

Abstract
Background
In eukaryotic cells, detection of replication stress results in the activation of the DNA replication checkpoint, a signaling cascade whose central players are the kinases ATR and Chk1. The checkpoint response prevents the accumulation of DNA damage and ensures cell viability by delaying progression into mitosis. However, the role and mechanism of the replication checkpoint transcriptional response in human cells, which is p53-independent, is largely unknown.
Results
We show that, in response to DNA replication stress, the regular E2F-dependent cell cycle transcriptional program is maintained at high levels and we establish the mechanisms governing such transcriptional upregulation. E2F6, a repressor of E2F-dependent G1/S transcription, replaces the activating E2Fs at promoters to repress transcription in cells progressing into S-phase in unperturbed conditions. Following replication stress, the checkpoint kinase Chk1 phosphorylates E2F6 leading to its dissociation from promoters. This promotes E2F-dependent transcription, which mediates cell survival by preventing DNA damage and cell death.
Conclusions
This work reveals, for the first time, that the regular cell cycle transcriptional program is part of the DNA replication checkpoint response in human cells and establishes the molecular mechanism involved. We show that maintaining high levels of G1/S cell cycle transcription in response to replication stress contributes to two key functions of the DNA replication checkpoint response, namely preventing genomic instability and cell death. Given the critical role of replication stress in oncogene transformation, a detailed understanding of the molecular mechanisms involved in the checkpoint response will contribute to a better insight into cancer development.
INTRODUCTION
To properly replicate the genome and prevent genomic instability, cells rely on the DNA integrity checkpoints, an evolutionarily conserved set of signalling pathways that constantly monitor for the loss of integrity of the DNA replication fork or DNA damage. The DNA integrity checkpoints are mediated by the evolutionarily conserved protein kinases ATM/ATR, acting through Chk1 and Chk2 [1–3]. These protein kinases transduce the checkpoint signal to the cell cycle and transcriptional machinery by phosphorylating protein targets [4]. The DNA replication and DNA damage checkpoints, ensure that DNA has been fully replicated and damage repaired before division [5]. The fundamental difference between the two checkpoints is that the DNA replication checkpoint is essential for preventing DNA damage in response to replication stress during S-phase, whereas the DNA damage checkpoint is required to detect and resolve DNA damage before entry into mitosis. Whereas both checkpoints delay progression into mitosis via largely overlapping mechanisms, they induce similar but distinct transcriptional responses [6]. The differences in mechanism and expression program are poorly established but are thought to reflect the intrinsically different aims of the replication and damage checkpoints, to prevent DNA damage or resolve DNA damage, respectively.
During DNA replication, cells are particularly vulnerable to genomic instability as replication forks are prone to stall and collapse when encountering replication blocks or damaged DNA templates [1, 7]. Replication stress is also a consequence of oncogene activation. A recent model of oncogenesis proposes that, following oncogene-induced replication stress, DNA damage ensues, which can lead to a number of mutations in key tumor suppressors, like TP53, and genomic instability [8]. It is therefore crucial to understand the cellular response to replication stress and its role in preventing the occurrence of DNA damage. The DNA replication checkpoint depends mainly on ATR and the downstream checkpoint protein kinase Chk1, although there is some degree of cross-talk with the DNA damage checkpoint protein kinases ATM and Chk2 [2]. Following phosphorylation by ATR, Chk1 becomes active and is released from chromatin to phosphorylate its substrates [9], which include cell cycle regulators, most notably Cdc25 [10], and proteins involved in DNA repair, including Rad51 [11].
Whereas the main regulator of DNA damage-inducible genes in G1 is the transcription factor p53, a target of both ATM and Chk2 [12], the molecular details of the largely p53-independent transcriptional response to replication stress have not been established [13]. Genome-wide expression analysis carried out in this study reveals that the G1/S cell cycle transcriptional program is regulated as part of the DNA replication checkpoint response in human cells. In mammalian cells, G1/S transcriptional regulation depends on the E2F family of transcription factors (E2F1-8) and their regulators, the pocket protein family members (pRb, p107, p130), which are well-established tumor suppressors (reviewed in [14–17]). Previous works established a pro-apoptotic role for the activator E2F, E2F1, following DNA damage checkpoint activation [18, 19] [20, 21]. However, this is independent of its role in the cell cycle transcriptional program. Here we report a role for the E2F-dependent cell cycle transcriptional program in the DNA replication checkpoint response in human cells. We establish the mechanism by which the G1/S transcriptional program is rewired by the DNA replication checkpoint and show that this regulation is required to cope with replication stress and avoid genomic instability and cell death. Our study highlights the inherent contrast between the DNA replication and DNA damage checkpoint responses, showing an intrinsically different role for E2F-dependent transcription in response to DNA replication stress; promoting genome stability and survival rather than cell death.
RESULTS
Replication stress induces E2F-dependent cell cycle transcripts
In order to investigate the DNA replication checkpoint transcriptional response, we analyzed the genome-wide expression profile of S-phase cells with and without replication stress induced by hydroxyurea (HU) treatment. HU, an inhibitor of ribonucleotide reductase, causes nucleotide depletion and is widely used to induce replication stress [22] [23]. It causes replication fork stalling after DNA replication origins have been licensed and fired, resulting in the activation of the checkpoint protein kinase ATR and its downstream target Chk1, inducing an intra S-phase arrest. Potential cell cycle positioning differences following replication stress were avoided by using synchronized human T98G cells. Serum starved cells were released in serum for 16 hours (G1/S), at which point cells were either treated with HU for 4 hours (20 h HU) or left untreated for 4 hours (20 h untreated; Experimental design is represented in Figure S1A). The short treatment with HU during S-phase induces DNA replication checkpoint activation, as detected by S345 phosphorylation of Chk1 (Figure S1B). Importantly, it does not induce significant DNA damage, as shown by the low levels of gamma-H2AX compared to the levels induced by the DNA damaging agent camptothecin (CPT) at the same time points. Together these data show that our experimental setup allows for specific activation of the DNA replication checkpoint without causing significant DNA damage and possible DNA damage checkpoint activation.
Expression profiles were established by microarray for untreated and HU-treated samples. Comparing expression levels resulted in a list of 846 genes significantly upregulated in response to HU treatment (Table SI). Gene ontology analysis of these revealed that the top enriched ontology categories include the DNA replication, DNA repair, and cell cycle groups (Table I). Since these groups of genes are mainly regulated during the G1-to-S transition of the cell cycle by the E2F transcription factors, we investigated whether there was an enrichment of E2F target genes among the HU-responsive gene list. A list of 459 cell cycle-regulated E2F targets from previous studies was compiled (Table SII)[24] [25] [26] [27]. We find significant enrichment of E2F targets in the HU-induced gene list (P=0.035) (Figure S1C/D, table SIII), suggesting that E2F-dependent cell cycle transcription could be part of the replication stress checkpoint transcriptional response.
Table I.
Enriched functional groups of the 846 genes induced by HU treatment, according to gene ontology analysis.
| Categories identified | Number of genes | % | P-Value |
|---|---|---|---|
| DNA metabolic process | 37 | 5 | 6.20E-05 |
| DNA repair | 22 | 3 | 1.30E-03 |
| Response to DNA damage stimulus | 26 | 3.5 | 1.90E-03 |
| Deoxynucleotide metabolic process | 6 | 0.8 | 2.40E-03 |
| DNA replication | 16 | 2.2 | 3.30E-03 |
| Defence response | 35 | 4.7 | 7.30E-03 |
| Macromolecular complex subunit organization | 39 | 5.3 | 8.00E-03 |
| Regulation of apoptosis | 43 | 5.8 | 8.30E-03 |
| Locomotor behaviour | 19 | 2.6 | 9.40E-03 |
| Regulation of transcription factor activity | 10 | 1.3 | 1.10E-02 |
| Cell cycle | 38 | 5.1 | 4.30E-02 |
To confirm and extend the genome-wide expression analysis, single gene expression analysis on a set of well-established G1/S E2F targets was carried out using quantitative PCR (qPCR). These data show that these are significantly upregulated during S-phase in response to replication stress (Figure 1A).
Figure 1.

HU causes activation of E2F transcription in a checkpoint-dependent manner. (A) Serum starved cells were released in serum with or without HU (2 mM) for 20 h. RNA levels of indicated genes were quantified by RT qPCR. Average +/− sd (n=4). All genes were significantly induced in HU treated cells with P<0.05. (B) T98G cells synchronized by serum starvation were stimulated with serum. 15 h after serum addition the cells were either treated with 2 mM HU or left untreated. UCN01 was added to the indicated samples 30 min before HU addition. At the indicated time points, cells were collected and RNA extracted. Relative RNA abundance for the indicated genes was evaluated by RT qPCR and normalized to actin. Graphs represent the average +/− sd (n=3). Black line HU treated, grey line untreated. (C) Repeat of the experiment in B. At the indicated time points, proteins were extracted in RIPA buffer, quantified and analyzed by western blot with the indicated antibodies. KU86 is a loading control. P-Ser345 of Chk1 is shown. Red boxes highlight the checkpoint activation; green box shows the checkpoint inhibition. See also Figure S1.
E2F-dependent cell cycle transcription is induced in response to replication stress in a Chk1-dependent manner
To establish the dynamics of E2F-dependent cell cycle transcriptional regulation in response to replication stress, expression analysis was carried out both in synchronized (Figure 1B, S1E) and asynchronous cells (Figure S1G). Expression levels of the well-established E2F G1/S cell cycle targets, cyclin E, RRM2 and CDC6 reveal that, while transcription is inactivated in a timely manner in untreated cell cultures, these transcripts remain active following HU treatment. Similar results were obtained with thymidine-induced replication block in T98G cells (Figure S1F) and in a primary human cell line treated with HU (Figure S1H). In agreement with the transcriptional data, the protein levels of Cyclin E and E2F1 remain high upon HU treatment (Figure 1C), while in control cells they peak at 18–20 hours and then decrease during cell cycle progression, as expected.
Since HU treatment causes an intra S-phase arrest through Chk1 activation [22, 23, 28], we tested whether the transcriptional response of E2F-targets is Chk1-dependent. Chemical inhibition of Chk1 function by UCN01 treatment shows that de-repression of G1/S transcripts and protein accumulation of E2F targets in response to HU treatment is Chk1-dependent (Figure 1B, C). Similar results were obtained with siRNA targeting Chk1 (Figure S1G). Altogether these results show that E2F-dependent G1/S transcription is maintained at high levels in response to replication stress via a Chk1-dependent mechanism.
The transcriptional repressor E2F6 binds to E2F-regulated promoters to inhibit G1/S transcription
Next we sought to establish the mechanism by which the checkpoint interferes with G1/S transcription. We hypothesized that the checkpoint could target a transcriptional repressor normally involved in turning off G1/S transcription during S-phase. A role for E2F6 in timely inactivation of E2F targets during S-phase has been suggested [29]. However, these studies, carried out in E2F6 knock-out mice fibroblasts, were inconclusive, likely due to a compensatory role of E2F4 [29]. To establish if E2F6 could be involved in turning off transcription during S-phase we first established the binding of E2F transcription factors to their target genes during the cell cycle. We carried out a cell cycle time course of chromatin immunoprecipitations (ChIP) using antibodies to E2F4, E2F6 and, as a representative activator E2F, E2F1.
E2F target promoters are mainly occupied by E2F4 in quiescence (Figure 2A) as previously reported [30]. Upon release into the cell cycle, E2F4 is replaced by E2F1, correlating with transcriptional activation, which, in turn, is replaced by E2F6 once cells progress into S-phase. E2F6 recruitment to promoters starts at G1/S and peaks in S-phase, when E2F1 is released, correlating with transcriptional repression, while E2F4 does not appear to be significantly recruited at this point (Figure 2A). To determine whether E2F6 plays a role in turning off G1/S transcription, we analyzed RNA and protein levels of E2F targets in synchronized cells where E2F6 had been depleted. We observe that acute loss of E2F6, by siRNA knockdown, leads to a persistently high level of G1/S transcription, relative to control transfected cells. This supports a role for E2F6 in the repression of E2F targets upon exit from G1 phase (Figure 2B, S2A). Transcriptional de-repression is also reflected in the sustained high levels of E2F-regulated proteins in the absence of E2F6 (Figure S2B, C). Whereas in control cells these proteins peak at the G1-to-S transition and then decrease with cell cycle progression, in E2F6 depleted cells they remain high. Collectively, these data suggest that E2F6 plays an important role in repressing G1/S transcription during G1 and S phases.
Figure 2.

E2F6 binding during S-phase correlates with G1/S transcription repression. (A) Time course of ChIP in starvation-synchronized T98G cells. At the indicated time points after serum addition, the samples were cross-linked and processed for ChIP with the reported antibodies. The relative binding for every time point was calculated as a percentage of the highest in each experiment. Graphs represent average +/− sd (n=4) of independent experiments. Red boxes highlight peak binding. (B) T98G cells were transfected in DMEM 0.1% FCS with the reported siRNA, after 48–72 h serum was added. At the indicated time points, RNA was extracted and the relative RNA abundance was calculated as previously described. Average +/− sd (n=3). Black lines E2F6 siRNA, grey lines control siRNA. Red boxes mark the times when checkpoint-dependent activation of these transcripts was observed in Figure 1. See also Figure S2.
E2F6 repressive function is inhibited in response to replication stress
E2F6’s function positions it as a potential target via which the replication checkpoint protein kinase Chk1 could keep E2F-regulated G1/S transcription active. If inhibiting E2F6 function is part of the DNA replication checkpoint response, the expression profile of HU-treated and E2F6-depleted cells should significantly overlap. Genome-wide expression analysis using bead-microarrays of synchronized S-phase cells either transfected with E2F6 or control siRNA, identified more than 700 targets as significantly induced following E2F6 depletion (Supplementary Table IV). Gene ontology analysis revealed enrichment for similar categories upon HU treatment, among which the most represented were the DNA replication and repair, and cell cycle groups (Supplementary Table V). Importantly, there is a significant overlap (P<10−4) between the ‘siE2F6 upregulated genes’ and ‘HU upregulated genes’ (Figure S3, Supplementary Table VI), indicating that a common gene set is regulated by E2F6 and in response to replication stress.
To confirm and extend our genome-wide expression analysis we tested if the E2F cell cycle targets induced in response to replication stress (Figure 1A) depend on E2F6 for their repression during S-phase. These data show that all of the 14 well-established E2F-dependent G1/S targets significantly upregulated in response to HU are also upregulated in response to E2F6 depletion (Figure 3A), suggesting that the replication checkpoint could inhibit E2F6-dependent repression.
Figure 3.

HU treatment reduces E2F6 binding to promoters and upregulates a group of E2F6-regulated transcripts. (A) T98G cells were transfected in DMEM 0.1% FCS with the reported siRNA, after 48–72 hours serum was added. RNA levels of indicated genes after 20 h were quantified by RT qPCR. Average +/− sd (n=4). All genes were significantly induced in siE2F6 treated cells with P<0.05. (B) Synchronized T98G cells were stimulated with serum; at 15 hours, 2 mM HU was added to half of the plates. ChIP was performed with the indicated antibodies. The relative binding for every time point was calculated as a percentage of the highest untreated in each experiment. Graphs represent average +/− sd (n=3) of independent experiments. Dark grey bars untreated, light grey bars HU treated. See also Figure S3.
We reasoned that one way of inhibiting E2F6 function in response to replication stress is via affecting its ability to bind, and repress, E2F target promoters. To test this, synchronized T98G cells were fixed at the indicated time points and the binding of E2F6 and E2F1 at two well-established E2F target promoters was analyzed by ChIP. While E2F6 is recruited to these promoters when cells enter S-phase (20 hours), this recruitment is significantly reduced in cells treated with HU (Figure 3B, S4A). Furthermore, the reduced recruitment of E2F6 to these promoters in response to HU treatment correlates with an enhanced binding of E2F1 compared to untreated cells. This suggests that E2F6 binding to promoters is impaired in response to nucleotide depletion.
Chk1 regulates E2F6 through direct phosphorylation
Overall, these data indicate that E2F6 could be a direct substrate of Chk1 kinase. An in vitro kinase assay, using immunoprecipitated E2F6 as a substrate and recombinant Chk1, confirmed that Chk1 can directly phosphorylate E2F6 in vitro (Figure 4A). Next we sought to identify the specific E2F6 site(s) targeted by Chk1. A deletion mutant lacking the first N terminal 60 amino acids identified that Chk1-dependent phosphorylation of E2F6 is mainly in the first 60 amino acids (Figure S4B). In the first N terminal 60 amino acids, two serine residues, Ser 12 and Ser 52, conform to a potential Chk1 consensus (Figure 4B) and an in vitro kinase assay peptide array identified both sites as Chk1 targets. Whereas Ser12 appeared to be the main target site based on in vitro kinase analysis of the single site mutants, the double site mutant (E2F6S12/52A) greatly reduces phosphorylation of the E2F6 protein by Chk1 in vitro (Figure 4C), suggesting that both sites can be targeted by Chk1. Phosphorylation of Ser12 upon HU treatment was also identified in vivo by a phospho-Ser12-specific antibody (Figure 4D) and by mass spectrometric analysis (data not shown). Inhibition of Chk1 with UCN01 reduces the extent of E2F6 phosphorylation on Ser12, confirming the role of Chk1 in the regulation of endogenous E2F6 (Figure S4C).
Figure 4.

Chk1-dependent phosphorylation of E2F6 causes its release from promoters and prevents DNA damage following HU treatment. (A) In vitro kinase assay (32P) using immunoprecipitated myc-tagged E2F6 as a substrate and recombinant Chk1 or buffer alone as a negative control. The immunoprecipitated E2F6 was split equally between the two assay reactions (buffer and Chk1) and an aliquot was used to check the protein by western blot (WB) with anti-E2F6 antibody. (B) Diagram representing E2F6 protein. The main domains are highlighted. Asterisks mark Ser12 and 52. (C) In vitro kinase assay (32P), with recombinant Chk1, of immunoprecipitated HA-tagged wild-type or mutant E2F6 as a substrate. Western blot (WB) of the immunoprecipitated proteins. (D) HEK293 cells transfected with HA-E2F6 were treated with HU (2mM) for 6 h or left untreated, then E2F6 was immunoprecipitated with an anti-HA antibody. Western blot was performed with the indicated antibodies. (E) Western blot showing the protein levels of transfected wild-type and mutant (12/52AA) E2F6 in synchronized T98G cells. (F) Serum-starved T98G cells were transfected with pcDNA-E2F6 wild type or mutant prior to release in serum for 20 h, with or without HU (2 mM). ChIP was performed with anti-HA antibody and the binding to p107 and Rad51 promoters was evaluated by qPCR. The graph represents the binding upon HU treatment relative to untreated. Average +/− sd (n=3). (G) HEK293 T-Rex cells stably expressing inducible wild-type or mutant E2F6 were treated with 0.5 mM HU for 24 h in the presence of the indicated concentrations of doxycycline, then lysates were prepared and the indicated proteins analyzed by western blot. See also Figure S4.
To determine the role of phosphorylation of E2F6 at Ser12/52 in response to replication stress, we assessed binding of E2F6 to E2F-regulated promoters and expression of E2F genes following HU treatment in synchronized cells expressing either ectopic wild-type E2F6, E2F6S12/52A or vector alone (Figure 4E). Maximal binding of wild type E2F6 to promoters during S-phase is reduced upon HU treatment (Figure 4F), as previously shown (Figure 3B). However, E2F6S12/52A does not appear to dissociate from promoters following HU addition, compared to the untreated samples (Figure 4F). The persistence of E2F6S12/52A at promoters correlates with reduced expression of E2F transcripts in response to checkpoint activation (Figure S4C). Altogether these results indicate that Chk1 phosphorylates E2F6 at serines 12 and 52 to regulate its function in response to DNA replication stress.
De-repressing G1/S transcription via E2F6 inactivation limits DNA damage upon replication stress
It has been well established that, in response to replication stress, checkpoint activation is required to stabilize replication forks [31] and activate other mechanisms to avoid the accumulation of DNA damage [11, 32]. When the checkpoint is inhibited, either by depletion of Chk1 or by treatment with inhibitors, increased DNA damage is detected. We therefore aimed to establish whether the activation of G1/S transcription was involved in limiting DNA damage in checkpoint-compromised cells exposed to replication stress. We reasoned that if inactivation of E2F6 by the checkpoint is required to prevent DNA damage, then ectopic expression of the checkpoint-insensitive E2F612/52 mutant should increase the extent of DNA damage following replication stress. Cells expressing doxycycline-inducible wild-type E2F6 or E2F612/52 were treated with a low concentration of HU for 24 hours in the presence of doxycycline, and DNA damage was analyzed by western blot with anti-gamma-H2AX antibody (Figure 4G). HU alone induces a barely detectable increase of gamma-H2AX, which could also be partially explained by the cell synchronization occurring upon HU treatment. Upon HU treatment, cells overexpressing wild-type E2F6 (2 and 4 ug/ml Doxy) show a slight increase in gamma-H2AX compared to non-induced conditions (0 Doxy). Strikingly, the extent of H2AX phosphorylation is greatly increased by overexpression of the checkpoint-insensitive E2F612/52 mutant (compare lane 9 with lanes 10 and 11), suggesting that DNA damage occurs mostly when cells cannot activate a proper transcriptional response to replication stress. Altogether these data support a role for checkpoint-dependent inactivation of E2F6 in preventing DNA damage following replication stress.
Inactivation of E2F6 is important for cell survival during replication stress
Our data point to an important role for Chk1 in regulating E2F6-dependent G1/S transcription. Next we investigated the biological significance of Chk1’s ability to keep G1/S transcription on. Since many G1/S genes encode proteins involved in DNA replication and repair [25, 33], we assessed whether G1/S transcriptional induction is an important part of the checkpoint response for cell viability. Chk1 is an essential gene whose inhibition causes cell death due to DNA damage accumulation during S-phase [34, 35]. We reasoned that, if Chk1-dependent inactivation of E2F6 is an important part of the checkpoint response, knocking down E2F6 in cells inhibited for Chk1 function should partially rescue cell death. Synchronized cells transfected with control or E2F6 siRNA were treated with Chk1 inhibitor UCN01, HU, or both and were analyzed for cell viability. We observe that depletion of E2F6 suppresses about 40% of the cell death induced by Chk1 inactivation, as assessed by accumulation of cells with a sub-G1 DNA content and caspase 3 cleavage (Figures 5A, S5A). This indicates that inactivation of E2F6 and, thereby, activation of G1/S transcription is important for cell survival in response to replication stress as a result of checkpoint activation. Based on this, we reasoned that the opposite, an inability to inactivate E2F6 and activate G1/S transcription, would be detrimental to cells upon replication stress. To investigate this we analyzed the survival of cells where E2F6 was overexpressed, compromising checkpoint inactivation, in a clonogenic survival assay. HEK293 T-Rex cells were treated with HU for 24 hours in the presence of various concentrations of doxycycline to induce E2F6 expression (Figure 5B), after which they were diluted and allowed to form colonies. Induction of E2F6 in these cells strongly reduces cell survival following HU treatment (Figure 5B, C), while increasing the extent of DNA damage compared to HU alone (Figure S5B). Collectively, these data support an important role for inactivation of E2F6 and thereby activation of G1/S transcription for cell survival following replication stress.
Figure 5.
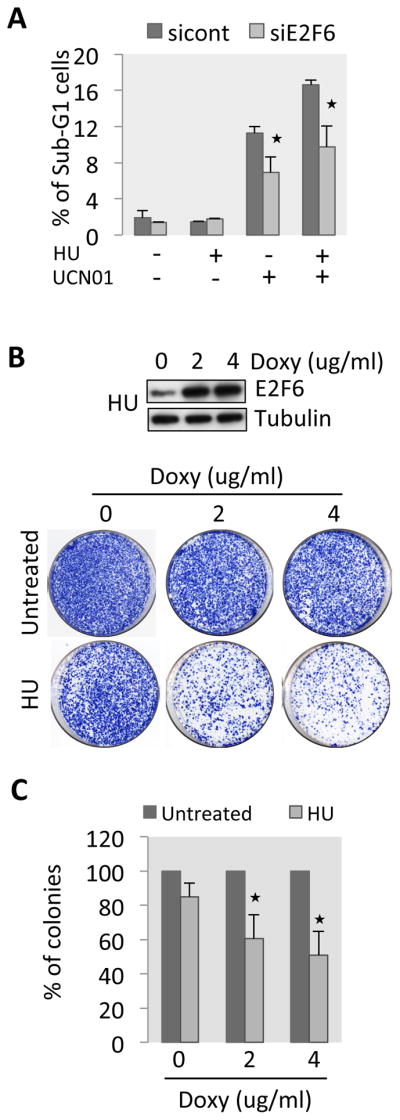
Inactivation of E2F6 enhances survival of HU treated cells. (A) Synchronized T98G cells transfected with control siRNA or E2F6 siRNA were treated with HU (2 mM) and UCN01 as indicated for 24–30 h. Cells were then collected, stained with propidium iodide and analyzed by flow cytometry. SubG1 population was evaluated using FlowJo. (B) Top panel: western blot showing E2F6 induction. Bottom panel: HEK293 T-Rex cells stably expressing inducible wild-type E2F6 were treated with 0.5 mM HU for 24 h in the presence of the indicated concentrations of doxycycline, then the cells were washed, diluted, plated on a 10 cm dish and allowed to grow for 12–15 days. The cells were then fixed and stained and colony density was scored. (C) Quantification of the colonies of the experiment shown in (B). Histogram representing the mean +/− sd of three independent experiments. The star represents significant difference between the non-doxycycline treated cells and doxycycline treated sample in HU. The quantification was performed with ImageJ. See also Figure S5.
DISCUSSION
In this study we show that, in response to DNA replication stress, the regular cell cycle transcriptional program is maintained at high levels in a Chk1-dependent manner and we have uncovered the mechanisms governing such transcriptional upregulation. We show that E2F6, a repressor of E2F-dependent G1/S transcription, replaces the activating E2Fs at promoters to repress transcription in cells progressing into S-phase. When cells experience replication stress, the DNA replication checkpoint kinase Chk1, which is activated by ATR, phosphorylates E2F6, leading to its dissociation from promoters. This promotes E2F-dependent transcription, including that of those genes involved in DNA repair and nucleotide synthesis, which mediates cell survival by preventing genomic instability and cell death.
There might be several reasons to maintain high expression levels of the cell cycle transcriptional program in response to replication stress. For one, many of the genes regulated during G1/S encode proteins required for DNA replication, including DNA polymerases and enzymes involved in the production of dNTPs. One reason for keeping G1/S transcription on during an intra S-phase arrest, therefore, might be to maintain the level of proteins required for DNA replication to complete replication once the checkpoint arrest has been relieved. In addition, many of the genes of the G1/S transcriptome have important roles in the stabilization of stalled replication forks and in DNA repair, hence their presence is required to prevent genomic instability. Chk1 itself is encoded by a G1/S gene and, because the activation of the checkpoint has been reported to increase Chk1 protein turnover [36], we speculate that enhanced Chk1 expression could be required to maintain an efficient checkpoint. Finally, maintaining the expression of genes that are required for subsequent cell cycle events, other than DNA replication, prepares the cell for resuming cell cycle progression once the checkpoint is turned off.
This study reveals that the DNA replication checkpoint interferes with the E2F6-dependent negative feedback loop to maintain high levels of G1/S transcription in response to replication stress. Other E2F family members might be involved in the checkpoint transcriptional response. For instance the activating E2Fs (E2F1, 2 and 3) are required for the expression of E2F target genes and might therefore also be the target of checkpoint regulation to maintain high levels of expression. E2F7 and 8 have been proposed to inactivate G1/S transcription during S-phase and recent studies have shown that the binding of those factors to the E2F1 promoter is positively regulated by the DNA damage checkpoint in order to reduce E2F1-dependent apoptosis [37, 38]. Consequently, depletion or deletion of both E2F7 and 8 increases DNA damage-induced cell death. It is likely that the various E2Fs involved in G1/S transcription regulation might be affected in a different way in response to particular forms of stress. This would allow cells to diversify their response to different stimuli and to titrate the biological response to the level and nature of the insult.
Activation of the regular periodic transcriptional program by replication stress has been demonstrated in Schizosaccharomyces pombe and in Saccharomyces cerevisiae [39], [40, 41]. However, the E2F family of transcription factors and their coregulators, the pocket proteins, share no sequence homology with their yeast counterparts. Remarkably, our study reveals that the simple yet elegant mechanism by which checkpoint activation can override the regular periodic transcriptional program is conserved from yeast to humans; the effector checkpoint protein kinase directly interferes with the auto-regulatory negative feedback loop involved in turning off G1/S transcription. This illustrates the importance of both the wiring of these transcriptional networks, and the capacity of those networks to be rewired in cells responding to stress.
A role for the activator E2F, E2F1, in the DNA damage checkpoint response has been well established [18, 19] [20, 21] [42]. Interestingly, in such studies, DNA damage induces E2F1 activity and thereby the expression of specific pro-apoptotic genes. These targets do not normally have a role in the cell cycle and are not part of the E2F-dependent cell cycle transcriptional program. Furthermore this mechanism of regulation depends on transcriptional induction while the seemingly modest upregulation of G1/S transcripts in response to replication stress results from the checkpoint merely maintaining peak expression, by interfering with timely repression, rather than inducing transcription. We speculate that since the G1/S transcriptional network comprises a large number of dosage-sensitive co-regulated genes this particular mechanism of regulation allows for a controlled high level of expression within the ‘normal’ range. Therefore this seemingly modest up-regulation represents the difference between ‘on’ and ‘off’ of G1/S genes expression during the cell cycle, which is undoubtedly of biological significance. In addition our study highlights an inherent difference between the DNA replication and DNA damage checkpoint responses, showing an intrinsically different role for E2F1 in response to DNA replication stress by promoting genome stability and survival rather than cell death. This emphasizes the contrasts between the diverse cellular responses to specific genotoxic stresses. Whereas very toxic DNA damage lesions such as double strand breaks can induce a strong apoptotic response, replication stress causes replication fork stalling, which only results in DNA damage when forks collapse after prolonged stress or in the absence of an efficient checkpoint [43]. Therefore the cellular response to replication stress aims to stabilize replication forks and prevent DNA damage, at least temporarily, rather than induce cell death.
We show that E2F6 antagonizes the function of E2F1 and depletion of E2F6 reduces the toxic effect of Chk1 inactivation, while overexpression of E2F6 reduces the survival of HU-treated cells with a concomitant increase in DNA damage. Collectively, our data support an important role for the expression of G1/S genes in cells responding to DNA replication stress, in addition to the well-established role in cell cycle progression. It highlights a critical role for this transcriptional response in preventing DNA damage and possible associated genomic instability, key events in oncogenic transformation. Future research will have to establish which E2F targets are of particular importance in responding to replication stress to prevent DNA damage and ensure cell survival.
EXPERIMENTAL PROCEDURES
Cell culture and transfection
T98G (p53 mutated) cells (ATCC) were cultured in DMEM with 10% fetal calf serum (FCS) supplemented with penicillin and streptomycin (Invitrogen). Synchronization was achieved by 48–72 hours culture in DMEM 0.1% FCS, and release in DMEM 10% FCS. HU was added as indicated, to serum-synchronized cells 15 hours after release. Transfection of siRNA was performed with Oligofectamine (Invitrogen).
ChIP and real time PCR
ChIP of synchronized cells was performed as previously reported [30]. RNA was isolated with the RNeasy kit from Qiagen and RT qPCR was performed with Eurogentec kit.
Kinase assays
HEK293 cells were transfected with pcDNA myc E2F6 [44] or pcDNA3 HA-E2F6 wild-type [45], the recombinant proteins were purified by immunoprecipitation and the kinase assay was performed as previously described using recombinant Chk1 (Activemotif). See Supplementary experimental procedures.
Supplementary Material
Highlights.
E2F-dependent G1/S cell cycle transcription is maintained at high levels in response to replication stress in a Chk1-dependent manner.
Chk1 protein kinase targets the G1/S transcriptional repressor E2F6, interfering with its binding to promoters.
Maintaining high levels of G1/S cell cycle transcription is required to prevent DNA damage during replication stress.
G1/S cell cycle transcription is important for preventing cell death upon replication stress.
Acknowledgments
We are grateful to Dr. K. Helin, Dr. S. Gaubatz, and Dr. J. Lees for kindly providing reagents, Umut Eser for analysis of the genome-wide expression data, Sarah Farmer for critical reading of the manuscript and to the de Bruin group for support and help. We thank Dr. J. Cooper and M. Begnis for help with PFGE. This work was supported by the MRC career development fellowship awarded to RdB (G0800297) and R01 GM059441 to CW.
Footnotes
AUTHOR CONTRIBUTION
C.B., R.d.B., C.W. and C.M.G. conceived the experiments; C.B. and R.d.B. performed the experimental work with the help of S.K.; C.B. and R.d.B. wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 2.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhind N, Russell P. Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J Cell Sci. 2000;113(Pt 22):3889–3896. doi: 10.1242/jcs.113.22.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 5.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 7.Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- 8.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 9.Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol. 2006;16:150–159. doi: 10.1016/j.cub.2005.11.066. [DOI] [PubMed] [Google Scholar]
- 10.Sorensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, Zhou BB, Bartek J, Lukas J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–258. doi: 10.1016/s1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 11.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 12.Carr AM. Cell cycle. Piecing together the p53 puzzle. Science. 2000;287:1765–1766. doi: 10.1126/science.287.5459.1765. [DOI] [PubMed] [Google Scholar]
- 13.Gottifredi V, Shieh S, Taya Y, Prives C. p53 accumulates but is functionally impaired when DNA synthesis is blocked. Proc Natl Acad Sci U S A. 2001;98:1036–1041. doi: 10.1073/pnas.021282898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9:785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–2826. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- 17.Trimarchi JM, Fairchild B, Verona R, Moberg K, Andon N, Lees JA. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc Natl Acad Sci U S A. 1998;95:2850–2855. doi: 10.1073/pnas.95.6.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- 19.Pediconi N, Ianari A, Costanzo A, Belloni L, Gallo R, Cimino L, Porcellini A, Screpanti I, Balsano C, Alesse E, et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol. 2003;5:552–558. doi: 10.1038/ncb998. [DOI] [PubMed] [Google Scholar]
- 20.Stevens C, Smith L, La Thangue NB. Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol. 2003;5:401–409. doi: 10.1038/ncb974. [DOI] [PubMed] [Google Scholar]
- 21.Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 2004;18:3041–3054. doi: 10.1101/gad.1221004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eser U, Falleur-Fettig M, Johnson A, Skotheim JM. Commitment to a cellular transition precedes genome-wide transcriptional change. Mol Cell. 2011;43:515–527. doi: 10.1016/j.molcel.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Bieda M, Jin VX, Rabinovich A, Oberley MJ, Green R, Farnham PJ. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 2007;17:1550–1561. doi: 10.1101/gr.6783507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karnani N, Dutta A. The effect of the intra-S-phase checkpoint on origins of replication in human cells. Genes Dev. 2011;25:621–633. doi: 10.1101/gad.2029711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giangrande PH, Zhu W, Schlisio S, Sun X, Mori S, Gaubatz S, Nevins JR. A role for E2F6 in distinguishing G1/S- and G2/M-specific transcription. Genes Dev. 2004;18:2941–2951. doi: 10.1101/gad.1239304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–816. [PMC free article] [PubMed] [Google Scholar]
- 31.Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- 32.Wang H, Powell SN, Iliakis G, Wang Y. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res. 2004;64:7139–7143. doi: 10.1158/0008-5472.CAN-04-1289. [DOI] [PubMed] [Google Scholar]
- 33.Polager S, Kalma Y, Berkovich E, Ginsberg D. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene. 2002;21:437–446. doi: 10.1038/sj.onc.1205102. [DOI] [PubMed] [Google Scholar]
- 34.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 35.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 2000;14:1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 37.Li J, Ran C, Li E, Gordon F, Comstock G, Siddiqui H, Cleghorn W, Chen HZ, Kornacker K, Liu CG, et al. Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev Cell. 2008;14:62–75. doi: 10.1016/j.devcel.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Westendorp B, Mokry M, Groot Koerkamp MJ, Holstege FC, Cuppen E, de Bruin A. E2F7 represses a network of oscillating cell cycle genes to control S-phase progression. Nucleic Acids Res. 2011;40:3511–3523. doi: 10.1093/nar/gkr1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bastos de Oliveira FM, Harris MR, Brazauskas P, de Bruin RA, Smolka MB. Linking DNA replication checkpoint to MBF cell-cycle transcription reveals a distinct class of G1/S genes. EMBO J. 2012;31:1798–1810. doi: 10.1038/emboj.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Bruin RA, Kalashnikova TI, Aslanian A, Wohlschlegel J, Chahwan C, Yates JR, 3rd, Russell P, Wittenberg C. DNA replication checkpoint promotes G1-S transcription by inactivating the MBF repressor Nrm1. Proc Natl Acad Sci U S A. 2008;105:11230–11235. doi: 10.1073/pnas.0801106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Travesa A, Kuo D, de Bruin RA, Kalashnikova TI, Guaderrama M, Thai K, Aslanian A, Smolka MB, Yates JR, 3rd, Ideker T, et al. DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 2012;31:1811–1822. doi: 10.1038/emboj.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang B, Liu K, Lin FT, Lin WC. A role for 14-3-3 tau in E2F1 stabilization and DNA damage-induced apoptosis. J Biol Chem. 2004;279:54140–54152. doi: 10.1074/jbc.M410493200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cartwright P, Muller H, Wagener C, Holm K, Helin K. E2F-6: a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene. 1998;17:611–623. doi: 10.1038/sj.onc.1201975. [DOI] [PubMed] [Google Scholar]
- 45.Gaubatz S, Wood JG, Livingston DM. Unusual proliferation arrest and transcriptional control properties of a newly discovered E2F family member, E2F-6. Proc Natl Acad Sci U S A. 1998;95:9190–9195. doi: 10.1073/pnas.95.16.9190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.