

Abstract
To identify further Mendelian causes of intellectual disability (ID), we screened a cohort of 996 individuals with ID for variants in 565 known or candidate genes by using a targeted next-generation sequencing approach. Seven loss-of-function (LoF) mutations—four nonsense (c.1195A>T [p.Lys399∗], c.1333C>T [p.Arg445∗], c.1866C>G [p.Tyr622∗], and c.3001C>T [p.Arg1001∗]) and three frameshift (c.2177_2178del [p.Thr726Asnfs∗39], c.3771dup [p.Ser1258Glufs∗65], and c.3856del [p.Ser1286Leufs∗84])—were identified in SETD5, a gene predicted to encode a methyltransferase. All mutations were compatible with de novo dominant inheritance. The affected individuals had moderate to severe ID with additional variable features of brachycephaly; a prominent high forehead with synophrys or striking full and broad eyebrows; a long, thin, and tubular nose; long, narrow upslanting palpebral fissures; and large, fleshy low-set ears. Skeletal anomalies, including significant leg-length discrepancy, were a frequent finding in two individuals. Congenital heart defects, inguinal hernia, or hypospadias were also reported. Behavioral problems, including obsessive-compulsive disorder, hand flapping with ritualized behavior, and autism, were prominent features. SETD5 lies within the critical interval for 3p25 microdeletion syndrome. The individuals with SETD5 mutations showed phenotypic similarity to those previously reported with a deletion in 3p25, and thus loss of SETD5 might be sufficient to account for many of the clinical features observed in this condition. Our findings add to the growing evidence that mutations in genes encoding methyltransferases regulating histone modification are important causes of ID. This analysis provides sufficient evidence that rare de novo LoF mutations in SETD5 are a relatively frequent (0.7%) cause of ID.
Main Text
The identification of over 100 rare but highly penetrant X chromosome genes in which mutations cause intellectual disability (ID) supports the hypothesis that the human genome contains more than 2,000 genes critical to normal intellectual development.1 When the causative variants are rare and when candidate genes are numerous, the interpretation of a single novel variant in a gene not previously associated with disease is challenging. The recent analysis by Piton et al. looked at evidence of pathogenicity for many of the X chromosome genes in which mutations are reported to cause ID and elegantly demonstrated how previously published evidence of disease causality needs careful review in the light of sequence data of large population sets.2 As our knowledge of rare variants in the normal population increases, there is a need to establish increasingly stringent criteria to evaluate whether a disease-causing variant has been identified to ensure the accurate translation of new knowledge into safe clinical practice.3
In order to identify further Mendelian causes of ID, we screened 996 ID-affected individuals for variants in previously associated genes and candidate genes for ID on the basis of current literature, in-house data, and sequence homology to genes previously implicated in ID. The appropriate ethical approval was obtained (research ethics committee reference 03/0/014), and parents or guardians provided written informed consent. We performed DNA sequence analysis by using next-generation sequencing methods to investigate the coding sequence of 565 genes (Table S1, available online) from 996 individuals with moderate to severe ID (all samples met DNA quality metrics). This was a subset of a large replication study of seven rare diseases and comprised a total of 2,812 individuals who were investigated within the UK10K study. The phenotypes studied were congenital heart disease, ciliopathy, coloboma, ID, neuromuscular disease, severe insulin resistance, and congenital thyroid disease; internal technical control samples were also included for comparison.
The GenomiPhi V2 DNA Amplification Kit (GE Healthcare) was used for whole-genome amplification of the DNA used for sequence analysis with the use of 1 μl of 10 ng/μl template DNA prior to pull-down. A custom-based targeted Agilent SureSelect pull-down array was designed with the SureDesign program (Agilent Technologies). This target was 3.4 Mb of sequence from the coding exons (GRCh37/hg19 human reference sequence, UCSC Genome Browser) of 1,189 genes, of which 565 were ID-related candidate or known genes. Target enrichment and amplification were performed with the HaloPlex Target Enrichment Kit (Agilent Technologies) according to the manufacturer’s instructions. The Illumina HiSeq 2000 platform was used to sequence the exons from the targeted regions. Reads were aligned to the reference genome (GRCh37/hg19) with the Burrows-Wheeler Aligner, and single-nucleotide variants (SNVs) and small indels were identified with SAMtools.4,5 For each sample, variants sites (SNVs and indels) were called with the Genome Analysis Toolkit Unified Genotyper.6 The calls were then annotated with vcf-annotate (VCFtools).7 Functional annotations were added with the Ensembl Variant Effect Predictor v.2.8 against Ensembl 70.8 Standard sequence quality-control criteria were applied to the called variants: variants with a Phred-scaled quality score > 40 and a mapping quality score > 50 were investigated further. The Integrative Genomics Viewer was used for visually inspecting the underlying sequencing data.9 In addition, only rare variants with a minor allele frequency < 1% in all of the following data sets were considered for downstream analyses: 1000 Genomes, the UK10K twins cohort, the NHLBI Exome Sequencing Project (ESP), a cohort of 2,172 individuals from whom whole exomes were sequenced at the same laboratory (UK10K), and the UK10K rare replication cohort itself (including all phenotypes). Furthermore, only putative loss-of-function (LoF) variants (nonsense, frameshift, and essential splice-site variants) were analyzed.
We selected the top ten genes with the highest number of rare LoF variants present at frequencies < 1% (Table S2). We then prioritized the genes for more detailed follow-up on the basis of the following information: (1) the number of different independent LoF variants identified in this cohort, (2) the presence of these variants in a candidate gene, and (3) the paucity of rare LoF variants in the candidate genes in controls according to frequencies in the NHLBI ESP. On the basis of the above, SET-containing-domain 5 (SETD5) was selected for further investigation.
Seven independent LoF variants were identified within the coding sequence of SETD5 in the ID cohort (Figure 1). All seven variants were observed only once within the whole UK10K replication cohort of 2,812 individuals with rare disease. The total number of SETD5 haplotypes sequenced was 5,624, which represents 1,992 alleles from individuals with ID and 3,632 alleles from individuals with other rare diseases not usually associated with ID. None of these seven LoF variants have been reported in PubMed, ClinVar, HGMD, dbSNP, or 1000 Genomes, and none were identified in the NHLBI ESP, where coverage of the respective exons was available for >4,000 European American individuals. Because none of the individuals within the cohorts contributing to the current data deposited in the NHLBI ESP were described as having intellectual impairment, this was used as an additional independent control set. The seven LoF variants within SETD5 (CCDS46741.1, RefSeq accession number NM_001080517.1) and their corresponding protein truncations (RefSeq NP_001073986.1) are the following: c.1195A>T (p.Lys399∗), c.1333C>T (p.Arg445∗), c.1866C>G (p.Tyr622∗), c.2177_2178del (p.Thr726Asnfs∗39), c.3001C>T (p.Arg1001∗), c.3771dup (p.Ser1258Glufs∗65), and c.3856del (p.Ser1286Leufs∗84) (Figure 1).
Figure 1.
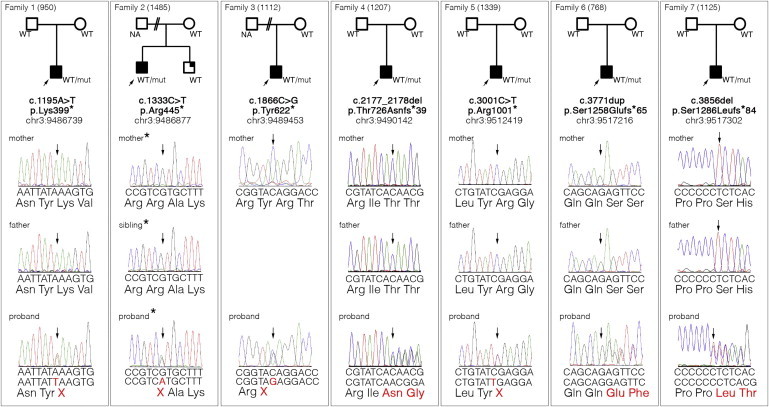
Families Affected by SETD5 Mutations
The pedigree for each family is shown at the top. Sanger sequencing electropherograms of the mutations are shown below the pedigrees. An arrow indicates the position of the mutation. The genomic coordinates are according to the GRCh37/hg19 human reference sequence. Abbreviations are as follows: NA, not available; WT, wild-type; and mut, mutation. Asterisks indicate that the Sanger sequencing illustration is of the reverse strand.
We confirmed all of the variants by Sanger sequence analysis by using stored nonamplified genomic DNA from the probands and performed familial segregation analysis to test a de novo hypothesis of disease (Figure 1). For 5/7, we were able to establish molecular evidence of a de novo variant in the proband. For the two families for which a paternal sample was unavailable, the variant was absent from the maternal sample and both parents were reported clinically to have normal intellect, suggesting that de novo inheritance was the most likely cause of disease in these families. In one of the families (family 2) with no paternal sample available, a sibling with a mild intellectual impairment was found not to carry the mutation. The number of de novo LoF variants expected to occur by chance in SETD5 in a cohort of this size was calculated with the use of the known exome mutation rate,10 the proportion of variants expected to be LoF,11 and the length of the coding sequence of SETD5 (4,329 bp). We compared this number to the observed number (five) of molecularly confirmed de novo LoF variants in SETD5 in our cohort and found that the probability that they occurred independently by chance was extremely low (p = 5.25 × 10−9, corrected for multiple testing). These results indicate that rare LoF mutations in SETD5 are a likely cause of ID.
The CCDS46741.1 transcript of the coding sequence of SETD5 is 4,329 bp long and encodes a protein of 1,442 amino acids. In this transcript, we found only one LoF variant listed in public databases: a 4 bp polymorphic indel (c.4277_4280del [p.Arg1426Profs∗82]) with genomic position chr3: 9,517,722. This LoF variant is located within the terminal 16 amino acids of the protein and is reported in 38/3,904 European American adults (from the NHLBI ESP). The seven SETD5 LoF variants present in the ID cohort are all within the CCDS46741.1 sequence and contribute to the major consensus transcript. The mutations are all located upstream of the single polymorphic LoF variant reported in the NHLBI ESP at the 3′ end of the gene. We then performed further analysis of the DNA sequence from the affected individuals to identify whether there were other more plausible variants that could account for disease. In 6/7 individuals, we did not identify further rare LoF or missense variants in any of the interrogated known genes in which mutations cause syndromic or nonsyndromic ID, nor were there rare LoF variants in candidate genes (565 genes in total). In the family 2 proband, we did identify an essential splice-site variant (c.2914+1G>A, genomic position chr3: 433,481, RefSeq NM_006614.3) in CHL1 (MIM 607416), a candidate gene not previously associated with ID. This variant was only present in the proband and was absent in the mother and mildly affected brother. In addition, in the family 2 proband, we observed a single missense variant (c.179A>G, RefSeq NM_004595.4) in SMS (MIM 300105), an X-linked gene in which mutations are reported to cause ID (Snyder-Robinson syndrome [MIM 309583]) in males. The variant was present in the unaffected mother and mildly impaired brother of the proband. The residue is not well conserved throughout evolution and is not located within a conserved domain of the protein. We concluded that this was not likely to be the primary cause of disease in the family. Furthermore, the phenotype of the mildly affected brother was not in keeping with Snyder-Robinson syndrome. Thus, it is uncertain whether this variant makes any additional contribution to the phenotype. The presence of the SETD5 variant in the proband and the absence of the variant in the younger brother are compatible with the more severe clinical features in the older brother. The additional contribution of the CHL1 variant in the proband remains uncertain.
On the basis of the genotypic similarity of the seven affected individuals, the clinical phenotype of each individual was collated from the recruiting physicians, who were blinded to the genotype for minimizing clinical bias in reporting (Figure 2). One family declined permission to publish photographs but permitted review by S.E.H. and F.L.R. Although the clinical features were variable, a number of common features other than ID included a similar facial morphology comprising brachycephaly and a prominent high forehead with striking eyebrows described as full, broad, straight, or with synophrys. The nose morphology was long, thin, and tubular. The morphology around the eyes was similar with long, narrow, and upslanting palpebral fissures; in addition, mild ptosis, unilateral amblyopia, nystagmus, and strabismus were described in single individuals. Ears tended to be large with fleshy lobes, long, and low set; one individual had a preauricular pit. The facial features of individual 2 (Figure 2) were slightly coarser than those of the other six individuals, which might reflect the additional sequence variants present. Feeding problems, particularly difficulties with swallowing and chewing, were noted by several families and physicians. Two children had congenital heart defects; one had a mitral valve prolapse, and the other had a ventricular septal defect with a patent ductus arteriosus (Table 1 and Table S3). Four of the seven children had either an inguinal hernia or hypospadias repaired at a young age. Also, 4/7 children had skeletal abnormalities that required varying degrees of intervention. Thoracic scoliosis, kyphosis, and lordosis were reported, and two children had a significant leg-length discrepancy (one of them also had talipes and hypoplasia of the left calf and required surgery). All children had intellectual impairment, although all were able to talk and communicate their needs. Speech, language, and motor developmental delay were noted in all individuals. Behavioral problems were a prominent feature of several of the children (5/7) and ranged from obsessive-compulsive disorder to hand flapping with ritualized behavior to features of autism. Involuntary movements and an exaggerated startle response were noted in several individuals, although in none were these a sustained feature over time. Older children had required special schooling because of their ID and behavioral problems, although some attended mainstream school at a young age but required educational statements and extra support. Growth parameters were within the normal range in all children, none had microcephaly or seizures, and all were born without antenatal or postnatal difficulties. Brain MRI was normal in one individual and was not performed in the remaining six individuals.
Figure 2.
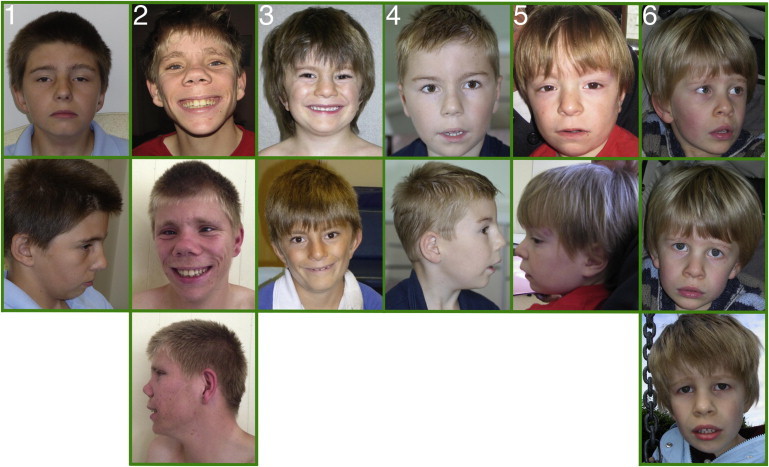
Facial Appearance of the Individuals with SETD5 Mutations
Columns numbered 1–6 correspond to families 1–6, respectively, in Figure 1. Photographs from family 7 were unavailable for publication.
Table 1.
Clinical Features of the Individuals with SETD5 Mutations and Comparison to Individuals with 3p25 Microdeletion Syndrome
| Clinical Features | Individuals with SETD5 LoF Mutations (n = 7) | Individuals with a 3p25 Deletion (n = 4) |
|---|---|---|
| Intellectual disability | 7 | 4 |
| Language delay and/or stammer | 6 | NA |
| Ritualized behavior and/or autism | 5 | NA |
| Seizures | 0 | 2 |
| Low birth weight and/or growth retardation | 0 | 2 |
| Microcephaly | 0 | 2 |
| Brachycephaly | 3 | NA |
| Low-set and/or malformed ears | 5 | 3 |
| Synophrys and/or abnormal eyebrows | 5 | 1 |
| Hypertelorism | 0 | 1 |
| Ptosis | 1 | 2 |
| Upslanting or downslanting palpebral fissures | 6 | 1 |
| Depressed nasal bridge | 3 | 3 |
| Abnormal nasal shape | 7 | NA |
| Long, smooth, and/or prominent philtrum | 5 | 3 |
| Thin upper lip | 5 | NA |
| Micrognathia | 3 | NA |
| Cleft palate | 0 | 1 |
| Postaxial polydactyly | 1 | 1 |
| Scoliosis or kyphosis | 4 | 1 |
| Leg-length discrepancy | 2 | NA |
| Feeding difficulties | 5 | NA |
| Congenital heart defects | 2 | 2 |
| Gastrointestinal and/or abdominal-wall anomalies | 5 | 1 |
Data for the 3p25 deletion were adapted from Kellogg et al.12 The following abbreviations are used: LoF, loss of function; and NA, not available.
Although multiple LoF variants in SETD5 have not been described previously in PubMed, this gene is one of three genes—THUMPD3, SETD5, and THUMPD3-AS1 (a non-protein-coding gene)—within the critical region for 3p25 microdeletion syndrome.12 Distal haploinsufficiency of chromosomal region 3p25 has long been associated with a clinical syndrome characterized by ID, low birth weight, microcephaly, telecanthus, ptosis, micrognathia, cleft palate, and congenital heart disease. Initially, the critical interval that defined the microdeletion syndrome was a 4.3 Mb region that was both large and gene dense.13 It was not clear whether deletion of a single dosage-sensitive gene within this region was sufficient to cause the syndrome or whether the phenotype was a composite of multiple gene losses. Defining the minimum common overlap of deletions in multiple individuals has reduced the critical region for 3p25 microdeletion syndrome to three genes within a 124 kb interval.12 The individuals with the smaller deletion within 3p25 have been reported to have a common phenotype of ID, hypotonia, a depressed nasal bridge, and a long philtrum. The presence of congenital heart disease and cleft palate is a more variable feature. Additional features also seen in the individuals reported with the smallest 3p25 microdeletion include synophrys, microcephaly, ptosis, abnormal palpebral fissures, postaxial polydactyly, scoliosis, cleft palate, gastrointestinal anomalies, and seizures.12,14–16 The similarity between these individuals with haploinsufficiency of 3p25 and the individuals reported here to have SETD5 truncating mutations is of note (Table 1). We suggest that similar to EHMT1 mutations in 9q34 for Kleefstra syndrome (MIM 610253) and KANSL1 mutations in 17q21 for Koolen-de Vries syndrome (MIM 610443), LoF mutations in SETD5 might be sufficient to cause many of the features of 3p25 microdeletion syndrome.
Further evidence of the potential pathogenicity of mutations in SETD5 is the observation of a single de novo LoF variant in an ID cohort and of de novo missense variants in two autism cohorts, although additional detailed phenotypic data have not been reported.10,17,18
SETD5 is a methyltransferase on the basis of sequence homology to other SET domain proteins.19 It is highly conserved throughout mammalian species, suggesting that it is functionally important, although little is known yet of its specific role. SETD5 is ubiquitously expressed, and especially high levels of SETD5 expression have been noted in the brain.20 On the basis of the other family members of this gene group, SETD5 is likely to be important in the control of histone modification of DNA and to act as a regulator of transcription. Genes encoding methyltransferases specifically and genes encoding histone modifiers in general are increasingly recognized to have a major contribution to the phenotype of ID.21 Genes encoding histone modifiers include MECP2 (MIM 300005), EHMT1 (MIM 607001), NSD1 (MIM 606681), KMT2D (MIM 602113), KDM6A (MIM 300128), and KDM5C (MIM 314690).22–28 These genes are all dosage sensitive, and haploinsufficiency alone is recognized to be sufficient to cause disease.21
Here, we present an analysis of children and young adults who were recruited to the Genetics of Learning Disability study with moderate to severe ID as the predominant clinical phenotype. This analysis provides sufficient evidence that loss of function of SETD5 is a relatively frequent cause of ID and occurs as a rare de novo mutational event. The high number of LoF mutations in this cohort (7/996 [0.7%]) suggests that SETD5 mutations, with a prevalence comparable to that of mutations in ARID1B,29 might be one of the more common causes of ID. The affected individuals showed phenotypic similarity to those previously reported with a deletion in the critical region of 3p25. Prior to mutation analysis, the clinical features alone were not sufficient or consistent for clinicians to delineate this syndrome. In none of the individuals we report was a 3p25 microdeletion syndrome clinically suspected. Genotype-driven syndrome recognition is likely to be increasingly used in the future as more subtle phenotypes emerge. This, however, poses concerns of overinterpreting the phenotypic features and the need for large data sets for distinguishing rare pertinent phenotypic associations from rare incidental findings.
Acknowledgments
We are indebted to all individuals who participated in the study. This study made use of data generated by the UK10K Project. Funding for the UK10K Project was provided by the Wellcome Trust under award WT091310. A full list of consortium members can be found at the UK10K Project website. This study was supported by grants from Action Medical Research, the Birth Defect Foundation, and the Cambridge National Institute for Health Research Biomedical Research Centre.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
Genetics Home Reference, Chromatin-modifying enzymes gene family, http://ghr.nlm.nih.gov/geneFamily/chromatinmodifyingenzymes
DECIPHER, http://decipher.sanger.ac.uk/
Ensembl Genome Browser, http://www.ensembl.org/index.html
Mutalyzer, https://mutalyzer.nl/index
NCBI HomoloGene, http://www.ncbi.nlm.nih.gov/homologene
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://omim.org
UCSC Genome Browser, http://genome-euro.ucsc.edu
UK10K Project, http://www.uk10k.org/
References
- 1.van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 2011;45:81–104. doi: 10.1146/annurev-genet-110410-132512. [DOI] [PubMed] [Google Scholar]
- 2.Piton A., Redin C., Mandel J.L. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 2013;93:368–383. doi: 10.1016/j.ajhg.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xue Y., Chen Y., Ayub Q., Huang N., Ball E.V., Mort M., Phillips A.D., Shaw K., Stenson P.D., Cooper D.N., Tyler-Smith C., 1000 Genomes Project Consortium Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 2012;91:1022–1032. doi: 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., 1000 Genomes Project Analysis Group The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauch A., Wieczorek D., Graf E., Wieland T., Endele S., Schwarzmayr T., Albrecht B., Bartholdi D., Beygo J., Di Donato N. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 11.Kryukov G.V., Pennacchio L.A., Sunyaev S.R. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellogg G., Sum J., Wallerstein R. Deletion of 3p25.3 in a patient with intellectual disability and dysmorphic features with further definition of a critical region. Am. J. Med. Genet. A. 2013;161A:1405–1408. doi: 10.1002/ajmg.a.35876. [DOI] [PubMed] [Google Scholar]
- 13.Shuib S., McMullan D., Rattenberry E., Barber R.M., Rahman F., Zatyka M., Chapman C., Macdonald F., Latif F., Davison V., Maher E.R. Microarray based analysis of 3p25-p26 deletions (3p- syndrome) Am. J. Med. Genet. A. 2009;149A:2099–2105. doi: 10.1002/ajmg.a.32824. [DOI] [PubMed] [Google Scholar]
- 14.Gunnarsson C., Foyn Bruun C. Molecular characterization and clinical features of a patient with an interstitial deletion of 3p25.3-p26.1. Am. J. Med. Genet. A. 2010;152A:3110–3114. doi: 10.1002/ajmg.a.33353. [DOI] [PubMed] [Google Scholar]
- 15.Peltekova I.T., Macdonald A., Armour C.M. Microdeletion on 3p25 in a patient with features of 3p deletion syndrome. Am. J. Med. Genet. A. 2012;158A:2583–2586. doi: 10.1002/ajmg.a.35559. [DOI] [PubMed] [Google Scholar]
- 16.Riess A., Grasshoff U., Schäferhoff K., Bonin M., Riess O., Horber V., Tzschach A. Interstitial 3p25.3-p26.1 deletion in a patient with intellectual disability. Am. J. Med. Genet. A. 2012;158A:2587–2590. doi: 10.1002/ajmg.a.35562. [DOI] [PubMed] [Google Scholar]
- 17.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.UniProt Consortium Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41(Database issue):D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagase T., Kikuno R., Hattori A., Kondo Y., Okumura K., Ohara O. Prediction of the coding sequences of unidentified human genes. XIX. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2000;7:347–355. doi: 10.1093/dnares/7.6.347. [DOI] [PubMed] [Google Scholar]
- 21.Berdasco M., Esteller M. Genetic syndromes caused by mutations in epigenetic genes. Hum. Genet. 2013;132:359–383. doi: 10.1007/s00439-013-1271-x. [DOI] [PubMed] [Google Scholar]
- 22.Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 23.Kleefstra T., Brunner H.G., Amiel J., Oudakker A.R., Nillesen W.M., Magee A., Geneviève D., Cormier-Daire V., van Esch H., Fryns J.P. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 2006;79:370–377. doi: 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurotaki N., Imaizumi K., Harada N., Masuno M., Kondoh T., Nagai T., Ohashi H., Naritomi K., Tsukahara M., Makita Y. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 2002;30:365–366. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- 25.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hannibal M.C., Buckingham K.J., Ng S.B., Ming J.E., Beck A.E., McMillin M.J., Gildersleeve H.I., Bigham A.W., Tabor H.K., Mefford H.C. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am. J. Med. Genet. A. 2011;155A:1511–1516. doi: 10.1002/ajmg.a.34074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lederer D., Grisart B., Digilio M.C., Benoit V., Crespin M., Ghariani S.C., Maystadt I., Dallapiccola B., Verellen-Dumoulin C. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 2012;90:119–124. doi: 10.1016/j.ajhg.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen L.R., Amende M., Gurok U., Moser B., Gimmel V., Tzschach A., Janecke A.R., Tariverdian G., Chelly J., Fryns J.P. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoyer J., Ekici A.B., Endele S., Popp B., Zweier C., Wiesener A., Wohlleber E., Dufke A., Rossier E., Petsch C. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 2012;90:565–572. doi: 10.1016/j.ajhg.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.