

Abstract
KCNQ1 (also known as KV7.1 or KVLQT1) is a voltage-gated potassium channel modulated by members of the KCNE protein family. Among multiple functions, KCNQ1 plays a critical role in the cardiac action potential. This channel is also subject to inherited mutations that cause certain cardiac arrhythmias and deafness. In this study, we report the overexpression, purification, and preliminary structural characterization of the voltage-sensor domain (VSD) of human KCNQ1 (Q1-VSD). Q1-VSD was expressed in Escherichia coli and purified into lyso-palmitoylphosphatidylglycerol micelles, conditions under which this tetraspan membrane protein yields excellent nuclear magnetic resonance (NMR) spectra. NMR studies reveal that Q1-VSD shares a common overall topology with other channel VSDs, with an S0 helix followed by transmembrane helices S1–S4. The exact sequential locations of the helical spans do, however, show significant variations from those of the homologous segments of previously characterized VSDs. The S4 segment of Q1-VSD was seen to be α-helical (with no 310 component) and underwent rapid backbone amide H–D exchange over most of its length. These results lay the foundation for more advanced structural studies and can be used to generate testable hypotheses for future structure–function experiments.
The human KCNQ1 channel belongs to the superfamily of voltage-gated potassium channels (KV) and plays critical roles in human physiology.1−4 For example, KCNQ1 is essential to both the cardiac action potential that mediates heartbeat and K+ homeostasis in the inner ear.1−4 KCNQ1 dysfunction has been linked to multiple diseases, including various cardiac arrhythmias, congenital deafness, and type II diabetes mellitus.5,6 Loss-of-function mutations in KCNQ1 can result in long QT and Romano-Ward syndromes, while gain-of-function mutations can result in atrial fibrillation and sudden infant death syndrome.1−4 Human KCNQ1 is a 676-residue protein consisting of a 100-residue N-terminal cytosolic domain, followed by an ∼260-residue channel domain containing six transmembrane helices, and a 300-residue cytosolic C-terminus. The first four transmembrane helices (S1–S4) form the voltage-sensor domain (VSD) that is linked to the pore domain (helices S5 and S6) by the S4–S5 linker.7 Roughly 40% of the >200 reported disease-related mutations in the KCNQ1 gene result in amino acid substitutions in the VSD4 (Figure 1A), making structural and functional studies of this domain important in unraveling molecular mechanisms in human pathophysiology.
Figure 1.

(A) Topology of the 100–243 construct of the KCNQ1 voltage-sensor domain (Q1-VSD), which spans helices S0–S4, as inferred on the basis of the results of this work. The sites for mutations linked to long QT syndrome and related disorders are colored pink, while sites for mutations linked to atrial fibrillation (AF) and related disorders are colored yellow. (B) Sequence alignment (by ClusterW2) of TM segments S2 and S4 of VSDs from various voltage-gated ion channels. (C) Sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel stained with Coomassie blue, showing that purified Q1-VSD runs as a monomer in SDS micelles, with an apparent molecular mass of 15 kDa.
KCNQ1 has been subjected to numerous mutagenesis-based electrophysiology studies. Moreover, several models for the channel domain have been developed on the basis of homology to potassium channels of known structure.7−13 Direct structural biological studies of KCNQ1 have also been conducted for water-soluble fragments of its large cytosolic C-terminal domain.14,15 However, neither the complete channel nor its substituent pore and voltage-sensor domains have been purified or subjected to high-resolution structural studies. This is despite tremendous progress over the past decade in experimental structural biology16−20 and experimentally restrained modeling21−24 of other KV channels. Here we report the overexpression, purification, and preliminary structural studies of the human KCNQ1 voltage-sensor domain.
Materials and Methods
Cloning and Overexpression of Q1-VSD
Segments of the human KCNQ1 cDNA starting at codon 100, 107, or 121 and stopping at codon 243 or 261 were cloned into a pET16b or pET21b expression vector (Novagen) with an added segment encoding either an N- or C-terminal His tag, respectively (N-terminal, MGHHHHHHG-; C-terminal, -LEHHHHHHHHHH). Escherichia coli expression vectors were also prepared using pQE32 [for expression in XL1-Blue and XL10-Gold strains (Qiagen)]. Expression of the Q1-VSD constructs was tested in several E. coli strains: BL21(DE3), Rosetta(DE3), C43, Rosetta/C43(DE3) (which contains the pRARE plasmid that encodes rare codon tRNAs for Arg, Gly, Ile, Leu, and Pro), XL1-Blue, and XL10-Gold. Successful transformants were cultured in M9 minimal medium with appropriate antibiotics and supplemented with a MEM vitamin (Mediatech) and ZnCl2 (50 μM). The culture was incubated at 22 °C and 230 rpm, and expression was induced at an OD600 of 0.8 by adding IPTG (1 mM), followed by continued rotary shaking at 22 °C for 24 h.
Q1-VSD Purification
Cells expressing Q1-VSD were harvested by centrifugation at 22000g for 20 min at 4 °C. The cell pellet was suspended in 20 mL of lysis buffer [70 mM Tris-HCl and 300 mM NaCl (pH 8.0)] per gram of wet cells, with 2 mM TCEP, lysozyme (0.2 mg/mL), DNase (0.02 mg/mL), RNase (0.02 mg/mL), PMSF (0.2 mg/mL), and magnesium acetate (5 mM). This mixture was tumbled for 30 min at room temperature (RT). The cell suspension was then sonicated (F550 sonic dismembrator, Misonix, Farmingdale, NY) at 4 °C for 5 min at a power level of 60 with 5 s pulses separated by 5 s. The lysate was centrifuged at 22000g for 20 min, yielding a pellet that contained inclusion bodies. The supernatant was collected and centrifuged at 100000g for 2 h to pellet the membrane fraction. The pellets of either membranes or inclusion bodies were homogenized using a glass homogenizer and diluted to a final volume 10 times the original cell pellet weight. Q1-VSD inclusion body fractions were solubilized with different detergents, including N,N-dimethyl-N-dodecylglycine [3% (v/v) (Empigen)], dihexanoylphosphatidylcholine {DHPC [0.5% (w/v)]}, dodecylphosphocholine {DPC [0.5% (w/v)]}, or 8 M urea containing 0.2% SDS (w/v; all handling of solutions containing SDS was conducted at RT). To solubilize Q1-VSD in the membrane-separated fraction, DPC was added to a final concentration of 0.5% (w/v). Extracts were tumbled overnight at 4 °C (except for solutions containing SDS, which were handled at RT). After detergent extraction, insoluble debris was removed by centrifugation at 22000g for 20 min at 4 °C. The supernatant was then incubated with Ni(II)-NTA Superflow resin (Qiagen) (1 mL/g of original cell pellet) for at least 45 min while the mixture was being tumbled at 4 °C.
To purify Q1-VSD, the resin was packed into a gravity-flow column and washed with 4–5 bed volumes of lysis buffer containing the extraction detergent. Impurities were eluted using a wash buffer [50 mM HEPES, 300 mM NaCl, and 60 mM imidazole (pH 7.8)] containing the extraction detergent until the A280 returned to baseline. The extraction detergent was then exchanged using 12 bed volumes of rinse buffer [50 mM Tris-HCl and 300 mM NaCl (pH 7.8)] containing one of the following detergents: 0.5% DPC, 0.5% β-n-decyl maltoside (DM), 0.5% DHPC, 0.2% n-tetradecylphosphocholine (TDPC), 0.2% n-hexadecylphosphocholine (HDPC), 0.2% lyso-myristoylphosphatidylcholine (LMPC), 0.2% lyso-palmitoylphosphatidylcholine (LPPC), 0.2% lyso-myristoylphosphatidylglycerol (LMPG), or 0.2% lyso-palmitoylphosphatidylglycerol (LPPG). All detergents were purchased from Avanti or Anatrace, unless otherwise stated. Q1-VSD was eluted with elution buffer [50 mM Tris-HCl, 300 mM NaCl, 500 mM imidazole, and 2 mM TCEP (pH 7.8)] containing the chosen detergent. The final concentration was determined by the A280 (0.59 OD unit per milligram per milliliter per centimeter). The protein was identified as Q1-VSD by trypsin digestion followed by mass spectrometry.
Preparation of Q1-VSD for Nuclear Magnetic Resonance (NMR) Spectroscopy
The Q1-VSD sample in detergent after purification and elution was centrifugally concentrated to ∼0.5 mL at 5000g using a 10 kDa molecular weight cutoff (MWCO) concentrator (Amicon Ultra-10, EMD Millipore) at 4 °C. The approximately 0.5 mL Q1-VSD samples were diluted with 5 mL of pH 5.5 buffer (50 mM MES, 0.5 mM EDTA, 2 mM TCEP, and 10% D2O) and then concentrated to 0.5 mL. This step was repeated three times to ensure complete buffer exchange. When needed, the concentrations of lysophospholipid detergent in [U-15N]Q1-VSD NMR samples were determined by 31P NMR. For this, 5 μL of the Q1-VSD NMR sample was mixed with 25 μL of 200 mM pyrophosphate and 170 μL of 50 mM MES buffer (pH 5.5) in a 3 mm NMR tube. The one-dimensional 31P NMR spectrum was then recorded on a Bruker Avance 500 or 600 MHz spectrometer, using a delay of 15 s between scans to ensure complete relaxation. The detergent concentration was then calculated by comparing the integrals of the lyso-phospholipid and pyrophosphate (standard) peaks in the NMR spectrum.25 The final protein and detergent concentrations in the NMR sample were adjusted to 0.5 and 60 mM [3% (w/v)], respectively.
For amino acid-selective isotopic labeling, a transaminase-deficient strain of E. coli, CT19, with genetic lesions of the aspC, avtA, ilvE, trpB, and tyrB genes, was used to reduce 15N-labeled amino acid scrambling. CT19 is well-suited for selective 15N labeling with Ala, Asp, Ile, Leu, Phe, Trp, Tyr, and Val.26 Briefly, cells were grown in 4 L of LB culture supplemented with 10 mg/L ampicillin, 100 mg/L kanamycin, and 20 mg/L tetracycline at 22 °C and were shaken at 230 rpm. Once the culture reached an OD600 of 0.6, cells were pelleted by centrifugation at 3000 rpm for 15 min and then resuspended in 1 L of M9 medium containing the same antibiotics, 0.2 g of the 15N-labeled amino acid of interest, and 0.5 g each of all the other amino acids with natural isotopic abundance amino acids. Reverse isotopic labeling was applied to identify Arg peaks by supplementing M9 medium with [15N]NH4Cl (1 g/L) and an excess of [14N]Arg (1.0 g/L M9). The subsequent 1H–15N TROSY spectrum of the protein exhibits peaks for all amino acid types except for Arg.
Q1-VSD Backbone NMR Resonance Assignments
NMR spectra were recorded at 50 °C on a Bruker Avance spectrometer at 600 MHz (14.4 T), 800 MHz (18.7 T), or 900 MHz (21.1 T) each equipped with a cryoprobe. Uniformly 2H-, 13C-, and 15N-labeled Q1-VSD was prepared in 60 mM LPPG micelles buffered at pH 5.5 with 50 mM MES, 0.5 mM EDTA, 2 mM TCEP, and 10% D2O. Proton chemical shifts were referenced to internal DSS, and 13C and 15N chemical shifts were referenced indirectly to DSS using absolute frequency ratios.27 The following series of three-dimensional (3D) experiments were used for sequential resonance assignments: TROSY-HNCO, TROSY-HN(CA)CO, TROSY-HNCA, TROSY-HN(CO)CA, TROSY-HNCACB, TROSY-HN(CA)CB, and NOESY-TROSY.18,28 Selective labeling of specific amino acids also aided the completion of assignments.
NMR Relaxation Experiments
TROSY-based pulse sequences were used to determine R1, R2, and 15N–{1H} heteronuclear NOEs (hetNOEs).18,28,29 Q1-VSD samples were uniformly 15N-labeled and concentrated to 0.25 mM in 50 mM MES buffer (pH 5.5) containing 0.5 mM EDTA, 2 mM TCEP, and 10% D2O. Relaxation experiments were performed at 800 MHz (18.7 T) and 50 °C. R1 values were determined from a series of inversion recovery spectra with 100, 200, 400, 600, 800, 1000, 1200, 1600, 2000, 4000, and 8000 ms relaxation delays. R2 values were obtained from Carr–Purcell–Meiboom–Gill data with 34, 51, 68, 85, 102, 119, 136, 153, 170, and 204 ms delays. Steady-state 1H–15N hetNOE values were determined from peak ratios between spectra collected with and without a 3 s proton presaturation pulse.29 All spectra were processed using NMRPipe and analyzed with Sparky.
H–D Exchange Experiments
Amide H–D exchange experiments were performed at 50 °C with uniformly 15N-labeled Q1-VSD (0.5 mM) in 60 mM LPPG and 50 mM MES buffer (pH 5.5) containing 0.5 mM EDTA, 2 mM TCEP, and 10% D2O; 500 μL this Q1-VSD solution was mixed with 5 mL of 100% D2O in 50 mM MES (pH 5.5) and concentrated back to 500 μL using a 10 MWCO concentrator to start the H–D exchange process. The 900 MHz 1H–15N TROSY spectra were recorded 4, 8, 12, 16, 20, and 24 h after the addition of D2O. Each spectrum was collected within 4 h, with 2048 × 256 complex points.
Near-UV and Far-UV Circular Dichroism (CD)
CD spectra (Figures S3 and S4 of the Supporting Information) were acquired using a Jasco J-810 spectrometer with temperature control. Near-UV and far-UV CD spectra were collected in quartz cuvettes with path lengths of 1 and 0.1 cm, respectively. Samples were purified as described above and desalted with an Econo-Pac 10DG column (Bio-Rad) into 50 mM MES buffer (pH 5.5, containing 0.5 mM EDTA, 2 mM TCEP, and 0.2% LPPG). Near-UV and Far-UV CD spectra were measured at 50 °C in the ranges of 320–260 and 260–190 nm at protein concentrations of 1.0 and 0.1 mg/mL, respectively.
Results
Optimization of Expression and Purification of Q1-VSD
N-Terminally His-tagged Q1-VSD constructs starting at residue 100, 107, or 121 and ending at residue 243 or 261 were tested for expression in several different strains of E. coli, including the XL10-Gold strain recently used to express the VSD from a voltage-sensitive phosphatase.30 The highest expression levels were observed in strain Rosetta/C43(DE3), which was employed in all subsequent work. Constructs lacking the complete S0-containing 100–121 segment was expressed poorly. The 100–243 fragment, which terminates at the end of S4, was expressed better (∼4 mg of Q1-VSD/L of M9 culture) than the 100–261 fragment, the latter of which includes the S4–S5 linker. Higher expression levels were observed for the N-terminally tagged protein than for the C-terminally tagged construct. The 100–243 domain was selected for structural characterization and is the protein hereafter in this paper termed Q1-VSD. Q1-VSD was expressed primarily in inclusion bodies, with <10% in E. coli membranes.
Q1-VSD could be extracted from inclusion bodies using Empigen, SDS–urea, DHPC, or DPC; however, only DPC extraction produced high yields and led to NMR samples exhibiting high-quality spectra following transfer of the samples into LPPG micelles (see below). Extraction of Q1-VSD by other detergents and/or denaturants resulted in either poor extraction efficiency or low-quality NMR spectra (even following transfer into LPPG micelles). One possible explanation is that harsh detergents like Empigen and SDS result in a denatured state of Q1-VSD from which refolding into a nativelike structure upon transfer back into a mild detergent is kinetically forbidden. This phenomenon has been encountered for other membrane proteins such as G protein-coupled receptors.31 On the other hand, DPC, considered to be moderate on the scale of detergent mildness and harshness, was efficient at solubilizing Q1-VSD from either plasma membranes or inclusion bodies and, following the transfer of the protein into LPPG micelles, led to samples yielding high-quality NMR spectra (see the following sections). Purified Q1-VSD migrated on SDS–PAGE as a monomer (>95%) with an apparent molecular weight of ∼15 kDa (Figure 1C).
Optimization of NMR Conditions for Q1-VSD
Q1-VSD was extracted from inclusion bodies using DPC and then purified into DPC, TDPC, HDPC, LLPC, LMPC, LPPC, or LPPG micelles. Most of these detergents yielded low-quality Q1-VSD NMR spectra. For example, the 1H–15N TROSY spectrum of Q1-VSD purified from inclusion bodies into DPC micelles exhibited only 60 of 151 expected peaks (Figure 2A), with only two of 12 Gly and three of four Trp side chain peaks being observed. Results were equally disappointing when NMR samples were prepared in DPC using Q1-VSD from isolated E. coli membranes (Figure 2B). Low-quality spectra resulted when Q1-VSD was purified into TDPC, HDPC, LLPC, LMPC, or LPPC micelles. On the other hand, the 1H–15N TROSY spectrum of Q1-VSD purified into LPPG micelles resulted in 134 of 151 expected amide peaks, including all expected Gly and Trp indole side chain peaks. The modest dispersion of resonances over a relatively narrow 1H chemical shift range (7–9 ppm) is consistent with a folded helical protein (Figure 2C,D). CD spectra support the notion that Q1-VSD in LPPG micelles is structured (Figure S1 of the Supporting Information): the far-UV CD spectrum of Q1-VSD is consistent with a highly α-helical content, while the nonbaseline near-UV CD spectrum indicates the presence of tertiary structure.
Figure 2.

600 MHz 1H–15N TROSY NMR spectra of the Q1-VSD that was first extracted using DPC and then purified in different detergent micelles at pH 6.5 and 45 °C. (A) Inclusion body-derived Q1-VSD in DPC micelles. (B) Membrane fraction-derived Q1-VSD in DPC micelles. (C) Inclusion body-derived Q1-VSD in LPPG micelles. (D) Membrane fraction-derived Q1-VSD in LPPG micelles.
Similar NMR results were obtained whether the Q1-VSD was purified into LPPG from isolated membranes or inclusion bodies (Figure 2C,D). In light of this and because the inclusion body fraction yielded more Q1-VSD than isolated membranes, all subsequent work was conducted using inclusion body-derived protein. It is interesting to note that the VSD of the hERG potassium channel has also been expressed in E. coli and subjected to NMR sample optimization, where lyso-myristoylphosphatidylglycerol (LMPG) micelles were found to yield the optimal NMR spectra.32 However, the quality of the best reported NMR spectrum of the hERG VSD (Figure 3C in ref (32)) is not nearly as high as for Q1-VSD (see especially Figure 3) in LPPG micelles, a fact that limited the number of backbone resonance assignments that could be made for the hERG VSD to 50%.
Figure 3.

(A) Assigned 900 MHz 1H–15N TROSY spectrum of the Q1-VSD in LPPG micelles. Main chain amide peaks have been assigned for 140 sites (of 151 non-Pro residues). The four Trp side chain peaks (Nε) have also been assigned. The spectrum was recorded at 50 °C on a 900 MHz spectrometer in 50 mM MES buffer (pH 5.5) containing 10% LPPG, 10% D2O, 2 mM TCEP, and 0.5 mM EDTA. The Gly residues from the N-terminal His tag are marked as G2 and G9; the His residues on the tag and the folded-in Arg side chain peaks are also marked. (B) Expanded view of the congested area within the dashed square of panel A.
Backbone NMR Resonance Assignments and Secondary Structure of Q1-VSD
NMR resonances were assigned using standard TROSY-based 3D triple-resonance and NOESY-TROSY amide proton–amide proton experiments,33 in combination with residue-specific labeling and site-directed mutagenesis (see Figure 3); 89% of backbone assignments were completed for Q1-VSD (Figure 3). Examples of the NMR data used to complete these assignments are shown in Figures S2–S4 of the Supporting Information. Backbone H(N), N, CO, Cα, and Cβ peak assignments have been deposited in the BioMagResBank (http://www.bmrb.wisc.edu) as accession number 19708.
Secondary structure was calculated by TALOS+ using backbone amide HN, 15N, 13CO, 13Cα, and 13Cβ chemical shifts.34 Nearly identical results were obtained using TALOS-N.35 The TALOS+-determined secondary structure of Q1-VSD in LPPG micelles (Figure 4) is dominated by α-helices (Figure 1A), consistent with its far-UV CD spectrum (Figure S1 of the Supporting Information). Significantly, the dihedral angles calculated with TALOS+33 using the NMR chemical shift data indicate that S4 of Q1-VSD in LPPG is α-helical with no 310-helical content.36,37
Figure 4.
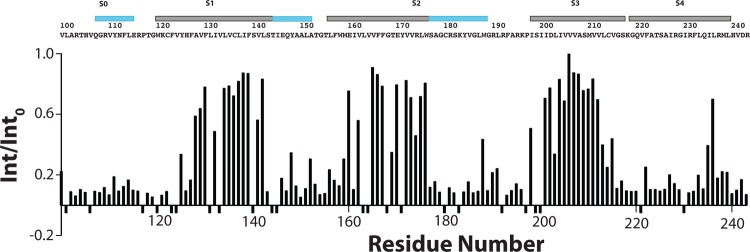
Backbone amide hydrogen–deuterium exchange data for Q1-VSD in LPPG micelles. Int/Into is the ratio between an observed backbone amide peak intensity in a spectrum that was acquired starting 2 h after mixing the Q1-VSD with excess D2O and the corresponding peak intensity in a matched but nonexchanged reference sample. The spectra on which these data are based are 900 MHz 1H–15N TROSY spectra that were recorded at 50 °C and pH 5.5. Sites showing negative intensity ratios are those for which it was not possible to make measurements because of peak absence or overlap.
Backbone Amide H–D Exchange of Q1-VSD
D2O incubation of Q1-VSD in LPPG micelles for 2 h followed by acquisition of a 1H–15N TROSY spectrum provided site-specific quantification of the degree of replacement of backbone amide protons with deuterium. These measurements (Figure 4) reveal that the transmembrane sections of helices S1–S3 exchange slowly, on the time scale of a couple of hours. On the other hand, S0, the water-exposed C-terminal end of S1, and the water-exposed end of S2 were seen to be rapidly exchanged, indicative of exposure to water. Interestingly, with the exception of one focal point centered at L236, most of the critical S4 segment underwent extensive H–D exchange. This is despite the high helicity (Figure 5) and rigidity (Figure 6) of this segment, except at its frayed C-terminus.
Figure 5.

Secondary structure of Q1-VSD and supporting NMR chemical shift data. (A) Results of TALOS+ analysis of the backbone chemical shifts, which identify the helical secondary structure elements within the Q1-VSD. Negative bars in panel A indicate that reliable chemical shift data are unavailable for that site, such that a TALOS+ determination of helical probability cannot be calculated. The sequence of Q1-VSD is shown above the TALOS+ output. The locations of the α-helical segments as determined by the TALOS+ results are indicated by the bars above the amino acid sequence. The TM segments are colored gray and the other helical segments (some of which are extramembrane extensions of TM helices) cyan. (B and C) Deviations of the observed chemical shift values for backbone 13Cα and 13CO resonances from corresponding random coil chemical shift values. Results of chemical shift index analysis66 of the data in panels B and C is generally consistent with the results of TALOS+ analysis.
Figure 6.

800 MHz 15N NMR relaxation measurements reveal the dynamic properties of 0.5 mM VSD in 10% LPPG micelles at pH 5.5 and 50 °C. R1 is the longitudinal relaxation rate. R2 is the transverse relaxation rate. 15N–{1H} NOE is the steady-state heteronuclear nuclear Overhauser effect (hetNOE). The displayed error bars illustrate the uncertainties associated with the fits of the raw relaxation data and an additional 20% of that uncertainty to account for other sources of error. Sites for which no values are reported represent residues for which assignments were not available or for which extensive resonance overlap hindered the extraction of reliable parameters. The dashed red line in the third panel indicates the position of a hetNOE of 0.7. The black line in the bottom panel indicates the position of the average R1R2 value, while the dashed red line indicates the R1R2 = 25 s–2 point. Residues displaying R1R2 values of >25 s–2 and hetNOE values of <0.7 are likely subject to intermediate exchange processes occurring on the microsecond to millisecond time scale.38
Backbone Dynamics of the Isolated Q1-VSD in LPPG Micelles
NMR 15N relaxation and 15N–{1H} hetNOE measurements were collected to assess Q1-VSD flexibility in the picosecond to nanosecond time scale range (Figure 6). As expected, the transmembrane helices exhibit generally high R2/R1 and hetNOE values. Reduced values of hetNOE and R2/R1 are seen at the N-terminus and (especially) C-terminus, and for the segments connecting S0 to S1, S1 to S2, S2 to S3, and S3 to S4.
After residues exhibiting hetNOE values of <0.7 had been excluded (dashed line in the third panel of Figure 6), the average R1R2 was calculated to be 16.5 s–2 (solid line in the bottom panel of Figure 6).38R1R2 analysis has the advantage over R2/R1 of significantly attenuating the effect of motional anisotropy. This approach identified four residues, T155, G168, V185, and V206, with R1R2 values significantly higher than the mean R1R2 value, potentially indicating intermediate exchange broadening as a result of microsecond to millisecond local backbone motions. Such intermediate time scale exchange may also explain why residues close to T155, G168, and V206 have broadened or missing peaks. This is particularly interesting for V206, which is located in S3 at a site homologous to a Val site in KvAP associated with a short break in S3 seen in several of the available structures of this microbial VSD (see Figure 7).
Figure 7.

Sequence alignments and secondary structures of Q1-VSD and of KV and NaV channels for which structures have been experimentally determined. Note that the MlotiK1 channel is not actually voltage-gated but does have a VSD-like domain. For Q1-VSD, the results of this work are presented and also the secondary structure observed in a composite homology/Rosetta model developed using previously published methods7 using the KV1.2/2.1 chimera crystal structure as a template. Protein Data Bank entries and references for the secondary structures presented in this figure are as follows: KV1.2/2.1 chimera (2R9R);16 KVAP, VSD-only, X-ray (1ORS);19 KVAP, mixed micelles, X-ray (2A0L);17 KVAP, micelles, X-ray (1ORQ);19 KVAP DPC, LDAO micelles, NMR;18 KVAP, D7PC micelles, NMR (2KYH);20 NaVRh (4DXW);67 NaVAb (3RYV);68 and MlotiK1 (3BEH).40 The depicted sequence alignment was generated manually, making heavy use of the available 3D structures as the basis for choosing between alternative alignment modes. A global multiple-sequence alignment for VSDs was recently generated using a sophisticated computational approach, as reported elsewhere,65 and differs in many details from that presented here.
Discussion
Comparison of Q1-VSD Secondary Structure to Other Experimental VSD Structures
Shown in Figure 7 is the alignment of the Q1-VSD sequence with the S0–S4 sequences of VSDs from potassium and sodium channels with experimentally determined structures. Also included in this figure are the locations of the α- and 310-helical segments observed in the various experimental structures. We have also included the secondary structure from a hybrid homology/Rosetta model for Q1-VSD that was developed in this lab using the KV1.2/2.1 chimera structure39 as a template. All of these VSD were structurally characterized in the absence of a transmembrane voltage potential and, with the exception of the MlotiK1 channel, are believed to populate a conformation that most closely resembles that of the activated conformational state.16−20 The MlotiK1 channel is not actually voltage-gated but has a VSD-like domain40 with a conformation very different from that observed for activated state VSDs.
It can be seen that Q1-VSD, with four transmembrane helices, shares the same general topology as the other VSDs (Figures 1A and 7). Q1-VSD also shares a S0 helix that is observed in the KV1.2/2.1 chimera, MlotiK1, the NaV channel, and two of the five KVAP channel structures. The high degree of variability seen in the length of S0 from channel to channel and structure to structure most likely reflects the modest stability of this helix. Its exact beginning and end, indeed, whether it is seen at all, is dependent on exact experimental conditions. This points to a more general observation that can be made for all of the VSD structures represented in Figure 7. While each of these structures has been enormously revealing, many of the details, such as the exact beginnings and ends of helices and the locations of short breaks in helices, should be taken with a grain of salt. With this in mind, we can nevertheless point out what appear to be meaningful differences between Q1-VSD and the other VSDs.
S1 appears to be significantly longer for Q1-VSD, beginning earlier and ending later in the sequence, than for other VSDs (Figures 1A and 7). Both ends of the Q1-VSD helix likely protrude out farther into the aqueous phase, as reflected by the higher rate of H–D exchange observed for those segments relative to the central transmembrane portion of S1 (Figure 4). The Q1-VSD S2 appears also be longer than in the other VSDs, with its extra residues being located at its intracellular C-terminal end, which extends into the aqueous phase (again, as reflected by the H–D exchange pattern in Figure 4). S3, on the other hand, resembles S3 of NaV in that it is just slightly shorter than the corresponding segment in the KV1.2/2.1 chimera and significantly shorter than that in KVAP. Helix S3 in Q1-VSD does not have any obvious break, unlike helix S3 in KVAP, which is punctuated differently from structure to structure by short breaks in the helices of this segment.
Segment S4 of Q1-VSD exhibits some fraying of its helix near its C-terminus. This is not seen for any of the other VSDs represented in Figure 7 and is almost certainly due to the fact that these other VSDs were not truncated at the end of S4 (as was the case for Q1-VSD) but continued at least partway into the S4–S5 linker. Except for its frayed C-terminus, both the NMR chemical shifts and the relaxation data for Q1-VSD indicate its S4 segment is a stable α-helix. It is interesting, therefore, that this helix was found to undergo rather rapid H–D exchange along its full length, except at Q235 and I236. A similar observation does not seem to have been made for KVAP S4 in either of the two fine NMR studies of this domain.18,20 We hypothesize that a combination of several phenomena may explain the generally rapid H–D exchange rate for the Q1-VSD S4 segment. First, some degree of conformational heterogeneity may be critical to the voltage-transducer function of S4. It is interesting to note that while S4 in the Q1-VSD is α-helical, the corresponding segment in the KV1.2/2.1 chimera crystallized as a 310-helix. The MlotiK1 and NaV channels also have considerable 310-helical content. It seems very possible that S4 segments may in some cases be able to populate both 310- and α-helical conformations23,24 depending on exact conditions and, most likely, depending on the state of VSD activation (or transition between states). This suggests that the energy barrier to backbone conformational switching of this segment is low. A second, nonexclusive possibility is that the location of S4 in LPPG micelles is closer to the micelle surface (or edge) than for S1–S3, where it would be exposed to higher D2O concentrations. This is reasonable in light of the higher polarity of S4 relative to the polarity of the other TM segments. Finally, it is believed that the membrane is thinned in the vicinity of the S4 transmembrane segment, such that water can permeate into what would normally be the plane of the membrane to hydrate the multiply charged residues of this segment,41−45 an effect that may also apply to micellar conditions and that would also be expected to contribute to enhanced H–D exchange rates.
As summarized above, there are significant differences in the secondary structure observed for Q1-VSD in LPPG micelles and the other potassium channel VSDs for which structures have been determined. For all other VSDs (except for the pseudo-VSD in the MlotiK1 channel), it is thought that the structures reflect the activated-state conformation. The NMR studies of this work were conducted at 0 V. Because only a single set of resonances were observed, Q1-VSD either populates only a single conformational state in LPPG micelles or is in rapid exchange (on the NMR time scale) between activated and resting states. For the intact channel, the open-state probability at 0 V favors the open channel by ∼4:1 relative to the resting state.46,47 This suggests the activated state of the VSD may be favored under the 0 V conditions of our NMR experiments, as might also be predicted on the basis of the fact that all available high-resolution VSD structures (all determined at 0 V) are thought to represent activated conformations. However, it should be noted that for the KCNQ1 tetramer there is an active debate about whether the transition of all, some, or any of the four VSDs to the activated state is needed for channel opening.48−55 Thus, we cannot infer the exact inactive-state versus active-state equilibrium constant for the VSD at 0 V (even assuming that excision of the VSD from the rest of the channel does not alter this equilibrium). In any case, it appears likely that the observed population-weighted average spectrum reflects primarily the intrinsic spectrum of the activated state, which would mean that the differences in secondary structure seen for Q1-VSD in this work as compared to the other VSDs further extend the previously documented conformational diversity seen among activated-state VSDs. It is possible that the differences in secondary structure observed for the KCNQ1 VSD are coupled to other known differences between it and the VSD of other channels. The S4 segment of KCNQ1 bears a lower net positive charge than S4 for any other eukaryotic voltage-gated ion channel.56,57 Moreover, KCNQ1 appears to always function under physiological conditions in concert with modulation by members of the KCNE family of single-span membrane proteins.58,59 It is now well-established that KCNE family members alter the electophysiological properties of the KCNQ1 channel in part through direct interaction with its VSD.8,56,57,60−64 The degree to which unique structural features of the Q1-VSD reflect adaptations that allow optimal interactions with KCNE family members will be an interesting topic for future study.
Conclusions
These studies of the structure of the human Q1-VSD represent a still-rare application of NMR methods to a mammalian multispan helical membrane protein. This work confirms the structural modularity of the voltage-sensor domain of KV channels and establishes an overall degree of similarity in structural topology between Q1-VSD and the VSD from other channels. However, significant differences are also observed in the sequential locations of the helices in Q1-VSD relative to the other VSDs characterized to date. These differences likely reflect both conformational diversity among members of the VSD superfamily and to some degree the fact that VSDs are intrinsically multiconformational. Structural plasticity in the face of the non-native environment of detergent micelles also cannot be ruled out as having an influence on some details of the observed secondary structure. The results of this work and of previous structural studies (as summarized in Figure 7) tend to emphasize structural variation among VSD domains from different channels. This is in juxtaposition to an elegant recent computational study that highlights important structural features that may be shared by all VSDs.65 Establishing which VSD traits are shared versus those that reflect more channel-specific adaptations is likely to be a major future area of study. In any case, as a first experimental probe of the structure of the KCNQ1 channel, the results of this study should prove useful both as an experimental platform for more advanced studies and as a source of structural hypotheses to guide experiments probing structure–function relationships of KCNQ1 in the future.
Acknowledgments
We thank Arina Hadziselimovic for help with preparative molecular biology, Prof. Markus Voehler for technical assistance with NMR experiments, and Prof. Alfred George for useful discussion and encouragement.
Supporting Information Available
Supporting Figures S1–S4. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
J.-H.K. and B.M.K. contributed equally to this work.
The authors declare no competing financial interest.
This work was supported by National Institutes of Health (NIH) Grants U54 GM094608 and RO1 DC007416. B.M.K. was supported by NIH Grant T32 NS007491.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Maljevic S.; Wuttke T. V.; Seebohm G.; Lerche H. (2010) KV7 channelopathies. Pfluegers Arch. 460, 277–288. [DOI] [PubMed] [Google Scholar]
- Jespersen T.; Grunnet M.; Olesen S.-P. (2005) The KCNQ1 potassium channel: From gene to physiological function. Physiology 20, 408–416. [DOI] [PubMed] [Google Scholar]
- Robbins J. (2001) KCNQ potassium channels: Physiology, pathophysiology, and pharmacology. Pharmacol. Ther. 90, 1–19. [DOI] [PubMed] [Google Scholar]
- Peroz D.; Rodriguez N.; Choveau F.; Baro I.; Merot J.; Loussouarn G. (2008) Kv7.1 (KCNQ1) properties and channelopathies. J. Physiol. 586, 1785–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q. M.; Song K.; Shen X. Z.; Cai Y. (2012) The association between KCNQ1 gene polymorphism and type 2 diabetes risk: A meta-analysis. PLoS One 7, e48578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki H.; Takahashi A.; Kawaguchi T.; Hara K.; Horikoshi M.; Andersen G.; Ng D. P.; Holmkvist J.; Borch-Johnsen K.; Jorgensen T.; Sandbaek A.; Lauritzen T.; Hansen T.; Nurbaya S.; Tsunoda T.; Kubo M.; Babazono T.; Hirose H.; Hayashi M.; Iwamoto Y.; Kashiwagi A.; Kaku K.; Kawamori R.; Tai E. S.; Pedersen O.; Kamatani N.; Kadowaki T.; Kikkawa R.; Nakamura Y.; Maeda S. (2008) SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 40, 1098–1102. [DOI] [PubMed] [Google Scholar]
- Smith J. A.; Vanoye C. G.; George A. L.; Meiler J.; Sanders C. R. (2007) Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry 46, 14141–14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Wang Y.; Meng X. Y.; Zhang M.; Jiang M.; Cui M.; Tseng G. N. (2013) Building KCNQ1/KCNE1 Channel Models and Probing their Interactions by Molecular-Dynamics Simulations. Biophys. J. 105, 2461–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J. R.; Pan H.; Wu D.; Nekouzadeh A.; Decker K. F.; Cui J.; Baker N. A.; Sept D.; Rudy Y. (2009) A multiscale model linking ion-channel molecular dynamics and electrostatics to the cardiac action potential. Proc. Natl. Acad. Sci. U.S.A. 106, 11102–11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gofman Y.; Shats S.; Attali B.; Haliloglu T.; Ben-Tal N. (2012) How does KCNE1 regulate the Kv7.1 potassium channel? Model-structure, mutations, and dynamics of the Kv7.1-KCNE1 complex. Structure 20, 1343–1352. [DOI] [PubMed] [Google Scholar]
- Strutz-Seebohm N.; Pusch M.; Wolf S.; Stoll R.; Tapken D.; Gerwert K.; Attali B.; Seebohm G. (2011) Structural basis of slow activation gating in the cardiac I Ks channel complex. Cell. Physiol. Biochem. 27, 443–452. [DOI] [PubMed] [Google Scholar]
- Haitin Y.; Yisharel I.; Malka E.; Shamgar L.; Schottelndreier H.; Peretz A.; Paas Y.; Attali B. (2008) S1 constrains S4 in the voltage sensor domain of Kv7.1 K+ channels. PLoS One 3, e1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A.; Lee A.; Campbell E.; Mackinnon R. (2013) Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. eLife 2, e00594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener R.; Haitin Y.; Shamgar L.; Fernandez-Alonso M. C.; Martos A.; Chomsky-Hecht O.; Rivas G.; Attali B.; Hirsch J. A. (2008) The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J. Biol. Chem. 283, 5815–5830. [DOI] [PubMed] [Google Scholar]
- Howard R. J.; Clark K. A.; Holton J. M.; Minor D. L. J. (2007) Structural insight into KCNO (Kv7) channel assembly and channelopathy. Neuron 53, 663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S. B.; Tao X.; Campbell E. B.; MacKinnon R. (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. [DOI] [PubMed] [Google Scholar]
- Lee S. Y.; Lee A.; Chen J.; MacKinnon R. (2005) Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. U.S.A. 102, 15441–15446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkarev Z. O.; Paramonov A. S.; Lyukmanova E. N.; Shingarova L. N.; Yakimov S. A.; Dubinnyi M. A.; Chupin V. V.; Kirpichnikov M. P.; Blommers M. J. J.; Arseniev A. S. (2010) NMR structural and dynamical investigation of the isolated voltage-sensing domain of the potassium channel KvAP: Implications for voltage gating. J. Am. Chem. Soc. 132, 5630–5637. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Lee A.; Chen J.; Ruta V.; Cadene M.; Chait B. T.; MacKinnon R. (2003) X-ray structure of a voltage-dependent K+ channel. Nature 423, 33–41. [DOI] [PubMed] [Google Scholar]
- Butterwick J. A.; MacKinnon R. (2010) Solution structure and phospholipid interactions of the isolated voltage-sensor domain from KvAP. J. Mol. Biol. 403, 591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. Ø.; Jogini V.; Borhani D. W.; Leffler A. E.; Dror R. O.; Shaw D. E. (2012) Mechanism of voltage gating in potassium channels. Science 336, 229–233. [DOI] [PubMed] [Google Scholar]
- Swartz K. J. (2008) Sensing voltage across lipid membranes. Nature 456, 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas E.; Yarov-Yarovoy V.; Khalili-Araghi F.; Catterall W. A.; Klein M. L.; Tarek M.; Lindahl E.; Schulten K.; Perozo E.; Bezanilla F.; Roux B. (2012) An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J. Gen. Physiol. 140, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarek M.; Delemotte L. (2013) Omega currents in voltage-gated ion channels: What can we learn from uncovering the voltage-sensing mechanism using MD simulations. Acc. Chem. Res. 46, 2755–2762. [DOI] [PubMed] [Google Scholar]
- Schlebach J. P.; Peng D.; Kroncke B. M.; Mittendorf K. F.; Narayan M.; Carter B. D.; Sanders C. R. (2013) Reversible folding of human peripheral myelin protein 22, a tetraspan membrane protein. Biochemistry 52, 3229–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang T.; Vishnivetskiy S. A.; Gurevich V. V.; Sanders C. R. (2010) Elucidation of inositol hexaphosphate and heparin interaction sites and conformational changes in arrestin-1 by solution nuclear magnetic resonance. Biochemistry 49, 10473–10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley J. L.; Bax A.; Arata Y.; Hilbers C. W.; Kaptein R.; Sykes B. D.; Wright P. E.; Wuthrich K. (1998) Recommendations for the presentation of NMR structures of proteins and nucleic acids: IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. Eur. J. Biochem. 256, 1–15. [DOI] [PubMed] [Google Scholar]
- Tian C.; Vanoye C. G.; Kang C.; Welch R. C.; Kim H. J.; George A. L.; Sanders C. R. (2007) Preparation, Functional Characterization, and NMR Studies of Human KCNE1, a Voltage-Gated Potassium Channel Accessory Subunit Associated with Deafness and Long QT Syndrome. Biochemistry 46, 11459–11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beel A. J.; Mobley C. K.; Kim H. J.; Tian F.; Hadziselimovic A.; Jap B.; Prestegard J. H.; Sanders C. R. (2008) Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): Does APP function as a cholesterol sensor?. Biochemistry 47, 9428–9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Jogini V.; Wanderling S.; Cortes D. M.; Perozo E. (2012) Expression, purification, and reconstitution of the voltage-sensing domain from Ci-VSP. Biochemistry 51, 8132–8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tastan O.; Dutta A.; Booth P.; Klein-Seetharaman J. (2013) Retinal proteins as model systems for membrane protein folding. Biochim. Biophys. Acta DOI: 10.1016/j.bbabio.2013.11.021. [DOI] [PubMed] [Google Scholar]
- Ng H. Q.; Kim Y. M.; Huang Q.; Gayen S.; Yildiz A. A.; Yoon H. S.; Sinner E. K.; Kang C. (2012) Purification and structural characterization of the voltage-sensor domain of the hERG potassium channel. Protein Expression Purif. 86, 98–104. [DOI] [PubMed] [Google Scholar]
- Riek R.; Pervushin K.; Wuthrich K. (2000) TROSY and CRINEPT: NMR with large molecular and supramolecular structures in solution. Trends Biochem. Sci. 25, 462–468. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Delaglio F.; Cornilescu G.; Bax A. (2009) TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Bax A. (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier L.; Couprie J.; le Maire A.; Guilhaudis L.; Milazzo-Segalas I.; Courcon M.; Moutiez M.; Gondry M.; Davoust D.; Gilquin B.; Zinn-Justin S. (2007) Solution structure of the region 51–160 of human KIN17 reveals an atypical winged helix domain. Protein Sci. 16, 2750–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. J.; Ikeya T.; Guntert P.; Chang L. S. (2013) NMR solution structure of a chymotrypsin inhibitor from the Taiwan cobra Naja naja atra. Molecules 18, 8906–8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller J.; Lu M.; Bracken C. (2002) An effective method for the discrimination of motional anisotropy and chemical exchange. J. Am. Chem. Soc. 124, 1852–1853. [DOI] [PubMed] [Google Scholar]
- Long S. B.; Campbell E. B.; MacKinnon R. (2005) Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903. [DOI] [PubMed] [Google Scholar]
- Clayton G. M.; Altieri S.; Heginbotham L.; Unger V. M.; Morais-Cabral J. H. (2008) Structure of the transmembrane regions of a bacterial cyclic nucleotide-regulated channel. Proc. Natl. Acad. Sci. U.S.A. 105, 1511–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepkiy D.; Mihailescu M.; Freites J. A.; Schow E. V.; Worcester D. L.; Gawrisch K.; Tobias D. J.; White S. H.; Swartz K. J. (2009) Structure and hydration of membranes embedded with voltage-sensing domains. Nature 462, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson H. P.; Baker O. S.; Dhillon D. S.; Isacoff E. Y. (1996) Transmembrane movement of the shaker K+ channel S4. Neuron 16, 387–397. [DOI] [PubMed] [Google Scholar]
- Unnerstale S.; Madani F.; Graslund A.; Maler L. (2012) Membrane-perturbing properties of two Arg-rich paddle domains from voltage-gated sensors in the KvAP and HsapBK K+ channels. Biochemistry 51, 3982–3992. [DOI] [PubMed] [Google Scholar]
- Freites J. A.; Tobias D. J.; von Heijne G.; White S. H. (2005) Interface connections of a transmembrane voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 102, 15059–15064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty T.; Su Y.; Hong M. (2010) High-resolution orientation and depth of insertion of the voltage-sensing S4 helix of a potassium channel in lipid bilayers. J. Mol. Biol. 401, 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franqueza L.; Lin M.; Shen J.; Splawski I.; Keating M. T.; Sanguinetti M. C. (1999) Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem. 274, 21063–21070. [DOI] [PubMed] [Google Scholar]
- Sanguinetti M. C.; Curran M. E.; Zou A.; Shen J.; Spector P. S.; Atkinson D. L.; Keating M. T. (1996) Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83. [DOI] [PubMed] [Google Scholar]
- Ruscic K. J.; Miceli F.; Villalba-Galea C. A.; Dai H.; Mishina Y.; Bezanilla F.; Goldstein S. A. (2013) IKs channels open slowly because KCNE1 accessory subunits slow the movement of S4 voltage sensors in KCNQ1 pore-forming subunits. Proc. Natl. Acad. Sci. U.S.A. 110, E559–E566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen J. D.; Gonzalez C.; Sampson K. J.; Iyer V.; Rebolledo S.; Larsson H. P.; Kass R. S. (2010) KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc. Natl. Acad. Sci. U.S.A. 107, 22710–22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteen J. D.; Barro-Soria R.; Robey S.; Sampson K. J.; Kass R. S.; Larsson H. P. (2012) Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc. Natl. Acad. Sci. U.S.A. 109, 7103–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choveau F. S.; Rodriguez N.; Abderemane Ali F.; Labro A. J.; Rose T.; Dahimene S.; Boudin H.; Le Henaff C.; Escande D.; Snyders D. J.; Charpentier F.; Merot J.; Baro I.; Loussouarn G. (2011) KCNQ1 channels voltage dependence through a voltage-dependent binding of the S4-S5 linker to the pore domain. J. Biol. Chem. 286, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel E.; Dvir M.; Haitin Y.; Giladi M.; Peretz A.; Attali B. (2012) KCNQ1 channels do not undergo concerted but sequential gating transitions both in the absence and presence of KCNE1. J. Biol. Chem. 287, 34212–34224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werry D.; Eldstrom J.; Wang Z.; Fedida D. (2013) Single-channel basis for the slow activation of the repolarizing cardiac potassium current. I(Ks). Proc. Natl. Acad. Sci. U.S.A. 110, E996–E1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choveau F. S.; Rodriguez N.; Abderemane Ali F.; Labro A. J.; Rose T.; Dahimène S.; Boudin H.; Le Hénaff C.; Escande D.; Snyders D. J.; Charpentier F.; Mérot J.; Baró I.; Loussouarn G. (2011) KCNQ1 channels voltage dependence through a voltage-dependent binding of the S4-S5 linker to the pore domain. J. Biol. Chem. 286, 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.-J.; Ohmert I.; Vardanyan V. (2011) Allosteric reatures of KCNQ1 gating revealed by alanine scanning mutagenesis. Biophys. J. 100, 885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Delaloye K.; Zaydman M. A.; Nekouzadeh A.; Rudy Y.; Cui J. (2010) State-dependent electrostatic interactions of S4 arginines with E1 in S2 during Kv7.1 activation. J. Gen. Physiol. 135, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G.; Abbott G. W. (2007) The role of S4 charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J. Gen. Physiol. 129, 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K.; Kubo Y. (2011) Nano-environmental changes by KCNE proteins modify KCNQ channel function. Channels 5, 397–401. [DOI] [PubMed] [Google Scholar]
- McCrossan Z. A.; Abbott G. W. (2004) The MinK-related peptides. Neuropharmacology 47, 787–821. [DOI] [PubMed] [Google Scholar]
- Kang C.; Tian C.; Sönnichsen F. D.; Smith J. A.; Meiler J.; George A. L.; Vanoye C. G.; Kim H. J.; Sanders C. R. (2008) Structure of KCNE1 and Implications for How It Modulates the KCNQ1 Potassium Channel. Biochemistry 47, 7999–8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Pan H.; Delaloye K.; Cui J. (2010) KCNE1 remodels the voltage sensor of Kv7.1 to modulate channel function. Biophys. J. 99, 3599–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. H.; Jiang M.; Xu X. L.; Hsu K. L.; Zhang M.; Tseng G. N. (2011) Gating-related molecular motions in the extracellular domain of the IKs channel: Implications for IKs channelopathy. J. Membr. Biol. 239, 137–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung D. Y.; Chan P. J.; Bankston J. R.; Yang L.; Liu G.; Marx S. O.; Karlin A.; Kass R. S. (2009) Location of KCNE1 relative to KCNQ1 in the I(KS) potassium channel by disulfide cross-linking of substituted cysteines. Proc. Natl. Acad. Sci. U.S.A. 106, 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P. J.; Osteen J. D.; Xiong D.; Bohnen M. S.; Doshi D.; Sampson K. J.; Marx S. O.; Karlin A.; Kass R. S. (2012) Characterization of KCNQ1 atrial fibrillation mutations reveals distinct dependence on KCNE1. J. Gen. Physiol. 139, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palovcak E.; Delemotte L.; Klein M. L.; Carnevale V. (2014) Evolutionary imprint of activation: The design principles of VSDs. J. Gen. Physiol. 143, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D. (1994) Chemical shifts as a tool for structure determination. Methods Enzymol. 239, 363–392. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Ren W.; DeCaen P.; Yan C.; Tao X.; Tang L.; Wang J.; Hasegawa K.; Kumasaka T.; He J.; Wang J.; Clapham D. E.; Yan N. (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J.; Scheuer T.; Zheng N.; Catterall W. A. (2011) The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.