

Abstract
Microhomology-mediated end joining (MMEJ) is a Ku and Ligase IV independent mechanism for repair of DNA double-strand breaks, which contributes to chromosome rearrangements. Here we used a chromosomal end-joining assay to determine the genetic requirements for MMEJ in Saccharomyces cerevisiae. We found that end resection influences the ability to expose microhomologies; however, it is not rate limiting for MMEJ in wild-type cells. The frequency of MMEJ increased by up to 350-fold in rfa1 hypomorphic mutants, suggesting that replication protein A (RPA) bound to the ssDNA overhangs formed by resection prevents spontaneous annealing between microhomologies. In vitro, the mutant RPA complexes were unable to fully extend ssDNA and were compromised in their ability to prevent spontaneous annealing. We propose the helix-destabilizing activity of RPA channels ssDNA intermediates from mutagenic MMEJ to error-free homologous recombination, thus preserving genome integrity.
INTRODUCTION
Chromosomal double-strand breaks (DSBs) are cytotoxic lesions that occur spontaneously during normal cellular processes, by treatment of cells with DNA damaging agents or as intermediates in programmed recombination events. Failure to repair DSBs or inappropriate repair can lead to chromosome loss, deletions, duplications or translocations. Two mechanistically distinct pathways have evolved to repair DSBs: homologous recombination (HR) and non-homologous end joining (NHEJ). HR relies on an intact homologous duplex to serve as a template for repair while NHEJ involves the direct ligation of DSB ends. Canonical NHEJ is defined as Ku and Ligase IV dependent and can occur with high fidelity or be associated with small deletions or insertions at the junctions1. NHEJ junctions exhibit either no homology or short (1–4 bp) microhomologies (MH). In the absence of Ku or Ligase IV, end joining occurs at a reduced frequency and is characterized by larger deletions with longer MH at the junctions2. There may be several distinct alternative end-joining pathways in mammals, but in budding yeast Ku-independent ligation is exclusively by microhomology-mediated end joining (MMEJ)3,4.
Mechanistically, MMEJ is similar to single-strand annealing (SSA), a pathway that can be used to repair DSBs formed between long direct repeats5. Both processes initiate by nucleolytic degradation of the 5′ strands of DSBs to yield 3′ single-stranded DNA (ssDNA) tails, a process referred to as 5′–3′ resection6. Resection is required to expose homologies internal to the DNA ends that are subsequently annealed resulting in loss of one of the repeats and sequence between them.
Studies in budding yeast have shown that the conserved Mre11-Rad50-Xrs2 (MRX) complex (Mre11-Rad50-Nbs1 in mammals), together with Sae2, initiates end resection while extensive processing of the 5′ strands requires the 5′–3′ exonuclease, Exo1, or the combined activities of the Sgs1 helicase and Dna2 endonuclease6. MRX and Sae2 can act directly to initiate resection by endonucleolytic cleavage of the 5′ strand resulting in limited end processing, or MRX acts indirectly by recruiting Sgs1, Dna2 and Exo17–9. Although Sae2 and the Mre11 nuclease activity are essential to remove covalent adducts from ends, they are dispensable for resection of endonuclease-induced DSBs and function only to accelerate resection initiation10. In contrast, Schizosaccharomyces pombe or mammalian cells depleted of the Sae2 ortholog, Ctp1 or CtIP, respectively, show greatly reduced end resection initiated from an endonuclease-induced DSB, similar to loss of the MRN complex11–13. Consistent with the requirement for end resection to reveal MH internal to DSB ends, short interfering RNA (siRNA) knockdown of CtIP, reduces the frequency of MMEJ14–16. Furthermore, siRNA knockdown of CtIP reduces the frequency of DSB-induced translocations in mouse cells, and the translocation breakpoints are associated with shorter deletions and with reduced MH usage17. In mouse and yeast cells elimination of Mre11 reduces both NHEJ and MMEJ, but the junctions recovered after Mre11 depletion in mouse cells do not show a change in the length of MH4,18–21.
Annealing between exposed homologies is critical for MMEJ and SSA. Rad52 catalyzes annealing of complementary ssDNA in vitro and is required for SSA in vivo22,23; however, its role in MMEJ is controversial. MMEJ between chromosomal MH of ≤14 bp was reported to be Rad52 independent24, whereas annealing of complementary ssDNA overhangs of >8 nt in a plasmid end-joining assay was shown to be partially Rad52 dependent25. A recent study in yeast showed the efficiency of MMEJ is exquisitely sensitive to the length of MH, and sequence composition and distance between the DSB and MH also influence repair efficiency24,25. These findings suggest annealing between MHs is spontaneous and driven by the thermal stability of the annealed sequence.
RPA is a heterotrimeric ssDNA binding protein (encoded by RFA1, RFA2 and RFA3 in S. cerevisiae), required for multiple DNA transactions that involve ssDNA26. RPA prevents spontaneous annealing between complementary ssDNA in vitro; however, this inhibitory effect can be overcome by Rad5227. The hypomorphic rfa1-D228Y allele was identified as a suppressor of the rad52Δ SSA defect28,29. Genetic screens identified several other RFA1 alleles (rfa1-t11, rfa1-t33 and rfa1-t48) that impart sensitivity to DNA damaging agents, and reduce mitotic and meiotic recombination30,31. Of the rfa1 alleles tested, none reduce end resection, but the rfa-t11 mutant is defective for SSA. A recent study showed that complete depletion of RPA from yeast cells prevents long-range resection by Exo1 and Sgs1-Dna2, and short (5–9 bp) inverted repeats within the partially resected ends anneal to form hairpin structures, suggesting one important function of RPA is to prevent spontaneous annealing between MH in vivo32.
Here we used a chromosomal end-joining assay, to examine the requirement for end resection and strand annealing during MMEJ. We found that resection influences the ability to expose MH but is not rate limiting, especially when MHs are close to the break ends. Furthermore, our studies revealed a role for RPA in preventing MMEJ, suggesting that annealing between MH is spontaneous in budding yeast and this critical step is inhibited by RPA bound to ssDNA.
RESULTS
Resection initiation prevents NHEJ repair of DSBs
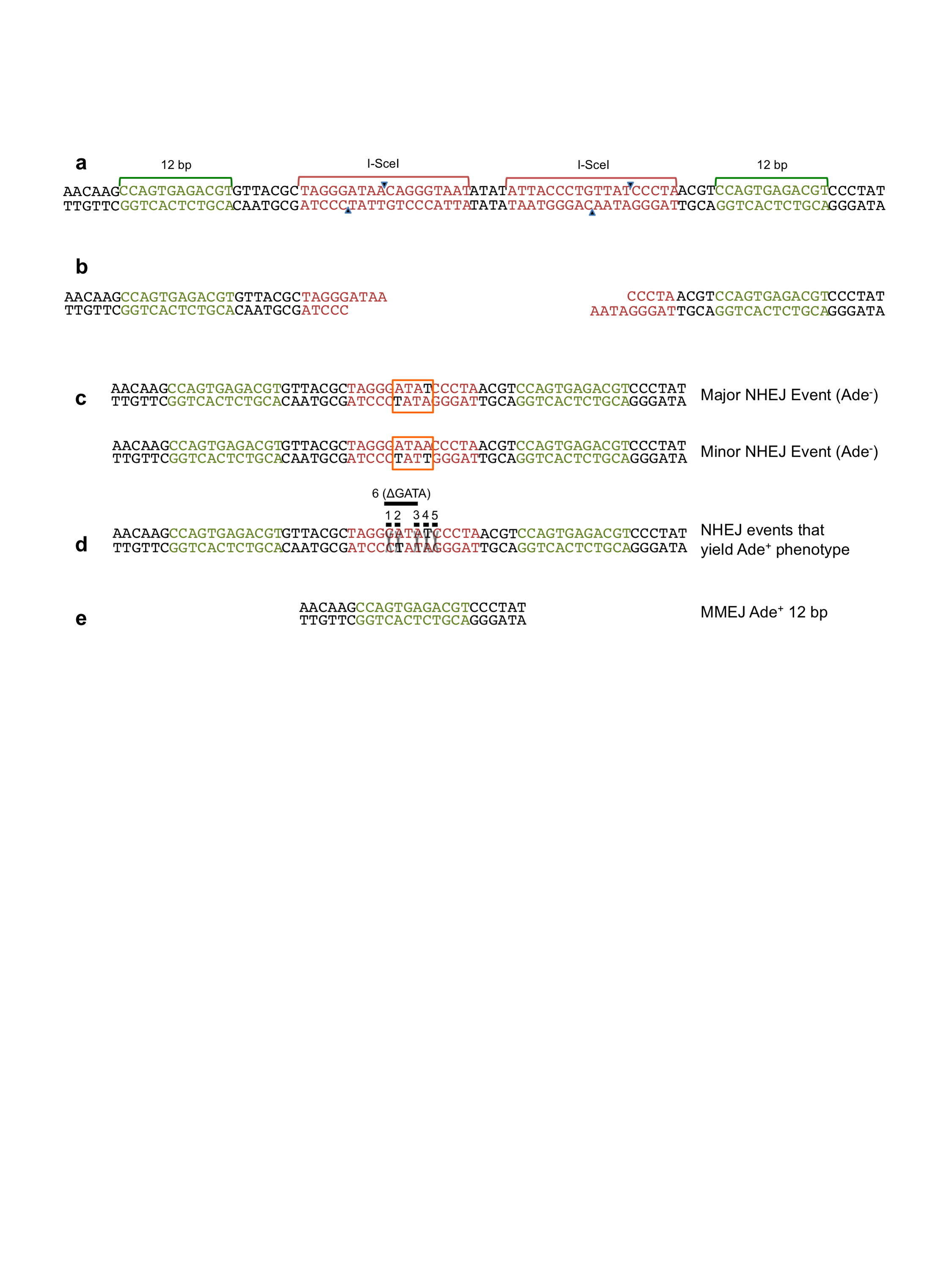
We developed a chromosomal end-joining assay to elucidate the role of resection initiation in directing repair by MMEJ or NHEJ in yeast. Two inverted 18-bp I-SceI endonuclease cleavage sites separated by a 4 bp linker and flanked by 12 bp direct repeats corresponding to ADE2 coding sequence were inserted within the ADE2 ORF, inactivating the gene. Inverted I-SceI cut sites were used to minimize multiple cycles of repair by accurate ligation and re-cleavage by I-SceI4. The 12 bp repeats were designed to restore the ADE2 coding region when repair occurs by MMEJ (Figure 1a). Yeast ade2 mutants accumulate a red pigment resulting in formation of red colonies, whereas wild type ADE2 cells form white colonies; thus, repair by MMEJ is directly scored by formation of white Ade+ colonies. Other types of end joining generate red Ade− colonies. The haploid strains used express the gene encoding I-SceI from the galactose inducible GAL1-10 promoter stably integrated at the lys2 locus. End joining is the only mechanism available to repair the I-SceI generated DSB because there is no homologous template and survival is measured by the plating efficiency of cells on medium containing galactose (I-SceI on) compared with medium containing glucose (I-SceI off).
Figure 1. Role of resection initiation and extensive resection in end-joining repair.

a, Schematic representation of the chromosomal end-joining assay. Most of the Ade+ survivors use the 12 bp direct repeats flanking the DSB to restore the ADE2 coding region. Ade− end joining products result from NHEJ, or from MMEJ between naturally occurring imperfect 16 bp repeats located 5 kb apart. b, Graphs showing survival frequencies (CFU galactose/CFU glucose) of the indicated strains. The upper panel shows all classes of events and the lower panel shows the distribution of MMEJ events. c, Graphs showing survival frequencies (left) and distribution of MMEJ products (right) in resection-initiation defective mutants. d, Graph showing survival frequency and distribution of end joined products for the exo1Δ and sgs1Δ mutants. Mean values are shown and error bars represent standard deviation (s.d.) from the mean, significance was determined by a 2-tailed Student’s t test. The number of trials for each strain is presented in Table 1. WT refers to wild type, other refers to Ade− MMEJ events which used MH other than the 16 bp interrupted MH (Supplementary Table 2).
The survival frequency of wild-type cells was 0.0036 and a majority (~98%) of survivors were Ade− (Figure 1b, Table 1). DNA sequencing revealed that 95% of the Ade− survivors repaired the DSB by NHEJ and most used the 2 bp MH within the 3′ ATAA overhangs produced by I-SceI cleavage (Supplementary Figure 1). The remaining Ade− events resulted from MMEJ between imperfect 16 bp repeats (2 mismatches within 18 bp) located 5 kb apart (Supplementary Table 1). Ade+ survivors were also sequenced and 31/37 used the 12 bp MH, but, unexpectedly, six events were due to NHEJ using the 2 bp MH within the 3′ overhangs associated with a frame-shift, or by deletion of 4 bp, to restore the ADE2 reading frame (Supplementary Figure 1). The Ade+ NHEJ events represent <1% of the total NHEJ events and are combined with the Ade− NHEJ class in Table 1.
Table 1.
Frequency of end joining in wild type and mutant strains
| Relevant genotype | Frequency NHEJ1 (x10−4) | Frequency Ade− MMEJ2 (x10−4) | Frequency Ade+ MMEJ (x10−4) | # trials |
|---|---|---|---|---|
| WT | 33.7±10.0 | 1.77 | 0.56±0.25 | 12 |
| yku70Δ | ND3 | 0.59 | 0.79±0.14 | 5 |
| dnl4Δ | ND | 0.63 | 0.45±0.14 | 4 |
| sae2Δ | 265±57.7 | ND | 0.96±0.58 | 6 |
| mre11-H125N | 165±43.0 | 2.86 | 0.87±0.40 | 4 |
| mre11Δ | ND | 0.55 | 3.22±0.95 | 5 |
| exo1Δ | 55.4±11.2 | 1.58 | 1.16±0.45 | 4 |
| sgs1Δ | 35.4±8.86 | 3.21 | 0.68±0.32 | 5 |
| exo1Δ sgs1Δ | 30.7±8.36 | ND | 13.7±3.23 | 4 |
| exo1Δ sgs1Δ yku70Δ | ND | ND | 15.4±3.53 | 3 |
| rad51Δ | 17.1±8.65 | 4.78 | 3.16±0.91 | 4 |
| rad52Δ | 13.1±5.35 | 2.72 | 1.90±0.75 | 4 |
| rad51Δ rad52Δ | 23.9±10.2 | 2.47 | 2.84±0.56 | 4 |
| rfa1-D228Y | 15.6±2.10 | 19.1 | 69.2±17.5 | 5 |
| rfa1-t33 | 23.3±4.07 | 23.3 | 47.5±16.5 | 5 |
| rfa1-t48 | 61.8±32.6 | 41.2 | 196±63.3 | 4 |
| rfa1-t11 | 24.0±6.10 | ND | 0.45±0.26 | 4 |
| rfa1-D228Y rad51Δ | 28.6±11.9 | 11.8 | 137±17.6 | 3 |
| rfa1-D228Y rad52Δ | 21.8±4.75 | 53.3 | 152±18.9 | 3 |
| rfa1-t33 sae2Δ | 137±19.3 | ND | 14.6±2.86 | 6 |
Ade− and Ade+ NHEJ events are combined
The frequency of Ade− MMEJ could not be accurately assessed in most strains because it represents a minor class of the Ade− survivors as determined by PCR analysis.
ND: Not detected.
Survival of yku70Δ and dnl4Δ mutants was reduced by >24-fold as compared to wild type. The Ade− and Ade+ events due to joining via the 2 bp MH within the I-SceI generated overhangs were eliminated in the yku70Δ and dnl4Δ mutants consistent with their formation by NHEJ. Although 53.0 and 39.1% of survivors in the yku70Δ and dnl4Δ mutants, respectively, were Ade+ due to MMEJ at the 12 bp MH, the absolute frequency of these events was the same as wild type (Figure 1b). The majority of Ade− survivors recovered from the yku70Δ and dnl4Δ mutants were formed by MMEJ using the 16 bp MH, but other MHs were identified at the junctions at a lower frequency (Supplementary Table 1). Consistent with previous studies4,19,33,34, we found the frequency of survivors to be greatly reduced in the mre11Δ mutant due to loss of NHEJ; however, the frequency of Ade+ MMEJ was higher than wild type (P=0.0001) (Figure 1b).
Survival of the sae2Δ mutant was increased by 7.4-fold relative to wild type due to an increased frequency of NHEJ. The absolute frequency of MMEJ using the 12 bp MH was similar to wild type, but no Ade− MMEJ events were detected. Only 5 of 100 Ade− survivors tested from wild type were due to MMEJ so the failure to detect one event among 120 analyzed from the sae2Δ mutant is not unexpected given the large increase in the NHEJ frequency. Similarly, survival and NHEJ increased by 4.7-fold in the mre11-H125N mutant (defective for the Mre11 nuclease activity) with no alteration in the frequency of MMEJ. End resection of endonuclease-induced DSBs is delayed in mre11Δ, mre11-H125N and sae2Δ mutants, but still occurs as a result of Exo1 and Sgs1-Dna2 activities. Since the triple mutants are inviable7, we could not assess the contribution of Sgs1-Dna2 and Exo1 to MMEJ in the absence of Mre11 or Sae2.
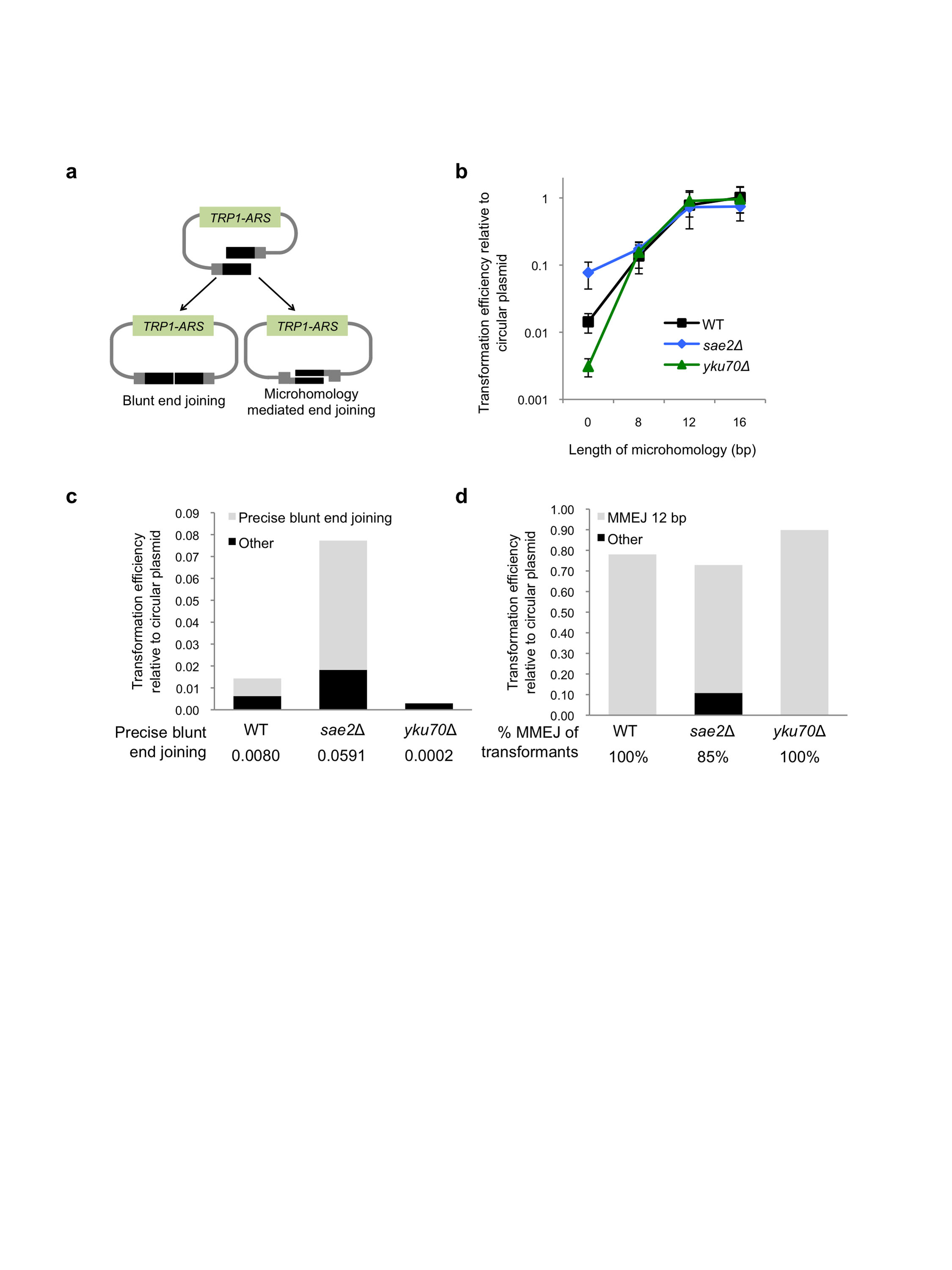
To further assess the role of Sae2 in MMEJ, a plasmid-based end-joining assay was used. Intra-molecular joining between the ends of a PCR-generated DNA fragment containing TRP1 and the ARS416 replication origin creates an autonomous replicon, detected by formation of Trp+ transformants (Supplementary Figure 2a). Primers were designed to create a substrate for blunt end joining (0 MH), or with an embedded direct repeat of 8, 12 or 16 bp to promote joining by MMEJ. We found blunt end joining is infrequent, but incorporation of repeats within the primers resulted in a length dependent increase in transformation frequency. The frequency of Trp+ transformants from the 0 MH substrate was increased by 5-fold (P=0.01) in the sae2Δ mutant compared with wild type (Supplementary Figure 2), but no significant change in the frequency of MMEJ was observed. Sequence analysis of junctions derived from the 12 MH substrate in the sae2Δ mutant revealed that 23/27 were formed by MMEJ, whereas all were due to MMEJ in wild type, indicating a subtle defect in MMEJ in the absence of Sae2 (Supplementary Figure 2d). Together, these results show that Sae2 prevents NHEJ, but is not essential for MMEJ in yeast.
Extensive resection is not required for proximal MMEJ
Exo1 and Sgs1-Dna2 act in parallel to degrade the 5′ ends of DSBs generating extensive tracts of ssDNA. In the absence of Exo1 and Sgs1-Dna2, resection by MRX-Sae2 removes nucleotides from the 5′ ends in increments of ~100 nt, but the 3′ ssDNA tails rarely exceed 700 nt7,9. Thus, we predicted the limited MRX-Sae2 dependent resection in the exo1Δ sgs1Δ mutant would be sufficient for MMEJ at the 12 bp repeats, but not for the Ade− MMEJ events. There was a small, but significant increase in both NHEJ and Ade+ MMEJ events in the exo1Δ mutant (P<0.05), but no alteration in the frequency or spectrum of events in the absence of Sgs1 (Figure 1d). The frequency of Ade+ MMEJ increased by 24-fold in the exo1Δ sgs1Δ mutant indicating that loss of extensive resection promotes MMEJ close to the DSB, and, as anticipated, no Ade− MMEJ events were recovered (Figure 1d). Because Ade− survivors are primarily due to NHEJ, we created a yku70Δ derivative of the exo1Δ sgs1Δ mutant to ensure no contribution from NHEJ and found all of the survivors from the triple mutant were Ade+ as a result of MMEJ using the 12 bp direct repeats (Table 1).
RPA suppresses MMEJ
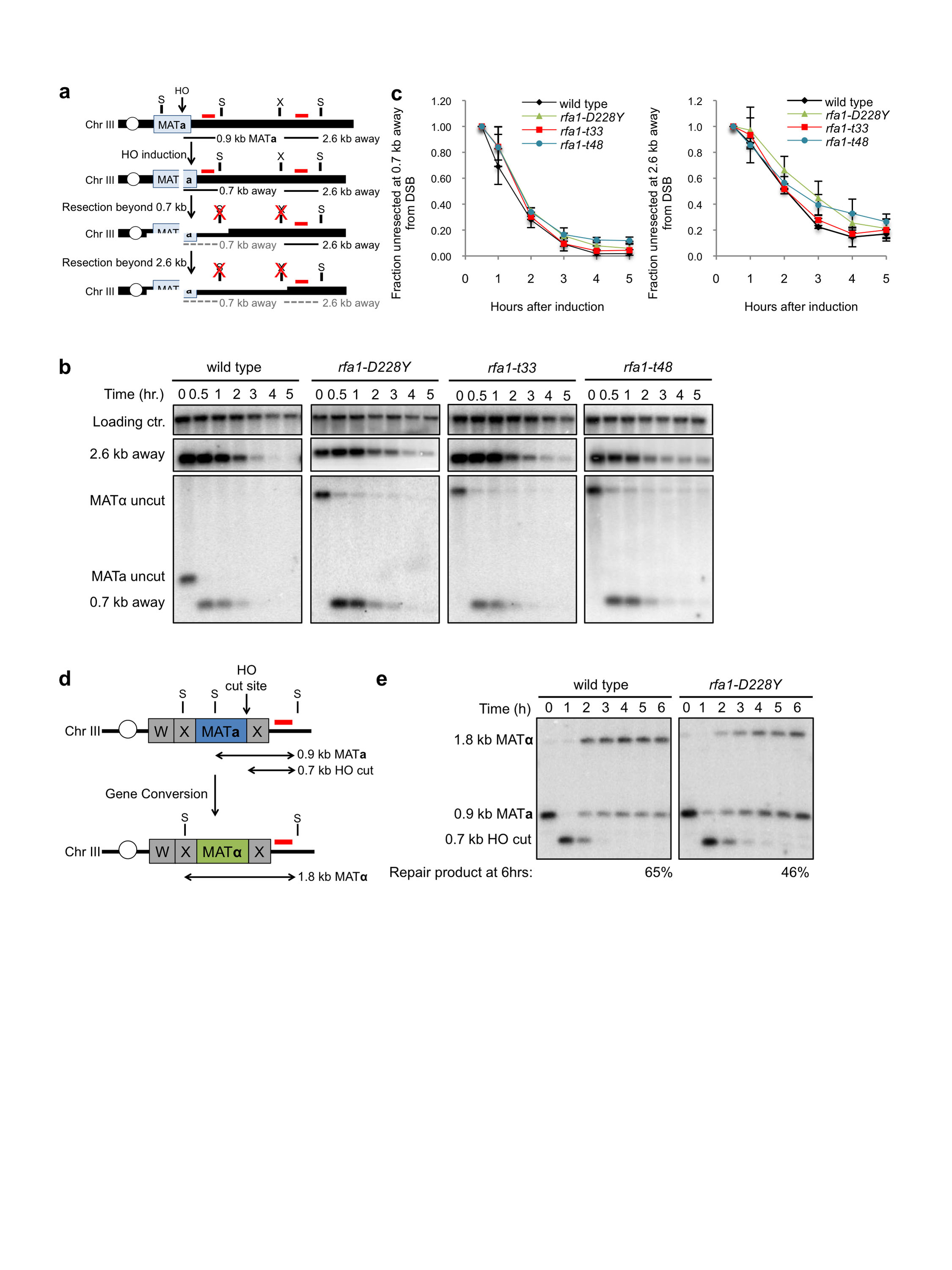
As resection is not limiting for end joining via the 12 bp MH, but the frequency of chromosomal MMEJ is very low, we considered the possibility that annealing is the limiting process. RPA removes secondary structures from ssDNA suggesting that it may prevent annealing between MH27,32. Furthermore, Smith and Rothstein identified a hypomorphic allele of RFA1 in a screen for suppressors of the SSA defect of the rad52Δ mutant, indicating that Rad52 catalyzed ssDNA annealing is no longer needed when RPA is defective28,29. Thus, we predicted the frequency of MMEJ would be elevated in the rfa1-D228Y mutant. Indeed, the frequency of Ade+ and Ade− MMEJ events increased by 124-fold and 14-fold, respectively, in the rfa1-D228Y background, resulting in higher cell survival in response to the DSB (Figure 2a and Table 1). The frequency of Ade− NHEJ remained at a similar level to wild type and no Ade+ NHEJ events were recovered.
Figure 2. Annealing between MH is prevented by RPA.

a, Graph showing survival frequencies and distribution of end joining events in rfa1 hypomorphic mutants. All NHEJ events detected in the rfa1-D228Y, rfa1-t33 and rfa1-t48 mutants were Ade−. b, Western blot of steady-state protein levels of the indicated rfa1 mutants. c, Graph showing survival frequencies and distribution of end joining events in strains over-expressing RFA1 or rfa1-D228Y in the rfa1-D228Y background. d, Viability and genotype of haploid spores derived from diploids heterozygous for sae2Δ and rfa1-D228Y. The genotype of dead spores is inferred by the segregation pattern of sae2Δ and rfa1-D228Y. e, Graph showing distribution of end joined products in the sae2Δ rfa1-t33 mutant. Mean values are shown and error bars represent s.d. from the mean, significance was determined by a 2-tailed Student’s t test. The number of trials for each strain is presented in Table 1.
The defect of the rfa1-D228Y mutant could be due to reduced abundance of the RPA(D228Y) complex or to an alteration in DNA binding by the mutant complex. Smith and Rothstein showed that over-expression of rfa1-D228Y could partially suppress the UV-sensitivity and hyper-recombination phenotype of the rfa1-D228Y mutant28. We verified that the steady state level of rfa1-D228Y is reduced relative to Rfa1 (Figure 2b and Supplementary Figure 3), but found that over-expression of the rfa1-D228Y allele in the rfa1-D228Y background did not reduce MMEJ, whereas over-expression of RFA1 resulted in a full suppression (Figure 2c).
Since rfa1-D228Y was identified in a screen for mutations that suppressed the rad52Δ direct repeat recombination defect, the increase in MMEJ could be due to a specific property of this allele rather than a general perturbation of RPA binding to ssDNA. The rfa1-t48 (L221P), rfa1-t33 (S373P), and rfa1-t11 (K45E) mutations were isolated by screening for RFA1 alleles that confer a temperature sensitive, UV- or MMS- sensitive phenotype31. Like rfa1-D228Y, rfa1-t48 and rfa1-t33 contain point mutations in the DNA binding domains, while rfa1-t11 has a mutation in the DNA Polα interaction domain. The rfa1-t48 and rfa1-t11 mutants exhibit recombination defects, and rfa1-t11 is also defective for SSA30,31. The SSA defect of the rfa1-t11 mutant is not due to decreased end resection, and in vitro studies demonstrated that Rad51 more slowly displaces RPA(t11) from ssDNA than wild-type RPA35,36. The rfa1-t33 and rfa1-t48 mutations increased the frequency of Ade+ MMEJ by 85 and 350-fold, respectively as compared to wild type, while MMEJ was unaffected by the rfa1-t11 mutation (Figure 2a). The steady state protein level of the mutant rfa1 proteins was 61–80% of wild type level and did not correlate with the MMEJ frequency (Figure 2b). Since the Ade− MMEJ events require resection of 5 kb, and this class of events also increased in the rfa1-D228Y, rfa1-t33 and rfa1-t48 mutants, it seems unlikely that the increased use of MHs in the rfa1 mutants is due to an extensive resection defect. However, to address this concern we measured end resection from an HO endonuclease-induced DSB in the rfa1-D228Y, rfa1-t33 and rfa1-t48 mutants by Southern blot hybridization, and found the mutants to be resection proficient (Supplementary Figure 4). These data suggest that the interaction between RPA and ssDNA is a critical determinant for repair by MMEJ.
In addition to MMEJ mediated by the 12 and 16 bp MHs, we identified other MHs used for repair in the rfa1 hyper-MMEJ mutants (Supplementary Table 1). In RFA1 cells, the only MMEJ events detected were mediated by the 12 and 16 bp MH; however, junctions with shorter MHs or MHs with more interruptions and mismatches were recovered from the rfa1-D228Y, rfa1-t33 and rfa1-t48 mutants. Use of other MH was also observed in the yku70Δ and dnl4Δ mutants but the frequency was >10-fold lower than found for the rfa1 mutants. Therefore, mutations that impair Rfa1 DNA binding allow for more promiscuous annealing.
Although end resection is not limiting for MMEJ in wild-type cells, we considered the possibility that Sae2 would be required if the constraint on MH annealing was reduced. Surprisingly, we were unable to generate an rfa1-D228Y sae2Δ double mutant (Figure 2d). The rfa1-D228Y rad51Δ and rfa1-D228Y rad52Δ mutants are viable indicating that the rfa1-D228Y sae2Δ lethality is not due to an increased need for HR in the rfa1-D228Y background. It is possible that promiscuous annealing in the rfa1-D228Y mutant leads to formation of secondary structures within ssDNA that require Sae2 for resolution37,38. The rfa1-t33 mutation confers a temperature-sensitive growth defect and we were able to generate an rfa1-t33 sae2Δ double mutant by germinating the spore clones at 23°. The Ade− and Ade+ MMEJ frequencies were both significantly lower for the rfa1-t33 sae2Δ double mutant than observed for the rfa1-t33 single mutant (P<0.001) indicating that resection becomes limiting when the barrier to annealing is lifted (Figure 2e).
The rfa1-D228Y mutant was reported to be defective for inter-chromosomal recombination raising the possibility that the increased frequency of MMEJ is due to reduced competition with HR (see below)28. To investigate whether rfa1-D228Y cells are defective for DSB-induced gene conversion, we used the well-characterized mating type switching system (Supplementary Figure 4). The repair efficiency of the rfa1-D228Y mutant was slightly reduced (71% of the level observed for wild type), indicating that the rfa1-D228Y mutant is largely proficient for Rad51 loading and gene conversion repair. Thus, we attribute the increased MMEJ frequency of the rfa1-D228Y mutant to be due to increased spontaneous annealing rather than defective HR.
RPA mutant complexes are defective for DNA binding in vitro
The increased MMEJ observed for the rfa1 mutants suggests the mutant complexes are compromised in their ability to prevent spontaneous annealing between short homologies. To test this hypothesis, the RPA(t33) and RPA(t48) mutant complexes were purified following expression in E. coli 39 and then tested for their ability to inhibit annealing of complementary oligonucleotides in vitro. We were unable to purify RPA(D228Y) due to instability of the complex. Strand annealing was quantified by formation of duplex product after incubating a 32P-labeled 48-mer oligonucleotide with an unlabeled complementary oligonucleotide in the absence or presence of RPA. After 8 minutes at 30°, 78% of the ssDNA oligonucleotide spontaneously annealed to form dsDNA product in the absence of added protein (Figure 3a). Consistent with a previous study27, less than 10% of the oligonucleotide annealed to its complement in the presence of wild-type RPA, whereas ~30% was annealed when incubated with RPA(t33) or RPA(t48).
Figure 3. RPA mutants are defective for ssDNA binding and disruption of secondary structure.

a, Polyacrylamide gel electrophoresis to separate substrates (ssDNA) and products (dsDNA) of the strand annealing reaction. Each gel shows time points (0–8 min) from the annealing reaction in the absence of protein or in the presence of 30 nM RPA, RPA(t33) or RPA(t48). b, Quantification of the annealed dsDNA product. Fraction of annealing was calculated as the radioactivity in the dsDNA band divided by the sum of two bands. Values are a mean of three trials and errors bars indicate s.d. c, Schematic of an ssDNA curtain assay showing ssDNA before and after RPA-eGFP binding. d, Kymographs showing ssDNA binding by RPA-eGFP, RPA(t33)-eGFP or RPA(t48)-eGFP (100 pM each), as indicated, followed by exchange with RPA-mCherry (100 pM). e, Graph of normalized signal intensity for both RPA-eGFP and RPA-mCherry over time. f, Graph comparing the dissociation kinetics of RPA-eGFP, RPA(t33)-eGFP as RPA(t48)-eGFP following the injection of RPA-mCherry (not shown). The solid black lines are single exponential fits approximating the dissociation of the eGFP-tagged RPA’s during exchange with RPA-mCherry, yielding rates of ~0.006 sec−1 for both RPA mutants and ~0.001 sec−1 for RPA-eGFP.
To further characterize the interaction of the mutant RPA complexes with ssDNA we used a DNA curtain assay and total internal reflection fluorescence microscopy (TIRFM) to directly visualize the binding of fluorescently-tagged RPA to long ssDNA molecules in real time (Figure 3c)39,40. Biotinylated ssDNA was generated by rolling circle replication and anchored to a lipid bilayer on the surface of a microfluidic sample chamber. The ssDNA was not fluorescent and remained highly compacted due to formation of extensive secondary structure. When eGFP-tagged wild-type RPA (RPA-eGFP) is injected it can bind to the tethered ssDNA, allowing it to be visualized and also causing an increase in observed contour length due to removal of secondary structure (Figure 3d, upper panel)39. RPA remains bound to the ssDNA with a half-life exceeding 2-hours when free RPA is not present in solution. However, ssDNA-bound RPA can also undergo much more rapid concentration-dependent turnover when free RPA is present in solution through a mechanism consistent with free RPA causing macroscopic dissociation of a microscopically dissociated RPA-ssDNA intermediate; this rapid protein exchange can be visualized as a change in fluorescence color of the ssDNA when switching between RPA-eGFP and RPA-mCherry (Figure 3d, e)40,41. The exchange of RPA-eGFP for RPA-mCherry does not coincide with a change in the observed extension of the ssDNA.
RPA(t33)-eGFP and RPA(t48)-eGFP both bound to the ssDNA, however, neither was able to extend the ssDNA to the same extent as was observed for the RPA-eGFP (Figure 3d, lower panels). This defect in removal of ssDNA secondary structure was revealed by allowing the binding reactions to continue for 20 minutes with the mutant RPAs, and then chasing with the same concentration of RPA-mCherry, which resulted in both rapid exchange of the mutant RPA for the RPA-mCherry and a corresponding increase in the extension of the ssDNA substrates. Exchange of RPA(t33)-eGFP and RPA(t48)-eGFP for RPA-mCherry resulted in a ~33% and ~84% increase in the observed ssDNA extension, respectively. The effect was most pronounced with RPA(t48)-eGFP, indicating that this mutant exhibits the most profound defect in secondary structure removal. In addition, quantification of the loss of eGFP signal upon injection of RPA-mCherry revealed that both RPA mutants dissociated from the ssDNA approximately 6-fold more rapidly than wild-type RPA-eGFP (Figure 3f). Taken together, the in vitro studies support the hypothesis that RPA(t33) and RPA(t48) allow more spontaneous annealing between MH in vivo as a consequence of defective ssDNA binding and removal of secondary structure.
HR competes with MMEJ repair
After resection initiation, the 3′ ssDNA coated by RPA is exchanged for Rad51 via the Rad52 mediator to initiate pairing and strand invasion with homologous duplex DNA5. If MMEJ results from delayed or failed initiation of HR we would predict the frequency of MMEJ to increase in rad51Δ and rad52Δ mutants. Consistent with this hypothesis, the rad51Δ, rad52Δ and rad51Δ rad52Δ mutants exhibited a 3 to 6-fold increase in Ade+ MMEJ (P=0.0001) (Table 1 and Figure 4a). Although a recent report showed MMEJ between repeats of >14 bp is partially RAD52 dependent24, we did not observe a decrease in the frequency of Ade− MMEJ in the rad52Δ and rad51Δ rad52Δ mutants.
Figure 4. HR and MMEJ are competing mechanisms.
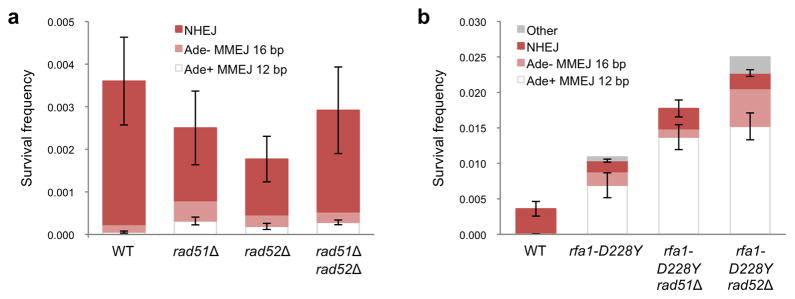
a, Graph showing survival frequency and distribution of end joining products for rad51Δ, rad52Δ and rad51Δ rad52Δ mutants. b, Frequencies of NHEJ and MMEJ in rfa1-D228Y derivatives. Mean values are shown and error bars represent s.d. from the mean, significance was determined by a 2-tailed Student’s t test. The number of trials for each strain is presented in Table 1.
Eliminating HR increases MMEJ by only 3 to 6-fold, whereas the rfa1-D228Y mutant exhibits a 124-fold increase in the frequency of Ade+ MMEJ indicating that the increase caused by dysfunctional RPA is not due simply to defective HR. If the slight HR defect of the rfa1-D228Y mutant contributed to the increased frequency of MMEJ we would predict the rfa1-D228Y mutation to be epistatic to rad51Δ and rad52Δ. The frequencies of MMEJ increased by 250 and 272-fold in the rad51Δ rfa1-D228Y and rad52Δ rfa1-D228Y double mutants, respectively, and were significantly higher than the rfa1-D228Y single mutant (P<0.005), consistent with independent functions.
DISCUSSION
Chromosomal DSBs can be repaired by several distinct mechanisms with different mutagenic potential. Interest in MMEJ has grown with the discovery that breakpoints of chromosome rearrangements frequently show MH implicating MMEJ as the underlying mechanism42–44. Here we used a chromosomal assay to monitor repair by NHEJ or by MMEJ in budding yeast and found the frequency of MMEJ to be very low (0.006%), even though a perfect 12 bp MH flanks the I-SceI cut site. Repair of a chromosomal DSB by homologous recombination, or SSA between long (>1 kb) repeats, occurs with close to 100% efficiency in S. cerevisiae45, indicating that MMEJ is rarely used to repair DSBs. By contrast, the frequency of MMEJ in mammalian cells using a substrate similar to the one we describe, but with only 8–9 bp repeats flanking the I-SceI cut site, occurs at a much higher frequency (0.5–1%) than we observe in yeast14,16. In a direct comparison of MMEJ and HR using a reporter that can detect both classes of events, the frequency of HR was only 5 to 10-fold higher than MMEJ, indicating that MMEJ plays a substantial role in DSB repair in mammalian cells16. The large difference in the frequency of MMEJ could be due to an active mechanism to prevent MMEJ in yeast, or to the presence of dedicated MMEJ synapsis and/or annealing proteins in mammalian cells. PARP-1 has been shown to synapse DNA ends in vitro and is required for end joining in the absence of Ku46–48. DNA ligase III functions with PARP-1 to catalyze alternative NHEJ in vitro, and promotes chromosome translocations in mouse cells46,49–51. PARP-1 and DNA ligase III are both absent from yeast and this could explain the low frequency of MMEJ observed. In addition, we propose that a critical step during MMEJ is annealing between MH, which is normally restricted by RPA.
In contrast to a previous report in budding yeast19, we found no MMEJ defect in the sae2Δ mutant. The system used by Lee and Lee detects MMEJ at naturally occurring MHs near the native HO cut site by sequencing the junctions present in colonies surviving HO induction19. In agreement with our findings they reported a 10-fold increase in the frequency of NHEJ in the sae2Δ mutant, but of the sequenced events none utilized MH of >5 nt. A later study using the same strain reported the same frequency of MMEJ in wild type and sae2Δ strains52. In the plasmid end-joining assay, similar frequencies of MMEJ were observed in sae2Δ and wild-type cells, but some aberrant products due to blunt end joining or partial loss of the MH were recovered from the sae2Δ mutant, whereas all of the products from wild type utilized the MH. Thus, a subtle MMEJ defect may be apparent in sae2Δ mutants depending on the assay used.
The most striking phenotype of the sae2Δ mutant is the increase in canonical NHEJ, a phenotype also observed following CtIP depletion from irradiated G2 cells53. Ku persists at DNA ends for longer in the absence of Ctp1/Sae2 suggesting that delayed resection initiation and presence of Ku allow more time for NHEJ8,12,54. An increased use of NHEJ to repair breaks in cis could potentially contribute to the decreased frequency of translocations reported for mouse cells depleted for CtIP. However, even in Ku deficient cells CtIP depletion results in reduced usage of MH at the junctions, consistent with the important role for CtIP in resection initiation and MMEJ in mammalian cells13,17.
As anticipated, we found that extensive resection is not required for MMEJ using the MH close to the break site but is needed for use of the distal MH. The frequency of Ade+ MMEJ increased by 24-fold in the exo1Δ sgs1Δ mutant suggesting that loss of extensive resection stabilizes the partially resected 3′ overhangs to provide increased opportunity for MMEJ to occur between the proximal MH. Ade+ MMEJ events were increased in the mre11Δ mutant, and the frequency of Ade− MMEJ was decreased relative to wild type, consistent with the requirement for the MRX complex to recruit the extensive resection nucleases8. If extensive resection and MMEJ via more distal MH, resulting in loss of essential genes, contributed to the low viability of wild type cells after DSB induction we might have expected an even higher frequency of survival for the exo1Δ sgs1Δ mutant. However, cell survival of the exo1Δ sgs1Δ mutant was only 0.44%, similar to wild type. Previous studies have shown that the DNA damage checkpoint is impaired in exo1Δ sgs1Δ cells, consequently, cell division might occur with an unrepaired DSB leading to mis-segregation of the acentric fragment9,55. De novo telomere addition at DSB ends is increased in the exo1Δ sgs1Δ mutant and could also contribute to cell death56.
A systematic study of MMEJ in S. cerevisiae revealed the end joining frequency to be sensitive to the length of MH24. Similarly, our results suggest that a critical step during MMEJ is the ability of MH to spontaneously anneal. RPA has been shown to inhibit annealing between oligonucleotides in vitro and to prevent the formation of DNA secondary structures in vivo27,32. By using hypomorphic alleles of RFA1 to perturb the interaction between RPA and ssDNA, we show the frequency of MMEJ can be increased by up to 350-fold, and purified RPA(t33) and RPA(t48) are less effective in removal of secondary structure from ssDNA and preventing spontaneous annealing in vitro. Furthermore, the rfa1 mutants exhibiting the greatest increase in MMEJ show a greater diversity in the sequences used for repair, with use of shorter MH and MH with more mismatches than observed in wild-type cells. Complete depletion of RPA does not prevent the initiation of end resection, but the ssDNA tails formed undergo intramolecular pairing between short inverted repeats to form hairpin capped ends32. If such structures formed in the rfa1 hypomorphic mutants they would not give rise to viable products. The only rfa1 allele tested that did not confer increased MMEJ frequency was rfa1-t11. In vitro RPA(t11) forms stable complexes with ssDNA and would be expected to inhibit strand annealing35.
A previous study showed rfa1 hypomorphic alleles confer greatly elevated rates of spontaneous gross chromosomal rearrangements (GCRs) as compared to wild type57,58. Moreover, most GCRs were due to chromosome truncation and de novo telomere addition in wild-type cells, while translocations and inversions mediated by MH were more frequently observed in the rfa1-t33 mutant. The GCR product spectrum was not determined for the rfa1-t11 and rfa1-t48 mutants. The rfa1-t33 and rfa1-t48 mutants both show elevated rates of spontaneous GCRs and increased frequencies of DSB-induced MMEJ. While the >100-fold increase in the GCR rate is most likely due to the generation of more initiating lesions when RPA is impaired, our results suggest the more promiscuous use of MMEJ contributes to the genome instability of rfa1 hypomorphic mutants. The rfa1-t48 allele, which confers a 350-fold increase in Ade+ MMEJ, causes embryonic lethality in the mouse when homozyogous and increased chromosome instability and cancer predisposition when heterozygous59. The breakpoints of chromosome rearrangements formed in the Rfa1+/t48 mouse have not been analyzed and it would be of interest to determine whether there is increased usage of MH.
In summary, we demonstrate that in wild-type cells, MMEJ is limited by RPA bound to ssDNA. The role of resection, previously thought to drive MMEJ, is necessary to reveal MH but does not guarantee annealing and completion of repair. Our studies reveal important roles for Sae2 and RPA in repair pathway choice (Figure 5). The initiation of end resection by Sae2 prevents repair by NHEJ by creating 3′ ssDNA overhangs that are poor substrates for Ku binding60, but are of sufficient length for RPA to bind and promote more extensive resection by Sgs1-Dna2 or Exo132. While ssDNA is essential for homology-directed repair, resection has the potential to reveal MH internal to the break site that can be used to align ends for mutagenic MMEJ repair. We suggest RPA plays an important role at this step to remove secondary structure from ssDNA, facilitating Rad51 nucleoprotein filament assembly, and at the same time prevents spontaneous annealing that can give rise to MMEJ. The role of RPA in binding ssDNA is highly conserved and is likely to play a role in preventing MMEJ and accompanying translocations in mammals59.
Figure 5. Regulation of repair pathway choice by Sae2 and RPA.
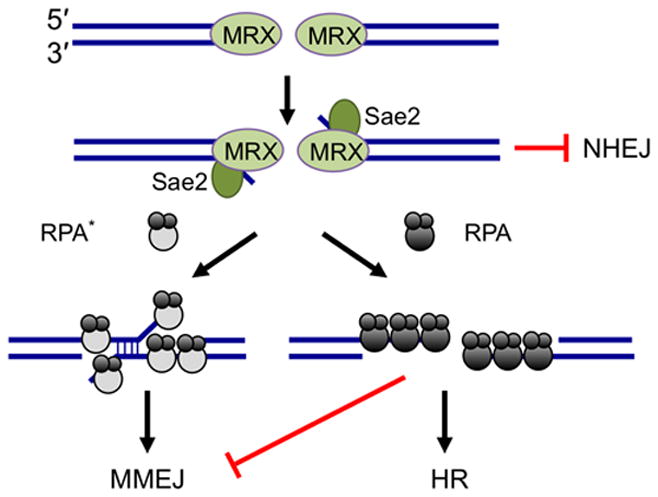
Sae2 is required for resection initiation and prevention of NHEJ repair in S and G2 phases of the cell cycle. End resection creates ssDNA, the substrate for HR and MMEJ. RPA bound to ssDNA inhibits MMEJ between MH internal to the break ends and promotes HR. When RPA binding to ssDNA is perturbed (RPA*) annealing between MH is increased and the efficiency of HR is reduced.
ONLINE METHODS
Yeast Strains
Saccharomyces cerevisiae strains used in this study are listed in Table S2. All strains are in the W303 background and were generated by PCR fragment-mediated gene targeting or by crossing appropriate haploid strains. To generate the MMEJ chromosomal system, a PCR generated ade2 fragment with two copies of the 18-bp I-SceI cut site in an inverse orientation flanked by a 12 bp MH (ade2-ISIR-12MH) was used to transform a strain with ADE2 replaced with K. lactis URA3, selection for 5-fluoroorotic acid (5-FOA) resistance and confirmed by DNA sequencing. The ade2-ISIR-12MH PCR fragment was synthesized by two-step PCR: the 5′ and 3′ fragments were amplified individually and then joined by overlap PCR. The strains used for the plasmid ligation assay were made by PCR-mediated gene replacement of the TRP1 locus of LSY1099 with kanMX6, removing sequences from 400 bp 5′ to the ORF, the complete coding region and associated ARS416 within the 3′ non-coding region.
The rfa1-t11, rfa1-t33 and rfa1-t48 mutations were introduced into LSY0678 or LSY0679 by transformation with NheI-linearized pRDK514, pRDK517 and pRS-t48/RDK4128, respectively30,57. Integration was selected for by the plasmid-borne URA3 marker, followed by selection on medium containing 5-FOA for clones that had lost URA3. Transformants that had replaced the RFA1 allele with desired mutant allele were identified by gamma-radiation sensitivity (rfa1-t11) or methyl methanesulfonate (MMS) sensitivity (rfa1-t33 and rfa1-t48). Positive clones were further confirmed by sequencing at the rfa1 locus. A clonNAT resistance marker (natMX4) was inserted 254 bp downstream of the RFA1 stop codon by transformation. Integration was selected by nourseothricin resistance and confirmed by PCR using primers internal to natMX4 cassette and RFA1. rfa1::natMX4 strains were then crossed to the ade2-ISIR-12MH strain. To ensure the natMX4 insertion does not interfere with RFA1 function, the untagged rfa1 alleles were also generated with the ade2-ISIR-12MH reporter and the resulting strains were shown to have a similar phenotype to the tagged versions (S.K.D., unpublished). RFA1 and rfa1-D228Y expressed from 2μ vectors (pWJ583 and pWJ585, respectively) were used for over-expression experiments.
Chromosomal MMEJ assay
Cells were grown to mid-log phase in YPL (1% yeast extract; 2% peptone; 3% lactic acid, pH 5.5), serial dilutions were then plated on YPL with 1% glucose or YPL with 1% galactose. Plates were incubated at 30°C for 3–5 days. The frequency of survival was determined by the number of colony forming units (CFU) on galactose containing plates divided by the number of CFUs on the glucose containing plates. Ade+ events were scored by the appearance of white colonies. DNA sequencing was initially used to distinguish between Ade+ MMEJ and NHEJ events and later a PCR assay was used because the two classes of events result in products of different lengths. Ade− MMEJ and NHEJ events were differentiated by three-primer PCR. PCR products were purified and verified by DNA sequencing. The other classes of Ade− end joined events recovered from the yku70Δ, dnl4Δ and rfa1 strains were amplified using nested primers flanking the ade2 locus and sequenced to identify the junctions. Because the Ade− MMEJ events could only be detected by PCR and represent only ~5% of the Ade− products in the wild-type strain we could not accurately calculate their frequency, except in the NHEJ defective and hyper-MMEJ rfa1 mutants, and no error bars are shown in Table 1 or the Figures. To compare the Ade− MMEJ events from strains with similar frequencies of Ade− survivors the Fisher exact test was used taking the total number of events analyzed from multiple trials.
For RFA1 and rfa1-D228Y over-expression, cells were grown in synthetic complete medium lacking leucine (SC-LEU) with 2% raffinose substituted for glucose and plated on SC-LEU + 2% glucose or SC-LEU + 2% galactose. Cells were grown for 3–5 days at 30°C. Classes of repair events were categorized as described above.
Plasmid end joining assay
The linear end joining substrate was amplified from the pLL111 plasmid using primers that anneal to sequences 320 bp upstream and 340 bp downstream of the TRP1 ORF and associated ARS41661. The primers were designed with the desired length of microhomology at the ends of the PCR fragment or with no homology at the ends62. Phusion polymerase (New England Biolabs) was used to avoid 3′ dA overhangs. PCR products were gel purified prior to transformation of yeast cells. 100 ng of each linear substrate or circular pLL111 was used to transform mid-log phase cells selecting for Trp+ transformants. The frequency of end joining was determined by the number of Trp+ transformants derived from the linear fragment relative to pLL111. The junctions formed by end joining were amplified by PCR from at least 16 independent Trp+ colonies from the blunt end or 12 bp MH substrates and then sequenced.
Mating type switching and end resection assays
The mating type switching assay was performed as previously described7. Briefly, HO-endonuclease expression was induced by addition of 2% galactose to cells grown in YPL. After one hour, cells were collected and resuspended in YPL + 2% glucose to prevent HO expression and allow repair by gene conversion. Samples were collected at indicated time points and genomic DNA was extracted and digested by StyI for Southern blot analysis. To detection of the HO-cut fragment and repaired products, a probe was generated by PCR amplification of MAT sequences distal to the HO-cut site (coordinates 201176–201580 on chromosome III sequence). The assays were performed twice and a representative gel shown in Supplementary Figure 3. End resection was measured by a similar protocol except the strains used lack the HML and HMR loci preventing gene conversion repair and HO was expressed continuously through the time course. The MAT-2.6 kb probe was used (coordinates 204184–204893 on chromosome III) was used to detect the fragment 2.6 kb distal to the HO-cut site, A POX1 probe (coordinates 108631–109001 on chromosome XII) was used for normalization of band intensities using Image J (NIH). DSB end resection for each time point was estimated as a ratio of the signal intensity corresponding to that before induction and represents the mean of 3 independent experiments.
Western blot analysis
Whole cell extracts prepared by TCA precipitation were analyzed by SDS-PAGE and western blot with anti-S.c.Rfa1 (Agrisera) and anti-α-tubulin (Sigma Aldrich) as a loading control.
Purification of wild type and mutant RPA
The rfa1-t33, rfa1-t48 and rfa1-D228Y mutations were generated using the Q5 site-directed mutagenesis kit (New England Biolabs) with RPA-eGFP (p11d-tscRPA_30gfphis6) and unlabeled RPA (p11d-tscRPA_30MxeHis6) plasmids as templates. The mutations were verified by DNA sequencing.
The RPA mutants, both eGFP and untagged versions, were expressed in BL21DE3 cells overnight at 16 °C. Pellets from 3L of cells were resuspended in Ni-Lysis buffer (50mM NaKPO4 pH 8.0, 150mM NaCl, 10mM Imidazole) and frozen at −80 °C. Purification of RPA-eGFP, RPA(t33)-eGFP and RPA(t48)-eGFP was performed as described39,40. Briefly, the clarified lysate after sonication and centrifugation was applied to a 8 ml Ni-NTA column and washed with 40 ml of Ni-Wash buffer (50mM NaKPO4 pH 8.0, 150 mM NaCI, 20 mM imidazole). The protein was eluted in approximately 25 ml of Ni-Elution buffer (50 mM NaKPO4 pH 8.0, 150 mM NaCl, 200 mM imidazole) and dialyzed overnight against 1L buffer (40 mM NaCl, 20 mM Tris-HCl pH 7.4, 1 mM DTT, 0.5 mM EDTA). The dialyzed protein was applied to MonoQ (5/50 GL: GE Healthcare) and developed with a gradient from 4–30% Buffer B (1 M NaCl, 20 mM Tris-HCl pH 7.4, 1 mM DTT, 0.5 mM EDTA) over 100 column volumes (CV). MonoQ fractions containing RPA were pooled and the MonoQ chromatography step repeated to increase protein purity. The pooled fractions corresponding to RPA-eGFP heterotrimeric complex were pooled, concentrated, dialyzed into storage buffer (100 mM NaCl, 20 mM Tris-HCl pH 7.4, 1 mM DTT, 50% glycerol), and aliquotted for storage at −80 °C.
Unlabeled RPA, RPA(t33) and RPA(t48) purification was identical to the RPA-eGFP tagged versions until the after Ni-NTA elution. The Ni-elution was directly applied to a 10 ml Chitin column, and washed with 4 CVs of chitin wash buffer (CWB: 20 mM Tris-HCl pH 8.0, 250 mM NaCl, 1 mM EDTA). The column was then exchanged into CWB containing 50 mM DTT and incubated overnight at 4 °C to allow for intein-mediated cleavage. The cleaved protein was collected as flow through and concentrated to approximately 5 ml. The concentrated protein was applied to a Superdex 200 16/60 column (GE Healthcare) and run with 40 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM DTT buffer. Fractions corresponding to pure heterotrimeric complex were pooled, concentrated, dialyzed into storage buffer, and aliquotted for storage at −80 °C. Concentrations of unlabeled RPA and RPA-eGFP variants were determined using the extinction coefficients 8.8 × 104 and 1.16 × 106, respectively, at 280 nM.
Strand Annealing Assays
Annealing of 32P-labeled 48-mer oligonucleotide (oligo-25) with the complementary unlabeled oligonucleotide (oligo-26) was performed as previously described27,63. Briefly, the reaction buffer contained 30 mM Tris-Cl (pH 7.5), 5 mM MgCl2, and 1 mM DTT. DNA concentrations were 200 nM and RPA concentration was 30 nM in all reactions. Reactions were initiated by the addition of unlabeled oligo-26 and quenched by the addition of excess unlabeled oligo 25. Annealing was monitored by separation through 12% polyacrylamide in 1x TBE buffer. Results were visualized by phosphoimager and quantified with the ImageJ software.
Flow Cells and DNA Curtains
Chromium barriers were fabricated on fused silica microscope slides using electron-beam lithography, as described previously39,64. Flowcells and lipid bilayers were prepared as described39,64. Single-stranded DNA substrates were generated by rolling circle replication, as described39. The ssDNA was coupled to the bilayer through a biotin-streptavidin linkage and aligned at the barriers by application of buffer flow.
DNA curtain experiments were performed using a prism-type TIRF microscope (Nikon) with two back-illuminated iXon EMCCDs (Andor Technology). Illumination was provided by a 200 mW, 488-nm laser and a 200 mW, 561-nm laser (Coherent, Inc.). Intensity at prism face was ~14 mW and ~25 mW for the 488-nm and 561-nm lasers, respectively. Fluorescence signals were separated by a filter cube equipped with a dichroic mirror (ZT561rdc), band pass filter (ET525/50m), and long pass filter (ET575lp)(Chroma Technology Corp.).
For visualizing the RPA-ssDNA complexes, RPA-eGFP or mutant RPAs (100 pM) were injected at 30 °C in HR buffer (30 mM Tris-acetate (pH 7.5), 5 mM Mg-acetate, 50 mM KCl, 1 mM DTT, 2.5 mM ATP and 200 μg ml−1 BSA). Reactions were allowed to continue for 20 minutes, and then chased with RPA-mCherry (100 pM). Throughout the experiments, 100 msec images were captured at 2-second intervals, and data collection continued for a period of 40 minutes. The resulting videos were used to generate kymographs, and integrated signal intensities were measured over the entire length of the ssDNA molecules. For quantitation, all data were normalized and corrected for background using a region of the slide surface without any ssDNA.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank R. Kolodner (Ludwig Institute for Cancer Research) and R. Rothstein (Columbia University) for gifts of yeast strains and plasmids, C. Mott for strain construction, and W.K. Holloman and members of the Symington lab for comments on the manuscript. This study was supported by grants from the US National Institutes of Health (R01 GM041784 (L.S.S), R01 GM074739 (E.C.G) and T32 CA009503 (S.K.D.)). This work was partially supported by the Nanoscale Science and Engineering Initiative of the US National Science Foundation under award No. CHE-0641523 to E.C.G., and by the New York State Office of Science, Technology, and Academic Research (NYSTAR). E.C.G. is supported as an Early Career Scientist with the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
S.K.D., B.G., E.C.G. and L.S.S. designed experiments and wrote the paper. Experiments in Figures 1 and 2, and Supplementary Figures were carried out by S.K.D., in Figure 3 by S.K.D. and B.G., and in Figure 4 by S.K.D and M.J.A.
References
- 1.Lieber MR. NHEJ and its backup pathways in chromosomal translocations. Nat Struct Mol Biol. 2010;17:393–5. doi: 10.1038/nsmb0410-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 4.Ma JL, Kim EM, Haber JE, Lee SE. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol Cell Biol. 2003;23:8820–8. doi: 10.1128/MCB.23.23.8820-8828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev. 2002;66:630–70. doi: 10.1128/MMBR.66.4.630-670.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 7.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–4. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shim EY, et al. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010;29:3370–80. doi: 10.1038/emboj.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–94. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mimitou EP, Symington LS. DNA end resection: many nucleases make light work. DNA Repair (Amst) 2009;8:983–95. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buis J, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135:85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langerak P, Mejia-Ramirez E, Limbo O, Russell P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011;7:e1002271. doi: 10.1371/journal.pgen.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–14. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol. 2011;18:75–9. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Truong LN, et al. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A. 2013;110:7720–5. doi: 10.1073/pnas.1213431110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol. 2011;18:80–4. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinkelmann M, et al. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–13. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee K, Lee SE. Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics. 2007;176:2003–14. doi: 10.1534/genetics.107.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–8. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rass E, et al. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–24. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 22.Mortensen UH, Bendixen C, Sunjevaric I, Rothstein R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci U S A. 1996;93:10729–34. doi: 10.1073/pnas.93.20.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugawara N, Haber JE. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol. 1992;12:563–75. doi: 10.1128/mcb.12.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villarreal DD, et al. Microhomology directs diverse DNA break repair pathways and chromosomal translocations. PLoS Genet. 2012;8:e1003026. doi: 10.1371/journal.pgen.1003026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daley JM, Wilson TE. Rejoining of DNA double-strand breaks as a function of overhang length. Mol Cell Biol. 2005;25:896–906. doi: 10.1128/MCB.25.3.896-906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 27.Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci U S A. 1998;95:6049–54. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith J, Rothstein R. A mutation in the gene encoding the Saccharomyces cerevisiae single-stranded DNA-binding protein Rfa1 stimulates a RAD52-independent pathway for direct-repeat recombination. Mol Cell Biol. 1995;15:1632–41. doi: 10.1128/mcb.15.3.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith J, Rothstein R. An allele of RFA1 suppresses RAD52-dependent double-strand break repair in Saccharomyces cerevisiae. Genetics. 1999;151:447–58. doi: 10.1093/genetics/151.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soustelle C, Vedel M, Kolodner R, Nicolas A. Replication protein A is required for meiotic recombination in Saccharomyces cerevisiae. Genetics. 2002;161:535–47. doi: 10.1093/genetics/161.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Umezu K, Sugawara N, Chen C, Haber JE, Kolodner RD. Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics. 1998;148:989–1005. doi: 10.1093/genetics/148.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen H, Lisby M, Symington L. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Molecular Cell. 2013;50:589–600. doi: 10.1016/j.molcel.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milne GT, Jin S, Shannon KB, Weaver DT. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:4189–98. doi: 10.1128/mcb.16.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–73. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kantake N, Sugiyama T, Kolodner RD, Kowalczykowski SC. The recombination-deficient mutant RPA (rfa1-t11) is displaced slowly from single-stranded DNA by Rad51 protein. J Biol Chem. 2003;278:23410–7. doi: 10.1074/jbc.M302995200. [DOI] [PubMed] [Google Scholar]
- 36.Lee SE, et al. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 37.Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–93. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 38.Rattray AJ, Shafer BK, Neelam B, Strathern JN. A mechanism of palindromic gene amplification in Saccharomyces cerevisiae. Genes Dev. 2005;19:1390–9. doi: 10.1101/gad.1315805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibb B, Silverstein TD, Finkelstein IJ, Greene EC. Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Anal Chem. 2012;84:7607–12. doi: 10.1021/ac302117z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibb B, et al. Concentration-dependent exchange of replication protein a on single-stranded DNA revealed by single-molecule imaging. PLoS One. 2014;9:e87922. doi: 10.1371/journal.pone.0087922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham JS, Johnson RC, Marko JF. Concentration-dependent exchange accelerates turnover of proteins bound to double-stranded DNA. Nucleic Acids Res. 2011;39:2249–59. doi: 10.1093/nar/gkq1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev. 2012;22:211–20. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephens PJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–10. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stephens PJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004;279:55117–26. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 47.Mansour WY, Rhein T, Dahm-Daphi J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010;38:6065–77. doi: 10.1093/nar/gkq387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang M, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–82. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang L, et al. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008;36:3297–310. doi: 10.1093/nar/gkn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simsek D, et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011;7:e1002080. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H, et al. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65:4020–30. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 52.Matsuzaki K, Terasawa M, Iwasaki D, Higashide M, Shinohara M. Cyclin-dependent kinase-dependent phosphorylation of Lif1 and Sae2 controls imprecise nonhomologous end joining accompanied by double-strand break resection. Genes Cells. 2012;17:473–93. doi: 10.1111/j.1365-2443.2012.01602.x. [DOI] [PubMed] [Google Scholar]
- 53.Shibata A, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–92. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mimitou EP, Symington LS. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010;29:3358–69. doi: 10.1038/emboj.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung WH, Zhu Z, Papusha A, Malkova A, Ira G. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 2010;6:e1000948. doi: 10.1371/journal.pgen.1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–5. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 58.Chen C, Umezu K, Kolodner RD. Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol Cell. 1998;2:9–22. doi: 10.1016/s1097-2765(00)80109-4. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, et al. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat Genet. 2005;37:750–5. doi: 10.1038/ng1587. [DOI] [PubMed] [Google Scholar]
- 60.Foster SS, Balestrini A, Petrini JH. Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol Cell Biol. 2011;31:4379–89. doi: 10.1128/MCB.05854-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langston LD, Symington LS. Gene targeting in yeast is initiated by two independent strand invasions. Proc Natl Acad Sci U S A. 2004;101:15392–7. doi: 10.1073/pnas.0403748101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Decottignies A. Microhomology-mediated end joining in fission yeast is repressed by pku70 and relies on genes involved in homologous recombination. Genetics. 2007;176:1403–15. doi: 10.1534/genetics.107.071621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davis AP, Symington LS. The yeast recombinational repair protein Rad59 interacts with Rad52 and stimulates single-strand annealing. Genetics. 2001;159:515–25. doi: 10.1093/genetics/159.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greene EC, Wind S, Fazio T, Gorman J, Visnapuu ML. DNA curtains for high-throughput single-molecule optical imaging. Methods Enzymol. 2010;472:293–315. doi: 10.1016/S0076-6879(10)72006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.