

Abstract
Many biological membranes consist of 50% or more (by weight) membrane proteins, which constitute approximately one-third of all proteins expressed in biological organisms. Helical membrane proteins function as receptors, enzymes, and transporters, among other unique cellular roles. Additionally, most drugs have membrane proteins as their receptors, notably the superfamily of G protein–coupled receptors with seven transmembrane helices. Determining the structures of membrane proteins is a daunting task because of the effects of the membrane environment; specifically, it has been difficult to combine biologically compatible environments with the requirements for the established methods of structure determination. There is strong motivation to determine the structures in their native phospholipid bilayer environment so that perturbations from nonnatural lipids and phases do not have to be taken into account. At present, the only method that can work with proteins in liquid crystalline phospholipid bilayers is solid-state NMR spectroscopy.
Keywords: solid-state NMR, isotopic labeling, proteoliposomes, membrane bilayers, transmembrane helices, GPCRs
1. INTRODUCTION
Membrane proteins are important targets for structure determination. Approximately one-third of all expressed polypeptides are recognized as membrane proteins by the presence of one or more transmembrane hydrophobic helices. This review emphasizes data for helical membrane proteins that contribute many essential physical, chemical, and structural properties to biological membranes. These proteins act as receptors, enzymes, and transporters, among a host of other functions. They also play roles in the assembly, fusion, and maintenance of cells, organelles, and viruses. They are important to medicine because human diseases result from mutations and truncations of these proteins (1). Additionally, most drugs, notably the superfamily of G protein–coupled receptors (GPCRs), utilize membrane proteins as their receptors (2). There are also examples of membrane proteins whose secondary structures are dominated by β-barrels.
NMR spectroscopy is particularly well suited for determining the three-dimensional structures of proteins and for describing their local and global motions in lipid environments, where they maintain their native conformation; NMR is the principal subject of this review. Membrane proteins can also be solubilized with detergents that form micelles or isotropic bicelles (3); however, there is always a risk of perturbing the structure due to the presence of detergents of nonnatural phases. A sample of protein-containing phospholipid bilayers enables the protein to be studied under near-native conditions, wherein it is fully active. Because the helical membrane proteins are immobilized in bilayers on the critical NMR timescale (~106 Hz), these samples can be studied with high-resolution solid-state NMR methods. Dramatic progress has recently been made with solid-state NMR approaches (4). This review also discusses complementary solution NMR studies of membrane proteins to provide context (5).
Protein structure determination has played a key role in biomedical research for more than 60 years (6, 7); however, significant limitations and gaps exist in experimental methods when applied to membrane proteins. Nearly all of these limitations stem from the use of detergents, instead of phospholipids in their liquid crystalline bilayer phase, in the samples. The first helical membrane protein to be crystallized was bacteriorhodopsin that was solubilized with the detergent octyl glucoside (8). The first structure of a membrane protein determined by X-ray diffraction, the photosynthetic reaction center of Rhodopseudomonas viridis (9), was crystallized from the detergent N,N-dodecyldimethylamine N-oxide, which contained additional amphiphilic compounds (10). Since 1996, the cubic phase of the detergent monoolein mixed with cholesterol (11) has had a major impact on the crystallization of membrane proteins, which has led to atomic-resolution structures (12), including those of GPCRs (13).
Initial attempts to obtain high-resolution 1H solution NMR spectra of protein-containing liposomes under physiological conditions yielded no signals from the protein or the lipids, demonstrating that membrane proteins are immobilized by their interactions within the phospholipid bilayer environment. For some time, investigators attempted to use sonicated liposomes, which do yield NMR signals from the lipids; however, these results were mired in controversy about the severe curvature affecting the packing of the lipids, and possible effects on associated proteins (14). It is uncertain whether any NMR signals have been detected from helical proteins in lipid bilayers, whether sonicated or not, by solution NMR methods. The controversy in the early to mid 1970s about the physical effects of sonication on the spectra is reminiscent of the current discussions about comparing the physical properties of membrane proteins in liposomes with those in micelles, isotropic bicelles, and nanodiscs.
The phospholipid bilayer environment affects the structures and dynamics of membrane proteins. This observation is a clear-cut example of Anfinsen’s (15, p. 223) conclusions about the relationship between the three-dimensional structure of a protein and its amino acid sequence: “[T]he native conformation is determined by the totality of interatomic interactions and hence by the amino acid sequence, in a given environment” (emphasis added; also see Reference 16). Consequently, the feasibility of a biologically relevant experimental structure determination depends on the environment provided by the sample, be it detergent micelles or phospholipid bilayers. These factors determine the feasibility of a particular experimental method, the resolution of the data, the structural resolution of the protein structure, and whether the structure is indeed representative of the native protein in its physiologically active conformation.
1.1. Membranes and the Formation of Life
Abiogenesis describes how life arose from inorganic matter following the formation of the Earth ~4.5 billion years ago. The monomeric subunits of biopolymers were formed by reactions that would have been possible with the chemical composition and environmental conditions of the young Earth (17). These precursors enabled the production of long biopolymers with defined sequences, such as DNA and proteins; evolution provided the means to store linear information in polynucleotides and its functional expression with folded proteins. Protobionts consist of organic molecules surrounded by a membrane-like barrier formed from prebiotic amphiphiles (18), which may resemble the amphiphilic compounds used for solubilization and crystallization of membrane proteins for structure determination. The emergence of the cell membrane played an important role in evolution by keeping the information-containing molecules in close proximity, separate from the environment, with controlled access to the precursors and products of the budding metabolic pathways. In addition to contributing to our understanding of the formation of life, prebiotic amphiphiles provide a starting point in the search for detergents that can solubilize hydrophobic membrane proteins. Samples used for both X-ray crystallography (8) and solution NMR spectroscopy (19) require the use of detergents as substitutes for phospholipids.
1.2. Protein-Containing Bilayers
Phospholipids consist of an amphipathic or charged head group attached to two long hydrocarbon chains through a trisubstituted glycerol backbone. A wide range of head groups, lengths of chains, numbers of double bonds, and other characteristics are found in the membranes of various organisms. Phospholipids have the unique ability to self-assemble into highly ordered bilayers in an aqueous solution. On the molecular scale, these bilayers are infinitely long in two dimensions but are only two molecules thick in the third dimension, providing the chemical barrier of the cell or organelle. Intramolecular motions add to the complexity of the membrane; in addition to their definitive gel to liquid crystalline phase transitions, lipids have different gradients of motion from the surface to the center of the bilayer (20).
Lipids and proteins undergo global motions in membranes, including rapid rotational diffusion about the bilayer normal (21, 22) and translational diffusion in the plane of the bilayer (20, 23). These large-scale motions strongly affect the feasibility of structural studies by diffraction, microscopic, and spectroscopic methods. They contributed to Singer & Nicholson’s (24) fluid mosaic model of the cell membrane and are now used as the foundation of a general NMR method for determining the structures of membrane proteins in liquid crystalline bilayer membranes (25). The goal is to replace artistically represented “blobular” protein structures (Figure 1) with atomic-resolution images that show all the details of the secondary and tertiary structures. There is no reason that the structural resolution of membrane proteins should be lower than that determined for globular, soluble proteins.
Figure 1.

Schematic three-dimensional and cross-sectional views of membrane proteins embedded in a lipid matrix, illustrating the fluid mosaic model of biological membranes. The proteins appear to be randomly distributed but may form specific aggregates in order to function. Modified from Reference 24.
Bacterial light-harvesting proteins are particularly amenable to spectroscopic and structural studies, perhaps because of their very high resistance to denaturation and added stability due to intermolecular interactions caused by close packing in biological membranes. Bacteriorhodopsin is the prototypical example, mostly because of the strong influence of its initial structure (Figure 2), which was obtained by Henderson & Unwin (26) by using electron microscopy. This observation was the first major step toward increasing the structural resolution of the membrane proteins in the fluid mosaic model. The initial results demonstrated the presence of seven transmembrane helices. Other proteorhodopsins have been used in both solution NMR (27–31) and magic angle spinning (MAS) solid-state NMR (18–24) studies because of their favorable physical characteristics. Although proteorhodopsins are unrelated to GPCRs (25), the prime targets for medicine, they have played valuable roles as model systems of membrane proteins with seven transmembrane helices for the development of new methods. Of course, photochemical processes are interesting in their own right, in physiology, and in capturing energy from the Sun.
Figure 2.

A model of a single-protein molecule in the membrane, viewed roughly parallel to the plane of the membrane. The top and bottom of the model correspond to the parts of the protein in contact with the solvent; the rest is in contact with the lipid. The most strongly tilted helices are in the foreground. Modified from Reference 26.
1.3. The Correlation Time Problem in NMR
The so-called correlation time problem reflects some of the most fundamental aspects of NMR spectroscopy; relaxation processes determine the rate at which nuclear spins reach equilibrium in the presence of a strong magnetic field. The correlation time problem is the reason that there is a size limit for structure determination of proteins and their complexes in aqueous solution. The extreme narrowing limit is defined by equal longitudinal (T1) and transverse (T2) relaxation times. It depends on the effective isotropic rotational correlation time of the molecule and the field strength of the magnet because these factors determine the resonance frequencies of the nuclei and the spans of the chemical shift interactions. In general, molecules with large-amplitude, rapid motions yield NMR spectra with narrow resonances. As the motions decrease in amplitude and/or frequency, the nuclear spins depart from the extreme narrowing limit as T1 correspondingly increases. Medium-to-large (<300-residue) globular proteins provide relatively broad resonance line widths, but individual resonances are still discernible, as in, for example, relatively small membrane proteins solubilized in detergent micelles, isotropic bicelles, and nanodiscs. Many helical membrane proteins behave in a predictable way in response to changes in the lipid composition of bicelles. When the overall rotational correlation time is relatively short, as for proteins with only one or two transmembrane helices in small micelles or isotropic bicelles at elevated temperatures, it is possible to observe backbone signals from all sites. However, as the protein becomes larger, or as the sample temperature is lowered, the signals become more difficult to detect.
The long rotational correlation times of the reorientation of solubilized membrane proteins in bicelles are almost feasible for solution NMR spectroscopy (32). However, as the q value of the bicelles increases, the signals broaden and become difficult to detect (33). It is generally feasible to find experimental conditions in which there are no signals from the structured helices and interhelical loops because of line broadening, and only signals from highly mobile terminal residues are observed. By the time magnetically alignable bicelles with q > 2.5 are formed, no signals can be observed in solution-phase NMR spectra.
When a protein is immobilized by interactions within the phospholipid bilayer, and the bilayer itself undergoes only slow reorientation, then it is necessary to utilize solid-state NMR methods to obtain high-resolution spectra (34, 35). Importantly, this is the case for proteoliposomes in which the protein is embedded in unoriented bilayers with no accompanying detergent molecules to induce motion or magnetic alignment (36, 37). As mentioned above, a limited number of residues, especially those near the N and C termini, have substantial local motion and can be identified because their signals are observable in solution NMR spectra or are missing in solid-state NMR spectra (37–40). Essentially all other residues, including those in helices and interhelical loops, are immobile on the NMR timescale of ~106 Hz. As a result, solution-phase NMR is complementary to solid-state NMR because the only resonances observable in the spectra are from the highly mobile residues, and the signals are missing from these same residues in solid-state NMR spectra because of the attenuation of the heteronuclear dipolar coupling (DC) needed for cross-polarization of the 13C or 15N signal.
There are two strategies for dealing with the broad lines associated with slowly reorienting membrane proteins in aqueous solution. One is to replace the bilayer and solubilize the protein with an amphipathic molecule that forms micelles, enabling the protein to undergo effectively isotropic reorientation fast enough to yield high-resolution spectra. The other is to utilize samples of the membrane protein in the phospholipid bilayers wherein it is immobilized and apply solid-state NMR methods to obtain the narrow resonances necessary for high-resolution spectroscopy. Both approaches have been successful, but the advantages of working under near-native conditions in bilayers suggest that future studies will be dominated by solid-state NMR of protein-containing phospholipid bilayer samples.
Context is important. Tanford & Reynolds (41) pioneered many of the concepts and methods used to solubilize membrane proteins for biophysical analysis. They emphasized the assembly of protein–lipid complexes, such as micelles, that contain a single protein, and they analyzed the pros and cons of a full range of detergents. One of their conclusions was that, under most conditions, sodium dodecyl sulfate (SDS) is the best choice for stabilizing the structures of membrane proteins, in contrast to its generally accepted use for partially denaturing globular proteins. Following these authors’ lead, other investigators attempted to optimize the choice of detergent(s) and experimental conditions for solution-phase NMR of solubilized membrane proteins (3, 42–47). A surprising observation is that detergent concentrations that are much higher than the critical micelle concentration are highly effective at providing single-line spectra with relatively narrow line widths.
Even when a detergent adequately solubilizes the protein, structural studies are further complicated by the observation of few or no interresidue nuclear Overhauser effects (NOEs) in helical membrane proteins. As a result, alternative parameters, such as residual dipolar couplings (RDCs) and paramagnetic relaxation enhancements (PREs), must be used for structure determination.
2. SOLUTION NMR STRUCTURE DETERMINATION OF SOLUBILIZED MEMBRANE PROTEINS
This review focuses on α-helical membrane proteins in near-native bilayer environments and surveys directly relevant studies of micelles and isotropic bicelles. Further structural information about all classes of membrane proteins can be obtained from the website maintained by Stephen White (http://blanco.biomol.uci.edu/mpstruc/listAll/list). Many peptides corresponding to transmembrane domains of membrane proteins have played seminal roles in the development of samples and experimental methods for structure determination. Particularly noteworthy are the structural studies of gramicidin, the glycophorin A transmembrane domain, the neuronal acetylcholine receptor M2 channel, the influenza M2 proton channel, the HIV Vpu channel, and the ErbB domains.
The first structure of a full-length protein containing a single transmembrane helix was determined by solution NMR for a detergent-solubilized coat protein of a filamentous bacteriophage. This protein exists in two different forms during the viral life cycle. After synthesis, this protein is stored in the cell membrane prior to viral assembly, which occurs at the membrane. In many ways, the membrane-bound form of fd coat protein is a typical helical membrane protein. It has one hydrophobic transmembrane helix and a large bend into a shorter, amphipathic surface helix. Because the membrane-bound form of fd coat protein contains only 50 residues and is readily purified in relatively large quantities from the bacteria it infects (19), it has provided a test bed for many sample-preparation and experimental methods of both solution NMR and solid-state NMR. Cross & Opella (19) initiated this investigation in 1976, and the modest initial goals of solubilizing the protein and obtaining spectra with resolved resonance required several years to attain because no membrane protein had been previously studied by NMR spectroscopy. Sample optimization allowed for observations of recognizable 1H and natural abundance 13C resonances by direct detection (48, 49). This research benefited from Charles Tanford’s crucial advice regarding the choice of SDS as the detergent to substitute for the phospholipids of the cell membrane.
The 13C NMR spectra in Figure 3 demonstrate a key difference between a detergent-solubilized membrane protein and a globular protein in an aqueous solution, in this case lysozyme. Figure 3 shows the α-carbon resonance region for both proteins; the lysozyme signals appear as expected for a fully structured polypeptide with uniform line widths spread over the entire isotropic frequency range of α-carbons. In contrast, the spectrum of fd coat protein in SDS micelles contains mainly very broad signals, but also some sharp, intense signals. Although the first step in obtaining these spectra was to find conditions for relatively rapid global isotropic reorientation to yield resolvable signals, the data immediately revealed the presence of at least two categories of backbone sites that are differentiated by their dynamics. Most of the α-carbons reorient with the same correlation time as the entire protein–micelle complex; however, some signals are associated with mobile residues, as demonstrated by their narrow, intense signals. Before structural studies can be performed, the global and local dynamics of membrane proteins must be characterized. We return to this topic in the discussion of solid-state NMR studies of membrane proteins, below; it provides an excellent demonstration of the complementarity of the two approaches.
Figure 3.

Comparison between α-carbon resonances in the 13C NMR spectra of two different proteins. (a) The membrane-bound form of fd coat protein solubilized in SDS (sodium dodecyl sulfate). (b) The enzyme lysozyme in an aqueous solution. Modified from Reference 48.
In general, the difficulty in solving the structures of these proteins is the near-total absence of long-range NOEs. Nonetheless, the structure of fd coat protein was determined with the standard suite of multidimensional solution NMR experiments, although a few ambiguities remained (50). The protein has an amphipathic α-helix (residues 7–16) and a hydrophobic α-helix (residues 27–44), which were determined to be approximately perpendicular on the basis of the few measurable NOEs among the residues that connect the two helices. The interhelical loop consists of two turns, and relaxation data indicate that some additional motions occur within the loop.
As of 2005, no three-dimensional structures of membrane proteins with more than one trans-membrane helix had been reported. This inability to determine structures in micellar environments reflected the difficulties encountered in resolving and assigning a sufficient number of long-range NOEs to calculate their three-dimensional folds. Fortunately, measurements and analyses of RDCs were then being developed for weakly aligned proteins (51–57). The secondary structure of a protein is defined by its regularity, and RDCs are very well suited for identifying helical residues on the basis of dipolar waves that reflect the regular oscillation of structural parameters as a function of residue number in helices. Also, the application of PRE reagents has been particularly useful in supplementing RDCs for the structure determination of solubilized membrane proteins.
Membrane proteins have evolved along with lipid membranes in all three domains of life (18). Archaeal membrane proteins with seven transmembrane helices have been studied with many biophysical methods, including NMR spectroscopy. Although they have no sequence relationship with GPCRs (58), these proteins provide convenient model systems for the development of methods for structure determination of membrane proteins with seven transmembrane helices. The prototypical example is bacteriorhodopsin, which has played important roles in the development of electron crystallography and microscopy (26, 59), X-ray crystallography (12), and both solution NMR and solid-state NMR of membrane proteins. This protein is relatively small, with 250 residues; can be isotopically labeled by expression; and is very stable. Even with high levels of deuteration and TROSY (transverse relaxation optimized spectroscopy) pulse sequences, bacteriorhodopsin in dodecylmaltoside detergent micelles (38, 60) yields solution NMR spectra that overlap significantly and probably display signals mostly from exterior loops and terminal regions.
Interestingly, retinal-binding membrane proteins with similar properties have been found in bacteria living in the ocean. These proteins are known as proteorhodopsins, and they exist in many variants and are sensitive to light at different wavelengths. More pertinently, they provide high-quality solution NMR spectra, enabling structure determination (28). The most thoroughly studied of these proteins is sensory rhodopsin II (29–31).
3. STRUCTURE DETERMINATION BY SOLID-STATE NMR
When detergent micelles are included in the correlation time calculations, it appears that the spectral quality of the proteorhodopsins and that of other fairly large oligomeric membrane proteins such as mammalian GPCRs should not differ. However, solution NMR has been notably unsuccessful with GPCRs; spectra obtained from micelles, bicelles, and nanodiscs show signals from only the mobile terminal residues (37, 61). The structure determination of a GPCR (62) demonstrates that it is feasible to analyze the structures of membrane proteins in their native phospholipid bilayer environments by solid-state NMR.
3.1. Line Narrowing of Solids
The essential concepts underlying high-resolution solid-state NMR are the averaging and separation of the splitting and broadening effects of the chemical shift and dipole–dipole coupling interactions, which can range from tens to hundreds of kilohertz, well beyond the range of any solution NMR instrument or experiments. The earliest, and one of the most important, contributions to the field was the 1948 observation and characterization of the angular dependence of the dipole–dipole interaction between two 1H nuclei (63). This classic experiment demonstrated that individual spin interactions can be separated from others, that the dipole–dipole interaction exists, and that this interaction depends on both angle and distance. Figure 4 shows three-dimensional spectra of both a powder sample (the Pake doublet) and a single crystal sample with two lines, whose splitting depended on the angle with respect to the direction of the magnetic field.
Figure 4.
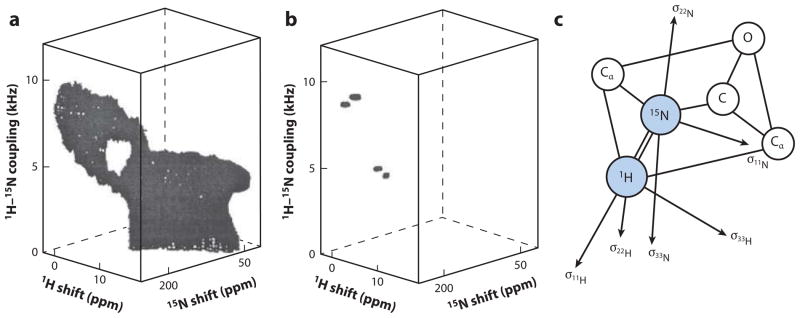
Experimental three-dimensional 1H chemical shift/15N–1H dipolar coupling/15N chemical shift correlation spectra of a model peptide. (a) Polycrystalline sample. (b) Single-crystal sample. (c) Diagram of tensors in a peptide plane. Modified from Reference 70.
The development of high-resolution solid-state NMR occurred in two major steps and many subsequent improvements. The first step involved decoupling homonuclear dipole–dipole interactions while leaving the chemical shift interaction available for measurements. Not only did the initial investigations conquer what is probably the most difficult impediment to obtaining high-resolution solid-state NMR spectra of solids—the narrowing of resonances whose breadth is due to homonuclear DCs—but also the accompanying theory of coherent averaging opened up the entire field of high-resolution solid-state NMR spectroscopy. Multiple-pulse (64) and cross-polarization double resonance (65) NMR, especially with the addition of MAS, made it possible to obtain high-resolution NMR spectra of single- and polycrystalline compounds (66). These techniques were initially used to determine chemical shift tensors and to investigate intramolecular motions in immobile molecules (67, 68).
Arguably, the most important chemical shift tensors are those in the peptide bond. The peptide bond strongly influences the structures of proteins; its alignment relative to an external axis can be deduced from the 1H chemical shift, the 15N chemical shift, and the 1H–15N chemical shift (69). Additional information becomes available when the 13C spectral parameters from the C′ and Cα sites are included. These principles are illustrated in Figures 4 and 5 (69, 70). The three-dimensional powder pattern of a sample of polycrystalline 15N-acetyl-leucine has intensity at all frequencies available from the three operative spin interactions at the 15N-labeled amide site. The three-dimensional powder pattern has an unusual shape because the principal elements of the three spin-interaction tensors are not collinear in the molecular frame of reference. When the three-dimensional powder pattern is viewed from the 15N chemical shift/1H–15N DC plane (Figure 5), it has a highly characteristic low-intensity region, demonstrating that the σ33 of the 15N chemical shift tensor points approximately 15° away from the N–H bond that causes DC. This difference between the angles in the molecular frame of reference is extremely valuable for obtaining resolution in multidimensional spectra and in determining the orientation of the peptide planes. Similar factors are also at play with the 13C chemical shift and 1H–13C interactions, which provide additional opportunities for resolution among sites and for measuring restraints for input into the structure calculations.
Figure 5.
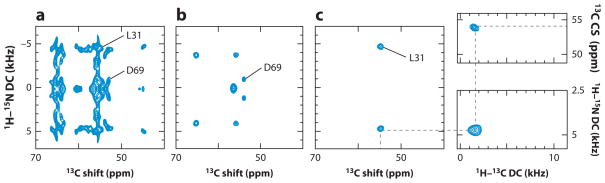
Example of spectroscopic data for residue L31 obtained from magic angle spinning solid-state NMR spectra of uniformly
13C/15N-labeled MerFt in DMPC (dimyristoyl phosphatidylcholine) proteoliposomes at 25°C. (a) Two-dimensional 1H–15N dipolar coupling (DC)/13C shift separated local field (SLF) spectrum. (b) Two-dimensional 1H–15N DC/13C shift SLF spectra plane selected from a three-dimensional spectrum at an isotropic 15N chemical shift frequency of 118.6 ppm. The 1H–15N DC motionally averaged powder pattern for residue L31 has a perpendicular edge frequency of 4.7 kHz, corresponding to a splitting of 9.4 kHz, and a DC value of 18.8 kHz. (c) Two-dimensional 1H–13CαDC/13C shift SLF spectral plane selected from a three-dimensional spectrum at an isotropic 15N chemical shift frequency of 54.6 ppm. All three spectral planes are associated with residue L31. The dashed line traces the correlation among the frequencies, which were obtained from three separate experiments. The DC frequencies in the spectra correspond to the perpendicular edge frequencies of the corresponding powder patterns. Modified from Reference 36.
There are three basic approaches to determining the structures of membrane proteins immobilized in phospholipid bilayers: (a) oriented sample (OS) solid-state NMR (71), which depends on the principles described above (Figure 5); (b) MAS solid-state NMR, wherein the powder patterns are averaged to their isotropic values by mechanical sample rotation (72, 73), after which distances are measured between peaks assigned to specific atoms in the amino acid residues (74) and dihedral angles are analyzed by conventional, statistical methods (75); and (c) rotationally aligned (RA) solid-state NMR (see the sidebar), in which the isotropic signals obtained with MAS solid-state NMR are used for resolution and assignment and the DC and chemical shift anisotropy (CSA) powder patterns are then recoupled in multidimensional experiments (25, 36, 62). The key principles of RA solid-state NMR are that membrane proteins undergo fast rotational diffusion about the bilayer and that the angles are measured from the frequency of the parallel edge of the motionally averaged powder pattern (25).
3.2. Structure Determination of Membrane Proteins by Solid-State NMR
The initial and highest-priority goal of designs of structural studies of membrane proteins is to meet the stringent sample requirements. Doing so has typically required the use of both nonnative detergent/lipid mixtures and, especially for X-ray crystallography, protein modifications that are so extensive as to cast doubt on the relevance of the structures. In some examples of all these methods, very low temperatures are used. Each factor can cause minor or major distortions in the structures, dynamics, and activities of the membrane proteins, especially in light of increasing evidence that many membrane proteins function through conformational fluctuations within the bilayer and that their structures are exquisitely sensitive to the details of the membrane environment, such as specific phospholipids and other membrane components, temperature, and pH.
PROTEIN STRUCTURE DETERMINATION BY ROTATIONALLY ALIGNED SOLID-STATE NMR.
A general method for determining the three-dimensional structures of membrane proteins in proteoliposomes is exemplified by the determination of the backbone structure of MerFt, the 60-residue helix–loop–helix core of the 81-residue mercury transporter MerF. The method merges OS solid-state NMR and MAS solid-state NMR techniques so that orientation restraints relative to a single external axis (the bilayer normal) can be measured from uniformly 13C/15N-labeled samples in unoriented phospholipid bilayers under physiological conditions. This technique relies on the fast (>106 Hz) rotational diffusion of a membrane protein about the bilayer normal to motionally average the static chemical shift anisotropy and heteronuclear dipole–dipole coupling powder patterns to axially symmetric powder patterns with reduced frequency spans. The frequencies associated with the parallel edges of the motionally averaged powder patterns can be used to measure the angle between the principal axis of the spin-interaction tensor and the bilayer normal. Significantly, the parallel edge of a rotationally averaged powder pattern has exactly the same frequency as the single-line resonance observed in the spectrum of a stationary sample aligned parallel to the magnetic field. Because the axis system is established by the properties of the sample, one of the most important advantages of OS solid-state NMR is retained; specifically, any experimental error or uncertainty in the magnitudes and orientations of the principal values of the static spin-interaction tensors has no cumulative effect on the calculation of the protein structure. That is, any errors tend to cancel out. This method provides three-dimensional, atomic-resolution structures of unmodified membrane proteins in their native phospholipid bilayer environment under physiological temperature and pH conditions.
Solid-state NMR is the only method that can obtain meaningful structures of membrane proteins in their native bilayer environments under physiological conditions. The instrumentation and experimental procedures allow one to obtain high-resolution spectra as well as precise and accurate structural restraints from proteins that are immobilized on the timescales (~106 Hz) of the chemical shift and heteronuclear dipole–dipole spin interactions. To date, all structure determinations of membrane proteins (http://www.drorlist.com/nmr.html) in liquid crystalline phospholipid bilayers have utilized OS solid-state NMR (40, 76–80), in a few cases in combination with data from solution NMR or MAS solid-state NMR methods (81–85). The samples consist of fully hydrated protein-containing bilayers of long-chain natural phospholipids aligned with their normals parallel or perpendicular to the direction of the applied magnetic field. Macroscopic bilayer alignment can be induced either magnetically (32) or mechanically by layering on glass slides (86, 87). Magnetically aligned bilayers yield spectra with the highest resolution and are generally more stable (35, 40, 88). However, magnetic alignment typically requires the addition of 20% to 30% nonnative short-chain lipids or detergents to provide orthogonal boundaries to the planar bilayer regions. Inevitably, some of these short-chain lipids or detergents are dissolved in the liquid crystalline long-chain lipid bilayers, as evidenced by a reduction-of-order parameter for both the lipids and the proteins from 1.0 in proteoliposomes to ~0.85 in magnetically aligned bilayers. This reduction may affect the structure, dynamics, and stability of the proteins.
The principal advantage of OS solid-state NMR is that it derives orientation restraints directly from the measurement of frequencies of single-line resonances (89, 90). The restraints are used both for determining the protein structure and for orienting it within the frame of the lipid bilayer membrane. However, in stationary, aligned samples, the dense network of 13C–13C homonuclear dipole–dipole couplings in uniformly 13C/15N-labeled proteins interferes with direct 13C detection (91) and with many other solid-state NMR experimental procedures, especially those that facilitate making resonance assignments.
A newly developed solid-state NMR approach eliminates the limitations on samples and improves the speed, accuracy, and precision of membrane protein structure determination. The principal advance is the use of proteins that are uniformly 13C/15N labeled and are in phospholipid bilayers under physiological temperature and pH conditions. This approach combines essential aspects of OS solid-state NMR and MAS solid-state NMR spectroscopies. An advantages of combining these methods, versus conventional OS solid-state NMR alone (89, 90), is that the ability to apply triple-resonance experiments to uniformly 13C/15N-labeled samples, which improves the sensitivity of the measurements through the use of 13C [instead of 15N (72)] detection, permits the implementation of systematic assignment schemes among proximate sites, and provides signals from essentially all backbone and side-chain sites for complete three-dimensional structure determination.
Mechanical rotation of the sample at the magic angle averages out the interfering 13C–13C homonuclear DCs in fully 13C/15N-labeled proteins. By using the appropriate recoupling pulse sequences, many of which have been developed for studies of polycrystalline peptides and proteins, one can measure the orientation-dependent frequencies from the motionally averaged powder patterns for the 1H–13Cα and 1H–15N amide heteronuclear DCs, as well as for the 13Cα, 13C′, and 15N amide CSAs. Complementary information about the dynamics of backbone and side-chain sites is available from these and related NMR experimental measurements.
Following their initial use in high-resolution solid-state NMR of proteins, MAS solid-state NMR (72, 73) and OS solid-state NMR (90) methods have progressed, for the most part, along separate paths. Here, we describe how they are reunited through the rotational alignment that results from the rapid diffusion of membrane proteins around the lipid bilayer normal (92, 93).
Using 31P NMR of phospholipids, McLaughlin et al. (94) became the first to demonstrate the equivalence between the single-line chemical shift frequency observed from a uniaxially oriented bilayer sample and the parallel edge of a rotationally averaged powder pattern from an unoriented bilayer sample. An early experiment by Griffin and colleagues (95) demonstrated that 13C′-labeled bacteriorhodopsin in unoriented phospholipid bilayers undergoes rotational diffusion around the bilayer normal and that lowering the sample temperature slows the diffusion sufficiently to enable the static powder pattern to be observed. Our research group (96) obtained similar results for a transmembrane helical polypeptide labeled with 15N at a single backbone amide site. Taken together, these results demonstrate that both phospholipids and proteins undergo rotational diffusion around the bilayer normal at a fast enough rate (>106 Hz) to motionally average the static CSA and DC powder patterns to axially symmetric powder patterns with reduced frequency spans. The sign and breadth of the powder patterns from individual sites reflect the angle between the principal axis of the relevant spin-interaction tensor and the lipid bilayer normal. We recently demonstrated experimentally that the orientation-dependent frequencies measured from samples of mechanically aligned, magnetically aligned, and protein-containing bilayers are the same (25). In additional studies, the rotational averaging of powder patterns has been used to ascertain the alignment of helices, sheets, and small peptides in bilayers (25). Building on the physical foundation of measuring orientation restraints from motionally averaged spin-interaction tensors, we developed an approach (97) to determining the three-dimensional structures of membrane proteins in phospholipid bilayers. We applied this approach to the backbone structure determination of MerFt, the 60-residue integral membrane core of the membrane protein transporter MerF from the bacterial mercury detoxification system. Previously, we determined the three-dimensional structure of MerFt in micelles by solution NMR (98) and in magnetically aligned bilayers by OS solid-state NMR (40). In addition, we used MerFt in the development of many experimental methods (34, 35, 40, 99–109).
Proteins become RA by fast rotational diffusion about the bilayer normal. The measurement of orientation-dependent frequencies from the parallel edges of the recoupled (110, 111), motionally averaged, axially symmetric powder patterns are identical to those measured from the single-line resonances observed in aligned samples (25). The recoupled powder pattern edge frequencies are measured in MAS solid-state NMR experiments that simultaneously perform homonuclear decoupling of the 13C–13C interactions and the averaging that yields resonances with isotropic chemical shift frequencies.
To verify that the membrane protein undergoes rapid uniaxial rotational diffusion around the bilayer normal and that this rotation can be switched from the slow to fast limits when the temperature is increased [from 10°C to 25°C for MerFt-containing dimyristoyl phosphatidylcholine (DMPC) bilayers], we monitored the effect of temperature on the 13C′ CSA powder pattern of uniformly 13C/15N-labeled MerFt (Figure 3). The 13C′ CSA powder pattern is particularly convenient for demonstrating that a helical membrane protein is undergoing fast rotational diffusion (95) because the static powder pattern is highly asymmetric and has a wide frequency span. In helical membrane proteins, a large fraction of the 13C′ bonds align along helix axes because they participate in hydrogen bonds with the amide NH groups (112). For transmembrane helices, the 13C′ bonds are roughly parallel to the bilayer normal (i.e., the axis of motional averaging); thus, when a protein undergoes fast rotational diffusion around this axis, the 13C′ powder pattern becomes axially symmetric and is significantly narrowed. MerFt in DMPC bilayers undergoes fast rotational diffusion at temperatures above ~18°C. As a result, the 13C′ powder pattern obtained at 25°C for uniformly 13C/15N-labeled MerFt in liposomes is fully rotationally averaged (Figure 3c), whereas the powder pattern obtained at 10°C spans the full width of the static spin interaction and shows no evidence of motional averaging (Figure 3d).
Because of the limited resolution among resonances in one-dimensional spectra, two- and three-dimensional spectra are required to resolve signals from individual sites and to serve as stepping stones for the crucial three-dimensional assignment and recoupling experiments. Typically, the first set of MAS solid-state NMR experiments is used to obtain two-dimensional 13C/13C dilute spin-exchange spectra (74, 113, 114) and 13C/15N heteronuclear correlation spectra (115).
Uniform 13C/15N isotopic labeling of the protein enables direct transfer of magnetization through dipole–dipole couplings in the backbone, providing a mechanism for correlating the isotropic chemical shift frequencies of directly bonded 13C and 15N nuclei. Systematic residue-by-residue assignment methods that involve the correlation of inter- and intraresidue resonances cannot be performed on static, aligned samples because of interference from the network of 13C–13C homonuclear DCs. However, because these couplings are effectively averaged out in MAS solid-state NMR experiments, we assigned the backbone resonances of MerFt in unoriented proteoliposomes by using triple-resonance experiments.
To characterize the rotationally averaged 1H–15N DC and 15N CSA powder patterns associated with each isotropic resonance, it is essential to implement three-dimensional MAS solid-state NMR recoupling experiments. The two-dimensional 1H–15N DC/13C shift separated local field (SLF) spectrum shown in Figure 5a contains resonances from all of the MerFt 13Cα sites. Although there is considerable spectral overlap, the individual resonances assigned to Leu31 and to Asp69 can be identified. Figure 6b,c depicts two-dimensional 1H–15N DC/13C shift SLF planes selected from a three-dimensional spectrum at the 15N frequencies corresponding to Asp69 (Figure 5b) and Leu31 (Figure 5c). Similarly 1H–13C heteronuclear DCs were measured in three-dimensional experiments correlating 13Cα and 15N amide isotropic chemical shifts. As an example, the strip plot for Leu31 is shown in Figure 5d,e.
Figure 6.
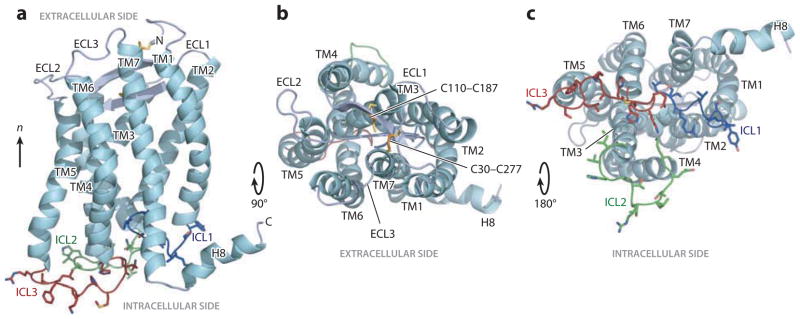
Three-dimensional structure of CXCR1. The backbone representation of CXCR1 shows helices (TM1–TM7 and H8) in aqua; extracellular loops (ECLs) in gray; and intracellular loops (ICLs) in blue (ICL1), green (ICL2), and red (ICL3). Disulfide-bonded cysteine (Cys) pairs (C30–C277 and C110–C187) are shown as sticks. (a) Side view. (b) View from the extracellular side. (c) View from the intracellular side. Modified from Reference 62.
Once the 13C/15N heteronuclear correlation spectrum of MerFt was assigned, the measured 1H–15N DCs were plotted as a function of residue number, and the values were fitted to a sinusoid of period 3.6, corresponding to the pitch of an α-helix. Fitting the data to a sine wave confidently identified residues in helices because of their periodicity. The measured spectroscopic parameters were also used to calculate de novo the three-dimensional structure of MerFt. The computational methods used to determine protein structures from orientation restraints obtained from OS solid-state NMR experiments with stationary, aligned samples (78, 102, 104, 116–126) are equally applicable to the frequencies measured from the edges of the rotationally averaged powder patterns. The 1H–15N and 1H–13Cα DC restraints and the 15N CSA restraints are sufficient to determine the orientation of the associated peptide plane relative to the axis of alignment.
3.3. Structure Determination of a G Protein–Coupled Receptor: CXCR1
Most therapeutic drugs are small molecules targeted to protein receptors that reside in cell membranes, the largest class of which is GPCRs. Determining the structure of the chemokine receptor CXCR1 and describing its interactions with interleukin (IL)-8, drugs, and G proteins will enable structure-based discovery of drugs that may affect inflammation, cancer metastasis, and other diseases.
CXCR1 is one of two high-affinity receptors for the CXC chemokine IL-8, a major mediator of immune and inflammatory responses that has been implicated in various disorders, including tumor growth (127–129). It is a ligand-activated, class A rhodopsin-like GPCR, a member of the largest class of integral membrane proteins that are responsible for cellular signal transduction and the largest class of drug receptors (130–133). The inhibition of CXCR by small-molecule drugs reduces breast cancer metastasis (134), making this receptor an attractive candidate for structure-based drug development.
When IL-8 is released in response to inflammatory stimuli, it binds to the extracellular side of CXCR1, activating intracellular signaling pathways that result in neutrophil migration to the site of inflammation (128). Despite its importance, its molecular mechanism is poorly understood, mostly because only limited structural information is available. Recently, investigators have advanced the structure determination of GPCRs by X-ray crystallography by tailoring the receptors to facilitate the formation of crystals by incorporating stabilizing mutations, inserting the protein T4 lysozyme, and truncating their amino acid sequences (135); also, stabilizing antibodies and small molecules were added to the samples (136) to enhance crystallization from cubic-phase monoolein mixtures (137). Previous function–structure studies showed that the intracellular loops (ICLs) of GPCRs are critical for G protein interactions (138) and suggested an activation mechanism that involves both the N-terminal residues and the extracellular loops of CXCR1 (128, 139, 140). Our previous NMR studies provided additional evidence for such a two-site mechanism and indicated that the binding of IL-8 to the receptor’s N terminus is mediated by the membrane; this finding underscores the importance of the phospholipid bilayer for the activity of membrane proteins (141). Here, we describe the three-dimensional structure of human CXCR1 determined in liquid crystalline phospholipid bilayers, without modification of its amino acid sequence, under physiological conditions. The NMR structure of CXCR1 has many similarities to, and some notable differences from, the crystal structure of CXCR4 (142). It also reveals information that is important for intracellular G protein activation and signal transduction.
GPCRs are nearly intractable to solution-phase NMR spectroscopy; spectra show resonance intensity only from mobile terminal residues, regardless of the detergent used to solubilize the protein. Moreover, the 15N NMR spectra are crowded in uniformly labeled samples and are difficult to assign without help from 13C labeling. These factors motivated the development of a new approach: RA solid-state NMR.
To examine the structure and function of CXCR1 in its native environment, we reconstituted the full-length, active receptor in phospholipid bilayers (proteoliposomes). Most data used to resolve, assign, and measure isotropic chemical shift frequencies from N, 13Cα, 13C′, and 13Cβ sites of residues 20–325 of CXCR1 were obtained from 13C-detected, three-dimensional, triple-resonance experiments. A total of 97% of the backbone resonances for CXCR1 residues 20–325 were assigned. All 15N and 13C signals from the mobile N and C termini (residues 1–19 and 326–350) were missing from the spectra, consistent with both our observation of appropriate signals in solid-state NMR experiments designed to detect only signals from mobile sites and our previous analysis of local and global motions by CXCR1 (61).
Three-dimensional 13C-detected SLF experiments (36) were used to measure the 1H–15N DC and 1H–13Cα DC frequencies that provide orientation restraints for structure determination. The protein backbone structure was calculated by a molecular fragment replacement approach. An initial structural model was generated from a set of molecular fragments generated with chemical shift ROSETTA (143) from the experimental chemical shifts and the amino acid sequence of CXCR1, together with the helical framework of the prototypical class A GPCR, bovine rhodopsin (144). This initial model was refined with experimental restraints, first from the implicit membrane potential of ROSETTA (145, 146), then by restrained simulated annealing with XPLOR-NIH (147).
The three-dimensional structure of CXCR1 (Figure 6) has the consensus fold of a GPCR, with seven transmembrane helices connected by three extracellular loops and three ICLs. The average backbone pair-wise root-mean-square deviation is 1.7 Å, and the experimentally measured 1H–15N DC and 1H–13Cα DC values correlate remarkably well with those calculated from the refined protein structure. Notably, the correlations improve dramatically following refinement of the initial structural model with the experimental data, demonstrating that the NMR structure of CXCR1 is determined by the experimentally measured backbone orientation restraints and dihedral angles.
Following the structure determination of rhodopsin from three- and two-dimensional crystals (144, 148), the structures of several class A ligand-activated GPCRs were determined by X-ray crystallography (132, 133). CXCR1 is now the first GPCR whose structure has been determined in liquid crystalline phospholipid bilayers, and it is the first ligand-activated GPCR whose structure has been determined without modification of its amino acid sequence. The structure of CXCR1 is significantly similar to that of CXCR4 (142), the only other chemokine receptor whose structure has been determined; however, there are also notable differences. Some reflect the modifications made to the sequence of CXCR4 (insertion of T4 lysozyme in ICL3, removal of 33 C-terminal residues, and mutation of L125W) that were required for crystallization; others may reflect sequence differences between the two proteins; still others may reflect the influence of the planar phospholipid bilayer and the rotational diffusion of the protein.
The CXCR1 helices are well defined by the spectroscopic data. The ICLs of GPCRs are critical for G protein interactions (138). Modification of ICL3 through the insertion of T4 lysozyme between TM5 and TM6 rendered CXCR4 incapable of activating G proteins (142). In contrast, unmodified CXCR1 is fully active with respect to both G protein activation and chemokine binding, and its three ICLs are structurally well defined. Notably, ICL3—which is important for CXCR1 coupling to G proteins and is involved in calcium mobilization, chemokine-mediated migration, and cell adhesion—extends from Thr228 to Gln236, connecting helices TM5 and TM6, both of which are one turn shorter than the corresponding helices in CXCR4. ICL3 protrudes into the cytoplasm, where it becomes available for G protein binding. Its sequence shares significant homology with that of other GPCRs; the ability to observe the structure of an intact GPCR, therefore, allows for the study of general mechanisms of G protein binding and activation.
4. PROGRESS, COMPARISONS, AND THE WAY FORWARD
Membrane protein structure determination by NMR is a rapidly advancing field. The best resource for an up-to-date view of the membrane protein structures determined by solution-phase NMR is the website maintained by Dror Warschawski (http://www.drorlist.com).
The primary advantage of the solid-state NMR method described herein is that it determines the structure of membrane proteins in their native phospholipid bilayer environment under physiological conditions. In particular, the use of detergents or nonnatural lipid assemblies that may distort the protein structure can be avoided; the composition of natural long-chain phospholipids can be varied over a wide range; other compounds of biological interest, such as cholesterol or drugs, can be readily added to the samples; and other parameters, such as temperature and pH, can be varied.
The most important spectroscopic advantage of combining OS solid-state NMR and MAS solid-state NMR methods is that doing so facilitates the use of uniformly 13C/15N-labeled protein samples, enabling various experiments to be implemented for the systematic assignment of resonances, the observation of signals from nearly all backbone and side-chain protein sites, and the measurement of numerous orientation and distance restraints for structure determination. The implementation of 13C detection provides higher sensitivity and circumvents the need for mechanical or magnetic sample alignment. Magnetic alignment, which yields the highest-resolution spectra, requires the addition of significant amounts of either nonnatural lipids or detergents. Significantly, this method retains the most important advantages of OS solid-state NMR, which are that both three-dimensional structure and integral membrane orientation are determined and that errors in measurements of orientation-dependent frequencies or uncertainties in the magnitudes and directions of the principal elements of the spin-interaction tensors do not accumulate. Each orientation restraint is determined independently relative to the single external axis; therefore, any errors or uncertainties are essentially random in direction relative to that axis.
This method for determining the structures of membrane proteins in phospholipid bilayers under physiological conditions has the potential to transform the field. It is no longer necessary to use membrane mimics of detergents of nonnatural lipids for crystallization or solubilization, to insert lysozyme or other extraneous segments into the sequence, or to require the presence of a stabilizing antibody fragment to facilitate stabilization or crystallization. As a result, it is possible to determine the three-dimensional structures of full-length, wild-type membrane proteins under native conditions; to study their interactions with ligands, drugs, and antibody fragments; and to characterize any conformational changes.
SUMMARY POINTS.
Membrane proteins represent approximately one-third of all expressed proteins. Because many of their functions are unique, mutations often cause disease.
Most drugs interact with membrane-associated receptors.
The largest class of drug receptors consists of GPCRs, which account for approximately half of all drugs currently used in medicine.
GPCRs have been important and challenging targets for all methods of structure determination because they require a phospholipid bilayer environment to be in their active conformation.
A new method, RA solid-state NMR, enables the determination of structures of GPCRs and other membrane proteins in phospholipid bilayers under physiological conditions.
FUTURE ISSUES.
Following the successful implementation of RA solid-state NMR to a 350-residue GPCR, the most important issue is to improve the sensitivity of the experiments so that this technique can become a high-throughput method.
Sample-preparation methods need to be generalized so that membrane proteins can be routinely refolded into phospholipid bilayers.
Acknowledgments
Many of the results and ideas described in this article resulted from research performed with former and current members of the Opella group. Specific topics mentioned here benefited from discussions with Bibhuti Das, Anna De Angelis, George Lu, Francesca Marassi, Sang Ho Park, and Ye Tian. The instrumentation was developed and maintained by Chin Wu and Christopher Grant. The research results described in this article were obtained with support from the National Institutes of Health through grants P41EB002031, RO1EB005161, RO1GM099986, RO1GM066978, and PO1AI074805.
Glossary
- GPCR
G protein–coupled receptor
- MAS
magic angle spinning
- DC
dipolar coupling
- OS
oriented sample
- RA
rotationally aligned
- CSA
chemical shift anisotropy
- DMPC
dimyristoyl phosphatidylcholine
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Sanders CR, Myers JK. Disease-related misassembly of membrane proteins. Annu Rev Biophys Biomol Struct. 2004;33:25–51. doi: 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- 2.Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–49. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 3.Page RC, Moore JD, Nguyen HB, Sharma M, Chase R, et al. Comprehensive evaluation of solution nuclear magnetic resonance spectroscopy sample preparation for helical integral membrane proteins. J Struct Funct Genomics. 2006;7:51–64. doi: 10.1007/s10969-006-9009-9. [DOI] [PubMed] [Google Scholar]
- 4.McDermott AE. Structural and dynamic studies of proteins by solid-state NMR spectroscopy: rapid movement forward. Curr Opin Struct Biol. 2004;14:554–61. doi: 10.1016/j.sbi.2004.09.007. Reviews MAS methods for studying proteins. Although it is an early review, many of the methods it describes are still in use. [DOI] [PubMed] [Google Scholar]
- 5.Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC. A three-dimensional model of the myoglobin molecule obtained by X-ray analysis. Nature. 1958;181:666. doi: 10.1038/181662a0. Presents the first structure of a protein determined by X-ray crystallography. [DOI] [PubMed] [Google Scholar]
- 7.Perutz MF, Rossmann MG, Cullis AF, Muirhead H, Will G, North ACT. Structure of haemoglobin: a three-dimensional Fourier synthesis at 5.5-Å resolution, obtained by X-ray analysis. Nature. 1960;185:416–22. doi: 10.1038/185416a0. [DOI] [PubMed] [Google Scholar]
- 8.Michel H, Oesterhelt D. Three-dimensional crystals of membrane proteins: bacteriorhodopsin. Proc Natl Acad Sci USA. 1980;77:1283–85. doi: 10.1073/pnas.77.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deisenhofer J, Epp O, Miki K, Huber R, Michel H. Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3- Å resolution. Nature. 1985;318:618–24. doi: 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- 10.Michel H. Three-dimensional crystals of a membrane protein complex. J Mol Biol. 1982;158:567–72. doi: 10.1016/0022-2836(82)90216-9. [DOI] [PubMed] [Google Scholar]
- 11.Landau EM, Rosenbusch JP. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci USA. 1996;93:14532–35. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 Å resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 13.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein–coupled receptor. Science. 2007;318:1258–65. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan SI. A physical chemist’s expedition to explore the world of membrane proteins. Annu Rev Biophys. 2009;38:1–27. doi: 10.1146/annurev.biophys.050708.133713. [DOI] [PubMed] [Google Scholar]
- 15.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–30. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 16.Xhou H-X, Cross TA. Influences of membrane mimetic environments on membrane protein structures. Annu Rev Biophys. 2013 doi: 10.1146/annurev-biophys-083012-130326. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller SL. A production of amino acids under possible primitive earth conditions. Science. 1953;117:528–29. doi: 10.1126/science.117.3046.528. [DOI] [PubMed] [Google Scholar]
- 18.Lombard J, López-García P, Moreira D. The early evolution of lipid membranes and the three domains of life. Nat Rev Microbiol. 2012;10:507–15. doi: 10.1038/nrmicro2815. [DOI] [PubMed] [Google Scholar]
- 19.Cross TA, Opella SJ. NMR of fd coat protein. J Supramol Struct. 1979;11:139–45. doi: 10.1002/jss.400110204. [DOI] [PubMed] [Google Scholar]
- 20.McConnell HM, Hubbell WL. Molecular motion in spin-labeled phospholipids and membranes. J Am Chem Soc. 1971;93:314–26. doi: 10.1021/ja00731a005. [DOI] [PubMed] [Google Scholar]
- 21.Cone RA. Rotational diffusion of rhodopsin in the visual receptor membrane. Nat New Biol. 1972;236:39–43. doi: 10.1038/newbio236039a0. [DOI] [PubMed] [Google Scholar]
- 22.Edidin M. Rotational and translational diffusion in membranes. Annu Rev Biophys Bioeng. 1974;3:179–201. doi: 10.1146/annurev.bb.03.060174.001143. [DOI] [PubMed] [Google Scholar]
- 23.Poo MM, Cone RA. Lateral diffusion of rhodopsin in the photoreceptor membrane. Nature. 1974;247:438–41. doi: 10.1038/247438a0. [DOI] [PubMed] [Google Scholar]
- 24.Singer SJ, Nicholson GL. The fluid mosaic model of the structure of cell membranes. Science. 1972;175:720–31. doi: 10.1126/science.175.4023.720. Presents a seminal model of a biological membrane that has motivated the field for 40 years. [DOI] [PubMed] [Google Scholar]
- 25.Park SH, Das BB, De Angelis AA, Scrima M, Opella SJ. Mechanically, magnetically, and “rotationally aligned” membrane proteins in phospholipid bilayers give equivalent angular constraints for NMR structure determination. J Phys Chem B. 2010;114:13995–4003. doi: 10.1021/jp106043w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henderson R, Unwin PR. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 27.Zerbe O. First solution structures of seven-transmembrane helical proteins. Angew Chem Int Ed. 2012;51:860–61. doi: 10.1002/anie.201107639. [DOI] [PubMed] [Google Scholar]
- 28.Reckel S, Gottstein D, Stehle J, Löhr F, Verhoefen MK, et al. Solution NMR structure of proteorhodopsin. Angew Chem Int Ed. 2011;50:11942–46. doi: 10.1002/anie.201105648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nietlispach D, Gautier A. Solution NMR studies of polytopic α-helical membrane proteins. Curr Opin Struct Biol. 2011;21:1–12. doi: 10.1016/j.sbi.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 30.Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach D. Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat Struct Mol Biol. 2010;17:768–74. doi: 10.1038/nsmb.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gautier A, Kirkpatrick JP, Nietlispach D. Solution-state NMR spectroscopy of a seven-helix trans-membrane protein receptor: backbone assignment, secondary structure, and dynamics. Angew Chem Int Ed. 2008;47:7297–300. doi: 10.1002/anie.200802783. [DOI] [PubMed] [Google Scholar]
- 32.Sanders CR, Hare B, Howard KP, Prestegard JH. Magnetically-oriented phospholipid micelles as a tool for the study of membrane-associated molecules. Prog Nucl Magn Reson Spectrosc. 1994;26:421–44. [Google Scholar]
- 33.Son WS, Park SH, Nothnagel HJ, Lu GJ, Wang Y, et al. “q-Titration” of long-chain and short-chain lipids differentiates between structured and mobile residues of membrane proteins studied in bicelles by solution NMR spectroscopy. J Magn Reson. 2012;214:111–18. doi: 10.1016/j.jmr.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Angelis AA, Jones DH, Grant CV, Park SH, Mesleh MF, Opella SJ. NMR experiments on aligned samples of membrane proteins. Methods Enzymol. 2005;394:350–82. doi: 10.1016/S0076-6879(05)94014-7. [DOI] [PubMed] [Google Scholar]
- 35.De Angelis AA, Nevzorov AA, Park SH, Howell SC, Mrse AA, Opella SJ. High-resolution NMR spectroscopy of membrane proteins in aligned bicelles. J Am Chem Soc. 2004;126:15340–41. doi: 10.1021/ja045631y. [DOI] [PubMed] [Google Scholar]
- 36.Das BB, Nothnagel HJ, Lu GJ, Son WS, Tian Y, et al. Structure determination of a membrane protein in proteoliposomes. J Am Chem Soc. 2012;134:2047–56. doi: 10.1021/ja209464f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian C, Breyer RM, Kim HJ, Karra MD, Friedman DB, et al. Solution NMR spectroscopy of the human vasopressin V2 receptor, a G protein–coupled receptor: erratum. J Am Chem Soc. 2006;128:5300. doi: 10.1021/ja051161b. [DOI] [PubMed] [Google Scholar]
- 38.Klein-Seetharaman J, Yanamala NVK, Javeed F, Reeves PJ, Getmanova EV, et al. Differential dynamics in the G protein–coupled receptor rhodopsin revealed by solution NMR. Proc Natl Acad Sci USA. 2004;101:3409–13. doi: 10.1073/pnas.0308713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein-Seetharaman J, Reeves PJ, Loewen MC, Getmanova EV, Chung J, et al. Solution NMR spectroscopy of [α-15N] lysine-labeled rhodopsin: The single peak observed in both conventional and TROSY-type HSQC spectra is ascribed to Lys-339 in the carboxyl-terminal peptide sequence. Proc Natl Acad Sci USA. 2002;99:3452–47. doi: 10.1073/pnas.052713999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. Structure determination of a membrane protein with two trans-membrane helices in aligned phospholipid bicelles by solid-state NMR spectroscopy. J Am Chem Soc. 2006;128:12256–67. doi: 10.1021/ja063640w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanford C, Reynolds JA. Characterization of membrane proteins in detergent solutions. Biochim Biophys Acta. 1976;457:133–70. doi: 10.1016/0304-4157(76)90009-5. [DOI] [PubMed] [Google Scholar]
- 42.Sanders CR, Prosser RS. Bicelles: a model membrane system for all seasons? Structure. 1998;6:1227–34. doi: 10.1016/s0969-2126(98)00123-3. [DOI] [PubMed] [Google Scholar]
- 43.Vinogradova O, Sonnichsen F, Sanders CR., 2nd On choosing a detergent for solution NMR studies of membrane proteins. J Biomol Nucl Magn Reson. 1998;11:381–86. doi: 10.1023/a:1008289624496. [DOI] [PubMed] [Google Scholar]
- 44.Kim HJ, Howell SC, Van Horn WD, Jeon YH, Sanders CR. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog Nucl Magn Reson Spectrosc. 2009;55:335–60. doi: 10.1016/j.pnmrs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tulumello DV, Deber CM. Efficiency of detergents at maintaining membrane protein structures in their biologically relevant forms. Biochim Biophys Acta. 2012;1818:1351–58. doi: 10.1016/j.bbamem.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Q, Tao H, Hong W. New amphiphiles for membrane protein structural biology. Methods. 2011;55:318–23. doi: 10.1016/j.ymeth.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem. 2006;44:S24–40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- 48.Cross TA, Opella SJ. Structural properties of fd coat protein in sodium dodecyl sulfate micelles. Biochem Biophys Res Commun. 1980;92:478–84. doi: 10.1016/0006-291x(80)90358-7. [DOI] [PubMed] [Google Scholar]
- 49.Cross TA, Opella SJ. Hydrogen-1 and carbon-13 nuclear magnetic resonance of the aromatic residues of fd coat protein. Biochemistry. 1981;20:290–97. doi: 10.1021/bi00505a010. [DOI] [PubMed] [Google Scholar]
- 50.Almeida FC, Opella SJ. fd coat protein structure in membrane environments: structural dynamics of the loop between the hydrophobic transmembrane helix and the amphipathic in-plane helix. J Mol Biol. 1997;270:481–95. doi: 10.1006/jmbi.1997.1114. [DOI] [PubMed] [Google Scholar]
- 51.Bax A. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 2003;12:1–16. doi: 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bax A, Grishaev A. Weak alignment NMR: a hawk-eyed view of biomolecular structure. Curr Opin Struct Biol. 2005;15:563–70. doi: 10.1016/j.sbi.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 53.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J Biomol Nucl Magn Reson. 2001;21:377–82. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 54.Chou JJ, Kaufman JD, Stahl SJ, Wingfield PT, Bax A. Micelle-induced curvature in a water-insoluble HIV-1 Env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J Am Chem Soc. 2002;124:2450–51. doi: 10.1021/ja017875d. [DOI] [PubMed] [Google Scholar]
- 55.Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–14. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 56.Prestegard JH, Al-Hashimi HM, Tolman JR. NMR structures of biomolecules using field oriented media and residual dipolar couplings. Q Rev Biophys. 2000;33:371–424. doi: 10.1017/s0033583500003656. [DOI] [PubMed] [Google Scholar]
- 57.Tolman JR, Al-Hashimi HM, Kay LE, Prestegard JH. Structural and dynamic analysis of residual dipolar coupling data for proteins. J Am Chem Soc. 2001;123:1416–24. doi: 10.1021/ja002500y. [DOI] [PubMed] [Google Scholar]
- 58.Soppa J. Sequence comparison does not support an evolutionary link between halobacterial retinal proteins including bacteriorhodopsin and eukaryotic G protein–coupled receptors. FEBS Lett. 1994;342:7–11. doi: 10.1016/0014-5793(94)80573-3. [DOI] [PubMed] [Google Scholar]
- 59.Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of the bacteriorhodopsin based on high-resolution electron cryomicroscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 60.Schubert M, Kolbe M, Kessler B, Oesterhelt D, Schmieder D. Heteronuclear multidimensional NMR spectroscopy of solubilized membrane proteins: resonance asignment of native bacteriorhodopsin. ChemBioChem. 2002;10:1019–23. doi: 10.1002/1439-7633(20021004)3:10<1019::AID-CBIC1019>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 61.Park SH, Casagrande F, Das BB, Albrecht L, Chi M, Opella SJ. Local and global dynamics of the G protein–coupled receptor CXCR1. Biochemistry. 2011;50:2371–80. doi: 10.1021/bi101568j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park SH, Das BB, Casagrande F, Nothnagel HJ, Chu M, et al. The structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–83. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pake GE. Nuclear resonance absorption in hydrated crystals: fine structure of the proton line. J Chem Phys. 1948;16:327–36. Uses the principles of the dipole–dipole interaction in the principal solid-state NMR method of structure determination. [Google Scholar]
- 64.Waugh JS, Huber LM, Haeberlen U. Approach to high-resolution NMR in solids. Phys Rev Lett. 1968;20:180–82. Demonstrates for the first time the line narrowing that enables chemical shift measurements in crystalline samples. These principles are used in many current solid-state NMR experiments. [Google Scholar]
- 65.Pines A, Gibby MG, Waugh JS. Proton-enhanced NMR of dilute spins in solids. J Chem Phys. 1973;59:569–90. Cleverly combines radio-frequency irradiations that increased sensitivity and resolution for “dilute” spins, such as 13C and 15N; these principles have been incorporated into essentially all current solid-state NMR experiments on proteins. [Google Scholar]
- 66.Schaefer J, Stejskal EO. Carbon-13 nuclear magnetic resonance of polymers spinning at the magic angle. J Am Chem Soc. 1976;98:1031–32. Combines, for the first time, MAS and cross-polarization double-resonance NMR; introduces solid-state NMR to chemical and biochemical applications. [Google Scholar]
- 67.Griffin RG, Pines A, Pausak S, Waugh JS. 13C chemical shielding in oxalic acid, oxalic acid dihydrate, and diammonium oxalate. J Chem Phys. 1975;63:1267–71. [Google Scholar]
- 68.Mehring M, Griffin RG, Waugh JS. 19F shielding tensors from coherently narrowed NMR powder spectra. J Chem Phys. 1971;55:746–55. [Google Scholar]
- 69.Wu CH, Ramamoorthy A, Gierasch LM, Opella SJ. Simultaneous characterization of the amide 1H chemical shift, 1H–15N dipolar, and 15N chemical shift interaction tensors in a peptide bond by three-dimensional solid-state NMR spectroscopy. J Am Chem Soc. 1995;117:6148–49. [Google Scholar]
- 70.Ramamoorthy A, Wu CH, Opella SJ. Three-dimensional solid-state NMR experiment that correlates the chemical shift and dipolar coupling frequencies of two heteronuclei. J Magn Reson B. 1995;107:88–90. doi: 10.1006/jmrb.1995.1063. [DOI] [PubMed] [Google Scholar]
- 71.Cross TA, Opella SJ. Protein structure by solid state nuclear magnetic resonance. Residues 40 to 45 of bacteriophage fd coat protein. J Mol Biol. 1985;182:367–81. doi: 10.1016/0022-2836(85)90197-4. [DOI] [PubMed] [Google Scholar]
- 72.Cross TA, DiVerdi JA, Opella SJ. Strategy for nitrogen NMR of biopolymers. J Am Chem Soc. 1982;104:1759–61. [Google Scholar]
- 73.Opella SJ, Frey MH, Cross TA. Detection of individual carbon resonances in solid proteins. J Am Chem Soc. 1979;101:5856–57. [Google Scholar]
- 74.Frey MH, Opella SJ. 13C spin exchange in amino acids and peptides. J Am Chem Soc. 1984;106:4942–45. [Google Scholar]
- 75.Shen Y, Delaglio F, Cornilescu F, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol Nucl Magn Reson. 2009;44:213–23. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ketchem RR, Hu W, Cross TA. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 1993;261:1457–60. doi: 10.1126/science.7690158. [DOI] [PubMed] [Google Scholar]
- 77.Opella SJ, Marassi FM, Gesell JJ, Valente AP, Kim Y, et al. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat Struct Biol. 1999;6:374–79. doi: 10.1038/7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marassi FM, Opella SJ. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 2003;12:403–11. doi: 10.1110/ps.0211503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Olbatt-Montal M, et al. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J Mol Biol. 2003;333:409–24. doi: 10.1016/j.jmb.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 80.Sharma M, Yi M, Dong H, Qin H, Peterson E, et al. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–12. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nishimura K, Kim S, Zhang L, Cross TA. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry. 2002;41:13170–7. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 82.Franzin CM, Teriete P, Marassi FM. Structural similarity of a membrane protein in micelles and membranes. J Am Chem Soc. 2007;129:8078–79. doi: 10.1021/ja0728371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hu J, Fu R, Cross TA. The chemical and dynamical influence of the anti-viral drug amantadine on the M2 proton channel transmembrane domain. Biophys J. 2007;93:276–83. doi: 10.1529/biophysj.106.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park SH, Opella SJ. Triton X-100 as the “short-chain lipid” improves the magnetic alignment and stability of membrane proteins in phosphatidylcholine bilayers for oriented-sample solid-state NMR spectroscopy. J Am Chem Soc. 2010;132:12552–53. doi: 10.1021/ja1055565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G. Structural topology of phospholamban pen-tamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc Natl Acad Sci USA. 2011;108:9101–6. doi: 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bechinger B, Kim Y, Chirlian LE, Gesell JJ, Neumann JM, et al. Orientations of amphipathic helical peptides in membrane bilayers determined by solid-state NMR spectroscopy. J Biomol Nucl Magn Reson. 1991;1:167–73. doi: 10.1007/BF01877228. [DOI] [PubMed] [Google Scholar]
- 87.Nicholson LK, Moll F, Mixon TE, LoGrasso PV, Lay JC, Cross TA. Solid-state 15N NMR of oriented lipid bilayer bound gramicidin A′. Biochemistry. 1987;26:6621–26. doi: 10.1021/bi00395a009. [DOI] [PubMed] [Google Scholar]
- 88.Casagrande F, Maier K, Kiefer H, Opella SJ, Park SH. Expression and purification of G protein–coupled receptors for NMR structural studies. In: Robinson A, editor. Production of Membrane Proteins: Strategies for Expression and Isolation. New York: Wiley; 2010. pp. 297–316. [Google Scholar]
- 89.Opella SJ, Waugh JS. Two-dimensional 13C NMR of highly oriented polyethylene. J Chem Phys. 1977;66:4919–24. [Google Scholar]
- 90.Cross TA, Opella SJ. Protein structure by solid state NMR. J Am Chem Soc. 1983;105:306–8. [Google Scholar]
- 91.Sinha N, Filipp FV, Jairam L, Park SH, Bradley L, Opella SJ. Tailoring 13C labeling for triple-resonance solid-state NMR experiments on aligned samples of proteins. Magn Reson Chem. 2007;45(Suppl 1):107–15. doi: 10.1002/mrc.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Edidin M. Rotational and translational diffusion in membranes. Annu Rev Biophys Bioeng. 1974;3:179–201. doi: 10.1146/annurev.bb.03.060174.001143. [DOI] [PubMed] [Google Scholar]
- 93.Cherry RJ. Protein mobility in membranes. FEBS Lett. 1975;55:1–7. doi: 10.1016/0014-5793(75)80943-4. [DOI] [PubMed] [Google Scholar]
- 94.McLaughlin AC, Cullis PR, Hemming MA, Hoult DI, Radda CK, et al. Application of 31P NMR to model and biological membrane systems. FEBS Lett. 1975;57:213–18. doi: 10.1016/0014-5793(75)80719-8. [DOI] [PubMed] [Google Scholar]
- 95.Lewis BA, Harbison GS, Herzfeld J, Griffin RG. NMR structural analysis of a membrane protein: bacteriorhodopsin peptide backbone orientation and motion. Biochemistry. 1985;24:4671–79. doi: 10.1021/bi00338a029. [DOI] [PubMed] [Google Scholar]
- 96.Park SH, Mrse AA, Nevzorov AA, De Angelis AA, Opella SJ. Rotational diffusion of membrane proteins in aligned phospholipid bilayers by solid-state NMR spectroscopy. J Magn Reson. 2006;178:162–65. doi: 10.1016/j.jmr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 97.Marassi FM, Das BB, Lu GJ, Nothnagel HJ, Park SH, et al. Structure determination of membrane proteins in five easy pieces. Methods. 2011;55:363–69. doi: 10.1016/j.ymeth.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Howell SC, Mesleh MF, Opella SJ. NMR structure determination of a membrane protein with two transmembrane helices in micelles: MerF of the bacterial mercury detoxification system. Biochemistry. 2005;44:5196–206. doi: 10.1021/bi048095v. [DOI] [PubMed] [Google Scholar]
- 99.Veglia G, Opella SJ. Lanthanide ion binding to adventitious sites align membrane proteins in micelles for solution NMR spectroscopy. J Am Chem Soc. 2000;122:11733–34. [Google Scholar]
- 100.Opella SJ, Ma C, Marassi FM. Nuclear magnetic resonance of membrane-associated peptides and proteins. Methods Enzymol. 2001;339:285–313. doi: 10.1016/s0076-6879(01)39319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.DeSilva TM, Veglia G, Porcelli F, Prantner AM, Opella SJ. Selectivity in heavy metal binding to peptides and proteins. Biopolymers. 2002;64:189–97. doi: 10.1002/bip.10149. [DOI] [PubMed] [Google Scholar]
- 102.Mesleh MF, Veglia G, DeSilva TM, Marassi FM, Opella SJ. Dipolar waves as NMR maps of protein structure. J Am Chem Soc. 2002;124:4206–7. doi: 10.1021/ja0178665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Opella SJ, DeSilva TM, Veglia G. Structural biology of metal-binding sequences. Curr Opin Chem Biol. 2002;6:217–23. doi: 10.1016/s1367-5931(02)00314-9. [DOI] [PubMed] [Google Scholar]
- 104.Mesleh MF, Lee S, Veglia G, Thiriot DS, Marassi FM, Opella SJ. Dipolar waves map the structure and topology of helices in membrane proteins. J Am Chem Soc. 2003;125:8928–35. doi: 10.1021/ja034211q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sinha N, Grant CV, Wu CH, De Angelis AA, Howell SC, Opella SJ. SPINAL modulated decoupling in high field double- and triple-resonance solid-state NMR experiments on stationary samples. J Magn Reson. 2005;177:197–202. doi: 10.1016/j.jmr.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 106.De Angelis AA, Howell SC, Opella SJ. Assigning solid-state NMR spectra of aligned proteins using isotropic chemical shifts. J Magn Reson. 2006;183:329–32. doi: 10.1016/j.jmr.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 107.De Angelis AA, Opella SJ. Bicelle samples for solid-state NMR of membrane proteins. Nat Protoc. 2007;2:2332–38. doi: 10.1038/nprot.2007.329. [DOI] [PubMed] [Google Scholar]
- 108.Diller A, Loudet C, Aussenac F, Raffard G, Fournier S, et al. Bicelles: a natural “molecular goniometer” for structural, dynamical and topological studies of molecules in membranes. Biochimie. 2009;91:744–51. doi: 10.1016/j.biochi.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Knox RW, Lu GJ, Opella SJ, Nevzorov AA. A resonance assignment method for oriented-sample solid-state NMR of proteins. J Am Chem Soc. 2010;132:8255–57. doi: 10.1021/ja102932n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wylie BJ, Franks WT, Rienstra CM. Determinations of 15N chemical shift anisotropy magnitudes in a uniformly 15N,13C-labeled microcrystalline protein by three-dimensional magic-angle spinning nuclear magnetic resonance spectroscopy. J Phys Chem B. 2006;110:10926–36. doi: 10.1021/jp060507h. [DOI] [PubMed] [Google Scholar]
- 111.Mou Y, Chen PH, Lee HW, Chan JC. Determination of chemical shift anisotropies of unresolved carbonyl sizes by C-α detection under magic-angle spinning. J Magn Reson. 2007;187:352–56. doi: 10.1016/j.jmr.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 112.Pauling L, Corey RB, Branson HR. The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA. 1951;37:205–11. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Szeverenyi NM, Sullivan MJ, Maciel GE. Observation of spin exchange by two-dimensional Fourier transform 13C cross-polarization magic angle spinning. J Magn Reson. 1982;47:462–75. [Google Scholar]
- 114.Bloembergen N. Relaxation effects in nuclear magnetic resonance absorption. Phys Rev. 1948;73:679–712. [Google Scholar]
- 115.Ladizhansky V, Jaroniec CP, Diehl A, Oschkinat H, Griffin RG. Measurement of multiple Ψ torsion angles in uniformly 13C,15N-labeled α-spectrin SH3 domain using 3D 15N–13C–13C–15N MAS dipolar-chemical shift spectroscopy. J Am Chem Soc. 2003;125:6827–33. doi: 10.1021/ja029082c. [DOI] [PubMed] [Google Scholar]
- 116.Tycko R, Stewart PL, Opella SJ. Peptide plane orientations determination by fundamental and overtone nitrogen 14 NMR. J Am Chem Soc. 1986;108:5419–25. [Google Scholar]
- 117.Opella SJ, Stewart PL, Valentine KG. Protein structure by solid-state NMR spectroscopy. Q Rev Biophys. 1987;19:7–49. doi: 10.1017/s0033583500004017. [DOI] [PubMed] [Google Scholar]
- 118.Opella SJ, Stewart PL. Solid-state nuclear magnetic resonance structural studies of proteins. Methods Enzymol. 1989;176:242–75. doi: 10.1016/0076-6879(89)76015-8. [DOI] [PubMed] [Google Scholar]
- 119.Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson. 2000;144:150–55. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marassi FM, Opella SJ. Using Pisa pies to resolve ambiguities in angular constraints from PISEMA spectra of aligned proteins. J Biomol Nucl Magn Reson. 2002;23:239–42. doi: 10.1023/a:1019887612018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nevzorov AA, Opella SJ. Structural fitting of PISEMA spectra of aligned proteins. J Magn Reson. 2003;160:33–39. doi: 10.1016/s1090-7807(02)00138-6. [DOI] [PubMed] [Google Scholar]
- 122.Nevzorov AA, Mesleh MF, Opella SJ. Structure determination of aligned samples of membrane proteins by NMR spectroscopy. Magn Reson Chem. 2004;42:162–71. doi: 10.1002/mrc.1320. [DOI] [PubMed] [Google Scholar]
- 123.Shealy P, Simin M, Park SH, Opella SJ, Valafar H. Simultaneous structure and dynamics of a membrane protein using REDCRAFT: membrane-bound form of Pf1 coat protein. J Magn Reson. 2010;207:8–16. doi: 10.1016/j.jmr.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Smurnyy Y, Opella SJ. Calculating protein structures directly from anisotropic spin-interaction constraints. Magn Reson Chem. 2006;44:283–93. doi: 10.1002/mrc.1761. [DOI] [PubMed] [Google Scholar]
- 125.Mahalakshmi R, Marassi FM. Orientation of the Escherichia coli outer membrane protein OmpX in phospholipid bilayer membranes determined by solid-state NMR. Biochemistry. 2008;47:6531–38. doi: 10.1021/bi800362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marassi FM. A simple approach to membrane protein secondary structure and topology based on NMR spectroscopy. Biophys J. 2001;80:994–1003. doi: 10.1016/S0006-3495(01)76078-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Holmes WE, Lee J, Kuang WJ, Rice GC, Wood WI. Structure and functional expression of a human interleukin-8 receptor. Science. 1991;253:1278–80. doi: 10.1126/science.1840701. [DOI] [PubMed] [Google Scholar]
- 128.Sallusto F, Baggiolini M. Chemokines and leukocyte traffic. Nat Immunol. 2008;9:949–52. doi: 10.1038/ni.f.214. [DOI] [PubMed] [Google Scholar]
- 129.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 130.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–86. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Goncalves JA, Ahuja S, Erfani S, Eilers M, Smith SO. Structure and function of G protein–coupled receptors using NMR spectroscopy. Prog Nucl Magn Reson Spectrosc. 2010;57:159–80. doi: 10.1016/j.pnmrs.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G protein–coupled receptors. Nature. 2009;459:356–63. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein–coupled receptor structures. Trends Pharmacol Sci. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Investig. 2010;120:485–97. doi: 10.1172/JCI39397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, et al. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science. 2007;318:1266–73. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 136.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, et al. Crystal structure of the human β2 adrenergic G protein–coupled receptor. Nature. 2007;450:383–87. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 137.Landau EM, Rosenbusch JP. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci USA. 1996;93:14532–35. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Oldham WM, Hamm HE. Heterotrimeric G protein activation by G protein–coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- 139.Crump MP, Gong JH, Loetscher P, Rajarantham K, Amara A, et al. Solution structure and basis for functional activity of stromal cell–derived factor 1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16:6996–7007. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rajagopalan L, Rajarathnam K. Ligand selectivity and affinity of chemokine receptor CXCR1. Role of N-terminal domain. J Biol Chem. 2004;279:30000–8. doi: 10.1074/jbc.M313883200. [DOI] [PubMed] [Google Scholar]
- 141.Park SH, Casagrande F, Cho L, Albrecht L, Opella SJ, et al. Interactions of interleukin-8 with the human chemokine receptor CXCR1 in phospholipid bilayers by NMR spectroscopy. J Mol Biol. 2011;414:194–203. doi: 10.1016/j.jmb.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu B, Chien ETY, Mol CD, Fenalti G, Liu W, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–71. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, et al. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA. 2008;105:4685–90. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, et al. Crystal structure of rhodopsin: a G protein–coupled receptor. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 145.Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 2008;77:363–82. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 146.Yarov-Yarovoy V, Schonbrun J, Baker D. Multipass membrane protein structure prediction using Rosetta. Proteins. 2006;62:1010–25. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The XPLOR-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 148.Krebs A, Edwards PC, Villa C, Li J, Schertler GF. The three-dimensional structure of bovine rhodopsin determined by electron cryomicroscopy. J Biol Chem. 2003;278:50217–25. doi: 10.1074/jbc.M307995200. [DOI] [PubMed] [Google Scholar]
RELATED RESOURCES
- 1.Drorlist. http://www.drorlist.com. Lists membrane proteins with structures determined by NMR spectroscopy.
- 2.Membrane Proteins of Known 3D Structure. http://blanco.biomol.uci.edu/mpstruc/listAll/list. Lists all membrane proteins with structures determined by all methods.
- 3.Protein Data Bank. http://www.rcsb.org. Lists all protein structures.
- 4.Biological Magnetic Resonance Data Bank. http://www.bmrb.wisc.edu. Contains NMR data for spectroscopic and structural analysis.