

Background: Otoferlin deficiency impairs hair cell exocytosis and causes deafness.
Results: Otoferlin binds Ca2+, regulating phosphatidylinositol 4,5-bisphosphate, t-SNARE, and intramolecular C2 interactions; otoferlin directly interacts with SNAP-25 and syntaxin-1B.
Conclusion: Low Ca2+ promotes interactions among otoferlin C2C, C2D, C2E, and C2F; high Ca2+ drives t-SNARE and lipid interactions.
Significance: Otoferlin may mediate novel modes of Ca2+-driven membrane fusion in hair cells.
Keywords: Calcium, Exocytosis, Hair Cell, Isothermal Titration Calorimetry, Mass Spectrometry (MS), Phosphoinositides, Snare, Surface Plasmon Resonance (SPR), C2 Domains, Otoferlin
Abstract
Mutations in otoferlin, a C2 domain-containing ferlin family protein, cause non-syndromic hearing loss in humans (DFNB9 deafness). Furthermore, transmitter secretion of cochlear inner hair cells is compromised in mice lacking otoferlin. In the present study, we show that the C2F domain of otoferlin directly binds calcium (KD = 267 μm) with diminished binding in a pachanga (D1767G) C2F mouse mutation. Calcium was found to differentially regulate binding of otoferlin C2 domains to target SNARE (t-SNARE) proteins and phospholipids. C2D–F domains interact with the syntaxin-1 t-SNARE motif with maximum binding within the range of 20–50 μm Ca2+. At 20 μm Ca2+, the dissociation rate was substantially lower, indicating increased binding (KD = ∼10−9) compared with 0 μm Ca2+ (KD = ∼10−8), suggesting a calcium-mediated stabilization of the C2 domain·t-SNARE complex. C2A and C2B interactions with t-SNAREs were insensitive to calcium. The C2F domain directly binds the t-SNARE SNAP-25 maximally at 100 μm and with reduction at 0 μm Ca2+, a pattern repeated for C2F domain interactions with phosphatidylinositol 4,5-bisphosphate. In contrast, C2F did not bind the vesicle SNARE protein synaptobrevin-1 (VAMP-1). Moreover, an antibody targeting otoferlin immunoprecipitated syntaxin-1 and SNAP-25 but not synaptobrevin-1. As opposed to an increase in binding with increased calcium, interactions between otoferlin C2F domain and intramolecular C2 domains occurred in the absence of calcium, consistent with intra-C2 domain interactions forming a “closed” tertiary structure at low calcium that “opens” as calcium increases. These results suggest a direct role for otoferlin in exocytosis and modulation of calcium-dependent membrane fusion.
Introduction
Mammalian cochlear inner hair cells transduce auditory stimuli and transmit signal to afferent neurons. Inner hair cells, due to their characteristic ribbon synapses, conduct glutamatergic neurotransmitter release with submillisecond accuracy to convey the signal from incoming complex sounds to the brain to decode and create the perception of sound. It is a general belief that the various forms of vesicle-membrane fusion share common molecular machinery, and therefore, the hair cell ribbon synapse is likely to share molecular features also attributed to neuronal secretion (1). In synaptic vesicle fusion, interaction of calcium and SNARE proteins plays a pivotal role. However, such fast, calcium-triggered exocytosis requires a protein calcium sensor to drive interaction of other SNARE-related molecules (2). Otoferlin has been an attractive candidate for calcium sensing in hair cells because 1) neuronal calcium sensors such as synaptotagmin-1 are absent in mature hair cells, 2) otoferlin contains calcium-binding C2 domains,2 3) otoferlin engages in calcium-dependent interaction with SNAREs and phospholipids, and 4) otoferlin-deficient hair cells lack exocytosis.
The otoferlin protein consists of six C2 domains and a C-terminal transmembrane domain (see Fig. 1). The OTOF gene is a member of the ferlin family of genes. Mutations of OTOF in humans, including protein truncation and amino acid substitutions, cause mild to profound non-syndromic hearing loss (3, 4). OTOF knock-out mice are profoundly deaf, manifest almost no hair cell exocytosis (5), and show subtle deficits in vestibular function (6). One of the remarkable changes in hair cell physiology with otoferlin deficiency is this lack of exocytosis despite intact, normal ribbon synapses and vesicle pools (5). Based on these observations, it has been suggested that otoferlin is a calcium-sensitive modulator of hair cell receptoneural secretion, and it has been shown to engage in calcium-dependent molecular interactions with the t-SNARE proteins syntaxin-1 and SNAP-25 (5, 7, 8). Moreover, otoferlin C2 domains bind calcium as detected by fluorescence measurements (5, 7, 8). Vesicle release in hair cells is both calcium- and otoferlin-dependent (5, 6, 9), and synaptotagmin-1, a neuronal calcium sensor, cannot replace otoferlin in otoferlin-deficient hair cells to enable exocytosis (10). Interestingly, otoferlin is the only protein candidate identified in hair cells so far that fits the molecular attributes of a calcium sensor. However, the exact role of otoferlin in modulating calcium-stimulated vesicle fusion in hair cells has yet to be elucidated.
FIGURE 1.
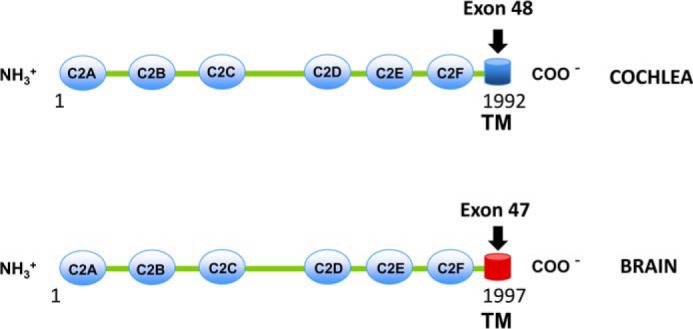
Otoferlin is alternatively spliced in cochlea and brain. Otoferlin is expressed in multiple tissues and organs (34). C2 domains A–F and the C-terminal transmembrane (TM) domains of cochlear and brain otoferlin are illustrated. Of the six C2 domains, four (C2C–C2F) are predicted to bind calcium. In the cochlea, an otoferlin transmembrane domain splice variant arises from exon 48, whereas in brain, it originates from exon 47. The amino acid sequence coded by exon 48 is 72% identical to that of exon 47. Neuronal synaptic vesicles do not contain otoferlin (32), whereas synaptosomal fractions from chick cochleas, which include hair cell synaptic vesicles, do contain otoferlin (33), possibly in parallel with the different transmembrane domains yielding different membrane inserts and localizations of the respective otoferlin molecules. In contrast to the differences in transmembrane sequence, the C2 domains of otoferlin for brain and cochlea are 100% identical.
Here, we present evidence that otoferlin is a Ca2+ sensor. Furthermore, otoferlin C2 domains were found to engage differentially in calcium-dependent interaction with phosphatidylinositol 4,5-bisphosphate (PIP2), syntaxin-1 SNARE domain, and SNAP-25, key components in calcium-driven exocytosis. Whereas the interaction of otoferlin C2 domains with t-SNARE proteins increased with increasing [Ca2+], we report for the first time that otoferlin C2 domains also interacted with each other and that these interactions were negatively impacted by [Ca2+]. These results are consistent with calcium serving as a molecular switch between “open” and “closed” forms of otoferlin that mechanistically underlie exocytosis.
EXPERIMENTAL PROCEDURES
Purification of Histidine-tagged C2 Domain Fusion Proteins
Escherichia coli BL21(DE3) cells were transformed with pRSET vector containing a selected C2 domain sequence or the syntaxin-1 SNARE motif and plated. A single colony was then cultured overnight in 100–500 ml of LB medium, overexpression was induced by addition of isopropyl 1-thio-β-d-galactopyranoside, and the cells were cultured for another 3–5 h. Cells were harvested by centrifugation, washed briefly in binding buffer (Qiagen His tag purification buffer or Clontech Talon purification buffer; both 10 mm phosphate, 1 mm Tris-HCl, pH 8.0, 300 mm NaCl), and resuspended in binding buffer containing 1× protease inhibitor (Sigma) and 1 mm imidazole. The cells were then treated with lysozyme (50 units/ml; Sigma) at room temperature for 30 min and ultrasonicated on ice using pulses of 30-s duration (five to six times). The lysate was centrifuged at 20,828 × g at 4 °C for 25 min. The clear supernatant was collected and placed on ice. Each C2 domain fusion protein was affinity-purified to homogeneity using nickel affinity columns as follows. Nickel-nitrilotriacetic acid spin columns (Qiagen) were equilibrated with the binding buffer at room temperature, the lysate was loaded, and the columns were centrifuged at 4 °C for 3 min at 1,233 × g. After removal of supernatant, the columns were washed three to five times at 4 °C with binding buffer containing 5 mm imidazole (Qiagen). Finally, the bound protein was eluted with 150–250 mm imidazole. The collected, purified protein was centrifuged at 4 °C in an Amicon Ultra centrifugation-dialysis column (Millipore Ireland) at 4,025 × g for 5–10 min to remove imidazole and change the buffer to HEPES-buffered saline (HBS), pH 7.4 containing 1× protease inhibitor mixture. Protein concentration was determined by the Qubit fluorescence assay (Invitrogen).
Purification of GST-C2 Domain Fusion Proteins
E. coli BL21(DE3) cells were transformed by pGEX6.1 vectors (GE Healthcare) containing desired C2 domains, and colonies were selected and cultured overnight in batches of 500 ml of LB medium containing ampicillin (100 mg/liter). The cells were harvested, washed once in phosphate-buffered saline (PBS) buffer, and resuspended in the same buffer containing 1× protease inhibitor mixture (Sigma). To this suspension, lysozyme (1 mg/ml) was added and incubated at room temperature for 30 min, and the lysis was completed by sonication five to seven times with 30-s pulses. Triton X-100 (Sigma) was added to a final concentration of 1%. The lysate was centrifuged at 20,828 × g for 25 min at 4 °C. The clear supernatant was collected and mixed with 100–500 μl 50% glutathione-agarose beads (GE Healthcare) pre-equilibrated in PBS containing 0.1% Tween 20 (PBST). The suspension was incubated in a mixer/inverter for 30 min at 4 °C with constant mixing.
The beads were collected by centrifugation (1,233 × g) at 4 °C for 3 min, washed two to three times in binding buffer containing 1 μm reduced glutathione for competitive removal of nonspecifically bound proteins, and resuspended in 1× cleavage buffer (50 mm Tris-HCl, pH 7.0, 150 mm NaCl, 1 mm EDTA, 1 mm DTT). The beads were then incubated for 5–6 h with 40 μl of PreScissionTM protease (80 units/ml; GE Healthcare) to cleave the purified C2 domain peptide from the GST moiety. The suspension was centrifuged (1,233 × g at 4 °C), and the supernatant containing the purified, free C2 domain was collected. The supernatant was once more mixed with glutathione-agarose to remove residual protease as well as GST. The beads were separated by centrifugation (1,233 × g at 4 °C for 3 min), and the supernatant containing the purified protein was dialyzed by centrifugation in an Amicon ultrafiltration system (Millipore Ireland). The buffer was exchanged with HBS containing 1× protease inhibitor mixture. Protein concentration was measured by the Qubit fluorescence method (Invitrogen).
Circular Dichroism Studies
Circular dichroism spectra from 190 to 280 nm were obtained with a Chirascan CD spectrometer (Applied Photophysics Ltd., Leatherhead, Surrey, UK). Affinity-purified C2 domain samples in HBS buffer (0.01 m HEPES, 0.15 m NaCl, 0.1% Tween 20, pH 7.4) were analyzed using a 1-cm path length cell. Spectra were acquired by sampling every 0.5 s. Sample ellipticity from an average of five readings was converted to mean residue molar ellipticity and plotted using Microsoft Excel software.
Light Scattering Studies
Dynamic light scattering experiments were conducted with a Zetasizer Nano ZS90 light scattering instrument (Malvern Instruments, Malvern, Worcestershire, UK). Purified C2F domain fusion proteins were prepared in HBS buffer at a concentration of 0.5 μg/μl. For analysis, 1 ml of protein solution was placed in a cuvette, and readings were taken at room temperature (25 °C) according to the user manual (Malvern Instruments). The data were analyzed using the Zetasizer software.
Protein/Protein Interaction: Surface Plasmon Resonance (SPR) Analysis
Purified otoferlin C2 domains and syntaxin-1 SNARE domain fusion proteins were dialyzed in calcium-free HBS, pH 7.4 and then immobilized on a CM5 chip using an amine coupling protocol (11). Purified C2 domains and SNARE proteins used as analyte samples were diluted to desired concentrations in calcium-free HBS. For binding analysis, samples were injected in triplicate with or without added calcium. Maximum response units for each group were averaged and plotted with Excel (Microsoft). For kinetic binding analysis, a minimum of five different concentrations were analyzed in replicate, and the binding constants were estimated using BIAevaluation software and a 1:1 Langmuir binding model (Biacore, GE Healthcare).
GST Pulldown Assays
Pulldown assays were performed in calcium-free HBS buffer. 30 μl of a 50% suspension of glutathione-agarose beads (GE Healthcare) was washed and resuspended in PBST (Sigma), pH 7.4. The beads were incubated with a bacterial lysate containing GST-C2F for 1 h at 4 °C in PBST plus protease inhibitor (PBST + PI) and briefly centrifuged at 950 × g, and the supernatant was discarded. The beads were washed once in cold PBS + PI and once in cold HBS + PI, the latter constituting the binding buffer. To this suspension, 500 μl of the bacterial lysate containing hexahistidine-tagged C2 domains, syntaxin-1, or syntaxin-1 SNARE motif fusion proteins were added and incubated in the presence or absence of calcium. Hexahistidine-tagged protein lysates were prepared in 1× calcium-free HBS buffer containing 1× protease inhibitor mixture. The assay solution was continuously agitated in a rotary mixer for 5 h at 4 °C and then centrifuged at 950 × g, the supernatant was removed, and pellets were washed in 1 ml of cold 1× HBS + PI five times. Finally, pellets were heat-denatured in SDS gel loading dye (Invitrogen), electrophoresed on a 4–20% NuPAGE SDS gel, and transferred by electroblotting onto a PVDF membrane. The interacting target fusion proteins were detected by Western analysis with anti-Xpress antibody (Invitrogen) using a chemiluminescence detection system (GE Healthcare). The blots were reprobed if required by anti-GST antibody (GE Healthcare) to verify equal loading.
Preparation of Otoferlin Antibody
Total RNA was isolated from 129/Sv mouse brain by homogenization in TRIzol (Invitrogen). cDNA was synthesized from total RNA using oligo(dT) as a primer with Superscript II (Invitrogen). Upstream AGGCGGCAGCAGCCAGAG and downstream ACGATCTTCTCTTCGGCCGC primers were utilized to amplify the cDNA with elongase enzyme mixture (Invitrogen). The cDNA extended from the 5′-UTR to nucleotide 5524 (bp 7–5524; GenBankTM accession number AF183184). Nested primers with adaptors were used for expression cloning (upstream, CGGATCCCTCCCCAACTGGAACACCACAG; downstream, ATAGTTTAGCGGCCGCATCTTATCTATGGGCCAGGGCTCTGGTTTT). The PCR product (bp 3815–4276) with adaptors and pGEX were digested with BamHI and NotI, together ligated, and used to transform BL-21 RP competent cells (Stratagene). Transformed cells were induced with isopropyl 1-thio-β-d-galactopyranoside to produce the recombinant GST-mOTOF peptide. This peptide was purified on glutathione-Sepharose 4B (Amersham Biosciences) and cleaved from GST with PreScission protease. A polyclonal antibody against the mOTOF peptide was produced in rabbits (ResGen, Invitrogen) and affinity-purified against the complete recombinant construct GST-mOTOF. The antigenic peptide, 154 amino acids in length, extended from amino acid 1238 to amino acid 1391 (GenBank accession number AAG12990).
Immunoprecipitation
Immunoprecipitation assays were carried out in calcium-free HBS. Rat brain tissue was lysed in HBS buffer containing 0.1% Tween 20 and protease inhibitor mixture (Promega) by brief sonication on ice as described (12). The lysate was centrifuged at 20,828 × g at 4 °C for 10 min. The cleared lysate was preincubated with protein A-agarose (Santa Cruz Biotechnology, Dallas, TX) for 1 h at 4 °C to remove proteins that bind nonspecifically, and the agarose beads were removed by centrifugation at 950 × g at 4 °C and discarded. The lysate containing ∼1 mg of total brain protein was incubated with mouse monoclonal anti-syntaxin-1A (Sigma, S-0664; 1:200) with and without added Ca2+ (100 μm) for 5 h at 4 °C. Rat brain lysate, similarly incubated, first with protein A-agarose beads and then with normal mouse IgG in place of the specific antibody, served as a negative control. After incubation, 30 μl of a 50% fresh suspension of protein A-agarose beads was added, incubated for 30 min at 4 °C, and centrifuged at 950 × g for 3 min, and the supernatant was removed. Pellets were then washed with 1 ml of cold HBS + PI binding buffer five times. 25 μl of 1× gel loading dye (NuPAGE lithium dodecyl sulfate, Invitrogen) was added to the final pellet and boiled for 10 min to release bound proteins. After centrifugation for 5 min at 20,828 × g, the supernatant was collected and loaded on a NuPAGE SDS gel (Invitrogen). Western blot detection was performed with the custom polyclonal antibody raised in rabbit against mouse otoferlin (13) (1:10,000) and donkey anti-rabbit secondary antibody (Santa Cruz Biotechnology; 1:6,000).
Conversely, for immunoprecipitation of otoferlin, a 1:1,000 dilution of the rabbit anti-otoferlin custom antibody was used in a 1-ml reaction containing 1 mg of total protein with 100 μm calcium as described above accompanied by a negative control rabbit IgG immunoprecipitation. For the associated Western detection of syntaxin-1, mouse anti-syntaxin-1A monoclonal antibody (S-0664, Sigma; 1:10,000) was the primary antibody, and HRP-conjugated donkey anti-mouse was the secondary antibody (Santa Cruz Biotechnology; 1:6,000). ECL reagents (GE Healthcare) were used for Western detection.
Proteomic Analysis
For proteomic analysis of the otoferlin interactome, a precleared rat brain lysate (10–25 mg of total protein) was incubated with the custom anti-otoferlin antibody as described above. After incubation and washing, the bound proteins were released, electrophoresed on an SDS-polyacrylamide gel, and stained with SYPRO Ruby according to the manufacturer's instructions (Invitrogen). Portions of the gel were sliced, washed, and digested with trypsin according to established methods (Proteomics Core Facility, Wayne State University). The digested peptides were then separated by reversed-phase chromatography (Magic C18 column, Bruker-Michrom, Auburn, CA), ionized with the ADVANCE ion source (Bruker-Michrom), and introduced into an LTQ-XL mass spectrometer (Thermo Electron, Waltham, MA). Data analysis was carried out using Proteome Discoverer 1.3 software (Thermo Electron), which incorporated a Mascot algorithm (Matrix Science, Boston, MA). The UniProt database was used against rat protein sequences, and secondary analysis was performed using Scaffold software (Proteome Software, Portland, OR). Minimum protein identification probability was set at ≥99% with two unique peptides at ≥95% minimum peptide identification probability.
Lipid Binding by Surface Plasmon Resonance
Purified C2F domain fusion protein was immobilized on a CM5 sensor chip using an amine coupling protocol (11). The reference surface was blocked with ethanolamine and used as control. PIP2 and phosphatidylinositol 3,4,5-trisphosphate (PIP3) diluted in HBS buffer minus Ca2+ were used as analytes. An appropriate amount of calcium or EGTA was added to the analyte solution. Each phospholipid was injected at 0.1 μm in triplicate in five sets of independent experiments. Binding was measured in terms of response units at the end of the association phase in each analysis. Response units were obtained from analyte binding on the ligand surface subtracted from analyte binding on the reference surface. There was little or no binding on the reference surface. Response unit values were averaged and plotted using the Microsoft Excel program. HBS buffer with calcium or EGTA was used as negative control.
Isothermal Titration Calorimetry (ITC)
Purified otoferlin C2F peptides with or without the pachanga mutation (C2Fpga) were dialyzed twice against ITC buffer (50 mm HEPES, pH 7.4, 250 mm NaCl, 5 mm 2-mercaptoethanol, 1× EDTA-free protease inhibitors). The buffer was equilibrated with Chelex-100 beads (Bio-Rad) for 2 h to remove residual calcium ions, the buffer pH was adjusted to 7.4, and the beads were removed by filtration. The ITC experiment was conducted on a Microcal ITC200 (GE Healthcare) at 25 °C with a stirring speed of 300 rpm. The peptide sample (50–100 μm) was introduced via the sample cell, and CaCl2 (5–10 mm) was separately injected in aliquots of 5–9 μl every 5 min over 30–50 injections, representing a total injection volume of 240 μl. To obtain the effective heat of binding, the heat of dilution measured by injecting the calcium chloride solution into the buffer was subtracted. Integrations of heat flow and curve fitting were performed using Microcal Origin software (GE Healthcare). Data were fitted to a two-site binding model.
Western Blot and Immunolocalization of Otoferlin in the Mouse Inner Ear
Cochleas were removed from CD-1, 129SV, or BALB/c mice (7–8 weeks of age) and extracted for protein with radioimmune precipitation assay buffer (30 μl/cochlea), which included a protease inhibitor (Sigma, P8340). The protein extracts were resolved on a Bio-Rad 4–20% SDS-polyacrylamide Ready Gel and transferred to a PVDF membrane. The membrane was blocked with 5% nonfat dry milk in TBS buffer and incubated in primary antibody for 1 h (1:1,000) followed by application of secondary antibody conjugated with horseradish peroxidase. Protein was visualized with the ECL Western blotting system (GE Healthcare).
For one set of immunofluorescence localizations of otoferlin in the mouse inner ear (see Fig. 2B), the bony cochleas were fixed with Bouin's fixative, decalcified, sectioned at 10–20 μm in a Reichert cryostat, freeze-thaw mounted onto glass slides, dried at 56 °C for 1 h, and stored desiccated at −20 °C until use. Otoferlin was detected with an affinity-purified rabbit polyclonal antibody targeting a 133-amino acid segment between C2 domains C2D and C2E (13). For immunofluorescence localization, the anti-otoferlin antibody was diluted at 1:3,000 with 1% goat serum and 0.3% Triton X-100 and incubated for 48 h at 4 °C. Alexa Fluor 546 goat anti-rabbit (Molecular Probes, Invitrogen) was used as the secondary antibody at 1:2,000 for 20 min. The slides were mounted in Vectashield HS DAPI (Vector Laboratories, Burlingame, CA) and visualized using a Zeiss LSM 510 META NLO scanning confocal microscope.
FIGURE 2.

Otoferlin antibody specifically labels hair cells in mouse inner ear and co-localizes with the voltage-gated calcium channel CaV1.3 in cochlear inner hair cells. A, Western blot detection of otoferlin (220–240 kDa) in mouse cochlea (arrow). Positions of standards are labeled. B, confocal immunofluorescence images of the inner ear using an anti-otoferlin antibody (1:1,000; see text) and anti-rabbit secondary antibody conjugated with Alexa Fluor 546 (magenta) (1:6,000) counterstained with DAPI (blue) to label cell nuclei for purposes of orientation. The Zeiss Tile Scan tool was used to create a montage from a 20-μm cryostat section. All inner hair cells in all cochlear turns are labeled by the otoferlin antibody. Otoferlin also labels a subpopulation of hair cells in the vestibular sensory epithelia. utr, utricular macula; sac, saccular macula; ssc, semicircular canal; ihc, inner hair cell. Arrowheads designate inner hair cells. Scale bar, 200 μm. C, mouse organ of Corti double labeled with a custom rabbit polyclonal antibody described in the present study targeting mouse otoferlin sequence (green) and a goat polyclonal antibody targeting mouse CaV1.3 antibody (red). Voltage-gated calcium channel CaV1.3 immunofluorescence (red) is found primarily at basal and apical sites of cochlear inner hair cells (apical turn). Otoferlin immunofluorescence (green) is concentrated at the base of cochlear inner hair cells. The merged image (yellow) indicates sites on the basolateral membrane of cochlear inner hair cells where immunofluorescence of CaV1.3 and otoferlin overlap, consistent with co-localization and results for protein/protein binding. Scale bar, 10 μm.
For a second set of otoferlin immunolocalizations (see Fig. 2C), 2-month-old CBAJ mice of either sex (The Jackson Laboratories, Bar Harbor, ME) were the source of bony cochleas fixed in 4% formaldehyde, 0.1% glutaraldehyde and decalcified with protocols described previously (14). Deparaffinized sections (4–5 μm) were incubated overnight at 4 °C with primary antibody (the custom polyclonal rabbit antibody targeting rat otoferlin diluted 1:1,500 in PBS containing chick serum). Alexa Fluor 488 chick anti-rabbit was used as the secondary antibody at 1:1,000 for 30 min at room temperature. The primary antibody for the L-type voltage-gated calcium channel was an affinity-purified goat polyclonal antibody targeting a C-terminal cytoplasmic domain of human voltage-gated calcium channel CaV1.3 that cross-reacts with the mouse sequence (sc-32072, Santa Cruz Biotechnology) used at 1:400–1:500 dilution overnight at 4 °C. The secondary antibody was Alexa Fluor 568 donkey anti-goat applied at 1:500 for 1 h at room temperature. Immunofluorescence was visualized with an Olympus BX60 microscope and photographed with a PM 20 camera system (Olympus), and the negatives were digitized.
RT-PCR
Rat organ of Corti cDNA (ACI black agouti rats, 4–6 weeks of age) was prepared as described previously (15). cDNA preparations from the apical and basal turns were used as templates in a 50-μl reaction mixture containing 1× Advantage polymerase, 1× PCR buffer (Clontech), 1 μm each of dNTPs, 10 μm forward and reverse primers (supplemental Table 1), and 1 μl of template cDNA. The reaction was carried out as follows: 1 min hold at 95 °C followed by 40 cycles of 30 s at 95 °C, 20 s at 62 °C, and 30 s at 72 °C and completed with a hold at 72 °C for 2 min. The PCR products were gel-purified using a Qiaex DNA gel purification kit (Qiagen), and the sequence was verified (DNA Sequencing Core, Wayne State University).
RESULTS
Otoferlin Splice Variants in Cochlea and Brain
In higher vertebrates, otoferlin is expressed mainly in brain and cochlea, each tissue with different exons coding for the transmembrane domain-containing region (Fig. 1). Exon 47 codes for the brain variant, and exon 48 codes for the cochlear variant (4). Within the transmembrane domain, these two variants share only 53% identity at the amino acid level (supplemental Fig. 1) compared with 98% for the remaining sequence containing the C2 domains.
Immunolocalization of Otoferlin in the Inner Ear
The rabbit custom polyclonal antibody targeting mouse otoferlin amino acid sequence labeled a protein on Western blots with the predicted molecular mass (227 kDa) for otoferlin in mouse cochlea and rat brain (Fig. 2A and supplemental Fig. 2, respectively). That the antibody would cross-react to rat was not unexpected given that rat otoferlin sequence is highly conserved relative to the targeted mouse epitope with 92% identity and 94% similarity. In the mouse inner ear, otoferlin was immunolocalized with this custom antibody primarily to hair cells. Fluorescence with the custom rabbit polyclonal antibody targeting mouse otoferlin sequence shows labeling of inner hair cells of the organ of Corti and hair cells of the utricle, saccule, and semicircular canals (Fig. 2B). This expression pattern is consistent with the observed auditory and vestibular defects in otoferlin-deficient mice (5, 6, 16) that affect hair cell exocytosis. Otoferlin immunoreactivity (Fig. 2C, green) in cochlear inner hair cells was found within the hair cell at both supranuclear and presynaptic sites. Compared with immunofluorescence for the major calcium channel of the hair cell, CaV1.3 (red), overlap (yellow) occurred both at basal presynaptic sites and apical subcuticular sites of the mouse inner hair cells (Fig. 2C). Co-localization at the basal plasma membrane would be anticipated for proteins constituting the hair cell synaptic complex.
Purity and Integrity of the C2 Protein
Fig. 3 shows electrophoresis gels, a circular dichroism (CD) spectrum and light scattering results for the C2F domain protein used in the present studies. Preparations of C2F (Fig. 3A) resulted in essentially pure bands (the faint satellite band probably represents a minor breakdown product). The CD spectrum (Fig. 3B) shows a negative peak around 215 nm reflecting a predominantly β-sheet structure typical of C2 domains (17, 18). The lower two panels in Fig. 3B show representative CD spectra from the literature (17) that are characteristic of β and α structure, respectively. The typical CD spectrum for the α structure clearly differs from that for β with two negative peaks at ∼207 and 222 nm in the α spectrum. Light scattering (Fig. 3C) indicates that the C2 proteins used exist in a non-aggregated state. The upper panel shows a C2F protein peak with a particle diameter of ∼6 nm (peak maximum), corresponding to a monomer with an estimated hydrodynamic molecular mass of 31 kDa and a calculated molecular mass of 25 kDa. In contrast, the lower panel shows light scattering for the same C2F protein intentionally denatured in 1 m urea. The broad peak represents particle size in the range of 1,000 nm, consistent with the presence of aggregates centering at 3,380-kDa molecular mass.
FIGURE 3.
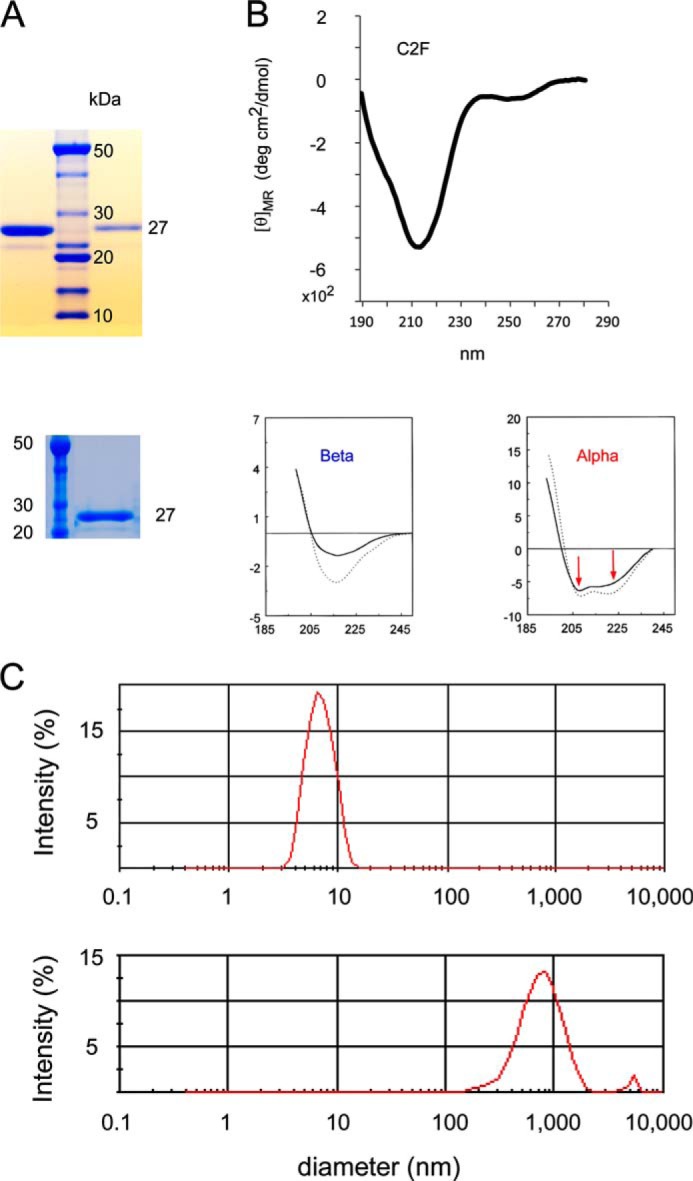
Characterization of otoferlin C2F domain protein by gel electrophoresis, circular dichroism, and light scattering. A, two SDS gels illustrating purity of otoferlin C2F domain of ∼27 kDa used in the present studies (upper gel, first lane, 2 μg of C2F protein; upper gel, third lane, 300 ng of C2F protein; lower gel, second lane, 1.5 μg of C2F protein) compared with molecular mass standards. GST-C2F fusion peptides were cleaved with PreScission protease to remove the GST tag, and the purified C2F domain was dialyzed via a dialysis centrifugation (Amicon) to exchange the buffer and concentrate the sample. An aliquot of each preparation was run in a SDS-polyacrylamide gel, and the protein was stained with SimplyBlueTM protein stain (Invitrogen). The faint satellite band probably represents a minor breakdown product. B, circular dichroism spectrum for purified C2F domain. The single minimum at ∼215 nm is characteristic of the β-sheet in contrast to two minima observed for α-helical proteins (see panels below spectrum from data originally published by Sreerama et al. (17)). Protein concentration was 0.5 μm. deg, degrees. C, C2F domain protein as assessed by light scattering. The upper panel shows the C2F peak with particle diameter of ∼6 nm (peak maximum), corresponding to a monomer with an estimated hydrodynamic molecular mass of 31 kDa and calculated molecular mass of 25 kDa. The plot indicates that the protein is non-aggregating under the solution conditions and sample concentrations used (0.5 μg/μl). As a comparison, the lower panel shows light scattering for the same C2F protein of the upper panel after its denaturation in 1 m urea. The broad peak represents a particle size in the range of 1,000 nm, consistent with the presence of aggregates centering at 3,380-kDa molecular mass.
Otoferlin Is a Calcium Sensor: the C2F Domain Binds Calcium
Isothermal titration calorimetry was utilized in the present investigation to directly characterize the molecular binding of Ca2+ by the otoferlin C2F domain. The raw thermal data plotted after subtracting the heat of dilution shows that the C2F domain binds calcium in an endothermic reaction (Fig. 4A). The raw data were fitted using Microcal Origin software (GE Healthcare) to evaluate binding parameters using a two-calcium binding model. Thermodynamic values for the major site were: enthalpy, ΔH = 3,476 ± 329 cal mol−1; entropy, ΔS = 16.7 cal K−1 mol−1; and KD = 2.67 ± 0.20 × 10−4 m.
FIGURE 4.

ITC measurement of otoferlin C2F domain calcium binding. Affinity-purified wild-type GST-C2F otoferlin protein and a fusion protein representing an otoferlin pachanga mutant (see text) were cleaved with PreScission protease (GE Healthcare) to remove the GST tag. Individual 5-μl volumes of a 5–10 mm CaCl2 solution prepared in ITC buffer were incrementally injected into an ITC chamber (Microcal ITC200) containing a 50–100 μm concentration of the purified C2F domain solution at 25 °C, and the heat change was recorded. Negative controls were either C2F samples injected with ITC buffer alone or ITC buffer injected with 10 mm CaCl2. A, raw data obtained in the titration of C2F domain by calcium. The CaCl2 solution was injected 50 times, and the response was recorded. The binding reaction was endothermic. Fitted data for the integrated heat change on injection of CaCl2 were plotted after subtraction from control values. B, curve fitting for A performed with Microcal Origin software using a two-calcium binding model. One main binding site was predicted with a KD of 267 μm. A second predicted site was in the mm range, which likely reflects nonspecific heat changes. C and D, raw data and corresponding curve fitting, respectively, obtained for the C2F pachanga point mutation (Asp to Gly). Experimental conditions were identical to those for the wild-type C2F. Data indicate a very small endothermic response with little reduction in the titration, suggesting either no binding or low affinity binding. Minimal changes in integrated heat did not allow determination of affinity values. E and F, raw data for control experiments. E, increments of ITC buffer alone (no added CaCl2) added to a 50 μm solution of wild-type C2F. No heat response was associated with the injections. F, ITC buffer alone incrementally injected with ITC buffer containing 10 mm CaCl2. A negligible endothermic response was observed for each injection, probably indicating the heat of calcium dilution.
The purified C2Fpga domain containing the pachanga mutation (D1767G), a missense mutation that causes deafness in mice, was titrated under similar conditions. There was very little thermal change as compared with the C2F. The raw data were analyzed as for C2F and indicated only weak calcium binding (Fig. 4C).
Calcium-dependent Interaction of Otoferlin C2F with Phosphoinositides
Otoferlin has six predicted C2 domains (Fig. 1) that are known to interact with phospholipids (8, 19). In the current study, we focused on the C2F domain of otoferlin that is closest to the C-terminal transmembrane domain and is important for hearing because many mutations in the C2F domain (Table 1) cause hearing loss in humans and mice (Refs. 16 and 20 and Table 1 references). Our aim was to detect and characterize direct binding of various phosphatidylinositols to the otoferlin C2F domain. SPR is suitable for determining protein/lipid interactions (7, 12) in part because lipid preparations tend to form vesicles of 40–100-nm diameter, giving sufficient binding response consistently.
TABLE 1.
Mutations in otoferlin C2 domains that cause hearing loss in humans and mice
Pathogenic, single nucleotide changes that cause missense mutations in the corresponding amino acid sequences are shown. Amino acid numbering is based on human and mouse sequences reported in the corresponding references. It is notable that DFNB9 hearing loss in humans is caused mainly by mutations in the C2C–C2F domains; human pathogenic mutations in the C2A and C2B domains have not been described. For mouse, the listed C2B and C2F domain mutations were induced.
| C2 domain | Pathogenic mutations | Deafness | Organism | Ref. |
|---|---|---|---|---|
| C2A | None | |||
| C2B | I318N | Deaf | Mouse | 63 |
| C2C | P490Q | Deaf | Human | 64 |
| I515T | Deaf | Human | 64 | |
| C2D | L1011P | Deaf | Human | 65 |
| C2E | R1520Q | Deaf | Human | 67 |
| R1607W | Deaf | Human | 67 | |
| C2F | E1733K | Deaf | Human | 68 |
| F1795C | Deaf | Human | 69 | |
| E1804dela | Deaf | Human | 66 | |
| P1852A | Deaf | Human | 20 | |
| R1856Q | Deaf | Human | 68 | |
| D1767G | Deaf | Mouse | 16 |
a Temperature-induced hearing loss.
In the present study, we immobilized affinity-purified C2F domain on a CM5 sensor chip using an amine coupling reaction as described (11). An adjacent flow cell on the CM5 chip was blocked with ethanolamine and served as the reference. Phospholipid binding was measured by subtracting the reference response from the ligand response. Phosphatidylinositols were diluted in calcium-free HBS buffer. For binding analysis, we used 100 nm phosphatidylinositol (PI), phosphatidylinositol 4-phosphate (PIP), PIP2, and PIP3. HBS buffer with or without Ca2+ served as the negative control.
Binding analysis revealed robust binding for PIP2 compared with phosphatidylinositol and PIP3 (Fig. 5, A and B). C2F/phospholipid binding occurred in the absence of calcium (Fig. 5A, lower trace) with almost a 40% increase in binding at 100 μm calcium (Fig. 5A, upper trace) as measured by the maximum binding response. Phosphatidylinositol, PIP, and PIP3 on the other hand showed no significant increase in binding when calcium was raised to 100 μm (Fig. 5B) as ascertained from the average binding in five independent experiments. It is clear from these studies that the C2F domain of otoferlin can bind phosphoinositides in the absence of calcium. However, calcium appears to increase the affinity of C2F for PIP2. These results indicate that the C2F domain is more likely to interact with PIP2 than with other phosphatidylinositols in the presence of calcium.
FIGURE 5.

Otoferlin C2 domain interacts with phosphatidylinositols. A, surface plasmon resonance analysis of otoferlin C2F interaction with PIP2. Purified C2F domain was immobilized on a CM5 sensor chip via an amine coupling reaction (11). 100 nm PIP2 (analyte) was diluted in HBS binding buffer containing no added Ca2+, and the solution was brought to 100 μm Ca2+ before injection. For binding assays lacking Ca2+, the analyte was adjusted to 1 mm EGTA. Samples were injected at a flow rate of 20 μl/min. Flow cells blocked with ethanolamine served as reference cells. Each sample was analyzed in five replicas, and all experiments were performed at least three times. There was approximately a 40% increase in binding for PIP2 in the presence of Ca2+. B, a 100 nm concentration of each of phosphatidylinositol (PI), PIP, PIP2, and PIP3 was analyzed on a C2F-immobilized sensor chip, and the maximum binding response at the end of the association phase (n = 5) was averaged. Error bars denote S.E. Among the different phosphoinositides and phosphatidylinositol, PIP2 showed the most robust binding to the C2F domain followed by phosphatidylinositol, PIP3, and PIP. PIP2 also showed a significantly higher response than the other phosphoinositols in the presence versus absence of 100 μm calcium. Phosphatidylinositol, PIP, and PIP3 did not show any remarkable increase in C2F binding upon addition of calcium. RU, response units.
Otoferlin Synaptic Protein Interactions for Full-length Proteins: Proteomic Analysis
Previously, we demonstrated that cloned otoferlin C2 domains individually interact with the synaptic proteins syntaxin-1 and SNAP-25 (7). Because these cloned C2 domains represent only portions of the otoferlin molecule, albeit active portions, we currently sought to detect these interactions in native tissue homogenate in which full-length proteins are expressed and are expected to be in their native conformation. The otoferlin C2 domains in rat brain and cochlea are 100% identical, and thus, immunoprecipitation experiments for otoferlin in brain serve as a model for hair cell otoferlin protein/protein interactions. Initially, with immunoprecipitation, we examined the interaction of otoferlin and syntaxin-1.
An antibody targeting syntaxin-1 applied to rat brain lysate did elicit immunoprecipitation of otoferlin with no added calcium as detected with the custom anti-otoferlin antibody (Fig. 6A, lane 3, arrow) and in the presence of 100 μm Ca2+ (Fig. 6B, lane 2, arrow). The band migrated above the 210-kDa molecular mass standard, a position comparable with that predicted for full-length otoferlin (227 kDa) (see supplemental Fig. 2). There were no similar bands in negative controls (mouse normal IgG; Fig. 6, A, lane 2, and B, lane 1).
FIGURE 6.

Interaction of full-length otoferlin with syntaxin-1 in immunoprecipitation reactions. A, immunoprecipitation of full-length otoferlin from rat brain lysate with syntaxin-1A antibody (Sigma) and no added calcium. Lane 1, standard; lane 2, mouse normal IgG immunoprecipitation control; lane 3, anti-syntaxin-1A antibody immunoprecipitation. Otoferlin co-immunoprecipitation was detected by Western blot using a custom anti-otoferlin antibody raised in rabbit (see “Experimental Procedures”). Lane 3 shows the otoferlin band at ∼220 kDa (arrow); the lower band represents a nonspecific reaction product that was minimally detectable in Western analysis (see supplemental Fig. 2). B, immunoprecipitation of full-length otoferlin from rat brain lysate with syntaxin-1A antibody in the presence of added calcium. Lane 1, mouse normal IgG control with 100 μm Ca2+; lane 2, immunoprecipitation of otoferlin with syntaxin-1A antibody in 100 μm Ca2+. The arrow indicates the otoferlin band of ∼220 kDa detected with the otoferlin antibody. C, immunoprecipitation of syntaxin-1 by anti-otoferlin antibody. Lane 1, anti-otoferlin antibody immunoprecipitation of syntaxin-1 with 100 μm Ca2+. Syntaxin-1 was detected by anti-syntaxin-1A antibody. In the sample immunoprecipitation (lane 1), a distinct band is visible around 37 kDa (arrow), the estimated molecular mass of intact syntaxin-1. Lane 2, anti-rabbit IgG control immunoprecipitation with 100 μm Ca2+. Additional bands in experimental and control samples represent unblocked IgG subunits.
The reciprocal result was obtained with immunoprecipitation of syntaxin-1: the rabbit anti-otoferlin antibody applied to rat lysate co-immunoprecipitated syntaxin-1 at 35 kDa at 100 μm Ca2+ (Fig. 6C, lane 1). No corresponding band was detected in negative control immunoprecipitation reactions with normal rabbit IgG (Fig. 6C, lane 2).
Given that syntaxin-1 isoforms share a high degree of amino acid identity and that the available antibodies for syntaxin cross-react between isoforms, we sought to identify the specific syntaxin-1 isoform present in an otoferlin protein complex via a proteomics approach utilizing mass spectrometry. For mass spectrometric identification of interacting proteins, we performed immunoprecipitation reactions using rat brain lysate and the custom rabbit antibody targeting otoferlin. We analyzed the products in an SDS-PAGE gel (Fig. 7A, lane 2). Identical experiments in which anti-otoferlin antibody was replaced with normal rabbit IgG served as a negative control (Fig. 7A, lane 1).
FIGURE 7.

Identification of t-SNARE proteins by proteomic analysis of the otoferlin interactome. A, rat brain lysate was used for immunoprecipitation with anti-otoferlin antibody. After washing steps, the co-precipitates were analyzed by SDS-PAGE on a 4–12% gel and stained with SYPRO Ruby (Invitrogen). Gel slices from selected areas of control immunoprecipitation and anti-otoferlin immunoprecipitation (in red boxes) were treated with trypsin and processed for liquid chromatography-mass spectrometry as described under “Experimental Procedures.” Slices 1–5 from the control reaction (lane 1) and slices 11–15 from the corresponding region of the anti-otoferlin reaction (lane 2) were analyzed. Lane 3 is a molecular mass marker (BenchMarkTM, Invitrogen). B, peptides characteristic of syntaxin-1B were detected with otoferlin immunoprecipitation. Five unique peptides with sequences representative of the syntaxin-1B isoform were identified (rows labeled “1B”). The latter are compared with the corresponding regions of syntaxin-1A obtained from the database (rows labeled “1A”). Amino acid alignments for at least four of the peptides show significant differences between the two isoforms, indicating that only syntaxin-1B (not any other syntaxin isoform) was detected even though brain is known to express many syntaxin isoforms, most of them of similar molecular mass. No syntaxin-like sequences were detected in control immunoprecipitations. C, SNAP-25 sequences detected by proteomic analysis of otoferlin interactions. Eight peptides exactly corresponding in sequence to SNAP-25 were detected in the otoferlin immunoprecipitation reactions but were not detected in control immunoprecipitations. D, expression of syntaxin-1B transcripts in the apical and basal turns of the rat cochlear organ of Corti. RT-PCR was carried out to detect expression of syntaxin-1B transcripts in organ of Corti cDNA preparations. Specific primers were designed to amplify syntaxin-1B sequences (see supplemental Table 1), targeting an estimated size of 210 bp. Lane 1 shows molecular mass standards (1 Kb Plus, Invitrogen). Lanes 2 and 3 show RT-PCR analysis of the apical and basal portions, respectively, of the rat organ of Corti. Lane 4 is a negative control (water blank). Strong bands can be seen at the level of the 200-bp standard, which is marked by the arrow. Primer pairs used in these experiments spanned at least one intron, allowing discrimination of mRNA message from genomic DNA. Organ of Corti RNA was treated with DNase before cDNA synthesis.
Because many bands in both the anti-otoferlin and control lanes appeared similar, we sliced the gel in areas where there was a substantial difference between the two samples (lane 1, slices 1–5, negative control, rabbit IgG; lane 2, slices 11–15, anti-otoferlin experiment). These gel slices were then processed and analyzed. A database search for the resulting peptides identified ∼127 proteins of which two were SNARE proteins. We identified the latter proteins as syntaxin-1B and SNAP-25 (Fig. 7, B and C, respectively) at confidence levels above 95%. No corresponding peptides were detected in the negative control experiment. Syntaxin-1B was identified from five unique peptide fragments that differed from the syntaxin-1A isoform by at least one amino acid (Fig. 7B). Syntaxin-1A would have co-precipitated if it had interacted with otoferlin, and consequently, syntaxin-1A if present would have been detected by mass spectrometry. However, no syntaxin-1A peptides were detected. Brain expresses syntaxin-1A and -1B equally, and they are both 35 kDa in mass. The other SNARE protein that we identified was SNAP-25 (Fig. 7C). Eight unique peptides corresponding to SNAP-25 were detected in the otoferlin immunoprecipitation reaction that were not detected in the control IgG immunoprecipitation. This result conclusively showed that otoferlin in its native state and in the presence of calcium at 100 μm interacts with syntaxin-1B and SNAP-25.
Expression of Syntaxin-1B in Rat Organ of Corti
Whether syntaxin-1B is expressed in the mammalian organ of Corti or in cochlear hair cells has not been addressed previously, although syntaxin-1B transcript has been identified in a purified hair cell preparation isolated from the trout saccule.3 For rat organ of Corti, we designed primers to selectively amplify syntaxin-1B cDNA (supplemental Table 1). With RT-PCR, we detected robust amplification of syntaxin-1B cDNA as a single band at 205 bp (Fig. 7D, arrow) from both the apical and basal turns of the rat cochlea (Fig. 7D, lanes 2 and 3, respectively). The primers used spanned an intron, and the cDNAs were DNase-treated, providing confirmation of the absence of genomic DNA amplification. The PCR band was sequence-verified as syntaxin-1B using the NCBI BLAST program.
Otoferlin/SNARE Motif Interaction Is Calcium-dependent
We investigated binding of individual otoferlin C2 domains to the SNARE domain portion of the syntaxin-1 molecule because the SNARE domain is the critical region necessary for SNARE protein interactions. Using GST pulldown assays, we found that C2C, C2D, C2E, and C2F domains interact with the SNARE domain fusion protein as detected by Western blot and anti-Xpress antibody (Fig. 8A, lanes 1, 2, 3, and 4, respectively). The estimated molecular mass of the syntaxin-1 SNARE-domain fusion protein is ∼16 kDa (Fig. 8A, arrow). We selected C2F as a model C2 domain, because of its documented biological importance, to examine calcium-dependent interaction specifically with the SNARE domain in pulldown assays.
FIGURE 8.

Interaction of syntaxin-1 SNARE motif with C2 domains of otoferlin determined by pulldown assay. A, GST pulldown assay of the syntaxin-1 SNARE domain with GST-C2 domain fusion proteins as probes. Lane 1, GST-C2C; lane 2, GST-C2D; lane 3, GST-C2E; lane 4, GST-C2F; lane 5, standard; lane 6, GST alone as a negative control. The SNARE fusion peptide was detected with anti-Xpress antibody (Invitrogen) in a chemiluminescence reaction (Promega). A band is visible around 16 kDa (arrow), the predicted molecular mass of the SNARE domain plus the hexahistidine tag. All C2 domains examined interacted with the SNARE motif construct in the presence of 100 μm calcium in HBS binding buffer. B, the C2F/SNARE domain (syntaxin-1) interaction is Ca2+-dependent. In pulldown reactions, we added increasing quantities of Ca2+ to the binding buffer. After incubation and washing steps, the SNARE fusion peptide was detected in Western blots with anti-Xpress antibody. Calcium concentrations in the samples were 0, 1, 10, 20, 50, 100, and 200 μm for lanes 1–7, respectively. Lane 8 is a negative control (GST). Lane 9 shows molecular mass standards, and lane 10 signifies an aliquot representing ∼5% of the volume of the total SNARE lysate used in each binding assay. Reactions were carried out in HBS buffer after removing Ca2+ by Chelex-100 (see “Experimental Procedures”). There was minimal binding in the absence of Ca2+ (lane 1; 100 μm EGTA buffer with no added Ca2+), and the band intensity increased as Ca2+ increased (lanes 2–7). No band was detected in the GST control (lane 8). C, graph quantitatively showing the intensity of the Western bands for the C2F/SNARE interaction illustrated in B. Band intensities were estimated and averaged with GelQuant.NET software (Biochem Lab Solutions). The averages and S.E. (error bars) were plotted using Excel (Microsoft). At 0 μm Ca2+, there was minimum binding to the SNARE motif, and as the calcium increased to 1 μm, almost a 4-fold increase in band intensity occurred. By 20 μm Ca2+, the band intensity had increased 12-fold. Further increases in calcium concentration to 50 and 100 μm did not result in any significant increase in binding, although at 200 μm, a further increase (∼17-fold) was observed.
Using GST-C2F as a probe, we detected that the SNARE interaction is calcium-dependent (Fig. 8B). There was small residual binding in the absence of calcium (100 μm EGTA in calcium-free HBS buffer; Fig. 8B, lane 1). Addition of 1 μm Ca2+ significantly increased the binding as measured by band intensity (Fig. 8B, lane 2) with a further increase in binding at 10, 20, 50, 100, and 200 μm Ca2+ (Fig. 8B, lanes 3, 4, 5, 6, and 7, respectively). No bands were observed in the negative control, GST (Fig. 8B, lane 8). Fig. 8B, lane 10, shows electrophoresis of 5% of the SNARE lysate used in each reaction. Overall, we found a 4-fold increase in binding intensity when calcium was added at 1 μm and a further 12-fold increase in binding at 20 μm compared with no calcium (Fig. 8C). The interaction did not further increase (significantly) from 20 up to 100 μm Ca2+. However, a further increase in intensity of the syntaxin-1 band (to 17-fold) was observed when calcium was raised to 200 μm (Fig. 8C).
We repeated the binding experiments with another method, SPR analysis, to examine whether the C2 domains could interact with the SNARE domain and whether the interaction was calcium-dependent (Fig. 9). Using purified C2D, C2E, and C2F domains as ligands, we tested binding to the syntaxin-1 SNARE motif (used as analyte) in HBS buffer containing 0–100 μm calcium. Our results showed that C2D, C2E, and C2F domains interact with the SNARE domain in similar ways in the presence of calcium (Fig. 9, A, C, and E, respectively). We also observed clear, but minimal, binding in the absence of calcium (Fig. 9, A, C, and E, 0 Ca2+). We found that the binding increased considerably in the presence of 20 μm Ca2+ (Fig. 9) similarly to the result in pulldown experiments. Interestingly, interaction with the SNARE domain decreased and probably stabilized beyond 20 μm Ca2+, remaining steady thereafter up to 100 μm Ca2+ (Fig. 9, B, D, and F).
FIGURE 9.

Calcium-dependent interaction of otoferlin C2 domains with the syntaxin-1 SNARE motif determined by surface plasmon resonance. A, otoferlin C2D domain (ligand) was immobilized on a CM5 sensor chip (Biacore) by the amine coupling reaction (11). Purified syntaxin-1 SNARE fusion protein was diluted in HBS buffer (analyte), and different amounts of calcium were added (indicated in A, C, and E). Maximum SPR response at the end of the injection (i.e. the end of the association phase) obtained from each binding curve shows the relative binding (ligand binding subtracted from reference binding). Maximum interaction of C2D with the SNARE motif protein was observed at 20 μm Ca2+ (top trace). Binding was reduced when 1 mm EGTA was added to the binding buffer (0 Ca2+). B, binding responses expressed graphically for C2D with the SNARE motif. Bars 1, 2, 3, 4, and 5 represent average maximum binding ±S.E. (error bars) for 0, 10, 20, 50, and 100 μm Ca2+, respectively. Three independent values were averaged for each condition. Bars 1–3 correspond to the plots shown in A. There was a significant increase in binding (∼6.6-fold) at 20 μm Ca2+ compared with 0 μm Ca2+. However, a further increase in calcium did not increase the binding; rather, a decline in binding was observed. C, SPR binding curves in a format similar to that shown in A for purified C2E domain as ligand and purified SNARE fusion protein as analyte. Again, maximum binding occurred when Ca2+ reached 20 μm. D, averaged maximum binding responses for C2E with the SNARE motif. The format is similar to that for B. There was a 5.6-fold increase in binding at 20 μm Ca2+ (bar 3) versus 0 μm Ca2+. E, similar SPR binding curves for C2F as ligand and SNARE fusion protein as analyte. F, similar averaged binding responses obtained for the C2F domain with the SNARE motif. A ∼5-fold increase in binding at 20 μm calcium versus 0 μm calcium was observed.
A comparison of interaction kinetics shows that C2D, C2E, and C2F bind the SNARE motif with rate constants of similar magnitude during the association phase in the presence or absence of calcium (Table 2, ka). Furthermore, the dissociation rates remained similar for C2D, C2E, and C2F when calcium was 0 μm (Table 2, kd). However, the dissociation rate decreased substantially when the calcium increased to 20 μm (Table 2), i.e. the interaction was stronger. These results indicate a mechanism by which calcium stabilizes the interaction between the C2 domains and SNARE motif. The results also suggest that there is a calcium threshold of ∼20 μm for syntaxin-1/SNARE interaction.
TABLE 2.
Otoferlin C2 domain-syntaxin-1 SNARE motif interactions
C2 domains D, E, and F were individually immobilized on a CM5 sensor chip (Biacore, GE Healthcare) and used as the ligands in SPR with a blank flow cell blocked with ethanolamine as reference. Purified syntaxin-1 SNARE motif (analyte) was diluted in HBS buffer containing 0.005% P20 (pH 7.4) for binding analysis. HBS buffer was treated with Chelex-100 to remove residual calcium as described under “Experimental Procedures.” The calcium-free HBS buffer was adjusted to 0 (1 mm EGTA), 10, 20, 50, 100, or 200 μm final calcium along with purified SNARE sample at 200 nm. Rate constants for association and dissociation phases for each interaction were estimated from the SPR sensorgrams using BIAevaluation software (Biacore, GE Healthcare). A Langmuir 1:1 binding model was used in curve fitting. Best fitting values are tabulated below. For the C2D, C2E, and C2F domains, association rate constants remained similar at the different calcium concentrations, whereas the dissociation rate decreased significantly at 20 μm calcium.
| Ligand | ka | kd | KA | KD |
|---|---|---|---|---|
| m−1 s−1 | s−1 | m−1 | m | |
| C2D (0 μm Ca2+) | 1.60 × 105 | 2.60 × 10−3 | 6.03 × 107 | 1.66 × 10−8 |
| C2D (20 μm Ca2+) | 1.38 × 105 | 6.58 × 10−4 | 2.09 × 108 | 4.79 × 10−9 |
| C2E (0 μm Ca2+) | 1.95 × 105 | 2.76 × 10−3 | 7.05 × 107 | 1.42 × 10−8 |
| C2E (20 μm Ca2+) | 1.57 × 105 | 8.64 × 10−4 | 1.82 × 108 | 5.49 × 10−9 |
| C2F (0 μm Ca2+) | 2.08 × 105 | 2.76 × 10−3 | 7.54 × 107 | 1.33 × 10−8 |
| C2F (20 μm Ca2+) | 1.58 × 105 | 9.60 × 10−4 | 1.64 × 108 | 6.10 × 10−9 |
Otoferlin C2 Domain Self-interaction
We conducted experiments to investigate whether otoferlin C2 domains could undergo self (C2/C2)-interactions. Using GST pulldown assays and surface plasmon resonance, we found that the C2 domains indeed interact with each other (Fig. 10, A–E and G–I). We performed GST pulldown assays with GST-C2F fusion protein as a probe and C2A, C2B, C2C, C2D, C2E, and C2F as targets (all expressed as hexahistidine fusion proteins and detected with anti-Xpress antibody). GST-C2F pulled down C2C, C2D, and C2E domains as detected with anti-Xpress (Fig. 10A). Furthermore, the binding was maximal when there was no calcium in the binding buffer (100 μm EGTA in zero calcium HBS buffer) (Fig. 10A). Addition of calcium diminished this interaction. C2F also interacted with C2A and C2B; however, the interaction was independent of Ca2+ (100 μm) (supplemental Fig. 3).
FIGURE 10.
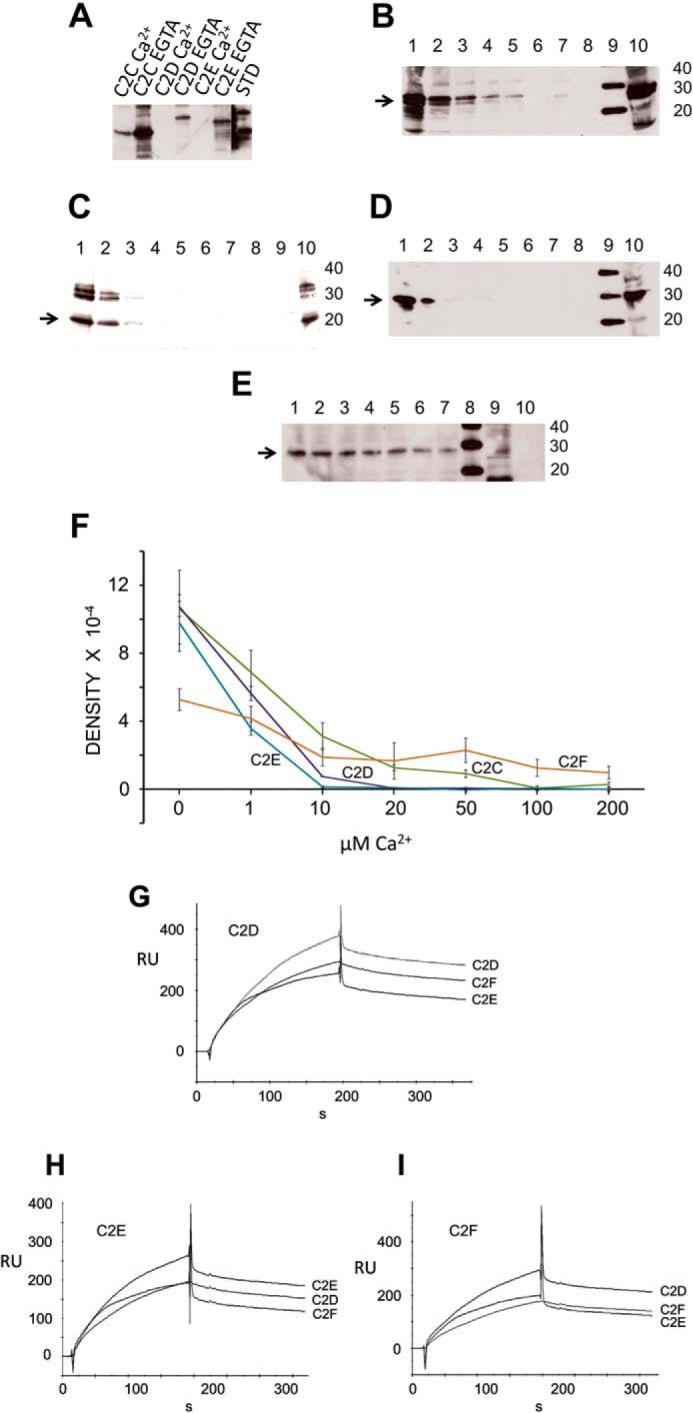
Otoferlin C2 domain self-interaction. A, otoferlin C2 domains interact with one another maximally at low calcium. In a GST pulldown assay utilizing GST-C2F as a probe, we observed that the C2C, C2D, and C2E domains each interacted with the C2F domain in the essential absence of calcium. Addition of 100 μm calcium significantly reduced the interactions. (Individual lanes are labeled. STD, standard.) B, otoferlin C2F/C2C interaction is diminished by calcium. The GST-C2F domain was used as a probe, and His-tagged C2C fusion protein was used as a target in pulldown assays with increasing amounts of calcium. C2C bands were detected by anti-Xpress antibody in a chemiluminescence reaction. The estimated molecular mass of the fusion product is 27 kDa (arrow). Lane 1, 0 μm Ca2+; lane 2, 1 μm Ca2+; lane 3, 10 μm Ca2+; lane 4, 20 μm Ca2+; lane 5, 50 μm Ca2+; lane 6, 100 μm Ca2+; lane 7, 200 μm Ca2+; lane 8, GST negative control; lane 9, molecular mass standard; lane 10, aliquot representing ∼5% of the volume of the total C2C domain lysate used in the binding assay. C, otoferlin C2F/C2D interaction is abolished by increasing calcium. Similarly to experiments in B, pulldown assays were performed in a series with increasing amounts of calcium, and the His-tagged C2D bands were detected in a chemiluminescence reaction using anti-Xpress antibody. The arrow indicates the C2D band (25 kDa). The format is the same as for B. D, otoferlin C2F/C2E interaction is abolished by an increase in calcium. Conditions and labeling are the same as those for B. The arrow indicates the C2E band, estimated to be 29 kDa. E, C2F/C2F interaction is moderately inhibited by calcium. Pulldown assays were performed with a calcium concentration series similar to that shown in B. F, plots of C2/C2 domain interactions. Intensities of the C2 domain bands in the pulldown experiments were estimated by GelQuant.NET (Biochem Lab Solutions), and the averages from three independent gels for each sample were plotted using Excel with calculated S.E. (error bars). Interaction of C2E, C2D, and C2C domains with C2F showed severe inhibition in the range of 1–10 μm Ca2+ with only small to negligible interaction beyond 10 μm calcium. A relatively protracted decline of interaction with an increase in calcium occurred for the C2F/C2F self-interaction. G, C2D interaction with C2D, C2E, and C2F by SPR. Purified C2D domain was immobilized on a CM5 sensor chip and used as ligand. Purified C2D, C2F, and C2E fusion peptides were diluted in calcium-free HSB buffer and used as analytes. H, C2E ligand interaction with C2E, C2D, and C2F as analytes performed in a manner similar to that shown for G. I, C2F ligand interaction with C2D, C2F, and C2E as analytes. RU, response units.
To better understand details of the C2 self-interaction behavior, we performed pulldown experiments at a series of calcium concentrations (0–200 μm Ca2+) (Fig. 10). C2A and C2B were not tested because our initial results indicated that their interactions are independent of calcium (supplemental Fig. 3). C2C, C2D, C2E, and C2F showed diminished binding with an increase in calcium concentration (Fig. 10, B–E). However, there was an observable difference in the calcium sensitivity of the individual interactions. C2C showed binding in the presence of Ca2+ up to 50 μm (Fig. 10B), whereas C2D and C2E showed no binding to the C2F domain beyond 10 μm Ca2+ (Fig. 10, C and D, respectively). C2F/C2F interaction on the other hand showed only mild inhibition in the presence of calcium (Fig. 10E). Overall, data averaged from independent pulldown assays showed a downward trend in binding as the calcium concentration increased in the binding buffer (Fig. 10F).
SPR binding experiments where C2D, C2E, and C2F domains were used as ligands and the same domains, respectively, were used as analytes showed interaction of these C2 domains in the absence of calcium (Fig. 10, G–I). A similar binding pattern could be observed in the sensorgrams for all C2 domains tested, suggesting a common mechanism of self-interaction among these domains.
DISCUSSION
Otoferlin is a C2 domain-containing protein that causes non-syndromic deafness in humans (3) and is crucial for hair cell exocytosis (5). In the inner ear, otoferlin is expressed predominantly in hair cells as detected by anti-otoferlin antibody reactivity (Fig. 2, B and C), overlapping the immunoreactivity for the presynaptic voltage-gated calcium channel CaV1.3 in cochlear inner hair cells (Fig. 2C). Otoferlin-interacting partners have been identified in the literature, including Rab8b GTPase (21), myosin VI (22, 23), CaV1.3 (7), and ERGIC-2 (24) in addition to the SNARE proteins syntaxin-1A and SNAP-25 (5, 7, 8), suggesting diverse roles in synaptic vesicle fusion, endocytosis, and regeneration.
The otoferlin protein/protein interactions may point to multiple functions of this protein. Membrane capacitance measurements show that exocytosis is otoferlin-dependent in auditory (5) and vestibular hair cells (6). However, vesicle pool and ribbon synapses appear normal in otoferlin-deficient mice (5), implying no obvious structural deficits. It is yet unclear exactly how otoferlin directs and regulates calcium-dependent vesicle fusion in hair cells. In the present study, we examined some of the molecular features of otoferlin that may contribute to regulation of hair cell exocytosis/receptoneural secretion.
C2 domains are modules for molecular interaction present in a diverse group of proteins that bind calcium. These domains interact with phospholipids as well as with proteins, modulating membrane-associated processes (25). Despite their general functional and structural similarity, C2 domains are varied in their amino acid sequences. Heterogeneity exists among the C2 domains for calcium binding and for their role as calcium sensors in cellular activity. For example, rat synaptotagmin-4 does not bind calcium (26) and inhibits SNARE-mediated membrane fusion (27) unlike the synaptotagmin-4 homologues in Drosophila that stimulate membrane fusion. Similarly, calcium binding properties vary between C2 domains within the same protein. For example, C2A and C2B domains of synaptotagmin-1 have different affinities for calcium, binding three and two calcium ions, respectively (28). In otoferlin, the C2A domain does not bind calcium (29) unlike the C2D (5), C2F (7), and other C2 domains (8).
C2 domain-containing proteins modulate membrane fusion largely due to their calcium-enhanced phospholipid binding. Membrane fusion during neuronal synaptic vesicle release is triggered by PIP2 interaction with calcium-bound synaptotagmin-1 that simultaneously mediates calcium-dependent SNARE interaction, activating SNARE-driven membrane apposition and membrane fusion (30). Thus, neuronal evoked exocytosis is SNARE-driven, rapid, synchronized, and calcium-dependent (1). Likewise, hair cell exocytosis has been demonstrated to be fast, synchronous, and calcium-dependent (5). However, whereas the basic machinery of vesicle fusion is similar in different fast synapses, the modulation of vesicle fusion in these synapses appears to differ (31).
As stated previously, hair cell calcium-dependent exocytosis is otoferlin-dependent (5). There are six predicted C2 domains in otoferlin, and at least four are assumed to bind calcium. Although otoferlin is expressed in brain, brain otoferlin is alternatively spliced to contain a transmembrane domain (coded by exon 47; Fig. 1) that is different from that of cochlear hair cell otoferlin (coded by exon 48; Fig. 1). Interestingly, brain synaptic vesicles apparently do not contain otoferlin (32), whereas mass spectroscopic analysis has identified otoferlin in synaptosomal fractions from chick cochleas, which would include hair cell synaptic vesicles (33). Immunofluorescence for otoferlin partially overlaps immunofluorescence for ribeye, the major protein component of the hair cell synaptic body in the ribbon synapse (33, 34). Immunogold labeling of otoferlin is circumferential with respect to the ribbon synaptic bodies (5) and corresponds to the predicted localization of synaptic vesicles. Because otoferlin deficiency affects exocytosis in the hair cell and not in brain, it is reasonable to argue that these two splice variants are targeted to different cellular membrane compartments due to the differences in their transmembrane domains.
The acidic phospholipid PIP2 is crucial for neuronal exocytosis (35) and is enriched in nanodomains of the presynaptic plasma membrane (36). PIP2 is also localized to the plasma membranes of hair cells (37). Otoferlin C2 domains have been found to interact with phosphatidylserine, phosphatidylcholine, and liposome preparations containing these phospholipids (38). In the current work, we found that the C2F domain particularly interacted with PIP2 and that this interaction was enhanced 40% upon addition of calcium to the binding buffer (Fig. 5). PIP and PIP3 did not bind as robustly as did PIP2 and did not show any significant increase in binding upon addition of calcium (Fig. 5). Calcium-dependent and -independent interactions of different affinity have been observed for synaptotagmin-1/PIP2 interaction (39). Interaction between synaptotagmin-1 and PIP2 via positively charged synaptotagmin lysine residues is an important step in neuronal vesicle fusion (40), and it has been shown that PIP2 enhances calcium binding by synaptotagmin-1 (28, 41, 42).
Light scattering results indicate that under the conditions of the present investigation otoferlin C2 domain proteins are properly folded and non-aggregating (Fig. 3), suggesting that the present binding studies are consistent with native configurations. Previous work has suggested that otoferlin C2 domains bind calcium, measured as a calcium-induced increase in intrinsic fluorescence (5, 7, 8). Circular dichroism measurements do not change significantly with addition of calcium (43), probably reflecting minimal changes in the secondary structures of C2 domains as also measured by NMR (44). In the current study, we also used isothermal titration calorimetry to directly ascertain calcium binding by otoferlin C2F. A D1767G mutation in the C2F domain (pachanga; Table 1) was found to nearly abolish calcium ion binding (Fig. 4). Synaptotagmin-1 C2 domains generally bind calcium with a KD in the range of 200 μm (28), which is similar in magnitude to that for otoferlin C2F. Thus, our experiments present further proof of the calcium binding nature of otoferlin, providing support for otoferlin acting as a calcium sensor.
ITC data for the wild-type otoferlin C2F domain indicate a possible two-calcium binding nature with one high affinity site (267 μm Ca2+) and one low affinity site (6–22 mm Ca2+). Many factors influence C2 domain calcium binding, including temperature, lipid environment, ionic strength, and pH, factors not considered in this study. Overall, our ITC results are consistent with the present observations of calcium-dependent protein/protein interaction of the C2F domain. The pachanga mutation in C2F (Asp to Gly) showed little Ca2+ binding. This mutation, originally created in mouse by N-ethyl-N-nitrosourea exposure (16), produces DFNB9 deafness with a profound hearing loss. Pachanga shows evoked exocytosis but with reduced amplitude compared with the wild type (10). The ribbon synapse of pachanga inner hair cells appears normal, but with a reduced number of synaptic vesicles. Because calcium is a factor in vesicle movement and the pachanga mutation appears to have deficits in vesicle recycling, exploring the calcium binding nature of the otoferlin C2F domain in this mutant is relevant to examining how calcium and otoferlin mediate and modulate hair cell exocytosis.
SNARE proteins such as syntaxin-1, SNAP-25, and synaptobrevin-1/2 have been reported to be expressed in cochlear hair cells of the guinea pig (45) and chick (33). Although the exact involvement of SNARE proteins in hair cell exocytosis has been the subject of controversy (46), the presence of SNAREs speaks to synaptic functionality. And otoferlin C2 domains, collectively (5) or individually (7, 8), have been found to interact with two SNARE proteins, syntaxin-1 and SNAP-25.
A database search of peptides obtained from mass spectrometric analysis showed that syntaxin-1B and SNAP-25 were the only SNARE proteins that co-immunoprecipitated with otoferlin in our studies where anti-otoferlin antibody was applied to a homogenate of rat brain. The finding of syntaxin-1B transcript in rat organ of Corti cDNA preparations by RT-PCR was consistent with the previous determination of syntaxin-1B mRNA expression in a teleost saccular hair cell preparation (47), furthering the likelihood that it is the syntaxin-1B isoform that interacts with otoferlin in hair cell exocytosis. We (7) and others (8) have previously determined that syntaxin-1 binds to cloned, individual C2 domains of otoferlin without distinguishing between syntaxin-1A and -1B.
The full sequence of the syntaxin-1B isoform is 84% identical to the full sequence of syntaxin-1A at the amino acid level with 94% identity within the SNARE motif. Generally, syntaxin-1A is thought to mediate SNARE-driven exocytosis in neuronal cells (48). A syntaxin-1A knock-out mouse, which still expresses syntaxin-1B, exhibited normal physiology and development (49) with no change in fast synaptic transmission in brain; however, this mouse exhibited changes in retinal layer organization (50). In contrast, a syntaxin-1B knock-out mouse showed severe reduction in evoked fast exocytosis and did not survive past 2 postnatal weeks (51). These findings indicate a functional diversity between the two syntaxin-1 isoforms, probably originating from differential targeting or modulation by other proteins.
Syntaxin and SNAP-25 contain motifs that are crucial for SNARE complex formation and calcium-dependent modulation (48). Binding of neuronal synaptotagmin-1 and complexin to the SNARE complex regulates SNARE protein interaction and controls synchronized, calcium-dependent fast exocytosis. In the present investigation, we studied the interaction of otoferlin specifically with the syntaxin-1 SNARE motif. We found that C2D, C2E, and C2F domains of otoferlin interact with the SNARE motif in pulldown assays and SPR analysis (Fig. 9). Moreover, these interactions were found to be calcium-dependent with a similar binding profile across different calcium concentrations. In pulldown assays, we observed small, yet detectable, interactions of the SNARE motif in the absence of calcium. As the calcium increased to 1 μm, there was a significant increase (∼4-fold) in binding, which rose further at 20 μm calcium (∼12-fold). However, the strength of binding did not change above 50 μm calcium and remained almost constant until calcium attained 200 μm (Fig. 8).
Kinetic measurements of SNARE motif binding show that C2D, C2E, and C2F domains bind with similar association rates irrespective of the calcium concentration, whereas a significant decrease in dissociation rate occurs when the calcium reaches 20 μm (Table 2). We attribute this increase in the observed maximum binding of the SNARE motif to stabilization of the interaction at the higher calcium. These results provide further evidence that otoferlin is capable of calcium-dependent binding, particularly to the SNARE motif, and suggest a function in modulation of vesicle fusion in hair cells. The levels of calcium required for maximum binding are in agreement with the 20–30 μm range required for efficient exocytosis in inner hair cells (52) and in immature outer hair cells (9).
In addition to interacting with other proteins, C2 domains are known to interact with each other. Synaptotagmin-1 C2 domains (53, 54) as well as dysferlin C2 domains (55) have been found to interact in this way. In the current study, we observed that otoferlin C2 domains exhibit C2/C2 domain interactions (Fig. 10, A–E), potentially causing the protein to fold into a closed tertiary structural configuration (Fig. 11). However, in contrast to synaptotagmin C2/C2 interactions, which optimally require 100 μm Ca2+ (54), and dysferlin C2/C2 interactions, which are Ca2+-independent (55), otoferlin C2/C2 domain interactions are inhibited by calcium beginning at low micromolar calcium concentrations (Fig. 10F). In pulldown experiments involving the GST-C2F domain and His-tagged C2C, C2D, C2E, and C2F domains, we consistently found that calcium increasingly inhibited the interactions at 1–200 μm (Fig. 10, B–E) but less severely for the C2F/C2F interaction. Both C2A and C2B bound C2F in a calcium-independent manner (supplemental Fig. 3). C2/C2 domain interaction was further confirmed by SPR binding analysis using C2D, C2E, and C2F as ligands and as analytes (Fig. 10, G–I).
FIGURE 11.
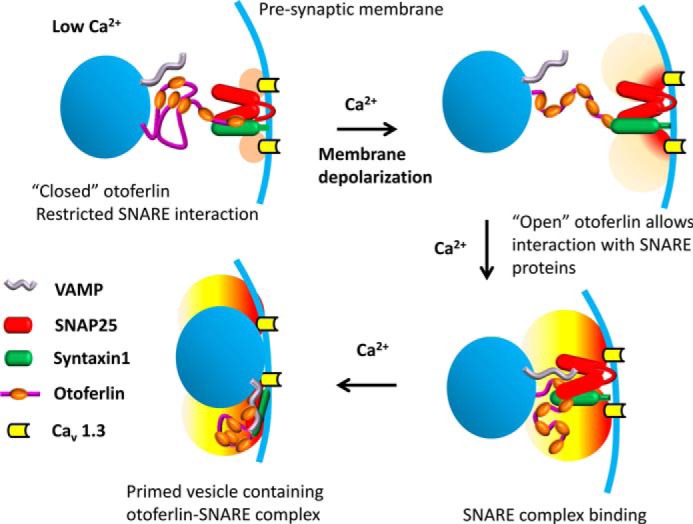
Model illustrating calcium regulation of otoferlin/SNARE interaction in the hair cell. It has been shown that otoferlin C2 domains interact with each other maximally in the absence of calcium, and when calcium levels increase beyond 10 μm, the interaction is reduced significantly (Fig. 10) except for domains C2A and C2B, which remain unaffected (supplemental Fig. 3). Based upon these observations, it is proposed that otoferlin may undergo a conformational shift from closed (top left) to open (top right) during a presynaptic increase in calcium. Closed otoferlin may be restricted in its interaction with other proteins, especially the C2C, C2D, C2E, and C2F interactions with t-SNARE proteins. As calcium increases, the opened otoferlin interacts with t-SNAREs (bottom right), facilitating vesicle-presynaptic membrane fusion (bottom left) (vesicles, blue spheres; presynaptic membrane, blue curve). The “halo” near the calcium channels indicates the calcium level with light orange suggesting low calcium (upper left), light red showing higher calcium entering (upper right), and yellow indicating the development of an intermediate calcium level attained with continuing calcium entry (lower diagrams). Consistent with this model, the predepolarization calcium concentration at the inner hair cell ribbon synapse is estimated to be ∼10 μm or less (56). There is little C2F interaction with the other C2 domains observed above 10 μm calcium (Fig. 10F). On the other hand, C2D, C2E, and C2F interactions with syntaxin-1 t-SNARE motif are stronger above 10 μm calcium, e.g. at 20 μm calcium (Table 2; KD = ∼10−9). The otoferlin/SNARE interaction at low calcium concentrations may correspond to baseline levels of exocytosis. VAMP, vesicle-associated membrane protein.
The differences in calcium dependence of otoferlin with respect to its intramolecular C2/C2 association and interaction with SNARE proteins may relate to calcium-driven folding and unfolding. Otoferlin C2 molecular interactions with SNARE proteins may be restricted at low calcium concentrations and enhanced by calcium influx. The calcium concentration in hair cells is thought to be regulated by depolarizations that activate clusters of CaV1.3 channels at the ribbon synapse (56) as well as by active buffering through calcium-binding proteins that capture free calcium ions rapidly, probably maintaining a constant calcium concentration within the calcium microdomain (57, 58). Thus, calcium concentration rises and falls within hundreds of milliseconds with an average free calcium level reaching 3 μm in the calcium microdomains (56) and ∼85 μm at calcium “hot spots” (59). It has been suggested that only a few CaV1.3 channels at the calcium nanodomains may be sufficient for effective exocytosis (60), indicating that even a submicromolar change in free calcium within the nanodomain may signal secretion.
Interestingly, a low range of Ca2+ concentration, 10–25 μm, is effective for normal neuronal neurotransmitter release (61). Cochlear inner hair cells operate within a similar range (7–40 μm Ca2+), exhibiting rapid, calcium-dependent exocytosis. A study that used changes in membrane capacitance in response to photorelease of caged calcium as a measure of afferent exocytosis (52) showed similar values of membrane capacitance within the range of 7–40 μm Ca2+ with a decrease in the time necessary to attain peak release with increasing calcium.
The picture that emerges is that the C2 domains of otoferlin act as molecular interaction modules directed by the level of free calcium. We found that C2 domains clearly interacted with SNARE motifs when the calcium concentration was only 1 μm, representing an interaction ∼4-fold higher than the small interaction at nominally zero calcium concentration (Fig. 8, B and C). There was also a corresponding decrease in the C2/C2 domain interactions starting at 1 μm Ca2+ and continuing with increasing calcium concentration (Fig. 10F). The interaction of otoferlin with SNAREs during stimulation at the periphery of the calcium microdomains where calcium is at the lower μm concentrations is expected to be initially restricted and limiting to the probability of calcium-evoked exocytosis in that region. Because C2 domain self-interactions and SNARE interactions show subtle variability with respect to calcium effect on binding, it is tempting to suggest that the different otoferlin C2 domains may interact differentially as the calcium level changes, enhancing the functional complexity and range of calcium dependence of exocytosis.
For example, the C2F/C2D and C2F/C2E interactions were strongly inhibited by calcium (Fig. 10, C and D), whereas the C2F/C2F interaction was only moderately inhibited (Fig. 10E). Thus, at the beginning of the stimulus when the ribbon synapse calcium concentration is at low micromolar levels (10 μm), the C2D or C2E domains may bind the SNARE motif more strongly relative to C2F or C2C. As the calcium concentration increases to levels above 10 μm within the submillisecond time span, C2C and C2F would also mediate exocytosis. It is possible that some of the C2 domains could act cooperatively as is the case for synaptotagmin-1 (62). Further studies are required to understand how otoferlin may fold and unfold at different calcium concentrations. A combination of cooperativity among the C2 domains and intermolecular interactions may occur, dictated by the prevailing calcium concentration.
In conclusion, our results support the role of otoferlin as a hair cell calcium sensor that preferentially interacts as a full-length, native protein with syntaxin-1B and SNAP-25, two proteins essential for fast exocytosis. The evidence also suggests that otoferlin can engage in two modes of molecular interaction (Fig. 11). With extremely low concentrations of calcium, C2A and C2B domains would interact with syntaxin-1B and SNAP-25 and with PIP2. Under these conditions, the C2D, C2E, and C2F domains would interact with each other. Then, with influx of calcium, otoferlin C2C, C2D, C2E, and C2F would separate and bind the syntaxin-1 SNARE domain with an optimal association at 20 μm calcium in the initial stages of vesicle exocytosis followed by other SNARE interactions. Thus, overall, our findings suggest a complex operative mechanism involving otoferlin, SNARE proteins, phospholipids, and most importantly calcium in the process of hair cell exocytosis.
Supplementary Material
Acknowledgments
We thank Dr. Stanley Terlecky, Wayne State University, for use of the Biacore 3000 SPR instrument; Dr. Raymond Trievel, University of Michigan, Ann Arbor, for isothermal calorimetric measurements; and Dr. Paul Stemmer, Wayne State University, for discussions regarding mass spectrometric protein identification.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DC000156 (to D. G. D.), R01 DC004076 (to M. J. D.), and R01 DC006907 (to B. J. M.). This work was also supported by the Hearing Health Foundation (to N. A. R.) and the Deafness Research Foundation (to N. A. R. and P. M. K.).

This article contains supplemental Figs. 1–3 and Table 1.
N. A. Ramakrishnan and D. G. Drescher, unpublished results.
- C2 domain
- protein structural domain for calcium binding and targeting proteins to cell membranes
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- NSF
- N-ethylmaleimide-sensitive fusion protein
- SPR
- surface plasmon resonance
- ITC
- isothermal titration calorimetry
- CaV1.3
- voltage-gated calcium channel α subunit isoform 1.3
- t-SNARE
- target SNARE
- v-SNARE
- vesicle SNARE
- HBS
- HEPES-buffered saline
- OTOF
- otoferlin
- mOTOF
- mouse OTOF
- PIP3
- phosphatidylinositol 3,4,5-trisphosphate
- PI
- protease inhibitor
- PIP
- phosphatidylinositol 4-phosphate.
REFERENCES
- 1. Jahn R., Fasshauer D. (2012) Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stein A., Radhakrishnan A., Riedel D., Fasshauer D., Jahn R. (2007) Synaptotagmin activates membrane fusion through a Ca2+-dependent trans-interaction with phospholipids. Nat. Struct. Mol. Biol. 14, 904–911 [DOI] [PubMed] [Google Scholar]
- 3. Yasunaga S., Grati M., Cohen-Salmon M., El-Amraoui A., Mustapha M., Salem N., El-Zir E., Loiselet J., Petit C. (1999) A mutation in OTOF, encoding otoferlin, a Fer-1-like protein, causes DFNB9, a non-syndromic form of deafness. Nat. Genet. 21, 363–369 [DOI] [PubMed] [Google Scholar]
- 4. Yasunaga S., Grati M., Chardenoux S., Smith T. N., Friedman T. B., Lalwani A. K., Wilcox E. R., Petit C. (2000) OTOF encodes multiple long and short isoforms: genetic evidence that the long ones underlie recessive deafness DFNB9. Am. J. Hum. Genet. 67, 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roux I., Safieddine S., Nouvian R., Grati M., Simmler M. C., Bahloul A., Perfettini I., Le Gall M., Rostaing P., Hamard G., Triller A., Avan P., Moser T., Petit C. (2006) Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell 127, 277–289 [DOI] [PubMed] [Google Scholar]
- 6. Dulon D., Safieddine S., Jones S. M., Petit C. (2009) Otoferlin is critical for a highly sensitive and linear calcium-dependent exocytosis at vestibular hair cell ribbon synapses. J. Neurosci. 29, 10474–10487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ramakrishnan N. A., Drescher M. J., Drescher D. G. (2009) Direct interaction of otoferlin with syntaxin 1A, SNAP-25, and the L-type voltage-gated calcium channel CaV1.3. J. Biol. Chem. 284, 1364–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johnson C. P., Chapman E. R. (2010) Otoferlin is a calcium sensor that directly regulates SNARE-mediated membrane fusion. J. Cell Biol. 191, 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beurg M., Safieddine S., Roux I., Bouleau Y., Petit C., Dulon D. (2008) Calcium- and otoferlin-dependent exocytosis by immature outer hair cells. J. Neurosci. 28, 1798–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pangrsic T., Lasarow L., Reuter K., Takago H., Schwander M., Riedel D., Frank T., Tarantino L. M., Bailey J. S., Strenzke N., Brose N., Müller U., Reisinger E., Moser T. (2010) Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat. Neurosci. 13, 869–876 [DOI] [PubMed] [Google Scholar]
- 11. Drescher D. G., Ramakrishnan N. A., Drescher M. J. (2009) Surface plasmon resonance (SPR) analysis of binding interactions of proteins in inner-ear sensory epithelia. Methods Mol. Biol. 493, 323–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramakrishnan N. A., Drescher M. J., Khan K. M., Hatfield J. S., Drescher D. G. (2012) HCN1 and HCN2 proteins are expressed in cochlear hair cells: HCN1 can form a ternary complex with protocadherin 15 CD3 and F-actin-binding filamin A or can interact with HCN2. J. Biol. Chem. 287, 37628–37646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kelley P. M., Morley B. J. (2006) Immunolocalization of otoferlin in mouse cochlear and vestibular hair cells. Abstr. Soc. Neurosci. 32, 140.4 [Google Scholar]
- 14. Ramakrishnan N. A., Drescher M. J., Barretto R. L., Beisel K. W., Hatfield J. S., Drescher D. G. (2009) Calcium-dependent binding of HCN1 channel protein to hair cell stereociliary tip link protein protocadherin 15 CD3. J. Biol. Chem. 284, 3227–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oh C. K., Drescher M. J., Hatfield J. S., Drescher D. G. (1999) Selective expression of serotonin receptor transcripts in the mammalian cochlea and its subdivisions. Brain Res. Mol. Brain Res. 70, 135–140 [DOI] [PubMed] [Google Scholar]
- 16. Schwander M., Sczaniecka A., Grillet N., Bailey J. S., Avenarius M., Najmabadi H., Steffy B. M., Federe G. C., Lagler E. A., Banan R., Hice R., Grabowski-Boase L., Keithley E. M., Ryan A. F., Housley G. D., Wiltshire T., Smith R. J., Tarantino L. M., Müller U. (2007) A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J. Neurosci. 27, 2163–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sreerama N., Venyaminov S. Y., Woody R. W. (1999) Estimation of the number of α-helical and β-strand segments in proteins using circular dichroism spectroscopy. Protein Sci. 8, 370–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sutton R. B., Davletov B. A., Berghuis A. M., Südhof T. C., Sprang S. R. (1995) Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 80, 929–938 [DOI] [PubMed] [Google Scholar]
- 19. Ramakrishnan N. A., Drescher M. J., Drescher D. G. (2010) Direct interaction of phosphatidylinositol 4,5-bisphosphate with otoferlin C2F domain. Assoc. Res. Otolaryngol. Abstr. 33, 31 [Google Scholar]
- 20. Migliosi V., Modamio-Høybjør S., Moreno-Pelayo M. A., Rodríguez-Ballesteros M., Villamar M., Tellería D., Menéndez I., Moreno F., Del Castillo I. (2002) Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. J. Med. Genet. 39, 502–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heidrych P., Zimmermann U., Bress A., Pusch C. M., Ruth P., Pfister M., Knipper M., Blin N. (2008) Rab8b GTPase, a protein transport regulator, is an interacting partner of otoferlin, defective in a human autosomal recessive deafness form. Hum. Mol. Genet. 17, 3814–3821 [DOI] [PubMed] [Google Scholar]
- 22. Heidrych P., Zimmermann U., Kuhn S., Franz C., Engel J., Duncker S. V., Hirt B., Pusch C. M., Ruth P., Pfister M., Marcotti W., Blin N., Knipper M. (2009) Otoferlin interacts with myosin VI: implications for maintenance of the basolateral synaptic structure of the inner hair cell. Hum. Mol. Genet. 18, 2779–2790 [DOI] [PubMed] [Google Scholar]
- 23. Roux I., Hosie S., Johnson S. L., Bahloul A., Cayet N., Nouaille S., Kros C. J., Petit C., Safieddine S. (2009) Myosin VI is required for the proper maturation and function of inner hair cell ribbon synapses. Hum. Mol. Genet. 18, 4615–4628 [DOI] [PubMed] [Google Scholar]
- 24. Żak M., Bress A., Brandt N., Franz C., Ruth P., Pfister M., Knipper M., Blin N. (2012) Ergic2, a brain specific interacting partner of otoferlin. Cell. Physiol. Biochem. 29, 941–948 [DOI] [PubMed] [Google Scholar]
- 25. Nalefski E. A., Falke J. J. (1996) The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 5, 2375–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dai H., Shin O. H., Machius M., Tomchick D. R., Südhof T. C., Rizo J. (2004) Structural basis for the evolutionary inactivation of Ca2+ binding to synaptotagmin 4. Nat. Struct. Mol. Biol. 11, 844–849 [DOI] [PubMed] [Google Scholar]
- 27. Wang Z., Chapman E. (2010) Rat and Drosophila synaptotagmin 4 have opposite effects during SNARE-catalyzed membrane fusion. J. Biol. Chem. 285, 30759–30766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Radhakrishnan A., Stein A., Jahn R., Fasshauer D. (2009) The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 284, 25749–25760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helfmann S., Neumann P., Tittmann K., Moser T., Ficner R., Reisinger E. (2011) The crystal structure of the C2A domain of otoferlin reveals an unconventional top loop region. J. Mol. Biol. 406, 479–490 [DOI] [PubMed] [Google Scholar]
- 30. Südhof T. C., Rizo J. (2011) Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 3, a005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ramakrishnan N. A., Drescher M. J., Drescher D. G. (2012) The SNARE complex in neuronal and sensory cells. Mol. Cell. Neurosci. 50, 58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takamori S., Holt M., Stenius K., Lemke E. A., Grønborg M., Riedel D., Urlaub H., Schenck S., Brügger B., Ringler P., Müller S. A., Rammner B., Gräter F., Hub J. S., De Groot B. L., Mieskes G., Moriyama Y., Klingauf J., Grubmüller H., Heuser J., Wieland F., Jahn R. (2006) Molecular anatomy of a trafficking organelle. Cell 127, 831–846 [DOI] [PubMed] [Google Scholar]
- 33. Uthaiah R. C., Hudspeth A. J. (2010) Molecular anatomy of the hair cell's ribbon synapse. J. Neurosci. 30, 12387–12399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schug N., Braig C., Zimmermann U., Engel J., Winter H., Ruth P., Blin N., Pfister M., Kalbacher H., Knipper M. (2006) Differential expression of otoferlin in brain, vestibular system, immature and mature cochlea of the rat. Eur. J. Neurosci. 24, 3372–3380 [DOI] [PubMed] [Google Scholar]
- 35. Aoyagi K., Sugaya T., Umeda M., Yamamoto S., Terakawa S., Takahashi M. (2005) The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J. Biol. Chem. 280, 17346–17352 [DOI] [PubMed] [Google Scholar]
- 36. van den Bogaart G., Meyenberg K., Risselada H. J., Amin H., Willig K. I., Hubrich B. E., Dier M., Hell S. W., Grubmüller H., Diederichsen U., Jahn R. (2011) Membrane protein sequestering by ionic protein-lipid interactions. Nature 479, 552–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirono M., Denis C. S., Richardson G. P., Gillespie P. G. (2004) Hair cells require phosphatidylinositol 4,5-bisphosphate for mechanical transduction and adaptation. Neuron 44, 309–320 [DOI] [PubMed] [Google Scholar]
- 38. Ramakrishnan N. A., Morley B. J., Drescher M. J., Kelley P. M., Drescher D. G. (2009) Secondary structure of otoferlin reflects calcium-dependent and -independent functions at the hair-cell synaptic complex. Assoc. Res. Otolaryngol. Abstr. 32, 16 [Google Scholar]
- 39. Kuo W., Herrick D. Z., Ellena J. F., Cafiso D. S. (2009) The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: two modes of C2B binding. J. Mol. Biol. 387, 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuo W., Herrick D. Z., Cafiso D. S. (2011) Phosphatidylinositol 4,5-bisphosphate alters synaptotagmin 1 membrane docking and drives opposing bilayers closer together. Biochemistry 50, 2633–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li L., Shin O. H., Rhee J. S., Araç D., Rah J. C., Rizo J., Südhof T., Rosenmund C. (2006) Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J. Biol. Chem. 281, 15845–15852 [DOI] [PubMed] [Google Scholar]
- 42. van den Bogaart G., Meyenberg K., Diederichsen U., Jahn R. (2012) Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J. Biol. Chem. 287, 16447–16453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shin O. H., Xu J., Rizo J., Südhof T. C. (2009) Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc. Natl. Acad. Sci. U.S.A. 106, 16469–16474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shao X., Fernandez I., Südhof T. C., Rizo J. (1998) Solution structures of the Ca2+-free and Ca2+-bound C2A domain of synaptotagmin I: does Ca2+ induce a conformational change? Biochemistry 37, 16106–16115 [DOI] [PubMed] [Google Scholar]
- 45. Safieddine S., Wenthold R. J. (1999) SNARE complex at the ribbon synapses of cochlear hair cells: analysis of synaptic vesicle- and synaptic membrane-associated proteins. Eur. J. Neurosci. 11, 803–812 [DOI] [PubMed] [Google Scholar]
- 46. Nouvian R., Neef J., Bulankina A. V., Reisinger E., Pangršič T., Frank T., Sikorra S., Brose N., Binz T., Moser T. (2011) Exocytosis at the hair cell ribbon synapse apparently operates without neuronal SNARE proteins. Nat. Neurosci. 14, 411–413 [DOI] [PubMed] [Google Scholar]
- 47. Ramakrishnan N. A., Drescher M. J., Sheikhali S. A., Khan K. M., Hatfield J. S., Dickson M. J., Drescher D. G. (2006) Molecular identification of an N-type Ca2+ channel in saccular hair cells. Neuroscience 139, 1417–1434 [DOI] [PubMed] [Google Scholar]
- 48. Jahn R., Scheller R. H. (2006) SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643 [DOI] [PubMed] [Google Scholar]
- 49. Fujiwara T., Mishima T., Kofuji T., Chiba T., Tanaka K., Yamamoto A., Akagawa K. (2006) Analysis of knock-out mice to determine the role of HPC-1/syntaxin 1A in expressing synaptic plasticity. J. Neurosci. 26, 5767–5776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaneko Y., Suge R., Fujiwara T., Akagawa K., Watanabe S. (2011) Unusual retinal layer organization in HPC-1/syntaxin 1A knockout mice. J. Mol. Histol. 42, 483–489 [DOI] [PubMed] [Google Scholar]
- 51. Mishima T., Fujiwara T., Sanada M., Kofuji T., Akagawa K. (2011) A study on the neuronal phenotype of syntaxin 1B knockout mice: implications of differential role of syntaxin 1A and syntaxin 1B in synaptic transmission. Jap. Neurosci. Res. Abstr. 71S, P2-a06 [Google Scholar]
- 52. Beutner D., Voets T., Neher E., Moser T. (2001) Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron 29, 681–690 [DOI] [PubMed] [Google Scholar]
- 53. Sugita S., Hata Y., Südhof T. C. (1996) Distinct Ca2+-dependent properties of the first and second C2 domains of synaptotagmin-1. J. Biol. Chem. 271, 1262–1265 [DOI] [PubMed] [Google Scholar]
- 54. Chapman E. R., An S., Edwardson J. M., Jahn R. (1996) A novel function for the second C2 domain of synaptotagmin. J. Biol. Chem. 271, 5844–5849 [DOI] [PubMed] [Google Scholar]
- 55. Xu L., Pallikkuth S., Hou Z., Mignery G. A., Robia S. L., Han R. (2011) Dysferlin forms a dimer mediated by the C2 domains and the transmembrane domain in vitro and in living cells. PLoS One 6, e27884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Frank T., Khimich D., Neef A., Moser T. (2009) Mechanisms contributing to synaptic Ca2+ signals and their heterogeneity in hair cells. Proc. Natl. Acad. Sci. U.S.A. 106, 4483–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Roberts W. M. (1993) Spatial calcium buffering in saccular hair cells. Nature 363, 74–76 [DOI] [PubMed] [Google Scholar]
- 58. Edmonds B., Reyes R., Schwaller B., Roberts W. M. (2000) Calretinin modifies presynaptic calcium signaling in frog saccular hair cells. Nat. Neurosci. 3, 786–790 [DOI] [PubMed] [Google Scholar]
- 59. Wu Y-C., Tucker T., Fettiplace R. (1996) A theoretical study of calcium microdomains in turtle hair cells. Biophys. J. 71, 2256–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brandt A., Khimich D., Moser T. (2005) Few CaV1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J. Neurosci. 25, 11577–11585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schneggenburger R., Neher E. (2005) Presynaptic calcium and control of vesicle fusion. Curr. Opin. Neurobiol. 15, 266–274 [DOI] [PubMed] [Google Scholar]
- 62. Bai J., Tucker W. C., Chapman E. R. (2004) PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 11, 36–44 [DOI] [PubMed] [Google Scholar]
- 63. Longo-Guess C., Gagnon L. H., Bergstrom D. E., Johnson K. R. (2007) A missense mutation in the conserved C2B domain of otoferlin causes deafness in a new mouse model of DFNB9. Hear. Res. 234, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mirghomizadeh F., Pfister M., Apaydin F., Petit C., Kupka S., Pusch C. M., Zenner H. P., Blin N. (2002) Substitutions in the conserved C2C domain of otoferlin cause DFNB9, a form of nonsyndromic autosomal recessive deafness. Neurobiol. Dis. 10, 157–164 [DOI] [PubMed] [Google Scholar]
- 65. Tekin M., Akcayoz D., Incesulu A. (2005) A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am. J. Med. Genet. A 138, 6–10 [DOI] [PubMed] [Google Scholar]
- 66. Marlin S., Feldmann D., Nguyen Y., Rouillon I., Loundon N., Jonard L., Bonnet C., Couderc R., Garabedian E. N., Petit C., Denoyelle F. (2010) Temperature-sensitive auditory neuropathy associated with an otoferlin mutation: deafening fever! Biochem. Biophys. Res. Commun. 394, 737–742 [DOI] [PubMed] [Google Scholar]
- 67. Rouillon I., Marcolla A., Roux I., Marlin S., Feldmann D., Couderc R., Jonard L., Petit C., Denoyelle F., Garabédian E. N., Loundon N. (2006) Results of cochlear implantation in two children with mutations in the OTOF gene. Int. J. Pediatr. Otorhinolaryngol. 70, 689–696 [DOI] [PubMed] [Google Scholar]
- 68. Choi B. Y., Ahmed Z. M., Riazuddin S., Bhinder M. A., Shahzad M., Husnain T., Riazuddin S., Griffith A. J., Friedman T. B. (2009) Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin. Genet. 75, 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rodríguez-Ballesteros M., Reynoso R., Olarte M., Villamar M., Morera C., Santarelli R., Arslan E., Medá C., Curet C., Völter C., Sainz-Quevedo M., Castorina P., Ambrosetti U., Berrettini S., Frei K., Tedín S., Smith J., Cruz Tapia M., Cavallé L., Gelvez N., Primignani P., Gómez-Rosas E., Martín M., Moreno-Pelayo M. A., Tamayo M., Moreno-Barral J., Moreno F., del Castillo I. (2008) A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum. Mutat. 29, 823–831 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.