

Abstract
Progressive microcephaly is a heterogeneous condition with causes including mutations in genes encoding regulators of neuronal survival. Here, we report the identification of mutations in QARS (encoding glutaminyl-tRNA synthetase [QARS]) as the causative variants in two unrelated families affected by progressive microcephaly, severe seizures in infancy, atrophy of the cerebral cortex and cerebellar vermis, and mild atrophy of the cerebellar hemispheres. Whole-exome sequencing of individuals from each family independently identified compound-heterozygous mutations in QARS as the only candidate causative variants. QARS was highly expressed in the developing fetal human cerebral cortex in many cell types. The four QARS mutations altered highly conserved amino acids, and the aminoacylation activity of QARS was significantly impaired in mutant cell lines. Variants p.Gly45Val and p.Tyr57His were located in the N-terminal domain required for QARS interaction with proteins in the multisynthetase complex and potentially with glutamine tRNA, and recombinant QARS proteins bearing either substitution showed an over 10-fold reduction in aminoacylation activity. Conversely, variants p.Arg403Trp and p.Arg515Trp, each occurring in a different family, were located in the catalytic core and completely disrupted QARS aminoacylation activity in vitro. Furthermore, p.Arg403Trp and p.Arg515Trp rendered QARS less soluble, and p.Arg403Trp disrupted QARS-RARS (arginyl-tRNA synthetase 1) interaction. In zebrafish, homozygous qars loss of function caused decreased brain and eye size and extensive cell death in the brain. Our results highlight the importance of QARS during brain development and that epilepsy due to impairment of QARS activity is unusually severe in comparison to other aminoacyl-tRNA synthetase disorders.
Introduction
Microcephaly refers to a condition in which an individual’s head circumference is significantly smaller than expected for age and gender, and it can manifest either congenitally or in early childhood.1–3 Autosomal-recessive primary microcephaly (MCPH) refers to congenital microcephaly with mild to moderate intellectual disability, relatively normal height, weight, appearance, and brain scan, and no other neurological findings.1 MCPH maps to at least nine genomic loci, and all reported causative mutations for MCPH are in genes that encode proteins associated with the centrosome or mitotic spindle, indicating that MCPH is caused by deficient mitosis of neural precursors.4 In contrast to MCPH, progressive microcephaly associated with diffuse cerebral-cerebellar atrophy represents a large collection of heterogeneous conditions and accompanies various neurodegenerative diseases.2,3 Genetic causes of progressive microcephaly tend to associate with defects in gene transcription5 or protein translation (see below). Progressive microcephaly with diffuse cerebral-cerebellar atrophy is classified as pontocerebellar hypoplasia (PCH) in cases in which the brain stem and pons are primarily affected.6 Out of eight genes previously reported to have causative mutations for progressive microcephaly and PCH, five encode proteins upstream of or involved in protein synthesis; of these five, three (TSEN54 [MIM 608755], TSEN2 [MIM 608753], and TSEN34 [MIM 608754])7 encode regulators of tRNA splicing, and two (SEPSECS [MIM 613009] and RARS2 [MIM 611524])8,9 encode enzymes for aminoacyl-tRNA formation.
Aminoacyl-tRNA synthetases (aaRSs) attach amino acids precisely to the correct tRNAs to maintain translational fidelity.10 There are 37 aaRSs encoded by the human genome: 18 of them function exclusively in the cytoplasm, 17 are localized in the mitochondria, and two are bifunctional.11,12 The first connection between an aaRS and human disease (Charcot-Marie-Tooth disease type 2D [MIM 601472] and distal spinal muscular atrophy type V [MIM 600794]) was identified a decade ago,13 and since then, disease-causing mutations have been reported in over a dozen aaRS-encoding genes.14,15 Mutations in mitochondrial aaRS2-encoding genes have been associated with a variety of human diseases,14,16 including mitochondrial myopathy (YARS2 [MIM 610957]),17 sensorineural deafness (HARS2 [MIM 600783] and LARS2 [MIM 604544]),18,19 spastic ataxia with leukoencephalopathy (MARS2 [MIM 609728]),20 and PCH with progressive cerebral atrophy (RARS2).9,21 In contrast, mutations in cytoplasmic aaRS-encoding genes14,16 are mainly associated with neurodegeneration of the peripheral nervous system or brain stem and spinal cord, such as in Charcot-Marie-Tooth neuropathies (GARS [MIM 600287], YARS [MIM 603623]), AARS [MIM 601065], and KARS [MIM 601421]),13,22–24 spinal muscular atrophy (GARS),13 brain stem and spinal cord hypomyelination (DARS [MIM 603084]),15 and nonsyndromic hearing loss (KARS).25 The surprisingly diverse human diseases associated with mutations in aaRS-encoding genes suggest diversified functions of individual aaRSs and mutant alleles and an exciting but poorly understood biology of protein translation during both development and degeneration of the human nervous system.
Here, we report a syndrome in two unrelated families characterized by progressive microcephaly, intractable seizures in infancy, diffuse atrophy of the cerebral cortex and cerebellar vermis, and considerably mild atrophy of the cerebellar hemispheres. We performed whole-exome sequencing in both families and identified mutations in QARS (glutaminyl-tRNA synthetase [MIM 603727]), encoding QARS, which functions in the cytoplasm, as the causative variants. Mosaic analyses in Drosophila have shown that QARS function is required for the dendritic and axonal terminal arborization during development,26 but no connection between QARS and human disease has been previously reported. We show that all four variants severely impaired QARS aminoacylation activity and that two substitutions appeared to make QARS susceptible to abnormal aggregation. Zebrafish homozygous mutants carrying a qars loss-of-function allele displayed small brain and neurodegenerative phenotypes similar to those of the human individuals carrying mutations in QARS. The distinctive features of loss of QARS activity in comparison to those of loss of other aaRSs highlight the essential and unique role of QARS during human and vertebrate brain development.
Material and Methods
Human Studies
All human protocols were reviewed and approved by the institutional review board of Boston Children’s Hospital, Hôpital Necker–Enfants Malades, and related local institutions. Informed consent was obtained from all subjects involved in this study or from parents of those who were less than 18 years old.
Whole-Exome Sequencing and Sanger Validation
For family I (MC30500, USA), DNA was extracted from peripheral-blood leukocytes, and genomic DNA was subjected to array capture with the SureSelect Human Exon Kit (Agilent Technologies) according to the manufacturer’s instructions. Adapters were ligated, and 2 × 76 bp paired-end sequencing was performed on an Illumina HiSeq 2000 at the Broad Institute, producing for each sample ∼10 Gb of sequence covering 86% of the target sequence at least 20 times. Sequencing reads were aligned to the reference human genome (hg19, UCSC Genome Browser) with the Burrows-Wheeler Aligner (v.0.5.7), consensus and variant bases were called with the Genome Analysis Toolkit, and variants were annotated with ANNOVAR. Annotated variants were entered into a MySQL database and filtered with custom queries.27
For family II (MC34900, France), DNA extracted from blood samples was captured with the SureSelect Human All Exon v.2 Kit (Agilent Technologies). Paired-end sequencing was carried out on an Illumina HiSeq 2000, which generated 100 bp reads. For each sample, 6–8 Gb of sequence was produced. The mean exome coverage was 52-fold, and 82% of the target sequence was covered at least 15 times.
To validate c.134G>T (p.Gly45Val), we amplified individual genomic DNA samples with primer pair CH91 and CH92 and Sanger sequenced them with CH90 and CH92 (see Table S1, available online, for oligonucleotide sequences). To validate c.1207C>T (p.Arg403Trp), we amplified individual genomic DNA samples with primer pair CH93 and CH94 and Sanger sequenced them with CH87 and CH93. To validate c.1543C>T (p.Arg515Trp) and c.169T>C (p.Tyr57His), we amplified genomic DNA samples and Sanger sequenced them with KCNT1-Ex1/2 F and R and KCNT1-Ex16/17 F and R, respectively.
Molecular Cloning
Wild-type (WT) human QARS coding sequence was PCR amplified from a cDNA clone (RefSeq accession number NM_005051.2, Origene SC320264) with primer pair CH379 and CH380 (see Table S1 for oligonucleotide sequences), cut with KpnI and XhoI, and ligated into pCDNA3.1A(+) for the generation of pCDNA3.1-Myc-FLAG-hsQARS-WT. Point mutations c.134G>T, c.1207C>T, c.169T>C, and c.1543C>T were introduced with the QuickChange II XL Site-Directed Mutagenesis Kit (Agilent) with primer pairs CH188 and CH189, CH190 and CH191, CH381 and CH382, CH383 and CH384, respectively. Equal amounts of five hsQARS expression vectors were transfected into Neuro2a cells (ATCC) or Cos7 cells (ATCC) with Lipofectamine 2000 (Life Technologies) for protein studies.
Immunoblotting, Coimmunoprecipitation, Immunostaining, and Microscopy
For immunoblotting, resuspended cell pellets were mixed with RIPA buffer (Pierce) and protease inhibitors (cOmplete Mini, Roche) and proteins were extracted. Immunoblots were carried out according to standard protocols. Protein immunoblots were scanned on a Li-Cor Odyssey imager.
For coimmunoprecipitation (coIP), recombinant Myc-FLAG-QARS proteins were transiently produced in human embryonic kidney 293T (HEK293T) cells (supplemented with 2.5 mM L-glutamine) and extracted with immunoprecipitation (IP) buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, and 5% glycerol supplemented with protease and phosphotase inhibitors [cOmplete Mini and PhosStop, Roche]). Cell lysates were cleared by centrifugation at 16,000 × g for 20 min at 4°C. Ten percent of the supernatant was saved as input, and the rest was incubated overnight with 20 μl anti-FLAG (M2) magnetic beads (Sigma-Aldrich) at 4°C. The beads were washed four times with 800 μl IP solution and boiled in 65 μl 1× laemmli SDS-PAGE loading buffer for 5 min. Eluted fractions, along with corresponding inputs, were subjected to SDS-PAGE and immunoblot.
Immunostaining was carried out according to standard protocols.28 Antigen retrieval (BD Retrievalgen) was performed on paraformaldehyde-fixed cryosections before staining. Alexa secondary antibodies (Life Technologies) were used, and all slides were counter stained with Hoechst33342. TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) assays were performed according to the manufacturer’s instructions (Click-iT, Life Technologies). Images were taken with an AxioVision confocal microscope (Zeiss) and processed with Adobe Photoshop CS5 and Illustrator CS5.
Antibodies used in this study include anti-QARS (SAB1406358, Sigma), anti-FLAG (M2, F3165, Sigma-Aldrich; 2368, Cell Signaling), anti-αTubulin (ab18251, Abcam), anti-Sox2 (sc-17320, Santa Cruz), anti-ERp72 (5503, Cell Signaling), anti-RCAS1 (11290, Cell Signaling), anti-phospho-histone H3 (H9908, Sigma-Aldrich), anti-Pax6 (PRB-278P, Covance), anti-HuC/D (A-21271, Life Technologies), anti-ASK1 (ab45178, Abcam), and anti-RARS (ab31537, Abcam).
Production and Purification of Recombinant Human QARS Proteins
Human QARS was codon optimized for E. coli strains (GeneArt) and cloned between NheI and XhoI sites of pET28a for the generation of an N-terminal His-tagged fusion protein with the use of the In-Fusion PCR Cloning System (Clontech). Point mutations c.134G>T, c.1207C>T, c.169T>C, and c.1543C>T were introduced with the QuickChange mutagenesis method. pET28a plasmids with WT QARS and mutant alleles were transformed into the Rosetta pLysS strain. QARS production was induced in cells that were growing in Terrific Broth II (MP) by 0.1 mM IPTG at 16°C overnight.
For purification of QARS proteins, cells from 500 ml culture were resuspended in 50 ml binding buffer (25 mM Tris, pH 8.0, 500 mM NaCl, 10% glycerol, 5 mM 2-mercaptoethanol, 0.1% Triton X-100, and 20 mM imidazole) and lysed by sonication. Cell lysate was clarified by centrifugation at 16,000 × g for 30 min. The supernatant was incubated with 1 ml Ni Sepharose High Performance beads (GE) at 4°C for 1 hr. The beads were washed with 50 ml wash buffer (25 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol, 5 mM 2-mercaptoethanol, 0.1% Triton X-100, and 50 mM imidazole) and eluted with a 10 ml elution buffer (25 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol, 5 mM 2-mercaptoethanol, 0.1% Triton X-100, and 250 mM imidazole). Eluted fractions were analyzed by SDS-PAGE, concentrated, and dialyzed twice against 1 liter of buffer (25 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% glycerol, and 5 mM 2-mercaptoethanol). Dialyzed proteins were used in aminoacylation activity assays.
Cell-Line Generation and Aminoacylation Assay
Immortalized lymphoblastoid cell lines were generated by transduction of individual blood samples with Epstein-Barr virus according to standard protocols. Lymphoblastoid cell lines were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum and antibiotics. Cells were collected, washed in 1× Dulbecco’s phosphate-buffered saline, and lysed in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM dithiothreitol, 0.5% Triton X-100, and a protease inhibitor at 4°C for 20 min. Cell debris were removed by centrifugation at 10,000 × g for 15 min. Total-protein concentration was determined with the bicinchoninic acid protein assay (Pierce) as described in the manufacture’s protocol. Then, 0.25–0.38 mg/ml of total protein was incubated with 5 μM human cytoplamic tRNAGln transcripts, 2 mM ATP, and 50 μM [3H] Gln (320 cpm/pmol) in 100 mM HEPES-NaOH, pH 7.2, 30 mM KCl, and 10 mM MgCl2 at 37°C. Aliquots were spotted on 3 MM Whatman paper discs presoaked with 5% trichloroacidic acid (TCA), washed three times with 5% TCA, dried, and scintillation counted.
Zebrafish Protocols
Zebrafish were maintained according to animal research guidelines at Boston Children’s Hospital. The qars mutant fish strain was generated by random insertion of a gene-trap cassette through retroviral infection of fish embryos.29 Insertion of the gene trap into the first intron of qars was confirmed by PCR amplification and Sanger sequencing.29 Fish genotypes were identified by PCR with primer pair CH184 and CH185 for the WT allele (350 bp) and with primer pair CH184 and CH186 for the mutant allele (315 bp).29 Male and female heterozygous fish were bred to each other, and their progeny’s tails were cut off at 2, 3, or 6 days postfertilization (dpf) for genotype determination. Fish heads of the same genotype were immediately fixed, pooled together after genotyping, and further processed for bright-field measurements, whole-mount staining, or cryosectioning. Images were taken on Zeiss microscopes, and eyes and brains were measured with AxioVision (Zeiss). Statistical analyses were carried out with GraphPad Prism.
Results
Clinical Findings in Two Families Affected by Progressive Microcephaly and Severe Epilepsy
Brothers I-1 and I-2 (family I, MC30500, Figure 1A), aged 5 years and 4 years, respectively, are from a European American nonconsanguineous family. Their key phenotypic features are presented here, and additional details are reported in Table S2. Their mother has Raynaud disease and scleroderma (MIM 613471), and their father has attention deficit hyperactivity disorder (MIM 143465). Neither parent has any history of neurological problems. The elder brother (I-1) was born at 38 weeks of gestation by normal spontaneous vaginal delivery (NSVD). At birth, he was noted to have a sloping forehead, an occipitofrontal circumference (OFC) of 27.9 cm (−3.5 SDs; below the mean for his age), normal height (50th percentile), and normal body weight (12th percentile). Individual I-1 showed severe developmental delay, and by 21 months of age, his OFC was 34 cm (−10.4 SDs below the mean), his height was 77 cm (−2.3 SDs below the mean), and his weight was 7.8 kg (−3.8 SDs below the mean).
Figure 1.
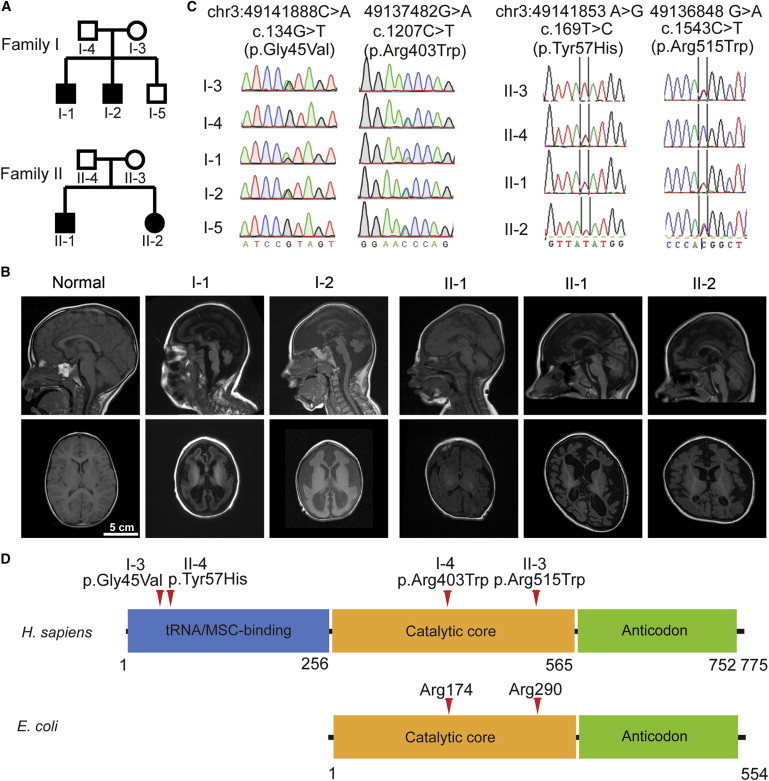
Microcephaly and Neurodegeneration in Two Nonconsanguineous Families and the Identification of QARS Mutations
(A) Pedigrees of microcephaly-affected families I (A) and II (B). Shaded symbols indicate individuals affected by microcephaly and diffuse cerebral-cerebellar atrophy.
(B) T1-weighted sagittal (top) and axial (bottom) brain MRI of normal and affected individuals. Common findings across the four affected individuals included microcephaly, enlarged subarachnoid space, enlarged lateral ventricles, thin corpus callosum, atrophy in cerebral cortex, and cerebellar vermis. Note that individuals I-1and I-2 displayed severely simplified gyral patterns and decreased contrast of white and gray matter. The individuals’ ages at examination are as follows: normal control, 2 years 8 months; I-1, 4 years 3 months; I-2, 3 years 1 month; II-1, 6 months; II-1, 5 years 4 months; and II-2, 3 years 7 months.
(C) Sanger sequencing traces confirmed that the QARS compound-heterozygous mutations in affected children were inherited separately from their mother (c.134G>T for family I; c.1543C>T for family II) or father (c.1207C>T for family I; c.169T>C for family II) and that the unaffected brother (I-5) did not have the c.134G>T mutation. Positions of variants are indicated on the genome (hg19), cDNA (RefSeq NM_005051.2), and protein (RefSeq NP_005042.1).
(D) Human QARS (RefSeq NP_005042.1) and E. coli GlnRS (RefSeq YP_488960.1) domains predicted by the NCBI and variants are indicated by red arrows.
Individual I-2 was noted to have microcephaly at 29 weeks of gestation and was born by NSVD at full term. At birth, he also had a sloping forehead, an OFC of 31.1 cm (−2.1 SDs below the mean), normal height (41th percentile), and normal body weight (25th percentile). He also exhibited profound developmental delay, and by 7 months of age, his OFC measured 34 cm (−7.8 SDs below the mean), his height was 63.5 cm (−2.1 SDs below the mean), and his weight was 6.6 kg (−2.1 SDs below the mean).
The elder brother (I-1) had seizure onset within the first hour of life and had hundreds of seizures per day and pharmacoresistant status epilepticus (SE). When he was 4 months of age, clinical reports showed 10–20 seizures per hour and persistent SE (epileptic episodes could last 10–12 hr). The seizures were described as either generalized tonic or focal clonic with either the right or the left side of the body involved and were accompanied by unresponsiveness. Trials of phenobarbital, levetiracetam, lorazepam, topiramate, oxcarbazepine, valproic acid, gabapentin, lamotrigine, and clonazepam were ineffective in controlling the seizure activity. At 18 months of age, twice per hour, he was noticed to have seizures with apnea lasting 30–60 s each time. Individual I-2 also had seizures on the first day of life; they were frequent, recurrent, and long lasting. He did not have electroencephalography (EEG) recordings and did not receive medication before 2 years of age because of parental preference and the lack of efficacy of seizure control in his brother. Individual I-2 was treated with clonazepam and gabapentin regularly for seizures after 2 years of age.
Notably, at 3 years of age, individual I-1 experienced a 4-month period of illness during which he was reported to have episodes characterized by sudden onset of constant kicking and thrashing, dehydration, pneumonia, and rhabdomyolysis with a peak creatine kinase level of >7,000 u/l. He had another similar period lasting 3 months around the age of 3.8 years. The younger brother (I-2) experienced similar episodes lasting 2 months at the age of 2 years and another one lasting 8 months at the age of 3 years. These episodes were marked by extreme agitation (especially when touched), sensitivity to sound, excessive sweating, arching, stiffness, swollen and puffy feet, thinning hair, hematemesis, and little to no sleeping. During these periods, the usual seizures were notably absent.
Individuals II-1 and II-2 (family II, MC34900, Figure 1A) are brother and sister and were born to healthy nonconsanguineous French parents of European descent. The elder brother (II-1) was born at 41 weeks of gestation after an uneventful pregnancy. At birth, he showed an OFC at −1 SD, normal body weight (tenth percentile), and normal height (51st percentile). Seizures started in the first hour of life and consisted of clonic movements of the right hemiface and lower limbs, drooling, and cyanosis. Seizures were polymorphic, long lasting, and pharmacoresistant. Ictal EEG showed a “migrating” pattern consistent with migrating partial seizures in infancy. He showed global hypotonia and lack of visual interaction. At 5.5 years of age, he showed profound psychomotor delay and microcephaly (−3 SDs) and had active epilepsy with weekly seizures that were resistant to antiepileptic drugs (AEDs).
The sister (II-2) was born at full term after an uneventful pregnancy and delivery. She had an OFC of 32 cm (−1 SD), normal height (40th percentile), and normal body weight (11th percentile). She started to have epilepsy at the age of 1 month, and her first seizures were clonic with apnea and cyanosis. Clusters of focal polymorphic seizures then occurred about twice per month and were poorly controlled despite many AED trials. When she was 5 months of age, seizures occurred in clusters of 20–30 several times per day. Clinical manifestations were variable but were often mild and accompanied by eye deviation, chewing, apnea, and cyanosis. Ictal EEG showed migrating focal seizures. When she was 15 months of age, her head circumference was below the third percentile and she had severe hypotonia with global psychomotor delay. At 3 years of age, she had weekly seizures, failed to gain further developmental skills, and had microcephaly (−2.5 SDs).
Neuroimaging Features of the Two Families
Brain MRI (Figure 1B) showed that by 5 years of age, the four affected children displayed very similar features, including microcephaly secondary to apparent neurodegeneration, hypomyelination or delayed myelination, thin corpus callosum and reduced white matter, moderately enlarged cerebral ventricles, small cerebellar vermis, and mild atrophy of the cerebellar hemispheres. Individuals I-1 and I-2 displayed more severe microcephaly (present at birth) and fewer gyral folds than did individuals II-1and II-2. In individual II-1, who had serial imaging, there was a marked increase in ventricular size and extra-axial fluid, suggesting that progressive atrophy had occurred in the interval between 6 months and 5 years. The syndrome is hereafter referred to as progressive microcephaly with diffuse cerebral-cerebellar atrophy.
Clinical Genetic Evaluation for Epilepsy and Microcephaly
Individuals I-1 and I-2 were evaluated with comparative genomic hybridization (CGH) arrays (Agilent) and SNP arrays (Affymetrix), but no clinically significant abnormalities in copy number or long continuous stretches of homozygosity were identified. Sequences of TUBA1A (MIM 602529), DCX (MIM 300121), LIS1 (MIM 607432), and ARX (MIM 300382), known genes that might have causative mutations for lissencephaly, were normal. For individual II-1, karyotype, CGH array, and sequencing of mitochondrial genes were all normal. Metabolic screening and studies of respiratory chain enzymes on muscle and liver did not identify a cause for the child’s condition. Sequencing of KCNT1 (MIM 608167), mutations in which cause migrating partial seizures,30 did not reveal any mutation.
Identification of QARS Mutations through Whole-Exome Sequencing
To identify the causative mutation(s) for individuals I-1 and I-2, we subjected genomic DNA samples from both affected children to whole-exome sequencing (WES) and aligned the sequencing reads to the reference human genome (see the Material and Methods for more details). We analyzed WES results under a recessive inheritance model, and the fact that no shared homozygous mutations were identified between the brothers is consistent with their parents’ nonconsanguinity. We then looked for possible compound-heterozygous mutations shared by individuals I-1 and I-2. After we filtered out variants that were noncoding, synonymous, and of low quality and had a high allele frequency in 1000 Genomes or the specific batch of sequenced samples, QARS and CFTR (cystic fibrosis transmembrane conductance regulator [MIM 602421]) were the only two candidate genes that remained. Because CFTR mutations are not associated with epilepsy and because the level of immunoreactive trypsinogen was normal at birth in individual I-1, we excluded CFTR and focused on QARS. Direct Sanger sequencing confirmed that the QARS mutations (c.134G>T [p.Gly45Val] and c.1207C>T [p.Arg403Trp]) were separately inherited from each parent and that the unaffected sibling inherited only one of these alleles (Figure 1C). These data suggest that the compound-heterozygous mutations in QARS are causative for the microcephaly and neurodegeneration symptoms.
For family II, blood DNA samples from both affected children and their parents were subjected to WES. Variants were filtered similarly as for family I (see the Material and Methods for details). We identified two compound-heterozygous missense variants (c.169T>C [p.Tyr57His] and c.1543C>T [p.Arg515Trp]) in QARS in both siblings, supporting the hypothesis of a recessive mode of inheritance. Both variants were confirmed by Sanger sequencing and were found to be inherited either from the father (c.169T>C) or from the mother (c.1543C>T) (Figure 1C). In addition, the two affected children also carried an apparent de novo missense variant (c.55G>A [p.Ala19Thr]) in ATP5G2 (MIM 603193), but Sanger sequencing revealed that this variant was actually inherited from the healthy father and was thus not further studied. No other novel variant compatible with a recessive or dominant mode of inheritance was identified.
Notably, none of the four parents had any history of neurological problems at the time of examination (I-3 at the age of 28 years, I-4 at 30, II-3 at 29, and II-4 at 43). Given the early age of onset for dominant phenotypes associated with other aaRSs,13,22 these healthy parents preclude the possibility that QARS-mutation-associated diseases are dominantly inherited.
QARS encodes QARS, a component of the multisynthetase complex (MSC) in human cells.31 All four variants (p.Gly45Val, p.Tyr57His, p.Arg403Trp, p.Arg515Trp) alter residues that are highly conserved across vertebrate and plant species, and Arg403 and Arg515 are conserved across the phylogenetic tree to yeast and bacteria (Figure S1). Variants p.Gly45Val and p.Tyr57His, each occurring in a different family, are located in the N-terminal domain that interacts with other aaRSs in the MSC32 and potentially interacts with glutamine tRNA,33 whereas p.Arg403Trp and p.Arg515Trp, also each occurring in a different family, reside in the aminoacylation catalytic domain (Figure 1D). All four amino acid substitutions are predicted to be damaging to protein function (Table S3).
QARS Is Widely Expressed in Fetal Human Brain during Early Development
To understand the function of QARS in the human brain, we analyzed published RNA-sequencing (RNA-seq) data34 on fetal human cerebral cortex and found that QARS mRNA was highly expressed and evenly distributed in the ventricular zone (VZ), inner subventricular zone (ISVZ), outer subventricular zone (OSVZ), and cortical plate (CP) (Figure 2A). Immunostaining on gestational week 15 normal fetal human brain sections showed high levels of QARS immunoreactivity in the VZ, ISVZ, OSVZ, and CP (Figure 2B and data not shown). Endogenous QARS was mainly distributed in the cytoplasm and showed a similar localization pattern to an endoplasmic reticulum (ER) protein, but not a Golgi protein (Figure S2A). Recombinant WT human QARS showed similar subcellular localization to endogenous monkey QARS in Cos7 cells, but we did not find any significant subcellular-localization change when each of the four substitutions was introduced (data not shown).
Figure 2.
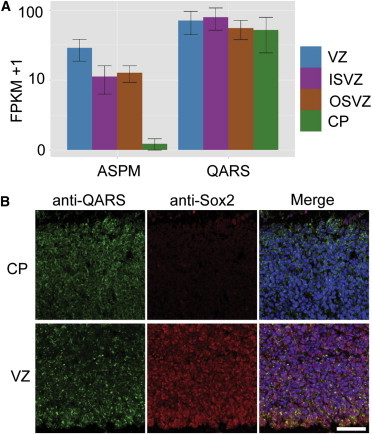
QARS Is Expressed in the Human Fetal Brain
(A) RNA-seq results from gestational week 15 (GW15) fetal human cortices were extracted from published data sets34 with the CummeRbund package in R. QARS was expressed at high levels in the VZ, ISVZ, OSVZ, and CP. ASPM expression is shown as a control.35 The mean of fragments per kilobase of transcript per million mapped reads (FPKM) ± SD is presented.
(B) Anti-QARS immunostaining results on a section of GW15 fetal human cerebral cortex show QARS amounts in the VZ and CP. The scale bar represents 200 μm.
Aminoacylation Activity of QARS Is Disrupted by Human Variants
To understand whether the function of QARS was impaired in the affected individuals, we examined glutamine aminoacylation activities by using cell extracts from lymphoblasts derived from human blood samples. We found that QARS activity was significantly lower in cell lines of individuals carrying variants in both families and mildly lower in cell lines derived from their parents than in normal control cell lines (Figure 3A–3C and Table 1). An initial aminoacylation experiment in the presence of 150 mM NaCl and 0.05% Triton X-100 (Figure 3A) revealed no significant difference in QARS activities between individuals I-1 (affected child) and I-3 (healthy mother). However, after passing the cell extracts through a G25 desalting column, we observed 30% lower QARS activity in I-1 than in I-3 (Figure 3C), suggesting that the additional variant (p.Arg403Trp) in I-1 is sensitive to misfolding at low salt concentrations.
Figure 3.
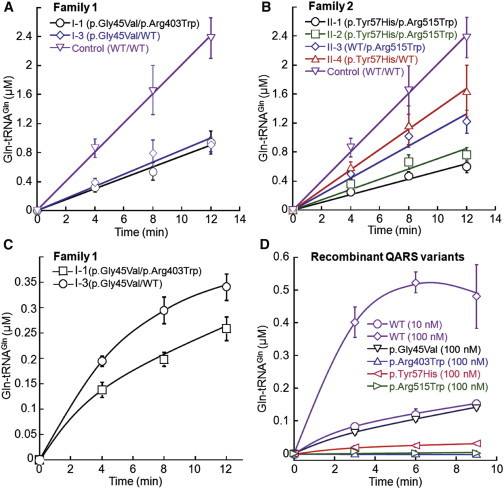
QARS Variants Impair tRNA Synthetase Activity
(A and B) Aminoacylation activity of QARS was lower in cell lines carrying compound-heterozygous QARS mutations from affected individuals I-1 (A) and II-1 and II-2 (B) than in a normal control cell line. Single substitution in p.Gly45Val (I-3), p.Tyr57His (II-4), and p.Arg515Trp (II-3) also impaired QARS aminoacylation activity. Mean values ± SD are presented.
(C) Under a desalted condition, aminoacylation activity of QARS was significantly lower in individual I-1 than in his unaffected mother (I-3). Mean values ± SD are presented.
(D) Recombinant QARS proteins were individually induced in E. coli and purified, and their aminoacylation activities are presented. Enzyme activity was undetectable in the p.Arg403Trp and p.Arg515Trp variants and was decreased to lower than 10% of WT QARS by p.Gly45Val and p.Tyr57His. Mean values ± SD are presented.
Table 1.
QARS Activity in Lymphoblast Cell Lines Derived from Healthy Controls and Individuals Carrying QARS Variants
|
Individuals with QARS Variants |
Healthy Controls |
|||||||
|---|---|---|---|---|---|---|---|---|
| 34901 | 34902 | 34903 | 34904 | 30501 | 30503 | 1403 | 2185 | |
| QARS variants | p.Tyr57His/ p.Arg515Trp |
p.Tyr57His/ p.Arg515Trp |
WT/ p.Arg515Trp |
p.Tyr57His/ WT |
p.Gly45Val/ p.Arg403Trp |
p.Gly45Val/ WT |
WT/ WT |
WT/ WT |
| Activity (nmol/min/mg)a | 0.24 ± 0.03 | 0.33 ± 0.05 | 0.51 ± 0.16 | 0.58 ± 0.11 | 0.28 ± 0.05 | 0.41 ± 0.08 | 0.83 ± 0.15 | 0.79 ± 0.19 |
QARS activity was calculated as nmol of product formation per min per mg of total protein from cell extracts. Mean values ± SD are presented.
To further explore the impact of individual human variants on QARS function, we purified each recombinant QARS variant and tested their aminoacylation activity (Figure 3D). Two variants (p.Arg403Trp and p.Arg515Trp) occurring in the catalytic domain completely disrupted QARS aminoacylation activity. The N-terminal domain variants (p.Gly45Val and p.Tyr57His) decreased QARS aminoacylation activity to less than 10% of WT QARS activity. Such a decrease in aminoacylation activity in the recombinant altered QARS proteins appears to be more drastic than that in the cell extracts, possibly because QARS is more stable in the cellular context. The addition of Triton X-100 in the cell extracts might also increase the solubility of altered QARS proteins. These results demonstrate that all four human QARS mutations cause severe loss of function.
Substitutions p.Arg403Trp and p.Arg515Trp Decrease Solubility of QARS
To determine whether reduced QARS aminoacylation activity was caused by reduced protein production, we directly tested QARS amounts in mutant cell lines. Immunoblotting showed that QARS amounts in the soluble and insoluble fractions of total protein were relatively normal in individuals I-1, II-1, and II-2 in comparison to those of their parents and control cell lines (Figure S2B).
To characterize individual variants, we cloned WT and mutant human QARS cDNAs into expression vectors and examined protein amounts in transiently transfected mouse Neuro2a cells. Interestingly, p.Arg403Trp or p.Arg515Trp altered QARS showed significantly reduced amounts in the soluble portion of total protein and increased amounts in the insoluble portion (Figure 4A). These results suggest that p.Arg403Trp and p.Arg515Trp might cause protein misfolding and aggregation.
Figure 4.
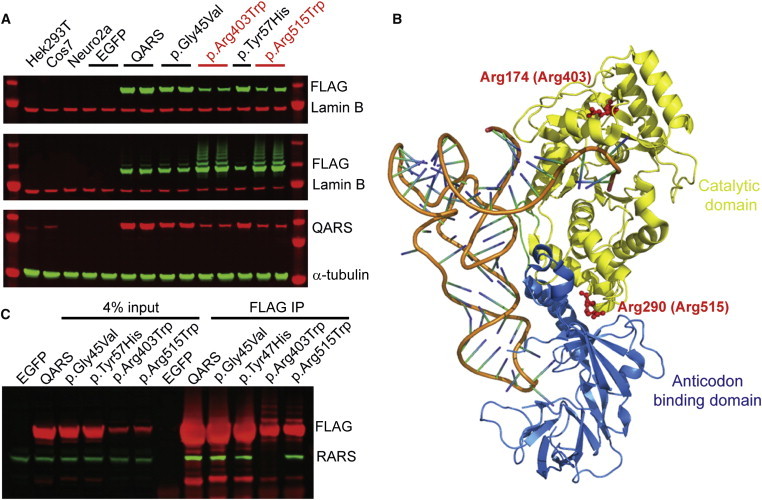
Effects of Individual Variants on QARS Solubility and Protein-Protein Interaction
(A) Immunoblot showing that the amounts of tagged p.Arg403Trp and p.Arg515Trp (anti-FLAG, green) were decreased in the supernatant (top) but increased in the pellet (middle). Rehybridization of the top membrane with anti-QARS (red) confirmed that p.Arg403Trp and p.Arg515Trp reduced the amount of QARS in the supernatant (bottom). The anti-QARS antibody was raised against human QARS. In the mouse cell line (Neuro2a), it detected recombinant human QARS, but not endogenous mouse QARS.
(B) E. coli Arg174 (equivalent to human Arg403, see Figure 1D) and Arg290 (equivalent to human Arg515) are shown on the 3D QARS structure (Protein Data Bank ID 1ZJW).36
(C) Individual recombinant QARS variants and WT QARS were immunoprecipitated (mouse anti-FLAG, clone M2) from a human cell line (HEK293T). Enrichment of recombinant proteins was confirmed by immunoblotting with a rabbit anti-FLAG antibody (red). Consistent interaction between recombinant QARS and endogenous RARS (green) was observed with all but the p.Arg403Trp variants.
Next, we analyzed the location of each variant by using the crystal structure of E. coli glutaminyl-tRNA synthetase (GlnRS),36 which is homologous to human QARS. Whereas the N-terminal domain of human QARS is absent from bacterial GlnRS, the catalytic and anticodon domains of human QARS are highly conserved in E. coli GlnRS (Figure 1D). Human positions Arg403 and Arg515 were mapped to E. coli Arg174 and Arg290, respectively. E. coli Arg174 (equivalent to human Arg403) is within the catalytic site, and E. coli Arg290 (human Arg515) is located at the interface of the catalytic and anticodon domains (Figure 4B). It is thus conceivable that these two Arg-to-Trp substitutions would disrupt the domain structure and overall folding of QARS.
Substitution p.Arg403Trp Disrupts the Interaction between QARS and RARS
The N-terminal domain of human QARS has been reported to directly interact with MARS, RARS, and IARS in the MSC.37 To examine whether p.Gly45Val and p.Tyr57His alter the interaction between QARS and MSC, we carried out coIP assays between QARS and RARS, a component of the MSC, in a HEK293T cell line. Although a clear interaction between WT QARS and RARS was detected, this interaction was not significantly affected by p.Gly45Val or p.Tyr57His (Figure 4C). However, the QARS-RARS interaction was severely disrupted by p.Arg403Trp (Figure 4C), suggesting again that the p.Arg403Trp change alters QARS confirmation.
qars Mutant Fish Display Small Eyes and Brains
To better understand the role of qars during brain development, we took advantage of a qars mutant fish that was previously reported in a large-scale genetic screen.29 A gene-trap cassette inserted into the first intron of qars is predicted to truncate the qars transcripts.29 We generated qars−/− embryonic fish by breeding qars+/− pairs and identified them at the expected Mendelian ratio at both 2 and 6 dpf. qars−/− fish demonstrated normal development up to 2 dpf, presumably because of compensation by maternal effects. At 3 dpf, qars homozygous mutant (qars−/−) fish displayed significantly smaller eyes and brains than did their qars+/− and qars+/+ siblings (Figures 5A–5D). Also starting from 3 dpf, qars−/− fish were smaller and less pigmented than their qars+/− and qars+/+ siblings. Compared to the well-balanced qars+/− and qars+/+ siblings at 5 dpf, the majority of hatched qars−/− fish were lying laterally, and when touched, they swam briefly in an unbalanced manner and settled down again on their sides. The small brain and uncoordinated movement of the qars−/− fish show striking parallels to the human symptoms.
Figure 5.
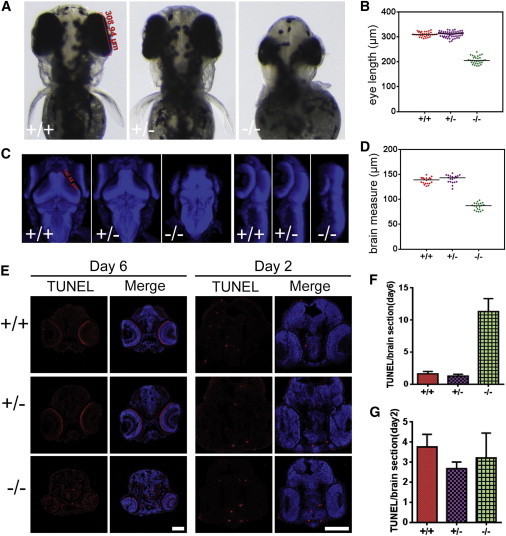
Neurodegenerative Phenotypes in the Eyes and Brains of qars Mutant Fish
(A and B) Bright-field images (A) and statistical analysis (B) show that 3 dpf qars mutant (−/−) fish displayed smaller eyes than did their heterozygous (+/−) and WT (+/+) siblings. The red bar and labeled value (A, left) show how the eye lengths were measured. Each dot (B) represents one measurement of one eye, and horizontal bars indicate the median values (309.7 μm for +/+, 312.9 μm for +/−, and 205.1 μm for −/−).
(C and D) Merged Z-stack nuclear stains (C, blue, top view in left panel and lateral view in right panel) and statistical analysis (D) show that 3 dpf qars mutant fish had smaller brains than their heterozygous (+/−) and WT (+/+) siblings. The red bar and labeled value counts (C, left) show as an example how midbrains were measured. Each dot (D) represents one measurement of one brain, and horizontal bars indicate the median values (139.3 μm for +/+, 143.3 μm for +/−, and 87.4 μm for −/−).
(E–G) TUNEL stains (E, red) and statistical analyses (F and G) of 6 dpf (left) and 2 dpf (right) fish sections show extensive cell death in qars mutant brains and eyes at 6 dpf. The scale bars represent 100 μm. Bar graphs (F and G) show that TUNEL-positive cell amounts on each section were higher in 6 dpf (F), but not 2 dpf (G), qars mutant (−/−) brains than in their +/+ and +/− siblings. Mean values ± SD are presented.
Neurodegeneration in qars−/− Fish Brains
To determine why qars−/− fish displayed small eyes and brains at 3 dpf, we examined neurogenesis and cell death in both organs before and after the defects appeared. At 2 dpf, the number of mitotic cells was relatively normal in qars−/− brains and slightly decreased in qars−/− eyes (Figure S3). Also at 2 dpf, the number of apoptotic cells in qars−/− brains was comparable to that in qars+/− and qars+/+ siblings (Figures 5E and 5G). In contrast, significantly more apoptotic cells were detected in 6 dpf qars−/− brains and eyes than in those of their qars+/− and qars+/+ siblings (Figures 5E and 5F). These results indicate that extensive cell death occurs in qars−/− brains and eyes at later stages of development. The cell-death phenotype again is consistent with the observations of progressive microcephaly and diffuse atrophy in human individuals carrying QARS mutations and suggests that QARS is essential for neuronal survival.
Discussion
Here, we have shown that loss-of-function mutations in QARS, encoding QARS, cause a syndrome of progressive microcephaly, intractable epilepsy in infancy, and diffuse brain atrophy in which the cerebral cortex and cerebellar vermis are most affected. Mutations were identified in two unrelated families affected by similar phenotypes, and loss of qars function in zebrafish caused strikingly similar defects. The mutations described thus far were all missense, and QARS variants were severely defective but appeared to retain residual biochemical activity, suggesting the possibility that the known mutations confer partial loss of function and that complete loss of QARS function might be even more severe, potentially lethal at birth or before.
GlnRS manifests as a monomer in E. coli38 and S. cerevisiae,39 and QARS manifests as a component of the MSC in human cells.40 The N-terminal domain of QARS is suggested to interact with arginyl-tRNA synthetase in the human MSC,37 and that of GlnRS contributes to the binding of tRNAGln in yeast.33 We have shown that the two variants (p.Gly45Val and p.Tyr57His) in the N-terminal domain lead to an over 10-fold decrease in aminoacylation efficiency (Figure 3C). However, the interactions with RARS do not appear to be severely affected by p.Gly45Val and p.Tyr57His, suggesting that the two variants might affect glutamine tRNA binding. Positions Arg403 and Arg515 are located in the catalytic domain; Arg403 is close to the tRNA acceptor binding site, and Arg515 is at the interface between the catalytic and anticodon domains (Figure 4B). It is highly likely that variants in the catalytic domain disrupt the domain structures and overall folding of QARS. In line with this notion, we observed that QARS variants p.Arg403Trp and p.Arg515Trp completely lost aminoacylation activity and appeared to be less soluble than WT QARS (Figure 4A). The p.Arg403Trp variant, but not p.Arg515Trp, disrupted the interaction between QARS and RARS, further supporting the idea of a protein conformation change (Figure 4C). The fact that p.Arg403Trp was more deleterious than p.Arg515Trp could relate to the more severe gyral defects in individuals I-1 and I-2 than in individuals II-1 and II-2 (Figure 1B). These results, as well as the aminoacylation activity tests on cell lines, suggest that all four individual substitutions severely impair QARS functions and that a fine threshold of QARS aminoacylation activity appears to be critical for the disease onset.
QARS has been reported to have glutamine-dependent antiapoptotic functions through binding to ASK1.41 To test the possibility that the human variants might impair this antiapoptotic function, we examined the interaction between QARS and ASK1 in the presence or absence of glutamine. In both cases, we detected a weak interaction between WT QARS and ASK1, but this interaction was not significantly weakened by p.Gly45Val, p.Tyr57His, p.Arg403Trp, or p.Arg515Trp (Figure S4 and data not shown). Thus, these variants most likely compromise QARS function by their direct effects on enzymatic activity.
Among the eight aaRSs of the MSC, aspartyl-tRNA synthetase (encoded by DARS) and lysyl-tRNA synthetase (encoded by KARS) have previously been associated with human diseases. KARS mutations were first identified in one person with Charcot-Marie-Tooth disease24 and were more recently uncovered in three independent consanguineous families affected by nonsyndromic hearing loss.25 Mutations in DARS have been reported to cause leg spasticity and hypomyelination in the brain stem and spinal cord.15 Surprisingly, the progressive microcephaly and diffuse cerebral-cerebellar atrophy caused by QARS mutations are distinct from the disorders caused by KARS or DARS mutations or by mutations in other genes encoding cytoplasmic tRNA synthetases (these latter disorders typically manifest as peripheral neuropathies;14,16 see the Introduction for more details), suggesting that mutations in QARS cause a different syndrome.
The progressive microcephaly and diffuse cerebral-cerebellar atrophy associated with QARS mutations show certain similarity to PCH with progressive microcephaly, caused by mutations in RARS2,9,21 TSEN54,7 and SEPSECS,8 all three of which encode proteins involved in or upstream of tRNA aminoacylation (see the Introduction for more details). However, the cerebral cortex and cerebellar vermis are primarily affected in individuals carrying QARS mutations, and only mild atrophy of cerebellar hemispheres has been observed; this is distinct from PCH, in which the cerebellum and brain stem are most affected, suggesting a syndrome specifically associated with mutations in QARS. Our results highlight the critical roles of QARS during CNS development and the diverse disease phenotypes caused by loss of function of tRNA synthetases in humans.
Acknowledgments
We would like to thank Nancy Mendelsohn and Janice Baker at Children’s Hospitals and Clinics of Minnesota for organizing clinical visits and information; Wen Fan Hu and Divya Jayaraman for editing the paper; and Aldo Rozzo, Byoung-Il Bae, Xuyu Cai, all C.A.W. lab members, and Chris Cho (Yale University) for helpful discussions and technical assistance. Human tissue was obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore. This project was supported by grants from the National Institute of Neurological Disorders and Stroke (R01 NS35129 to C.A.W. and K23 NS069784 to A.P.), the Manton Center for Orphan Disease Research (to C.A.W.), and Foundation Maladies Rares (to R.N.). G.H.M. is the recipient of a research grant from F. Hoffmann-La Roche Ltd. L.I.Z. is a founder and stockholder of Fate Therapeutics Inc. and Scholar Rock. C.A.W. is a Distinguished Investigator of the Paul G. Allen Foundation and a Howard Hughes Medical Institute Investigator.
Contributor Information
Christopher A. Walsh, Email: christopher.walsh@childrens.harvard.edu.
Rima Nabbout, Email: rima.nabbout@nck.aphp.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
CummeRbund, http://compbio.mit.edu/cummeRbund/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
SIFT, http://sift.jcvi.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Protein Data Bank (PDB), http://www.rcsb.org/pdb/home/home.do
References
- 1.Woods C.G., Bond J., Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet. 2005;76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barkovich A.J., Guerrini R., Kuzniecky R.I., Jackson G.D., Dobyns W.B. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012;135:1348–1369. doi: 10.1093/brain/aws019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abuelo D. Microcephaly syndromes. Semin. Pediatr. Neurol. 2007;14:118–127. doi: 10.1016/j.spen.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Megraw T.L., Sharkey J.T., Nowakowski R.S. Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol. 2011;21:470–480. doi: 10.1016/j.tcb.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufmann R., Straussberg R., Mandel H., Fattal-Valevski A., Ben-Zeev B., Naamati A., Shaag A., Zenvirt S., Konen O., Mimouni-Bloch A. Infantile cerebral and cerebellar atrophy is associated with a mutation in the MED17 subunit of the transcription preinitiation mediator complex. Am. J. Hum. Genet. 2010;87:667–670. doi: 10.1016/j.ajhg.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Namavar Y., Barth P.G., Poll-The B.T., Baas F. Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet J. Rare Dis. 2011;6:50. doi: 10.1186/1750-1172-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budde B.S., Namavar Y., Barth P.G., Poll-The B.T., Nürnberg G., Becker C., van Ruissen F., Weterman M.A., Fluiter K., te Beek E.T. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat. Genet. 2008;40:1113–1118. doi: 10.1038/ng.204. [DOI] [PubMed] [Google Scholar]
- 8.Agamy O., Ben Zeev B., Lev D., Marcus B., Fine D., Su D., Narkis G., Ofir R., Hoffmann C., Leshinsky-Silver E. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am. J. Hum. Genet. 2010;87:538–544. doi: 10.1016/j.ajhg.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ibba M., Söll D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 11.Ling J., Reynolds N., Ibba M. Aminoacyl-tRNA synthesis and translational quality control. Annu. Rev. Microbiol. 2009;63:61–78. doi: 10.1146/annurev.micro.091208.073210. [DOI] [PubMed] [Google Scholar]
- 12.Nagao A., Suzuki T., Katoh T., Sakaguchi Y., Suzuki T. Biogenesis of glutaminyl-mt tRNAGln in human mitochondria. Proc. Natl. Acad. Sci. USA. 2009;106:16209–16214. doi: 10.1073/pnas.0907602106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonellis A., Green E.D. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- 15.Taft R.J., Vanderver A., Leventer R.J., Damiani S.A., Simons C., Grimmond S.M., Miller D., Schmidt J., Lockhart P.J., Pope K. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 2013;92:774–780. doi: 10.1016/j.ajhg.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao P., Fox P.L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med. 2013;5:332–343. doi: 10.1002/emmm.201100626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riley L.G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., Lim S.C., Thorburn D., Ryan M.T., Giegé R. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am. J. Hum. Genet. 2010;87:52–59. doi: 10.1016/j.ajhg.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierce S.B., Chisholm K.M., Lynch E.D., Lee M.K., Walsh T., Opitz J.M., Li W., Klevit R.E., King M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA. 2011;108:6543–6548. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierce S.B., Gersak K., Michaelson-Cohen R., Walsh T., Lee M.K., Malach D., Klevit R.E., King M.C., Levy-Lahad E. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 2013;92:614–620. doi: 10.1016/j.ajhg.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bayat V., Thiffault I., Jaiswal M., Tétreault M., Donti T., Sasarman F., Bernard G., Demers-Lamarche J., Dicaire M.J., Mathieu J. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012;10:e1001288. doi: 10.1371/journal.pbio.1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cassandrini D., Cilio M.R., Bianchi M., Doimo M., Balestri M., Tessa A., Rizza T., Sartori G., Meschini M.C., Nesti C. Pontocerebellar hypoplasia type 6 caused by mutations in RARS2: definition of the clinical spectrum and molecular findings in five patients. J. Inherit. Metab. Dis. 2013;36:43–53. doi: 10.1007/s10545-012-9487-9. [DOI] [PubMed] [Google Scholar]
- 22.Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- 23.Latour P., Thauvin-Robinet C., Baudelet-Méry C., Soichot P., Cusin V., Faivre L., Locatelli M.C., Mayençon M., Sarcey A., Broussolle E. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2010;86:77–82. doi: 10.1016/j.ajhg.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLaughlin H.M., Sakaguchi R., Liu C., Igarashi T., Pehlivan D., Chu K., Iyer R., Cruz P., Cherukuri P.F., Hansen N.F., NISC Comparative Sequencing Program Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet. 2010;87:560–566. doi: 10.1016/j.ajhg.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santos-Cortez R.L., Lee K., Azeem Z., Antonellis P.J., Pollock L.M., Khan S., Irfanullah, Andrade-Elizondo P.B., Chiu I., Adams M.D., University of Washington Center for Mendelian Genomics Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am. J. Hum. Genet. 2013;93:132–140. doi: 10.1016/j.ajhg.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chihara T., Luginbuhl D., Luo L. Cytoplasmic and mitochondrial protein translation in axonal and dendritic terminal arborization. Nat. Neurosci. 2007;10:828–837. doi: 10.1038/nn1910. [DOI] [PubMed] [Google Scholar]
- 27.Yu T.W., Mochida G.H., Tischfield D.J., Sgaier S.K., Flores-Sarnat L., Sergi C.M., Topçu M., McDonald M.T., Barry B.J., Felie J.M. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet. 2010;42:1015–1020. doi: 10.1038/ng.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harlow E., Lane D. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1998. Using Antibodies: A Laboratory Manual. [Google Scholar]
- 29.Amsterdam A., Nissen R.M., Sun Z., Swindell E.C., Farrington S., Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barcia G., Fleming M.R., Deligniere A., Gazula V.R., Brown M.R., Langouet M., Chen H., Kronengold J., Abhyankar A., Cilio R. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 2012;44:1255–1259. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaminska M., Havrylenko S., Decottignies P., Gillet S., Le Maréchal P., Negrutskii B., Mirande M. Dissection of the structural organization of the aminoacyl-tRNA synthetase complex. J. Biol. Chem. 2009;284:6053–6060. doi: 10.1074/jbc.M809636200. [DOI] [PubMed] [Google Scholar]
- 32.Norcum M.T., Warrington J.A. Structural analysis of the multienzyme aminoacyl-tRNA synthetase complex: a three-domain model based on reversible chemical crosslinking. Protein Sci. 1998;7:79–87. doi: 10.1002/pro.5560070108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grant T.D., Snell E.H., Luft J.R., Quartley E., Corretore S., Wolfley J.R., Snell M.E., Hadd A., Perona J.J., Phizicky E.M., Grayhack E.J. Structural conservation of an ancient tRNA sensor in eukaryotic glutaminyl-tRNA synthetase. Nucleic Acids Res. 2012;40:3723–3731. doi: 10.1093/nar/gkr1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fietz S.A., Lachmann R., Brandl H., Kircher M., Samusik N., Schröder R., Lakshmanaperumal N., Henry I., Vogt J., Riehn A. Transcriptomes of germinal zones of human and mouse fetal neocortex suggest a role of extracellular matrix in progenitor self-renewal. Proc. Natl. Acad. Sci. USA. 2012;109:11836–11841. doi: 10.1073/pnas.1209647109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bond J., Roberts E., Mochida G.H., Hampshire D.J., Scott S., Askham J.M., Springell K., Mahadevan M., Crow Y.J., Markham A.F. ASPM is a major determinant of cerebral cortical size. Nat. Genet. 2002;32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 36.Gruic-Sovulj I., Uter N., Bullock T., Perona J.J. tRNA-dependent aminoacyl-adenylate hydrolysis by a nonediting class I aminoacyl-tRNA synthetase. J. Biol. Chem. 2005;280:23978–23986. doi: 10.1074/jbc.M414260200. [DOI] [PubMed] [Google Scholar]
- 37.Rho S.B., Kim M.J., Lee J.S., Seol W., Motegi H., Kim S., Shiba K. Genetic dissection of protein-protein interactions in multi-tRNA synthetase complex. Proc. Natl. Acad. Sci. USA. 1999;96:4488–4493. doi: 10.1073/pnas.96.8.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rould M.A., Perona J.J., Söll D., Steitz T.A. Structure of E. coli glutaminyl-tRNA synthetase complexed with tRNA(Gln) and ATP at 2.8 A resolution. Science. 1989;246:1135–1142. doi: 10.1126/science.2479982. [DOI] [PubMed] [Google Scholar]
- 39.Ludmerer S.W., Wright D.J., Schimmel P. Purification of glutamine tRNA synthetase from Saccharomyces cerevisiae. A monomeric aminoacyl-tRNA synthetase with a large and dispensable NH2-terminal domain. J. Biol. Chem. 1993;268:5519–5523. [PubMed] [Google Scholar]
- 40.Kaminska M., Havrylenko S., Decottignies P., Le Maréchal P., Negrutskii B., Mirande M. Dynamic Organization of Aminoacyl-tRNA Synthetase Complexes in the Cytoplasm of Human Cells. J. Biol. Chem. 2009;284:13746–13754. doi: 10.1074/jbc.M900480200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ko Y.G., Kim E.Y., Kim T., Park H., Park H.S., Choi E.J., Kim S. Glutamine-dependent antiapoptotic interaction of human glutaminyl-tRNA synthetase with apoptosis signal-regulating kinase 1. J. Biol. Chem. 2001;276:6030–6036. doi: 10.1074/jbc.M006189200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.