

Abstract
DNA transposases catalyze the movement of transposons around genomes by a cut-and-paste mechanism related to retroviral integration. Transposases and retroviral integrases share a common RNaseH-like domain with a catalytic DDE/D triad that coordinates the divalent cations required for DNA cleavage and integration. The anti-retroviral drugs Raltegravir and Elvitegravir inhibit integrases by displacing viral DNA ends from the catalytic metal ions. We demonstrate that Raltegravir, but not Elvitegravir, binds to Mos1 transposase in the presence of Mg2+ or Mn2+, without the requirement for transposon DNA, and inhibits transposon cleavage and DNA integration in biochemical assays. Crystal structures at 1.7 Å resolution show Raltegravir, in common with integrases, coordinating two Mg2+ or Mn2+ ions in the Mos1 active site. However, in the absence of transposon ends, the drug adopts an unusual, compact binding mode distinct from that observed in the active site of the prototype foamy virus integrase.
Transposons and viruses are mobile genetic elements that survive and propagate by integrating into their hosts’ genomes. DNA transposons are cut out from one genomic location and pasted into another by a DNA transposase, often encoded within the transposon sequence. This genetic rearrangement provides a driving force for genomic variation and evolution but can also generate genomic instability. Some transposons have become domesticated within their host’s genome and provide useful new functions: for example the V(D)J recombination system, which generates antibody diversity, and the methyltransferase-DNA transposase fusion protein SETMAR involved in DNA repair.1,2
The mechanism of DNA transposition is closely related to the integration of retroviruses, such as human immunodeficiency virus 1 (HIV-1). DNA transposases specifically recognize short inverted repeat (IR) sequences that mark the transposon ends. Excision of the transposon and its integration at a new genomic site is coordinated within a nucleoprotein complex, the transpososome, in which the two transposon ends are paired. Likewise, viral DNA ends contain long terminal repeat (LTR) sequences that are recognized specifically by a retroviral integrase and are brought together in a nucleoprotein complex, the intasome. The integrase cleaves two nucleotides from the reactive DNA strand before joining the processed viral ends irreversibly to the host’s genome.
The mechanistic similarities of DNA transposases and retroviral integrases are reflected in common active site architectures and similar structural features.3,4 The catalytic core domains of these enzymes adopt a RNase-H like fold5 bringing together a triad of catalytic acidic amino acids: the DDE/D motif. The carboxylate oxygens coordinate the Mg2+ or Mn2+ ions required for DNA cleavage and integration.6 A number of crystal structures of isolated catalytic core domains of DNA transposases and integrases have been determined: these include the active mariner family transposase Mos1 (from Drosophila mauritiana(7)), the closely related mariner transposase catalytic domain of SETMAR (human8), and the retroviral integrase HIV-19,10 (shown in Figure 1). Furthermore there are crystal structures of the Mos1,11 Tn5,12 and Mu13 transpososomes and the Spumavirus Prototype Foamy Virus (PFV) intasome,14 each of which contains the full length enzyme in a synaptic complex with two cognate DNA ends.
Figure 1.

The catalytic domains of mariner DNA transposases and retroviral integrases adopt a common Rnase-H like fold. Catalytic core domain structures of (a) Mos1 transposase (PDB ID: 2F7T), (b) SETMAR transposase domain (PDB ID: 3K9J), (c) HIV-1 integrase (PDB ID: 1BIS), and (d) PFV Integrase in the intasome complex (PDB ID: 3S3M). Residues of the DDD/N or DDE active site triads are labeled, along with the coordinated metal ions and conserved Tyr and Pro residues.
The Mos1 and human SETMAR mariner transposases show a higher degree of structural similarity compared with integrases (Figure 1 and Supplementary Figure 1). The active sites of HIV-1 and PFV integrase contain DD-35-E motifs, whereas the mariner family DNA transposase Mos1 active site has a DD-34-D triad. The SETMAR mariner transposase catalytic domain has a DD-34-N motif, which supports DNA cleavage and integration,15,16 and shares 38.7% sequence identity and 48.4% sequence similarity to Mos1. In all four enzymes the loop preceding the third catalytic residue contains conserved Tyr and Pro residues; these are Try276 and Pro278 in Mos1 Transposase (Tnp). In the Mos1 Tnp and SETMAR catalytic domain crystal structures, this loop is ordered due to its stabilizing interactions with the N- and C-terminal capping helices, α1 and α7 respectively (Figure 1). As a result the active sites are fully structured without DNA. By contrast, in the crystal structure of the isolated HIV-1 integrase catalytic core domain,10 the loop was disordered. NMR relaxation measurements indicated that loop residues are dynamic, moving between several distinct conformational clusters.17 This is consistent with the proposal that the integrase active site does not adopt a well-defined conformation, capable of binding divalent metal ions and inhibitor, until the integrase has assembled on viral ends.18
The DNA integration step of the retroviral life cycle has been targeted for the development of anti-retroviral therapies. Currently, several integrase strand transfer inhibitors (INSTIs) are available or in development for the treatment of HIV-1 infections, including Raltegravir19 and Elvitegravir.20 Attempts to crystallize the HIV-1 integrase with viral DNA ends have so far been unsuccessful, and surrogate models have been sought in order to better understand how INSTIs act. The Tn5 transposase was considered as a model system, because of the wealth of structural information on its interactions with inverted repeat DNA. Several diketoacid HIV-1 integrase inhibitors were identified using Tn5 transposase as the target in a chemical library screen;21 these affected cleavage, synapsis, or integration steps of Tn5 transposition.22 More recently, the PFV integrase, a closer relative of the HIV-1 enzyme, has provided an amenable model system.23−25 Co-crystal structures of the PFV intasome with Raltegravir or Elvitegravir revealed that the drugs inhibit viral DNA integration by coordinating two divalent metal ions bound to the DDE motif carboxylates and displacing the reactive viral DNA end from the active site.14,26
Recently it was shown that Raltegravir and Elvitegravir can also inhibit the nuclease activity of the transposase domain within the human fusion protein SETMAR.27 We asked if Raltegravir and Elvitegravir could bind to and inhibit the DNA cleavage and integration activities of Mos1,28 an active mariner family transposase closely related to the transposase domain of SETMAR.2 Here we show that Raltegravir binds to Mos1 transposase and that divalent metal ions, but not transposon IR DNA, are required for binding. By contrast Elvitegravir did not bind to Mos1 Tnp. We have determined crystal structures of Raltegravir bound in the active site of the Mos1 Tnp catalytic domain in the presence of Mg2+ or Mn2+, to a resolution of 1.7 Å. The drug adopts a distinct, compact, and curved binding mode that contrasts markedly with the conformation of the drug in the viral DNA-bound PFV intasome structures. Furthermore, Raltegravir inhibits Mos1 in vitro DNA cleavage and integration. Our structural results for Mos1-Raltegravir interactions can potentially be used as a surrogate model for development of drugs to target the transposase domain of SETMAR. Inhibiting the DNA repair functions of SETMAR in this way could be used to augment current chemotherapies.
Results and Discussion
Mos1 Transposase Is Stabilized by Divalent Metal Ions
In previous crystallographic analyses7 of the Mos1 catalytic domain, we observed one metal ion bound in the active site when 5 mM MgCl2 or MnCl2 was included in the crystallization conditions; this was coordinated by the carboxylate oxygen atoms of Asp 156 and Asp 249, in site 1. When we increased the MnCl2 concentration to 20 mM, a second Mn2+ was bound to Asp 156 and Asp 284, in site 2.7 To establish the extent of stabilization of Mos1 Tnp by divalent metal ions, we performed thermal denaturation assays29 to determine the melting temperatures (Tm) of Mos1 Tnp at different metal ion concentrations (Figure 2a). In the absence of divalent metal ions, the Tm of Mos1 Tnp was 40.5 °C. Upon addition of MgCl2 or MnCl2 (5 mM, 20 mM or 50 mM) the Tm increased by up to 10.0 °C to a maximum of 50.5 °C in 20 mM MnCl2. These results reflect stabilization of transposase by the metal ions. A concentration-dependent increase in transposase thermal stability was observed for the Mg2+ series. The larger increases in Tm measured with MnCl2, compared to equivalent concentrations of MgCl2, indicate that Mn2+ stabilizes Mos1 transposase to a greater extent than Mg2+.
Figure 2.

Thermal stability of Mos1 transposase. (a) Mos1 transposase is stabilized by MgCl2 and MnCl2. The melting temperature (Tm) and standard deviations (s.d.) were calculated as the mean of three measurements. The chemical structures of (b) Raltegravir and (c) Elvitegravir from http://www.chemspider.com. (d) Effect of increasing Raltegravir or Elvitegravir concentrations on the Tm of Mos1 transposase. Reactions contained 20 mM MgCl2 or MnCl2 as indicated.
Raltegravir Binds to Mos1 Transposase
Next, we investigated the effect of Raltegravir (Figure 2b) and Elvitegravir (Figure 2c) on the thermal stability of Mos1 Tnp. Raltegravir (5 μM to 1 mM) or Elvitegravir (5 μM to 100 μM) was incubated with Mos1 (4 μM) in the presence or absence of either MgCl2 (20 mM) or MnCl2 (20 mM). The addition of 1 mM Raltegravir and 20 mM MnCl2 increased the Tm of Mos1 Tnp to 61 °C (Figure 2d). However, Raltegravir did not induce a change in the Tm in the absence of MgCl2 or MnCl2. These data indicate that Raltegravir binds to Mos1 Tnp and that the interaction requires divalent cations. By contrast, Elvitegravir induced little or no change in the Tm of Mos1 Tnp, suggesting there is no binding in the conditions tested.
Raltegravir Binds Mos1 in an Unusual Compact Conformation
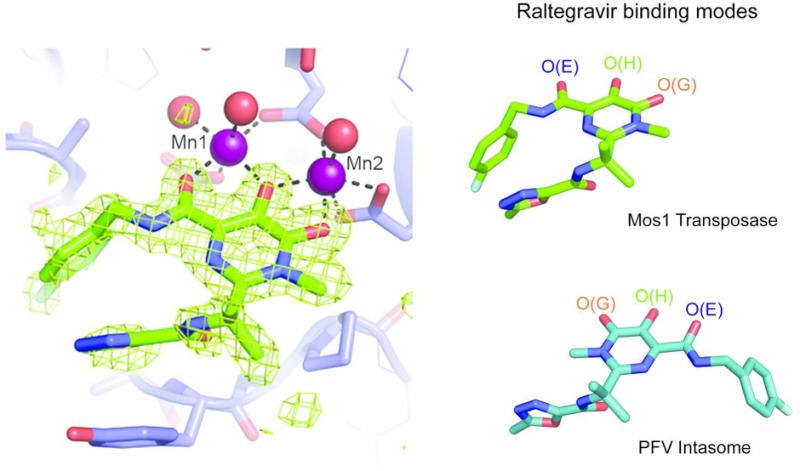
To establish the conformation of Raltegravir bound to Mos1 transposase, we soaked crystals of the Mos1 transposase catalytic domain, grown in either 10 mM MgCl2 or 10 mM MnCl2, with 1 mM Raltegravir, as detailed in Methods. Crystals diffracted X-rays to 1.7 Å resolution (Table 1), and structures were determined by molecular replacement using the structure of the Mos1 catalytic domain (PDBID: 2F7T) as the model. The initial 2Fo – Fc map contained clear additional electron density near the active site into which Raltegravir was built. The structures were refined to a final Rfree of 24.1% and 24.2% for the structures containing Mg2+ and Mn2+, respectively (Figure 3a–c, Table 1). We also collected diffraction data from crystals grown in MnCl2 at the Mn K-edge (λ = 1.896 Å, Table 1), and the peaks in an anomalous difference map confirmed the positions of the two Mn2+ ions in the active site (Figure 3b).
Table 1. X-ray Data Collection, Scaling, and Refinement Statistics.
| crystal |
||||||
|---|---|---|---|---|---|---|
| Mos1 + Raltegravir (10 mM MgCl2 ) | Mos1 + Raltegravir (10 mM MnCl2 ) | |||||
| PDB ID | 4MDB | 4MDA | ||||
| space group | P4(1)2(1)2 | P4(1)2(1)2 | ||||
| cell dimensions | a,b = 44.2, c = 206.3 Å | a,b = 44.6, c = 209.6 Å | ||||
| wavelength (Å) | 0.9795 | 0.9795 | 1.8961 | |||
| overall | outer shell | overall | outer shell | overall | outer shell | |
| resolution (Å) | 44.2–1.7 | 1.8–1.7 | 43.6–1.7 | 1.79–1.7 | 43.6–2.13 | 2.25–2.13 |
| Rmerge | 0.065 | 0.302 | 0.071 | 0.248 | 0.066 | 0.138 |
| total observations | 129702 | 17666 | 166470 | 24181 | 106759 | 15206 |
| unique observations | 23539 | 3320 | 24487 | 3466 | 12671 | 1794 |
| ⟨I⟩/σ⟨I⟩ | 14.5 | 4.7 | 16.7 | 6.6 | 21.9 | 13.0 |
| completeness (%) | 99.7 | 99.9 | 100.0 | 100.0 | 99.9 | 100.0 |
| multiplicity | 5.5 | 5.3 | 6.8 | 7.0 | 8.4 | 8.5 |
| anomalous completeness (%) | 99.9 | 100.0 | ||||
| anomalous multiplicity | 4.6 | 4.5 | ||||
| Rwork | 0.197 | 0.197 | ||||
| Rfree | 0.241 | 0.242 | ||||
| rmsd from ideality | ||||||
| bond length (Å) | 0.019 | 0.019 | ||||
| bond angle (deg) | 2.08 | 2.05 | ||||
| chirality (Å) | 0.232 | 0.259 | ||||
| Ramachandran plot | ||||||
| preferred (%) | 97.9 | 96.7 | ||||
| allowed (%) | 2.1 | 3.3 | ||||
| outliers (%) | 0 | 0 | ||||
| av B factor (Å2) | 25.7 | 19.6 | ||||
| no. of metal ions | 2 | 2 | ||||
Figure 3.

X-ray crystal structures of Raltegravir bound to the Mos1 transposase catalytic domain. (a) Active site of Mos1 transposase (blue) with Raltegravir (green sticks) bound to Mn2+ ions (purple spheres). The omit map of electron density (green mesh) is contoured at 3σ. Water molecules coordinated to the metal ions are displayed as red spheres. (b) Octahedral coordination of manganese ions in the active site, with anomalous difference electron density map contoured at 2.5σ (pink mesh). (c) Active site of the magnesium ion bound structure, with magnesium ions shown as black spheres. Conformations of Raltegravir bound to (d) Mos1 transposase catalytic domain and (e) the PFV intasome (PDB ID: 3OYA). Rotation about the CBC–CBF bond (indicated by a black arrow) would interconvert these two conformations. (f) Conformation of Elvitegravir bound to the PFV intasome (PDB ID: 3LSU).
Raltegravir adopts a compact, curved conformation in the Mos1 active site (Figure 3a). The fluorobenzyl ring is oriented on the same side as the methyl-oxadiazole group and fills a hydrophobic pocket on Mos1 Tnp lined by Ala 251 and Pro 252. The six-membered fluorobenzyl ring tops a four-tiered aromatic ring stack, which also includes the five-membered oxadiazole moiety of Raltegravir and the side chains of Tyr 276 and His 122 of Mos1 Tnp (Figure 3a). In addition, the Raltegravir isopropyl group makes hydrophobic contacts with the conserved residue Pro 278. A similar “folded-over” binding mode was predicted for Raltegravir docked in the HIV-1 integrase active site in the absence of viral DNA, using computational methods.30
Our crystal structures show that the carboxylate side chains of the active site aspartic acid triad coordinate two divalent metal ions (Figure 3a–c). Three coplanar oxygen atoms of the Raltegravir diketo acid moiety chelate Mg2+ or Mn2+ ions in the active site, explaining the requirement for metal ions for drug binding. The central oxygen, O(H), bridges both metal ions, whereas O(E) coordinates the site 1 metal ion and O(G) coordinates the metal ion in site 2 (Figure 3b,c). Both metal ions exhibit octahedral coordination geometry. Coordination of the site 1 metal ion is completed by monodentate interactions with Asp 249 and Asp 156 side chains, with additional coordinating atoms from two water molecules. The second carboxylate oxygen of Asp 156 chelates the metal in site 2, and the coordination geometry is completed by bidentate interactions with the carboxylates of Asp 284 and one water molecule.
Previously both Raltegravir and Elvitegravir were found to be inactive against the Tc1 transposase Sleeping Beauty.31 Comparison of the sequences of Mos1 and Sleeping Beauty transposases reveals that key residues of the Mos1 catalytic domain that are involved in stacking interactions of the Raltegravir oxidiazole group in our crystal structure are not conserved in Sleeping Beauty transposase (Supplementary Figure 1). These include Tyr 276, His 122, and Ala 251 in Mos1, which are Gln 270, Pro 121, and Asp245 at the equivalent positions in the Sleeping Beauty transposase sequence. This suggests a molecular explanation for the observed inability of Raltegravir and Elvitegravir to inhibit Sleeping Beauty transposition.31
Raltegravir Has a Different Binding Mode in Mos1 Tnp Compared with the PFV Intasome
The Mos1-bound conformation of Raltegravir (Figure 3d) contrasts markedly with its elongated conformation when bound to PFV integrase with viral DNA ends14 (Figure 3e). In the PFV intasome bound conformation, the fluorobenzyl group is distant from the oxadiazole group and occupies a tight pocket created by displacement of the 3′ adenosine of the viral DNA strand where it stacks with the penultimate base-pair of the viral DNA duplex. Elvitegravir adopts a similar conformation when bound to the PFV intasome (Figure 3f).
The two distinct conformations of Raltegravir bound to Mos1 or the PFV intasome (Figure 3d–e) are related by a rotation of ∼180° about the Raltegravir CBC–CBF bond, as shown by the arrow in Figure 3e. This rotation flips the position of the fluorobenzyl ring with respect to the oxadiazole and isopropyl groups, which are in similar positions in both structures. These groups make stacking and hydrophobic interactions with the side chains of the conserved Tyr and Pro loop residues in both binding modes: Tyr 276 and Pro 278 of Mos1 and Tyr 212 and Pro 214 of PFV integrase (Figure 4).
Figure 4.
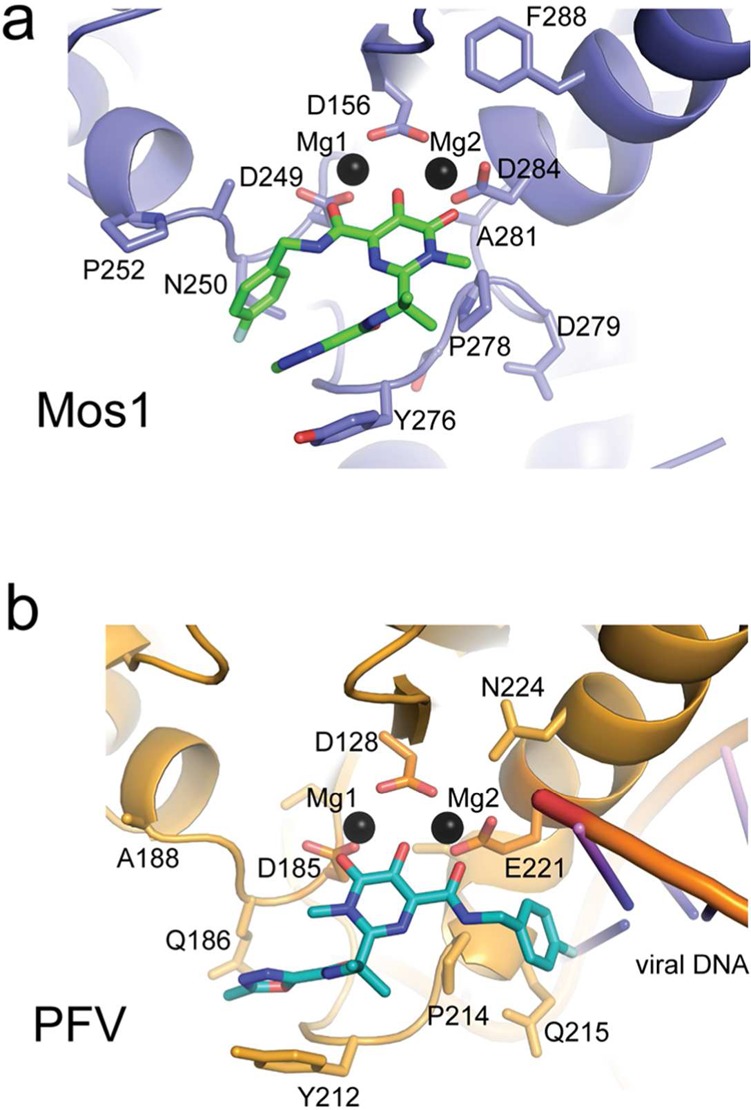
Comparison of the binding modes of Raltegravir in (a) the active site of the Mos1 catalytic domain and (b) the PFV intasome.
Rotation about the Raltegravir CBC–CBF bond also swaps the relative positions of the three metal-chelating oxygen atoms. However, their spatial arrangement is intact, enabling coordination of the two divalent metal ions in the active site of Mos1 transposase in a manner similar to that observed in the PFV intasome co-crystal structures, despite the drug adopting a distinctly different conformation. Thus, the rotational freedom within Raltegravir allows it to adopt a previously unappreciated bound conformation, highlighting that a drug can have distinct binding modes in subtly different molecular environments.
The chemical structure of Elvitegravir (Figure 2c) is inherently less flexible than that of Raltegravir (Figure 2b), and there is a different separation of the three, rigidly coplanar chelating oxygen atoms. It also lacks the oxadiazole group that forms key stacking interactions in the Raltegravir-bound Mos1 structure. Taken together, these factors may explain our observation that Elvitegravir did not bind to Mos1 transposase.
Raltegravir Binds to the Mos1 Paired-End Complex
Mos1 transposase recognizes specific IR sequences at each transposon end and brings them together in a paired-end complex (PEC) for DNA cleavage, before inserting the cleaved ends into TA dinucleotide sites on target DNA. To test if Raltegravir could also interact with and inhibit Mos1 Tnp bound to IR DNA, we first performed thermal denaturation assays using the Mos1 PEC. This complex was prepared using ‘precleaved’ IR DNA substrates as before.32 Similarly to Mos1 Tnp, the Mos1 PEC was stabilized by divalent metal ions (Figure 5a); the stabilization was greater with 20 mM MnCl2 (Tm = 55.5 °C) than with 20 mM MgCl2 (Tm = 49 °C). Raltegravir (100 or 400 μM) bound the Mos1 PEC in the presence of Mg2+ or Mn2+, increasing the Tm up to a maximum of 62.5 °C with 400 μM Raltegravir and 20 mM MnCl2. We also tested if Elvitegravir could bind to the Mos1 PEC. A small increase, of 1.75 °C, in the Tm of the Mos1 PEC in the presence of 400 μM Elvitegravir and 20 mM MgCl2 indicated that Elvitegravir may bind weakly in these conditions. However, no change in thermal stability of the PEC was observed with Elvitegravir in 20 mM MnCl2. Thus, Raltegravir binds to the Mos1 PEC in the presence of divalent metal ions.
Figure 5.

Raltegravir stabilizes the Mos1 PEC and inhibits Mos1 cleavage in vitro. (a) The melting temperature (Tm) of the Mos1 PEC measured in a thermal denaturation assay, without or with 20 mM MgCl2 or MnCl2, and in the absence or presence of Raltegravir or Elvitegravir at the concentrations indicated. The Tm and standard deviations (s.d.) were calculated as the mean of three measurements. (b) Agarose gel of the products of Mos1 IR plasmid cleavage reactions: without Mos1 transposase (lane 4), with Mos1 (lanes 5–10), and with increasing concentrations of Raltegravir (lanes 6–10). Supercoiled (sc) and open circle (oc) plasmids are indicated, and supercoiled plasmid is shown in lane 3. The plasmid linearized by restriction digestion is shown in lane 2. (c) Graph of the percentage of sc and oc plasmid in lanes 4–10 of the gel in panel b. (d) Agarose gel of the products of Mos1 IR plasmid cleavage reactions; without Mos1 transposase (lane 3), with Mos1 (lanes 4–9), and with increasing concentrations of Elvitegravir (lanes 5–9). (e) Graph of the percentage of sc and oc plasmid in lanes 3–9 of the gel in panel d.
Raltegravir Inhibits Mos1 Cleavage and Strand Transfer
To establish if Raltegravir or Elvitegravir inhibit the DNA cleavage activity of Mos1 transposase, we performed a plasmid cleavage assay.33 The 5.6 kb supercoiled plasmid contains Mos1 transposon IRs. After incubation with Mos1 transposase, ∼70% of the supercoiled plasmid is relaxed or linearized by transposase DNA cleavage (Figure 5b, lane 5). Mos1 cleavage activity is inhibited by 10 to 500 μM Raltegravir (Figure 5b, lanes 6–10) with an IC50 of 60–70 μM as estimated from quantification of the % of supercoiled plasmid in each reaction (Figure 5c). By contrast, the addition of Elvitegravir over the same concentration range (Figure 5d, lanes 5–9) had no significant effect on Mos1 cleavage activity or the % of supercoiled and open circle plasmid in each reaction (Figure 5e).
Next, we tested the effect of Raltegravir on Mos1 strand transfer by monitoring the integration of fluorescently labeled, precleaved transposon IR DNA substrates into a 50-mer target DNA duplex containing one TA dinucleotide (Figure 6a). Integration into the top or bottom strand of the target duplex produces two major strand transfer products, of 68 and 40 nt, respectively, which are then detected by denaturing PAGE (Figure 6b, lane 3). Other minor products result from integration into the IR DNA substrate, the sequence of which contains two TA dinucleotides. When Raltegravir is added (in the concentration range 1 to 100 μM), integration is inhibited (Figure 6b, lanes 4–10) with an IC50 of ∼2 μM (Figure 6c). This result implies that Raltegravir binds more tightly to the Mos1 PEC than to the transposase in the absence of DNA. The tighter binding could reflect additional interactions between precleaved IR DNA and the drug and/or a different Raltegravir binding mode; a co-crystal structure of Raltegravir and the Mos1 PEC would shed light on these possibilities.
Figure 6.

Raltegravir inhibits Mos1 strand transfer. (a) Schematic of the strand transfer assay. (b) Denaturing polyacrylamide gel showing the effect of increasing concentrations of Raltegravir on strand transfer activity of Mos1 transposase. Lane 1 shows oligonucleotide markers of length indicated to the left of the lane. The control reaction in lane 2 did not contain Mos1 transposase. (c) Graph of intensity of the product bands as a function of Raltegravir concentration (band intensities are normalized to the intensity of the 40 nt product without Raltegravir).
Both Raltegravir and Elvitegravir inhibit the in vitro DNA cleavage activity of the human DNA repair component fusion protein SETMAR,27 by binding to the active site of the mariner transposase catalytic domain. SETMAR is overexpressed in malignant cells34 (e.g., leukemia) and enhances the efficiency and accuracy of DNA repair by non-homologous end-joining.2 It has been proposed that targeting the nuclease activity of SETMAR with small molecules could augment current chemotherapies27 by inhibiting DNA repair. We have shown here that Raltegravir can also inhibit the in vitro DNA cleavage and integration of the mariner transposase Mos1. Given the wealth of structural and mechanistic understanding of the Mos1 DNA transposase and the close sequence and structural similarities between Mos1 and the transposase domain of SETMAR, the Mos1 transposase may provide an ideal model system to aid the development of drugs to target SETMAR.
Methods
Sequence Alignments
Structure-based sequence alignments of the transposase and integrase catalytic domains were performed using Expresso (35) and displayed using ESPript2.2.36
Purification of Mos1 Transposase
Mos1 transposase containing the solubilizing mutation T216A (referred to as Mos1 throughout) was expressed and purified as previously described37 with some changes. The protein was extracted from resuspended cells in a cell disruptor (Constant Systems, Ltd.) at 27 kPsi. After cation exchange chromatography of cleared lysate, the protein was further purified by hydrophobic interaction chromatography. Ammonium sulfate was added (to 1 M) before loading Transposase onto a HiTrap Phenyl HP column (GE Healthcare). Transposase was eluted using a decreasing ammonium sulfate gradient (1 to 0 M), then exchanged into 50 mM Tris pH7.5, 0.35 M KCl, 1 mM DTT, and concentrated using a 6 mL Vivaspin column (10 kDa cutoff). The purity of the protein was assessed by SDS-PAGE.
Annealing of IR DNA Duplex
Oligonucleotides were synthesized by Integrated DNA Technologies and dissolved in water prior to annealing. Duplex IR DNA containing the right inverted repeat sequence and with a 3 base overhang, corresponding to an excised transposon end, was prepared by annealing a 28-mer (5′-AAACGACATTTCATACTTGTACACCTGA-3′) and a complementary 25-mer (5′-GGTGTACAAGTATGAAATGTCGTTT-3′). For the target integration assays the 28-mer had an IRDye 700 fluorescent tag at the 5′ end.
Paired-End Complex Preparation
For paired-end complex formation, transposase and duplex IR DNA were mixed in the molar ratio 1 to 1.1, in buffer containing 25 mM Tris (pH 7.5) and 0.25 M NaCl. The protein was added in 10 μL aliquots to the DNA and mixed thoroughly between additions. Final concentrations of the PEC ranged from 20 to 200 nM.
Thermal Denaturation Assays
The thermal stability of Mos1 transposase and the Mos1 PEC was measured in an iCycler IQ5 Real Time Detection System (BioRad) thermal cycler. Each sample was prepared to a final volume of 50 μL in a 96-well plate and contained 4 μM purified Mos1 transposase or 6 μM PEC, 5x Sypro Orange (Sigma Aldrich), 50 mM Tris pH 7.5, 0.35 M KCl, 1 mM DTT, and metal ions (MgCl2 or MnCl2) at 0, 5, 20 or 50 mM. Thermal denaturation was also performed with samples containing Raltegravir (at 5, 20, 100, 500 μM or 1 mM) or Elvitegravir (at 5, 20, or 100 μM) with 20 mM MgCl2 or 20 mM MnCl2. Control reactions, containing 100 μM Raltegravir or 100 μM Elvitegravir, without metal ions were also measured.
Samples were heated from 20 to 80 °C in increments of 0.5 °C every 30 s. The fluorescence of Sypro Orange was excited at 485 nm and measured at 575 nm in relative fluorescence units (RFU). The sensitivity of Sypro Orange to the hydrophobicity of the chemical environment results in an increase in fluorescence intensity as the sample unfolds and hydrophobic residues are exposed. The transition unfolding temperature (Tm) of each sample was taken as the minimum value of the derivative −δRFU/δTemp. Each condition was measured in triplicate and an average Tm for each condition calculated.
Crystallization and Crystal Soaks
Crystals of the Mos1 catalytic domain were grown by hanging drop vapor diffusion as previously described.37 Purified Mos1 transposase at 8 mg mL–1 was mixed in the ratio 1:1 with well solution containing MgCl2 (5 mM) or MnCl2 (5 mM), 22% (w/v) PEG 4000, 100 mM Tris pH 6.8. Crystals appeared after incubation at 290 K for 10 days. For soaking experiments, crystals were transferred to a solution containing 10 mM MgCl2 or MnCl2, 22% (w/v) PEG 4000, 100 mM Tris pH 6.8, 20% (v/v) glycerol, and 1 mM Raltegravir and incubated over the well solution for 18 h. Crystals were then flash frozen in liquid nitrogen prior to data collection.
Structure Determination and Refinement
X-ray diffraction data were collected at beamline I02 at the Diamond Light Source. Images were collected on a ADSC Q315r detector, integrated with iMosflm, and scaled using SCALA within the CCP4 suite.38 Phases were calculated by molecular replacement in PHASER using the coordinates of the Mos1 catalytic domain (PDB ID: 2F7T) as the search model. Structure refinement was performed with REFMAC and the final data collection and refinement statistics are shown in Table 1. Stereoviews of the final Mg2+ (PDB ID: 4MDB) or Mn2+ (PDB ID: 4MDA) containing structures showing final 2Fo – Fc electron density maps are shown in Supplementary Figure 2 and Supplementary Figure 3, respectively.
Plasmid Cleavage Assays
Assays were performed in 20 μL reactions containing 500 ng of plasmid pEPMosRR (containing a 1.3 kB Kanamycin resistance gene flanked by Mos1 IRs, with a 4.3 kB pEP185.2 backbone), 48 nM Mos1 transposase, 25 mM Hepes pH 7.5, 0.1 M NaCl, 10% (v/v) glycerol, 12.5 μg mL–1 acetylated BSA, 2 mM DTT, and 10 mM MnCl2. Reactions were incubated at 30 °C for 20 min, stopped by the addition of EDTA to 10 mM, and separated on a 1% (w/v) agarose gel in 1x TAE buffer at 70 V for 2 h. DNA was stained using SafeView (NBS Biologicals) and visualized and quantified on a GelDoc EZImager (BioRad).
Target Integration Assays
A 50-mer target DNA substrate containing one TpA dinucleotide was prepared by annealing the 50 nt sequence 5′ AGCAGTCCACTAGTGCACGACCGTTCAAAGCTTCGGAACGGGACACTGTT with its complementary strand. Annealed target and IR DNA oligonucleotides were purified by HPLC. Assays were performed in 20 μL reactions containing 15 nM 50-mer target DNA, 1.5 nM IR DNA, and 15 nM Mos1 Transposase in buffer containing 25 mM Hepes pH 7.5, 50 mM potassium acetate, 10% (v/v) glycerol, 0.25 mM EDTA, 1 mM DTT, 10 mM MgCl2, 50 μg mL–1 BSA, and 20% (v/v) DMSO. Raltegravir was added to reactions as indicated. Reactions were incubated for 2 h at 30 °C, and the products were separated on an 8% denaturing polyacrylamide gel as described previously.11 To visualize the products, the IRDye 700 was excited at 680 nm and detected on a LI-COR Odyssey system. The fluorescence intensities of the product bands were quantified using Image Studio software.
Acknowledgments
X-ray data were collected at the Diamond Light Source (beam line I02), and we thank the beam line staff for their help and expertise. J.M.R. is funded by a Wellcome Trust University Award (085176/Z/08/Z), M.T. was supported by a Darwin Trust PhD studentship, and E.R.M. is funded by the BBSRC (BB/J000884/1). We thank A. Cook for critical comments on the manuscript.
Supporting Information Available
Supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The molecular coordinates of the Mg2+ and Mn2+ containing crystal structures of Raltegravir bound to the Mos1 transposase catalytic domain have been deposited in the protein data bank (PDB) with accession codes 4MDB and 4MDA, respectively.
The authors declare no competing financial interest.
Supplementary Material
References
- Robertson H. M.; Zumpano K. L. (1997) Molecular evolution of an ancient mariner transposon, Hsmar1, in the human genome. Gene 205, 203–217. [DOI] [PubMed] [Google Scholar]
- Shaheen M.; Williamson E.; Nickoloff J.; Lee S. H.; Hromas R. (2010) Metnase/SETMAR: a domesticated primate transposase that enhances DNA repair, replication, and decatenation. Genetica 138, 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano S. P.; Rice P. A. (2011) Moving DNA around: DNA transposition and retroviral integration. Curr. Opin. Struct. Biol. 21, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman A. B.; Chandler M.; Dyda F. (2010) Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit Rev. Biochem. Mol. Biol. 45, 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P. A.; Baker T. A. (2001) Comparative architecture of transposase and integrase complexes. Nat. Struct. Biol. 8, 302–307. [DOI] [PubMed] [Google Scholar]
- Nowotny M.; Gaidamakov S. A.; Crouch R. J.; Yang W. (2005) Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 121, 1005–1016. [DOI] [PubMed] [Google Scholar]
- Richardson J. M.; Dawson A.; O’Hagan N.; Taylor P.; Finnegan D. J.; Walkinshaw M. D. (2006) Mechanism of Mos1 transposition: insights from structural analysis. EMBO J. 25, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin K. D.; He H.; Imasaki T.; Lee S. H.; Georgiadis M. M. (2010) Crystal structure of the human Hsmar1-derived transposase domain in the DNA repair enzyme Metnase. Biochemistry 49, 5705–5713. [DOI] [PubMed] [Google Scholar]
- Dyda F.; Hickman A. B.; Jenkins T. M.; Engelman A.; Craigie R.; Davies D. R. (1994) Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 266, 1981–1986. [DOI] [PubMed] [Google Scholar]
- Goldgur Y.; Dyda F.; Hickman A. B.; Jenkins T. M.; Craigie R.; Davies D. R. (1998) Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc. Natl. Acad. Sci. U.S.A. 95, 9150–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson J. M.; Colloms S. D.; Finnegan D. J.; Walkinshaw M. D. (2009) Molecular architecture of the Mos1 paired-end complex: the structural basis of DNA transposition in a eukaryote. Cell 138, 1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies D. R.; Goryshin I. Y.; Reznikoff W. S.; Rayment I. (2000) Three-dimensional structure of the Tn5 synaptic complex transposition intermediate. Science 289, 77–85. [DOI] [PubMed] [Google Scholar]
- Montano S. P.; Pigli Y. Z.; Rice P. A. (2012) The mu transpososome structure sheds light on DDE recombinase evolution. Nature 491, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Gupta S. S.; Valkov E.; Engelman A.; Cherepanov P. (2010) Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H.; Oshige M.; Durant S. T.; Rasila K. K.; Williamson E. A.; Ramsey H.; Kwan L.; Nickoloff J. A.; Hromas R. (2005) The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl. Acad. Sci. U.S.A. 102, 18075–18080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Bischerour J.; Siddique A.; Buisine N.; Bigot Y.; Chalmers R. (2007) The human SETMAR protein preserves most of the activities of the ancestral Hsmar1 transposase. Mol. Cell. Biol. 27, 1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzkee N. C.; Masse J. E.; Shen Y.; Davies D. R.; Bax A. (2010) Solution conformation and dynamics of the HIV-1 integrase core domain. J. Biol. Chem. 285, 18072–18084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeseth A. S.; Felock P.; Wolfe A.; Witmer M.; Grobler J.; Anthony N.; Egbertson M.; Melamed J. Y.; Young S.; Hamill T.; Cole J. L.; Hazuda D. J. (2000) HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc. Natl. Acad. Sci. U.S.A. 97, 11244–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B. Y.; Isaacs R. D.; Teppler H.; Leavitt R. Y.; Sklar P.; Iwamoto M.; Wenning L. A.; Miller M. D.; Chen J.; Kemp R.; Xu W.; Fromtling R. A.; Vacca J. P.; Young S. D.; Rowley M.; Lower M. W.; Gottesdiener K. M.; Hazuda D. J. (2011) Raltegravir: the first HIV-1 integrase strand transfer inhibitor in the HIV armamentarium. Ann. N.Y. Acad. Sci. 1222, 83–89. [DOI] [PubMed] [Google Scholar]
- Wills T.; Vega V. (2012) Elvitegravir: a once-daily inhibitor of HIV-1 integrase. Expert. Opin. Invest. Drugs 21, 395–401. [DOI] [PubMed] [Google Scholar]
- Ason B.; Knauss D. J.; Balke A. M.; Merkel G.; Skalka A. M.; Reznikoff W. S. (2005) Targeting Tn5 transposase identifies human immunodeficiency virus type 1 inhibitors. Antimicrob. Agents Chemother. 49, 2035–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czyz A.; Stillmock K. A.; Hazuda D. J.; Reznikoff W. S. (2007) Dissecting Tn5 transposition using HIV-1 integrase diketoacid inhibitors. Biochemistry 46, 10776–10789. [DOI] [PubMed] [Google Scholar]
- Krishnan L.; Li X.; Naraharisetty H. L.; Hare S.; Cherepanov P.; Engelman A. (2010) Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc. Natl. Acad. Sci. U.S.A. 107, 15910–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Smith S. J.; Metifiot M.; Jaxa-Chamiec A.; Pommier Y.; Hughes S. H.; Cherepanov P. (2011) Structural and functional analyses of the second-generation integrase strand transfer inhibitor Dolutegravir (S/GSK1349572). Mol. Pharmacol. 80, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metifiot M.; Maddali K.; Johnson B. C.; Hare S.; Smith S. J.; Zhao X. Z.; Marchand C.; Burke T. R. Jr.; Hughes S. H.; Cherepanov P.; Pommier Y. (2013) Activities, crystal structures, and molecular dynamics of dihydro-1H-isoindole derivatives, inhibitors of HIV-1 integrase. ACS Chem. Biol. 8, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov P. (2010) Integrase illuminated. EMBO Rep. 11, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson E. A.; Damiani L.; Leitao A.; Hu C.; Hathaway H.; Oprea T.; Sklar L.; Shaheen M.; Bauman J.; Wang W.; Nickoloff J. A.; Lee S. H.; Hromas R. (2012) Targeting the transposase domain of the DNA repair component Metnase to enhance chemotherapy. Cancer Res. 72, 6200–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson A.; Finnegan D. J. (2003) Excision of the Drosophila mariner transposon Mos1. Comparison with bacterial transposition and V(D)J recombination. Mol. Cell 11, 225–235. [DOI] [PubMed] [Google Scholar]
- Lo M. C.; Aulabaugh A.; Jin G.; Cowling R.; Bard J.; Malamas M.; Ellestad G. (2004) Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 332, 153–159. [DOI] [PubMed] [Google Scholar]
- Perryman A. L.; Forli S.; Morris G. M.; Burt C.; Cheng Y.; Palmer M. J.; Whitby K.; McCammon J. A.; Phillips C.; Olson A. J. (2010) A dynamic model of HIV integrase inhibition and drug resistance. J. Mol. Biol. 397, 600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh Y.; Matreyek K. A.; Engelman A. (2011) Differential sensitivities of retroviruses to integrase strand transfer inhibitors. J. Virol. 85, 3677–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson J. M.; Finnegan D. J.; Walkinshaw M. D. (2007) Crystallization of a Mos1 transposase-inverted-repeat DNA complex: biochemical and preliminary crystallographic analyses. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 63, 434–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Dawson A.; Finnegan D. J. (2001) DNA-binding activity and subunit interaction of the mariner transposase. Nucleic Acids Res. 29, 3566–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray J.; Williamson E. A.; Sheema S.; Lee S. H.; Libby E.; Willman C. L.; Nickoloff J. A.; Hromas R. (2009) Metnase mediates chromosome decatenation in acute leukemia cells. Blood 114, 1852–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armougom F.; Moretti S.; Poirot O.; Audic S.; Dumas P.; Schaeli B.; Keduas V.; Notredame C. (2006) Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 34, W604–W608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P.; Courcelle E.; Stuart D. I.; Metoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 15, 305–308. [DOI] [PubMed] [Google Scholar]
- Richardson J. M.; Zhang L.; Marcos S.; Finnegan D. J.; Harding M. M.; Taylor P.; Walkinshaw M. D. (2004) Expression, purification and preliminary crystallographic studies of a single-point mutant of Mos1 mariner transposase. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 962–964. [DOI] [PubMed] [Google Scholar]
- Bailey S. (1994) The Ccp4 Suite–programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.