

Abstract
Cytolytic activity by CD8+ cytotoxic T lymphocytes (CTLs) is a powerful strategy for the elimination of intracellular pathogens and tumor cells. The destructive capacity of CTLs is progressively dampened during chronic infection, yet the environmental cues and molecular pathways that influence immunological `exhaustion' remain unclear. Here we found that CTL immunity was regulated by the central transcriptional response to hypoxia, which is controlled in part by hypoxia-inducible factors (HIFs) and the von Hippel–Lindau tumor suppressor VHL. Loss of VHL, the main negative regulator of HIFs, led to lethal CTL-mediated immunopathology during chronic infection, and VHL-deficient CTLs displayed enhanced control of persistent viral infection and neoplastic growth. We found that HIFs and oxygen influenced the expression of pivotal transcription, effector and costimulatory-inhibitory molecules of CTLs, which was relevant to strategies that promote the clearance of viruses and tumors.
During the response to infection1 and malignancy2, CD8+ T cells traffic through a broad range of tissue microenvironments, including those with low oxygen tension. Oxygen availability regulates both developmental processes and the responses to tissue damage, infection and neoplastic growth3,4. The hypoxia-inducible factors (HIFs) are heterodimeric transcription factors that are constitutively degraded under normal oxygen tension by a process dependent on the VHL (von Hippel–Lindau) complex. VHL function has been extensively studied since it was identified as a tumor suppressor, and loss of VHL function via spontaneous and inherited mutations leads to renal and other specific cancers5; however, the additional role of HIFs in immunity raises the possibility that VHL may affect immune responses as well. The subunits HIF-1α and HIF-2α do not interact with VHL complexes under conditions of low oxygen (hypoxia), which results in the accumulation of HIF-1α and HIF-2α, heterodimerization with HIF-1β and subsequent localization to the nucleus; that results in increased transcription of target genes that allow functional and metabolic adaptations to hypoxic microenvironments6. Notably, Hif1a and Epas1 mRNA and the proteins they encode (HIF-1α and HIF-2α, respectively) can also increase in response to additional extracellular inputs, such as signals mediated by T cell antigen receptors (TCRs), cytokines, Toll-like receptors and the metabolic checkpoint kinase mTOR, even under normal oxygen tension7–9. In the context of innate immunity, HIF-1α promotes inflammation, bactericidal activity, infiltration and cytokine production by macrophages and neutrophils10. Cells of the adaptive immune system have also been shown to use HIF activity to regulate the balance between CD4+ regulatory T cells and lymphocytes of the TH17 subset of helper T cells and the function of regulatory T cells; thus, HIF activity influences T cell–mediated autoimmunity11–13. Glycolysis and HIF-1β have been linked to control of the expression of effector molecule–encoding genes by cytotoxic T lymphocytes (CTLs)14. However, the role of HIF-1α and HIF-2α in the differentiation and function of CD8+ T cells in vivo during the response to infection is poorly understood.
After antigen recognition, CD8+ effector T cells induce apoptosis of host cells via targeted release of cytotoxic granules containing granzymes and perforin; they also produce proinflammatory cytokines, including tumor-necrosis factor (TNF) and interferon-γ (IFN-γ), that promote pathogen clearance15. However, during persistent viral infections such as those caused by hepatitis B virus, hepatitis C virus and human immunodeficiency virus type I, the immune response of CD8+ T cells becomes attenuated, probably as a mechanism for protecting key tissues from destruction by cells of the immune system16,17. Cancer results in similar chronic antigen stimulation and dysfunction of CTLs18,19. Such `exhaustion' of CTLs is characterized by deletion and progressive functional impairment of antigen-specific T cells20. Lymphocytic choriomeningitis virus (LCMV) is a natural mouse pathogen of the genus Arenaviridae; the strain LCMV clone 13 establishes persistent infection that models many aspects of the exhaustion observed during persistent viral infection in humans17, whereas the closely related LCMV Armstrong strain produces acute infection that is cleared within 5–7 d.
Here we studied how enhanced HIF activity, due to loss of VHL, influenced CD8+ T cell immunity in the context of persistent viral and tumor antigens. We found that deletion of VHL altered the differentiation of effector and memory CD8+ T cells, and hypoxia modulated the expression of pivotal transcription factors, effector molecules, costimulatory receptors and activation-induced inhibitory receptors in an HIF-1α- and HIF-2α-dependent fashion. Our work defines a key role for HIF-mediated transcription in modulating the adaptive immune responses of CD8+ T cells to persistent infection and, in turn, has broad relevance for therapeutic strategies to promote viral and tumor clearance.
RESULTS
Elevated HIF results in mortality in persistent infection
To explore the role of the VHL-HIF pathway in adaptive immunity, we deleted the gene encoding VHL (Vhl), the principal negative regulator of the subunits HIF-1α and HIF-2α, in T cells and observed the outcome of acute or chronic infection with LCMV. In a published study examining the result of deletion of Vhl early in thymic development by Cre recombinase driven by the proximal promoter of the gene encoding the tyrosine kinase Lck, few T cells survived to populate the periphery21. In our study, we used Cre driven by the distal promoter of Lck (dLck) to allow the thymic development of T cells and accumulation of naive T cells, albeit in reduced numbers (Supplementary Fig. 1); this permitted us to explore the role of enhanced HIF activity in peripheral T cells. Mice with loxP-flanked Vhl alleles (Vhlfl/fl)22 and Cre driven by that distal Lck promoter23 underwent deletion of Vhl in mature T cells (Vhlfl/fldLck; called `VHL-deficient' here) (Supplementary Fig. 1). All mice lacking VHL in CD8+ T cells (VHL-deficient mice) succumbed to chronic infection with LCMV clone 13, unlike their VHL-sufficient counterparts (Fig. 1a). In contrast, both VHL-deficient and VHL-sufficient mice survived acute infection with LCMV Armstrong (Fig. 1a), which suggested that VHL-mediated control of HIF activity was particularly relevant for the T cell response to chronic viral infection induced by LCMV clone 13. Both strains of LCMV are noncytopathic; the tissue damage that accompanies infection is caused by the host immune response rather than by direct viral damage. To determine if the observed mortality was mediated by antigen-specific CD8+ T cells, we generated VHL-deficient CD8+ T cells from mice of the P14 strain, which use a TCR specific for a peptide fragment of LCMV glycoprotein (gp33) presented by the major histocompatibility complex molecule H-2Db. We transferred VHL-sufficient or VHL-deficient P14 cells into C57BL/6J (B6) hosts, which we then infected with LCMV clone 13. Mice that received VHL-deficient CD8+ T cells showed greater mortality (80%) in the context of chronic infection with LCMV clone 13 than that of hosts that received VHL-sufficient P14 T cells (5%) or that of mice infected with LCMV Armstrong (0%) (Fig. 1a). Histological analysis revealed that LCMV clone 13–infected (chronic infection) recipients of VHL-deficient CD8+ T cells had consolidated, edematous lungs, in contrast to control mice that received VHL-sufficient cells (Fig. 1b), which indicated substantial CD8+ T cell–mediated immunopathology. Although HIF-1α (encoded by Hif1a) has been reported to mediate the hypoxic response in T cells7, in certain cell types and conditions, HIF-2α (encoded by Epas1) mediates hypoxic responsiveness24. We obtained CD8+ T cells from wild-type mice, Vhlfl/fldLck mice, mice with deletion of loxP-flanked Hif1a alleles mediated by Cre expressed from the T cell–specific Cd4 promoter (Hif1afl/flCd4-Cre mice)25 or mice with deletion of loxP-flanked Epas1 alleles mediated by Cre expressed from the endothelial cell–specific Tie2 promoter (Epas1fl/flTie2-Cre mice), activated the cells in vitro with anti-CD3 plus anti-CD28 or left them unstimulated, obtained nuclear extracts of those cells and analyzed them by immunoblot. Under normal oxygen conditions, stimulation via the TCR was sufficient to result in the accumulation of HIF-1α and HIF-2α in wild-type cells; unstimulated cells did not exhibit detectable HIF-1α or HIF-2α during normoxia (Fig. 1c). VHL-deficient cells exhibited enhanced amounts of HIF-1α and HIF-2α protein after activation during normoxia relative to that in wild-type cells (Fig. 1c). To assess the role of HIF-1α and HIF-2a in the mortality of VHL-deficient mice during chronic infection, we generated mice with T cell–specific triple deficiency in Vhl, Hif1a and Epas1 (Vhlfl/flHif1afl/flEpas1fl/fldLck; called `VHL–HIF-1α–HIF-2α–deficient' here). Unlike the VHL-deficient mice, none of the VHL–HIF1α-HIF2α–deficient mice suffered any mortality after infection with LCMV clone 13, despite the presence of ample virus-specific cells (Fig. 1d and Supplementary Fig. 2). Similarly, VHL–HIF-1α–HIF-2α-deficient P14 CD8+ T cells transferred into B6 hosts did not mediate death during infection with LCMV clone 13 (Fig. 1d, right). Notably, CTL effector function during chronic infection with LCMV clone 13 is already markedly compromised by day 9 after infection26,27. Therefore, given the substantial immunopathology induced by noncytopathic infection, our results suggested that `always-on' HIF signaling induced by Vhl deletion augmented the effector capacity of CTLs beyond the attenuated levels observed for wild-type cells during chronic infection and resulted in lethal immunopathology.
Figure 1.

VHL-deficient CD8+ T cells mediate HIF-1α–HIF-2α–dependent death during persistent viral infection. (a) Survival of wild-type (Vhlfl/fl) mice (WT) or VHL-deficient (Vhlfl/fldLck) mice (VHL-KO) after acute infection with LCMV Armstrong (+ Arm) or chronic infection with LCMV clone 13 (+ cl13) (far left) and core body temperature of those mice after chronic infection with LCMV clone 13 (middle left), as well as survival (middle right) and core body temperature (far right) of wild-type host mice given ~1 × 104 VHL-sufficient (WT) or VHL-deficient (VHL-KO) P14 CD8+ T cells (P14→WT), then infected 1 d later as at left. Pooled sample size (host mice): survival (left), n = 15 (wild type) or 11 (VHL-deficient); survival (right), n = 9 (wild-type cells) or 9 (VHL-deficient cells); body temperature, n ≥ 3 per time point. *P =0.003 and **P < 0.001, wild-type versus mutant during chronic infection (log-rank (Mantel-Cox) test). (b) Perimortal lung pathology in wild-type host mice (n = 4) given no cells and left uninfected (UI) or given 1 × 104 VHL-sufficient P14 CD8+ T cells (WT P14→WT) or VHL-deficient P14 CD8+ T cells (VHL-KO P14→WT), then infected 1 d later with LCMV clone 13 and assessed 9 d later. Scale bar, 100 μm. (c) Immunoblot analysis of HIF-1α, HIF-2α and lamin B (loading control throughout) in nuclear extracts of purified wild-type, VHL-deficient (VHL-KO), Hif1afl/flCd4-Cre (HIF-1α-KO) or Epas1fl/flTie2-Cre (HIF-2α-KO) CD8+ T cells left unstimulated (US) or activated for 55 h in vitro with anti-CD3 plus anti-CD28. (d) Survival and core body temperature as in a, but with wild-type and VHL–HIF-1α–HIF-2α-deficient (VHL–HIF-1α–HIF-2α-KO) mice (left) or in wild-type hosts after transfer of wild-type P14 or VHL–HIF-1α–HIF-2α-deficient P14 cells (P14WT; right). Pooled sample size (mice): survival (left), n = 3 (wild type) or 3 (VHL–HIF-1α–HIF-2α-deficient); survival (right), n = 5 (wild-type cells) or 8 (VHL–HIF-1α–HIF-2α-deficient cells); body temperature, n ≥ 3 mice per time point. Data are pooled from three experiments (a,d; error bars, s.e.m.) or are representative of two experiments (b,c).
Altered CTL differentiation by VHL-deficient CD8+ T cells
During acute infection, the differentiation of CD8+ T cells into effector or memory T cell subsets can be predicted by expression of the phenotypic marker KLRG1 and CD127 (the interleukin 7 (IL-7) receptor). KLRG1 marks CTLs that are terminally differentiated and will rapidly wane after infection28, whereas CD127 is re-expressed in the CTL population that contains precursors of memory T cells29, which will survive and sustain long-lived immunity. Shortly after activation, CD8+ T cells transit through a CD127loKLRG1lo stage; these cells have cytotoxic function yet are thought to be uncommitted to a terminal-effector or memory-precursor state30. To evaluate the immune response to persistent infection in more detail, we used VHL-sufficient and VHL-deficient P14 cells, which had similar naive phenotypes and responsiveness to antigen (Supplementary Fig. 1). We transferred equal numbers of those cells into B6 host mice, which we then infected with LCMV clone 13 (Fig. 2a). This strategy allowed us to compare the phenotypic and functional differences between VHL-sufficient (wild-type) and VHL-deficient CD8+ T cells in the same host and ensured equivalent inflammatory and antigenic environments. Initially, fewer VHL-deficient cells than VHL-sufficient cells accumulated; however, at later time points, we recovered these cells at similar abundance in the spleen and, to a lesser extent, in some other tissues (Fig. 2b,c and data not shown). Distinct phenotypic differences between the VHL-sufficient and VHL-deficient effector cells were evident; whereas VHL-sufficient effector cells formed a sizable KLRG1hi population, the majority of VHL-deficient cells did not express KLRG1 (Fig. 2d), which suggested that terminal CTL differentiation was delayed or inhibited in the absence of VHL. We found that VHL-deficient P14 cells also poorly formed KLRG1hi subsets in response to acute infection (Supplementary Fig. 3), which suggested that the altered differentiation due to VHL deficiency was not specific to persistent infection. To determine if alterations observed in VHL-deficient cells during infection were dependent on HIF-1α and HIF-2α, we monitored VHL-HIF-1α-HIF-2α-sufficient and VHL-HIF-1α-HIF-2α-deficient P14 CD8+ T cells in a mixed-transfer infection experiment. Here, the presence of a KLRG1hi effector population was restored for VHL-HIF-1α-HIF-2α- deficient CTLs (Fig. 2e), which showed that the altered effector differentiation observed for VHL-deficient cells was dependent on HIF-1α and HIF-2α. Thus, enhanced HIF activity seemed to inhibit or delay the terminal differentiation of CD8+ effector cells.
Figure 2.
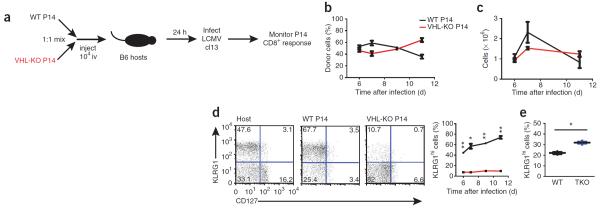
Increased HIF-1α and HIF-2α activity alters CD8+ T cell differentiation during persistent infection. (a) Experimental protocol for b–d: congenically distinct VHL-sufficient P14 CD8+ T cells (WT P14) and VHL-deficient P14 CD8+ T cells (VHL-KO P14) were injected intravenously (iv) together (1 × 104 of each cell type, mixed at a ratio of 1:1) into wild-type B6 hosts (n = 3), followed by infection of the hosts 24 h later with LCMV clone 13. (b) Abundance of transferred VHL-sufficient and VHL-deficient donor cells in the peripheral blood of host mice. (c) Absolute number of VHL-sufficient and VHL-deficient P14 CD8+ T cells recovered from spleen of host mice. (d) Expression of CD127 and KLRG1 by host CD8+ T cells (Host) and by VHL-sufficient or VHL-deficient P14 donor CD8+ cells on day 7 of infection, assessed by flow cytometry (left), and frequency of KLRG1hi donor cells in host mice (right). Numbers in quadrants (left) indicate percent cells in each. *P = 0.0004 and **P < 0.0001 (unpaired Student's t-test). (e) Frequency of KLRG1hi donor cells in hosts given cotransfer of VHL–HIF-1α–HIF-2α-sufficient (WT) and VHL–HIF-1α–HIF-2α-deficient (TKO) P14 CD8+ T cells, followed by infection with LCMV clone 13 (as in a) and analysis 7 d later. Each symbol represents an individual mouse; small horizontal lines indicate the mean. *P = 0.002 (unpaired Student's t-test). Data are representative of three experiments with similar results (error bars, s.e.m.).
HIF activity drives an augmented CTL effector signature
To better understand how the response of VHL-deficient T cells to persistent versus acute infection resulted in such different outcomes (death versus resolution of infection), we examined the effects of VHL deletion on global gene expression during infection (Fig. 3). We transferred VHL-sufficient and VHL-deficient P14 T cells into wild-type host mice and infected the hosts with LCMV (as in Fig. 2a). As the VHL-deficient cells did not form a KLRG1hi population (Fig. 1d), we sorted KLRG1lo VHL-deficient and VHL-sufficient P14 CD8+ effector cells responding in the same host on day 7 of infection with LCMV clone 13 to minimize differences resulting from the presence or absence of terminally differentiated KLRG1hi effector cells. We found significant differences in gene expression: 582 genes displayed a difference in expression of more than twofold (Fig. 3a). Notably, in response to either acute infection or chronic infection, the differences in gene expression in VHL-deficient P14 CD8+ T cells versus VHL-sufficient cells were largely conserved; >96% of genes up- or downregulated during infection with LCMV clone 13 were coordinately regulated during infection with LCMV Armstrong (Fig. 3b). Together these data suggested that the mortality associated with Vhl deletion in CD8+ T cells was dependent on sustained antigen rather than on markedly different transcriptional programs induced by persistent versus acute infection.
Figure 3.

Gene expression by VHL-deficient CTLs reveals an augmented effector phenotype with increased glycolysis, activation-associated receptors and alterations of key transcription factors. (a,b) Microarray analysis of gene expression (mean) by KLRG1lo VHL-sufficient (WT) or VHL-deficient (VHL-KO) P14 CD8+ T cells sorted from a host mouse (n = 2) after cotransfer (as in Fig. 2a), assessed on day 7 of infection of the recipient with LCMV clone 13 (a) or on day 6 of acute infection of the recipient with LCMV Armstrong (b); numbers in plots indicate total genes upregulated (red) or downregulated (blue) in VHL-deficient cells relative to their expression in wild-type cells (a; with a cutoff of a twofold change in expression, and coefficient of variation of 0.8) or frequency of genes regulated as in a (b). (c) Expression of transcripts involved in the glycolytic pathway and negative regulation of oxidative phosphorylation (Neg reg oxphos) in VHL-deficient cells relative to their expression in VHL-sufficient cells, both sorted from a host mouse (after cotransfer) on day 7 of infection with LCMV clone 13 (as in a). (d) Proton production (ECAR), oxygen consumption (OCR) and OCR/ECAR ratio of wild-type and VHL-deficient CD8+ T cells (n = 4 mice per genotype) activated with anti- CD3 and anti-CD28 and then incubated with IL-2 in vitro. *P = 0.0005 and **P < 0.0001 (Student's unpaired t-test). (e) Expression of transcripts involved in various pathways (below plot) in VHL-deficient cells relative to their expression in VHL-sufficient cells, both sorted from a host mouse (after cotransfer) on day 7 of infection with LCMV clone 13 (as in a). (f–h) Expression of granzyme B and perforin (f), 4-1BB and GITR (g), and PD-1, CD244, CTLA-4 and LAG-3 (h) by KLRG1lo VHL-deficient and VHL-sufficient P14 cells from host mice (n = 3) on day 6 of infection with LCMV clone 13, assessed by flow cytometry and presented as geometric mean fluorescence intensity (gMFI); Naive (h), naive (CD44lo) wild-type CD8+ T cells from the same host. *P < 0.0001 and **P = 0.008 (f), *P < 0.0001 and **P = 0.002 (g) and *P < 0.0001 and **P = 0.0003 (h) (all unpaired Student's t-test). (i) Expression of transcription factors by P14 CD8+ T cells after cotransfer into host mice (n = 3) and infection of the hosts for 7 d with LCMV clone 13 as in a, assessed by intracellular immunostaining followed by flow cytometry with gating on KLRG1lo cells. *P = 0.02, **P < 0.0001 and ***P = 0.002 (unpaired Student's t-test). Data are from one experiment (a–c,e; error bars (c), range) or are representative of three experiments (d,f–i; error bars, mean and s.e.m.).
Naive T cells substantially upregulate glycolysis after antigen-driven activation31, which is tightly correlated with lymphocyte activation and function32, yet the role of HIFs in this process during the CTL response to in vivo infection is unknown. Consistent with the function of HIFs reported for other cell types, increased HIF activity in VHL-deficient P14 cells responding to chronic viral infection of host mice in vivo led to potent induction of transcripts involved in the glycolytic pathway above that in VHL-sufficient P14 cells after cotransfer of the cells into host mice (Fig. 3c), with upregulation of mRNA encoding molecules that facilitate many steps in glycolytic metabolism. In accordance with HIF-mediated induction of glycolysis, metabolic measurements demonstrated that the glycolytic activity of VHL-deficient CTLs was enhanced compared with that of wild-type CTLs (Fig. 3d). We activated wild-type or VHL-deficient T cells in vitro and measured their consumption of oxygen and proton-production rate, which are molecular surrogates of oxidative phosphorylation and lactate production from glycolysis, respectively. The VHL- deficient CTLs demonstrated a lower oxygen consumption rate (OCR) and more proton production per cell (extracellular acidification rate (ECAR)), and the ratio of those metabolic determinations (Fig. 3d) confirmed a high glycolysis-low oxidative phosphorylation metabolic profile for VHL-deficient CTLs.
The abundance of mRNA encoding molecules essential for CTL effector function (including various members of the granzyme family, perforin and TNF) was also elevated after deletion of Vhl (Fig. 3e); we also found that expression of the HIF target Vegf (which encodes vascular endothelial growth factor) was elevated. Further analysis of the data set revealed that VHL-deficient cells had increased expression of transcripts encoding molecules associated with activation of T cells, including the costimulatory and activating receptors SLAMF7 (Slamf7), OX40 (Tnfrsf4), 4-1BB (Tnfrsf 9) and GITR (Tnfrsf18) (Fig. 3e). Paradoxically, given the immunopathology suffered by VHL-deficient mice after chronic infection, we also found upregulation of several transcripts induced after T cell activation that encode molecules with characterized inhibitory roles; i.e., Ctla4, Lag3, Havcr2 (TIM-3) and Cd244 (Fig. 3e). Additionally, VHL-deficient cells showed altered expression of various transcriptional regulators known to have central or emerging roles in CTL differentiation (Fig. 3e). Therefore, apart from the expected increased expression of mediators of glycolytic activity, HIF activity affected the expression of genes whose products are responsible for a wide variety of CTL functions, including effector molecules, costimulatory receptors, activation and inhibitory receptors and key transcriptional regulators of effector and memory cell differentiation.
We next verified at the protein level several of the transcriptional changes observed in VHL-deficient cells during infection. In a mixed transfer of VHL-sufficient and VHL-deficient P14 CD8+ T cells assessed on day 7 of infection of the host with LCMV clone 13, we found a substantially greater amount of intracellular granzyme B and perforin in VHL-deficient cells than in VHL-sufficient cells (Fig. 3f). The costimulatory molecules 4-1BB and GITR (of the TNF receptor superfamily) also showed greater induction on VHL-deficient P14 CD8+ T cells than on VHL-sufficient P14 CD8+ T cells (Fig. 3g). Surface expression of activation-induced inhibitory receptors further reflected the transcriptional data, with substantially higher expression of CD244, CTLA-4, LAG-3 (Fig. 3h) and TIM-3 (Supplementary Fig. 4a) in VHL-deficient cells than in VHL-sufficient cells. In contrast, expression of Pdcd1 mRNA and the PD-1 protein it encodes (another key regulator of CTL exhaustion and activation) in VHL-deficient CTLs after infection was significantly higher than that in their naive counterparts but lower than that in virus-specific VHL-sufficient cells (Fig. 3e,h and Supplementary Fig. 4b). The diminished PD-1 expression was dependent on VHL but independent of HIF-1α and HIF-2α (Supplementary Fig. 4c,d); VHL-HIF-1α-HIF-2α-deficient CTLs had lower surface expression of PD-1 than did wild-type CTLs, but VHL-HIF-1α-HIF-2α-deficient mice showed no morbidity during chronic infection, with accumulation of virus-specific cells, survival and core body temperature similar to that of wild-type mice (Fig. 1d and Supplementary Fig. 2). Thus, we concluded that the moderate diminution in PD-1 was unlikely to be the cause of pathology observed in the absence of VHL expression by CD8+ T cells.
Transcriptional regulators control the acquisition of effector function, memory capacity and the exhaustion phenotype of CD8+ T cells. We found that the expression of Tbx21, Eomes and Tcf7 was diminished while Prdm1 expression was elevated in CD8+ cells lacking VHL relative to wild-type cells (Fig. 3e,i). That HIF-induced alteration in transcription factor expression did not correlate with a specific known CTL subset. However, a lower abundance of T-bet may be consistent with the absence of a KLRG1hi population and the accumulation of KLRG1loCD127lo effector cells observed among VHL-deficient cells28. Furthermore, these results suggested that higher expression of Prdm1 (which encodes Blimp-1), associated with the terminal differentiation of effector cells, alone was not sufficient to drive the formation of this population. These data showed that elevated HIF activity modulated the expression of many transcriptional regulators that regulate differentiation into effector and memory CD8+ T cell populations.
Hypoxia and HIF regulate the expression of key CTL molecules
We next used an in vitro system to determine how oxygen tension, HIF-1α, HIF-2α and cytokines were involved in mediating alterations in the expression of key transcription factors and exhaustion-associated receptors. We obtained CD8+ T cells from the spleens of uninfected wild-type mice, Hif1afl/flCd4-Cre mice25, Epas1fl/flTie2-Cre mice or Vhlfl/fldLck mice, activated the cells in vitro with antibody to CD3 (anti-CD3) and anti-CD28, expanded their populations in medium containing IL-2 and then incubated them in normoxic (21%) or hypoxic (1%) conditions. Here Cre was expressed under the control of different promoters that all allowed deletion in T cells; subsequently, we found similar results when we used dLck-driven Cre to induce deletion of the target genes (data not shown). Immunoblot analysis revealed the accumulation of a moderate amount of HIF-1α protein in the activated wild-type CTLs under conditions of normoxia, which was increased further by hypoxic incubation (Fig. 4a). As expected, we found more HIF-1α and HIF-2α in VHL-deficient cells than in wild-type cells during normoxia (Fig. 4a). There was more granzyme B, an essential effector molecule, and the activation-associated costimulatory receptors 4-1BB, GITR and OX40 in wild-type cells exposed to hypoxia than in their normoxic control counterparts, but these were poorly induced in HIF-1α-deficient CTLs by hypoxia (Fig. 4b,c). Also reflective of our in vivo observations, surface expression of the inhibitory receptors LAG-3 and CTLA-4 was also increased in wild-type CD8+ T cells exposed to hypoxia in an HIF-1α-dependent manner, as shown by the ablation of the hypoxic induction of these markers in Hif1afl/flCd4-Cre cells (Fig. 4b,c). VHL-deficient T cells also had high expression of those molecules, which was minimally potentiated by hypoxia, consistent with the observed increase in HIF-1α and HIF-2α during normoxia (Fig. 4a). The expression of T-bet and TCF-1 protein was decreased in VHL-deficient cells and by hypoxic incubation of wild-type cells but not by hypoxic incubation of Hif1afl/flCd4-Cre cells (Fig. 4b,c), which demonstrated the ability of oxygen tension, acting through the HIF pathway, to modulate the expression of transcription factors central to CD8+ T cell differentiation. Hif1afl/flCd4-Cre cells had higher TCF-1 expression and lower granzyme B expression than that of wild-type cells, even under normal oxygen tension (Fig. 4b,c), consistent with the proposal that cytokine stimulation or activation was sufficient for HIF accumulation and had a role in CTLs even in conditions of abundant oxygen. Together these data showed that HIFs served as potent upstream modulators of multiple aspects of CD8+ T cell function, an effect that was further potentiated by hypoxia.
Figure 4.

Oxygen, HIF-1α and HIF-2α regulate essential effector, activation-inhibitory and differentiation-associated proteins of T cells. (a) Immunoblot analysis of HIF-1α, HIF-2α and lamin B in nuclear extracts of wild-type, Hif1afl/flCd4-Cre, Epas1fl/flTie2-Cre and VHL-deficient CD8+ T cells activated in vitro with anti-CD3 plus anti-CD28, followed by population expansion for 96 h in IL-2 and incubation for 6 h in normoxia (ambient air; ~21% O2) or hypoxia (1% oxygen). (b) Flow cytometry of cell-surface or intracellular proteins in wild-type, Hif1afl/flCd4-Cre and VHL-deficient cell populations activated and expanded as in a, followed by 36 h (Gzmb, LAG-3, T-bet and TCF-1) or 12 h (4-1BB, GITR and OX40) of normoxic or hypoxic incubation (as in a). (c) Geometric mean fluorescence intensity of the results in b (n = 3 mice per genotype). *P < 0.01 and **P < 0.001 (two-way analysis of variance with Bonferroni's post-hoc test). (d) Expression of Gzmb, GITR, LAG-3 and T-bet in wild-type CD8+ splenocyte populations (n = 3 mice) activated and expanded as in a, then incubated for 48 h in 1% oxygen with 1 mM 2-deoxy-d-glucose (+2DG), presented relative to that of cells not treated with 2-deoxy-d-glucose (UT), set as 100%. (e) Immunoblot analysis of nuclear extracts of CD8+ T cells activated as in a, followed by incubation with IL-2 and IL-4 for population expansion and then culture for 17 h in normoxia or hypoxia. (f) Expression of granzyme B in cells (n = 3 mice per genotype) activated as in a, followed by incubation for 24 h in IL-2 alone or IL-2 plus IL-4, with or without hypoxia (top), and induction of granzyme B expression during hypoxia relative to its expression during normoxia (bottom). Data are representative of three experiments (a, and b,c (Gzmb, LAG-3, T-bet and TCF-1)) or two experiments (b,c (4-1BB, GITR and OX40) and d; mean and s.e.m. in c; error bars (d), s.e.m.) or three experiments with similar results (e,f; error bars, s.e.m.).
A greater abundance of HIFs resulted in augmented glycolysis by activated T cells and enhancement of many parameters of effector- and activation-associated molecules (Fig. 3f–h) and modulated key transcriptional regulators(Fig. 3i). We used the glucose analog 2-deoxy-d-glucose, which is efficiently taken up by cellular glucose transporters but cannot be metabolized and this competitively inhibits hexokinase and subsequent glycolytic flux, to examine how HIF- and hypoxia-mediated regulation of key molecules was affected by glycolytic activity. The addition of 2-deoxy-d-glucose (at the relatively low concentration of 1 mM) to previously activated T cells markedly blunted the hypoxia-mediated induction of multiple effector- and activation-associated molecules (Fig. 4d).
The induction of HIF-1α relative to that of HIF-2α can be regulated by cytokines in macrophages9. As early T cell activation induced detectable HIF-2α protein (Fig. 1c), yet the amount seemed to be lower after the addition of IL-2 to the stimulation system (Fig. 4a), we analyzed by immunoblot analysis and flow cytometry cells treated as described above (Fig. 4a–c) but with the addition of IL-4, which drives HIF-2α accumulation in macrophages9. Indeed, the addition of IL-4 was sufficient to induce HIF-2α together with HIF-1α in wild-type CTLs exposed to hypoxia (Fig. 4e). Reflective of the amount of each isoform (Fig. 4e), we found that the cytokine environment determined which member of the HIF family was responsible for the hypoxic upregulation of granzyme B in CTLs. The CTL hypoxic response that drove granzyme B expression was completely HIF-1α dependent in cells cultured with IL-2, whereas in cultures that included IL-2 and IL-4, both HIF-1α and HIF-2α had a role in the hypoxic induction of granzyme B expression (Fig. 4f). Therefore, the cytokine milieu and abundance of the individual HIF isoform determined the contribution of HIF-1α or HIF-2α to mediating the hypoxic response for CTLs.
Elevated HIF sustains CTL effector function
To better understand how VHL-deficient CD8+ T cells ultimately resulted in mortality during a persistent infection that is typically tolerated without substantial morbidity, we further characterized the course of infection with LCMV clone 13 in wild-type B6 hosts. First we used a `single-transfer' experimental design with which we could isolate the effects of VHL-sufficient or VHL-deficient P14 CD8+ T cells on viral load. We determined that viral titers were lower in mice that received VHL-deficient P14 CD8+ T cells (Fig. 5a,b). Similar to the findings reported above (Fig. 3), we determined that VHL-deficient effector cells had higher expression of granzyme B and TNF, and this trend was sustained or enhanced over the course of the infection relative to that of VHL-sufficient cells responding in the same host (Fig. 5c). These data were particularly notable, as antigen-specific CD8+ T cells typically undergo progressive loss of production of granzyme B and TNF during infection with LCMV clone 13 (ref. 25); thus, VHL-deficient cells maintained the potential for greater effector function during the early stages of a persistent infection. The in vivo increase in granzyme B in VHL-deficient cells was dependent on HIF-1α and HIF-2α (data not shown). Notably, analysis of plasma at day 6 after infection did not show substantial changes in the abundance of common cytokines (Supplementary Fig. 5a), which suggested CTL-driven immunopathology rather than systemic dysregulation of cytokines. Thus, deletion of Vhl resulted in higher expression of effector molecules (granzyme B and TNF) by antigen-specific CD8+ effector cells, accompanied by lower viral titers (Fig. 5a–d) and increased immunopathology, as revealed by histology (Fig. 1b).
Figure 5.

CD8+ T cells with enhanced HIF activity sustain expression of effector molecules, are refractory to exhaustion and demonstrate superior control of persistent viral infection. (a) Quantitative PCR analysis of the abundance of mRNA encoding LCMV glycoprotein (LCMV gp) in spleen and liver tissue from wild-type B6 hosts given transfer of 1 × 104 VHL-sufficient or VHL-deficient P14 CD8+ T cells, followed by infection with LCMV clone 13 and analysis 7 d later, presented relative to that of control mRNA encoding HPRT. Sample size: n = 10 (host mice given VHL-sufficient P14 cells) or 9 (host mice given VHL-deficient P14 cells). Each symbol represents an individual mouse; small horizontal lines indicate the mean (± s.e.m.). *P < 0.0001 (unpaired Student's t-test). (b) Immunofluorescence microscopy of LCMV antigen (green) and the DNA-intercalating dye DAPI (blue) in liver sections after transfer and infection as in a (n = 5 host mice per cell genotype). Scale bar, 100 μm. (c,d) Expression of granzyme B (c) and TNF (d) in B6 hosts (n = 3) given cotransfer of VHL-sufficient and VHL-deficient P14 CD8+ T cells (1 × 104 cells of each genotype), followed by infection with LCMV clone 13 and analysis on days 6–11 of infection immediately after isolation (c) or after 5–6 h of in vitro culture with a LCMV glycoprotein peptide of amino acids 33–41 (d), assessed by flow cytometry (left) and presented as average gMFI in VHL-deficient cells relative to that in VHL-sufficient cells (right). *P < 0.0001, **P = 0.0003, ***P = 0.002, †P = 0.03 and ‡P = 0.007 (unpaired Student's t-test). (e) Expression of IFN-γ and TNF in cells from B6 hosts given transfer of VHL-sufficient or VHL-deficient P14 CD8+ T cells, followed by infection with LCMV clone 13 and analysis 21 d later (far left) and frequency of cells producing IFN-γ and/or TNF (middle left); expression of IFN-γ and TNF in donor P14 cells obtained from the spleens of surviving host mice and stimulated in vitro with a peptide of LCMV glycoprotein (as above; middle right); and expression of granzyme B (top, far right) and abundance of viral RNA in the spleen (bottom, far right). Sample size (host mice): n = 3 (VHL-sufficient cells) or 5 (VHL-deficient cells). *P = 0.0004, **P = 0.0007, ***P = 0.003 and †P = 0.02 (unpaired Student's t-test). (f) Expression of IFN-γ and TNF in P14 donor cells from B6 hosts (n = 3) given cotransfer of VHL-sufficient and VHL-deficient P14 CD8+ T cells, followed by infection with LCMV clone 13 and analysis 17 d later (far left) and frequency of donor P14 cells producing IFN-γ and/or TNF (middle left); expression of IFN-γ and TNF in donor P14 cells obtained from surviving host mice and stimulated as in e (middle right); and expression of granzyme B in cells immediately after isolation (top, far right). *P = 0.02, **P < 0.0001 and ***P = 0.05 (unpaired Student's t-test). Data are representative of three experiments (a,c,d) or two experiments (b,e,f; error bars (c–f), s.e.m.).
One hallmark of exhaustion is loss of the ability of CTLs to produce both TNF and IFN-γ following antigenic challenge, an effect that becomes progressively more severe as the antigen persists. To assess CTL responsiveness at later time points, we analyzed surviving mice after transfer of either VHL-sufficient P14 cells or VHL-deficient P14 cells (at day 21 of infection of host mice with LCMV clone 13; Fig. 5e) or transfer of a mixture of VHL-sufficient and VHL-deficient P14 cells (at day 17 of infection of host mice with LCMV clone 13; Fig. 5f). We found that VHL-deficient cells were refractory to exhaustion; more of those cells produced both TNF and IFN-γ, and those that did displayed higher intracellular protein expression of these cytokines. VHL-deficient P14 cells also had much more intracellular granzyme B than did VHL-sufficient P14 cells in both the single- and mixed-transfer experiments (Fig. 5e,f). Consistent with an enhanced effector function, VHL-deficient cells were also more efficient than VHL-sufficient cells were in an in vivo cytoxicity assay (Supplementary Fig. 5c).
In vivo, we found that VHL-deficient cells responding to infection with LCMV clone 13 upregulated many genes associated with exhausted wild-type cells26 (Supplementary Fig. 6). However, despite that similarity, ~30% of the genes displayed an inverse association; i.e., for these genes (including Eomes, which identifies a terminally differentiated population of T cells that has poor cytokine production during chronic infection33), the VHL-deficient cells were more like acute infection–generated, functional effector cells than exhausted cells. These data suggested VHL-deficient cells may not differentiate to this end stage of exhaustion. Thus, loss of Vhl expression by CD8+ T cells led to a sustained effector state in which cells did not express transcription factors or cell-surface markers associated exclusively with terminal differentiation or memory formation. The poor formation of virus-specific KLRG1hi CTLs during acute infection and the early stages of chronic infection further suggested that deletion of Vhl limited terminal CTL differentiation.
Persistent viral infection and cancer are both characterized by sustained antigens and loss of CD8+ T cell function19. We next assessed the ability of VHL-deficient CTLs to control the growth of a B16 mouse melanoma tumor line that expresses ovalbumin. VHL-deficient CTLs expressing the OT-I TCR, which is specific for ovalbumin peptide presented by H-2Kb, demonstrated superior control of established tumors and prolonged the time to reach a specific tumor volume relative to that of VHL-sufficient OT-I CTLs (Fig. 6). Moreover, 5 of 20 mice that received VHL-deficient OT-I CD8+ T cells had no detectable masses at the end of the study (50 d), while 0 of 19 mice that received VHL-sufficient OT-I CD8+ T cells and 0 of 25 mice that received no OT-I T cells were tumor free (Fig. 6b). These data suggested that enhanced activity of HIF-1α and HIF-2α may be a potent strategy with which to sustain the effector function of CD8+ T cells in two clinically important settings in which prolonged antigen exposure dampens CTL function: persistent viral infection and cancer.
Figure 6.
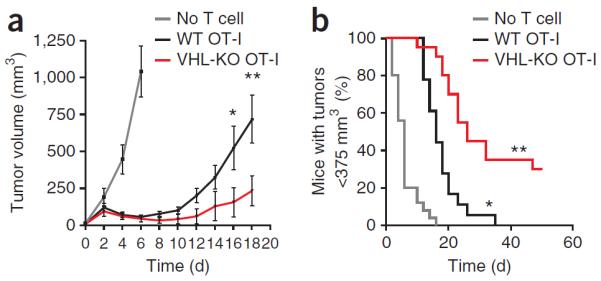
VHL-deficient CTLs exhibit enhanced control of experimental melanoma. Tumor volume (a) and frequency of mice with a tumor volume of <375 mm3 (b) among mice given subcutaneous injection of 1 × 106 ovalbumin-expressing B16 melanoma cells, followed 7 d later by intravenous adoptive transfer of no T cells or ovalbumin peptide–activated VHL-sufficient or VHL-deficient OT-I CD8+ T cells (3 × 106). (a) *P = 0.04 and **P = 0.01, VHL-sufficient versus VHL-deficient (unpaired Student's t-test). (b) *P < 0.0001, no T cells versus VHL-sufficient T cells, and **P = 0.0001, VHL-sufficient versus VHL-deficient (log-rank (Mantel-Cox) test). Sample size (host mice): n = 25 (no T cell transfer), 19 (VHL-sufficient) or 20 (VHL-deficient). Data are representative of two independent experiments (error bars, s.e.m.).
DISCUSSION
Many viruses and pathogenic bacteria have evolved a strategy of persistence to optimize transmission in specific host populations34. For this class of pathogen, the immune system is ineffective in clearance, yet an ongoing immune response has the potential to cause severe or even lethal immunopathology16. The ability of the host to survive infection can be dependent on dampening of the cytotoxic immune response in the face of sustained pathogen burden35. Loss of VHL in virus-specific CTLs led to a failure in the tolerance-exhaustion adaptation to persistent infection. Elevated HIF activity in CD8+ T cells increased glycolytic metabolism and augmented effector capacity and the expression of effector molecules and activation-associated costimulatory and inhibitory receptors. CTLA-4 (ref. 36), LAG-3 (ref. 37) and TIM-3 (ref. 38) all had high expression on VHL-deficient cells, and each has been shown to have a role in inhibiting CTLs in the context of tumors and/or persistent infection. The high expression of inhibitory receptors may have reflected the enhanced activation state of the VHL-deficient cells. Nevertheless, mice infected with LCMV clone 13 succumbed to persistent infection when Vhl was deleted from virus-specific CTLs, which demonstrated that the aggregate program induced by the VHL-HIF pathway can bypass signals from multiple inhibitory receptors and act to maintain effector function. The costimulatory receptors induced in CTLs by hypoxia in a HIF-dependent fashion (4-1BB, OX40 and GITR) have been shown to enhance and sustain T cell effector function; 4-1BB, specifically, induces granzyme B in CTLs, and many members of the TNF receptor superfamily are being investigated for their ability to modulate the antipathogen and antitumor activity of T cells39. Further, mRNA encoding soluble factors, including the cytokines TNF, IFN-γ and VEGFA, was also more abundant in VHL-deficient T cells than in wild-type T cells. Although these molecules did not show a difference in abundance in the plasma at day 6 in the polyclonal post-infection model, these factors may serve local roles either in the immunopathology observed or through paracrine signaling to other cell types. While the expression of various transcription factors linked to CTL function was altered in VHL-deficient cells, including lower expression of Tbx21, Eomes and Bcl6, these may not be expected to account for enhanced effector activity; however, higher expression of Prdm1 could drive some aspects of the VHL-deficient phenotype40. Of note, in response to infection, VHL-sufficient P14 cells and VHL-deficient P14 cells had similar expression of Rorc, Il17, IL23r and Il10 at the mRNA level and had ample IFN-γ production and cytotoxic capacity and thus did not resemble the `Tc17' cells that mediate pathology during infection in mice doubly deficient in Tbx21 and Eomes or the IL-10-producing CTL subset associated with Blimp-1 (refs. 40,41). Blocking glycolytic activity with 2-deoxy-d-glucose inhibited the hypoxic induction of activation-effector molecules, consistent with the proposal that augmented glycolysis enables, at least in part, the enhanced effector-activation program induced by deletion of Vhl.
Spontaneous mutations in VHL that result in loss or impairment of function can give rise to familial kidney cancers and other cancers through HIF-dependent and HIF-independent mechanisms; thus, VHL acts as a classic tumor suppressor5,42. It has also been found that specific partial loss-of-function mutations in VHL can cause familial polycythemias. A spontaneously occurring mutation in the gene encoding the VHL protein R200W results in an impaired ability to degrade HIF and globally elevated HIF function; people homozygous for that mutation suffer from Chuvash polycythemia43. Although some reports have suggested that heterozygosity for that mutation results in an advantage in adaptating to the hypoxia of high altitude through increased red blood cell production44, initial studies have also indicated altered cytokine production by lymphocytes from patients with Chuvash polycythemia45. Notably, distinct mutations have been found in other populations that result in a polycythemic phenotype similar to that of Chuvash polycythemia46; thus, understanding how VHL-HIF affects CD8+ T cell–mediated immunity will be of considerable interest in understanding these patient populations.
In vivo, activated CTLs traffic to sites of infection, where hypoxia from tissue damage and/or disruption coincides with the recognition of pathogen-infected cells and, ultimately, cytolytic activity. In this context, both hypoxia and TCR-mediated signals can lead to an increased abundance of HIF-1α and HIF-2α. Therefore, the ability of HIF signaling to locally limit the terminal differentiation of CTLs and enhance effector function in such zones, despite increased expression of inhibitory receptors, may serve to tie pathogen load and tissue damage to proliferative potential and effector function. That balance is disrupted by the enhanced HIF activity that results from Vhl deletion. Our study did not address whether persistent infection with a noncytopathic virus such as LCMV clone 13 increases tissue hypoxia. However, because TCR signaling and hypoxia together induce maximal HIF transactivation, which in turn drives effector molecules, we hypothesize that this pathway serves to augment T cell function in scenarios such as cytopathic viral infection, during which local tissue damage induces hypoxia and containment of the pathogen even at the expense of local immunopathology would be beneficial to the host. An exhaustion-induced reduction in cytotoxic function may be beneficial in averting immunopathology during persistent viral infection; however, therapeutically overriding the exhaustion of CD8+ T cells can also lead to the resolution of infection47 or, in the context of antitumor responses, enhanced elimination of tumors18,19.
The observation that the HIF pathway promoted the effector function of CD8+ T cells is consistent with a proinflammatory role for HIF in myeloid lineages10. HIF activity in macrophages suppresses localized T cell responses48, which suggests that microenvironmental cues specific to infected tissues may provide a homeostatic mechanism whereby myeloid cells and T cells balance effector function to suppress excessive tissue damage. This has relevance to cancer and infection; in both of those settings, the balance of the infiltration of T cells and that of myeloid cells in a hypoxic setting can lead to clearance of the diseased tissue or immunosuppression and advancement of the pathology. A nuanced approach using compounds to manipulate the hypoxic response could therefore induce activation or inhibition of immunity, depending on the cellular composition of the microenvironment. Thus, our work suggests novel strategies for either promoting antitumor and antipathogen responses or dampening immunopathology.
ONLINE METHODS
Mice and experimental design
Mice were bred and housed in specific pathogen–free conditions in accordance with the Institutional Animal Care and Use Guidelines of the University of California San Diego. The following mice have been described: Vhlfl/fl mice22, Hif1afl/fl mice32, and Epas1fl/fl mice24. Deletion of those loxP-flanked genes in T cells was achieved by crossing of mice with loxP-flanked alleles to dLck mice23, mice expressing Cre from the T cell–specific Cd4 promoter49 or Tie2-Cre mice to obtain mice with homozygous loxP-flanked alleles without Cre or hemizygous for Cre. P14 mice, which recognize an immunodominant epitope of the LCMV glycoprotein common to LCMV Armstrong strain and clone 13 (ref. 50) were bred to the appropriate lines with loxP-flanked alleles. All mice were backcrossed over ten generations to the B6 background. Randomization and steps to reduce experimental bias were done as follows: after initial identification and selection of wild-type and mutant mice, processing of samples and was routinely done by identification code rather than genotype until data-acquisition stages. For the tumor assay (Fig. 6), wild-type and mutant T cells were transferred in mixed order into random tumor-bearing mice and the genotype of the transferred cells was not indicated on cages or adjacent to measurements during monitoring of tumor size.
Infection and cell transfer
Mice were infected47 with LCMV Armstrong strain (2 × 105 plaque-forming units, injected intraperitoneally) or LCMV clone 13 (2 × 106 plaque-forming units, injected intravenously). For adoptive transfer, 1 × 104 to 5 × 104 Vα2+CD8+ P14 cells were injected intravenously into B6 recipient mice, which were then infected with the relevant pathogen 1 d later.
In vitro and hypoxic incubation
Samples from mouse spleens were enriched for CD8+ T cells by negative selection. Incubation with biotinylated anti-B220 (RA3-6B2), anti-CD19 (1D3), anti-Ter-119 (Ter119), anti-CD4 (GK1.5), anti-NK1.1 (PK136) and anti-CD11b (M1/70; all from eBiosceince) was followed by incubation with streptavidin-labeled magnetic beads and depletion on a MACS column. Purified CD8+ T cells were then activated for 48–72 h with plate bound anti-CD3 (145-2C11; University of California, San Francisco, Antibody Core) and soluble anti-CD28 (37.51; University of California, San Francisco, Antibody Core) in RPMI-1640 medium containing 10% FCS, 25 mM HEPES, pH 7.2, 1% penicillin-streptomycin-glutamine and 55 μM β-mercaptoethanol. Cells were then pelleted and replated in fresh medium supplemented with 100 U/ml human IL-2 alone or additionally with 20 ng/ml mouse IL-4, followed by incubation for 24–48 h. Cells were then split into normoxic conditions (standard incubator) or 1% oxygen (Thermo Scientific-Hera Cell incubator equipped to replace oxygen with nitrogen), followed by incubation for various times. Flasks were then removed, placed immediately on ice and processed for flow cytometry or immunoblot analysis. For analysis of cellular metabolism, CD8+ T cells were purified on columns, activated and cultured in IL-2 as described above and then were plated and analyzed on a Seahorse XF24–3 according to the manufacturer.s instructions. 1 mM 2-deoxy-d-glucose (Sigma) was added to the medium where appropriate.
Immunoblot analysis
Nuclear extracts were isolated (NE-PER kit; Pierce) and ~15 μg protein was loaded in each lane (Novex). HIF-1α and HIF-2α were detected with NB100–449 and NB100–122, respectively (Novus). Lamin B was used as a loading control (sc-6217; Santa Cruz Biotechnology).
Flow cytometry and sorting
Cells were immunostained and acquired on a BD FACSCalibur or Fortessa, and were sorted (where needed) on a BD FACSAria. In addition to the antibody clones noted above for depletion, the following fluorophore-conjugated antibodies were used for flow cytometry and sorting: anti-4-1BB (17B5), anti-CD8α (53–6.7), anti-CD25 (PC61.5), anti-CD44 (IM7), anti-CD45.1 (A20), anti-CD45.2 (104), anti-CD62L (MEL-14), anti-CD69 (H1.2F3), anti-CD127 (A7R34), anti-CD244 (244F4), anti-GITR (DTA-1), anti-Eomes (Dan11mag), anti-IFN-γ (XMG1.2), anti-KLRG1 (2F1), anti-LAG-3 (C9B7W), anti-OX40 (OX86), anti-T-bet (4B10), anti-TNF (MPGXT22), anti-PD-1 (J43), anti-perforin (OMAK-D), anti-TIM-3 (RMT3–23) and anti-V2 (B20.1; all from eBioscience); anti-CTLA-4 (UC10-4F10-11; BD), anti-granzyme B (MHGB05; Life Technologies); and anti-TCF1 (C63D9; Cell Signaling Technologies). Phycoerythrin-conjugated H-2Db tetramers loaded with a peptide with the sequence KAVYNFATC were from Beckman Coulter.
Gene expression
For microarray studies, transferred P14 cells were sorted on ice from pooled spleens on day 6–7 after infection, as CD8α+CD4−KLRG1−Vα2+ cells with the appropriate CD45 congenic marker. 3 × 104 cells were double-sorted into TRIzol to a purity of >95%, then RNA was amplified twice (MessageAmp RNA Amplification kit; Ambion), labeled with biotin (BioArray HighYield RNA Transcript Labeling kit; Enzo Diagnostics) and purified (RNeasy Mini kit; Qiagen). The resulting cRNA was hybridized to GeneChip Mouse Gene 1.0 ST arrays. Data were normalized by the robust multi-array average method and analyzed with the GenePattern software suite.
Tumor challenge
1 × 106 ovalbumin-expressing B16 melanoma cells were injected subcutaneously into B6 mice. Lymphocytes from Vhlfl/fl OT-I or Vhlfl/fldLck OT-I donor lymph nodes were cultured for 60 h at a density of 1 × 106 cells per ml with 1 μg/ml ovalbumin peptide. Activated OT-I cells (3 × 106) were injected intravenously 7 d after the injection of tumor cells, and tumor size was monitored by measurement in two dimensions with digital calipers. Volume was then estimated by the formula a2 × b, where a was the larger dimension.
Statistical analysis
Two-group comparisons were assessed with an unpaired Student's t-test, survival data were assessed with the log-rank (Mantel-Cox) test, and grouped data were assessed by two-way analysis of variance followed by Bonferroni post-test (to adjust for multiple comparisons). P values of less than 0.05 were considered significant. No data-point-exclusion criteria were used, and the normality and variance of the distribution of the data was not assessed.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Zuniga Laboratory at the University of California, San Diego, for technical advice, discussions and reagents; V. Jhaveri for technical assistance; A. Best for assistance with bioinformatics; the Immunological Genome Project for microarray reagents, processing and assistance; members of the Hedrick Laboratory (University of California, San Diego) for human IL-2; and E. Zuniga, S. Hedrick, G. Barton and J. Harker for discussions and review of the manuscript.
Supported by the US National Institutes of Health (A.W.G. and R.S.J.), the Pew Scholars Program and the Leukemia Lymphoma Society (A.W.G.), the US National Institutes of Health and University of California, San Diego, Cancer Biology Fund (A.L.D.), the University of California, San Diego, and US National Institutes of Health Cell (A.T.P.) and the Austrian Science Fund (M.H.S.).
Footnotes
Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS A.L.D. designed and did experiments, analyzed the data and wrote the paper; A.T.P. designed and did experiments, analyzed data and assisted in writing the paper; M.H.S. did histological analysis, provided advice for the design and analysis of pathophysiological experiments and assisted in writing the paper; J.K.F. and J.V.N. did and analyzed viral titer experiments and assisted with animal work; E.Y. did and analyzed immunofluorescence experiments; R.S.J. provided reagents, provided advice for experimental design and wrote the paper; and A.W.G. supervised the project, designed the experiments, analyzed the data and wrote the paper.
COMPETING FINANCIAL INTERESTS The authors declare competing financial interests: details are available in the online version of the paper.
Accession codes. GEO: microarray data, GSE50158.
References
- 1.Via LE, et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect. Immun. 2008;76:2333–2340. doi: 10.1128/IAI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaupel P. Hypoxia in neoplastic tissue. Microvasc. Res. 1977;13:399–408. doi: 10.1016/0026-2862(77)90106-6. [DOI] [PubMed] [Google Scholar]
- 3.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N. Engl. J. Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rankin EB, Giaccia AJ. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008;15:678–685. doi: 10.1038/cdd.2008.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Latif F, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 6.Semenza GL. HIF-1 and human disease: one highly involved factor. Genes Dev. 2000;14:1983–1991. [PubMed] [Google Scholar]
- 7.Nakamura H, et al. TCR engagement increases hypoxia-inducible factor-1α protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J. Immunol. 2005;174:7592–7599. doi: 10.4049/jimmunol.174.12.7592. [DOI] [PubMed] [Google Scholar]
- 8.Blouin CC, Page EL, Soucy GM, Richard DE. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1α. Blood. 2004;103:1124–1130. doi: 10.1182/blood-2003-07-2427. [DOI] [PubMed] [Google Scholar]
- 9.Takeda N, et al. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cramer T, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dang EV, et al. Control of TH17/Treg balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi LZ, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McNamee EN, Korns Johnson D, Homann D, Clambey ET. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol. Res. 2013;55:58–70. doi: 10.1007/s12026-012-8349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finlay DK, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012;209:2441–2453. doi: 10.1084/jem.20112607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harty JT, Tvinnereim AR, White DW. CD8+ T cell effector mechanisms in resistance to infection. Annu. Rev. Immunol. 2000;18:275–308. doi: 10.1146/annurev.immunol.18.1.275. [DOI] [PubMed] [Google Scholar]
- 16.Frebel H, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med. 2012;209:2485–2499. doi: 10.1084/jem.20121015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 18.Matsuzaki J, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA. 2010;107:7875–7880. doi: 10.1073/pnas.1003345107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baitsch L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wherry EJ. T cell exhaustion. Nat. Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 21.Biju MP, et al. Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol. Cell Biol. 2004;24:9038–9047. doi: 10.1128/MCB.24.20.9038-9047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc. Natl. Acad. Sci. USA. 2001;98:1583–1588. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang DJ, et al. Selective expression of the Cre recombinase in late-stage thymocytes using the distal promoter of the Lck gene. J. Immunol. 2005;174:6725–6731. doi: 10.4049/jimmunol.174.11.6725. [DOI] [PubMed] [Google Scholar]
- 24.Gruber M, et al. Acute postnatal ablation of Hif-2α results in anemia. Proc. Natl. Acad. Sci. USA. 2007;104:2301–2306. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaech SM, et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 30.Jameson SC, Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity. 2009;31:859–871. doi: 10.1016/j.immuni.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J. Immunol. 2004;172:4661–4665. doi: 10.4049/jimmunol.172.8.4661. [DOI] [PubMed] [Google Scholar]
- 32.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paley MA, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338:1220–1225. doi: 10.1126/science.1229620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oldstone MB. Viral persistence. Cell. 1989;56:517–520. doi: 10.1016/0092-8674(89)90573-4. [DOI] [PubMed] [Google Scholar]
- 35.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamoto N, et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog. 2009;5:e1000313. doi: 10.1371/journal.ppat.1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blackburn SD, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin HT, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA. 2010;107:14733–14738. doi: 10.1073/pnas.1009731107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snell LM, Lin GH, McPherson AJ, Moraes TJ, Watts TH. T-cell intrinsic effects of GITR and 4–1BB during viral infection and cancer immunotherapy. Immunol. Rev. 2011;244:197–217. doi: 10.1111/j.1600-065X.2011.01063.x. [DOI] [PubMed] [Google Scholar]
- 40.Welsh RM. Blimp hovers over T cell immunity. Immunity. 2009;31:178–180. doi: 10.1016/j.immuni.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Intlekofer AM, et al. Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science. 2008;321:408–411. doi: 10.1126/science.1159806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaelin WG., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- 43.Ang SO, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002;32:614–621. doi: 10.1038/ng1019. [DOI] [PubMed] [Google Scholar]
- 44.Miasnikova GY, et al. The heterozygote advantage of the Chuvash polycythemia VHLR200W mutation may be protection against anemia. Haematologica. 2011;96:1371–1374. doi: 10.3324/haematol.2011.045609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niu X, et al. Altered cytokine profiles in patients with Chuvash polycythemia. Am. J. Hematol. 2009;84:74–78. doi: 10.1002/ajh.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomasic NL, et al. The phenotype of polycythemia due to Croatian homozygous VHL (571C>G:H191D) mutation is different from that of Chuvash polycythemia (VHL 598C>T:R200W) Haematologica. 2013;98:560–567. doi: 10.3324/haematol.2012.070508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 48.Doedens AL, et al. Macrophage expression of hypoxia-inducible factor-1α suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70:7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee PP, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 50.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.