this allostery and conformational flexibility is important for targeting IgE’s receptor interactions for therapeutic benefit.
We have shown here that aεFab inhibits FcεRI binding both sterically and allosterically (Fig 5). Since extended IgE-Fc conformations cannot bind FcεRI, it may be possible to exploit the intrinsic capacity of the molecule to unbend and enhance the dissociation of IgE from the receptor. Indeed, the recent demonstration that accelerated dissociation of IgE from FcεRI can occur upon binding of a DARPin45 may be explained in terms of the flexibility that we have identified in IgE-Fc. The reported structure of this DARPin complexed with a disulphide-constrained Fcε3-445 (lacking Cε2 domains) leaves its mechanism of action upon IgE unclear. We do know however that the binding of the anti-IgE omalizumab Fab causes a partial unbending of IgE-Fc as monitored by FRET16, which may contribute to its mechanism of action. In any event, the discovery that IgE can flex between bent and transiently occupied extended conformations provides a new framework for understanding IgE function in allergen recognition, and offers a more complete description of the structure of this important therapeutic target.
ONLINE METHODS
Protein expression and purification
Anti-human IgE-Fc antibody was supplied by UCB and isolated V-region genes sub-cloned in the human IgG1 Fab format. aεFab was expressed by transient transfection in CHO cells, cultured in CD-CHO medium with the addition of 10 mM glutamine at 37°C with 8% CO2 and a rotation of 140 rpm. 2×108 cells/mL were resuspended in Earles Balance Salt Solution before 400 μg of DNA were added. Cells were electroporated and resuspended in 100 mL of CD-CHO medium and incubated for 24 hours. Incubation continued at 32 °C for 13 days and at 4 days post-transfection sodium butyrate (3 mM final concentration) was added to the culture. On day 14 post-transfection, cell culture supernatants were harvested by centrifugation (400 × g for 1 hour) for purification.
aεFab was purified by Protein G affinity chromatography (GE Healthcare) and bound aεFab eluted in 100mM glycine-HCl, pH 2.7 and fractions neutralized with 1/25th fraction volume of 2 M Tris-HCl, pH 8.5. aεFab was further purified by size exclusion chromatography (SEC) on a Superdex S200 column (GE Healthcare) in 25 mM Tris-HCl, 20 mM NaCl, 0.05% (w/v) NaN3, pH 7.5.
IgE-Fc(N265Q,N371Q) secreted from a stable NS-0 cell line was purified from tissue culture supernatant by cation exchange. Supernatant was buffer-exchanged into 50 mM sodium acetate pH 6.0, 75 mM NaCl and loaded onto a SPHP cation-exchange column (GE Healthcare). IgE-Fc(N265Q,N371Q) was eluted with a 10 × column volume gradient into 50 mM sodium acetate, pH 6.0, 1 M NaCl. Eluted fractions were pooled, concentrated and further purified by SEC on a Superdex S200 column (GE Healthcare) in PBS, pH 7.4.
aεFab:IgE-Fc complex was prepared by mixing aεFab with IgE-Fc and purification to homogeneity by SEC as described above.
sFcεRIα (Val1-Lys176) was engineered as a C-terminal His6 tagged protein and transiently expressed in CHO cells as described for the aεFab protein. Harvested tissue culture supernatant was buffer exchanged into 50 mM NaH2PO4 pH 7.4, 0.3 M NaCl, 10 mM Imidazole and the sFcεRIα-His purified by affinity chromatography on a Ni-NTA column (Qiagen) eluting the bound protein on a linear gradient from 50 mM NaH2PO4 pH 7.4, 0.3 M NaCl, 10 mM Imidazole into 50 mM NaH2PO4 pH 7.4, 0.3 M NaCl, 250 mM Imidazole. Pooled fractions were further purified by SEC on a Superdex S200 column (GE Healthcare) in PBS, pH 7.4.
Crystallisation
Sitting drop vapor diffusion crystallization experiments were set up with a protein complex concentration of 5 mg/mL in 20 mM NaCl, 25 mM Tris-HCl pH 7.5, and 0.05% sodium azide. Crystals were grown at 18 °C using 12–22% PEG3350, 0.25 M sodium citrate, and 0.1 M Bis-Tris Propane pH 7.5–9.0 as precipitant. Drops were microseeded with crystals grown under identical crystallization conditions in earlier trials. Crystals were flash-cooled in liquid nitrogen using 4 M trimethylamine N-oxide as cryoprotectant.
Data collection and structure determination
Diffraction data were collected at beamline I03, Diamond Light Source (Harwell, U.K.). Xia247 was used to index, integrate, and merge data to 2.9 Å resolution48-54. The phases were solved using Phaser molecular replacement55. To generate the aεFab search model the RCSB PDB protein sequence search engine was used to find 3QHZ, from which non-conserved residues were removed using CHAINSAW56,57. IgE-Fc search models were generated by splitting the coordinates from the high resolution IgE-Fc structure (PDB 2WQR14), into (Cε2)2 and (Cε3-4)2 fragments. Molecules were located in the asymmetric unit sequentially: aεFab1 was followed by (Cε3-4)2, aεFab2, and finally (Cε2)2. The structure was initially rebuilt using the autobuild wizard of PHENIX58, and then refined using iterative cycles of PHENIX58, with 5% of reflections set aside from refinement for calculation of Rfree. Between refinement cycles, the structure was manually built into 2Fo – Fc and Fo – Fc electron density maps using COOT59. Composite omit maps were generated using the autobuild wizard in PHENIX to prevent model bias58. Carbohydrate and water molecules were manually built into the structure. MolProbity60 and CARP61 were used to assess protein and carbohydrate geometry respectively. 93.6% of residues are within the Ramachandran favoured region, and 0.2% are outliers. PISA62, CONTACT and NCONT as part of the CCP4 program suite54 were used for analysis of protein-protein interfaces, and DynDom63 was used to calculate the domain motion involved in the conformational changes. Structure morphs for movies were calculated using the UCSF CHIMERA package64, and videos made using PyMOL65.
Enhanced molecular dynamics
The bent crystal structure (PDB 1O0V13) was used as the starting point for molecular dynamics simulation, after adding hydrogen atoms and removing terminal residues to symmetrise the two chains. Protonation states were predicted with Maestro (Schrödinger LLC). After initial exploratory simulations of unbending, manual checks were made for possible changes to protonation states in the unbent conformations. No changes in protonation states were found that could be justified, and thus the initial states were retained for the final simulation. The AMBER ff99SB-ildn and GLYCAM force fields were used for protein and carbohydrate respectively. The structure was solvated in a truncated octahedron (dimensions 123 × 123 × 123 Å) such that no protein atom of the extended IgE-Fc conformation was within 8 Å of the edge after pressure equilibration. Monatomic ions were added to a salt concentration of 0.15 M. The final simulation system had 129,459 atoms.
Simulations were carried out with NAMD 2.866 patched with Plumed-1.346. Particle mesh Ewald67 was used for long-range electrostatics along with 9 Å cut-offs for Coulomb and Lennard-Jones potential functions. A preliminary 500 ns unbiased simulation was used to extract two collective variables (CVs) through principal component analysis (PCA). Only every other α-carbon was included in the PCA CVs. An exploratory metadynamics simulation then used these PCA CVs to explore unbending for 1,200 ns with Gaussians of height 8 kJ/mol and sigma of 0.5 Å added every 4 ps. This exploratory metadynamics run then provided new PCA CVs for a final simulation, hence the final CVs were extracted from a simulation that fully sampled unbending. The final multiple-walker metadynamics simulation converged after 3,755 ns using Gaussians of height 4 kJ/mol added every 4 ps. Convergence of this calculation was gauged by the reduction of the Gaussian heights being added (50–100 times smaller than at the beginning) and a convergence in the relative depths of the lowest free energy wells. Additionally, the converged simulation walkers were able to move through CV space relatively freely suggesting the suitability of the CVs.
To confirm the results of the metadynamics simulation, a short (250 ns) unbiased simulation starting from the extended crystal conformation of IgE-Fc was performed (Supplementary Fig. 3d).
An estimation of the rate of transition from a bent to an unbent conformation was made assuming a single rate-limiting barrier according to equation 16 of Whitford et al. 2012 (ref 26)
| (Eq.1) |
where kt is the transition rate, C is the “attempt frequency” for barrier crossing and ΔG is the barrier height. A value for C was taken as 10 μs−1 since many protein folding and functional transitions have been found to range from ≈ 1 – 10 μs−1 (ref 26) and the barrier height was taken as 18 kBT (Figure 2c).
Isothermal titration calorimetry
Experiments were carried out using a Microcal iTC200 calorimeter (GE Healthcare) at 20 °C in PBS buffer pH 7.4. Depending on the final ratio required, 25–30 μM of protein were used in the cell with 10–20 times higher concentrations in the syringe. The number and volume of injections were varied as appropriate. Heats of dilution were subtracted from the data before analysis. Analyses were carried out using MicroCal Origin, using a 1:1 binding model when IgE-Fc or Fcε3-4 was titrated into aεFab, or a sequential 2:1 (non-identical) binding model when aεFab was titrated into IgE-Fc or Fcε3-4. The non-identical 2:1 binding model was selected based on prior knowledge of the crystal structure, and observation that the curve clearly deviated from an identical 2:1 binding mode.
Stopped-flow fluorescence
Experiments were carried out using a Chirascan Plus (Applied Photophysics Ltd) with a stopped-flow attachment at 20 °C in PBS buffer pH 7.4 and with pseudo-first order protein concentrations varied as required. Fluorescence was excited at 280 nm (1 nm slit width) and emitted fluorescence above 305 nm detected with a long-band pass. 6–10 runs were averaged for each experiment. Data were collected and analyzed using supplied software according to the manufacturer’s instructions. Experimental transients were fitted either to single-exponential (Eq.2; [IgE-Fc] or [Fcε3-4] > [aεFab] and only aεFab1 binding observable) or double-exponential (Eq.3; [aεFab] > [IgE-Fc] or [Fcε3-4] and binding of both aεFab1 and aεFab2 observable) equations:
| (Eq.2) |
| (Eq.3) |
where F is the observed fluorescence, ΔFn is the fluorescence amplitude change for the nth transient, kobsn is the pseudo-first order rate constant for the nth step and Fe is the end-point fluorescence. The bimolecular association rate constants for aεFab1 (k+1) and aεFab2 (k+2) binding were determined by fitting the linear concentration dependences of kobs1 and kobs2 to Eq. 4:
| (Eq.4) |
where kobsn is the pseudo-first order rate constant for the nth transient at the ranges of ligand concentrations used, k+n is the association rate constant for the nth aεFab binding event and k−n is the dissociation rate constant for the nth Fab binding.
FRET
Intramolecular FRET was carried out using IgE-Fc(E289C)-BirA (IgE-Fc with the BirA recognition motif added to the C-terminus and biotinylated according to the manufacturer’s instructions (Avidity)), labeled with thiol-reactive terbium chelate (Invitrogen) and monovalent streptavidin68 labeled with amine-reactive Alexa488 (Invitrogen), each according to the manufacturer’s instructions. Terbium labeled IgE-Fc and Alexa488 labeled streptavidin were mixed in equi-molar ratios (final assay concentration 25 nM) with aεFab titrated to 30 μM in PBS and incubated for 120 minutes at room temperature. FRET was measured on an Analyst LJL-HT with excitation 330 nm (80 nm slit width), emission 485 (20 nm slit width) and 520 nm (10 nm slit width). The FRET ratio ((E520/E485) × 104) was plotted as a function of aεFab concentration. Data was fit to a sequential, two-step binding mechanism.
Surface plasmon resonance
SPR experiments were carried out on a Biacore T200 instrument (GE Healthcare). Specific binding surfaces were prepared by coupling aεFab through amine coupling. Coupling density was limited to <500 resonance units. IgE-Fc was injected over the sensor chip at a flow rate of 25 μl min−1, for 180 s at concentrations of 78, 156, 313, 625, 1,250, 2,500, and 5,000 nM in running buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 5 mM CaCl2, and 0.005% (v/v) Surfactant P-20). For sandwich assays, IgE-Fc at 100 nM in running buffer was injected over the sensor chip for 180 s at a flow rate of 25 μl min−1. 45 s after completion of IgE-Fc injection, aεFab (at concentrations of 0, 78, 156, 313, 625, 1250, 2,500, and 5,000 nM) or sFcεRIα (at concentrations of 0, 31.2, 62.5, 125, 250, 500, and 1,000 nM) were injected over the chip at a flow rate of 25 μl min−1 for 120 s. Measurements were performed at 25 °C. Double referencing data subtraction methods were performed69.
Molecular modeling of whole, extended IgE
FPMOD70 was used to model whole, extended IgE molecules. The Fab domains from an IgE-like antibody (PDB 2R56) were fused onto the extended IgE-Fc structure, and allowed to move rigidly about a flexible linker until 2,000 models had been generated.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
The authors thank the Medical Research Council, UK (G1100090; B.J.S) and The Wellcome Trust for grant funding (076343; B.J.S.) and support for the King’s Biomolecular Spectroscopy Centre (085944).
The work was carried out with the support of the Diamond Light Source (Harwell, UK).
Footnotes
ACCESSION CODES: Coordinates and structure factors for aεFab1–IgE-Fc–aεFab2 have been deposited in the RCSB Protein Data Bank (www.rcsb.org) with accession code 4J4P.
COMPETING FINANCIAL INTERESTS: The authors declare no competing financial interests.
References
- 1.Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat. Rev. Immunol. 2008;8:205–217. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 2.Holgate ST, Djukanovic R, Casale T, Bousquet J. Anti-immunoglobulin E treatment with omalizumab in allergic diseases: an update on anti-inflammatory activity and clinical efficacy. Clin. Exp. Allergy. 2005;35:408–416. doi: 10.1111/j.1365-2222.2005.02191.x. [DOI] [PubMed] [Google Scholar]
- 3.Nezlin RS, Zagyansk Ya, Kaivarai Ai, Stefani DV. Properties of Myeloma Immunoglobulin E(Yu) chemical, fluorescence polarisation and spin-labeled studies. Immunochemistry. 1973;10:681–688. doi: 10.1016/0019-2791(73)90211-5. [DOI] [PubMed] [Google Scholar]
- 4.Sykulev YK, Nezlin RS. Spin labeling of Immunoglobulin-M and Immunoglobulin-E carbohydrates. Immunol. Lett. 1982;5:121–126. doi: 10.1016/0165-2478(82)90095-5. [DOI] [PubMed] [Google Scholar]
- 5.Oi VT, et al. Correlation between segmental flexibility and effector function of antibodies. Nature. 1984;307:136–140. doi: 10.1038/307136a0. [DOI] [PubMed] [Google Scholar]
- 6.Zheng Y, Shopes B, Holowka D, Baird B. Dynamic conformations compared for IgE and IgG1 in solution and bound to receptors. Biochemistry. 1992;31:7446–7456. doi: 10.1021/bi00148a004. [DOI] [PubMed] [Google Scholar]
- 7.Holowka D, Baird B. Structural studies on the membrane-bound Immunoglobulin E-receptor complex. 2. Mapping of distances between sites on IgE and the membrane-surface. Biochemistry. 1983;22:3475–3484. doi: 10.1021/bi00283a025. [DOI] [PubMed] [Google Scholar]
- 8.Holowka D, Conrad DH, Baird B. Structural mapping of membrane-bound Immunoglobulin-E receptor complexes - use of monoclonal anti-IgE antibodies to probe the conformation of receptor-bound IgE. Biochemistry. 1985;24:6260–6267. doi: 10.1021/bi00343a033. [DOI] [PubMed] [Google Scholar]
- 9.Zheng Y, Shopes B, Holowka D, Baird B. Conformations of IgE bound to its receptor Fc-Epsilon-RI and in solution. Biochemistry. 1991;30:9125–9132. doi: 10.1021/bi00102a002. [DOI] [PubMed] [Google Scholar]
- 10.Padlan EA, Davies DR. A model of the Fc of Immunoglobulin-E. Mol. Immunol. 1986;23:1063–1075. doi: 10.1016/0161-5890(86)90005-2. [DOI] [PubMed] [Google Scholar]
- 11.Davis KG, Glennie M, Harding SE, Burton DR. A model for the solution conformation of rat IgE. Biochem. Soc. Trans. 1990;18:935–936. doi: 10.1042/bst0180935. [DOI] [PubMed] [Google Scholar]
- 12.Beavil AJ, Young RJ, Sutton BJ, Perkins SJ. Bent domain-structure of recombinant human IgE-Fc in solution by x-ray and neutron-scattering in conjunction with an automated curve-fitting procedure. Biochemistry. 1995;34:14449–14461. doi: 10.1021/bi00044a023. [DOI] [PubMed] [Google Scholar]
- 13.Wan T, et al. The crystal structure of IgE Fc reveals an asymmetrically bent conformation. Nat. Immunol. 2002;3:681–686. doi: 10.1038/ni811. [DOI] [PubMed] [Google Scholar]
- 14.Holdom MD, et al. Conformational changes in IgE contribute to its uniquely slow dissociation rate from receptor Fc epsilon RI. Nat. Struct. Mol. Biol. 2011;18:571–576. doi: 10.1038/nsmb.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holowka D, Wensel T, Baird B. A nanosecond fluorescence depolarization study on the segmental flexibility of receptor-bound Immunoglobulin-E. Biochemistry. 1990;29:4607–4612. doi: 10.1021/bi00471a015. [DOI] [PubMed] [Google Scholar]
- 16.Hunt J, et al. A fluorescent biosensor reveals conformational changes in human Immunoglobulin E Fc: Implications for mechanisms of receptor binding, inhibition, and allergen recognition. J. Biol. Chem. 2012;287:17459–17470. doi: 10.1074/jbc.M111.331967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonnell JM, et al. The structure of the IgE C epsilon 2 domain and its role in stabilizing the complex with its high-affinity receptor Fc epsilon Rl alpha. Nature Struct. Biol. 2001;8:437–441. doi: 10.1038/87603. [DOI] [PubMed] [Google Scholar]
- 18.Frey G, et al. Distinct conformational states of HIV-1 gp41 are recognized by neutralizing and non-neutralizing antibodies. Nat. Struct. Mol. Biol. 2010;17:1486–1491. doi: 10.1038/nsmb.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wurzburg BA, Jardetzky TS. Conformational flexibility in Immunoglobulin E-Fc(3-4) revealed in multiple crystal forms. J. Mol. Biol. 2009;393:176–190. doi: 10.1016/j.jmb.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barducci A, Bussi G, Parrinello M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008;100:020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 21.Laio A, Gervasio FL. Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008;71:126601. [Google Scholar]
- 22.Crespo Y, Marinelli F, Pietrucci F, Laio A. Metadynamics convergence law in a multidimensional system. Phys. Rev. E. 2010;81:055701. doi: 10.1103/PhysRevE.81.055701. [DOI] [PubMed] [Google Scholar]
- 23.Barducci A, Bonomi M, Parrinello M. Metadynamics. WIREs Comput. Mol. Sci. 2011;1:826–843. [Google Scholar]
- 24.Best RB, Chen YG, Hummer G. Slow protein conformational dynamics from multiple experimental structures: The helix/sheet transition of arc repressor. Structure. 2005;13:1755–1763. doi: 10.1016/j.str.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Whitford PC, Miyashita O, Levy Y, Onuchic JN. Conformational transitions of adenylate kinase: switching by cracking. J. Mol. Biol. 2007;366:1661–1671. doi: 10.1016/j.jmb.2006.11.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitford PC, Sanbonmatsu KY, Onuchic JN. Biomolecular dynamics: order-disorder transitions and energy landscapes. Rep. Prog. Phys. 2012;75:076601. doi: 10.1088/0034-4885/75/7/076601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fenley AT, Muddana HS, Gilson MK. Entropy-enthalpy transduction caused by conformational shifts can obscure the forces driving protein-ligand binding. Proc. Natl. Acad. Sci. USA. 2012;109:20006–20011. doi: 10.1073/pnas.1213180109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monod J, Wyman J, Changeux JP. On nature of allosteric transitions - a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 30.Wlodarski T, Zagrovic B. Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin. Proc. Natl. Acad. Sci. USA. 2009;106:19346–19351. doi: 10.1073/pnas.0906966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garman SC, Wurzburg BA, Tarchevskaya SS, Kinet JP, Jardetzky TS. Structure of the Fc fragment of human IgE bound to its high-affinity receptor Fc epsilon RI alpha. Nature. 2000;406:259–266. doi: 10.1038/35018500. [DOI] [PubMed] [Google Scholar]
- 32.Roux KH, Strelets L, Brekke OH, Sandlie I, Michaelsen TE. Comparisons of the ability of human IgG3 hinge mutants, IgM, IgE, and IgA2, to form small immune complexes: A role for flexibility and geometry. J. Immunol. 1998;161:4083–4090. [PubMed] [Google Scholar]
- 33.Teplyakov A, Zhao Y, Malia TJ, Obmolova G, Gilliland GL. IgG2 Fc structure and the dynamic features of the IgG CH(2)-CH(3) interface. Mol. Immunol. 2013;55:131–139. doi: 10.1016/j.molimm.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 34.Dhaliwal B, et al. Crystal structure of IgE bound to its B-cell receptor CD23 reveals a mechanism of reciprocal allosteric inhibition with high affinity receptor Fc epsilon RI. Proc. Natl. Acad. Sci. USA. 2012;109:12686–12691. doi: 10.1073/pnas.1207278109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feinstein A, Munn EA. Conformation of the free and antigen-bound IgM antibody molecules. Nature. 1969;224:1307–1309. doi: 10.1038/2241307a0. [DOI] [PubMed] [Google Scholar]
- 36.Perkins SJ, Nealis AS, Sutton BJ, Feinstein A. Solution structure of human and mouse Immunoglobulin M by synchrotron X-ray scattering and molecular graphics modelling. A possible mechanism for complement activation. J. Mol. Biol. 1991;221:1345–1366. doi: 10.1016/0022-2836(91)90937-2. [DOI] [PubMed] [Google Scholar]
- 37.Holowka DA, Cathou RE. Conformation of Immunoglobulin-M .2. Nanosecond fluorescence depolarization analysis of segmental flexibility in anti-epsilon-1-dimethylamino-5-naphthalenesulfonyl-L-lysine anti-immunoglobulin from horse, pig, and shark. Biochemistry. 1976;15:3379–3390. doi: 10.1021/bi00660a033. [DOI] [PubMed] [Google Scholar]
- 38.Czajkowsky DM, Shao Z. The human IgM pentamer is a mushroom-shaped molecule with a flexural bias. Proc. Natl. Acad. Sci. USA. 2009;106:14960–14965. doi: 10.1073/pnas.0903805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Müller R, et al. High-resolution structures of the IgM Fc domains reveal principles of its hexamer formation. Proc. Natl. Acad. Sci. USA. 2013;110:10183–10188. doi: 10.1073/pnas.1300547110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tolar P, Sohn HW, Liu W, Pierce SK. The molecular assembly and organization of signaling active B-cell receptor oligomers. Immunol. Rev. 2009;232:34–41. doi: 10.1111/j.1600-065X.2009.00833.x. [DOI] [PubMed] [Google Scholar]
- 42.Tolar P, Pierce SK. A conformation-induced oligomerization model for B cell receptor microclustering and signaling. Curr. Top. Microbiol. Immunol. 2010;340:155–169. doi: 10.1007/978-3-642-03858-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radaev S, et al. Structural and functional studies of Ig alpha beta and its assembly with the B cell antigen receptor. Structure. 2010;18:934–943. doi: 10.1016/j.str.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitford PC, Onuchic JN, Sanbonmatsu KY. Connecting energy landscapes with experimental rates for aminoacyl-tRNA accommodation in the ribosome. J. Am. Chem. Soc. 2010;132:13170–13171. doi: 10.1021/ja1061399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim B, et al. Accelerated disassembly of IgE-receptor complexes by a disruptive macromolecular inhibitor. Nature. 2012;491:613–617. doi: 10.1038/nature11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonomi M, et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009;180:1961–1972. [Google Scholar]
- 47.Winter G. Xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010;43:186–190. [Google Scholar]
- 48.Kabsch W. Automatic processing of rotational diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 49.Kabsch W. Evaluation of single-crystal x-ray-diffraction data from a position-sensitive detector. J. Appl. Crystallogr. 1988;21:916–924. [Google Scholar]
- 50.Kabsch W. Automatic-indexing of rotation diffraction patterns. J. Appl. Crystallogr. 1988;21:67–71. [Google Scholar]
- 51.Sauter NK, Grosse-Kunstleve RW, Adams PD. Robust indexing for automatic data collection. J. Appl. Crystallogr. 2004;37:399–409. doi: 10.1107/S0021889804005874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evans P. Scaling and assessment of data quality. Acta Crystallogr. D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Z, Sauter NK, van den Bedem H, Snell G, Deacon AM. Automated diffraction image analysis and spot searching for high-throughput crystal screening. J. Appl. Crystallogr. 2006;39:112–119. [Google Scholar]
- 54.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCoy AJ, et al. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwarzenbacher R, Godzik A, Grzechnik SK, Jaroszewski L. The importance of alignment accuracy for molecular replacement. Acta Crystallogr. D. 2004;60:1229–1236. doi: 10.1107/S0907444904010145. [DOI] [PubMed] [Google Scholar]
- 57.Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr. 2008;41:641–643. [Google Scholar]
- 58.Adams PD, et al. The Phenix software for automated determination of macromolecular structures. Methods. 2011;55:94–106. doi: 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lutteke T, Frank M, von der Lieth CW. Carbohydrate Structure Suite (CSS): analysis of carbohydrate 3D structures derived from the PDB. Nucleic Acids Res. 2005;33:D242–D246. doi: 10.1093/nar/gki013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 63.Hayward S, Berendsen HJC. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins. 1998;30:144–154. [PubMed] [Google Scholar]
- 64.Pettersen EF, et al. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 65.Schrodinger, LLC . The PyMOL Molecular Graphics System, Version 1.5. 2010. [Google Scholar]
- 66.Phillips JC, et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Darden T, Perera L, Li LP, Pedersen L. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999;7:R55–R60. doi: 10.1016/s0969-2126(99)80033-1. [DOI] [PubMed] [Google Scholar]
- 68.Howarth M, Ting AY. Imaging proteins in live mammalian cells with biotin ligase and monovalent streptavidin. Nat. Protoc. 2008;3:534–545. doi: 10.1038/nprot.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Myszyka DG. Improving biosensor analysis. J. Mol. Recognit. 1999;12:279–284. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 70.Pham E, Chiang J, Li I, Shum W, Truong K. A computational tool for designing FRET protein biosensors by rigid-body sampling of their conformational space. Structure. 2007;15:515–523. doi: 10.1016/j.str.2007.03.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.