

Abstract
Kinases are among the most intensively pursued enzyme superfamilies as targets for anti-cancer drugs. Large data sets on inhibitor potency and selectivity for more than 400 human kinases became available recently, offering the opportunity to design rationally novel kinase-based anti-cancer therapies. However, the expression levels and activities of kinases are highly heterogeneous among different types of cancer and even among different stages of the same cancer. The lack of effective strategy for profiling the global kinome hampers the development of kinase-targeted cancer chemotherapy. Here, we introduced a novel global kinome profiling method, based on our recently developed isotope-coded ATP-affinity probe and a targeted proteomic method using multiple-reaction monitoring (MRM), for assessing simultaneously the expression of more than 300 kinases in human cells and tissues. This MRM-based assay displayed much better sensitivity, reproducibility, and accuracy than the discovery-based shotgun proteomic method. Approximately 250 kinases could be routinely detected in the lysate of a single cell line. Additionally, the incorporation of iRT into MRM kinome library rendered our MRM kinome assay easily transferrable across different instrument platforms and laboratories. We further employed this approach for profiling kinase expression in two melanoma cell lines, which revealed substantial kinome reprogramming during cancer progression and demonstrated an excellent correlation between the anti-proliferative effects of kinase inhibitors and the expression levels of their target kinases. Therefore, this facile and accurate kinome profiling assay, together with the kinome-inhibitor interaction map, could provide invaluable knowledge to predict the effectiveness of kinase inhibitor drugs and offer the opportunity for individualized cancer chemotherapy.
Protein phosphorylation, one of the most important types of post-translational modifications (PTMs)1, is catalyzed by protein kinases (collectively referred to as the kinome), which are encoded by over 500 genes in higher eukaryotes (1). Aberrant expression and/or activation/deactivation of kinases have been implicated as among the major mechanisms through which cancer cells escape normal physiological constraints of cell growth and survival (2). Additionally, dynamic kinome reprogramming has been found to be closely associated with resistance toward cancer chemotherapy (3). Owing to their crucial roles in cancer development, kinases have become one of the most intensively pursued enzyme superfamilies as drug targets for cancer chemotherapy and more than 130 distinct kinase inhibitors have been developed for phase 1–3 clinical trials (4). Recently, inhibitor potency and selectivity for more than 400 kinases have been reported, which provided a comprehensive target-inhibition profile for the majority of the human kinome (5–7). Therefore, the kinome-inhibitor interaction networks coupled with comprehensive profiling of global kinome expression and activity associated with certain types of cancer could be invaluable for understanding the mechanisms of carcinogenesis and for designing rationally novel kinase-directed anti-cancer chemotherapies.
Unfortunately, currently there is no optimal strategy for profiling the expression levels of the entire kinome at the protein level. Traditional methods for measuring kinase expression rely primarily on antibody-based immunoassays because of their high specificity and sensitivity (8). The immunoassays, however, are limited by the availability of high-quality antibodies; therefore, these methods are only useful for assessing a small number of kinases in low-throughput. Recent advances in MS instrumentation and bioinformatic tools enable the identification and quantification of a significant portion of the human proteome from complex samples (9). However, proteomic studies of global kinome by MS are still very challenging, which is largely attributed to the fact that, similar as other regulatory enzymes, protein kinases are generally expressed at low levels in cells (10). This analytical challenge is further aggravated in shotgun proteomics approach where even more complex mixtures of peptides instead of proteins from whole cell or tissue extracts are analyzed (11). Therefore, selective enrichment of protein kinases from cellular extracts is essential for the comprehensive identification and quantification of the global kinome.
Affinity columns immobilized with kinase inhibitors have been employed as capture ligands for the enrichment of kinases, and ∼200 protein kinases could be identified and quantified by subsequent LC-MS/MS analyses (3, 10, 12). Recently, we and others reported the application of biotin-conjugated acyl nucleotide probes for the enrichment and identification of kinases from complex protein mixtures (13–17). This enrichment technique, in combination with multi-dimensional LC-MS platform, facilitates the identification of ∼200 protein kinases (15). Despite these advances, such large-scale kinome studies are often performed in the data-dependent acquisition (DDA) mode, where typically 10–20 most abundant ions found in MS are subsequently selected for fragmentation in MS/MS to enable peptide identification (18). Although this discovery-mode (or shotgun) proteomic approach provides the potential to uncover novel protein targets, sample complexity, together with inherent variation in automated peak selection, results in compromised sensitivity and reproducibility for protein quantification. As a result, only partially overlapping sets of proteins can be identified even from substantially similar samples (11). The inadequate sensitivity and reproducibility of these kinome detection strategies hamper their utility in biomarker discovery and clinical studies.
Targeted proteomics technique, which relies on multiple-reaction monitoring (MRM) on triple quadrupole mass spectrometers, has become increasingly used in quantitative proteomics studies (19). In the MRM mode, mass filtering of both the precursor and product ions is employed to provide high specificity for the quantification of target proteins. Additionally, this MRM-based targeted MS analysis permits rapid and continuous monitoring of specific ions of interest, which enhances the sensitivity for peptide detection by up to 100-fold relative to MS analysis in DDA-based discovery mode (20). Therefore, the MRM-based targeted proteomic approach may enable global kinome profiling with high specificity, sensitivity, throughput, and reproducibility.
Here, we developed the first MRM-based platform to support the multiplexed, reproducible, and sensitive quantification of ∼300 protein kinases in the human kinome. Aside from conventional MRM-based assay design, we selectively label and enrich kinases from complex human proteome prior to MRM analysis with the use of desthiobiotin-based isotope-coded ATP-affinity probe (ICAP) (21) to attain high specificity and sensitivity. We demonstrated that this MRM-based kinome detection strategy coupled with ICAP reagent is applicable for clinical samples that are not amenable to metabolic labeling. Additionally, this MRM-based kinome assay is easily transferable between instruments and laboratories, rendering it a facile and universal strategy for global kinome detection.
MATERIALS AND METHODS
Kinase Labeling with Nucleotide Affinity Probe Followed by Affinity Purification
Protein lysates were generated from cultured cells or tissue samples using previously described procedures (see supplementary Materials) (21). HeLa-S3 cells were purchased from the National Cell Culture Center (Minneapolis, MN), and IMR-90, K562, WM-115, and WM-266–4 cells were obtained from ATCC (Manassas, VA). Human lung tumor and paired adjacent normal tissues were purchased from National Disease Research Interchange (NDRI, Philadelphia, PA).
The desthiobiotin-conjugated nucleotide affinity probes were prepared previously (15, 21). Approximately 1 mg cell lysate was treated separately with light and heavy labeled desthiobiotin-ATP affinity probes at a final concentration of 100 μm. Labeling reactions were carried out with gentle shaking at room temperature for 1.5 h. After the reaction, the remaining probes in the cell lysates were removed by buffer exchange with 25 mm NH4HCO3 (pH 8.5) using Amicon Ultra-4 filters (10,000 NMWL, Millipore).
After addition of 8 m urea for protein denaturation, and dithiothreitol and iodoacetamide for cysteine reduction and alkylation, the labeled proteins were digested with modified sequencing-grade trypsin (Roche Applied Science) at an enzyme/substrate ratio of 1:100 in 25 mm NH4HCO3 (pH 8.5) at 37 °C for overnight. The peptide mixture was subsequently dried in a Speed-vac and redissolved in 1 ml PBS buffer (100 mm potassium phosphate and 0.15 m NaCl, pH 7.5), to which solution was subsequently added 200 μl avidin-agarose resin (Sigma-Aldrich). The mixture was then incubated at 25 °C for 1 h with gentle shaking. The agarose resin was washed sequentially with 3 ml PBS buffer and 3 ml H2O to remove unbound peptides, and the desthiobiotin-conjugated peptides were subsequently eluted with 1% TFA in CH3CN/H2O (7:3, v/v) at 65 °C. Enriched peptides were first analyzed on an LTQ-Orbitrap Velos mass spectrometer for discovery-mode proteomic analysis (see supplementary Materials).
LC-MRM Analysis
All LC-MRM experiments were carried out on a TSQ Vantage triple quadrupole mass spectrometer equipped with a nanoelectrospray ionization source coupled to an Accela HPLC with customized split-flow configuration or an EASY-nLC II system (Thermo Scientific). Samples were automatically loaded onto a 4-cm trapping column (150 μm i.d.) packed with 5 μm 120 Å reversed-phase C18 material (ReproSil-Pur 120 C18-AQ, Dr. Maisch) at 3 μl/min. The trapping column was coupled to a 20-cm fused silica analytical column (PicoTip Emitter, New Objective, 75 μm i.d.) with 3 μm C18 beads (ReproSil-Pur 120 C18-AQ, Dr. Maisch). The peptides were then separated with a 130-min linear gradient of 2–35% acetonitrile in 0.1% formic acid and at a flow rate of 250 nL/min. The spray voltage was 1.9 kV. Q1 and Q3 resolutions were 0.7 Da and the cycle time was 4–5 s depending on the maximum number of transitions per cycle.
To enable automated, multiplexed MRM analyses, we first generated an interactive Skyline spectral library file containing tandem mass spectra of all desthiobiotin-conjugated kinase peptides along with their normalized retention time score (iRT) (22), which were acquired from discovery mode analysis on an LTQ Orbitrap Velos mass spectrometer, using Skyline (version 1.4.0.4421, see Supplementary Materials) (23). Collision energies were calculated using a linear equation that was specific to the TSQ Vantage instrument according to the Skyline default setting. BSA standard mixtures were analyzed in unscheduled MRM-mode prior to the analysis of the enriched desthiobiotin-conjugated peptides. The linear predictor of empirical RT from iRT for targeted kinase peptides was then determined by the linear regression of RTs of BSA standard peptides obtained for the current chromatography setup. This linear predictor was rechecked between every four MRM sample analyses by injecting another BSA standard mixture. In identification experiment, four transitions were monitored for each light desthiobiotin-labeled peptide, whereas three pairs of transitions were monitored for each light/heavy desthiobiotin-labeled peptide pair in quantification experiments. These targeted transitions were monitored in four separate injections for each sample in scheduled MRM mode with a retention time window of 10 min.
All raw files were processed using Skyline (version 1.4.0.4421) for generation of extracted ion chromatograms and peak integration. The targeted peptides were first manually checked to ensure the overlaid chromatographic profiles of multiple fragment ions derived from light and heavy forms of the same peptide. The data were then processed to ensure that the distribution of the relative intensities of multiple transitions associated with the same precursor ion correlates with the theoretical distribution from kinome MS/MS spectral library entry. Finally, the remaining transitions from both forward and reverse ICAP labeling experiments were exported and filtered using AuDIT algorithm (24), where t test was performed to evaluate whether the relative product ion intensities of the light-labeled peptides to the heavy labeled peptides are similar. The significant difference (p value <0.1) of the relative product ion intensities indicates the presence of interference or imprecision of the corresponding transition and these transitions were therefore discarded (24). The sum of peak area from all transitions of light- or heavy-labeled peptides was used for quantification.
Other experimental details including Western blot and MTT cell proliferation assay are described in supplementary Materials.
RESULTS
1. Global Kinome Profiling Based on Isotope-Coded ATP-Affinity Probe (ICAP) and MRM
The MRM-based kinome assay is built upon our previously described ICAP-based strategy for the simultaneous enrichment and isotopic labeling of ATP-binding proteins in cell lysates (21). The ICAP reagent harbors three components (Fig. 1A), namely, a binding moiety (ATP), an isotope-coded linker present in light (contains six hydrogens) or heavy form (contains six deuterons), and an enrichment moiety (i.e., desthiobiotin), where the former two are conjugated through an acyl phosphate linkage. Upon binding to kinases, the acyl-phosphate component of the ICAP reacts with the ε-amino group of the P-loop lysine residue in kinases to yield a stable amide bond, which results in the covalent attachment of desthiobiotin together with light or heavy isotope-coded linker to the lysine residue (supplementary Fig. S1). Therefore, the ICAP labeling of a kinase relies on its expression level and ATP binding affinity, which renders the method useful for profiling the expression and ATP-binding affinities of kinases at the global proteome scale.
Fig. 1.
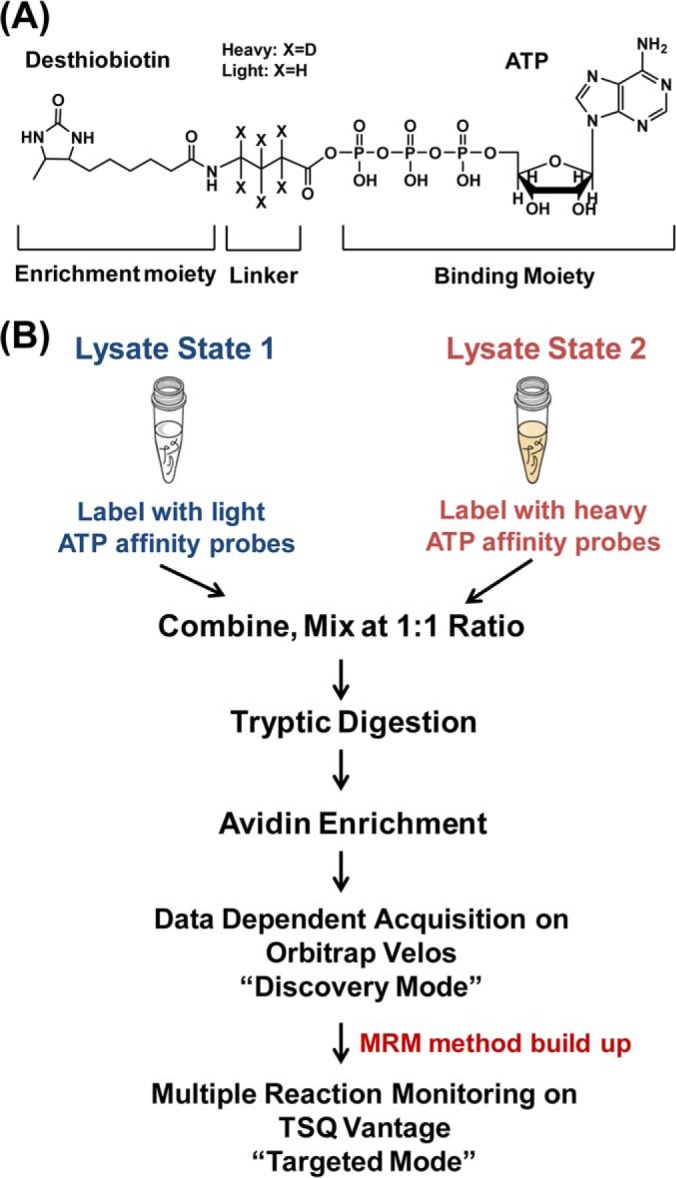
Global kinome profiling with the use of isotope-coded ATP affinity probe (ICAP) and multiple-reaction monitoring (MRM). A, The structure of the ICAP probe. B, A schematic diagram showing the general workflow for MRM analysis of global kinome using ICAP.
The general experimental procedures for the MRM-based kinome assay with the use of ICAP encompass the following steps (Fig. 1B): (1) Kinases from protein samples representing two experimental states are labeled individually with the isotopically light and heavy forms of the ATP affinity probe; (2) The two protein samples are combined and digested with trypsin, and the resultant light/heavy desthiobiotin-labeled peptide pairs are enriched with avidin agarose; (3) The affinity-purified peptides with the desthiobiotin tag are analyzed by LC-MS/MS in DDA mode to generate tandem mass spectra of interested kinase peptides, which allowed for the construction of MRM kinome library; (4) Quantitative analysis of global kinome was performed on a triple quadruple mass spectrometer in MRM mode using the established MRM kinome library.
2. Development of an MRM Assay for Human Kinome Profiling
The design of MRM-based kinome assay requires a priori tandem mass spectral information for the interested desthiobiotin-modified kinase peptides (20). The primary sources for building the MRM library of human kinome were large-scale discovery-based shotgun MS experiments conducted in our laboratory. Human kinases from whole cell lysates of K562, IMR-90, HeLa-S3, Jurkat-T, WM-115, and WM-266–4 cells were enriched using either light or heavy labeled ICAP reagents and the resulting desthiobiotin-labeled peptides were analyzed on an LTQ Orbitrap Velos mass spectrometer in DDA mode, which together included more than 100 LC-MS/MS runs. The tandem mass spectra of all desthiobiotin-modified peptides from human kinases together with their retention time information were imported into Skyline to establish the primary kinome library. A maximum of five peptides were used for each protein. Transitions were chosen from the three or four most abundant y-ions based on reference spectra acquired from the shotgun approach.
Peptide Selection
Owing to its relatively high reactivity, the ICAP probe may react with, aside from the lysine residue(s) located at the ATP-binding site, other lysine residues in kinases or other proteins through electrostatic interactions (15, 21). The probe labeling emanating from such nonspecific interactions does not reflect the ATP-binding affinities of the kinases. Moreover, the probe-labeled peptides stemming from the nonspecific interaction are generally less abundant than those arising from specific nucleotide binding, which may compromise the sensitivity for kinase quantification. Thus, we reason that the quantification of kinome based on the pre-selected representative peptides would generate more reliable results than the quantification based on all the peptides identified in the DDA mode. To improve the specificity of our MRM-based kinome assay, these targeted peptides in our final kinome library were further divided into two categories depending on the local amino acid sequences surrounding the desthiobiotin-modified lysine. In the category with the highest confidence (class I), target peptides represent the confirmed ATP-binding sites on kinases and must satisfy one of the following two criteria: (A) Peptides cover at least one of the three ATP-binding motifs; discovered in our previous studies, i.e., HRDxKxxN, VAxK or GxxxxGK (15, 21); (B) Peptides contain the ATP-binding sites discovered in our previous ATP-affinity profiling assay even though they do not reside in any of the conserved motif sequences (21). For instance, although FLSGLELVK#QGAEAR (K# represents the probe-labeled lysine), a peptide from TP53-regulating kinase, does not belong to any of the aforementioned conserved motifs, it displays strong ATP binding affinity in our previous report (21). The class-II peptides include those that were identified at least five times in our shotgun proteomics experiment. Although these peptides do not reside in any of the conserved ATP binding sites and their involvement in ATP binding remains undefined, their high frequencies of identification in shotgun proteomic studies render these peptides good candidates for determining the quantities of their corresponding kinases. Our current MRM peptide list includes 386 peptides, with 265 and 121 being class-I and class-II peptides, respectively (supplementary Table S1).
Peptide Uniqueness
In our enrichment strategy for human kinases using ICAP reagents, a large portion of protein kinases were labeled on the reactive lysine close to the ATP-binding sites, which share highly conserved sequence (1). Therefore, some targeted peptides in the final MRM peptide list may be assigned to multiple protein kinases. We manually inspected the uniqueness of each targeted peptide using Skyline (supplementary Table S1). Among the 386 peptides in the library, 320 (83%) are unique and belong only to single protein sequences, whereas the rest 66 (17%) can be assigned to multiple kinases. Accordingly, these 386 peptides can be mapped to 313 human kinases and 242 (77%) of these kinases contained at least one unique targeted peptide to unambiguously identify the targeted protein isoform. Unlike the cross-activity of antibody-based immunoblots (25), the advantage of this MRM kinome assay lies in its capability in identifying unequivocally the potential cross-talk targets. Our current MRM kinome library contains a total of 386 peptides from 313 human kinases, including 292 protein kinases, 14 lipid kinases, and 7 metabolic kinases bearing GxxxxGK motif (supplementary Table S1). Placement of these protein kinases to the human kinome map (1) revealed that our kinome library spreads over all major categories of the human kinome (Fig. 2A).
Fig. 2.

Targeted protein kinases mapped in the dendrogram of the human kinome and linearity of iRT versus measured RT on different instruments and experimental platforms. A, Mapping of the identified protein kinases to the human kinome dedrogram. B, iRT values predict measured RT in on-line 2D experiment on Orbitrap Velos (180 min linear gradient) with an excellent correlation coefficient (R2 = 0.999). C, iRT values predict measured RT in MRM experiment on TSQ Vantage (130 min linear gradient) with an excellent correlation coefficient (R2 = 0.999).
Retention Time Calibration
Our kinome peptide list encompasses 386 peptides, which involves the monitoring of more than 2000 MRM transitions in quantitative measurements. To achieve this level of multiplexed detection, it is essential to perform scheduled MRM analysis in which the mass spectrometer is programmed to detect only a limited number of peptides in pre-defined retention time windows. Therefore, accurate retention time (RT) prediction for these kinase peptides becomes essential for our multiplexed MRM-based kinome assays. To this end, we calculated the iRT for each peptide on our target list following a previously described method (22). Based on previous retention time information of targeted peptides analyzed on an LTQ Orbitrap Velos coupled with EASY-nLC II system, we used 10 BSA peptides as standards to successfully convert empirically determined retention times of 94% (362 out of 386) of targeted peptides into normalized iRT scores, which reflect their conserved elution order. As shown in Fig. 2B, peptides from human kinases detected in one of our LC-MS/MS runs on the Orbitrap system show linear correlation (R2 > 0.999) between the assigned iRT values and empirically measured RT values following the Skyline MS1 filter workflow. As reported previously (22), we found that this iRT value is very stable, and the iRT score and empirically measured RT exhibit a linear relationship across different LC configurations, including online 2D-LC and 1D-LC configurations with different column lengths (supplementary Fig. S2). Moreover, because the iRT value represents an inherent attribute of the hydrophobicity of a peptide, it can also serve as another parameter for the validation of the quantification results from MRM assay, where any outliers deviated from the linear plot of iRT versus measured RT could be attributed to false-positive detection.
Based on the above MRM-based assay design, we set out to test the sensitivity and reproducibility of this kinome detection assay using lysates of IMR-90 cells, where four transitions were monitored for each of the 386 peptides on the target list. This MRM-based kinome analysis led to the detection of 242 peptides from the kinase list corresponding to 227 human kinases, which include 210 protein kinases covering all major categories of the human kinome (supplementary Table S2). This result demonstrates the superior sensitivity of the MRM-based kinome assay. Moreover, we found that the linearity between the iRT value and measured RT remains for each peptide detected in this MRM assay, indicating that our MRM-based kinome assay can be easily transferred among different instrument configurations and laboratories (Fig. 2C).
3. Quantitative Profiling of the Global Kinomes of WM-115 and WM-266–4 Human Melanoma Cells
We next applied the MRM-based kinome assay to assess the differential expression of kinases in a pair of human melanoma cell lines, WM-115 and WM-266–4, which were initially derived from the primary and metastatic melanoma sites of the same patient (26). These two cell lines are particularly interesting because dasatinib, a tyrosine kinase inhibitor, was found to inhibit the growth and reduce the migration and invasion of WM-115, but not WM-266–4 cells (27). However, conventional immunoblot assay for a limited number of kinases did not reveal the target kinase(s) conferring the distinct sensitivities of these two cell lines (27). We reason that quantitative global kinome profiling of these two cell lines may uncover the target kinases of dasatinib in WM-115 cells. In addition, such a study may provide important new knowledge about kinome reprogramming during cancer progression. To this end, we employed our ICAP probes, along with the aforementioned MRM-based assay, and assessed the differential expression of kinases in these two cell lines (Fig. 1). For comparison, we also injected the same samples into the LTQ Orbitrap Velos instrument for shotgun proteomics analysis (supplementary Table S3).
A total of 246 kinases in the library, including 228 protein kinases, 14 lipid kinases, and 4 other metabolic kinases, were quantified in MRM-based targeted kinome assay, whereas only 136 protein kinases were quantified by shotgun proteomics strategy even with multi-dimensional LC separation (Fig. 3A). In addition, 10 and 120 protein kinases were only quantified in DDA-based discovery mode and MRM-based targeted mode, respectively. Accordingly, from the total list of 386 kinase peptides, 242 were successfully quantified by the MRM method, whereas only 168 were quantified from shotgun proteomics analysis (Fig. 3B). The much better kinome coverage provided by the MRM assay than the shotgun proteomic approach is attributed to the superior sensitivity of the former method. In this vein, our ATP probe enriches kinases along with other ATP-binding proteins. Therefore, the precursor ion intensities for the probe-labeled peptides from some low-abundance kinases are likely to be too weak to trigger collisional activation, thereby preventing their identification and quantification in the DDA-based shotgun proteomics approach. Monitoring constantly the target peptides in the MRM mode, however, provided substantially improved sensitivity that is needed for the quantification of kinases of low abundance.
Fig. 3.
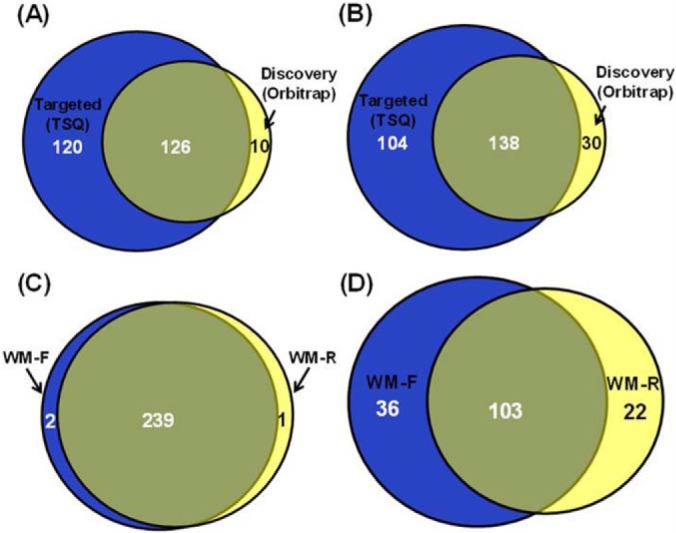
MRM-based kinome assay exhibits better sensitivity, reproducibility compared with data-dependant shotgun proteomics. A, The Venn diagrams showing the overlap of quantified kinases from cell lysates of two melanoma cells obtained from MRM analysis and shotgun proteomics. B, The Venn diagrams showing the overlap of quantified kinase peptides from cell lysates of two melanoma cells by MRM analysis and shotgun proteomics. C, The Venn diagrams showing the overlap of quantified kinase peptides from cell lysates of two melanoma cells from two replicates of MRM analysis. D, The Venn diagrams showing the overlap of quantified kinase peptides from cell lysates of two melanoma cells from two replicates of shotgun proteomics experiments.
Aside from inferior sensitivity, inadequate reproducibility is another major disadvantage for shotgun proteomics. Along this line, 99% (239 out of 242) of the kinase peptides were successfully quantified in both forward and reverse labeling experiments in MRM-based targeted analysis, whereas only 64% (103 out of 161) was quantified with the same samples using the DDA-based shotgun proteomics approach (Figs. 3C and 3D). Thus, the MRM-based targeted approach provided much better reproducibility than the DDA-based shotgun approach for kinome profiling analysis in which multiple experimental replicates are generally required.
Our results also revealed that the quantification accuracy for the MRM method is better than the shotgun approach. As displayed in Fig. 4A, we observed a very good correlation (R2 = 0.92) for the quantification results of 71 peptides of high abundance (peak intensity in MRM assay >100000) commonly quantified by these two modes of analyses. Nevertheless, there are a few discrepant results obtained by these two methods for some low-abundance kinase peptides (peak intensity <100,000 in MRM assay). For example, a unique peptide ETSVLAAAK#VIDTK from SLK kinase exhibited conflicting results based on these two modes of analysis (RatioMRM = 0.47; RatioDDA = 2.63). In MRM assay, the peptide shows consistent ratios in forward and reverse experiments (Fig. 4B), which revealed the up-regulation of the SLK kinase in WM-266–4 cells. Additionally, distribution of the peak area observed for each monitored transition from light- and heavy-labeled peptides was consistent with the theoretical distribution derived from MS/MS spectra stored in MRM kinome library, further validating the identity of this unique peptide. In DDA-based shotgun proteomic analysis, we only identified this peptide once in the reverse experiment, and the chromatogram displays very poor signal-to-noise ratio, suggesting the lack of reliability for the quantification of this peptide (supplementary Fig. S3). Moreover, the consistent quantification results from both MRM and DDA analyses (RatioMRM = 0.50; RatioDDA = 0.56) for another peptide DLK#AGNILFTLDGDIK from the same kinase further substantiated our MRM quantification results for the peptide ETSVLAAAK#VIDTK. Likewise, the MRM method offers more robust quantification for the peptide FLEDDTSDPTYTSALGGK#IPIR from Ephrin type-B receptor 2 than DDA mode (supplementary Fig. S4). Together, the MRM-based targeted analysis offers better sensitivity, reproducibility and accuracy than DDA-based shotgun proteomics for kinome profiling.
Fig. 4.
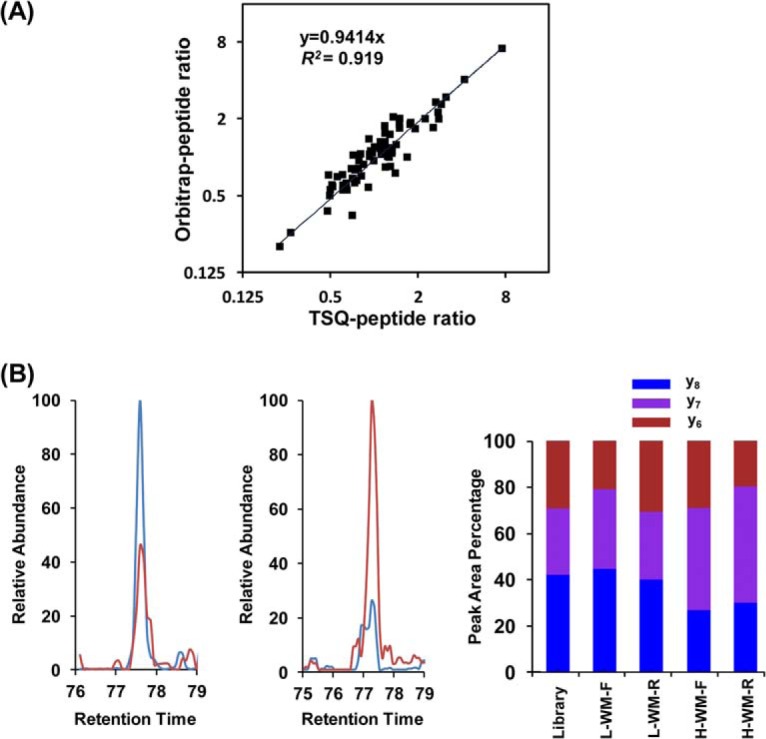
MRM-based kinome assay provided robust quantification results. A, Linear regression comparing quantification results of abundant kinase peptides from two melanoma cells obtained by MRM assay and shotgun proteomics analysis. B, Quantitative results by MRM assay for peptide ETSVLAAAK#VIDTK from SLK kinase: Extracted-ion chromatograms for three transitions monitored for light-labeled (Red) and heavy-labeled (Blue) peptides ETSVLAAAK#VIDTK in forward (Left) and reverse (Middle) labeling reactions; The consistent distribution of the peak area observed for each monitored transition from light- and heavy- labeled peptides in both forward and reverse labeling reaction along with the theoretical distribution derived from MS/MS spectra store in MRM kinome library (Right).
By combining MRM and DDA analyses, we quantified a total of 256 kinases in WM-115 and WM-266–4 cells (supplementary Table S3). Kinases from all seven major kinome groups as well as other and atypical kinases were covered in our analysis. Although a large portion of the kinome is similar in WM-115 and WM-266–4 cells, the two lines of melanoma cells exhibit distinct kinome expression profiles (Fig. 5). A total of 48 protein kinases display substantially different expression profiles (R115/266–4 > 2 or R115/266–4 < 0.5); except for PDK1, the rest 47 protein kinases show consistent expression ratios in MRM-based kinome assay for samples obtained from forward and reverse ICAP labeling experiments (supplementary Fig. S5).
Fig. 5.

Quantitative comparison of kinome expression for WM-115 and WM-266–4 cells. Blue bar denotes kinase that is up-regulated in WM-266–4 cells; red bar denotes kinase that is up-regulated in WM-115 cells.
The tyrosine kinase group exhibits the most distinct expression profiles between these two melanoma cell lines and more than 30% of tyrosine kinases show different expression levels (supplementary Fig. S6). Moreover, significantly more kinases in AGC, STE, and tyrosine kinase groups were up-regulated in WM-115 cells than those that were up-regulated in WM-266–4 cells. For example, in the AGC group, GPRK5, MAST3, PKCα, PKCβ, PKCγ, PDK1, and RSKL1 are up-regulated in WM-115 cells, whereas only MSK1, MSK2, and PKN1 are up-regulated in WM-266–4 cells. The distinct kinome expression profiles between these two cell lines suggest considerable kinome reprogramming during melanoma progression.
Among the differentially expressed tyrosine kinases, multiple receptor tyrosine kinases in the Eph family are up-regulated in WM-115 cells. For instance, EphA2, EphB2 and EphB3 are expressed at much higher levels in WM-115 than WM-266–4 cells (Fig. 6A). On the other hand, CMGC is the only kinase group where significantly more members are up-regulated in WM-266–4 than WM-115 cells. In this vein, CDK1 and CDK2 are expressed at levels that are 4.5- and 3.6-fold higher in the former than latter cells (Fig. 6A). The differential expression of EphA2 and CDK2 in these two cell lines was also validated by Western analysis (Fig. 6B).
Fig. 6.
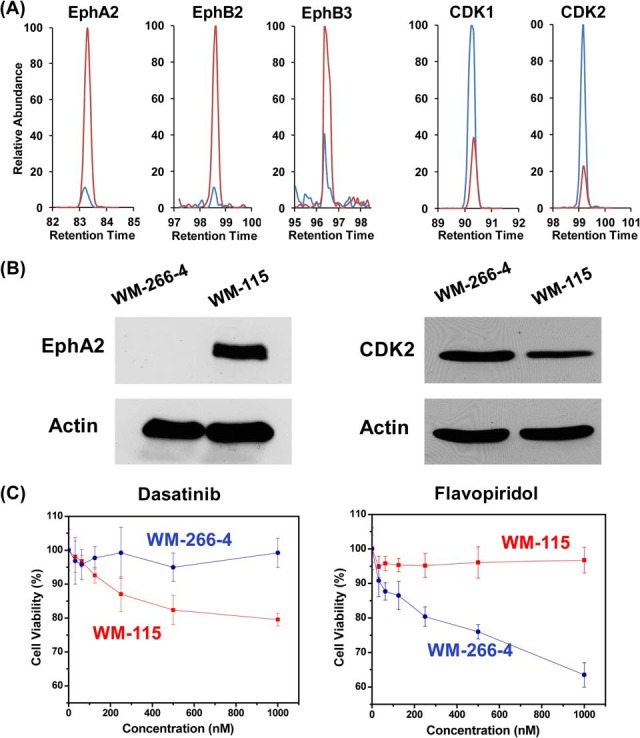
Eph tyrosine family kinases and CDKs are differentially expressed in WM-115 and WM-266-4 cells, which confers distinct sensitivities of the two lines of cells toward kinase inhibitors. A, Representative quantitative results by MRM assay in forward labeling reaction for peptide VLEDDPEATYTTSGGK#IPIR from EphA2, FLEDDTSDPTYTSALGGK#IPIR from EphB2, and FLEENSSDPTYTSSLGGK#IPIR from EphB3; peptide DLK#PNLLIDDK from CDK1, and DLK#PQNLLINTEGAIK from CDK2. Extracted ion chromatograms of light-labeled peptides from WM-115 cells are depicted in red and extracted ion chromatograms of heavy-labeled peptides from WM-266–4 cells are depicted in blue. B, Total cell lysates of WM-115 and WM-266–4 were immunoblotted with antibodies recognizing EphA2 (left) and CDK2 (right), where actin served as a loading control. C, Cell viability of WM-266–4 (blue line) and WM-115 cells (red line) when treated with dasatinib (left panel) and flavopiridol (right panel).
The differential expression of the aforementioned kinases in these two melanoma cell lines may confer different sensitivities of these two lines of cells toward the inhibitors for these kinases. In this regard, dasatinib is a potent inhibitor for EphA2, EphB3, EphB4, with dissociation constants (Kd) being < 1 nm; dasatinib, however, is a relatively poor inhibitor for those tyrosine kinases that are up-regulated in WM-266–4 cells, which included FES (Kd > 10 μm) and FGFR1 (Kd > 3.7 μm) (5). We assessed the relative sensitivities of WM-115 and WM-266–4 cells toward dasatinib, and our results revealed that WM-115 cells are markedly more sensitive toward this tyrosine kinase inhibitor than WM-266–4 cells (Fig. 6C). This finding is reminiscent of a previous study showing that dasatinib could inhibit the growth and reduce the migration and invasion of WM-115 cells, but not WM-266–4 cells (27).
We reason that the substantially higher levels of expression of CDK1 and CDK2 in the WM-266–4 than WM-115 cells may render WM-266–4 cells more sensitive toward CDK inhibitors than WM-115 cells. To test this, we examined whether flavopiridol, which is the first cyclin-dependent kinase inhibitor in human clinical trials and could potently inhibit CDKs 1, 2, and 4 (28), can preferentially inhibit the proliferation of WM-266–4 over WM-115 cells. It turned out that flavopiridol can effectively inhibit the growth of WM-266–4 cells at nm concentration range and ∼40% cell death was observed at 1 μm (Fig. 6C). In contrast, no appreciable sensitivity toward flavopiridol was observed for WM-115 cells, even at the highest concentration tested (1 μm, Fig. 6C). Together, our results demonstrate that the anti-proliferative effects of kinase inhibitors are correlated with the expression or ATP-binding affinity of their targeted kinases.
4. Application of MRM-based Kinome Profiling for Human Tissues
An advantage of the ICAP-based strategy lies in its potential application for kinome profiling for any biological samples, including clinical samples that are not amenable to metabolic labeling (29). To exploit this, we employed our MRM-based kinome assay to assess the differential kinase expression between human lung tumor and adjacent normal lung tissues from the same patient.
We successfully quantified 98 unique kinase peptides, which represent 124 kinases on our list (supplementary Table S4). In addition, results from our MRM kinome assay for human tissue samples again exhibit excellent reproducibility among three independent labeling experiments. In this vein, 96 out of the 98 (98%) kinase peptides were quantified in all three labeling experiments (supplementary Fig. S7), and most quantified kinases exhibit consistent trends in all three experiments (Fig. 7A). Additionally, most kinases display elevated expression in lung tumor tissue than adjacent normal tissue (Fig. 7A), which is a common phenomenon for tumor tissues (3). For instance, a unique peptide from MAPK3 (Erk1), i.e., DLK#PSNLLINTTCDLK, showed a higher level of expression in lung tumor than adjacent normal lung tissue in all three labeling experiments (Fig. 7B). Although this proof-of-concept experiment for a single pair of human lung tissue samples does not allow us to draw a general conclusion about the implication of this and other aberrantly expressed kinases in lung cancer, our results clearly demonstrate the potential of our MRM kinome assay in quantitative study of tissue samples.
Fig. 7.

MRM-based global kinome profiling revealed differential expression of kinases in lung tumor and adjacent normal lung tissue. A, A heatmap showing the differential expression of kinases from tumor and adjacent normal lung tissue based on Rtumor/normal ratio in two forward and one reverse labeling reactions. Dark red and white boxes designate those kinases that are up-regulated in tumor tissue and normal tissue, respectively, as indicated by the scale bar. B, Quantitative results by MRM assay for peptide DLK#PSNLLINTTCDLK from MAPK3 kinase: (Left and Middle) Extracted ion chromatograms for three transitions monitored for light-labeled (Red) and heavy-labeled (Blue) peptides in both forward and reverse labeling reaction; (Right) the consistent distribution of the peak area observed for each monitored transition from light- and heavy- labeled peptides in both forward and reverse labeling reaction along with the theoretical distribution derived from MS/MS spectra stored in MRM kinome library.
It is of note that the number of kinases quantified for the human tissue sample was much less than those for lysates of cultured human cells. By analyzing the same tissue sample in DDA mode on the LTQ Orbitrap Velos, we found that the signal is dominated with highly abundant blood proteins, especially the different isoforms of human hemoglobin and albumin. The compromised performance of ICAP reagents for human tissue sample could be attributed to the inherent design of this probe. The ICAP reagents react with proteins that can bind to ATP; as a result, the ICAP probes target not only kinases, but also any other ATP-binding proteins. Unfortunately, highly abundant proteins including albumin and hemoglobin in human tissue and plasma also possess strong ATP-binding affinity (30, 31). These proteins, therefore, compete favorably with low-abundance kinases in almost every step of the sample processing and analysis, such as probe binding and conjugation, avidin enrichment and MS detection. Nevertheless, we reason that depletion of highly abundant proteins from tissue samples, as has been widely used for improving the coverage of low-abundance proteins for biomarker discovery using human tissue or plasma samples (32), should enable the profiling of much more kinases from tissue lysates.
CONCLUSIONS
Kinases are among the most intensively pursued superfamilies of enzymes as targets for anti-cancer drugs. Recently large data sets on inhibitor potency and selectivity for more than 400 human kinases became available (5–7), which provided the opportunity to design rationally novel kinase-directed anti-cancer therapies. However, the kinase expression and activity are highly heterogeneous among different types of cancer and even among different stages of the same cancer, which point to the needs for global kinome expression profile in the development of kinase-targeted cancer therapy. Here, we introduced an MRM-based kinome profiling assay for more than 300 human protein and lipid kinases by monitoring specifically targeted peptides located at the ATP-binding sites of the kinases. In particular, we demonstrated that ∼ 250 protein kinases (50% of the entire human kinome and more than 80% of human kinome in a single cell line) could be routinely quantified without extensive separation using multi-dimensional chromatography. Our on-going discovery-based analysis of kinases labeled using our ICAP reagents for different cell lines will further expand our kinome library. Moreover, our studies showed that the MRM-based kinome assay exhibits superior sensitivity, reproducibility and accuracy over discovery-based shotgun proteomics. Importantly, the incorporation of iRT into MRM kinome library renders our MRM kinome assay easily transferrable across different chromatographic systems and laboratories. This MRM-based approach for kinome expression profiling of two melanoma cell lines derived from the same patient revealed that the anti-proliferative effects of kinase inhibitor drugs are correlated with the expression levels of their targeted kinases. Therefore, this facile and accurate kinome profiling assay, together with the kinome-inhibitor interaction map, may provide invaluable knowledge to predict the effectiveness of kinase inhibitor drugs and offer the opportunity for individualized therapy in cancer treatment.
We also demonstrated that this MRM-based kinome assay is applicable to clinical samples that are not amenable to metabolic isotope labeling. We believe that the depletion of highly abundant proteins from tissue samples prior to the ICAP labeling will further improve kinome coverage for tissue samples. It can be envisaged that our method could be used for future global kinome profiling of human tumor biopsy samples, which may guide the design of kinase-targeted, individualized anti-cancer therapy (33, 34).
Supplementary Material
Acknowledgments
We thank the National Disease Research Interchange (NDRI) for supplying the lung tissue samples.
Footnotes
Author contributions: Y.X. and Y.W. designed research; Y.X. and L.G. performed research; Y.X., L.G., and Y.W. analyzed data; Y.X. and Y.W. wrote the paper.
* This work was supported by the National Institutes of Health (R01 DK082779 to Y.W.) and Y.X. was supported by a Dissertation Research Award from the California Tobacco-Related Disease Research Program (20DT-0040).
 This article contains supplemental Figs. S1 to S7 and Tables S1 to S4.
This article contains supplemental Figs. S1 to S7 and Tables S1 to S4.
1 The abbreviations used are:
- PTM
- post-translational modifications
- MRM
- multiple reation monitoring
- RT
- retention time
- iRT
- normalized retention time.
REFERENCES
- 1. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 2. Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 3. Duncan James S., Whittle Martin C., Nakamura K., Abell Amy N., Midland Alicia A., Zawistowski Jon S., Johnson Nancy L., Granger Deborah A., Jordan Nicole V., Darr David B., Usary J., Kuan P.-F., Smalley David M., Major B., He X., Hoadley Katherine A., Zhou B., Sharpless Norman E., Perou Charles M., Kim William Y., Gomez Shawn M., Chen X., Jin J., Frye Stephen V., Earp H. S., Graves Lee M., Johnson Gary L. (2012) Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang J., Yang P. L., Gray N. S. (2009) Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39 [DOI] [PubMed] [Google Scholar]
- 5. Karaman M. W., Herrgard S., Treiber D. K., Gallant P., Atteridge C. E., Campbell B. T., Chan K. W., Ciceri P., Davis M. I., Edeen P. T., Faraoni R., Floyd M., Hunt J. P., Lockhart D. J., Milanov Z. V., Morrison M. J., Pallares G., Patel H. K., Pritchard S., Wodicka L. M., Zarrinkar P. P. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotech. 26, 127–132 [DOI] [PubMed] [Google Scholar]
- 6. Fabian M. A., Biggs W. H., Treiber D. K., Atteridge C. E., Azimioara M. D., Benedetti M. G., Carter T. A., Ciceri P., Edeen P. T., Floyd M., Ford J. M., Galvin M., Gerlach J. L., Grotzfeld R. M., Herrgard S., Insko D. E., Insko M. A., Lai A. G., Lelias J.-M., Mehta S. A., Milanov Z. V., Velasco A. M., Wodicka L. M., Patel H. K., Zarrinkar P. P., Lockhart D. J. (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotech. 23, 329–336 [DOI] [PubMed] [Google Scholar]
- 7. Davis M. I., Hunt J. P., Herrgard S., Ciceri P., Wodicka L. M., Pallares G., Hocker M., Treiber D. K., Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotech. 29, 1046–1051 [DOI] [PubMed] [Google Scholar]
- 8. Pulford K., Lamant L., Morris S. W., Butler L. H., Wood K. M., Stroud D., Delsol G., Mason D. Y. (1997) Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 89, 1394–1404 [PubMed] [Google Scholar]
- 9. Cox J., Mann M. (2011) Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 80, 273–299 [DOI] [PubMed] [Google Scholar]
- 10. Oppermann F. S., Gnad F., Olsen J. V., Hornberger R., Mann M., Daub H. (2009) Large-scale proteomics analysis of the human kinome. Mol. Cell. Proteomics 8, 1751–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marx V. (2013) Targeted proteomics. Nat. Meth. 10, 19–22 [DOI] [PubMed] [Google Scholar]
- 12. Daub H., Olsen J. V., Bairlein M., Gnad F., Oppermann F. S., Stemmann O., Mann M. (2008) Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell. 31, 438–448 [DOI] [PubMed] [Google Scholar]
- 13. Villamor J. G., Kaschani F., Colby T., Oeljeklaus J., Zhao D., Kaiser M., Patricelli M. P., van der Hoorn R. A. L. (2013) Profiling protein kinases and other ATP binding proteins in arabidopsis using Acyl-ATP Probes. Mol. Cell. Proteomics 12, 2481–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ansong C., Ortega C., Payne Samuel H., Haft Daniel H., Chauvignè-Hines Lacie M., Lewis Michael P., Ollodart Anja R., Purvine Samuel O., Shukla Anil K., Fortuin S., Smith Richard D., Adkins Joshua N., Grundner C., Wright Aaron T. (2013) Identification of widespread adenosine nucleotide binding in mycobacterium tuberculosis. Chem. Biol. 20, 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xiao Y., Guo L., Jiang X., Wang Y. (2013) Proteome-wide discovery and characterizations of nucleotide-binding proteins with affinity-labeled chemical probes. Anal. Chem. 85, 3198–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patricelli M. P., Szardenings A. K., Liyanage M., Nomanbhoy T. K., Wu M., Weissig H., Aban A., Chun D., Tanner S., Kozarich J. W. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 46, 350–358 [DOI] [PubMed] [Google Scholar]
- 17. Patricelli Matthew P., Nomanbhoy Tyzoon K., Wu J., Brown H., Zhou D., Zhang J., Jagannathan S., Aban A., Okerberg E., Herring C., Nordin B., Weissig H., Yang Q., Lee J.-D., Gray Nathanael S., Kozarich John W. (2011) In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 18, 699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olsen J. V., Schwartz J. C., Griep-Raming J., Nielsen M. L., Damoc E., Denisov E., Lange O., Remes P., Taylor D., Splendore M., Wouters E. R., Senko M., Makarov A., Mann M., Horning S. (2009) A dual pressure linear ion trap orbitrap instrument with very high sequencing speed. Mol. Cell. Proteomics 8, 2759–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Picotti P., Aebersold R. (2012) Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Meth. 9, 555–566 [DOI] [PubMed] [Google Scholar]
- 20. Lange V., Picotti P., Domon B., Aebersold R. (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol., 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiao Y., Guo L., Wang Y. (2013) Isotope-coded ATP probe for quantitative affinity profiling of ATP-binding proteins. Anal. Chem. 85, 7478–7486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M. J., Rinner O. (2012) Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 12, 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abbatiello S. E., Mani D. R., Keshishian H., Carr S. A. (2010) Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin. Chem. 56, 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. James L. C., Tawfik D. S. (2003) The specificity of cross-reactivity: Promiscuous antibody binding involves specific hydrogen bonds rather than nonspecific hydrophobic stickiness. Protein. Sci. 12, 2183–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Westermark B., Johnsson A., Paulsson Y., Betsholtz C., Heldin C. H., Herlyn M., Rodeck U., Koprowski H. (1986) Human melanoma cell lines of primary and metastatic origin express the genes encoding the chains of platelet-derived growth factor (PDGF) and produce a PDGF-like growth factor. Proc. Natl. Acad. Sci. U.S.A. 83, 7197–7200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eustace A. J., Dowling P., Henry M., Doolan P., Meleady P., Clynes M., Crown J., O'Donovan N. (2011) 2D-DIGE analysis of phospho-enriched fractions from dasatinib-treated melanoma cell lines. J. Proteomics 74, 490–501 [DOI] [PubMed] [Google Scholar]
- 28. Senderowicz A. (1999) Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest. New Drugs 17, 313–320 [DOI] [PubMed] [Google Scholar]
- 29. Mann M. (2006) Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 7, 952–958 [DOI] [PubMed] [Google Scholar]
- 30. Bauer M., Baumann J., Trommer W. E. (1992) ATP binding to bovine serum albumin. FEBS Lett. 313, 288–290 [DOI] [PubMed] [Google Scholar]
- 31. Costello A. J. R., Marshall W. E., Omachi A., Henderson T. O. (1977) ATP binding to human hemoglobin in the presence and absence of magnesium ions investigated with 31P NMR spectroscopy and ultrafiltration. Biochim. Biophys. Acta 491, 469–472 [DOI] [PubMed] [Google Scholar]
- 32. Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A. (2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chene P. (2002) ATPases as drug targets: learning from their structure. Nat. Rev. Drug Discov. 1, 665–673 [DOI] [PubMed] [Google Scholar]
- 34. Garman K. S., Nevins J. R., Potti A. (2007) Genomic strategies for personalized cancer therapy. Hum. Mol. Genet. 16, R226–R232 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.