

Abstract
The limited size of the germline antibody repertoire has to recognize a far larger number of potential antigens. The ability of a single antibody to bind multiple ligands due to conformational flexibility in the antigen-binding site can significantly enlarge the repertoire. Among the six hyper-variable complementarity determining regions (CDRs) that comprise the binding site, the CDR H3 loop is particularly flexible. Computational protein design studies showed that predicted low energy sequences compatible with a given backbone structure often have considerable similarity to the corresponding native sequences of naturally occurring proteins, indicating that native protein sequences are close to optimal for their structures. Here, we take a step forward to determine whether conformational flexibility, believed to play a key functional role in germline antibodies, is also central in shaping their native sequence. In particular, we use a multi-constraint computational design strategy, along with the Rosetta energy function, to propose that the native sequences of CDR H3 loops from germline antibodies are nearly optimal for conformational flexibility. Moreover, we find that antibody maturation may lead to sequences with a higher degree of optimization for a single conformation, while disfavoring sequences that are intrinsically flexible. In addition, this computational strategy allows us to predict mutations in the CDR H3 loop to stabilize the antigen-bound conformation, a computational mimic of affinity maturation, that may increase antigen binding affinity by pre-organizing the antigen binding loop. In vivo affinity maturation data are consistent with our predictions. The method described here can be useful to design antibodies with higher selectivity and affinity by reducing conformational diversity.
Keywords: antibody flexibility, computational structural biology, computational design, multi-constraint design, affinity maturation
INTRODUCTION
Antibodies recognize and neutralize antigens through interactions mediated by the variable domains VH and VL. The antigen binding site is primarily composed of six hyper-variable loops known as the complementarity determining regions (CDRs), with each VH and VL contributing three loops, called H1, H2, H3 and L1, L2, L3, respectively1,2. The broad range of binding specificities exhibited by antibodies is the result of the diversity in sequence, length and conformational flexibility of the CDRs3–6. The limited size of the germline antibody repertoire has to recognize a far larger number of potential antigens. Even though gene rearrangements broaden the spectrum of binding specificities, additional mechanisms for increasing antibody cross-reactivity have been hypothesized to overcome the limits imposed by the available B cell receptors7–11. In particular, structural and biochemical studies have shown that germline antibodies often possess flexible binding sites, which frequently undergo loop conformational changes and side-chain rearrangements upon antigen binding, with the most prominent changes occurring in the CDR H3 loop12–18. Conformational flexibility, defined as the ability to adopt multiple conformations, of germline antibodies could thus provide alternative ways of presenting the binding site to accommodate structurally unrelated ligands19. This flexibility-derived multi-specificity might be achieved at the expense of a relative weak strength of binding12,13. Antibody maturation could then act by increasing the affinity of an antigen-antibody complex, often by reducing flexibility and stabilizing the antibody binding site in a conformation pre-organized for the interaction with the targeted antigen12,13,15–17,20. This decrease in conformational flexibility might in turn reduce any potential cross-reactivity that resulted from conformational diversity19,21.
Computational protein design methods have progressed considerably22–26, advancing our understanding of the relationship between protein sequence and three-dimensional structure. Recently, a computational design method was used to increase antibody-antigen binding affinity mainly through modulation of electrostatic interactions27. Moreover, a variety of methods for designing protein variants with altered conformational flexibility have been implemented with considerable success. These approaches consider simultaneously several protein conformers during the design simulation (multi-constraint design28) and explicitly stabilize one conformation over alternative input conformers29,30, or all input conformations simultaneously31. In the latter case, the output sequences generated by the design simulation are likely to represent a compromise between the different preferences of all conformers considered.
It has been shown that low energy designed protein sequences for a given protein backbone structure often have considerable similarity to the corresponding native sequences of naturally occurring proteins, suggesting that native protein sequences are close to optimal for their structures32–38. This prompted us to hypothesize that, if conformational flexibility is an intrinsic property of the germline antigen-binding site, then antibody native sequences, particularly those of the CDR H3 loops, should show a compromise between the sequence preferences of alternative conformations adopted by the loop. Here, we use a multi-constraint computational design strategy39, based on the Rosetta design algorithm35 and scoring function24,40,41, to suggest that the native sequences of CDR H3 loops in germline antibodies known to adopt several conformations are close to optimal for conformational flexibility. While the computational design of surface-exposed and loop regions is challenging, the Rosetta algorithm has been applied successfully to engineer a protein loop in good agreement with the crystal structure of the designed protein42, indicating that, although still difficult, high-resolution design of protein loops is becoming possible. By generating sequence profiles from the design simulations, we predict mutations in CDR H3 loops to preferentially stabilize the bound conformation, and show that our predictions agree with existing experimental data on antibody affinity maturation. The strategy used in this study can serve to design antibodies with increased specificity and affinity by reducing antibody conformational diversity.
MATERIALS AND METHODS
Creation of a dataset of pairs of free and antigen-bound antibody structures with CDR H3 loops adopting alternative conformations
We used a combined approach that included an exhaustive search of the Protein Data Bank43 as well as a literature search to identify all pairs of germline antibody X-ray structures that have been crystallized in both the bound and free conformations. The final set of germline antibodies is shown in Table I, rows 1 to 6. The sequence and length of the CDR loops was determined using the SACS database44. The Cα RMSD for each of the CDR loops was calculated by local superimposition of the bound and free forms of each of the CDR loops independently (Cα loop atoms were used for the superimposition and RMSD calculations). Among all six CDR loops, only CDR H3 loop showed considerable differences (at least 0.6 Å Cα RMSD) between the bound and free forms for all germline antibodies in our dataset (Table SI). Similarly, we then identified all mature antibodies crystallized in both the antigen-bound and free forms that differ in their CDR H3 loop conformations. We ensured that germline and mature antibody sets had comparable characteristics, by enforcing in all cases the following criteria: (i) CDR H3 loop Cα RMSD ≥ 0.6 Å between bound and free conformations, and (ii) CDR H3 loop length ranging from 5 to 12 amino acid residues. Note that all Fv antibody bound-free pairs share 100% sequence identity, except antibody 50.1, in which one position differs (residue 5 in the H chain is Lys in the bound form, but Gln in the free form)45,46; this mutation is not in contact with the CDR H3 loop or its surrounding shell47. In cases in which the same antibody was crystallized bound to different molecules, the structure containing the molecule against which the antibody was raised was chosen (e.g. structure 1n7m for 7g12, 1q9q for s25-2, or 1oau for spe7). This choice was important for mature antibodies and when variants of an antibody with varying degrees of affinity maturation were compared (see next paragraph). When several structures of the same antibody form were available, the one with the highest resolution was selected (Table I).
Table I.
Dataset of germline and mature antibody structures
| Antibody Name |
Structures (PDB code) |
Resolution (Å) |
CDR H3 loop RMSD (Å) |
CDR H3 loop No. of residues |
||
|---|---|---|---|---|---|---|
| Bound | Free | Bound | Free | |||
| 7g12 | 1n7m | 1ngz | 1.8 | 1.6 | 1.0 | 5 |
| 28b4 | 1fl6 | 1fl5 | 2.8 | 2.1 | 1.8 | 8 |
| az-28 | 1dv6 | 1d5i | 2.0 | 2.0 | 1.2 | 11 |
| 36–65 | 2a6i | 2a6j | 2.5 | 2.7 | 0.6 | 12 |
| s25-2 | 1q9q | 1q9k | 1.5 | 2.0 | 2.2 | 11 |
| 48g7 | 1a7j | 2rcs | 2.1 | 2.1 | 0.8 | 5 |
| pc282 | 1kcs | 1kcv | 2.5 | 1.8 | 1.6 | 6 |
| 3f4 | 1cu4 | 1cr9 | 2.9 | 2.0 | 1.0 | 5 |
| 50-1 | 1ggi | 1ggc | 2.8 | 2.8 | 1.7 | 5 |
| nc6-8 | 2cgr | 1cgs | 2.2 | 2.6 | 0.8 | 7 |
| bv04 | 1cbv | 1nbv | 2.7 | 2.0 | 1.1 | 10 |
| 17-9 | 1ifh | 1hil | 2.8 | 2.0 | 2.0 | 11 |
| spe7 | 1oau | 1oaq | 1.8 | 1.5 | 1.5 | 11 |
| mn12h2 | 1mpa | 1mnu | 2.6 | 2.5 | 0.8 | 12 |
The CDR H3 loop of these antibodies shows conformational changes in bound and free conformations according to X-ray data.
Antibodies are classified as germline (rows 1 to 6) and mature (rows 7 to 14) according to published data.
Creation of a dataset of X-ray structures of pairs of antibodies differing only in their degree of maturation
We created a dataset of X-ray structures of pairs of antibodies that differ only in their degree of maturation, using the same methodology described above. To minimize structural differences within pairs that could arise from the absence or presence of different binding partners, we looked for structures crystallized in the same form (e.g. both in the free form or, in the case of antigen-bound forms, both bound to the same epitope). The final dataset is composed of the following pairs of structures (the resolution in Angstroms is shown in brackets and the germline antibody (or the antibody isolated after a “short period” of exposure to the antigen) is listed first for each pair): 1n7m(1.0)-1ngw(2.6), 1fl6(2.8)-1kel(1.9), 1dv6(2.0)-1axs(2.6), 1aj7(2.1)-1gaf(2.0), 1q9q(1.5)-1q9w(1.8), 1mlc(2.5)-1p2c(2.0), 1ndm(2.1)-1ndg(1.9), 1ngz(1.8)-1ngy(2.2), 1fl5(2.1)-1kem(2.2), 1d5i(2.0)-1d5b(2.8), 2a6j(2.7)-1jfq(1.9), 2rcs(2.1)-1hkl(2.7), 1q9k(2.0)-1q9o(1.8), 1mlb(2.1)-2q76(2.0). Pairs 1 to 7 correspond to bound states and pairs 8 to 14 to free states.
Antibody CDR H3 loop design
Before starting the design protocol, PDB structures were prepared as in39. Briefly, all antigens, hetero-atoms (including water molecules), and hydrogen atoms present in the original PDB file were excluded. Then, hydrogen atoms were added using the procedure described in41. Finally, side-chain torsion angle minimization was performed using the Rosetta scoring function (cysteine side chains were kept fixed to avoid interfering with native disulfide bonds).
The Rosetta design method, described in35, and full-atom scoring function24,40,41 was used in all simulations and implemented as in39. Briefly, the Rosetta design score is dominated by a Lennard-Jones potential, an explicit hydrogen-bonding term41, and an implicit solvation model48; the total score results from a trade off among the different terms present in the scoring function. Side chains from a rotamer library (including the native amino acid residue PDB conformation), and with additional rotamers around the chi 1 and chi 2 angles, were sampled on a fixed backbone using a Monte Carlo simulated annealing optimization protocol24. Sequences were optimized for a single structure or for a set of input structures by using single- and multi-constraint protocols, respectively. Single-constraint simulations serve to identify lowest score sequences for each of the target conformations separately, whereas multi-constraint simulations serve to identify lowest score sequences compatible with multiple target conformations. Similar to the method described in39, for multi-constraint simulations the score was a sum of the scores of a given amino acid sequence calculated for both conformations. Simulations designed all positions of the CDR H3 loop. Residues for which, based on the native sequence and structure, at least one side chain heavy atom was located within 4 Å of a heavy atom of any residue in the H3 loop were chosen for design were repacked (allowed to change rotamer conformation while keeping the amino acid residue type fixed). Native cysteine residues were excluded from designing or repacking. Each single- or multi-constraint optimization allowed all amino acid residues (except cysteine) to be substituted at each position selected for design. All simulations used a genetic algorithm to generate and propagate putative sequences. An initial random population of 2000 sequences was allowed to propagate for 70–150 generations. Lowest scoring sequences were taken after the score in sequence simulations using the genetic algorithm remained approximately constant over several generations (not more than 0.7 Rosetta units difference in score; on average, convergence defined by this criterion was observed after 50–100 generations). For a more detailed description of the method refer to39. It should be noted that both search methods applied here, Monte Carlo simulated annealing for rotamer optimization and the genetic algorithm for sequence optimization, do not guarantee to find the globally optimal solution. Therefore, we compared the genetic algorithm design predictions to exhaustive sequence enumerations for the single- and multi-constraint design simulations for the germline antibody 7g12 (where design on 5 loop positions yielded a tractable number of 19^5 total possible sequences per simulation). The designed output low-energy sequences obtained using the Rosetta genetic algorithm ranked 1st (for free and multi-constraint design) and 4th (for single-constraint design on the bound conformation) among all designed sequences from the exhaustive search. In the latter case, the score was within 0.5 Rosetta units (approximating kcal/mol) of that of the global minimum design, and the sequences only differed in one position (WWHMF and WWHMW). Thus, we expect the results obtained using the genetic algorithm to be close to the global minimum (although these results do not exclude the possibility that search problems are more severe for longer designed sequences).
Generation of sequence profiles
Lower Rosetta scores correspond to predicted increase in stability; therefore, for each simulation the most stable designed sequences had lower negative values than the native sequence (all cases showed values smaller than zero). Profiles were created by including all designed sequences that scored lower than a “delta” value from the lowest score obtained in the simulation. The value of delta was dependent on the extent of the optimization and defined as follow: (native score – lowest score) *0.25 and denoted “lowest scoring 25%”. This criterion was used consistently for all antibodies and simulations (single- and multi-constraint). Varying the delta value resulted in qualitatively similar profiles.
Binomial test
We determined the statistical significance of the observed differences in native sequence recovery between multi- and single-constraint methods using the Binomial test49. We considered that each position had two options: recover or not recover the native amino acid residue, and, as an approximation, that native sequence recovery in one position was independent of the outcome at any other position. The binomial probability p0 of recovery was estimated by averaging the recovery observed in the three analyzed cases (multi-constraint and single-constraint for bound and free forms). The null hypothesis assumes that, regardless of the design protocol, the percentage of native sequence recovery is the same. Then, we evaluated if our cases satisfy the following inequality to determine if a “large sample test” could be performed:
where n is the number of designed positions and p0 the probability of recovery of the native amino acid residue type. This inequality was satisfied both by the germline and mature antibody sets. Thus, we tested H0:p = p0 versus H1 :p > p0 ; where p is the probability of recovery in the multi-constraint simulations, by evaluating:
Note that×is the number of positions recovered in the multi-constraint protocol. Once z was known, we calculated the P-value and determined whether H0 should be rejected or not. In all cases, P-values lower than 0.05 were considered significant.
RESULTS
Rationale and computational strategy
The structural flexibility of the germline antigen-binding site, in particular of the CDR H3 loop, led us to the following hypotheses: if conformational diversity is an intrinsic property of germline CDR H3 loops, then their native sequences may be compromises between the sequence preferences for several conformations. It may then follow that, when flexibility is reduced during antibody affinity maturation, the sequences of mature antibodies should instead be closer to optimal for single conformations.
To assess whether CDR H3 loop sequences are optimal for any of the alternative conformations they adopt or, on the contrary, are compromises between the sequences preferred by each of the experimentally observed conformations, we used a computational design method as implemented in the Rosetta design algorithm and all-atom scoring function24,35,40,41. We applied a Rosetta-based multi-constraint protein design methodology31,39 to a dataset of 28 structures of 14 pairs of germline and mature antibodies with CDR H3 loops that adopt two alternative conformations in the bound and free forms (Table I). Single-constraint optimization minimized the folding score for a single conformation, while multi-constraint design aimed at searching for low energy CDR H3 loop amino acid sequences that are simultaneously consistent with both input antibody structures (minimizing the sum of the calculated folding scores over both bound and free conformations). Sequences optimized in this way for stability compatible with both bound and free conformations (multi-constraint design) are then compared to the designed sequences optimized for each conformation separately (single-constraint design), as well as to the “native” (or wild type) sequence. In this manner, we sought to determine the degree of predicted optimality of each native sequence with respect to its two known alternative conformations (Fig. 1) (it should be noted that our analysis does not necessarily rely on the assumption that conformations similar to the bound structure are populated to a significant extent in the unbound state, see Discussion). In addition, we reasoned that this analysis should reveal candidate positions for modulating flexibility which, when altered by mutagenesis, could result in less flexible antibodies with higher binding affinities.
Figure 1.

Computational strategy for estimating the degree of optimization of the native sequence for conformational flexibility. The CDR H3 loop of germline antibodies is expected to show higher native sequence recovery in multi-constraint simulations (case 2) than in each of the single-constraint simulations (case 1).
The native CDR H3 loop sequences of germline antibodies are optimized to adopt alternative conformations
For clarity, we will first present a simple example of the computational strategy, shown in Table II, for the CDR H3 loop of the germline 7g12 antibody17 depicted in Fig. 2. Table II lists the native sequence, the predicted lowest scoring sequence obtained from the multi-constraint design simulation, as well as the lowest scoring sequences obtained for each of the single-constraint design simulations for the free and bound conformations (while the sequence optimization methods used here are stochastic and thus do not guarantee obtaining the global minimum23, we tested for convergence in the simulations, see Methods). In this simple example, for three out of the five designed positions the native amino acid residue was recovered by the multi-constraint design simulation. In contrast, native amino acid residues were recovered at none or two native positions when the single-constraint design strategy was applied to the bound and free structures, respectively (the antigen is omitted in all design simulations). The designed sequence for the single constraint bound conformation of antibody 7g12 shown in Table II had a substantially hydrophobic character. To assess our design prediction with an alternative method, we used the ERIS server for stability estimation50. ERIS predicts the WWHMF sequence to be about 2.8 kcal/mol more stable than the wild-type sequence, consistent with our results. Similar hydrophobic sequence stretches can also be present in H3 loops of naturally occurring antibodies51 (see Supplementary Materials).
Table II.
An example of the computational strategy
| Protocol (Design Targets) | Sequence Position | Native sequence recovery |
||||
|---|---|---|---|---|---|---|
| 99 | 100 | 101 | 102 | 103 | ||
| Native sequence | 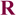 |
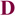 |
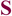 |
|
 |
|
| Multi-constraint design (Bound & Free structures) | |
F | A | |
|
3/5 |
| Single-constraint design (Bound structure) | W | W | H | M | F | 0/5 |
| Single-constraint design (Free structure) | |
F | A | H | |
2/5 |
The lowest scoring sequences selected in single- and multi-constraint simulations using the bound and free conformations of the germline antibody 7g12 and its native sequence. The CDR H3 loop positions shown were subjected to design.
Figure 2.

Superimposition of the VH domain of the germline 7g12 antibody. Bound (pdb: 1n7m) and free (pdb: 1ngz) forms are shown in green and magenta, respectively.
The results of the single- and multi-constraint analysis, applied to the 28 antibody structures in our dataset (Table I), are shown in Fig. 3. In general, considering the two observed alternative structures for each antibody simultaneously during the design simulation leads to modeled sequences that more closely resemble the native antibody sequences. This observation is substantially more pronounced in germline than in mature antibodies. We use the term “native sequence recovery” to measure the fraction of all design positions at which the native amino acid residue was present in the lowest scoring designed sequence. Germline antibodies have a lower native sequence recovery than mature antibodies when the designs were performed using any of the single structures as inputs (free or bound conformations), but a larger recovery than mature antibodies when both conformations were used as inputs simultaneously. In order to assess the statistical significance of these observations, we performed a Binomial test (Table SII)49. The null hypothesis assumes that the binomial probability of recovering the native amino acid residue for a given position is identical for any of the three procedures applied (multi-constraint, single-constraint for the bound structure, and single-constraint for the free structure) and, as an additional approximation, independent of the output in other positions. In this way, we calculated the probabilities of native sequence recovery in the multi-constraint simulation to be H0 (null hypothesis): p = 0.436; H1 (test hypothesis): p > 0.436 for germline antibodies and H0: p = 0.485; H1: p > 0.485 for mature antibodies. The resulting P-values were 0.01 and 0.11 for germline and mature antibodies, respectively. These results thus indicate that the multi-constraint design protocol leads to a significantly larger native sequence recovery with respect to the single-constraint design strategy for germline antibodies, but not for mature antibodies. We conclude that the native CDR H3 loop sequences of germline antibodies are compromises between the sequence preferences of at least each of the individual bound and free conformational states analyzed. We observed similar trends when, instead of considering only the sequence with the lowest score (the designed sequence with predicted highest stability, according to the Rosetta scoring function), we examined the top three or five unique sequences with the lowest scores (data not shown). This indicates that our observations are independent of the precise number of lowest score designed sequences analyzed.
Figure 3.

Average native sequence recovery for CDR H3 loops in germline and mature antibodies. The following design simulations were performed: single-constraint design for the bound conformation (white bar), the free conformation (grey bar) and multi-constraint design for both conformations (black bar) for germline and mature antibodies crystallized in different bound and free conformations (Table I). The star indicates that there is a statistically significant difference (as determined by a Binomial test) between the native sequence recovery obtained from multi- and single-constraint design simulations for germline antibodies.
The native sequence recovery for each individual antibody in our dataset is shown in Fig. S1. The higher native sequence recovery obtained by the multi-constraint design strategy applies to all germline antibodies, even though the relative recovery for different antibodies spans a range. Conversely, for mature antibodies the sequence recovery patterns are case-dependent, with some showing better native sequence recovery in multi-state simulations, some in single-constraint simulations for the bound conformation, and some for the unbound conformation (see Figures S1, S2).
The extent of sequence optimization of the CDR H3 loop is related to the degree of exposure to the antigen (antibody maturation)
The higher degree of sequence optimization of the individual CDR H3 loop conformations in mature antibodies is also reflected in the larger recovery observed for mature compared to germline antibodies when the designs were performed using any of the individual structures as input (Fig. 3). This observation prompted us to compare the extent of native sequence recovery in CDR H3 loop positions for a set consisting of pairs of corresponding antibodies that differ only in their degree of exposure to the same antigen epitope. Therefore, to minimize structural changes that result just from the absence or presence of different binding partners, we applied the single-constraint design strategy to the 14 pairs of corresponding germline and mature antibody structures shown in Table III that were crystallized in the same form (either both in the free form or both bound to the same antigen epitope; see Methods). Using this dataset, we find that antibody maturation correlates with an increase in the percentage of overall native sequence recovery from 35.5% (for germline antibodies or antibodies isolated after a “short period” of exposure to the antigen) to 53.6% for more mature forms (Fig. 4). The larger native sequence recovery observed for the more mature antibodies is not a consequence of systematically higher crystallographic resolution of mature antibody structures (see Methods) or presence of the antigen in the simulations, which is omitted in all design runs. We assessed the statistical significance of the difference in sequence recovery with a Binomial test, with the null hypothesis assuming that there is no difference in the native sequence recovery between the germline (or “short exposure” to antigen) antibodies and more mature forms. The resulting P-value was 0.03 (see Table SIII for details), suggesting that longer exposures to the antigen select sequences with a higher degree of optimization for the corresponding single conformation, likely at the expense of sequences that are intrinsically flexible. The relative recovery within the germline and mature antibody groups spans a range (Figure S3).
Table III.
Dataset of antibodies differing in their degree of maturation after exposure to the same antigen.
| Antibody | Antigen | Germline | Mature | ||
|---|---|---|---|---|---|
| Bound-free | Cα RMSD (Å) | Bound-free | Cα RMSD (Å) | ||
| 7g12 | MMP | 1n7m-1ngz | 1.0 | 1ngw-1ngy | 0.2 |
| 28b4 | AAH | 1fl6-1fl5 | 1.8 | 1kel-1kem | 0.2 |
| az-28 | HOP | 1d6v-1d5i | 1.2 | 1axs-1d5b | 0.7 |
| s25-2/s45-18 | KDO-trisaccharide1 | 1q9q-1q9k | 2.2 | 1q9w-1q9o | 0.3 |
| 48g7 | NPE | 1aj7-2rcs | 0.8 | 1gaf-1hkl | 0.1 |
| 36–65 | dodecapeptide | 2a6i-2a6j | 0.6 | N.A.-1jfq | N.A. |
| d44.1/f10.6.62 | Lysozyme | 1mlc-1mlb | 0.3 | 1p2c-2q76 | 0.1 |
| hyhel-26/hyhel-82 | Lysozyme | 1ndm-N.A. | N.A. | 1ndg-N.A. | N.A. |
The ligand in the mature form contains the KDO-trisaccharide epitope as part of a pentasaccharide
Antibodies isolated after different extents of exposure to the antigen (1mlc,1mlb and 1ndm have a shorter exposure than 1p2c, 2q76 and 1ndg, respectively)
N.A. not applicable
Abbreviations used: MMP, N-methylmesoporphyrin; AAH, 1-[n-4'-nitrobenzylcarboxybutylamino] methylphosphonic acid; HOP, (1s,2s,5s)2-(4-glutaridylbenzyl)-5-phenyl-1-cyclohexanol; NPE, 5-(para-nitrophenyl phosphonate)-pentanoic acid
Figure 4.

Average native sequence recovery for CDR H3 loops in germline antibodies and their corresponding mature forms. The single-constrain design strategy was used in all cases. Note that for this analysis we grouped as germline the antibody state with no or short-term exposure to antigen (which was omitted in all simulations) and as mature the corresponding antibody after longer term exposure to the same antigen. In each case, a corresponding pair consists of structures that are both either bound to the same epitope or free (Table III). The star indicates that the observed difference is statistically significant (as determined by a Binomial test).
Available biochemical data for the unbound state of the mature antibody d44.1 and its more mature form, named f.10.6.6, indicate that f.10.6.6 is more stable, both in circular dichroism and fluorescence studies52. Longer antigen exposure of this antibody resulted in two mutations in the CDR H3 loop: Asn102 to Phe and Gly104 to Val. Interestingly, the lowest score sequence predicted in the single-constraint simulation by our design algorithm for the free d44.1 antibody indicates that positions 102 and 104 could be further stabilized, as these positions did not recover the native amino acids; instead, the simulations predicted non-native amino acid residues as optimal (Table SIV). In particular, for position 102, the design algorithm predicted a Phe residue to improve atomic packing between the CDR H3 loop and the rest of the Fv domain. The atomic packing is similar to the structure of the more mature f.10.6.6 antibody that, in fact, acquired a Phe at this position (see Fig. 5). Moreover, reduced antibody flexibility upon antigen exposure is also consistent with available structural data that indicate that somatic mutations often lead to a decrease in antibody conformational entropy by pre-organizing the antigen binding site13,53,54. Structural comparisons for free and bound forms of antibodies crystallized in different maturation stages indicate that, for a given antibody, the conformational differences between the bound and free states of the CDR H3 loops (as measured by Cα RMSD) are larger in germline than in mature forms (Table III). For position 104 in antibody d44.1, our method predicts Lys, even though a Gly is the native residue and Val is found in the more mature form. This may be explained by the sampling protocol we applied here: Structural inspection suggests that the mutation of position 55 outside the H3 loop in the light chain from Ser in antibody d44.1 to Met in antibody f.10.6.6 would have a steric overlap with a Lys at position 104. In our simulations, the design is restricted to residues within the H3 loop and therefore does not consider the effects that mutations in positions outside the H3 loop could have.
Figure 5.

Substitution of Asn by Phe in position 102 improves atomic packing in the d44.1 antibody. The CDR H3 residue 102 and its neighbors (all heavy atoms within 5Å of the residue 102 side chain) are shown in space-fill representation. Residues 46, 49 and 50 belong to the light chain and residues 100,102 and 104 belong to the heavy chain CDR H3 loop. Panels A, B, C correspond to the native d44.1 antibody (pdb: 1mlb), a model of the designed d44.1 antibody and native f10.6.6 antibody (pdb: 2q76); (note that the NZ atom of Lys 49 is not seen in panel A because is located further than 5Å from the Asn 102 side chain).
Identification of CDR H3 loop positions modulating flexibility
Our analysis of native sequence recovery indicates that germline antibodies are optimized for conformational flexibility, which in turn suggests that mutations could stabilize the CDR H3 loop in a particular conformation. Thus, we next sought to identify CDR H3 loop positions important for flexibility that, if mutated, could lead to the stabilization of the CDR H3 loop in one particular conformation. Towards this goal, we generated sequence profiles to determine the preference at a certain designed position for a given amino acid residue when each alternative CDR H3 loop structure is considered alone, or when both are considered simultaneously. Specifically, instead of retrieving only the sequence with the lowest Rosetta score from each simulation, we generated sequence profiles (for the multi- and each single-constraint design protocol) by retrieving the lowest (best) scoring 25% of all sequences that scored better than the native sequence (see Methods). As a simple approximation, we assume independence of all designed positions. To facilitate analysis, “amino acid residue classes” for each of the designed positions were defined according to their chemical properties and size55, as follows: Aliphatic =[V,L,I], Aromatic =[F,W,H,Y], Met=[M], Small =[S,T,A,G], Polar =[N,Q], Basic =[K,R], Acidic =[D,E], and Pro=[P].
Analysis of the designed sequence profiles enabled us to define two types of amino acid positions in the CRD H3 loops. First, “constrained” positions, where the amino acid residue predicted to be optimal in multi-constraint design is also optimal in single constraint design (i.e. positions at which a residue class is favored in one or both single conformations) (see Table SV). Second, “compromised” are those positions where residues are only predicted to be native (or native-like) in multi-constraint design. In other words, these are positions at which both alternative conformations would prefer another amino acid residue class, but a compromise is chosen to accommodate both conformations simultaneously. In particular, we are interested in cases in which further sequence optimization for the bound conformation can be predicted. This may be the case for compromised or constrained positions in which the multi- and single-constraint simulations for the free form share similar profiles, but differ from the profile obtained for the single-constraint protocol applied to the bound form.
Analysis of the sequence profiles for the 52 positions in the CDR H3 loops of the germline antibodies in our dataset indicated that 30 positions are constrained by both alternative conformations (they share similar amino acid residue class preferences), and in 25 of the 30 cases the representative amino acid residues were native or native-like (as defined by the similarity classes listed above). In addition, we identified eight positions constrained by a single conformation. Five of these positions are predicted to be candidates for stabilization of the bound conformation upon mutation (here the free conformation and the multi-constraint optimized sequences share the same preference), while the remaining three are candidates for stabilization of the free conformation. In addition, we found one “compromised” (Table SV) position predicted to be a good target for stabilization of either the bound or free conformation. Most of the remaining thirteen positions (out of the 52 positions in our dataset) appear plastic for at least one of the conformations (they did not show particular amino acid residue class preferences). Thus, in total our sequence profile analysis identified nine positions (~20% of all designed positions) predicted to be relevant for modulation of CDR H3 loop flexibility.
Particularly interesting are the six cases for which we predict that further optimization of the bound conformation might be possible (Table IV). Heavy chain position 101 in antibody 7g12 is known to mutate during affinity maturation19. Consistent with this observation, our analysis predicts that mutation of position 101 in 7g12 could lead to the stabilization of the CDR H3 loop in the bound conformation (discussed in more detail below). Four of the six positions predicted to stabilize the bound conformation, if mutated, have Ser as the native amino acid residue. Previous studies have shown that Ser frequently mutates during the affinity maturation process56. For all six positions shown in Table IV, the sequence profiles obtained for the multi-constraint design and the single-constraint designs for the free form are similar and include mostly small residues. In contrast, the sequence profile obtained for the bound form is enriched in large hydrophobic amino acid residues. Four out of the six positions predicted for stabilization of the bound form are located at least at 5.5 Å away47 from the crystallized ligand (defined by the closest distance between two heavy atoms on the protein and ligand, respectively47) (Table IV). This suggests that at least some mutations in positions located in the CDR H3 loop may be amenable to a design approach aiming to stabilize the desired conformation without directly affecting ligand contacts. The remaining two predicted positions are at least 3.7Å away from the ligand, making it difficult to predict the effect of the mutation on ligand binding. However, for one of them (position 101 in 7G12), in vivo affinity maturation data available17 validate our prediction (see next paragraph).
Table IV.
Predicted positions for stabilization of the bound conformation in germline antibodies
| Position class | constrained | compromised | ||||
|---|---|---|---|---|---|---|
| Antibody | 7g12 | 7g12 | 28b4 | 36–65 | s25-2 | s25-2 |
| CDR H3 loop position | 99 | 101 | 97 | 105 | 100D | 100B |
| Native amino acid |  |
 |
|
|
|
|
| Ligand distance (Å) | 3.7 | 3.7 | 5.5 | - | - | - |
| Ligand-contacting surface (Å2)1 | 106 | 23 | 2 | - | - | - |
| Bound&Free (multi-constraint)2 | |
A,
|
T,
|
A,,D,E |
A,G | |
| Bound (single-constraint)2 | W,F,Y | H,F,M | H,W,F,Y,K,Q,E | H,R,K,E,M | H | H |
| Free (single-constraint)2 |
,E |
A,
|
T,
|
A,,T,G,N,D,E,V |
A,G | G |
| Bound Score (lowest-native) 2,3 | −3.3 | −1.4 | −1.3 | −0.9 | −0.8 | −0.6 |
Contact surface area between the ligand and the antibody residue calculated using the LPC software46
Sequence profiles obtained considering the lowest scoring 25% of the designed sequences
Predicted delta score (designed sequence with best score – native) upon mutation (Rosetta score, in approximate units of kcal/mol)
Germline 7g12 antibody: a case in which somatic mutations increase hapten affinity by stabilizing the CDR H3 loop in the bound conformation
The germline 7g12 antibody is the best structurally characterized example of somatic mutations leading to an increase in hapten binding affinity through the stabilization of the CDR H3 loop in the antigen bound conformation17,19. Table V shows the predicted sequence profiles obtained for the CDR H3 loop of germline 7g12. In this case, the multi-constraint design profiles recover native amino acid residues at four out of the five CDR H3 loop native positions, whereas the single-constraint simulations for the bound or free forms recover native amino acid residues only at two and three positions, respectively. This is the case even though the number of predicted different sequences in the profile for the bound form (168) is substantially larger than those of the free and multi-constraint profiles (3 and 2, respectively). From the 7g12 profile, we identified two positions, 99 and 101, in which multi-constraint as well as single-constraint design (for the free form) share similar characteristics: charged residues are selected for position 99 (Arg, Glu) and small amino acid residues for position 101 (Ala, Ser), respectively, recovering the native residues Arg and Ser. In contrast, the amino acid residues present in the simulated profile for the single-constraint bound form are large aromatics at both positions. Moreover, the differences in score between the predicted best sequence and the native sequence for positions 99 and 101 in the bound form are substantial, suggesting that both positions could be further optimized. This example illustrates how positions that are “constrained” by the free structure could be good candidates for mutations to stabilize the CDR H3 loop in the conformation of the bound form, likely increasing antibody-antigen affinity. Interestingly, we found that CDR H3 loops from antibodies (extracted from the ArchDB database57), that adopt conformations similar to that of the germline 7g12 bound form, have a Trp at position 99 (Fig. S4). This is consistent with our prediction that large aromatic residues (including Trp, see Table V) may stabilize the bound conformation. Another example is position 101, which undergoes a somatic mutation during affinity maturation. Here, the single-constraint profile for the structure of the hapten-bound form contains exclusively Phe, His and Met. Therefore, our algorithm predicts that substitution of the native Ser in position 101 by a large hydrophobic residue should stabilize the CDR H3 loop in the desired hapten-binding conformation. Notably, Met has been selected in that position by in vivo affinity maturation19, consistent with our predictions.
Table V.
Sequence profiles obtained in single- and multi-constraint simulations for the germline antibody 7g12.
| Protocol (Design targets) |
Sequence Position | Native sequence recovery |
No. of unique sequences |
||||
|---|---|---|---|---|---|---|---|
| 99 | 100 | 101 | 102 | 103 | |||
| Native Sequence | |
|
|
|
|
||
| Multi-constraint (Bound & Free structures) | |
F | A,
|
|
|
4/5 | 2 |
| Single-constraint (Bound structure) | W,F,Y | W,F,Y,H | F,M,H | M,V,T,R,K,Q,,E |
F,,W,R,H |
2/5 | 168 |
| Single-constraint (Free structure) |
,E |
F | A,
|
H | |
3/5 | 3 |
Residues in the CDR H3 loop region were subjected to design.
The profile is composed of the lowest scoring 25% of the designed sequences that scored better than the native sequence.
DISCUSSION
Numerous studies have shown that germline antibodies display conformational flexibility, in particular in their CDR H3 loop regions12–18,58. Furthermore, flexible antibodies have been shown to bind multiple antigens in vitro, often through alternative conformations19,21. Thus, germline antibody conformational flexibility has been proposed to be beneficial, as it may enlarge the conformational repertoire available to the immune system7,8,11. In this work, we investigated whether we can identify sequence signatures in native germline antibodies responsible for CDR H3 loop flexibility. Towards this goal, we used computational protein design to determine the extent to which native CDR H3 loop sequences are optimized for their structures, in germline and mature antibodies whose free and bound forms show different CDR H3 loop conformations. Computational protein design has previously shown that, for most proteins, the low energy sequences for a given structure obtained from computational re-design are close to the native protein sequences32–38,59. Our hypothesis was that, if the native sequences of germline antibodies are optimized for flexibility, then Rosetta-based multi-constraint design using multiple conformations as inputs should lead to a high recovery of the germline native sequences. Indeed, we observed that for germline antibodies the CDR H3 loop native sequence recovery is significantly higher when both conformations are considered simultaneously in the design simulation, than when each of the single conformations is used separately. In contrast, using the same design test we found no significant differences in native sequence recovery for CDR H3 loops of mature antibodies able to adopt at least two alternative conformations. Our results indicate that the CDR H3 loop native sequences of germline antibodies represent compromises between the sequence preferences of each of the individual conformational states analyzed. Our findings suggest that germline CDR H3 loop sequences might be selected for flexibility.
Proteins sample an ensemble of conformations, even in their “native” states60,61. Hence, using just two observed conformations, as in our simulations, is a substantial simplification that is likely to underestimate the true flexibility. However, as this flexibility is not directly accessible experimentally, we are limited to an analysis of the experimentally characterized conformational states, which nevertheless yields considerable agreement between designed multi-constraint and native sequences. That our method only identifies few positions predicted to be involved in controlling flexibility may be explained by the fact that other residues may be required for alternative conformations or sparsely-populated higher-energy conformations that need to be sampled in transitions from one conformation to another, not modeled here, as well as inaccuracies in design methods. By our method, some mature antibodies also seem to show some evidence for a preference for flexibility (although statistically not significant, P= 0.11). Again, as our analysis is restricted to two experimentally observed conformations, it does not test the possibility that mature antibodies sample a more restricted ensemble of solution conformations than germline antibodies. A related point concerns the question of whether altered protein conformations observed in different bound or functional states62–65 are already populated in the unbound state. Our analysis describes which low energy sequences are consistent with a given conformation, but, as discussed above, does not evaluate conformational transitions or populations of conformations in structural ensembles. In other words, we predict low energy sequences given a target structure, but do not determine the inverse, the specificity or population of a structure given its sequence. Therefore and in turn, our analysis does not require the assumption that the bound structures are populated to a significant extent in the unbound state.
Although the Rosetta full-atom energy function has been parameterized to recover native sequences given the native backbone structure, the validity of the conclusions drawn here is supported by several lines of evidence: First, the parameterization uses a large dataset that should average out native bias for individual structures. This conclusion is consistent with the finding that native sequence recovery on an independent test set is essentially the same as on the training set24. In addition, native sequence recovery is also considerable when side chains surrounding a designed position are redesigned simultaneously24. Second, the Rosetta full-atom energy function has also been used to more directly assess the specificity of a structure given the sequence. In applications in both ab initio structure prediction66 and model refinement67, the same full-atom energy function originally parameterized for sequence design has been able to successfully guide the sampling and identification of near-native protein structures. An extension of our current study would be to carry out refinement simulations starting with native and designed sequences to more directly test the specificity of the designed sequence for the target (native) structure. Third, our study provides an “internal control” showing significant differences in sequence recovery for germline and mature antibodies. Taken together, we believe these findings support the applicability of the RosettaDesign energy function to the question addressed here.
Even though conformational flexibility could enlarge the conformational repertoire available to respond to foreign antigens, the intrinsic flexibility of germline antibodies has to be selected before the antibodies actually encounter foreign antigens for the first time. If so, what are the selective pressures leading to native sequences of germline binding sites, in particular CDR H3 loops, with intrinsic flexibility? During B cell development, clones expressing antibodies that are either too reactive or not reactive at all against self-antigens are negatively selected68,69. This eliminates, on one hand, B cell clones that could potentially lead to autoimmune responses, and, on the other hand, clones leading to defective B-cell receptors. Indeed, evidence indicates that clones capable of low avidity interactions with self-ligands have the highest likelihood of maturation and survival68. Thus, flexibility of germline antibodies serves two purposes: by sampling alternative conformations, germline antibodies have higher chances to find binding partners; at the same time, by being intrinsically flexible, they are less likely to bind any partner with too high an affinity due to the entropic costs of ordering flexible regions upon binding. In this way, an optimal intermediate affinity range can be achieved allowing survival of the B cell clone. A consequence of that flexibility is then the ability to bind, again with a limited number of possible antibody sequences, a larger number of antigens, even though this is not the property that had been selected for originally.
Generation of sequence profiles for each of the multi- and single-constraint simulations lead us to propose amino acid mutations along the CDR H3 loops that could increase the rigidity of the CDR H3 loop bound conformation, reducing overall conformational flexibility. For most of the proposed cases, the replacements suggested are unlikely to interfere with ligand binding (see Results). Affinity maturation data available for the germline antibody 7g1217 are consistent with our predictions. Our strategy can serve to engineer antibodies with higher affinity and specificity by designing mutations that preferentially stabilize a desired conformation. Thus, identifying sequence determinants of conformational flexibility based on a comparison of single- and multi-constraint design simulations computationally mimics the reduction in flexibility often resulting from affinity maturation. Similar mechanisms reducing H3 loop flexibility may explain the effect of other known mutations that, despite being located away from the antibody-antigen interaction interface, cause affinity maturation and cannot easily be rationalized using fixed backbone methods70,71. Furthermore, as the sequence diversity sampled by computational methods is not restricted by the genetic mechanisms that generate antibody diversity72, it is possible to explore areas of sequence space that are otherwise not accessible to the natural antibody repertoire.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Vladimir Potapov for sharing his structural superimposition algorithm, Francisco Quintana, Dan Tawfik, Marvin Edelman, Vladimir Sobolev, Elisabeth Humphris, Greg Kapp and Javier Ángel Velázquez-Muriel for helpful comments and critical reading of the manuscript, Richard Oberdorf and Elisabeth Humphris for helping with statistical tests and data analysis, and members of the Kortemme lab for stimulating discussions. This work was supported by the NIH Roadmap (PN2EY016525) and an NSF CAREER award to T.K. (MCB 0744541).
Abbreviations
- VL, VH
variable domains of light chain and heavy chain, respectively
- Fv
variable domains of immunoglobulin
REFERENCES
- 1.Poljak RJ, Amzel LM, Avey HP, Chen BL, Phizackerley RP, Saul F. Three-dimensional structure of the Fab' fragment of a human immunoglobulin at 2,8-A resolution. Proc Natl Acad Sci U S A. 1973;70(12):3305–3310. doi: 10.1073/pnas.70.12.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196(4):901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 3.Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321(6069):522–525. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- 4.Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, Sheriff S, Padlan EA, Davies D, Tulip WR, et al. Conformations of immunoglobulin hypervariable regions. Nature. 1989;342(6252):877–883. doi: 10.1038/342877a0. [DOI] [PubMed] [Google Scholar]
- 5.Wu TT, Johnson G, Kabat EA. Length distribution of CDRH3 in antibodies. Proteins. 1993;16(1):1–7. doi: 10.1002/prot.340160102. [DOI] [PubMed] [Google Scholar]
- 6.Vargas-Madrazo E, Lara-Ochoa F, Almagro JC. Canonical structure repertoire of the antigen-binding site of immunoglobulins suggests strong geometrical restrictions associated to the mechanism of immune recognition. J Mol Biol. 1995;254(3):497–504. doi: 10.1006/jmbi.1995.0633. [DOI] [PubMed] [Google Scholar]
- 7.Pauling L. A theory of the structure and process of formation of antibodies. J Am Chem Soc. 1940;62:2643–2657. [Google Scholar]
- 8.Foote J, Milstein C. Conformational isomerism and the diversity of antibodies. Proc Natl Acad Sci U S A. 1994;91(22):10370–10374. doi: 10.1073/pnas.91.22.10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joyce GF. Evolutionary chemistry: getting there from here. Science. 1997;276(5319):1658–1659. doi: 10.1126/science.276.5319.1658. [DOI] [PubMed] [Google Scholar]
- 10.James LC, Tawfik DS. Conformational diversity and protein evolution--a 60-year-old hypothesis revisited. Trends Biochem Sci. 2003;28(7):361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 11.Mariuzza RA. Multiple paths to multispecificity. Immunity. 2006;24(4):359–361. doi: 10.1016/j.immuni.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 12.Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Structural insights into the evolution of an antibody combining site. Science. 1997;276(5319):1665–1669. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]
- 13.Manivel V, Sahoo NC, Salunke DM, Rao KV. Maturation of an antibody response is governed by modulations in flexibility of the antigen-combining site. Immunity. 2000;13(5):611–620. doi: 10.1016/s1074-7613(00)00061-3. [DOI] [PubMed] [Google Scholar]
- 14.Mundorff EC, Hanson MA, Varvak A, Ulrich H, Schultz PG, Stevens RC. Conformational effects in biological catalysis: an antibody-catalyzed oxy-cope rearrangement. Biochemistry. 2000;39(4):627–632. doi: 10.1021/bi9924314. [DOI] [PubMed] [Google Scholar]
- 15.Yin J, Mundorff EC, Yang PL, Wendt KU, Hanway D, Stevens RC, Schultz PG. A comparative analysis of the immunological evolution of antibody 28B4. Biochemistry. 2001;40(36):10764–10773. doi: 10.1021/bi010536c. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen HP, Seto NO, MacKenzie CR, Brade L, Kosma P, Brade H, Evans SV. Germline antibody recognition of distinct carbohydrate epitopes. Nat Struct Biol. 2003;10(12):1019–1025. doi: 10.1038/nsb1014. [DOI] [PubMed] [Google Scholar]
- 17.Yin J, Andryski SE, Beuscher AEt, Stevens RC, Schultz PG. Structural evidence for substrate strain in antibody catalysis. Proc Natl Acad Sci U S A. 2003;100(3):856–861. doi: 10.1073/pnas.0235873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sethi DK, Agarwal A, Manivel V, Rao KV, Salunke DM. Differential epitope positioning within the germline antibody paratope enhances promiscuity in the primary immune response. Immunity. 2006;24(4):429–438. doi: 10.1016/j.immuni.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Yin J, Beuscher AEt, Andryski SE, Stevens RC, Schultz PG. Structural plasticity and the evolution of antibody affinity and specificity. J Mol Biol. 2003;330(4):651–656. doi: 10.1016/s0022-2836(03)00631-4. [DOI] [PubMed] [Google Scholar]
- 20.Zimmermann J, Oakman EL, Thorpe IF, Shi X, Abbyad P, Brooks III CL, Boxer SG, Romesberg FE. Antibody evolution constrains conformational heterogeneity by tailoring protein dynamics. Proc Natl Acad Sci U S A. 2006;103(37):13722–13727. doi: 10.1073/pnas.0603282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299(5611):1362–1367. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 22.Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278(5335):82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 23.Pokala N, Handel TM. Review: protein design--where we were, where we are, where we're going. J Struct Biol. 2001;134(2–3):269–281. doi: 10.1006/jsbi.2001.4349. [DOI] [PubMed] [Google Scholar]
- 24.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302(5649):1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 25.Looger LL, Dwyer MA, Smith JJ, Hellinga HW. Computational design of receptor and sensor proteins with novel functions. Nature. 2003;423(6936):185–190. doi: 10.1038/nature01556. [DOI] [PubMed] [Google Scholar]
- 26.Kortemme T, Baker D. Computational design of protein-protein interactions. Curr Opin Chem Biol. 2004;8(1):91–97. doi: 10.1016/j.cbpa.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25(10):1171–1176. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Havranek JJ, Harbury PB. Automated design of specificity in molecular recognition. Nat Struct Biol. 2003;10(1):45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 29.Shimaoka M, Shifman JM, Jing H, Takagi J, Mayo SL, Springer TA. Computational design of an integrin I domain stabilized in the open high affinity conformation. Nat Struct Biol. 2000;7(8):674–678. doi: 10.1038/77978. [DOI] [PubMed] [Google Scholar]
- 30.Summa CM, Rosenblatt MM, Hong JK, Lear JD, DeGrado WF. Computational de novo design, and characterization of an A(2)B(2) diiron protein. J Mol Biol. 2002;321(5):923–938. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 31.Ambroggio XI, Kuhlman B. Computational design of a single amino acid sequence that can switch between two distinct protein folds. J Am Chem Soc. 2006;128(4):1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 32.Shakhnovich EI, Gutin AM. A new approach to the design of stable proteins. Protein Eng. 1993;6(8):793–800. doi: 10.1093/protein/6.8.793. [DOI] [PubMed] [Google Scholar]
- 33.Shakhnovich E, Abkevich V, Ptitsyn O. Conserved residues and the mechanism of protein folding. Nature. 1996;379(6560):96–98. doi: 10.1038/379096a0. [DOI] [PubMed] [Google Scholar]
- 34.Koehl P, Levitt M. De novo protein design. II. Plasticity in sequence space. J Mol Biol. 1999;293(5):1183–1193. doi: 10.1006/jmbi.1999.3212. [DOI] [PubMed] [Google Scholar]
- 35.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000;97(19):10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dokholyan NV, Shakhnovich EI. Understanding hierarchical protein evolution from first principles. J Mol Biol. 2001;312(1):289–307. doi: 10.1006/jmbi.2001.4949. [DOI] [PubMed] [Google Scholar]
- 37.Koehl P, Levitt M. Protein topology and stability define the space of allowed sequences. Proc Natl Acad Sci U S A. 2002;99(3):1280–1285. doi: 10.1073/pnas.032405199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaramillo A, Wernisch L, Hery S, Wodak SJ. Folding free energy function selects native-like protein sequences in the core but not on the surface. Proc Natl Acad Sci U S A. 2002;99(21):13554–13559. doi: 10.1073/pnas.212068599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humphris EL, Kortemme T. Design of multi-specificity in protein interfaces. PLoS Comput Biol. 2007;3(8):e164. doi: 10.1371/journal.pcbi.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A. 2002;99(22):14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kortemme T, Morozov AV, Baker D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J Mol Biol. 2003;326(4):1239–1259. doi: 10.1016/s0022-2836(03)00021-4. [DOI] [PubMed] [Google Scholar]
- 42.Hu X, Wang H, Ke H, Kuhlman B. High-resolution design of a protein loop. Proc Natl Acad Sci U S A. 2007;104(45):17668–17673. doi: 10.1073/pnas.0707977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allcorn LC, Martin AC. SACS--self-maintaining database of antibody crystal structure information. Bioinformatics. 2002;18(1):175–181. doi: 10.1093/bioinformatics/18.1.175. [DOI] [PubMed] [Google Scholar]
- 45.Rini JM, Stanfield RL, Stura EA, Salinas PA, Profy AT, Wilson IA. Crystal structure of a human immunodeficiency virus type 1 neutralizing antibody, 50.1, in complex with its V3 loop peptide antigen. Proc Natl Acad Sci U S A. 1993;90(13):6325–6329. doi: 10.1073/pnas.90.13.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stanfield RL, Takimoto-Kamimura M, Rini JM, Profy AT, Wilson IA. Major antigen-induced domain rearrangements in an antibody. Structure. 1993;1(2):83–93. doi: 10.1016/0969-2126(93)90024-b. [DOI] [PubMed] [Google Scholar]
- 47.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15(4):327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 48.Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35(2):133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 49.Larsen RJ, Marx LM. An Introduction to mathematical statistics and its application. 2000:375–379. [Google Scholar]
- 50.Yin S, Ding F, Dokholyan NV. Eris: an automated estimator of protein stability. Nat Methods. 2007;4(6):466–467. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 51.Martin AC. Accessing the Kabat antibody sequence database by computer. Proteins. 1996;25(1):130–133. doi: 10.1002/(SICI)1097-0134(199605)25:1<130::AID-PROT11>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 52.Acierno JP, Braden BC, Klinke S, Goldbaum FA, Cauerhff A. Affinity maturation increases the stability and plasticity of the Fv domain of anti-protein antibodies. J Mol Biol. 2007;374(1):130–146. doi: 10.1016/j.jmb.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 53.Sagawa T, Oda M, Ishimura M, Furukawa K, Azuma T. Thermodynamic and kinetic aspects of antibody evolution during the immune response to hapten. Mol Immunol. 2003;39(13):801–808. doi: 10.1016/s0161-5890(02)00282-1. [DOI] [PubMed] [Google Scholar]
- 54.Thorpe IF, Brooks CL., 3rd Molecular evolution of affinity and flexibility in the immune system. Proc Natl Acad Sci U S A. 2007;104(21):8821–8826. doi: 10.1073/pnas.0610064104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor WR. The classification of amino acid conservation. J Theor Biol. 1986;119(2):205–218. doi: 10.1016/s0022-5193(86)80075-3. [DOI] [PubMed] [Google Scholar]
- 56.Clark LA, Ganesan S, Papp S, van Vlijmen HW. Trends in antibody sequence changes during the somatic hypermutation process. J Immunol. 2006;177(1):333–340. doi: 10.4049/jimmunol.177.1.333. [DOI] [PubMed] [Google Scholar]
- 57.Espadaler J, Fernandez-Fuentes N, Hermoso A, Querol E, Aviles FX, Sternberg MJ, Oliva B. ArchDB: automated protein loop classification as a tool for structural genomics. Nucleic Acids Res. 2004;32(Database issue):D185–D188. doi: 10.1093/nar/gkh002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Furukawa K, Shirai H, Azuma T, Nakamura H. A role of the third complementarity-determining region in the affinity maturation of an antibody. J Biol Chem. 2001;276(29):27622–27628. doi: 10.1074/jbc.M102714200. [DOI] [PubMed] [Google Scholar]
- 59.Xia Y, Levitt M. Simulating protein evolution in sequence and structure space. Curr Opin Struct Biol. 2004;14(2):202–207. doi: 10.1016/j.sbi.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 60.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254(5038):1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 61.Vendruscolo M, Domany E. Efficient dynamics in the space of contact maps. Fold Des. 1998;3(5):329–336. doi: 10.1016/S1359-0278(98)00045-5. [DOI] [PubMed] [Google Scholar]
- 62.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313(5793):1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 63.Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KF, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320(5882):1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 64.Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson MA, Petsko GA, Karplus M, Hubner CG, Kern D. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450(7171):838–844. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- 65.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291(5512):2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 66.Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309(5742):1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 67.Qian B, Ortiz AR, Baker D. Improvement of comparative model accuracy by free-energy optimization along principal components of natural structural variation. Proc Natl Acad Sci U S A. 2004;101(43):15346–15351. doi: 10.1073/pnas.0404703101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cancro MP, Kearney JF. B cell positive selection: road map to the primary repertoire? J Immunol. 2004;173(1):15–19. doi: 10.4049/jimmunol.173.1.15. [DOI] [PubMed] [Google Scholar]
- 69.Rashedi I, Panigrahi S, Ezzati P, Ghavami S, Los M. Autoimmunity and apoptosis--therapeutic implications. Curr Med Chem. 2007;14(29):3139–3151. doi: 10.2174/092986707782793952. [DOI] [PubMed] [Google Scholar]
- 70.Sivasubramanian A, Maynard JA, Gray JJ. Modeling the structure of mAb 14B7 bound to the anthrax protective antigen. Proteins. 2008;70(1):218–230. doi: 10.1002/prot.21595. [DOI] [PubMed] [Google Scholar]
- 71.Harvey BR, Georgiou G, Hayhurst A, Jeong KJ, Iverson BL, Rogers GK. Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc Natl Acad Sci U S A. 2004;101(25):9193–9198. doi: 10.1073/pnas.0400187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bond CJ, Wiesmann C, Marsters JC, Jr, Sidhu SS. A structure-based database of antibody variable domain diversity. J Mol Biol. 2005;348(3):699–709. doi: 10.1016/j.jmb.2005.02.063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.