

Abstract
Dot1/DOT1L catalyzes the methylation of histone H3 lysine 79 (H3K79), which regulates diverse cellular processes, such as development, reprogramming, differentiation, and proliferation. In regards to these processes, studies of Dot1/DOT1L-dependent H3K79 methylation have mainly focused on the transcriptional regulation of specific genes. Although the gene transcription mediated by Dot1/DOT1L during the cell cycle is not fully understood, H3K79 methylation plays a critical role in the progression of G1 phase, S phase, mitosis, and meiosis. This modification may contribute to the chromatin structure that controls gene expression, replication initiation, DNA damage response, microtubule reorganization, chromosome segregation, and heterochromatin formation. Overall, Dot1/DOT1L is required to maintain genomic and chromosomal stability. This review summarizes the several functions of Dot1/DOT1L and highlights its role in cell cycle regulation.
Keywords: DOT1L, H3K79, cell cycle, differentiation, histone methyltransferase, meiosis, mitosis, proliferation, replication, senescence
Introduction
Histones undergo post-translational covalent modifications, including acetylation, phosphorylation, ubiquitination, sumoylation, and methylation.1 Multiple modifications on the same or different residues and crosstalk between modifications orchestrate the regulation of chromatin structure, which affects cellular processes involving DNA, such as replication, recombination, transcription, repair, and chromosome segregation.2 Therefore, the tight regulation of histone modifications is critically involved in development, differentiation, proliferation, and disease.3
Among histone modifications, histone methylation occurs at 3 basic residues, including lysine (K), arginine (R), and histidine (H), although histidine methylation is rarely observed.4 Histone methylation occurs using S-adenosylmethionine (SAM or AdoMet) as a methyl group donor.5 Histone lysine is mono-, di-, or tri-methylated, whereas arginine is mono- or di-methylated.6 The enzymes that catalyze histone methylation are categorized into 3 families: the PRMT (protein arginine N-methyltransferase) family, the SET (Su[var]3–9, Enhancer of Zeste, Trithorax)-domain-containing family, and the non-SET domain proteins.4 In particular, lysine methylation at H3K4, H3K9, H3K27, H3K36, and H4K20 is mediated by lysine methyltransferases (KMTs) that contain a SET domain.7 By contrast, H3K79 is methylated by a methyltransferase that lacks a SET domain.8 H3K79 methylation is also unique in that the residue is not located in the histone tail, but in a globular domain of histone H3 that is exposed on the nucleosomal surface.9 The H3K79 methyltransferase regulates diverse cellular processes (Fig. 1). This review summarizes the features and functions of the H3K79 methyltransferase in the cell cycle.
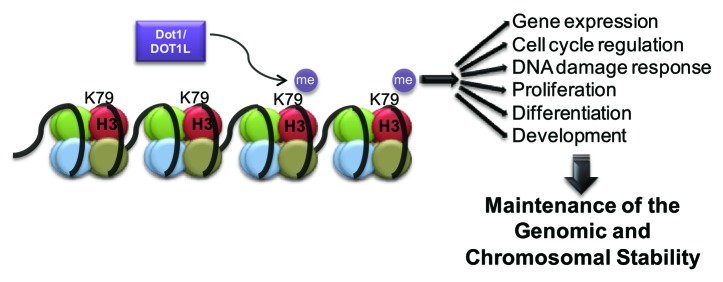
Figure 1. The functions of Dot1/DOT1L-dependent H3K79 methylation in organisms.
Dot1/DOT1L-Mediated H3K79 Methylation
The lysine methyltransferase responsible for H3K79 methylation (KMT4) is called Dot1 (disruptor of telomeric silencing 1) in lower eukaryotes.10-13 The homologous protein conserved in mammals is called Dot1-like protein (DOT1L).14,15 Dot1/DOT1L is believed to be the sole enzyme mediating non-processive methylation of H3K79 in Saccharomyces cerevisiae, Drosophila melanogaster, and humans (Fig. 2).12-14 However, Trypanosoma brucei possesses 2 homologous DOT1 proteins, DOT1A and DOT1B, that are responsible for mono- and di-methylation (DOT1A) and tri-methylation (DOT1B) of H3K76 (the residue homologous to H3K79 in other organisms).11 DOT1B can trimethylate H3K76 without the prerequisite of dimethylation by DOT1A.16 In Caenorhabditis elegans, 5 putative methyltransferases (DOT-1.1-DOT1–1.5) have been identified.10 In mice, 5 splicing variants of DOT1L (Dot1a-Dot1e) have been predicted;15 however, it is not known whether the variants have different effects on H3K79 methylation. By contrast, H3K79 methylation has not been found in Schizosaccharomyces pombe or Arabidopsis thaliana.17,18

Figure 2. The structure of Dot1/DOT1L proteins in several organisms. The catalytic domain of Dot1/DOT1L is conserved among species. Binding motifs with diverse interacting proteins are shown.
Dot1/DOT1L catalyzes the sequential mono-, di-, and tri-methylation of H3K79, while other SET domain-containing methyltransferases mediate methylation in a processive manner.19 This methylation occurs only in the nucleosomal histone H3, not in free/soluble histone H3, indicating that Dot1/DOT1L methylates K79 when histone H3 is incorporated into chromatin.11,13,20-22 One exception is that S. cerevisiae Dot1 (Dot1p), which it is preincubated with DNA, methylates recombinant H3.22 It has recently been reported that besides H3K79, the K349 residue of the androgen receptor is a novel substrate of human DOT1L methyltransferase.23 The identification of non-histone substrates of the DOT1L enzyme would help in understanding DOT1L-mediated cellular functions.
Factors Affecting Dot1/DOT1L Expression and H3K79 Methylation
The level of DOT1L mRNA declines during early preimplantation development in mice;24 however, DOT1L mRNA is ubiquitously expressed in mouse organs.15 Expression of DOT1L gene is regulated in response to certain signals. The level of DOT1L mRNA in immortalized mouse inner medullary collecting duct cells increases upon exposure to dimethyl sulfoxide; however, the mechanism by which transcription factors upregulate the transcription of mouse DOT1L is unknown.15 The level of DOT1L mRNA decreases in an aldosterone-sensitive manner in mouse renal collecting duct cells.25 During metamorphosis in Xenopus tropicalis, DOT1L is transcriptionally activated through a T3 response element (TRE) bound to the liganded thyroid receptor.26 In addition, the expression of DOT1 gene in S. cerevisiae peaks at G1, suggesting that Dot1 is involved in cell cycle regulation.27
Furthermore, the levels of mono-, di-, and tri-methylated H3K79 (H3K79me1, H3K79me2, and H3K79me3) are dynamically dependent on the chromatin region, the cell cycle phase, and developmental stage as described in this review. This dynamic level may be regulated by the following mechanisms.
Histone crosstalk affects the level of H3K79 methylation. Dot1/DOT1L-mediated H3K79 di- and tri-methylation depend on the monoubiquitination of H2B K120 (mammal)/K123 (yeast) and H2B K34.28-33 The interaction between the ubiquitin of H2B and the lysine-rich region (amino acids 101–140) of S. cerevisiae Dot1 is required for H3K79 di- and tri-methylation.34,35 A putative consensus sequence for the ubiquitin-interacting motif (UIM; amino acids 361~380) of human DOT1L has recently been suggested.36 In an alternative mechanism for ubiquitination-dependent H3K79 trimethylation, COMPASS complex or proteasomal ATPases Rpt4 and Rpt6 connect ubiquitinated H2B and H3K79.37,38 For transcriptional elongation, the Paf1 complex, which is associated with elongating RNA polymerase II, is required for H2B K123 monoubiquitination, which results in H3K79 methylation.39 Some components of the Paf1 complex, such as Rtf1 and Paf1, also interact with COMPASS and Dot1.39,40 In addition, human DOT1L interacts with phosphorylated RNA polymerase II, possibly leading to H3K79 methylation around transcriptional elongation regions.41 Overall, the transcriptional elongation complex enhances Dot1/DOT1L-mediated H3K79 di- and tri-methylation. The other possible explanation is that ubiquitinated H2B promotes an open chromatin structure to increase substrate accessibility.42 Meanwhile, the ubiquitination of H2A K119 does not affect DOT1L activity in vitro.32 Another histone crosstalk mechanism affecting the level of H3K79me1, H3K79me2, and H3K79me3 in S. cerevisiae is the interaction between the N-terminal short basic patch of histone H4 and the acidic patch of Dot1.21,43 Sir3 and Dot1 compete for binding to the short basic patch of histone H4. K16-acetylated histone H4 disrupts the binding of Sir3 to the H4 tail and instead stimulates Dot1-dependent H3K79 methylation. In addition, the local level of H3K79me2 is decreased by overexpression of the H4K16 deacetylase SIRT1 in human cells, suggesting that the silencing protein and DOT1L compete for binding to histone H4.44 By contrast, the N-terminal tail of histone H4 is not required for di- or tri-methylation of H3K76 in T. brucei.16
Several genes are critical for maintaining H3K79 methylation levels. In S. cerevisiae, Swi4 (regulatory subunit) and Swi6 (DNA binding component), which are components of the transcription complex SCB-binding factors (SBF), are required to maintain the level of H3K79me2, but not the level of H3K79me3.45 By contrast, Ard1 (N-terminal acetyltransferase A complex catalytic subunit) is required to maintain the levels of H2B-monoubiquitination and H3K79me3, not H3K79me2.46
Based on the results of mass spectrometry, the nonmethylated form of H3K79 is more abundant than the methylated form. H3K79me1 is detected at a higher rate than H3K79me2, whereas H3K79me3 is rarely detected in mice and humans.47-49 Because no specific demethylase has been identified, the mechanism governing the direct conversion of methylated H3K79 to one-fewer-methylated H3K79 has not been proposed. Instead, the multiple forms of H3K79 methylation suggest several regulatory mechanisms as follows. First, Dot1 catalyzes the transition from mono- to di- and from di- to tri-methylation, which requires Bre1-dependent H2B K123 ubiquitination.50-52 Second, the level of H3K79me1 decreases concurrently with the increase in H3K79me2 during gene expression, indicating that this modification contributes to active transcription.53 Consistent with this, H3K79me2 is decreased during heterochromatin formation.54 However, the methylation levels do not correlate with the expression levels, possibly because histone replacement occurs during transcription on a highly transcribed region.55 In this regard, the accumulation of H3K79me3 at centromeric regions could be due to the slow histone replacement in heterochromatic regions.56,57 Third, H3K79 methylation occurs on histones newly incorporated into nucleosomes as well as on pre-existing histones.49,52 H3K79 methylation is affected by the histone dilution that occurs during cell division. In other words, replication-dependent histone exchange explains the removal of H3K79 methylation.54 However, following fertilization, loss of H3K79 methylation occurs independently of DNA synthesis.57 Lastly, because 90% of H3K79 is methylated in budding yeast,13 it has been suggested that demethylase activity is masked by efficient methylation in this organism.54,58 The identification of the demethylase to remove H3K79 methylation requires further investigation.
H3K79-Methylated Regions in Chromatin
H3K79me3 is detected in open reading frames (ORFs) and H3K79me2 is enriched in intergenic regions, including promoters as well as ORFs in budding yeast.45 Because most of genome in the budding yeast is characterized as euchromatin, 90% of the budding yeast genome is methylated at H3K79; however, H3K79me2 and H3K79me3 overlap in only 2% of the genome.13,45 Except for budding yeast, while H3K79 is hypomethylated in silenced loci such as telomeres, H3K79me2 and H3K79me3 are more abundant in actively transcribed regions in D. melanogaster, mice, and humans.53,59-63 Consistent with this notion, methylated H3K79 is higher in the H3.3 variant, which is deposited in the euchromatic region, than in canonical H3, such as H3.1 and H3.2.49,59 By contrast, although H3K79me3 is associated with transcriptional activation, the high levels of H3K79me3 in the promoter and coding regions of marginally expressed or silent genes are also detected in human T cells.60 The plausible cause is a slow histone exchange as described above. In addition, although the relationship between H3K76 methylation and transcription in T. brucei has not been studied, H3K76me1 and H3K76me2 have been detected at certain transcription start sites and termination sites.61 Overall, the data indicate that H3K79 methylation is associated with transcriptional activation and is a critical transcriptional regulator.
In contrast to the role of Dot1/DOT1L in transcriptional activation, S. cerevisiae DOT1 and the Drosophila ortholog grappa are genes whose mutation, deletion, or overexpression disrupts silencing at telomeric regions, mating type loci, or rDNA.9,12,62-64 Although H3K79 is hypomethylated in silenced loci, Dot1-dependent transcriptional silencing at telomeres is regulated through blocking access of silencing factors such as Sir2 and Sir3 to H3K79-methylated subtelomeric regions.9,13,46 In addition, DOT1L-deleted mouse ES cells show a defect in H3K9me2 and H4K20me3 heterochromatin marks at telomeres and centromeres.48 Overall, H3K79 methylation may be important for transcriptional silencing in heterochromatic regions. However, in contrast to previous findings, a recent study showed that Dot1 is not required for transcriptional activation at euchromatic regions, but is required for the local derepression of heterochromatin in a K79 methylation-dependent or independent manner.65 This result suggests that the Dot1-dependent transcriptional silencing needs to be determined in a specific chromatic region.
DOT1L-mediated methylated H3K79 is localized to the heterochromatic region in mouse and human cells. While H3K79me2 is detected in the entire genome, H3K79me3 is enriched in the highly condensed DNA adjacent to regions stained with the anti-centromere/kinetochore antibody CREST and co-localizes with HP1β in mouse somatic cells and oocytes.57 This indicates that H3K79 is tri-methylated on pericentromeric heterochromatin in the mouse. The localization of methylated H3K79 in heterochromatic regions in oocytes also implicates the role of DOT1L in the organization of heterochromatin during mouse preimplantation development.24 In addition; the centromeric localization of H3K79 methylation has been suggested in human HeLa cells. Survival motor neuron (SMN) containing a Tudor domain relocates to damaged interphase centromeres upon transfection of the viral E3 ubiquitin ligase ICP0 from herpes simplex virus type 1 and interacts with H3K79me1 and H3K79me2, but not H3K79me3.66 However, without DOT1L, SMN is not recruited to the damaged centromeres, demonstrating that SMN might recognize H3K79me1 and H3K79me2 in the lesion. These findings indicate that methylated H3K79 might be enriched at centromeres in human cells and be involved in the interphase centromere damage response. Therefore, H3K79 methylation may be an epigenetic modification marking the centromeric and/or pericentromeric region.
Regulation of the Cell Cycle by Dot1/DOT1L-Mediated H3K79 Methylation
The cell cycle is a unidirectional and irreversible process for ensuring proper cell division. This transitional process from one cell cycle phase to the next is tightly controlled by several factors, including cyclin/cyclin-dependent kinase (CDK) complexes and CDK inhibitors.67,68 Beyond these well-known cell cycle regulators, diverse histone modifications are emerging as critical factors for cell cycle progression. Histone modifications dynamically change during the cell cycle and govern cell cycle progression, including replication timing and mitosis/meiosis.69-71 In addition, some modifications transcriptionally regulate the expression of cell cycle regulators such as CDK inhibitors.72,73 Therefore, as described below, the relevance of Dot1/DOT1L methyltransferase to the cell cycle sheds light on the importance of H3K79 methylation as a critical epigenetic mark for coordination of the cell cycle.
Level of H3K79 Methylation During the Cell Cycle
The level of H3K79 methylation is dynamically regulated during the cell cycle (Fig. 3). The level of H3K79me2 is low in G1 phase, begins to increase in S phase, and peaks at the G2/M phase, while the level of H3K79me3 remains constant during the cell cycle in S. cerevisiae.45,74 However, in slowly growing, G1- or G2/M-arrested budding yeast cells, H3K79me3 is more abundant than H3K79me2.52 Similarly, in T. brucei, the level of H3K76me3 does not change during the cell cycle; however, H3K76me2 is only detected in mitosis, not in G1 or G2 phase, and H3K76me1 is only detected in G2 and mitosis.11,61 During mouse preimplantation development, the level of H3K79me2 is higher in M phase than in interphase before the blastocyst stage; however, H3K79me3 is not detected in interphase or M phase.57 By contrast, in human A549 lung cancer and HeLa cervical cancer cells, the level of H3K79me2 is highest at the G1/S transition, begins to decrease in S phase, and is lowest in G2/M phase.14,75 In addition, while the level of H3K79me3 remains constant in S. cerevisiae and T. brucei during the cell cycle, H3K79me3 is regulated similarly to H3K79me2 in human A549 cancer cells;75 however, another study showed that the overall level of H3K79me2 does not change during the cell cycle in human K562 leukemia cells.76 Although cell cycle-dependent regulation of H3K79 methylation is not conserved across species, this dynamic change implicates the role of H3K79 methylation in cell cycle regulation (Fig. 4).

Figure 3. The levels of H3K79 methylation during the mitotic cell cycle in several organisms. H3K79me1, H3K79me2, and H3K79me3 are shown as a green, blue, and red line, respectively. H3K76me1, H3K76me2, and H3K76me3 are also shown.

Figure 4. The functions of H3K79 methylation during the mitotic cell cycle. Well-defined or expected functions are listed for each cell cycle phase.
Role of H3K79 Methylation in the G1/S Transition
Budding yeast Dot1 deletion mutants, DOT1L siRNA-transfected human lung cancer cells, DOT1L-knockout mouse yolk sac-derived erythroid progenitors, and DOT1L-knockout cells transformed with MLL (mixed lineage leukemia)-fusion proteins undergo G1 cell cycle arrest.45,75,77-79 These results suggest that Dot1/DOT1L-mediated methylation is required for the G1/S transition; however, the level of methylated H3K79 at the G1/S boundary differs across species. As described in the above section, during the G1/S transition, global levels of H3K79me2 are low in S. cerevisiae and T. brucei but high in human cancer cell lines including HeLa and A549.11,14,45,74,75 Although the reason for this difference is unclear, a possible mechanism for Dot1/DOT1L depletion-mediated G1 arrest is as follows.
In S. cerevisiae, the MBF (MCB binding factor) and SBF (SCB binding factor) complex bind to the MluI-cell cycle box (MCB) promoter and Swi4/6-dependent cell cycle box (SCB) promoter, respectively, and activate gene transcription during the G1/S transition. The promoter of the budding yeast DOT1 gene includes an MCB element, suggesting that DOT1 expression is cell cycle-regulated.80 Indeed, microarray analysis reveals that the level of DOT1 expression peaks at G1 phase.27 In contrast to the upregulation of DOT1 gene in G1, global level of H3K79me2 begins to increase in S phase. However, the promoters of SBF-regulated genes such as cyclins in G1 and S phases are enriched with H3K79me2 during M phase.45 H3K79me2 is then downregulated by Nrm1 and Whi3, which are negative regulators of the MBF and SBF complexes, for entry into S phase. These facts suggest that the decoration of H3K79me2 at SBF-regulated genes acts as an epigenetic memory for the regulation of cell cycle-regulatory gene expression. In addition, slowly growing or G1-arrested cells in budding yeast have more H3K79me3 and less H3K79me2 than proliferating log-phase cells, because H3K79 methylation accumulates on aging histones.52 Pre-existing histones also undergo mono- and di-methylation.49 This accumulation of H3K79 methylation could be an indicator of the period of the cell cycle before the new histones are deposited during replication in S phase. However, although H3K79me2 is associated with the control of cell cycle-regulatory genes, and H3K79me3 is associated with a slow proliferation rate, the direct evidence for Dot1-dependent cell cycle regulation has not been forthcoming in budding yeast. A more detailed investigation of the mechanisms is required.
In mammals, DOT1L-dependent methylation is suggested as a critical factor to regulate cell cycle progression at the G1/S transition. Aged rat lung tissues have lower levels of H3K79me3 than young rat lung tissues; therefore, hypomethylated H3K79 may be a histone mark that is associated with senescence, an irreversible cell cycle arrest at G0/G1 phase.75 However, in contrast to the H3K79 hypomethylation in aged rat lung tissues, H3K79me1 and H3K79me2 are upregulated in the brain of senescence-accelerated-prone mouse (SAMP).81 One possible explanation is that the effect of H3K79 methylation on senescence differs depending on the organism. Alternatively, upregulation of H3K79 methylation in SAMP may be a consequence of senescence model.
In human NCI-H1299 and A549 lung cancer cells, DOT1L depletion leads to upregulation of CDK inhibitors such as p18INK4c and p21CIP1/WAF1, respectively.75 These CDK inhibitors inactivate CDK2, leading to an irreversible G1 arrest called senescence. DOT1L-deficiency-induced senescence might be independent of DNA damage or telomere shortening. Rather, DOT1L-deficient human cancer cells show low levels of histone H2AX phospho-S139 (γH2AX), a marker of DNA strand breaks, and DOT1L-knockout mouse embryonic stem (ES) cells exhibit aberrant telomere elongation.48,75 In addition, loss of DOT1L also induces G1 arrest of MLL-fusion protein-transformed mouse cells via the upregulation of p16INK4a.77 Last, DOT1L-knockout yolk sacs grown in erythroid growth medium exhibit significant G1 arrest and apoptosis.78 Overall, Dot1/DOT1L is an epigenetic regulator of G1/S progression. This information suggests that downregulation of H3K79 methylation is a potential therapeutic approach in cancer treatment.
Role of H3K79 Methylation During S Phase
During S phase, the levels of H3K76me1 and H3K76me2 are not detectable in T. brucei; however, the level of H3K79me2 is low in S. cerevisiae and relatively high in human cells.11,45,61,74,75 Because the levels of H3K79 methylation differ across species, the role of H3K79 methylation in the regulation of replication during S phase might be different among budding yeast, trypanosomes, and humans.
In S. cerevisiae, the level of H3K79me2 starts to increase in S phase45,74; however, the regulatory role of Dot1 in replication has not been fully defined. The CAF-1 (chromatin assembly factor 1) complex, which functions as a histone chaperone for H3–H4 dimers during DNA replication and DNA repair, is associated with K79 methylated-histone H3 in budding yeast.74 Although H3K79 methylation is not required for the interaction between CAF-1 and H3, the replication-dependent deposition of methylated histone H3 by CAF-1 during S phase might affect nucleosome formation during S phase. However, this suggestion is in contrast to other findings that Dot1-mediated methylation occurs in nucleosomes, not in free histone H3.
In T. brucei, DOT1A deficiency induces the formation of hypoploidy and inhibits replication.11,61 By contrast, DOT1A overexpression leads to the formation of aneuploid cells, such as enucleated cells or cells containing more than 4N DNA content.61 The formation of hyperploid cells is the result of continuous replication without chromosome segregation and karyokinesis. Indeed, DOT1A overexpression results in the early appearance of H3K76me2 in S phase, whereas wild-type cells show no appearance of H3K76me2.61 Although the association of H3K76me2 with replication initiation sites is not clear yet, the methylated loci during G2 and M phase may license the replication origin to permit one round of replication during S phase. Overall, the programmed fluctuation of H3K76me1 and H3K76me2 during the cell cycle is critical for maintaining proper replication and cell cycle progression in T. brucei.
Meanwhile, DOT1L deficiency in HCT116 human cancer cells leads to the re-initiation of replication and results in apoptotic cells, hyperploid cells, or non-replicating S phase cells.76 H3K79me2 is indeed enriched in the replication initiation sites of the human genome in G1 and G2 phase, and H3K79me2-associated chromatin regions expand during S phase. H3K79 methylation prohibits re-replication, although it does not affect the rate of replication initiation and elongation. These data suggest that DOT1L-mediated H3K79 methylation is critical for replication timing and acts as a modification that marks the replicated chromatin, presumably by preventing premature initiation of replication or re-replication in human cells. However, because the effect of H3K79 methylation in replication has only been studied in specific cancer cell line, it should be investigated in other human cell lines or normal cells.
In summary, DOT1A overexpression in T. brucei leads to over-replication, but DOT1L deficiency in human cancer cells results in over-replication. Although H3K76 methylation in T. brucei and H3K79 methylation in human cancer cells show opposite effects on replication, these epigenetic marks are critical for the regulation of replication initiation.
Role of H3K79 Methylation in Mitosis
Dot1 may play a role in mitotic exit in S. cerevisiae.82 The Cdc14 phosphatase is released from the nucleolus at the onset of anaphase and induces the subsequent inactivation of CDK, finally triggering mitotic exit.83 Release of Cdc14 from nucleolar sequestration is defective in budding yeast Dot1-deletion mutants. A possible mechanism is that Dot1-mediated H3K79 methylation leads to a structural change in rDNA chromatin, which releases Cdc14 from the nucleolus. According to the previous report, overexpressed Dot1-GFP has been detected in the nucleolus in budding yeast.80 In addition; affinity purification showed that the human DOT1L-associated complex contains nucleolar proteins, including nucleophosmin (NPM1) and nucleolar RNA helicase 2.84 It would be interesting to investigate the role of Dot1/DOT1L in the nucleolus.
In mouse fibroblasts, H3K79me3 is also localized at chromosome boundaries during mitosis, suggesting that it is enriched in the pericentromeric region.57 The specific localization of H3K79me3 during mitosis in mouse somatic cells indicates that this methylation specifically functions during the mitotic phase. By contrast, H3K79me2 and H3K79me3 are lower in mitosis than in interphase in human A549 cancer cells.75 Because H3K79 methylation is enriched in the euchromatic region, its low level during mitosis, when transcription is repressed, is expected.53,85 Although methylated H3K79 has been suggested to be distributed around centromeres during interphase in human cells,66 the role of H3K79 methylation during mitosis has not been investigated in mammals. Specific localization at centromeric and/or pericentromeric regions indicates that H3K79 methylation might function at this heterochromatic region.
Several reports showing that DOT1L deficiency induces aneuploidy in mouse stem cells and human cancer cells raise the possibility that DOT1L is required for proper cell division.48,75,76,86 One study showed that DOT1L-deficient mouse ES cells undergo mitotic arrest, not cell death, with increased levels of histone H3 phospho-S10, and show polyploidy.86 This polyploidy results from the aberrant formation of mitotic spindles. However, another study showed that DOT1L-deficient mouse ES cells display polyploidy and undergo apoptosis.48 Although the effect of DOT1L deficiency on cell death varies depending on the study, DOT1L appears to be essential for proper mitotic progression. In human cancer cells including HCT116, A549, and NCI-H1299, DOT1L deficiency induces aneuploidy, including hypo- and hyper-ploidy.75,76 To explain the induction of aneuploidy by DOT1L siRNA transfection, Fu et al. suggest re-replication whereas Kim et al. demonstrate a failure of mitotic spindle formation.75,76 In addition, the type of aneuploidy observed in DOT1L-deficient A549 and NCI-H1299 cells is multinucleation, in which a cell contains multiple nuclei, owing to unsuccessful cytokinesis.75 This finding suggests that DOT1L affects the reorganization of the cytoskeletal structure. In any case, the findings indicate that H3K79 methylation in mammals is required for 2N chromosomes to be passed to the daughter cells. Meanwhile, although DOT1L deficiency does not induce more than 4N DNA content in chicken DT40 cells, it results in a slightly increased level of cell death and a slow proliferation rate, suggesting that DOT1L is critical for cell survival.87 Transfection of mouse fibrosarcoma cells with shRNA against the H2B K123 monoubiquitin ligase Bre1, which reduces H3K79 methylation, also results in apoptosis and genomic instability, such as double-strand breaks (DSBs) and anaphase bridges.88
Overall, although the role of H3K79 methylation in mitotic progression has not been fully addressed in several organisms, it appears important in maintaining chromosomal stability and proper cell division.
Role of H3K79 Methylation in Meiosis
In S. cerevisiae, diploid cells undergo meiosis, producing 4 haploid nuclei to form spores in response to nitrogen starvation and glucose absence. The global levels of H3K79me1, H3K79me2, and H3K79me3 in meiotic cells are similar to levels found in vegetative cells.89 Dot1 mutants show similar spore viability and crossover compared with wild-type strains, indicating that Dot1 is not required for sporulation.80,90 However, Dot1 is essential for the meiotic pachytene checkpoint,80,89 a surveillance mechanism that ensures proper chromosome separation during meiosis progression, including meiotic recombination and chromosome synapsis, by blocking the completion of meiotic division in the presence of unrepaired or unsynapsed recombination intermediates.91 In zip1 mutants, in which chromosomes are homologously paired but fail to synapse, the absence of Dot1 alleviates meiotic arrest but induces chromosome missegregation and aneuploidy, resulting in low spore viability.80 In dmc1 mutants arrested in meiosis with unrepaired DSBs, the absence of Dot1 repairs DSBs and triggers sporulation but results in low spore viability.80 In addition to Dot1 mutants, H3K79 mutants are also defective in the meiotic checkpoint.89 Dot1-mediated H3K79 methylation requires the localization and phosphorylation of the adaptor protein Hop1 and then Mek1 kinase for full activation of the meiotic checkpoint.89 Hop1-mediated Mek1 activation occurs at least in part through the localization of meiotic checkpoint protein pch2 at the unsynapsed nucleolar rDNA region from synapsed chromosomes.89
H3K79me2 and H3K79me3 are localized in the whole genome and at pericentromeric regions, respectively, during meiotic maturation in mouse oocytes.57 The status of H3K79 methylation has also been suggested to play a specific role in mouse spermatogenesis.56 During meiotic progression in the male mouse, the levels of DOT1L, H3K79me2, and H3K79me3 begin to increase in pachynema and persist throughout prophase I, while H3K79me1 is present at a constant low level throughout prophase I. However, H3K79me2 and H3K79me3 show differences in the subnuclear distribution in spermatocytes, as shown previously in oocytes.57 Whereas H3K79me2 is distributed on the chromatin of all chromosomes except for sex chromosomes, H3K79me3 is enriched at heterochromatic centromeres and sex chromosomes. Following the replacement of histone H3.1 and H3.2 by the H3.3 variant in the XY body during pachynema, the level of H3K79me3 begins to increase at histone H3.3 during diplonema, possibly to maintain silencing of the XY chromosome. Meanwhile, the widespread distribution of H3K79me2 during pachynema might be a cause or consequence of transcriptional reactivation in autosomes. Taken together, these results suggest that DOT1L-mediated H3K79 methylation is involved in meiotic events in mouse oocyte and spermatocytes.
Cell Cycle Checkpoint Following DNA Damage
The Dot1-deletion mutant, the H3K79 mutant, and the H2B-K123 mutant, in which H3K79 methylation is lost, show radiation sensitivity in budding yeast.92-95 The yeast Dot1 deletion mutant decreases and/or retards the phosphorylation of the 53BP1 ortholog Rad9 and the Chk2 homolog Rad53 kinase to activate the G1- and intra-S checkpoint following DNA damage induced by ultraviolet (UV), X-ray, and ionizing radiation (IR).94,96,97 Dot1 is required to recruit Rad9 to DSBs, although whether the Tudor domain of Rad9 directly interacts with methylated H3K79 remains a matter of debate.96,98-100 Furthermore, loss of Rad9 binding to histone H3 increases DSB resection, which occurs during homologous recombination, thereby generating single-stranded DNA (ssDNA).101 The accumulation of replication protein A (RPA)-coated ssDNA leads to the activation of Mec1 kinase, an ortholog of mammalian ATR. In fact, loss of Dot1 hyperactivates the Mec1–Ddc2 kinase complex after DSB formation, indicating that Dot1 is important for the precise regulation of Mec1 activity. Dot1 also promotes efficient sister chromatid recombination during repair of DSBs through loading of cohesin to the damaged sites.102 Overall, budding yeast Dot1 plays a critical role in the repair of DSBs.
Budding yeast Dot1 repairs UV-induced DNA lesions through nucleotide excision repair, recombination repair, or post-replication repair.93,103 In addition, mouse DOT1L participates in survival following UV irradiation. However, mouse DOT1L is not involved in nucleotide excision repair, which removes UV-induced DNA photoproducts; rather, it promotes binding of RNA polymerase II to the promoters of UV-repressed genes to reactivate transcription initiation following UV irradiation.104 In contrast to the survival effect of Dot1/DOT1L following UV irradiation, budding yeast Dot1 has the opposite effect after exposure to alkylating agents. Dot1 inhibits translesion synthesis, which allows error-prone polymerases to bypass DNA lesions and enhances survival following DNA damage.105 Therefore, the Dot1 mutant is more resistant than wild-type cells to the alkylating agent methyl methanesulfonate (MMS), but promotes MMS-induced mutagenesis.105-107 This effect of the Dot1 mutant requires the full activation of Rad53 and controls the binding of the error-prone polymerase Polζ/Rev1 to chromatin.107 Consequently, a Dot1 mutation increases the mutagenesis frequency. Taken together, these observations show that Dot1/DOT1L is required to maintain genomic stability without mutagenesis.
Beyond yeast data, DOT1L-siRNA-transfected U2OS human osteosarcoma cells show the prolonged presence of γH2AX, a marker of DNA strand breaks, following IR.36 DOT1L-knockout mouse embryonic fibroblasts and DOT1L siRNA-treated HEK293 cells also show low viability after IR.47 Thus, mammalian DOT1L is required for the repair of DSBs; however, the requirement of DOT1L for the recruitment of 53BP1, the mammalian ortholog of budding yeast Rad9, has been debated. Studies from Huyen et al. and Wakeman et al. showed that human 53BP1 recruitment is dependent on H3K79 methylation.36,108 Deletion of human DOT1L abolishes 53BP1 formation following IR, likely because it results in the loss of the interaction between the UIM of DOT1L and the ubiquitin-like motif (UBL) of HLA-B-associated transcript 3 (Bat3).36 By contrast, Botuyan et al. demonstrated that H4K20 methylation, not H3K79 methylation, is indispensable for the recruitment of mouse and human 53BP1.109 Even in S. pombe in which methylated H3K79 is absent, the localization of Crb2, an ortholog of Rad9, to the damaged site, is dependent on H4K20 methylation, not on H3K79 methylation.110 However, DOT1L−/− DT40 chicken cells do not show any defect in 53BP1 foci formation or sensitivity to IR.87 Overall, the different systems have yielded different results regarding the requirement of H3K79 methylation for 53BP1 recruitment to the damaged site. The mechanism by which mammalian DOT1L-mediated H3K79 methylation regulates repair and survival following DSBs requires further investigation. In summary, it suggests that Dot1/DOT1L is required to maintain genomic stability following genotoxic stress.
Regulation of Differentiation and Reprogramming by DOT1L-Mediated H3K79 Methylation
In addition to proliferation, DOT1L also participates in the regulation of differentiation. First, DOT1B, which is responsible for H3K76 trimethylation in T. brucei, is essential for differentiation.11 Second, DOT1L-deficient mouse ES cells undergo slow proliferation, G2/M arrest, and hyperploidy following differentiation induced by retinoic acid, suggesting that DOT1L is required for the initial stages of differentiation.86 Third, DOT1L plays a critical role in differentiation during mouse hematopoiesis.78,79,111,112 Loss of DOT1L impairs erythroid differentiation in the yolk sac of the germline DOT1L-knockout mouse.78 This impairment results from the aberrant expression of target genes, i.e., downregulated GATA2 and upregulated PU.1/Sfpi1, and blocks erythroid formation in early embryonic hematopoiesis.78 In addition, DOT1L plays an important role in normal adult hematopoiesis. Mice with conditional knockout of DOT1L develop postnatal anemia and pancytopenia.79,112 DOT1L-deficient bone marrow also fails to maintain a terminally differentiated myeloid and lymphoid population, hematopoietic stem cells, and progenitor cells.79,111,112
DOT1L might contribute to gene reprogramming during development. DOT1L and H3K79me3 disappear in oocytes immediately after fertilization until the 2-cell stage; however, their levels increase at the blastocyst stage.24,57 During this process, H3K79 methylation is thought to be a critical epigenetic mark that regulates gene expression during genome remodeling after fertilization. In addition to the role of DOT1L in fertilization, DOT1L-mediated H3K79 methylation plays a role in the reprogramming of adult somatic cells to obtain induced pluripotent stem cells (iPSCs). Suppression of DOT1L through shRNA or the small-molecule inhibitor EPZ004777 enhances the reprogramming efficiency of human fibroblasts, mouse embryonic fibroblasts, and mouse peripheral blood cells.113 DOT1L suppression can reprogram human fibroblasts into iPSCs only in the presence of OCT4 and SOX2, indicating that the absence of DOT1L replaces the requirement for KLF4 and c-Myc. This reprogramming requires increased levels of NANOG and LIN28. During reprogramming induced by DOT1L inhibition, mesenchymal genes such as SNAI1, SNAI2, ZEB1, ZEB2, and TGFB2 are suppressed, while epithelial genes such as CDH1 and OCLN are upregulated. Indeed, ChIP sequencing (chromatin immunoprecipitation followed by DNA sequencing) analysis revealed that H3K79me2 is lost in genes that need to be silenced for reprogramming. Overall, the above findings suggest that DOT1L-dependent H3K79 methylation acts as a barrier to reprogramming. In contrast to the phenomenon shown in iPSCs, DOT1L and H3K79me3 is enriched in pluripotency-related genes such as Nanog and Oct4 in human embryonic carcinoma NCCIT cells.41 Transfection of NCCIT cells with DOT1L siRNA reduces the level of H3K79me3 in the gene body of Nanog and Oct4 and decreases their expression. Moreover, H3K79me2 is enriched in Oct4 and Sox2 in mouse ES cells, although their expression is not affected by DOT1L deficiency.86 Therefore, DOT1L-mediated H3K79 methylation plays a role in gene reprogramming. However, the mechanism needs to be investigated further.
Control of DOT1L-Dependent Diseases through the Regulation of DOT1L Activity
As described above, DOT1L deficiency prohibits normal cell cycle progression and finally induces abnormal proliferation; however, the consequences of this abnormal proliferation, which include G1 arrest, mitotic arrest, senescence, aneuploidy, and apoptosis, depend on the context and the species. In human cancer cells, inactivation or depletion of DOT1L prevents proliferation. DOT1L-deficient human lung cancer cells undergo senescence, which may be a way to eradicate aggressively proliferating cancer cells.75 DOT1L deficiency also results in the death of Ls174T colorectal carcinoma cells, but not of HEK293T cells.114 MLL-fusion protein-induced transformed cells, which exhibit local hypermethylation of H3K79, undergo G1 arrest or apoptosis in the absence of DOT1L.77,79,115 Specific DOT1L inhibitors, such as EPZ5676, EPZ004777, and SGC0946, induce apoptosis in leukemia cells induced by MLL-fusion proteins.116-120 By contrast, one of leukemogenic fusion protein, CALM-AF10, leads to the dissociation of DOT1L from chromatin and a global reduction in H3K79me2.47 This global hypomethylation contributes to the accumulation of genomic instability. Overall, DOT1L is a potential cancer therapeutic target because the absence of DOT1L activity prohibits the proliferation of cancer cells.
However, targeting DOT1L may be toxic to normal cells, because several DOT1L-knockout phenotypes suggest its critical role in the diverse biological functions, including development, differentiation, and organ function. First, DOT1L is required for embryonic development, as germline DOT1L knockout causes embryonic lethality at day 10.5, possibly due to reduced angiogenesis in the yolk sac, embryonic anemia, and an enlarged heart.48,109 Second, cardiac-specific DOT1L knockout in mice triggers dilated cardiomyopathy and postnatal lethality.121 DOT1L depletion in the heart enhances cell proliferation, indicating that DOT1L inhibits cardiac cell proliferation. The downregulation of dystrophin, a key transcriptional target of DOT1L in the regulation of heart function, would be a cause of cardiomyopathy in cardiac-specific DOT1L-knockout mice. Third, conditional DOT1L-knockout mice show a reduced level of H3K79me2 at the 5′-regulatory region of endothelin-1 in kidney tissues.25 DOT1L-knockout mice also display polyuria, possibly because of the upregulation of Aqp5, a potential target of DOT1L.122 These data suggest that therapeutic drugs inhibiting DOT1L activity could contribute to the development of kidney injury. Fourth, DOT1L is required for normal hematopoiesis as described earlier. Overall, DOT1L-mediated H3K79 methylation plays a critical role in development, hematopoiesis, and maintenance of organ function, suggesting that therapeutic DOT1L inhibition must be carefully considered. However, according to Daigel et al., treatment with the DOT1L inhibitor EPZ004777 kills MLL-rearranged leukemic cells, but not non-MLL-rearranged leukemic cells or hematopoietic stem cells.118 This discrepancy may provide an effective therapeutic window in MLL-fusion leukemia that needs to be assessed in other solid tumors to control cell proliferation.
Discussion
Dot1/DOT1L-mediated H3K79 methylation plays a role in transcription, reprogramming, development, differentiation, and proliferation. Among these cellular processes, proliferation is tightly regulated by cell cycle progression. As mentioned in this review, the fluctuating pattern of H3K79me1, H3K79me2, and H3K79me3 suggests that they play roles in cell cycle regulation. However, the changes in H3K79 methylation during the cell cycle are not evolutionarily conserved. In addition, H3K79 methylation-dependent functions in cell cycle regulation are not conserved. In other words, the role of Dot1/DOT1L-mediated H3K79 methylation in cell proliferation is dependent on the species or nature of the tissues under investigation. The depletion of Dot1/DOT1L results in reduced cell proliferation in budding yeast, trypanosomes, mouse embryonic stem cells, and human cancer cells. DOT1A deletion results in haploidy in trypanosomes; the deletion of Dot1/DOT1L in budding yeast, human cancer cells, and mouse erythroid progenitors induces G1 arrest, and DOT1L-depleted mouse ES cells undergo G2/M arrest following differentiation. By contrast, the cardiac-specific deletion of DOT1L leads to increased proliferation of heart tissues. The specific function of Dot1/DOT1L in cell proliferation requires further elucidation.
Dot1/DOT1L is responsible for the mono-, di-, and tri-methylation of H3K79. It is intriguing that the same methyltransferase mediates these 3 types of methylation, despite the fact that these methylations have different functions. One possible explanation for the different functions is that other histone modifications that might crosstalk with differentially methylated H3K79 (i.e., H3K79me1, H3K79me2, and H3K79me3) influence the final functions of these methylations in certain contexts. Alternatively, the different effects of each type of H3K79 methylation might be mediated by transcriptional regulation of specific target genes. Indeed, DOT1L regulates the transcription of diverse genes that are involved in differentiation and reprogramming. However, the exact mechanism by which Dot1/DOT1L controls the expression of cell cycle-associated genes has not been elucidated. Methylated H3K79-dependent target genes that control cell proliferation must be identified to understand how Dot1/DOT1L regulates cell cycle progression. Meanwhile, the global and local levels of H3K79 methylation are differentially regulated. Even if the global levels of H3K79 methylation do not fluctuate during the cell cycle, its local level in a specific chromatin region could change. Therefore, the local level of H3K79 methylation should be determined during each cell cycle phase, depending on the euchromatic/heterochromatic regions and on cell cycle-regulatory genes. Taken together, H3K79me1, H3K79me2, and H3K79me3 might act as differential markers for Dot1/DOT1L-mediated cellular functions, including cell cycle regulation. In addition, the discrepancy in global and/or local levels of each H3K79 methylation during the cell cycle might reflect differences in Dot1/DOT1L functions between organisms.
DOT1L controls the development of diseases including leukemia, cardiac dysfunction, and kidney injury, as well as normal cellular processes. Therefore, regulation of DOT1L activity may be a useful therapeutic approach for multiple diseases. For instance, DOT1L inhibition may be beneficial in MLL-fusion-induced leukemia. In addition, because DOT1L-mediated H3K79 methylation is a critical regulator of cell cycle progression, modulation of DOT1L activity can halt the proliferation of aggressive cancer cells in addition to leukemia cells. Specific inhibitors of DOT1L should be tested in other tumors. In addition, although DOT1L is thought to mediate methylation only at histones, one non-histone substrate for DOT1L has recently been identified. The discovery of other non-histone substrates for DOT1L will enable the elucidation of the mechanisms by which DOT1L is involved in diverse cellular functions including cell cycle regulation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2012R1A2A2A01011164 and No.2011-0030072).
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/28104
References
- 1.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 2.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005;15:163–76. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–57. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta. 2009;1789:45–57. doi: 10.1016/j.bbagrm.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell. 2002;109:801–6. doi: 10.1016/S0092-8674(02)00798-5. [DOI] [PubMed] [Google Scholar]
- 7.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–49. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 8.Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003;112:711–23. doi: 10.1016/S0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 9.Ng HH, Feng Q, Wang H, Erdjument-Bromage H, Tempst P, Zhang Y, Struhl K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16:1518–27. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cecere G, Hoersch S, Jensen MB, Dixit S, Grishok A. The ZFP-1(AF10)/DOT-1 complex opposes H2B ubiquitination to reduce Pol II transcription. Mol Cell. 2013;50:894–907. doi: 10.1016/j.molcel.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janzen CJ, Hake SB, Lowell JE, Cross GA. Selective di- or trimethylation of histone H3 lysine 76 by two DOT1 homologs is important for cell cycle regulation in Trypanosoma brucei. Mol Cell. 2006;23:497–507. doi: 10.1016/j.molcel.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 12.Shanower GA, Muller M, Blanton JL, Honti V, Gyurkovics H, Schedl P. Characterization of the grappa gene, the Drosophila histone H3 lysine 79 methyltransferase. Genetics. 2005;169:173–84. doi: 10.1534/genetics.104.033191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–56. doi: 10.1016/S0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 14.Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12:1052–8. doi: 10.1016/S0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- 15.Zhang W, Hayashizaki Y, Kone BC. Structure and regulation of the mDot1 gene, a mouse histone H3 methyltransferase. Biochem J. 2004;377:641–51. doi: 10.1042/BJ20030839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frederiks F, van Welsem T, Oudgenoeg G, Heck AJ, Janzen CJ, van Leeuwen F. Heterologous expression reveals distinct enzymatic activities of two DOT1 histone methyltransferases of Trypanosoma brucei. J Cell Sci. 2010;123:4019–23. doi: 10.1242/jcs.073882. [DOI] [PubMed] [Google Scholar]
- 17.Nishida H. Evolutionary conservation levels of subunits of histone-modifying protein complexes in fungi. Comp Funct Genomics. 2009;•••:379317. doi: 10.1155/2009/379317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang K, Sridhar VV, Zhu J, Kapoor A, Zhu JK. Distinctive core histone post-translational modification patterns in Arabidopsis thaliana. PLoS One. 2007;2:e1210. doi: 10.1371/journal.pone.0001210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frederiks F, Tzouros M, Oudgenoeg G, van Welsem T, Fornerod M, Krijgsveld J, van Leeuwen F. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat Struct Mol Biol. 2008;15:550–7. doi: 10.1038/nsmb.1432. [DOI] [PubMed] [Google Scholar]
- 20.Lacoste N, Utley RT, Hunter JM, Poirier GG, Côte J. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem. 2002;277:30421–4. doi: 10.1074/jbc.C200366200. [DOI] [PubMed] [Google Scholar]
- 21.Fingerman IM, Li HC, Briggs SD. A charge-based interaction between histone H4 and Dot1 is required for H3K79 methylation and telomere silencing: identification of a new trans-histone pathway. Genes Dev. 2007;21:2018–29. doi: 10.1101/gad.1560607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawada K, Yang Z, Horton JR, Collins RE, Zhang X, Cheng X. Structure of the conserved core of the yeast Dot1p, a nucleosomal histone H3 lysine 79 methyltransferase. J Biol Chem. 2004;279:43296–306. doi: 10.1074/jbc.M405902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, Merkurjev D, Ohgi KA, Meng D, Zhang J, et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013;500:598–602. doi: 10.1038/nature12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ooga M, Suzuki MG, Aoki F. Involvement of DOT1L in the remodeling of heterochromatin configuration during early preimplantation development in mice. Biol Reprod. 2013;89:145. doi: 10.1095/biolreprod.113.113258. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Q, Liu K, Wu H, Chen L, Pouranan V, Yuan M, Xiao Z, Peng W, Xiang A, Tang R, et al. Spironolactone rescues Dot1a-Af9-mediated repression of endothelin-1 and improves kidney injury in streptozotocin-induced diabetic rats. PLoS One. 2012;7:e47360. doi: 10.1371/journal.pone.0047360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsuura K, Fujimoto K, Das B, Fu L, Lu CD, Shi YB. Histone H3K79 methyltransferase Dot1L is directly activated by thyroid hormone receptor during Xenopus metamorphosis. Cell Biosci. 2012;2:25. doi: 10.1186/2045-3701-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–97. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakanishi S, Lee JS, Gardner KE, Gardner JM, Takahashi YH, Chandrasekharan MB, Sun ZW, Osley MA, Strahl BD, Jaspersen SL, et al. Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J Cell Biol. 2009;186:371–7. doi: 10.1083/jcb.200906005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng HH, Xu RM, Zhang Y, Struhl K. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem. 2002;277:34655–7. doi: 10.1074/jbc.C200433200. [DOI] [PubMed] [Google Scholar]
- 30.Wu L, Zee BM, Wang Y, Garcia BA, Dou Y. The RING finger protein MSL2 in the MOF complex is an E3 ubiquitin ligase for H2B K34 and is involved in crosstalk with H3 K4 and K79 methylation. Mol Cell. 2011;43:132–44. doi: 10.1016/j.molcel.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chatterjee C, McGinty RK, Fierz B, Muir TW. Disulfide-directed histone ubiquitylation reveals plasticity in hDot1L activation. Nat Chem Biol. 2010;6:267–9. doi: 10.1038/nchembio.315. [DOI] [PubMed] [Google Scholar]
- 32.Whitcomb SJ, Fierz B, McGinty RK, Holt M, Ito T, Muir TW, Allis CD. Histone monoubiquitylation position determines specificity and direction of enzymatic cross-talk with histone methyltransferases Dot1L and PRC2. J Biol Chem. 2012;287:23718–25. doi: 10.1074/jbc.M112.361824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGinty RK, Kim J, Chatterjee C, Roeder RG, Muir TW. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature. 2008;453:812–6. doi: 10.1038/nature06906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oh S, Jeong K, Kim H, Kwon CS, Lee D. A lysine-rich region in Dot1p is crucial for direct interaction with H2B ubiquitylation and high level methylation of H3K79. Biochem Biophys Res Commun. 2010;399:512–7. doi: 10.1016/j.bbrc.2010.07.100. [DOI] [PubMed] [Google Scholar]
- 35.Cheng X, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct. 2005;34:267–94. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wakeman TP, Wang Q, Feng J, Wang XF. Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. EMBO J. 2012;31:2169–81. doi: 10.1038/emboj.2012.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JS, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, Bhaumik SR, Shilatifard A. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131:1084–96. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- 38.Ezhkova E, Tansey WP. Proteasomal ATPases link ubiquitylation of histone H2B to methylation of histone H3. Mol Cell. 2004;13:435–42. doi: 10.1016/S1097-2765(04)00026-7. [DOI] [PubMed] [Google Scholar]
- 39.Wood A, Schneider J, Dover J, Johnston M, Shilatifard A. The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J Biol Chem. 2003;278:34739–42. doi: 10.1074/jbc.C300269200. [DOI] [PubMed] [Google Scholar]
- 40.Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, et al. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell. 2003;11:721–9. doi: 10.1016/S1097-2765(03)00091-1. [DOI] [PubMed] [Google Scholar]
- 41.Kim SK, Jung I, Lee H, Kang K, Kim M, Jeong K, Kwon CS, Han YM, Kim YS, Kim D, et al. Human histone H3K79 methyltransferase DOT1L protein [corrected] binds actively transcribing RNA polymerase II to regulate gene expression. J Biol Chem. 2012;287:39698–709. doi: 10.1074/jbc.M112.384057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol. 2011;7:113–9. doi: 10.1038/nchembio.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altaf M, Utley RT, Lacoste N, Tan S, Briggs SD, Côté J. Interplay of chromatin modifiers on a short basic patch of histone H4 tail defines the boundary of telomeric heterochromatin. Mol Cell. 2007;28:1002–14. doi: 10.1016/j.molcel.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 45.Schulze JM, Jackson J, Nakanishi S, Gardner JM, Hentrich T, Haug J, Johnston M, Jaspersen SL, Kobor MS, Shilatifard A. Linking cell cycle to histone modifications: SBF and H2B monoubiquitination machinery and cell-cycle regulation of H3K79 dimethylation. Mol Cell. 2009;35:626–41. doi: 10.1016/j.molcel.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takahashi YH, Schulze JM, Jackson J, Hentrich T, Seidel C, Jaspersen SL, Kobor MS, Shilatifard A. Dot1 and histone H3K79 methylation in natural telomeric and HM silencing. Mol Cell. 2011;42:118–26. doi: 10.1016/j.molcel.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin YH, Kakadia PM, Chen Y, Li YQ, Deshpande AJ, Buske C, Zhang KL, Zhang Y, Xu GL, Bohlander SK. Global reduction of the epigenetic H3K79 methylation mark and increased chromosomal instability in CALM-AF10-positive leukemias. Blood. 2009;114:651–8. doi: 10.1182/blood-2009-03-209395. [DOI] [PubMed] [Google Scholar]
- 48.Jones B, Su H, Bhat A, Lei H, Bajko J, Hevi S, Baltus GA, Kadam S, Zhai H, Valdez R, et al. The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 2008;4:e1000190. doi: 10.1371/journal.pgen.1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sweet SM, Li M, Thomas PM, Durbin KR, Kelleher NL. Kinetics of re-establishing H3K79 methylation marks in global human chromatin. J Biol Chem. 2010;285:32778–86. doi: 10.1074/jbc.M110.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shahbazian MD, Zhang K, Grunstein M. Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol Cell. 2005;19:271–7. doi: 10.1016/j.molcel.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 51.Foster ER, Downs JA. Methylation of H3 K4 and K79 is not strictly dependent on H2B K123 ubiquitylation. J Cell Biol. 2009;184:631–8. doi: 10.1083/jcb.200812088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Vos D, Frederiks F, Terweij M, van Welsem T, Verzijlbergen KF, Iachina E, de Graaf EL, Altelaar AF, Oudgenoeg G, Heck AJ, et al. Progressive methylation of ageing histones by Dot1 functions as a timer. EMBO Rep. 2011;12:956–62. doi: 10.1038/embor.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–39. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katan-Khaykovich Y, Struhl K. Heterochromatin formation involves changes in histone modifications over multiple cell generations. EMBO J. 2005;24:2138–49. doi: 10.1038/sj.emboj.7600692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sawado T, Halow J, Im H, Ragoczy T, Bresnick EH, Bender MA, Groudine M. H3 K79 dimethylation marks developmental activation of the beta-globin gene but is reduced upon LCR-mediated high-level transcription. Blood. 2008;112:406–14. doi: 10.1182/blood-2007-12-128983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ontoso D, Kauppi L, Keeney S, San-Segundo PA. Dynamics of DOT1L localization and H3K79 methylation during meiotic prophase I in mouse spermatocytes. Chromosoma. 2013 doi: 10.1007/s00412-013-0438-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ooga M, Inoue A, Kageyama S, Akiyama T, Nagata M, Aoki F. Changes in H3K79 methylation during preimplantation development in mice. Biol Reprod. 2008;78:413–24. doi: 10.1095/biolreprod.107.063453. [DOI] [PubMed] [Google Scholar]
- 58.Tu S, Bulloch EM, Yang L, Ren C, Huang WC, Hsu PH, Chen CH, Liao CL, Yu HM, Lo WS, et al. Identification of histone demethylases in Saccharomyces cerevisiae. J Biol Chem. 2007;282:14262–71. doi: 10.1074/jbc.M609900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McKittrick E, Gafken PR, Ahmad K, Henikoff S. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc Natl Acad Sci U S A. 2004;101:1525–30. doi: 10.1073/pnas.0308092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 61.Gassen A, Brechtefeld D, Schandry N, Arteaga-Salas JM, Israel L, Imhof A, Janzen CJ. DOT1A-dependent H3K76 methylation is required for replication regulation in Trypanosoma brucei. Nucleic Acids Res. 2012;40:10302–11. doi: 10.1093/nar/gks801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. Proc Natl Acad Sci U S A. 2003;100:1820–5. doi: 10.1073/pnas.0437846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Osborne EA, Dudoit S, Rine J. The establishment of gene silencing at single-cell resolution. Nat Genet. 2009;41:800–6. doi: 10.1038/ng.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998;150:613–32. doi: 10.1093/genetics/150.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stulemeijer IJ, Pike BL, Faber AW, Verzijlbergen KF, van Welsem T, Frederiks F, Lenstra TL, Holstege FC, Gasser SM, van Leeuwen F. Dot1 binding induces chromatin rearrangements by histone methylation-dependent and -independent mechanisms. Epigenetics Chromatin. 2011;4:2. doi: 10.1186/1756-8935-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sabra M, Texier P, El Maalouf J, Lomonte P. The Tudor protein survival motor neuron (SMN) is a chromatin-binding protein that interacts with methylated lysine 79 of histone H3. J Cell Sci. 2013;126:3664–77. doi: 10.1242/jcs.126003. [DOI] [PubMed] [Google Scholar]
- 67.Hochegger H, Takeda S, Hunt T. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol. 2008;9:910–6. doi: 10.1038/nrm2510. [DOI] [PubMed] [Google Scholar]
- 68.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–69. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 69.Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell. 2012;48:491–507. doi: 10.1016/j.molcel.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu D, Bai J, Duan Q, Costa M, Dai W. Covalent modifications of histones during mitosis and meiosis. Cell Cycle. 2009;8:3688–94. doi: 10.4161/cc.8.22.9908. [DOI] [PubMed] [Google Scholar]
- 71.Sawicka A, Seiser C. Histone H3 phosphorylation - a versatile chromatin modification for different occasions. Biochimie. 2012;94:2193–201. doi: 10.1016/j.biochi.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He J, Kallin EM, Tsukada Y, Zhang Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b) Nat Struct Mol Biol. 2008;15:1169–75. doi: 10.1038/nsmb.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenführ M, Maertens G, Banck M, Zhou MM, Walsh MJ, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23:1177–82. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou H, Madden BJ, Muddiman DC, Zhang Z. Chromatin assembly factor 1 interacts with histone H3 methylated at lysine 79 in the processes of epigenetic silencing and DNA repair. Biochemistry. 2006;45:2852–61. doi: 10.1021/bi0521083. [DOI] [PubMed] [Google Scholar]
- 75.Kim W, Kim R, Park G, Park JW, Kim JE. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J Biol Chem. 2012;287:5588–99. doi: 10.1074/jbc.M111.328138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu H, Maunakea AK, Martin MM, Huang L, Zhang Y, Ryan M, Kim R, Lin CM, Zhao K, Aladjem MI. Methylation of histone H3 on lysine 79 associates with a group of replication origins and helps limit DNA replication once per cell cycle. PLoS Genet. 2013;9:e1003542. doi: 10.1371/journal.pgen.1003542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117:6912–22. doi: 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feng Y, Yang Y, Ortega MM, Copeland JN, Zhang M, Jacob JB, Fields TA, Vivian JL, Fields PE. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood. 2010;116:4483–91. doi: 10.1182/blood-2010-03-276501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jo SY, Granowicz EM, Maillard I, Thomas D, Hess JL. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood. 2011;117:4759–68. doi: 10.1182/blood-2010-12-327668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.San-Segundo PA, Roeder GS. Role for the silencing protein Dot1 in meiotic checkpoint control. Mol Biol Cell. 2000;11:3601–15. doi: 10.1091/mbc.11.10.3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang CM, Tsai SN, Yew TW, Kwan YW, Ngai SM. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology. 2010;11:87–102. doi: 10.1007/s10522-009-9231-5. [DOI] [PubMed] [Google Scholar]
- 82.Hwang WW, Madhani HD. Nonredundant requirement for multiple histone modifications for the early anaphase release of the mitotic exit regulator Cdc14 from nucleolar chromatin. PLoS Genet. 2009;5:e1000588. doi: 10.1371/journal.pgen.1000588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.De Wulf P, Montani F, Visintin R. Protein phosphatases take the mitotic stage. Curr Opin Cell Biol. 2009;21:806–15. doi: 10.1016/j.ceb.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 84.Park G, Gong Z, Chen J, Kim JE. Characterization of the DOT1L network: implications of diverse roles for DOT1L. Protein J. 2010;29:213–23. doi: 10.1007/s10930-010-9242-8. [DOI] [PubMed] [Google Scholar]
- 85.Im H, Park C, Feng Q, Johnson KD, Kiekhaefer CM, Choi K, Zhang Y, Bresnick EH. Dynamic regulation of histone H3 methylated at lysine 79 within a tissue-specific chromatin domain. J Biol Chem. 2003;278:18346–52. doi: 10.1074/jbc.M300890200. [DOI] [PubMed] [Google Scholar]
- 86.Barry ER, Krueger W, Jakuba CM, Veilleux E, Ambrosi DJ, Nelson CE, Rasmussen TP. ES cell cycle progression and differentiation require the action of the histone methyltransferase Dot1L. Stem Cells. 2009;27:1538–47. doi: 10.1002/stem.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.FitzGerald J, Moureau S, Drogaris P, O’Connell E, Abshiru N, Verreault A, Thibault P, Grenon M, Lowndes NF. Regulation of the DNA damage response and gene expression by the Dot1L histone methyltransferase and the 53Bp1 tumour suppressor. PLoS One. 2011;6:e14714. doi: 10.1371/journal.pone.0014714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chernikova SB, Razorenova OV, Higgins JP, Sishc BJ, Nicolau M, Dorth JA, Chernikova DA, Kwok S, Brooks JD, Bailey SM, et al. Deficiency in mammalian histone H2B ubiquitin ligase Bre1 (Rnf20/Rnf40) leads to replication stress and chromosomal instability. Cancer Res. 2012;72:2111–9. doi: 10.1158/0008-5472.CAN-11-2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ontoso D, Acosta I, van Leeuwen F, Freire R, San-Segundo PA. Dot1-dependent histone H3K79 methylation promotes activation of the Mek1 meiotic checkpoint effector kinase by regulating the Hop1 adaptor. PLoS Genet. 2013;9:e1003262. doi: 10.1371/journal.pgen.1003262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morohashi N, Mitchell AP, Shimizu M. Effect of histone methyltransferase gene mutations on sporulation in S. cerevisiae. Nucleic Acids Symp Ser (Oxf) 2005;•••:325–6. doi: 10.1093/nass/49.1.325. [DOI] [PubMed] [Google Scholar]
- 91.Roeder GS, Bailis JM. The pachytene checkpoint. Trends Genet. 2000;16:395–403. doi: 10.1016/S0168-9525(00)02080-1. [DOI] [PubMed] [Google Scholar]
- 92.Game JC, Williamson MS, Baccari C. X-ray survival characteristics and genetic analysis for nine Saccharomyces deletion mutants that show altered radiation sensitivity. Genetics. 2005;169:51–63. doi: 10.1534/genetics.104.028613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bostelman LJ, Keller AM, Albrecht AM, Arat A, Thompson JS. Methylation of histone H3 lysine-79 by Dot1p plays multiple roles in the response to UV damage in Saccharomyces cerevisiae. DNA Repair (Amst) 2007;6:383–95. doi: 10.1016/j.dnarep.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 94.Giannattasio M, Lazzaro F, Plevani P, Muzi-Falconi M. The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem. 2005;280:9879–86. doi: 10.1074/jbc.M414453200. [DOI] [PubMed] [Google Scholar]
- 95.Evans ML, Bostelman LJ, Albrecht AM, Keller AM, Strande NT, Thompson JS. UV sensitive mutations in histone H3 in Saccharomyces cerevisiae that alter specific K79 methylation states genetically act through distinct DNA repair pathways. Curr Genet. 2008;53:259–74. doi: 10.1007/s00294-008-0182-1. [DOI] [PubMed] [Google Scholar]
- 96.Wysocki R, Javaheri A, Allard S, Sha F, Côté J, Kron SJ. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol. 2005;25:8430–43. doi: 10.1128/MCB.25.19.8430-8443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Puddu F, Granata M, Di Nola L, Balestrini A, Piergiovanni G, Lazzaro F, Giannattasio M, Plevani P, Muzi-Falconi M. Phosphorylation of the budding yeast 9-1-1 complex is required for Dpb11 function in the full activation of the UV-induced DNA damage checkpoint. Mol Cell Biol. 2008;28:4782–93. doi: 10.1128/MCB.00330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Toh GW, O’Shaughnessy AM, Jimeno S, Dobbie IM, Grenon M, Maffini S, O’Rorke A, Lowndes NF. Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair (Amst) 2006;5:693–703. doi: 10.1016/j.dnarep.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 99.Grenon M, Costelloe T, Jimeno S, O’Shaughnessy A, Fitzgerald J, Zgheib O, Degerth L, Lowndes NF. Docking onto chromatin via the Saccharomyces cerevisiae Rad9 Tudor domain. Yeast. 2007;24:105–19. doi: 10.1002/yea.1441. [DOI] [PubMed] [Google Scholar]
- 100.Lancelot N, Charier G, Couprie J, Duband-Goulet I, Alpha-Bazin B, Quémeneur E, Ma E, Marsolier-Kergoat MC, Ropars V, Charbonnier JB, et al. The checkpoint Saccharomyces cerevisiae Rad9 protein contains a tandem tudor domain that recognizes DNA. Nucleic Acids Res. 2007;35:5898–912. doi: 10.1093/nar/gkm607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lazzaro F, Sapountzi V, Granata M, Pellicioli A, Vaze M, Haber JE, Plevani P, Lydall D, Muzi-Falconi M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008;27:1502–12. doi: 10.1038/emboj.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Conde F, Refolio E, Cordón-Preciado V, Cortés-Ledesma F, Aragón L, Aguilera A, San-Segundo PA. The Dot1 histone methyltransferase and the Rad9 checkpoint adaptor contribute to cohesin-dependent double-strand break repair by sister chromatid recombination in Saccharomyces cerevisiae. Genetics. 2009;182:437–46. doi: 10.1534/genetics.109.101899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tatum D, Li S. Evidence that the histone methyltransferase Dot1 mediates global genomic repair by methylating histone H3 on lysine 79. J Biol Chem. 2011;286:17530–5. doi: 10.1074/jbc.M111.241570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oksenych V, Zhovmer A, Ziani S, Mari PO, Eberova J, Nardo T, Stefanini M, Giglia-Mari G, Egly JM, Coin F. Histone methyltransferase DOT1L drives recovery of gene expression after a genotoxic attack. PLoS Genet. 2013;9:e1003611. doi: 10.1371/journal.pgen.1003611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Conde F, San-Segundo PA. Role of Dot1 in the response to alkylating DNA damage in Saccharomyces cerevisiae: regulation of DNA damage tolerance by the error-prone polymerases Polzeta/Rev1. Genetics. 2008;179:1197–210. doi: 10.1534/genetics.108.089003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lévesque N, Leung GP, Fok AK, Schmidt TI, Kobor MS. Loss of H3 K79 trimethylation leads to suppression of Rtt107-dependent DNA damage sensitivity through the translesion synthesis pathway. J Biol Chem. 2010;285:35113–22. doi: 10.1074/jbc.M110.116855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Conde F, Ontoso D, Acosta I, Gallego-Sánchez A, Bueno A, San-Segundo PA. Regulation of tolerance to DNA alkylating damage by Dot1 and Rad53 in Saccharomyces cerevisiae. DNA Repair (Amst) 2010;9:1038–49. doi: 10.1016/j.dnarep.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 108.Huyen Y, Zgheib O, Ditullio RA, Jr., Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–11. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 109.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–73. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sanders SL, Portoso M, Mata J, Bähler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–14. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 111.Nguyen AT, He J, Taranova O, Zhang Y. Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 2011;21:1370–3. doi: 10.1038/cr.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Marcarci BO, Unternaehrer J, Gupta PB, et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mahmoudi T, Boj SF, Hatzis P, Li VS, Taouatas N, Vries RG, Teunissen H, Begthel H, Korving J, Mohammed S, et al. The leukemia-associated Mllt10/Af10-Dot1l are Tcf4/β-catenin coactivators essential for intestinal homeostasis. PLoS Biol. 2010;8:e1000539. doi: 10.1371/journal.pbio.1000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chang MJ, Wu H, Achille NJ, Reisenauer MR, Chou CW, Zeleznik-Le NJ, Hemenway CS, Zhang W. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. 2010;70:10234–42. doi: 10.1158/0008-5472.CAN-10-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen L, Deshpande AJ, Banka D, Bernt KM, Dias S, Buske C, Olhava EJ, Daigle SR, Richon VM, Pollock RM, et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia. 2013;27:813–22. doi: 10.1038/leu.2012.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–25. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, Dias S, Chang J, Olhava EJ, Daigle SR, et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013;121:2533–41. doi: 10.1182/blood-2012-11-465120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, Marineau JJ, Qi J, Barsyte-Lovejoy D, Yi J, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- 121.Nguyen AT, Xiao B, Neppl RL, Kallin EM, Li J, Chen T, Wang DZ, Xiao X, Zhang Y. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 2011;25:263–74. doi: 10.1101/gad.2018511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wu H, Chen L, Zhang X, Zhou Q, Li JM, Berger S, Borok Z, Zhou B, Xiao Z, Yin H, et al. Aqp5 is a new transcriptional target of Dot1a and a regulator of Aqp2. PLoS One. 2013;8:e53342. doi: 10.1371/journal.pone.0053342. [DOI] [PMC free article] [PubMed] [Google Scholar]