

Background: The P23H opsin mutant causes the blinding human disease, retinitis pigmentosa.
Results: Molecular properties of bovine P23H mutant opsin were characterized in both in vitro and a transgenic C. elegans model.
Conclusion: Thermally unstable P23H isorhodopsin containing correct disulfide bond can be slowly regenerated in transgenic C. elegans.
Significance: This study produced novel information about the disease-causing P23H mutant opsin.
Keywords: G Protein-coupled Receptors, Retina, Retinal Degeneration, Vision, Vitamin A
Abstract
The P23H opsin mutation is the most common cause of autosomal dominant retinitis pigmentosa. Even though the pathobiology of the resulting retinal degeneration has been characterized in several animal models, its complex molecular mechanism is not well understood. Here, we expressed P23H bovine rod opsin in the nervous system of Caenorhabditis elegans. Expression was low due to enhanced protein degradation. The mutant opsin was glycosylated, but the polysaccharide size differed from that of the normal protein. Although P23H opsin aggregated in the nervous system of C. elegans, the pharmacological chaperone 9-cis-retinal stabilized it during biogenesis, producing a variant of rhodopsin called P23H isorhodopsin. In vitro, P23H isorhodopsin folded correctly, formed the appropriate disulfide bond, could be photoactivated but with reduced sensitivity, and underwent Meta II decay at a rate similar to wild type isorhodopsin. In worm neurons, P23H isorhodopsin initiated phototransduction by coupling with the endogenous Gi/o signaling cascade that induced loss of locomotion. Using pharmacological interventions affecting protein synthesis and degradation, we showed that the chromophore could be incorporated either during or after mutant protein translation. However, regeneration of P23H isorhodopsin with chromophore was significantly slower than that of wild type isorhodopsin. This effect, combined with the inherent instability of P23H rhodopsin, could lead to the structural cellular changes and photoreceptor death found in autosomal dominant retinitis pigmentosa. These results also suggest that slow regeneration of P23H rhodopsin could prevent endogenous chromophore-mediated stabilization of rhodopsin in the retina.
Introduction
Retinitis pigmentosa (RP)2 is a progressive retinal degenerative disease caused by heterogeneous genetic defects that affect more than a million people worldwide (1–4). The disease is characterized by night blindness and progressive loss of peripheral vision due to rod photoreceptor cell death. Secondary cone photoreceptor cell death then causes legal blindness at later stages of RP. According to its inheritance type, RP is classified as autosomal dominant (ad), autosomal recessive (ar), and X-linked. Among more than 44 RP causal genes, mutations in the RHO gene encoding rhodopsin, the rod-specific G protein-coupled receptor, account for 25% of adRP cases (5, 6). P23H RHO is the first and most frequently reported mutation in adRP cases (∼10%) in North America (5). Therefore, this mutation has been extensively studied in cellular and animal models to assess its pathobiology (6–14).
The molecular properties of P23H opsin were initially investigated in mammalian cells. Wild type (WT) rhodopsin can be readily regenerated with cis-chromophores, but regeneration of P23H mutant was inefficient in vitro (9, 15, 16). Chromophore incorporation and generation of P23H isorhodopsin occurred only when living cells were treated with 9-cis-retinal (9). Pharmacological chaperones such as 9-cis-retinal stabilized P23H opsin and protected cultured cells from apoptosis (8, 17). 9-cis-Retinal is more stable in tissue culture than the endogenous chromophore both in vitro and in a transgenic C. elegans model 11-cis-retinal. Thus, it comprises a good substitute for 11-cis-retinal, because 9-cis-retinal's mode of photoactivation is also similar for both rhodopsin and isorhodopsin (18, 19).
Overexpressed P23H opsin aggregated in the endoplasmic reticulum (ER) of mammalian cultured cells and activated the unfolded protein response (UPR), leading to its proteasomal and lysosomal degradation (7–9, 17). Instability of P23H opsin was also seen in P23H transgenic (TG) rats resulting in activation of UPR signaling (20). Gene delivery of the chaperone proteins preserved retinal structure and function in P23H TG rats (21, 22). Interestingly, a single low dose of the heat-shock protein 90 (Hsp90) inhibitor HSP990 enhanced visual function and delayed photoreceptor degeneration in P23H TG rats, most likely by promoting degradation of the mutant protein (23). More recently, a P23H knock-in mouse model was developed to more faithfully recapitulate the genotype and phenotype of adRP patients. No significant inner segment opsin accumulation was observed in these mice, and the P23H opsin was rapidly degraded (24).
The P23H mutant role in disease pathology is unclear. One unknown aspect is whether P23H opsin-induced UPR is the major factor triggering photoreceptor death, because P23H rhodopsin can be expressed in cell culture without cellular destabilization (8, 9). Also, homozygous P23H knock-in mice form discs at the ciliary protrusion of photoreceptors, suggesting that P23H rhodopsin can escape ER quality control and reach the rod outer segments (ROS) (25). Even though heterozygous P23H knock-in mice had less than 10% P23H rhodopsin, they still exhibited aberrant internal membrane structures in their ROS (25). This suggests that P23H rhodopsin residing in ROS could be toxic to photoreceptors. In homozygous young P23H knock-in mice with cone-signaling genetically ablated, electroretinogram recordings clearly demonstrated that P23H rhodopsin could initiate phototransduction (25).
Because of the instability of P23H opsin in existing expression systems, isolation of P23H rhodopsin was difficult (8, 9). Therefore, knowledge of P23H rhodopsin's biochemical properties is limited. Is P23H rhodopsin folded or misfolded? Is it correctly disulfide bonded? Is the mutant pigment protein functional, i.e. can it regenerate efficiently and initiate phototransduction? How does it differ from WT rhodopsin? In this study, we investigated these questions by in vitro and in vivo assays of P23H bovine isorhodopsin generated by Caenorhabditis elegans.
C. elegans is a transparent free-living nematode with a life span of 2–3 weeks (26). The adult worm contains 302 neurons that form a simple nervous system (27). TG C. elegans expressing heterologous vertebrate rhodopsin has been generated and functionally evaluated (28–30). Isorhodopsin can be purified from large cultures of TG worms for biochemical characterization. C. elegans lack vision and do not express any endogenous visual pigments. However, TG expression of bovine opsin produced a 9-cis-retinal-dependent coupling of light exposure to the loss of locomotion in worms due to activation of the endogenous Gi/o signaling cascade by photoactivated isorhodopsin (28–30). Here, we sought to further characterize the P23H mutant opsin to obtain molecular insights into the mechanism of photoreceptor degeneration driven by this adRP mutation. P23H bovine opsin was expressed in the nervous system of C. elegans, and its localization and molecular properties were characterized. Our observations suggest a novel molecular mechanism for P23H rhodopsin-induced photoreceptor death in adRP.
EXPERIMENTAL PROCEDURES
C. elegans Strains and Maintenance
The Bristol N2 strain of C. elegans used for this study was maintained on nematode growth medium (NGM) (0.25% peptone, 51 mm NaCl, 25 mm K3PO4, 5 μg/ml cholesterol, 1 mm CaCl2, and 1 mm MgCl2) plates seeded with OP50 at room temperature. Loss-of-function mutants of Gi/o and Gq, goa-1(sa734) and egl-30(md186), respectively, were obtained from the Caenorhabditis Genetics Center (University of Minnesota, Minneapolis). Primers used to identify homozygous mutants and mutated segments were goa-1(sa734), GCTGCACCACATACAGTGAGTGA (forward) and ACGAAATATTCGGACG TTCTATGG (reverse) with an early stop mutation in codon 52. Standard methods were employed for C. elegans cryostorage and recovery (31).
WT and P23H Bovine Rhodopsin Constructs and Generation of TG Worm Lines
For the pan-neuronal bovine opsin ((b)opsin) expression construct (Genscript, Piscataway, NJ), codon-optimized (32) (b)opsin cDNA (sequence reference for (b)opsin, NP_001014890) was driven by the H20 promoter in a pBluscript KS(+) vector (28, 30, 33). The pan-neuronal bovine opsin P23H expression construct itself was generated from WT (b)opsin vector by site-directed mutagenesis. TG worm lines were created by injecting WT or P23H opsin constructs (10 ng/μl) and DNA encoding the coral-derived red fluorescent protein (DsRed) (3 ng/μl) under control of the same promoter (PH20). Worms transiently expressing WT opsin or P23H (b)opsin were selected based on DsRed fluorescence. To integrate opsin cDNA into the worm genome, TG worm lines were exposed to 350 × 100 1 μJ/cm2 ultraviolet light (Spectrolinker XL-1500; Spectronics Corp., Westbury, NY), and F3 progeny of integrated TG lines were screened. TG worm lines with the highest expression of WT or P23H (b)opsin were then selected for further experiments.
Real Time PCR
C. elegans samples were prepared according to a published protocol (34). Total RNA was extracted with TRI Reagent (from MRC). One μg of RNA was reverse-transcribed with M-MLV reverse transcriptase (AB Applied Sciences). Real time-PCR was performed with TaqDNA polymerase (Roche Applied Science). Quantitative RT-PCR was carried out with SYBR® Master Mix (Invitrogen). Specificities of the quantitative PCR analyses were verified by denaturing curve analyses. Expression levels of particular gene transcripts were calculated based on a standard curve normalized to β-tubulin (tbb-1) levels. Primers used were as follows: P23H rhodopsin, CAATGCTCTAGAATGAACGGAACCGAGG (forward) and CGAAGACCATGAAGAGGTAGG (reverse); tbb-1, TGGTTCCATTCCCACGTC (forward) and TCGTCAACCTCTCTCATGGA (reverse).
Immunoblotting
C. elegans samples were prepared as described previously (34). Briefly, worms were homogenized by sonication and centrifuged to remove debris. Supernatants were centrifuged at 17,000 × g for 15 min, and pellets were mixed in electrophoresis loading buffer, vortexed, and centrifuged briefly, and samples were resolved by SDS-PAGE in 10% Tris/glycine polyacrylamide gels. Immunoblotting was then done with 1D4 mouse monoclonal antibody recognizing the C-terminal peptide of bovine opsin followed by horseradish peroxidase (HRP)-conjugated secondary antibody. Quantification of immunoblot signals was accomplished with National Instruments Vision Assistant 8.5 software. Band intensities were measured and compared among samples loaded on the same gel.
Immunohistochemistry (IHC)
Age-synchronized larva stage 4 (L4) or day 1 animals from TG worm lines were sandwiched between two cover glasses, buried in dry ice for 30 min, and then fixed with 100% methanol (10 min), followed by 100% acetone (10 min). These worms were washed with PBS (137 mm NaCl, 2.7 mm KCl, 8.1 mm Na2HPO4, and 1.76 mm KH2PO4, pH 7.4) for 0.5 h and incubated overnight at 4 °C with PBS containing Alexa-488-conjugated anti-rhodopsin 1D4 antibody and 0.1% Triton X-100. Stained worms were washed three times with PBS before their examination by confocal microscopy. All imaging was done with a Leica TCS SP2 confocal microscope (Leica Microsystems, Bannockburn, IL). Either live worms immobilized with 10 mm NaN3 on 2% agarose pads or methanol/acetone-fixed worms were used. Stains employed were DsRed (λex = 543/λem = 580–630 nm) and Alexa-488 (λex = 488/λem = 510–530 nm).
Neuronal Degeneration
Age-synchronized L4 animals from TG worm lines were cultured in the dark on foil-covered NGM plates for 13 days with or without 10 μm 9-cis-retinal or DMSO. Because this stain was integrated together with WT or P23H opsin constructs, DsRed was constitutively expressed in the nervous system of TG worms. On day 13, DsRed images were obtained through an upright confocal microscope to analyze the nervous system structure.
Large Scale Worm Cultures for Purification of P23H and WT Isorhodopsin
TG worm lines expressing the highest amounts of P23H or WT opsin were selected and cultured for purification of isorhodopsin. This procedure was modified from that previously reported for WT isorhodopsin (28). Briefly, TG worms were cultured for two generations (5–6 days) in 10 150-mm high growth medium (HGM) culture dishes seeded with HB101 Escherichia coli bacteria and then transferred into a 10-liter fermenter filled with autoclaved S medium (31). Worms then were cultured for two generations at room temperature, pH 7.0, with 50% dissolved oxygen and 300 rpm agitation, until their density reached ∼10 adult worms/μl. For P23H opsin worms, 50 mg of 9-cis-retinal was added to the culture 24 h before harvest, and the entire culture was incubated in the dark for biosynthesis of P23H isorhodopsin. Worm cultures were collected and centrifuged at 382 × g (JA-10; Beckman Instruments, Brea, CA) for 10 min, and the pellet was suspended in minimum S medium. Thirty-ml aliquots of worm suspension were then carefully loaded onto 300 ml of ice-cold 35% sucrose solution and centrifuged at 170 × g (JA-10; Beckman Instruments) for 5 min. The top layer containing live worms was collected, diluted with 1 liter of H2O and centrifuged at 382 × g for 10 min. The worm pellet was washed again with H2O and suspended in 50 mm Bis-tris propane (BTP) buffer, pH 7.5, 150 mm NaCl with protease inhibitor mixture (Complete Mini, EDTA-free, Roche Applied Science) prior to lysis. When worms were collected for disulfide bond analysis of isorhodopsin, 100 mm N-ethylmaleimide (NEM, Sigma) was added to the lysis and the wash and elution buffers to modify the free Cys residues. The worm suspension then was lysed with a microfluidizer (M-110Y Microfluidizer Processor, Microfluidic, Newton, MA) at 120 p.s.i. four times, and the debris was pelleted by a 300 × g centrifugation for 10 min. The resulting worm lysate was incubated with 0.7 mg of 9-cis-retinal (greater than a 100:1 chromophore/opsin ratio) to obtain either ground state WT isorhodopsin or stabilize P23H isorhodopsin. Then 100 units of bee venom phospholipase A2 (Sigma) with 1 mm CaCl2 was stirred in at 4 °C for 30 min. After membrane proteins were solubilized with 20 mm n-dodecyl-β-d-maltoside (DDM) under gentle rotation for 1 h at 4 °C, solubilized membrane proteins were separated by ultracentrifugation at 195,000 × g (Optima L-90K Ultracentrifuge, Beckman Coulter, Pasadena, CA) for 30 min. The supernatant was incubated with agarose-immobilized anti-rhodopsin 1D4 antibody (200–400-μl bed volumes) for 1 h at 4 °C. This mixture was passed through a 0.5-cm diameter column and washed with more than 20 bed volumes of wash buffer (50 mm BTP, pH 7.5, 300 mm NaCl, and 1 mm DDM). Isorhodopsin was eluted with 5 bed volumes of elution buffer (50 mm BTP, pH 7.5, 300 mm NaCl, 2 mm DDM, and 1 mg/ml 1D4 competing peptide). WT isorhodopsin was either used immediately or frozen at −80 °C, whereas P23H isorhodopsin was used promptly after elution due to its instability.
Gt Activation Assays
Gt was purified according to a procedure previously described (35). The functionality of WT isorhodopsin or P23H isorhodopsin heterologously expressed and purified from worms was evaluated by a Gt activation fluorescence assay. Isorhodopsin/Gt and P23H isorhodopsin/Gt samples were used at concentrations of 25 and 250 nm, respectively, to achieve a molar ratio of isorhodopsin to Gt of 1:10. Protein samples were diluted in 20 mm BTP buffer, pH 7.0, containing 120 mm NaCl, 2 mm MgCl2, and 1 mm DDM and then exposed to light for 15 s from a fiber light covered with a bandpass wavelength filter (480–520 nm). The reaction was carried out at 20 °C in a continuously stirred cuvette. After 300 s of incubation, 5 μm GTPγS was added. The intrinsic fluorescence increase emanating from Gtα was measured with an L55 luminescence spectrophotometer (PerkinElmer Life Sciences) at excitation and emission wavelengths of 300 and 345 nm, respectively (36–38). A pseudo-first order kinetic rate constant (k) was derived from the following function: A(t) = Amax(1 − exp−kt), where Amax is the maximal fluorescence change of Gtα, and A(t) is the relative fluorescence change at time t. No changes in Trp fluorescence were detected in control experiments without GTPγS.
Meta II Decay Rate
All measurements were performed with 25 nm WT isorhodopsin or P23H isorhodopsin diluted with buffer composed of 10 mm BTP, 100 mm NaCl, and 1 mm DDM, pH 6.0, which favored the formation of Meta II. An LS 55 luminescence spectrophotometer (PerkinElmer Life Sciences) was used to measure the intrinsic Trp fluorescence increase that correlates with the decrease in protonated Schiff base concentration (39–42). Isorhodopsins were illuminated with a fiber light covered with a bandpass wavelength filter (480–520 nm) for 15 s immediately before the fluorescence measurements. Bleaching was carried out at a distance of 10 cm to prevent heat accumulation, and a thermostat was applied to stabilize the temperature of the cuvette at 20 °C. Spectrofluorometer slit width settings were 5.0 nm at 295 nm for excitation and 8.0 nm at 330 nm for emission.
Photosensitivity of Visual Pigments
Immunoaffinity purified WT or P23H isorhodopsin samples were bleached for 5, 15, 30, 60, 120, 180, or 240 s with a fiber light through a bandpass wavelength filter (480–520 nm) from a distance of 15 cm. UV-visible spectra were recorded immediately after each bleaching procedure at room temperature. Assuming 100% absorption at the initial measurement point, the percentage of isorhodopsin remaining after each bleaching procedure was calculated from its absorption at 488 nm for WT isorhodopsin and at 479 nm for P23H isorhodopsin and plotted as a function of time. The t½ of chromophore release was calculated from this plot.
Thermal Stability
Freshly purified WT or P23H isorhodopsin samples were incubated at 37 °C and monitored for changes in their UV-visible absorption spectra. Absorbance at 488 nm for WT isorhodopsin and at 479 nm for P23H isorhodopsin was recorded at the following time intervals: 0, 2, 4, 6, 8, 10, 12, 14, 16, 18, and 20 min. Results were plotted assuming 100% absorption at the initial time point.
Disulfide Bond Analysis of Isorhodopsin by Mass Spectrometry (MS) After Protein Digestion
Freshly purified NEM-treated WT isorhodopsin or P23H isorhodopsin was denatured in SDS sample buffer and loaded onto 12% polyacrylamide gels for SDS-PAGE. Gels were stained with Coomassie Blue, and the ∼37-kDa bands were cut out for in-gel digestion with sequencing grade modified chymotrypsin (Roche Applied Science) as described previously (43). Briefly, pieces excised from an SDS-polyacrylamide gel were first destained in 50% acetonitrile containing 50 mm ammonium bicarbonate and then dehydrated with acetonitrile. Proteins were reduced with 20 mm dithiothreitol (DTT) at room temperature for 1 h, alkylated with 50 mm iodoacetamide in 50 mm ammonium bicarbonate for 30 min in the dark, followed by proteolytic digestion overnight. Resulting peptides were extracted from the gel with 5% formic acid in 50% acetonitrile and then suspended in 0.1% formic acid after being completely dried under vacuum.
High performance liquid chromatography with tandem mass spectrometry (LC-MS/MS) was performed with an Orbitrap Elite Hybrid Mass Spectrometer (Thermo Electron, San Jose, CA) equipped with a Waters nanoAcquity UPLC system (Waters, Taunton, MA). A full scan at ×120,000 resolution was obtained in the Orbitrap spectrometer for eluted peptides in the 300-1 800 atomic mass unit range, followed by 20 MS/MS scans with a normalized collision energy of 35%. Peptides were identified with Mascot Daemon (version 2.3.0, Matrix Science), and the data were searched against the rhodopsin primary sequence. The mass tolerance was set at 10 ppm for precursor ions and at 0.8 Da for product ions. Carbamidomethylation (CAM) or NEM labeling of Cys residues and oxidation of Met residues were set as variable modifications.
In Vivo Light Response Assays
One day before these experiments, L4 animals raised at 20 °C were transferred onto NGM plates seeded with 100 μl of OP50 bacteria containing either DMSO vehicle control (no retinal) or 10 μm 9-cis-retinal. The resulting plates were wrapped with aluminum foil and stored in a cardboard box overnight at 20 °C. Light-response experiments were performed in a dark room at 22 °C using a Zeiss Stemi SV11-Apo microscope (Carl Zeiss, Oberkochen, Germany) mounted with a Kramer Universal Stereo Fluorescence Attachment and Cubes unit (Kramer Scientific, Amesbury, MA), an Andor iXon DV897 electron-multiplying charge-coupled device camera (Andor, South Windsor, CT), and a ProScan II H117 motorized stage (Prior Scientific, Rockland, MA). A ×1.6 objective lens was used along with a ×2.5 magnifying lens and 7 lux of transmitted white light for (b)opsin-expressing worms. For each light response assay, a day 1 worm with an embedded platinum wire (an L4 worm raised overnight becomes a day 1 young adult) was placed on an unseeded NGM tracking plate. Worms crawled vigorously under these conditions.
To measure the motor response to light of worms expressing opsin in neurons, blue light (488 ± 20 nm) was delivered after 10 s of control imaging to animals from a metal halide short arc bulb housed in an EXFO X-Cite 120PC-Q unit (Lumen Dynamics, Mississauga, Ontario, Canada) through a Kramer Universal Stereo Fluorescence Attachment and Cubes unit for 1 s, and animals were continuously imaged for another 30 s. Worm locomotion before and after illumination was recorded in AVI format movies at 30 Hz with a custom software package to capture images, control the onset and duration of illumination, and integrate the resulting information. Light intensity output of the EXFO unit was calibrated to reach ±5% of targeted intensity at the microscopic plane measured with a Macam L203 Photometer (MacamPhotometrics, Livingston, UK). Worm locomotion velocities were computed with a previously published algorithm (44), and images were analyzed frame by frame to score the degree of motor activity loss. The light-response index was defined as follows: 5 = complete lack of motion >10 s; 4 = complete lack of motion >10 s except for head shaking; 3 = lack of motion 2–10 s; 2 = lack of motion <2 s; 1 = changed locomotion speed or direction; and 0 = no change noted in motor activity.
To measure the recovery of TG worms from complete paralysis after exposure to light, blue light (488 ± 20 nm, 500 lux for WT opsin worms, and 2000 lux for P23H opsin worms) was delivered to TG animals, and locomotion was recorded as described above. Images were analyzed frame by frame to score the degree of locomotion recovery. The recovery index was defined as follows: 5 = normal locomotion; 4 = uncoordinated locomotion; 3 = noticeable movement; 2 = body wiggling; 1 = head shaking; 0 = no movement.
Chemical Treatment of Worms
Age-synchronized L1 C. elegans from TG worm lines were cultured on NGM plates for 1 day. Animals were supplemented with 10 μm 9-cis-retinal at 1 and 16 h. Added chemicals included bortezomib (final concentration 30 nm) and cycloheximide (final concentration 50 μm), which were co-administered with 9-cis-retinal for 1 h.
RESULTS
Expression Level of P23H Opsin
The Pro to His residue substitution in the N-terminal intradiscal loop of opsin causes adRP (Fig. 1A). We generated a TG C. elegans line constitutively expressing P23H bovine opsin in the nervous system by co-injecting a DNA vector encoding P23H bovine opsin driven by the neuron-specific PH20 promoter (Fig. 1B) together with a DNA vector encoding DsRed fluorescent protein under the same promoter. Selective genomic integration was indicated by DsRed fluorescence in the nervous system (28, 30), and the TG worm line with the greatest P23H opsin expression was used for this study. Previously described TG C. elegans expressing WT bovine opsin (WT worms) were used as controls (28, 30). As quantified by quantitative RT-PCR, expression of WT opsin and P23H opsin in the two TG worm lines was comparable at the mRNA level (Fig. 2A). However, as shown by immunoblots, the P23H opsin level was much lower than that of WT opsin (Fig. 2B). This result agrees with previous findings in P23H knock-in mice (24) suggesting that P23H opsin is largely degraded. Moreover, the P23H opsin tended to form SDS- and DTT-insoluble opsin aggregates in the protein lysate that migrated as dimers or even higher order oligomers in SDS-polyacrylamide gels, whereas WT opsin migrated largely as monomers (Fig. 2E). The apparent molecular mass of P23H opsin was larger than that of WT opsin (Fig. 2, B and C). This was primarily due to differences in protein glycosylation as peptide:N-glycosidase F (PNGase F) treatment abrogated this band shift (Fig. 2C). Aberrant glycosylation of P23H opsin has also been observed in P23H knock-in mice and in tissue culture, although the molecular masses of the mutant opsin varied across species (8, 9, 24). 9-cis-Retinal is an analog of the natural 11-cis-retinal chromophore known to stabilize P23H opsin in cell culture experiments (8, 9). To test the effect of pharmacological chaperones on P23H opsin in the TG nematode, both WT and P23H TG C. elegans were treated with 9-cis-retinal. Overnight exposure of P23H TG worms to 9-cis-retinal increased the amount of P23H opsin protein without changing its apparent molecular mass, suggesting glycosylation-independent stabilization of the P23H opsin (Fig. 2D).
FIGURE 1.

Generation of a stable C. elegans line expressing P23H bovine opsin. A, two-dimensional topology of the bovine opsin sequence (black letters) compared with human (green) and mouse (blue). Mutations in adRP cases are shown with a black background and those in arRP cases are shown with gray. The P23H mutation is featured with red background as indicated by a black arrow. B, shown are the P23H opsin and DsRed expression constructs injected into C. elegans for expression of bovine P23H opsin and DsRed in neurons. Both constructs contain a neuronal driving PH20 promoter (blue block) followed by the codon-optimized cDNA of bovine opsin with a Pro → His mutation at codon 23 (green block) or the cDNA of DsRed (red block).
FIGURE 2.

Characterization of P23H opsin expression in C. elegans. A, comparison of opsin transcripts expressed in WT and P23H opsin worms normalized with β-tubulin. B, immunoblots of bovine opsin in stable C. elegans expressing WT opsin or P23H opsin with non-TG worms used as a control. β-Tubulin was the loading control. Because of the low abundance of P23H opsin in each worm, 30 μg of worm lysate protein were loaded for P23H TG worm lines, but only 3 μg of lysate were used to generate a comparable WT band. Quantified band intensities normalized by β-tubulin are shown in the right panel. C, immunoblots of WT opsin and P23H opsin from nematode lysates with or without peptide:N-glycosidase F (PNGase-F) treatment for deglycosylation. Positions of the bands for untreated WT (open arrow) and P23H (black arrow) opsin are indicated. The difference in band positions between WT opsin and P23H opsin was due to altered glycosylation because the bands of deglycosylated WT opsin and P23H opsin occupied similar positions after treatment with peptide:N-glycosidase F (thick black arrow). D, effect of 9-cis-retinal pretreatment on WT opsin and P23H nematode opsin. Freshly prepared samples were immediately loaded on SDS-PAGE. Both WT (∼35 kDa) and P23H opsin (∼40 kDa) were observed as monomers. β-Tubulin was used as a loading control. E, immunoblots of WT and P23H opsin from worm lysates frozen at −80 °C for 1 day. Worms were homogenized and frozen immediately for SDS-PAGE on the next day. P23H opsin was present primarily as a dimer at ∼70 kDa, whereas WT opsin existed primarily as a monomer at ∼35 kDa.
Localization of P23H Opsin Expressed in C. elegans
To characterize the localization of P23H opsin, IHC was performed on L4 or day 1 young adult C. elegans. P23H and WT animals were fixed and stained with anti-rhodopsin antibody. WT opsin-expressing worms showed strong homogeneous staining along the ventral and dorsal neuronal cord of the worm (Fig. 3A). In contrast, immunostained P23H opsin-expressing worms showed much weaker and more punctate fluorescence (Fig. 3C), and P23H opsin aggregation was clearly visible in neuronal cell bodies (Fig. 3C, inset). Reduced expression of P23H opsin is consistent with the immunoblotting results (Fig. 2B) and with previous reports that demonstrated rapid degradation of P23H opsin in various models (16, 17, 24, 45). Interestingly, a significant increase in fluorescence intensity together with a decrease in P23H opsin aggregation was observed after treatment of TG worms with 9-cis-retinal (Fig. 3E).
FIGURE 3.
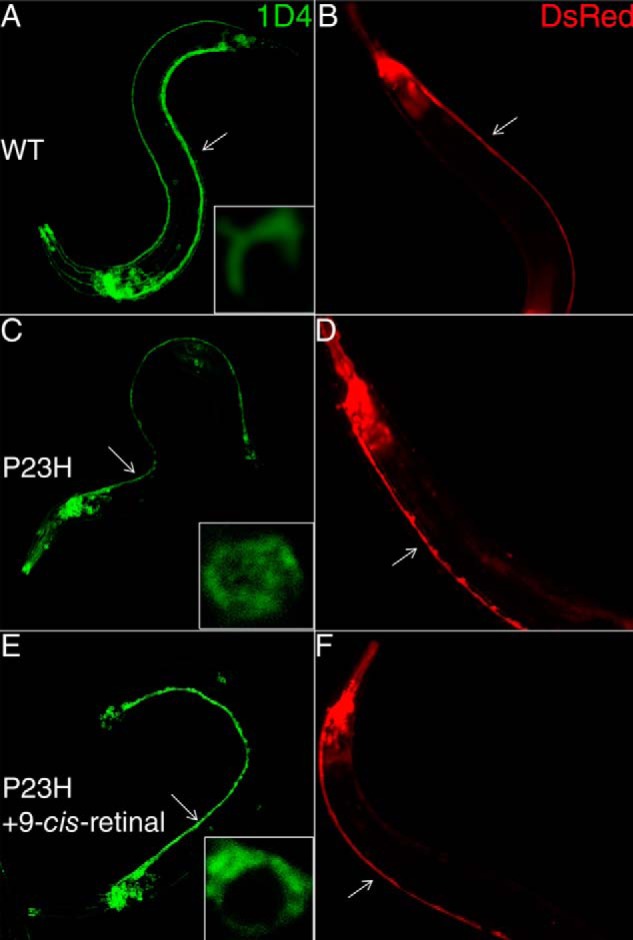
P23H localization in young adult C. elegans and neuronal morphology in older adult C. elegans. Left panels (A, C, and E) show typical immunofluorescent images of day 1 C. elegans expressing WT opsin or P23H opsin stained with Alexa-488-conjugated 1D4 antibody. Insets are representative single cell images that reveal the cellular localization of WT and mutant opsin. Right panels (B, E, and F) are fluorescent images from DsRed expressed in the nervous system of day 13 adult C. elegans that display morphological changes. A and B are from C. elegans expressing WT opsin without treatment; C and D are from C. elegans expressing P23H opsin without treatment; and E and F are from P23H opsin nematodes preincubated with 10 μm 9-cis-retinal in the dark. Ventral cords are marked with white arrows.
To test whether the constitutive expression of P23H opsin affects the nervous system of TG C. elegans, we imaged the DsRed fluorescence in the nervous system of day 13 WT and P23H opsin TG worms. WT opsin worms showed continuous staining of the ventral cord (Fig. 3B), whereas P23H opsin worms evidenced disruptions in such staining (Fig. 3D) consistent with neurodegeneration. Upon pretreatment with 9-cis-retinal, DsRed fluorescence was continuous along the ventral cords of P23H opsin worms (Fig. 3F) similar to its appearance in WT opsin-expressing worms (Fig. 3B). This suggests that pharmacological chaperones could alleviate the neuronal stress caused by continuous degradation and aggregation of P23H opsin.
Biochemical Properties of P23H Isorhodopsin Expressed in C. elegans
To characterize the biochemical properties of P23H isorhodopsin, we incubated TG C. elegans with 9-cis-retinal and purified the resulting isorhodopsin by 1D4-immunoaffinity chromatography. Because previous studies indicated that WT bovine isorhodopsin purified from TG worms had properties similar to native rhodopsin purified from bovine retina (34), we used WT isorhodopsin purified from TG worms as a control. According to previous studies, P23H rhodopsin could only be generated in vivo, whereas WT rhodopsin can be generated in vitro (8, 9, 15). Therefore, a bulk culture of P23H opsin worms was treated with 17 μm 9-cis-retinal for 3 days in the dark before harvest, whereas WT isorhodopsin was generated in the homogenized worm lysate to reduce 9-cis-retinal consumption. Both WT and P23H isorhodopsin were purified from 10 liters of TG worm culture under dim red light at 4 °C. Because of its instability, only ∼21 μg of purified P23H isorhodopsin was obtained as compared with 200 μg of WT isorhodopsin. The purity and identity of P23H and WT isorhodopsin were confirmed by SDS-PAGE followed by Coomassie Blue staining or immunoblotting (Fig. 4, A and B). Biochemical characterization of P23H isorhodopsin was performed immediately after purification, whereas WT isorhodopsin was analyzed either promptly after purification or after storage at −80 °C (28, 30). No biochemical changes were noted in WT isorhodopsin after weeks at −80 °C. Absorption spectra of both pigments are shown in Fig. 4C. The protonated Schiff-base linkage of 9-cis-retinal in WT isorhodopsin caused a maximum absorption at around 488 nm, whereas P23H isorhodopsin had a maximum absorption at 479 nm, a blue shift of about 9 nm relative to WT isorhodopsin. The λ280 nm/λ488 nm absorption ratio for WT isorhodopsin was 2.24, whereas the λ280 nm/λ479 nm ratio for P23H isorhodopsin was 2.57 indicating a comparable purity and suggesting that purified P23H isorhodopsin was properly folded. We also measured the photosensitivity and thermal stability of purified WT isorhodopsin and P23H isorhodopsin. First, samples with similar protein concentrations were illuminated over a range of time intervals with absorption spectra recorded after each illumination. A gradual decrease of the maximum absorption peak at 479 nm for P23H isorhodopsin and at 488 nm for WT isorhodopsin, together with an increase of the peak at 380 nm, was observed (Fig. 4, D and E). These changes were attributed to a 9-cis to all-trans chromophore isomerization and deprotonation of the Schiff base linkage upon photoactivation (46). Changes in absorption at the maxima of λ488 nm and λ479 nm then were plotted as a function of time and fitted to an exponential decay formula, yielding half-lives for WT isorhodopsin and P23H isorhodopsin of 5 and 15 s, respectively (Fig. 4F). Thus, equal amounts of P23H isorhodopsin needed three times more photons than its WT counterpart to become 50% bleached, indicating that the P23H mutant is less photosensitive than WT isorhodopsin. Next, we tested the thermal stability of WT and P23H pigments at physiological temperature. Similar amounts of protein were incubated at 37 °C, and their rates of thermal decay were calculated from the decrease in maximum absorption at 488 and 479 nm, respectively (Fig. 4, G and H). Although WT isorhodopsin remained nearly 100% unbleached, 50% of the P23H isorhodopsin decayed after 20 min at 37 °C (Fig. 4I), indicating a lower thermal stability for the P23H rhodopsin mutant. This result is consistent with previous studies showing that P23H rhodopsin purified from stable mammalian cells also displayed thermal instability (8, 9). To characterize the kinetics of isorhodopsin decay after illumination, a Meta II decay assay was performed by measuring the change in Trp fluorescence at 330 nm from Meta II isorhodopsin during its decay to free opsin as a function of time (Fig. 5A). Here, the half-lives of WT and P23H Meta II decay were similar (33.2 ± 1.2 and 36.6 ± 1.5 min, respectively). To test whether P23H isorhodopsin could activate the rod G protein, transducin, a fluorescence-based Gt activation assay was performed by monitoring the guanine nucleotide exchange-induced conformational change of the Gtα subunit via its Trp fluorescence at 345 nm. WT isorhodopsin activated Gt coupling normally as noted previously (28, 30). Surprisingly, P23H isorhodopsin did not show any Gt activation (Fig. 5B). This lack of activity could result from an inherent inability of P23H isorhodopsin to activate G protein or instability of this protein in detergent. This question was addressed by the in vivo C. elegans light response assay described below.
FIGURE 4.

Biochemical characterization of WT and P23H isorhodopsins purified from C. elegans. Purified WT isorhodopsin and P23H isorhodopsin in Coomassie Blue-stained SDS-polyacrylamide gels (A) and immunoblots (B) are indicated by open and black arrows, respectively. C, absorption spectra of WT (black line) and P23H (red line) isorhodopsin. The maximum absorptions of WT isorhodopsin and P23H isorhodopsin were at 487.5 and 478.8 nm, respectively. D and E show photosensitivity assay results for WT isorhodopsin and P23H isorhodopsin, respectively. Absorption spectra were taken from isorhodopsin illuminated by a fiber light through a bandpass filter (480–520 nm) for different periods of time. F, comparison of WT (solid circles) and P23H (open circles) isorhodopsin photosensitivities plotted as a function of illumination time in minutes. Isorhodopsin concentrations were measured by absorption at 488 nm for WT and at 479 nm for P23H isorhodopsin and normalized to the isorhodopsin concentration before illumination. G and H, absorption spectra of WT isorhodopsin (G) and P23H isorhodopsin (H) after incubation at 37 °C for specified amounts of time reflect their thermal stability. I, thermal stability of WT isorhodopsin (solid circles) and P23H isorhodopsin (open circles) was compared by plotting the percentage of isorhodopsin remaining as a function of time at 37 °C, normalized by their initial concentrations.
FIGURE 5.
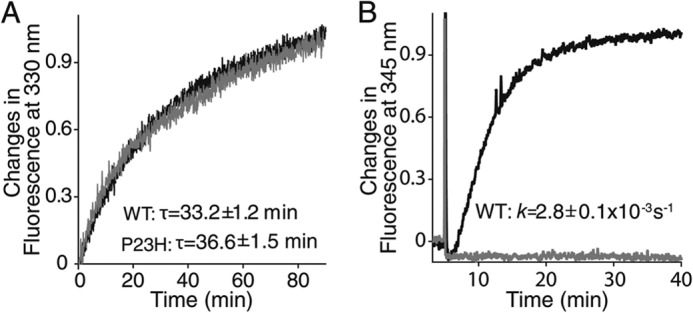
Meta II decay and Gt activation assays of purified WT (black line) and P23H (gray line) isorhodopsin in detergent. A, Meta II decay of isorhodopsin is shown by the time course of changes in Trp fluorescence at 330 nm. Decay rates (τ) of WT isorhodopsin and P23H Meta II isorhodopsin are listed in the insets of A. B, results of a Gt activation assay of isorhodopsin are shown by the time course of Trp fluorescence at 345 nm due to guanylyl nucleotide exchange. The pseudo-first order kinetic rate (k) of Gt activation (derived from the function A(t) = Amax(1 − exp−kt)), where Amax is the maximal Gt fluorescence change, and A(t) is the relative fluorescence change at time (t) for WT isorhodopsin shown in the inset of B. Purified P23H isorhodopsin in detergent did not activate Gt.
Disulfide Bond Analysis of P23H Isorhodopsin
The three-dimensional structure of rhodopsin shows that Pro-23, located within the flexible N-terminal loop, is close to the Asn-15 glycosylation site. In addition, Pro-23 is also near the Cys-110–Cys-187 disulfide bond, the only disulfide bond in rhodopsin (47). To test whether the P23H mutation disrupted this disulfide bond, we analyzed the disulfides of WT isorhodopsin and P23H isorhodopsin by protein digestion and LC-MS/MS. Briefly, free Cys residues were labeled by NEM, and disulfide-bonded Cys residues were labeled with CAM. After chymotrypsin digestion, peptides containing all 10 Cys residues were detected in both WT and P23H isorhodopsin. Among these, Cys-185 and Cys-187 as well as Cys-322 and Cys-323 were present in the same peptide. Overall, WT and P23H isorhodopsin showed similar results for CAM and NEM labeling of peptides containing Cys-110 and those containing Cys-185 and Cys-187. For both WT isorhodopsin and P23H isorhodopsin, all Cys-110-containing peptides were labeled only with CAM (Fig. 6A), suggesting that Cys-110 is involved in a disulfide bond (Table 1). The Cys-185- and Cys-187-containing peptides were then mix-labeled with both NEM and CAM or double-labeled with CAM. Upon further deconvolution of these peptides, it was found that all NEM alkylation occurred at Cys-185 and that CAM alkylation was found solely at Cys-187 (Fig. 6B), which suggested that Cys-187 was originally in a disulfide form. Thus, Cys-110–Cys-187 form a disulfide bond in both WT isorhodopsin and P23H isorhodopsin. Notably, weak CAM labeling of Cys-185 was observed in both WT and P23H samples, in comparison with Cys-187 singly labeled peptides. If this was due to disulfide bonding between Cys-185 and Cys-187, then Cys-110 labeled with NEM should also be found. Because such mixed labeling with both alkylation agents was also observed for other Cys residues, we conclude that CAM labeling of Cys-185 was probably due to incomplete NEM labeling of this residue under native conditions. Because prolonged rhodopsin denaturation by SDS causes artificial dimerization, isorhodopsin samples were only briefly denatured by SDS added just before SDS-PAGE.
FIGURE 6.

Mass spectrometry-based disulfide bond analysis of P23H isorhodopsin. A, tandem mass spectrum of P23H isorhodopsin peptide VFGPTGCNLEGFF with the precursor ion of 722.83(2+) (m/zcharge). The series b or y ions from b5 to b12 and y2 to y8 clearly show CAM labeling at Cys-110. B, tandem mass spectrum of P23H isorhodopsin peptide IPEGMQCSCGIDYY with the precursor ion of 888.85(2+). The series b ions from b5 to b13 clearly show NEM labeling at Cys-185 and CAM labeling at Cys-187. Met-193 is oxidized.
TABLE 1.
Mass spectrometry of chymotrypsin-digested P23H isorhodopsin and WT isorhodopsin purified from C. elegans for disulfide bond analysis
Proteins were purified and characterized as described under “Experimental Procedures.”
| Peptide sequence | Location | Cys in peptide | WT label |
P23H label |
Note | ||
|---|---|---|---|---|---|---|---|
| NEM | CAM | NEM | CAM | ||||
| VFGPTGCNL | 104–112 | Cys-110 | − | + | − | + | CAM |
| VFGPTGCNLEGFF | 104–116 | Cys-110 | − | + | − | + | CAM |
| GPTGCNL | 106–112 | Cys-110 | − | + | − | + | CAM |
| GPTGCNLEGFF | 106–116 | Cys-110 | − | + | − | + | CAM |
| GPTGCNLEGFFATL | 106–119 | Cys-110 | − | + | − | + | CAM |
| SRYIPEGMQCSCGIDYY | 176–192 | Cys-185, Cys-187 | − | + | − | + | 2 CAM |
| SRYIPEGMQCSCGIDYY | Cys-185, Cys-187 | + | + | + | + | 1NEM and 1CAM | |
| IPEGMQCSCGIDY | 179–191 | Cys-185, Cys-187 | − | + | − | + | 2 CAM |
| IPEGMQCSCGIDY | Cys-185, Cys-187 | + | + | + | + | 1NEM and 1CAM | |
| IPEGMQCSCGIDYY | 179–192 | Cys-185, Cys-187 | − | + | − | + | 2 CAM |
| IPEGMQCSCGIDYY | Cys-185, Cys-187 | + | + | + | + | 1NEM and 1CAM | |
| QCSCGIDYY | 184–192 | Cys-185, Cys-187 | − | + | − | + | 2 CAM |
Light Responses of P23H Isorhodopsin in TG C. elegans
As a soil-inhabiting nematode, C. elegans lacks vision and has no rhodopsin ortholog. But TG C. elegans expressing WT opsin in the nervous system exhibit a quantifiable light-induced and 9-cis-retinal-dependent sudden and temporary paralysis, although no such behavior is seen in non-TG worms (30). Light illumination of isorhodopsin triggers activation of the endogenous Gi/o protein signaling pathway in the worm nervous system leading to profound whole body muscle relaxation evidenced by complete loss of motion. Thus, this physiological phenotype constitutes a useful tool to characterize photoactivation of WT rhodopsin and P23H mutant rhodopsin in vivo. Phototransduction of P23H isorhodopsin in this worm was characterized as described previously (30). Because of the absence of food on the tracking plate, worms moved rapidly at first and then slowed down slightly. After 10 s of locomotion recorded in dim light, the NGM plate was exposed to light and changes in both WT and P23H opsin-expressing worm locomotion were continuously recorded (supplemental Movies 1 and 2). Before illumination, the P23H TG worms moved rapidly, but upon illumination, their locomotion suddenly and almost completely stopped; only the head displayed small movements. After about 15 s, a slow recovery of body movement was noted, which lasted until the end of the recording (1.2 min). In contrast, movement of control WT opsin TG worms was completely blocked upon illumination, and this lasted until the end of the recording. The locomotion of non-TG worms is not affected by illumination (30). Thus, both WT and P23H isorhodopsin activated phototransduction in vivo but with different sensitivity to light exposure. We used a response index to quantify the locomotion changes of worms upon illumination (see “Experimental Procedures”). Briefly, a higher response index number correlates with a longer duration and more complete paralysis of the worm (30). To compare the photosensitivity of WT and P23H worms, they were maintained in 10 μm 9-cis-retinal for 24 h, and their locomotion was recorded after illumination with flashes of blue light of varying intensity. As low as 300 lux light elicited a maximum response for WT opsin-expressing worms, whereas P23H worms required a 2000 lux illumination to achieve a maximum response (Fig. 7A). Possible explanations for the lower light sensitivity of P23H worms compared with WT worms include the following: 1) less P23H isorhodopsin protein compared with WT isorhodopsin as shown by immunoblots (Fig. 2B) and IHC (Fig. 3); 2) reduced photosensitivity of P23H opsin (Fig. 4F); and 3) inefficient G protein coupling of P23H isorhodopsin. The last is difficult to test in vitro due to the thermal instability of P23H isorhodopsin (Figs. 4I and 5B).
FIGURE 7.

Characterization of P23H TG C. elegans responses to light. After incubation with 9-cis-retinal overnight, P23H and WT TG C. elegans became paralyzed upon illumination with bright light. (The degree of light-induced paralysis was scored by the light response index described under “Experimental Procedures.”) A, light sensitivity of P23H and WT worms is indicated by plotting their light response index scores as a function of light intensity. Response scores for WT and P23H opsin worms were plotted as solid and open circles, in A, B, D, and E. B, spectra of light sensitivity exhibited by P23H and WT worms are compared by plotting their light response index scores as a function of illumination wavelength at 300 lux for 1 s. C, light response of P23H worms (left bar) was abolished if they were crossed with Goα mutant worms (middle bar), whereas crossing them with Gqα mutant worms did not affect the light response (right bar). D, recovery index values (described under “Experimental Procedures”) for both P23H and WT worms are plotted as a function of time. P23H worms needed more time to recover from light-induced paralysis than WT worms. At time = 0, worms were illuminated with either 500 lux light (WT opsin-expressing worms) or 2000 lux light (P23H opsin-expressing worms), the lowest light intensities that caused full paralysis for each line. E, biosynthesis of P23H opsin and WT opsin was estimated in C. elegans by plotting their light response index values as a function of 9-cis-retinal incubation time. F, bar graphs show the effect of cycloheximide (a protein synthesis/translation inhibitor), bortezomib (a protein degradation inhibitor), or both on light responses of WT (black) and P23H (gray) worms after a 1-h incubation with 9-cis-retinal in the dark. Worms incubated for 16 h with 9-cis-retinal served as positive controls. Worms in C, E, and F were illuminated with 1000 lux light with a maximum emission at 488 nm for 1 s.
Purified P23H isorhodopsin exhibited a 9-nm blue shift of its maximum absorption wavelength (Fig. 4C). To test whether this shift had behavioral consequences, we recorded the locomotion changes of 9-cis-retinal-treated worms exposed to illumination with light at different wavelengths. WT opsin worms were maximally sensitive to 488 nm light, whereas P23H opsin worms showed similar responses after illumination to either 430 or 488 nm light (Fig. 7B). This result confirms the different spectral properties of WT and P23H mutant rhodopsin molecules.
As shown previously (30), light-induced paralysis of WT opsin worms involves activation of the endogenous Gi/o protein signaling cascade by light-illuminated WT isorhodopsin. Crossing of WT isorhodopsin worms with a Go loss-of-function strain (goa-1) led to loss of light responses, but crossing with Gs(gsa-1), G12(gpa-12), or Gq(egl-30) loss-of-function mutants did not affect light response in WT opsin worms (30). To test whether the light response of P23H opsin worms is coupled with activation of the endogenous Gi/o cascade, we crossed a Go mutant (goa-1) C. elegans strain with P23H opsin worms. As a control, a Gq mutant (egl-30) also was crossed with P23H opsin worms. The light response of P23H opsin worms was abolished if Gi/o signaling was genetically ablated, whereas loss of Gq signaling failed to affect the light response of TG P23H worms. Thus, similar to WT isorhodopsin, P23H isorhodopsin acts through the Gi/o signaling cascade (Fig. 7C). Next, we characterized the recovery of TG worms from light-induced paralysis. Locomotion of P23H and WT worms was recorded after 1-s illuminations with blue light (488 nm) at intensities sufficient to produce a maximum light response (2000 lux for P23H worms and 500 lux for WT worms), and their recovery was quantified by an index described under “Experimental Procedures.” Thus, a higher recovery index number correlated with locomotion and less body paralysis. P23H isorhodopsin worms needed 90 min and WT isorhodopsin worms needed 60 min to fully recover from complete loss of motor function (Fig. 7D), much longer periods compared with the millisecond recovery times measured in the eye (48). Such prolonged recovery of TG worms after illumination may be due to the lack of rhodopsin kinase and rhodopsin-specific arrestin, which cause continuous activation of G protein. Although WT isorhodopsin and P23H Meta II isorhodopsin evidenced similar decay kinetics, P23H worms recovered more slowly than WT opsin worms, probably due to slower isorhodopsin regeneration.
P23H isorhodopsin can only be generated in live cells or animals (8, 9), whereas WT isorhodopsin can be generated in vitro by incubation of opsin-containing isolated membranes with 9-cis-retinal (28, 49). Reasons why isorhodopsin synthesis could be enhanced in vivo include the presence of chaperone proteins that might stabilize P23H opsin, promoting its folding and 9-cis-retinal incorporation. Alternatively, 9-cis-retinal could be co-translationally incorporated into P23H opsin. To differentiate between these possibilities, we recorded the light response of P23H and WT worms after treatment with 9-cis-retinal for different periods. WT worms achieved a maximum light response within only 1 h of 9-cis-retinal treatment, but P23H worms required 10 h of chromophore incubation to attain their maximum light response (Fig. 7E). To test whether a decrease in protein synthesis affects isorhodopsin generation, WT and P23H worms were co-treated for 1 h with 9-cis-retinal and cycloheximide, an inhibitor of protein synthesis that acts at the translational level. This reduced the light response of P23H isorhodopsin worms, but not as much as that of WT isorhodopsin worms. If P23H isorhodopsin was generated only during translation, inhibiting protein degradation should not affect the cycloheximide-induced reduction of light responsiveness. To test this, we co-incubated P23H or WT isorhodopsin with the proteasome inhibitor bortezomib along with cycloheximide and 9-cis-retinal, and we then monitored light-induced paralysis of worms (Fig. 7F). Inhibitory effects of cycloheximide on protein synthesis (50, 51) and bortezomib on proteasomes (52, 53) were confirmed in C. elegans. We found that inhibiting protein degradation with bortezomib prevented the decreased light recovery response induced by cycloheximide. This means that P23H isorhodopsin generation reduced by interference with protein synthesis can be counteracted by reducing protein degradation, suggesting that P23H isorhodopsin can be generated during and after translation of opsin. Failure of P23H opsin to incorporate chromophore in vitro could result mainly from instability of the opsin protein, which requires chaperone proteins for stabilization and more time for chromophore binding and proper folding.
DISCUSSION
In this study, we characterized the P23H isorhodopsin in TG C. elegans. The C. elegans model system allowed us to unambiguously relate insights derived from in vivo functional assays with the results of in vitro biochemical assays performed on a purified protein isolated from the same source. Our data provide several conclusions as follows: 1) P23H isorhodopsin is folded with a correct disulfide bond; 2) P23H isorhodopsin is activated by light and undergoes normal Meta II decay as does WT isorhodopsin; 3) P23H isorhodopsin can initiate phototransduction in vivo; and 4) in the presence of 9-cis-retinal, the biogeneration of P23H isorhodopsin is much slower than that of WT isorhodopsin. These results reveal an additional molecular mechanism that contributes to P23H opsin cytotoxicity and photoreceptor death in adRP.
We used bovine rather than human opsin due to its well documented structural and biochemical profile. As there are only minimal differences between these orthologs, it appears likely that our findings will translate to the human protein. Mammalian rhodopsins are highly conserved. Bovine and human rhodopsin protein sequences (both have 348 residues) differ by only 23 residues, 20 of which are chemically similar. The remaining three residues are all located within a flexible loop region, and none of them are correlated with RP, suggesting these variations do not cause major structural or functional changes in rhodopsin (Fig. 1A).
In the crystal structure of bovine rhodopsin (Fig. 8A), Pro-23 is located in the N-terminal loop on the extracellular/intradiscal side of the membrane surrounded by a hydrophobic cluster of residues, including Phe-103, Val-11, Phe-24, Phe-9, Pro-180, Gln-28, and Gln104 (54). This cluster sits on top of the anti-parallel β-plug, which stabilizes the 11-cis-retinal binding pocket. Because Pro is the only common amino acid residue with a five-carbon ring moiety that locks the free rotation of a backbone C–N bond, it is often found in turns within a protein structure. In rhodopsin, Pro-23 forms a sharp turn in the backbone loop inserted in the middle of the above hydrophobic cluster (Fig. 8A). The Pro to His mutation changes the side chain size and hydrophobicity, and it also releases the structural restriction on the backbone conformation (Pro → His replacement by Coot in Fig. 8B). This, in turn, is likely to cause a local structural rearrangement around the N-terminal loop.
FIGURE 8.
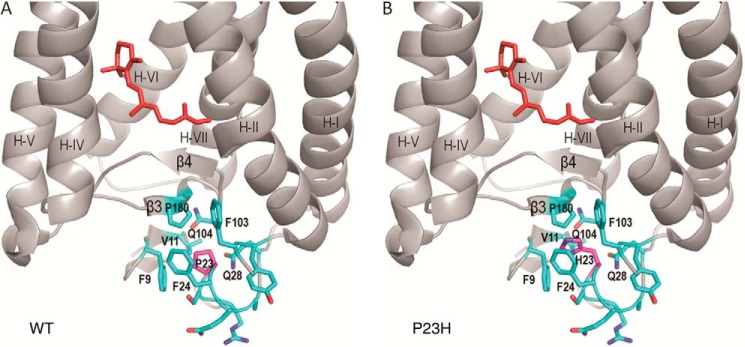
Structural comparison of WT and P23H rhodopsin molecules. A, local structure around Pro-23 in WT rhodopsin (Protein Data Bank code 1F88 (54)). B, structural model of the P23H mutant opsin showing that Pro replacement by a His residue could disrupt the hydrophobic cluster around residue 23, which is next to the anti-parallel β-plug covering the chromophore binding pocket of rhodopsin. 11-cis-Retinal is shown in red.
Rhodopsin mutations associated with RP are classified according to their molecular properties in mammalian cell culture expression systems (7, 15, 16, 55). The P23H mutation was placed in class IIa by Sung et al. (15, 16) or class III by Kaushal et al. (70), both of which are characterized by the following: 1) low rhodopsin yield; 2) retention in the ER; 3) aggregation in SDS-PAGE; and 4) inefficient regeneration with 11-cis-retinal. Pharmacological chaperones such as 9-cis-retinal and its analogs are known to stabilize P23H opsin (8, 9, 56). In C. elegans, P23H opsin was expressed in neurons with no endogenous chromophore supply. The observations of P23H opsin aggregation, ER retention, and enhanced degradation were similar to what was previously detected in mammalian cell culture. TG expression of P23H opsin in C. elegans caused incomplete ventral cord imaging based on DsRed co-expressed in the neurons of aged P23H worms, suggesting neuronal damage. The P23H opsin was stabilized by treatment with 9-cis-retinal, indicating that the chromophore is protective against neuronal damage caused by P23H expression. In animals with vision, the natural chromophore, 11-cis-retinal, is continuously regenerated by the retinoid visual cycle (57). Genetic ablation of retinoid storage by knocking out lecithin retinal acyltransferase in P23H/+ knock-in mice significantly accelerates the progression of retinal degeneration due to the P23H mutation (24). Despite the protective effect of endogenous chromophore, all previous animal models, including P23H TG Drosophila (14), Xenopus (10, 58), rats (12), mice (11, 59–61), pigs (13), and knock-in mice (24, 25), undergo various degrees of retinal degeneration that are also reported in adRP patients carrying this mutation (24, 62). This suggests that the presence of the endogenous chromophore does not suffice to prevent P23H-induced adRP. Therefore, the 9-cis-retinal, which stabilizes P23H opsin, could be needed to treat P23H adRP. Other than supplemental chromophore analogs, new pharmacological chaperones are needed to test whether alleviating UPR (4, 63, 64) and/or proteasome overload (65) can rescue P23H adRP.
Structural disruption around mutated His-23 may affect glycosylation of the nearby Asn-15 (Fig. 1A). Glycosylation is one of the quality control markers used to evaluate proper protein folding in the ER (66). Thus, disruption of P23H opsin glycosylation may underlie the ER retention and degradation observed in different model systems. In mammalian cells, P23H opsin showed altered endoglycosidase H-sensitive glycosylation, suggesting that P23H opsin is glycosylated in the ER (16). Similarly, glycosylation of P23H opsin in knock-in mice also showed endoglycosidase H susceptibility, whereas WT rhodopsin was resistant to this enzyme (24). Consistent with these other models, TG C. elegans also revealed a different molecular mass for P23H and WT opsin due to altered glycosylation. All models indicate that the P23H mutation affects glycosylation, but the molecular masses for P23H opsin/rhodopsin vary in different models (15, 16, 24). This could result from species-specific differences in glycosylation factors. Both Asn-2 and Asn-15 are glycosylated in rhodopsin (67–69), and a mutation of Asn-15 causes adRP, suggesting that Asn-15 glycosylation could be important in stabilizing the rhodopsin structure as suggested previously (70, 71). Whether one or two glycosylation sites are affected by the P23H rhodopsin mutation is still unclear. A mass spectrometry analysis could address this question if sufficient P23H isorhodopsin can be purified.
The only disulfide bond observed in the bovine rhodopsin structure, Cys-110–Cys-187, is near Pro-23 (7.9 Å away). This disulfide bond is essential for stabilizing rhodopsin's structure, because mutation of either Cys-110 or Cys-187 in cell cultures leads to defects in rhodopsin regeneration, whereas mutating other Cys residues has no effect (7, 72). Moreover both Cys-110 and Cys-187 single mutations have been found in adRP cases (Fig. 1), further confirming the importance of this disulfide for rhodopsin stability. Other RP mutations near the rhodopsin disulfide bond, including H211P and L125R, contain an aberrant Cys-185–Cys-187 disulfide bond (73). The hypothesis that the P23H mutation could cause a similar aberrant linkage was evaluated previously in a cell culture model (74). Mutation of Cys-185 to Ala, which would eliminate a potential Cys-185–Cys-187 disulfide, failed to restore rhodopsin regeneration in the P23H mutant background. Using TG C. elegans, we purified enough P23H isorhodopsin to directly analyze its disulfide bonding for the first time. Our data suggest that the Cys-110–Cys-187 disulfide bond was correctly formed in folded P23H isorhodopsin. Whether disruption of the disulfide bond occurs only in misfolded and aggregated P23H opsin still needs to be addressed.
Reports about the localization of P23H opsin/rhodopsin are inconsistent in animal models. Olsson et al. (11) reported that P23H rhodopsin accumulates in the inner segments of retina in several lines of P23H TG mice, a finding confirmed by others (75). However, TG mice expressing VPP rhodopsin bearing a triple mutation P23H/V20G/P27L showed that mutant rhodopsin predominantly localized in the ROS, with residual mislocalization noted in the outer plexiform layer (76). Frederick et al. (60) then generated a P23H TG mouse on a rhodopsin knock-out background, and they reported that GHL (P23H)/Rho−/− mice failed to form ROS and that P23H rhodopsin mainly accumulated in the outer nuclear layer. P23H TG rat line-3 is the most commonly used animal model for gene therapy and drug testing (12, 21, 22, 77–83), but immunochemistry showed rhodopsin is localized in the ROS of this TG rat (84). The P23H transgene has also been expressed in Xenopus, revealing an inner segment accumulation (10). The P23H-EGFP fusion protein expressed in Xenopus showed both ROS and inner segment accumulation, but cross-section imaging of ROS showed P23H-EGFP aggregates distributed unevenly on the disc membrane (58). In TG C. elegans, we expressed the P23H opsin in neurons with no endogenous visual function. We found punctate immunostaining of aggregated P23H opsin along the ventral cord. Upon treatment with 9-cis-retinal, P23H opsin staining became more homogeneous but was still not as smooth as WT opsin. The discrepancy in P23H opsin/rhodopsin localization in TG animals could be due to differences in their expression at the mRNA level. Generation of P23H/+ knock-in mice replaced one allele of the WT RHO gene with the P23H mutated gene, which recapitulated the genetic change occurring in P23H adRP patients. P23H/rhodopsin-EGFP knock-in mice showed P23H rhodopsin staining in the ROS (24), and P23H/P23H knock-in mice showed that P23H rhodopsin can form extended membranous structures (immature discs) at the ciliary protrusion (25), further confirming the capability of P23H rhodopsin to successfully escape retention in the ER and form discs.
Previously, due to its instability, only residual amounts of P23H isorhodopsin were purified from cell culture, limiting its biochemical characterization (8, 9). The small body size and short life cycle of C. elegans make this worm a useful model for production of G protein-coupled receptors (28). Here, we confirmed the biochemical properties of P23H isorhodopsin reported before and extended the biochemical profile of this rhodopsin mutant by utilizing this novel protein expression system. Compared with its WT counterpart, P23H isorhodopsin displays an ∼9-nm blue shift in its absorption spectrum, a reduced photosensitivity, and decreased thermal stability. These properties of P23H isorhodopsin were also reported for cell culture-based expression systems (8, 9), inferring that the expression system does not markedly influence the biochemical characteristics of this protein. In addition, we showed that the P23H isorhodopsin Meta II state decays at the same rate as that of WT isorhodopsin. However, our P23H isorhodopsin did not activate Gt in vitro. The photobleaching and Meta II decay of P23H isorhodopsin clearly indicate that this mutant is folded. Inability to activate Gt in vitro could be due to thermal instability of the mutant isorhodopsin because our in vivo light response assay clearly showed that P23H isorhodopsin can activate the worm's endogenous G protein signaling.
Previously, in vivo detection of phototransduction initiated by P23H rhodopsin was difficult to document in TG and knock-in vertebrate animals, because of the low abundance of the mutant rhodopsin and interference from endogenous WT rhodopsin. Recently, employing Gnat2−/− and P23H homozygous knock-in mice, Sakami et al. (25) used electroretinograms to identify visual phototransduction responses from P23H rhodopsin, although the signals were weak due to the lack of functional ROS and low levels of the mutant rhodopsin. The absence of endogenous visual pigments in P23H TG C. elegans allowed us to apply an in vivo assay by exposing the worms to 9-cis-retinal chromophore. We have developed this assay using the WT TG C. elegans, which showed a light-induced response due to the coupling of photoactivated isorhodopsin with the worm's endogenous Go protein cascade (30). Using the same assay, we found that P23H isorhodopsin can also initiate phototransduction by activating the Go protein of the worm, a result that, in agreement with the findings of Sakami et al. (25), indicates that P23H rhodopsin is a functional visual pigment. By comparing light responses of WT and P23H isorhodopsin in C. elegans, we showed that P23H worms are less sensitive to light (Fig. 7). This could be due to the following: 1) P23H isorhodopsin being less photosensitive, as supported biochemically (Fig. 4); 2) P23H isorhodopsin being present at lower amounts relative to WT isorhodopsin as shown by immunoblotting (Fig. 2); or 3) inefficient G protein coupling of P23H isorhodopsin, which is difficult to address in vitro due to the thermal instability of P23H isorhodopsin (Figs. 4I and 5B). The light response assay also suggested a blue shift in light sensitivity of P23H worms compared with WT animals, although this effect was more clearly documented biochemically with purified P23H isorhodopsin. Agreement between in vivo and biochemical assays suggests that the molecular properties observed in vitro support the characteristics of P23H rhodopsin noted in vivo. Using the in vivo assay, we showed for the first time that the biogenesis of P23H isorhodopsin takes longer than that of WT isorhodopsin, suggesting that the mutant opsin has difficulty incorporating the chromophore even in vivo.
Altogether, our studies in TG C. elegans showed that P23H opsin has aberrant glycosylation and is unstable, which leads to a tendency for it to aggregate in the nervous system of the worm. Although supplementation with 9-cis-retinal did not correct the aberrant glycosylation, it did support slow formation of P23H isorhodopsin. The mutant pigment was found to have the correct disulfide bond, to fold, and be photoactivable. Folded P23H isorhodopsin at least was partially able to escape ER retention and degradation, thereby alleviating neuronal stress associated with protein misfolding and aggregation. Compared with WT isorhodopsin, P23H isorhodopsin had a lower photosensitivity and was thermally less stable. Thermal bleach or photobleach led to generation of P23H opsin in situ. Considering that regeneration is markedly slowed for P23H opsin, and that it tends to aggregate, the latter process could occur in the disc membranes of the ROS in the retina, causing disruption of disc organization. Thus, irregular disc orientations have been reported in P23H/+ knock-in mice (24). In addition, aggregated punctate fluorescence was observed in the discs of P23H-EGFP TG Xenopus, but not WT-rhodopsin-EGFP TG Xenopus, indicating that P23H opsin aggregation could occur in the ROS discs (58). Possible strategies for treatment could involve: 1) efficient supplementation of the chromophore with its analog; 2) novel compounds that accelerate pigment regeneration by stabilizing P23H opsin via an allosteric binding site and accelerate pigment regeneration; 3) novel compounds that enhance degradation of P23H opsin, to promote the maintenance of the ROS structure and visual function by the remaining WT allele; and 4) some combination of the above.
In addition to characterization of P23H rhodopsin, our study also indicates that P23H TG C. elegans is a useful efficient tool for validation of novel compounds aimed at treating individuals with P23H-associated adRP. This idea has a solid basis in the literature. Application of C. elegans drug discovery has been reported (85). For example, a C. elegans-based high throughput screening identified inhibitors of SKN-1, the master regulator of detoxification, from full Molecular Libraries Small Molecule Repository (∼364,000 compounds) (86).With the set of developed in vitro and in vivo assays featured in this study, the amount, localization, and phototransduction properties of P23H rhodopsin as well as its neuropathological effects can be quickly and easily examined to evaluate potential therapeutics. Thus, initial drug validation in the scalable TG C. elegans model can save both time and expense as compared with more demanding mouse models.
Supplementary Material
Acknowledgments
We are grateful to Dr. Xiongying Tu for preparation of Fig. 8. We thank Dr. Leslie T. Webster, Jr., and members of the Palczewski laboratory for helpful comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants EY008061 and EY019478. This work was also supported by a grant from the Foundation Fighting Blindness.

This article contains supplemental Movies 1 and 2.
- RP
- retinitis pigmentosa
- ad
- autosomal dominant
- CAM
- carbamidomethylation
- DDM
- n-dodecyl-β-d-maltoside
- DsRed
- coral-derived red fluorescent protein
- ER
- endoplasmic reticulum
- IHC
- immunohistochemistry
- NGM
- nematode growth medium
- NEM
- N-ethylmaleimide
- ROS
- rod outer segment
- TG
- transgenic
- UPR
- unfolded protein response
- BTP
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- (b)opsin
- bovine opsin
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate.
REFERENCES
- 1. Hartong D. T., Berson E. L., Dryja T. P. (2006) Retinitis pigmentosa. Lancet 368, 1795–1809 [DOI] [PubMed] [Google Scholar]
- 2. Rattner A., Sun H., Nathans J. (1999) Molecular genetics of human retinal disease. Annu. Rev. Genet. 33, 89–131 [DOI] [PubMed] [Google Scholar]
- 3. Berson E. L. (1996) Retinitis pigmentosa: unfolding its mystery. Proc. Natl. Acad. Sci. U.S.A. 93, 4526–4528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Athanasiou D., Aguilà M., Bevilacqua D., Novoselov S. S., Parfitt D. A., Cheetham M. E. (2013) The cell stress machinery and retinal degeneration. FEBS Lett. 587, 2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Daiger S. P., Bowne S. J., Sullivan L. S. (2007) Perspective on genes and mutations causing retinitis pigmentosa. Arch. Ophthalmol. 125, 151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mendes H. F., van der Spuy J., Chapple J. P., Cheetham M. E. (2005) Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol. Med. 11, 177–185 [DOI] [PubMed] [Google Scholar]
- 7. Kaushal S., Khorana H. G. (1994) Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry 33, 6121–6128 [DOI] [PubMed] [Google Scholar]
- 8. Noorwez S. M., Kuksa V., Imanishi Y., Zhu L., Filipek S., Palczewski K., Kaushal S. (2003) Pharmacological chaperone-mediated in vivo folding and stabilization of the P23H-opsin mutant associated with autosomal dominant retinitis pigmentosa. J. Biol. Chem. 278, 14442–14450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Noorwez S. M., Malhotra R., McDowell J. H., Smith K. A., Krebs M. P., Kaushal S. (2004) Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J. Biol. Chem. 279, 16278–16284 [DOI] [PubMed] [Google Scholar]
- 10. Tam B. M., Moritz O. L. (2006) Characterization of rhodopsin P23H-induced retinal degeneration in a Xenopus laevis model of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 47, 3234–3241 [DOI] [PubMed] [Google Scholar]
- 11. Olsson J. E., Gordon J. W., Pawlyk B. S., Roof D., Hayes A., Molday R. S., Mukai S., Cowley G. S., Berson E. L., Dryja T. P. (1992) Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron 9, 815–830 [DOI] [PubMed] [Google Scholar]
- 12. Lewin A. S., Drenser K. A., Hauswirth W. W., Nishikawa S., Yasumura D., Flannery J. G., LaVail M. M. (1998) Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat. Med. 4, 967–971 [DOI] [PubMed] [Google Scholar]
- 13. Ross J. W., Fernandez de Castro J. P., Zhao J., Samuel M., Walters E., Rios C., Bray-Ward P., Jones B. W., Marc R. E., Wang W., Zhou L., Noel J. M., McCall M. A., DeMarco P. J., Prather R. S., Kaplan H. J. (2012) Generation of an inbred miniature pig model of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 53, 501–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Griciuc A., Aron L., Roux M. J., Klein R., Giangrande A., Ueffing M. (2010) Inactivation of VCP/ter94 suppresses retinal pathology caused by misfolded rhodopsin in Drosophila. PLoS Genet. 6, e1001075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sung C. H., Davenport C. M., Hennessey J. C., Maumenee I. H., Jacobson S. G., Heckenlively J. R., Nowakowski R., Fishman G., Gouras P., Nathans J. (1991) Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 88, 6481–6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sung C. H., Schneider B. G., Agarwal N., Papermaster D. S., Nathans J. (1991) Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 88, 8840–8844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saliba R. S., Munro P. M., Luthert P. J., Cheetham M. E. (2002) The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J. Cell Sci. 115, 2907–2918 [DOI] [PubMed] [Google Scholar]
- 18. Palczewski K. (2006) G protein-coupled receptor rhodopsin. Annu. Rev. Biochem. 75, 743–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palczewski K. (2010) Retinoids for treatment of retinal diseases. Trends Pharmacol. Sci. 31, 284–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin J. H., Li H., Yasumura D., Cohen H. R., Zhang C., Panning B., Shokat K. M., Lavail M. M., Walter P. (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gorbatyuk M. S., Knox T., LaVail M. M., Gorbatyuk O. S., Noorwez S. M., Hauswirth W. W., Lin J. H., Muzyczka N., Lewin A. S. (2010) Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. U.S.A. 107, 5961–5966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gorbatyuk M. S., Gorbatyuk O. S., LaVail M. M., Lin J. H., Hauswirth W. W., Lewin A. S. (2012) Functional rescue of P23H rhodopsin photoreceptors by gene delivery. Adv. Exp. Med. Biol. 723, 191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aguila M., Bevilacqua D., McCulley C., Schwarz N., Athanasiou D., Kanuga N., Novoselov S. S., Lange C. A., Ali R. R., Bainbridge J. W., Gias C., Coffey P. J., Garriga P., Cheetham M. E. (2013) Hsp90 inhibition protects against inherited retinal degeneration. Hum Mol. Genet. 10.1093/hmg/ddt613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakami S., Maeda T., Bereta G., Okano K., Golczak M., Sumaroka A., Roman A. J., Cideciyan A. V., Jacobson S. G., Palczewski K. (2011) Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J. Biol. Chem. 286, 10551–10567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sakami S., Kolesnikov A. V., Kefalov V. J., Palczewski K. (2014) P23H opsin knock-in mice reveal a novel step in retinal rod disc morphogenesis. Hum. Mol. Genet. 23, 1723–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Edgar R. S., Wood W. B. (1977) The nematode Caenorhabditis elegans: a new organism for intensive biological study. Science 198, 1285–1286 [DOI] [PubMed] [Google Scholar]
- 27. White J. G., Southgate E., Thomson J. N., Brenner S. (1986) The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1–340 [DOI] [PubMed] [Google Scholar]
- 28. Salom D., Cao P., Sun W., Kramp K., Jastrzebska B., Jin H., Feng Z., Palczewski K. (2012) Heterologous expression of functional G-protein-coupled receptors in Caenorhabditis elegans. FASEB J. 26, 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jastrzebska B., Salom D., Jin H., Cao P., Sun W., Palczewski K., Feng Z. (2013) Expression of mammalian G protein-coupled receptors in Caenorhabditis elegans. Methods Enzymol. 520, 239–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cao P., Sun W., Kramp K., Zheng M., Salom D., Jastrzebska B., Jin H., Palczewski K., Feng Z. (2012) Light-sensitive coupling of rhodopsin and melanopsin to G(i/o) and G(q) signal transduction in Caenorhabditis elegans. FASEB J. 26, 480–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stiernagle T. (2006) Maintenance of C. elegans. WormBook, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stenico M., Lloyd A. T., Sharp P. M. (1994) Codon usage in Caenorhabditis elegans: Delineation of translational selection and mutational biases. Nucleic Acids Res. 22, 2437–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yabe T., Suzuki N., Furukawa T., Ishihara T., Katsura I. (2005) Multidrug resistance-associated protein MRP-1 regulates dauer diapause by its export activity in Caenorhabditis elegans. Development 132, 3197–3207 [DOI] [PubMed] [Google Scholar]
- 34. Salom D., Wu N., Sun W., Dong Z., Palczewski K., Jordan S., Salon J. A. (2008) Heterologous expression and purification of the serotonin type 4 receptor from transgenic mouse retina. Biochemistry 47, 13296–13307 [DOI] [PubMed] [Google Scholar]
- 35. Goc A., Angel T. E., Jastrzebska B., Wang B., Wintrode P. L., Palczewski K. (2008) Different properties of the native and reconstituted heterotrimeric G protein transducin. Biochemistry 47, 12409–12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fahmy K., Sakmar T. P. (1993) Regulation of the rhodopsin-transducin interaction by a highly conserved carboxylic acid group. Biochemistry 32, 7229–7236 [DOI] [PubMed] [Google Scholar]
- 37. Farrens D. L., Altenbach C., Yang K., Hubbell W. L., Khorana H. G. (1996) Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274, 768–770 [DOI] [PubMed] [Google Scholar]
- 38. Heck M., Hofmann K. P. (2001) Maximal rate and nucleotide dependence of rhodopsin-catalyzed transducin activation: initial rate analysis based on a double displacement mechanism. J. Biol. Chem. 276, 10000–10009 [DOI] [PubMed] [Google Scholar]
- 39. Farrens D. L., Khorana H. G. (1995) Structure and function in rhodopsin. Measurement of the rate of metarhodopsin II decay by fluorescence spectroscopy. J. Biol. Chem. 270, 5073–5076 [DOI] [PubMed] [Google Scholar]
- 40. Schadel S. A., Heck M., Maretzki D., Filipek S., Teller D. C., Palczewski K., Hofmann K. P. (2003) Ligand channeling within a G-protein-coupled receptor. The entry and exit of retinals in native opsin. J. Biol. Chem. 278, 24896–24903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heck M., Schädel S. A., Maretzki D., Bartl F. J., Ritter E., Palczewski K., Hofmann K. P. (2003) Signaling states of rhodopsin. Formation of the storage form, metarhodopsin III, from active metarhodopsin II. J. Biol. Chem. 278, 3162–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fahmy K., Sakmar T. P. (1993) Light-dependent transducin activation by an ultraviolet-absorbing rhodopsin mutant. Biochemistry 32, 9165–9171 [DOI] [PubMed] [Google Scholar]
- 43. Tsybovsky Y., Wang B., Quazi F., Molday R. S., Palczewski K. (2011) Posttranslational modifications of the photoreceptor-specific ABC transporter ABCA4. Biochemistry 50, 6855–6866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang S., Jin W., Huang Y., Su W., Yang J., Feng Z. (2011) Profiling a Caenorhabditis elegans behavioral parametric dataset with a supervised K-means clustering algorithm identifies genetic networks regulating locomotion. J. Neurosci. Methods 197, 315–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Illing M. E., Rajan R. S., Bence N. F., Kopito R. R. (2002) A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J. Biol. Chem. 277, 34150–34160 [DOI] [PubMed] [Google Scholar]
- 46. Ridge K. D., Palczewski K. (2007) Visual rhodopsin sees the light: structure and mechanism of G protein signaling. J. Biol. Chem. 282, 9297–9301 [DOI] [PubMed] [Google Scholar]
- 47. Karnik S. S., Khorana H. G. (1990) Assembly of functional rhodopsin requires a disulfide bond between cysteine residues 110 and 187. J. Biol. Chem. 265, 17520–17524 [PubMed] [Google Scholar]
- 48. Palczewski K., Van Hooser J. P., Garwin G. G., Chen J., Liou G. I., Saari J. C. (1999) Kinetics of visual pigment regeneration in excised mouse eyes and in mice with a targeted disruption of the gene encoding interphotoreceptor retinoid-binding protein or arrestin. Biochemistry 38, 12012–12019 [DOI] [PubMed] [Google Scholar]
- 49. Zhu L., Imanishi Y., Filipek S., Alekseev A., Jastrzebska B., Sun W., Saperstein D. A., Palczewski K. (2006) Autosomal recessive retinitis pigmentosa and E150K mutation in the opsin gene. J. Biol. Chem. 281, 22289–22298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Burkewitz K., Choe K. P., Lee E. C., Deonarine A., Strange K. (2012) Characterization of the proteostasis roles of glycerol accumulation, protein degradation and protein synthesis during osmotic stress in C. elegans. PLoS One 7, e34153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Epstein H. F., Liu F. (1995) Proteins and protein assemblies. Methods Cell Biol. 48, 437–450 [DOI] [PubMed] [Google Scholar]
- 52. Segref A., Torres S., Hoppe T. (2011) A screenable in vivo assay to study proteostasis networks in Caenorhabditis elegans. Genetics 187, 1235–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Glover-Cutter K. M., Lin S., Blackwell T. K. (2013) Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genet. 9, e1003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., Miyano M. (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 55. Jacobson S. G., Kemp C. M., Sung C. H., Nathans J. (1991) Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am. J. Ophthalmol. 112, 256–271 [DOI] [PubMed] [Google Scholar]
- 56. Mendes H. F., Cheetham M. E. (2008) Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 17, 3043–3054 [DOI] [PubMed] [Google Scholar]
- 57. Kiser P. D., Golczak M., Maeda A., Palczewski K. (2012) Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta 1821, 137–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Haeri M., Knox B. E. (2012) Rhodopsin mutant P23H destabilizes rod photoreceptor disk membranes. PLoS One 7, e30101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Naash M. I., Hollyfield J. G., al-Ubaidi M. R., Baehr W. (1993) Simulation of human autosomal dominant retinitis pigmentosa in transgenic mice expressing a mutated murine opsin gene. Proc. Natl. Acad. Sci. U.S.A. 90, 5499–5503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frederick J. M., Krasnoperova N. V., Hoffmann K., Church-Kopish J., Rüther K., Howes K., Lem J., Baehr W. (2001) Mutant rhodopsin transgene expression on a null background. Invest. Ophthalmol. Vis. Sci. 42, 826–833 [PubMed] [Google Scholar]
- 61. Goto Y., Peachey N. S., Ripps H., Naash M. I. (1995) Functional abnormalities in transgenic mice expressing a mutant rhodopsin gene. Invest. Ophthalmol. Vis. Sci. 36, 62–71 [PubMed] [Google Scholar]
- 62. Berson E. L., Rosner B., Sandberg M. A., Dryja T. P. (1991) Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His). Arch. Ophthalmol. 109, 92–101 [DOI] [PubMed] [Google Scholar]
- 63. Haeri M., Knox B. E. (2012) Endoplasmic reticulum stress and unfolded protein response pathways: potential for treating age-related retinal degeneration. J. Ophthalmic Vis. Res. 7, 45–59 [PMC free article] [PubMed] [Google Scholar]
- 64. Lin J. H., Lavail M. M. (2010) Misfolded proteins and retinal dystrophies. Adv. Exp. Med. Biol. 664, 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lobanova E. S., Finkelstein S., Skiba N. P., Arshavsky V. Y. (2013) Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc. Natl. Acad. Sci. U.S.A. 110, 9986–9991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chakrabarti A., Chen A. W., Varner J. D. (2011) A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 108, 2777–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fukuda M. N., Papermaster D. S., Hargrave P. A. (1979) Rhodopsin carbohydrate. Structure of small oligosaccharides attached at two sites near the NH2 terminus. J. Biol. Chem. 254, 8201–8207 [PubMed] [Google Scholar]
- 68. Fukuda M. N., Papermaster D. S., Hargrave P. A. (1982) Structural analysis of carbohydrate moiety of bovine rhodopsin. Methods Enzymol. 81, 214–223 [DOI] [PubMed] [Google Scholar]
- 69. Liang C. J., Yamashita K., Muellenberg C. G., Shichi H., Kobata A. (1979) Structure of the carbohydrate moieties of bovine rhodopsin. J. Biol. Chem. 254, 6414–6418 [PubMed] [Google Scholar]
- 70. Kaushal S., Ridge K. D., Khorana H. G. (1994) Structure and function in rhodopsin: the role of asparagine-linked glycosylation. Proc. Natl. Acad. Sci. U.S.A. 91, 4024–4028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Murray A. R., Fliesler S. J., Al-Ubaidi M. R. (2009) Rhodopsin: the functional significance of Asn-linked glycosylation and other post-translational modifications. Ophthalmic Genet. 30, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Karnik S. S., Sakmar T. P., Chen H. B., Khorana H. G. (1988) Cysteine residues 110 and 187 are essential for the formation of correct structure in bovine rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 85, 8459–8463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hwa J., Klein-Seetharaman J., Khorana H. G. (2001) Structure and function in rhodopsin: Mass spectrometric identification of the abnormal intradiscal disulfide bond in misfolded retinitis pigmentosa mutants. Proc. Natl. Acad. Sci. U.S.A. 98, 4872–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McKibbin C., Toye A. M., Reeves P. J., Khorana H. G., Edwards P. C., Villa C., Booth P. J. (2007) Opsin stability and folding: the role of Cys-185 and abnormal disulfide bond formation in the intradiscal domain. J. Mol. Biol. 374, 1309–1318 [DOI] [PubMed] [Google Scholar]
- 75. Roof D. J., Adamian M., Hayes A. (1994) Rhodopsin accumulation at abnormal sites in retinas of mice with a human P23H rhodopsin transgene. Invest. Ophthalmol. Vis. Sci. 35, 4049–4062 [PubMed] [Google Scholar]
- 76. Wu T. H., Ting T. D., Okajima T. I., Pepperberg D. R., Ho Y. K., Ripps H., Naash M. I. (1998) Opsin localization and rhodopsin photochemistry in a transgenic mouse model of retinitis pigmentosa. Neuroscience 87, 709–717 [DOI] [PubMed] [Google Scholar]
- 77. Rossmiller B., Mao H., Lewin A. S. (2012) Gene therapy in animal models of autosomal dominant retinitis pigmentosa. Mol. Vis. 18, 2479–2496 [PMC free article] [PubMed] [Google Scholar]
- 78. LaVail M. M., Yasumura D., Matthes M. T., Drenser K. A., Flannery J. G., Lewin A. S., Hauswirth W. W. (2000) Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc. Natl. Acad. Sci. U.S.A. 97, 11488–11493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nunes A. F., Amaral J. D., Lo A. C., Fonseca M. B., Viana R. J., Callaerts-Vegh Z., D'Hooge R., Rodrigues C. M. (2012) TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 45, 440–454 [DOI] [PubMed] [Google Scholar]
- 80. Fernández-Sánchez L., Lax P., Pinilla I., Martín-Nieto J., Cuenca N. (2011) Tauroursodeoxycholic acid prevents retinal degeneration in transgenic P23H rats. Invest. Ophthalmol. Vis. Sci. 52, 4998–5008 [DOI] [PubMed] [Google Scholar]
- 81. Wang X., Venable J., LaPointe P., Hutt D. M., Koulov A. V., Coppinger J., Gurkan C., Kellner W., Matteson J., Plutner H., Riordan J. R., Kelly J. W., Yates J. R., 3rd, Balch W. E. (2006) Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127, 803–815 [DOI] [PubMed] [Google Scholar]
- 82. Fernández-Sánchez L., Lax P., Esquiva G., Martín-Nieto J., Pinilla I., Cuenca N. (2012) Safranal, a saffron constituent, attenuates retinal degeneration in P23H rats. PLoS One 7, e43074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vasireddy V., Chavali V. R., Joseph V. T., Kadam R., Lin J. H., Jamison J. A., Kompella U. B., Reddy G. B., Ayyagari R. (2011) Rescue of photoreceptor degeneration by curcumin in transgenic rats with P23H rhodopsin mutation. PLoS One 6, e21193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chrysostomou V., Stone J., Stowe S., Barnett N. L., Valter K. (2008) The status of cones in the rhodopsin mutant P23H-3 retina: light-regulated damage and repair in parallel with rods. Invest. Ophthalmol. Vis. Sci. 49, 1116–1125 [DOI] [PubMed] [Google Scholar]
- 85. Artal-Sanz M., de Jong L., Tavernarakis N. (2006) Caenorhabditis elegans: a versatile platform for drug discovery. Biotechnol. J. 1, 1405–1418 [DOI] [PubMed] [Google Scholar]
- 86. Leung C. K., Wang Y., Malany S., Deonarine A., Nguyen K., Vasile S., Choe K. P. (2013) An ultra high-throughput, whole-animal screen for small molecule modulators of a specific genetic pathway in Caenorhabditis elegans. PLoS One 8, e62166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.