

Abstract
OBJECTIVES
Chronic rhinosinusitis with nasal polyps (CRSwNP) is a disorder characterized by eosinophilic inflammation and local Th2 cytokine production. Innate lymphoid cells that elaborate Th2 cytokines have recently been characterized within nasal polyps. These cells can be activated by the epithelial cell-derived cytokine IL-33. The objective of this study is to determine whether two molecules associated with tissue damage (HMGB-1 and ATP) elicit expression of IL-33 in sinonasal epithelial cells (SNEC) derived from recalcitrant CRSwNP patients.
METHODS
Ethmoid tissue was obtained from 8 recalcitrant CRSwNP and 9 control subjects during ESS. Tissue was prepared for immunohistochemistry and for SNEC air-liquid interface culture. After exposure to either HMGB1 or ATP in vitro, SNEC were processed for mRNA extraction and immunocytochemistry. IL33 levels were determined by real-time PCR and by immunochemical staining with anti-IL-33 antibody.
RESULTS
Intranuclear IL-33 is normally expressed in basal epithelial cells, but is present in more apical cells and outside the nucleus in CRSwNP. Exposure of SNEC to HMGB-1 or ATP resulted in a statistically significant increase in IL-33 mRNA expression in SNEC derived from recalcitrant CRSwNP patients. This increase was reflected at the protein level by immunochemical staining of IL-33.
CONCLUSIONS
Tissue damage is a non-specific trigger of epithelial IL-33 production in treatment-recalcitrant polyps, which may be responsible for perpetuating eosinophilic inflammation in CRSwNP. This common pathway may help explain why multiple environmental and infectious agents have been implicated in association with CRSwNP exacerbation.
Keywords: IL-33, Chronic Rhinosinusitis, Innate immunity, Nasal polyps, DAMPs
INTRODUCTION
Chronic rhinosinusitis with nasal polyps (CRSwNP) is an inflammatory disorder of unknown etiology characterized by eosinophilic inflammation and a Th2 cytokine preponderance.1 While some forms of chronic sinus disease are driven by ostiomeatal obstruction or persistent infection, CRSwNP appears to be principally an inflammatory disorder of the mucosa.2 As such, it cannot be reversed by surgery or by antimicrobial therapy, and is therefore a frustrating condition for patients and physicians alike. Multiple theories of pathogenesis have been advanced, citing microbial triggers that may stimulate a Th2 inflammatory response.3–7 To date, no specific pathogen or exogenous agent has been directly linked to the development of nasal polyps. Among the hypotheses of CRSwNP pathogenesis, a common element has been the concept that an abnormal host immune response underlies the perpetual inflammatory process.8
The mucosal immune system of the upper airway is comprised of integrated innate and adaptive mechanisms that provide homeostasis under healthy conditions. There is evidence that over-expression of Th2 cytokines such as IL-4, IL-5, and IL-13, plays a key role in eosinophilic airway inflammation.9, 10 While T cells are believed to be the major source of Th2 cytokines in CRSwNP, an additional population of cells called innate lymphoid cells, or nuocytes, has recently been described as an additional producer of these mediators.11, 12 Pathways of Th2 lymphocyte activation are reasonably well understood, and involve cell-mediated presentation of antigenic molecules and stimulation by particular types of cytokines. Activation of nuocytes is not yet well characterized, but seems to occur via the ST2 receptor, whose ligand is the IL-1 family cytokine IL-33.13 A primary source of IL-33 in airway epithelium is epithelial cells.14–17
Epithelial cells exist at the interface of the host and the environment and are the first line of defense against potential airborne threats.18 In addition to providing innate immune protection via mucus, mucociliary clearance, and production of anti-microbial effectors, epithelial cells express a wide array of signaling molecules and cytokines that modulate the activity of the local mucosal immune system.19, 20 Sinonasal epithelial cells (SNEC) are capable of detecting “danger” at the mucosal surface via a variety of pattern recognition receptors specific to microbial products, as well as to endogenous molecules associated with cellular damage.18 Under homeostatic conditions, cytokines produced by SNEC play a role in the orchestration of controlled local immune responses and their subsequent resolution. Bidirectional communication between SNEC and other resident cell populations, such as lymphocytes and innate immune cells, is essential to maintaining homeostasis. Disorders of these mechanisms that lead to Th2 cytokine expression may underlie CRS with or without nasal polyps.
When tissue damage occurs, a wide assortment of molecules are released that are normally sequestered intracellularly or within the extracellular matrix. This class of molecules, termed “damage-associated molecular patterns”, or DAMPs, has been recognized to broadly elicit innate immune responses.21 Analogous to the stimulation of the innate immune system by pathogen-associated molecular patterns (PAMPs), DAMPs are believed to act as alarm signals to local cell populations at the site of injury to initiate host repair and defense mechanisms.22 The prototypical DAMP is a constitutively-expressed nuclear DNA-binding protein called high mobility group box-1 (HMGB-1), which was first described over a decade ago as a key mediator of endotoxic shock.23, 24 Among the non-protein DAMPs, purine metabolites such as ATP, when present at high concentrations in the extracellular space, have been implicated as important innate immune stimuli. Other examples of DAMPs include DNA, RNA, S100 molecules, and uric acid. In addition to passively exiting dying or injured cells, DAMPs may also be actively synthesized and secreted by injured cells to act as “alarmins” warning the host of danger.25 Increased expression of HMGB1 protein has recently been described in CRSwNP. 26
The processes of mucosal repair and inflammation are tightly linked, and activation of mucosal innate immune pathways likely elicits both concurrently.27 In this study, we investigate the concept that IL-33 is an “alarmin” induced in SNEC in the setting of tissue damage, which promotes Th2 inflammation in CRSwNP. We have previously demonstrated that CpG, a bacterial PAMP that is a ligand for toll-like receptor 9, induces IL-33 expression by SNEC derived from recalcitrant CRSwNP patients.26 This finding suggests a mechanism by which environmental triggers such as bacterial infection could exacerbate Th2 inflammation in CRSwNP. Here, we examine whether IL-33 expression by CRSwNP SNEC occurs more broadly in response to molecular patterns associated with cellular damage.
MATERIALS AND METHODS
Human Subjects
10 patients with CRS with nasal polyps and 9 patients without sinusitis were enrolled in the study. The research protocol was approved through the Johns Hopkins Institutional Review process, and all subjects gave signed informed consent. Inclusion criteria included continuous symptoms of rhinosinusitis as defined by the AAO-HNS Chronic Rhinosinusitis Task Force for greater than 12 weeks, computed tomography of the sinuses revealing isolated or diffuse sinus mucosal thickening or air-fluid levels, and nasal polyps visible on diagnostic endoscopy. None of the subjects had a history of tobacco use, cystic fibrosis, ciliary dyskinesia, systemic inflammatory or autoimmune disease, or immunodeficiency. The CRSwNP group consisted of 5 males and 3 females, mean age 44.2 years, and the control group consisted of 6 males and 3 females with a mean age of 41.9 years. All CRSwNP subjects had a history of asthma. Although most had symptoms consistent with allergic rhinitis and had been treated with allergy therapy, allergy skin testing was not performed. Control subjects underwent endonasal endoscopic approaches for cerebrospinal fluid leak, orbital decompression, or benign sinus lesions. These subjects did not have a history of asthma or active allergic rhinitis symptoms. Prior to surgery, control and sinusitis patients received 1 week of oral methylprednisolone. All tissue specimens were taken from the resected uncinate process or the anterior ethmoid sinus. Gross polyps were not taken as specimens. The tissue was immediately placed in saline and processed for cell culture as described below in the “Sinonasal epithelial cell culture at the air-liquid interface” section, and either grown immediately or stored frozen at −80°C. All patients received an identical pre-operative medication regimen the week prior to surgery, including topical and systemic corticosteroids and antibiotics.
Patients undergoing surgery for CRSwNP were followed clinically for at least 6 months postoperatively. Those that demonstrated recurrence of nasal polyps despite surgery and a postoperative regimen including topical steroids and nasal saline irrigations, at minimum, were classified as recalcitrant CRSwNP. SNEC that had been collected at the time of surgery and frozen after growth in P0 culture, were thawed and grown at the air-liquid interface for use in the studies. Control SNEC were selected from frozen P0 cells derived from patients that had undergone surgery in a similar time frame.
Sinonasal epithelial cell culture at the air-liquid interface (ALI)
The technique of air-liquid interface cultures has been previously published by our group.28 Epithelial cells were isolated from tissue samples by enzymatic digestion and grown in cell culture. Once confluent, the cells were trypsinized, suspended in BEGM media containing DMSO, and frozen at −80 degrees. After thawing, the cells were replated onto human type IV placental collagen (Sigma, Type VI) coated 6-well Falcon filter inserts (0.4μm pore size; Becton Dickinson, Franklin Lakes, NJ). The P1 cells were grown to confluence with BEGM above (1 ml) and below (2 ml) the filter inserts. When confluent, medium was removed from above the cultures and the medium below the inserts were changed to ALI medium consisting of LHC Basal Medium:DMEM-H (Gibco) (50:50). Each set of cultures came from a separate patient source and was maintained at the air-liquid interface with the apical surfaces remaining free of medium for at least 3 weeks prior to study.
Treatment of cells with High Mobility Group Box 1 (HMGB-1) or ATP
In preliminary studies, a range of doses and durations of HMGB-1 and ATP exposure were trialed in order to determine optimal conditions for the experiments (data not shown). For HMGB-1 challenge, epithelial cells at the ALI were exposed to 100 ng/ml HMGB-1 (Sigma, St Louis, MO) in 250 microliters of media applied to the apical surface for 48 hours. For ATP challenge, epithelial cells at the ALI were exposed to 100 uM ATP (Sigma, St Louis, MO) in 250 microliters of media applied to the apical surface for 8 hours.
RNA Extraction/Reverse Transcription
Total RNA was isolated with the RNeasy Mini kit (Qiagen, Valencia, CA) the using manufacturer’s protocol. RNA was quantified spectrophotometrically and absorbance ratios at 260/280 nm were > 1.80 for all samples studied. Five hundred nanograms of total RNA was reverse transcribed in a 20 μL volume with random hexamer primers (Invitrogen), 20 U of RNase inhibitor (Applied Biosystems, Foster City, CA), and the Omniscript RT kit (Qiagen) under conditions provided by the manufacturer.
Real Time Polymerase Chain Reaction
Real time PCR was performed in a Light-Cycler 1.2 (Roche) using the SYBR Green PCR Kit (Qiagen). Primers were commercially synthesized by Invitrogen: 18S (sense 5′ GTAACCCGTTGAACCCCATT-3′; antisense 5′-CCATCCAATCGGTAGTAGCG-3′) and IL-33 (sense 5′ CATGCCAACAACAAGGAACA-3′, antisense 5′ AGGACAAAGAAGGCCTGGTC-3′). The cycle parameters used were 95°C for 15 minutes, then 45 cycles of 94°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds. Amplicon expression in each sample was normalized to its 18S RNA content. The level of expression of target mRNA was determined as the delta CT (Δ CT), as previously described 16. The difference between stimulated and unstimulated IL-33 expression was represented by the change in Δ CT; i.e. the Δ Δ CT. Fold-change was calculated as 2 raised to the Δ Δ CT power. Negative controls, consisting of reaction mixtures containing all components except target RNA, were included with each PCR run. Amplified products were sequenced to verify authenticity.
Immunofluorescence
Adherent cells at the ALI were washed and fixed with ice-cold 4% paraformaldehyde for 15 minutes at 4°C. The filter membranes were cut from the inserts and divided into 4 quadrants that were placed separately in 24-well plates. After permeabilization with 0.3% Triton-X (EMD Chemicals, Gibbstown, NJ) and blocking of nonspecific binding sites with 10% goat serum, inserts were incubated (4°C, overnight) with rabbit anti-ZO-1(Millipore, Billerica, MA) and mouse anti-IL33 monoclonal antibody (Enzo Life Sciences, Plymouth Meeting, PA). Inserts were then washed and incubated for 90 minutes with Alexa 568-conjuagted goat anti-rabbit IgG (Invitrogen, Eugene, OR). Each sample was counterstained by the nuclear stain, DAPI (Vector Labs, Burlingsgame, CA). After washing, the filter membranes were mounted on glass slides with Cytoseal mounting medium (Richard-Allen Scientific, Kalamazoo, MI). A negative control was performed by omitting primary antibody. The cells were observed under a Zeiss 510 meta confocal microscope equipped with epifluorescence illumination (Carl Zeiss, Thornwood, NY). Images were displayed as Z stacks with a 0.5 micron slice thickness. IL-33 staining was quantified as the percentage of IL-33 immunofluorescence per total nuclear area (DAPI-positive). Whole ethmoid sinus tissue samples obtained from patients undergoing sinus surgery and fixed in 4% paraformaldehyde overnight, rinsed in PBS and embedded in Tissue-Tek. Cryostat sections were blocked for 1 hour in phosphate-buffered saline (PBS) containing 5% normal secondary serum and then incubated overnight at 4°C in 5% normal serum containing primary antibody to IL-33 (Enzo Life Sciences). Primary antibodies were detected using Alexa Fluor 488-conjugated F(ab′)2 fragment of Goat anti Mouse IgG (Invitrogen, Grand Island, NY). Each sample was counterstained by the nuclear stain, DAPI (Vector Labs). Images were viewed using a LSM510 confocal microscope (Carl Zeiss Micro-imaging).
Statistical Analysis
Raw data from real-time PCR were entered into a spreadsheet (Excel; Microsoft Corp, Redmond, Wash). Statistical analysis was performed using a software program (GraphPad Prism; GraphPad Software, Inc, LaJolla, CA). Data are expressed as mean ± SEM. Statistical significances of differences in IL-33 mRNA expression were determined using the Wilcoxon signed rank test for paired data or unpaired t-test assuming unequal variances. Statistical analysis of IL-33 immunostaining of cultured cells was performed using repeated measures one-way ANOVA. Differences were considered statistically significant at P<0.05.
RESULTS
Expression of IL-33 in normal and polypoid sinonasal epithelium
In sinus mucosa obtained from control subjects, IL-33 staining was observed within the nucleus of the majority of epithelial cells in the basal layer of the epithelium. In one control subject a limited number of apically-located epithelial cells were observed to have IL-33 staining. In recalcitrant CRSwNP subjects, nuclear staining was also present in the majority of basal epithelial cells, but was more prevalent in epithelial cells located in more apical positions (6 of the 10 CRSwNP subjects), with IL-33 immunolabeling extending to the cytoplasm in a subset of those cells (figure 1). Diffuse extracellular IL-33 staining was also observed in CRSwNP tissue, which was absent in control mucosa.
Figure 1.
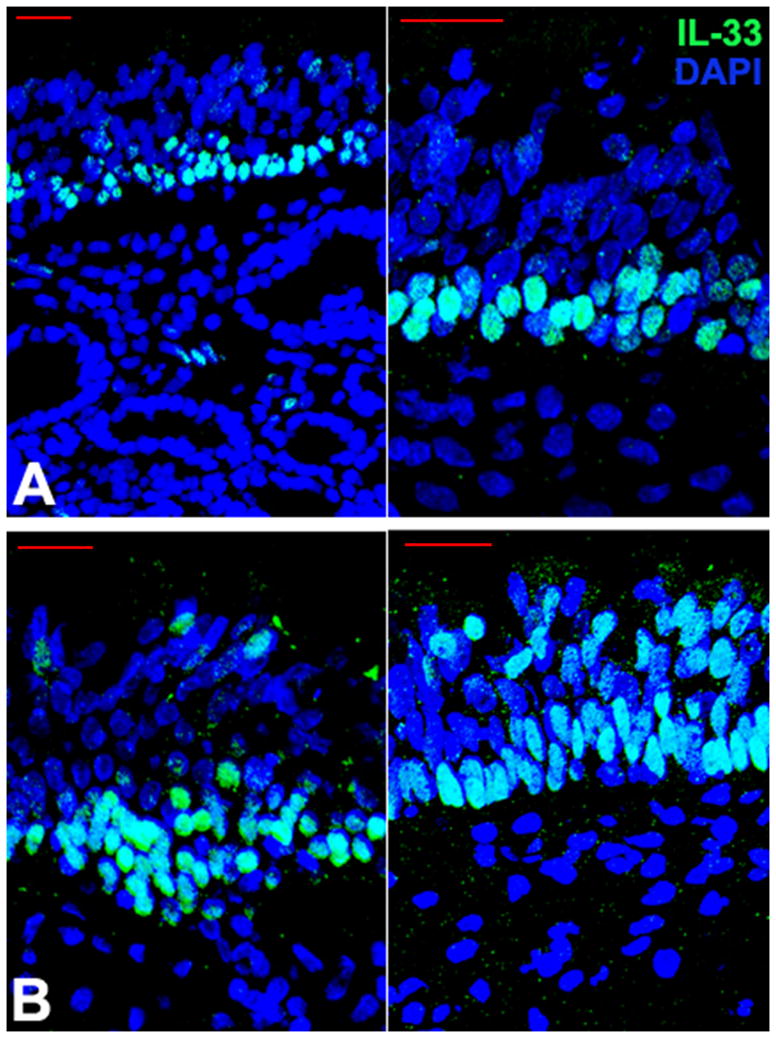
Localization of IL-33 protein in ethmoid mucosa in control and CRSwNP subjects. In control ethmoid tissue, IL-33 is expressed primarily by epithelial cells located in a basal location in the epithelium, where it is retained in the nucleus (upper panels, A). In recalcitrant CRSwNP, IL-33 is also expressed largely in the nucleus of basal epithelial cells, but also extends to cells in apical portions of the epithelium and labeling occurs outside the nucleus in many cases, both in the cytoplasm and extracellularly (lower panels, B). Scale bar=25 microns.
Treatment with damage-associated molecular patterns increases expression of IL-33 in epithelial cells derived from patients with recalcitrant CRSwNP
SNEC from 8 subjects with recalcitrant CRSwNP and 9 subjects without CRS were exposed to either 100 ng/ml HMGB-1 for 48 hours or 100 μM ATP for 8 hours. In pairwise comparisons of individual control subject SNECs, these DAMPs did not elicit a statistically significant increase in IL-33 mRNA expression. In contrast, in CRSwNP subject SNECs, HMGB-1 increased expression of IL-33 by a mean cycle threshold of 1.2 cycles (p=0.005) and ATP increased IL-33 mRNA expression by a mean of 1.0 cycle (p=0.003). Comparing mean control and CRSwNP group SNEC responses, HMGB-1 induced a 2.8-fold increase in IL-33 mRNA in polyp SNEC as compared to a 1.2 fold increase in controls (p=0.046). Similarly, ATP induced a 2.2-fold mean increase in IL-33 mRNA in CRSwNP, and a 1.2-fold increase in controls (p=0.033) (figure 2). These findings were confirmed at the protein level using immunohistochemical staining for IL-33 in differentiated SNEC grown at the ALI (figure 3). There is overall increase in nuclear staining for IL-33, although this did not achieve statistical significance by repeated measures one-way ANOVA.
Figure 2.

The damage-associated molecules, HMGB-1 and ATP induce expression of IL-33 mRNA in sinonasal epithelial cells derived from recalcitrant CRSwNP subjects. Data is expressed as fold increase over unstimulated condition within individual subjects. * p= 0.046; **p=0.033
Figure 3.

Stimulation of sinonasal epithelial cell expression of IL-33 with HMGB-1 and ATP. Sinonasal epithelial cells grown at the air-liquid interface were exposed to the damage-associated molecules, HMGB-1 (100 ng/ml) for 24 hours, or ATP (100 μM) for 8 hours. A marked increase in nuclear expression of IL-33 protein was observed in recalcitrant CRSwNP subjects, although the mean increase in nuclear immunofluoresence did not achieve statistical significance for the group as a whole. (A/B: pre- and post-HMGB-1 exposure; C/D: pre- and post-ATP exposure). E: Graphical representation of increase in mean IL-33 immunostaining following exposure to HMGB-1 and ATP, across all subjects (p=ns).
DISCUSSION
The sinonasal epithelium is subject to numerous microbial and environmental insults that inflict damage. As the first line of defense, a critical role of SNEC is to maintain a physical and immune barrier at the mucosal surface. SNEC also communicate via cytokines with other local cell types to coordinate adaptive immune responses and ultimately resolve inflammation to allow repair of injuries. While inflammatory and repair mechanisms remain in a homeostatic balance in health, CRSwNP research has suggested that dysregulation of these functions may underlie chronic Th2-mediated eosinophilic inflammation. The present study provides evidence that abnormal expression of the pro-Th2 cytokine IL-33 by SNEC may be a feature of recalcitrant CRSwNP contributing to persistent inflammation. This may suggest a pathogenic pathway in CRSwNP in which SNEC-derived IL-33 targets innate lymphoid cells, resulting in ongoing local production of IL-4, IL-5, and IL-13.
IL-33 is an evolutionarily conserved molecule that drives a variety of cell types (e.g. Th2 cells, mast cells, basophils, eosinophils, natural killer cells, and innate lymphoid cells) to produce cytokines and chemokines.29–34 It is constitutively expressed and stored by cells at barrier surfaces, which strongly implies a function in mucosal immunity.35 Two recent large genome-wide association studies have identified polymorphisms in the IL-33 gene as significantly associated with asthma 36, 37, and increased expression of IL-33 is present in the airways of subjects with severe asthma.38 Given the histopathologic similarities between asthma and CRSwNP, it is reasonable to postulate a similar role for IL-33 in inflammatory eosinophilic sinus disease. As we have demonstrated in SNEC, lower airway epithelial cells display strong IL-33 nuclear staining and release IL-33 when stimulated by extracellular ATP.15, 39 Features shared by asthma and CRSwNP include not only eosinophilic infiltration and tissue remodeling, but also damage of the airway epithelium, likely with substantial release of DAMPs. Even though the mechanisms of IL-33 expression, processing and secretion are not yet clear, IL-33 is of great interest as a link between innate and adaptive Th2-type immune responses, acting on novel non-T/non-B cell population such as nuocytes to produce Th2 cytokines..11, 40, 41
In the present study, while DAMPs significantly increased IL-33 mRNA expression in vitro, a corresponding statistically significant increase in IL-33 protein could not be demonstrated by immunofluorescence. This may be a reflection of the time points chosen, which were based on the time course of mRNA expression rather than protein translation. A more detailed examination using longer time points and a more quantifiable technique for protein assessment might reveal correlating differences in IL-33 protein expression. It also remains possible that the DAMP-induced IL-33 mRNA in CRSwNP SNEC is not translated into protein. The mechanism of the differential response to DAMPs by CRSwNP SNEC is unclear, but may relate to epigenetic alterations that occur in a setting of chronic inflammation. Alternatively, CRSwNP SNEC may be intrinsically abnormal in their innate immune activity, perhaps predisposing the mucosa to developing persistent eosinophilic inflammation.
Our present findings of elevated IL-33 expression by CRSwNP sinonasal epithelial cells in response to DAMP stimulation, taken together with previous similar observations with TLR9 activation, suggests that a broad dysregulation of IL-33 expression by SNEC may contribute to recalcitrant CRSwNP. IL-33 appears to be normally retained in the nucleus of basal epithelial cells, consistent with its role as an “alarmin” released during necrotic cell death and associated with infection or tissue injury.42–44 In CRSwNP, IL-33 expression extends to more apically-positioned SNEC where its sub-cellular localization demonstrates release from the nucleus. While typically considered pathologic in the context of human diseases, Th2 inflammation is an important mechanism of anti-parasitic defense and likely also plays a normal role in balancing Th1 immune responses in healthy mucosa. Because Th2 cytokines have anti-inflammatory properties, they may be essential to resolution and repair after mucosal injury or infection.45 Future studies with human tissue and animal models will be necessary to demonstrate the normal function of IL-33 and elucidate patterns of abnormal regulation that potentially underlie chronic eosinophilic sinonasal inflammation.
CONCLUSIONS
The present study demonstrates that sinonasal epithelial cells derived from recalcitrant CRSwNP patients express the cytokine IL-33 in response to damage-associated molecular patterns in vitro. Moreover, while the expression of IL-33 is largely confined to the nuclei of basal epithelial cells in control subject ethmoid mucosa, tissue samples from recalcitrant CRSwNP frequently display IL-33 protein expression in the nuclei of more apically-located cells with extension into the cytoplasm. Because IL-33 has the capacity to activate Th2 cytokine-producing innate lymphoid cells residing in the sinus mucosa, dysregulation of this pathway may play a part in perpetuating eosinophilic inflammation in CRSwNP patients who are recalcitrant to medical and surgical therapy.
Acknowledgments
Research supported by NIH AI072502 (A.P.L.).
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Presented at the Annual Meeting of the American Rhinologic Society, September 8, 2012, Washington, DC.
References
- 1.Fokkens WJ, Lund VJ, Mullol J, et al. EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012. A summary for otorhinolaryngologists. Rhinology. 2012 Mar;50(1):1–12. doi: 10.4193/Rhino12.000. [DOI] [PubMed] [Google Scholar]
- 2.Van Crombruggen K, Zhang N, Gevaert P, Tomassen P, Bachert C. Pathogenesis of chronic rhinosinusitis: Inflammation. J Allergy Clin Immunol. 2011 Oct;128(4):728–732. doi: 10.1016/j.jaci.2011.07.049. [DOI] [PubMed] [Google Scholar]
- 3.Palmer JN. Bacterial biofilms: do they play a role in chronic sinusitis? Otolaryngol Clin North Am. 2005 Dec;38(6):1193–1201. viii. doi: 10.1016/j.otc.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Orlandi RR, Marple BF. Fungus and chronic rhinosinusitis: weighing the evidence. Otolaryngol Head Neck Surg. 2010 Nov;143(5):611–613. doi: 10.1016/j.otohns.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Bachert C, van Zele T, Gevaert P, De Schrijver L, Van Cauwenberge P. Superantigens and nasal polyps. Curr Allergy Asthma Rep. 2003 Nov;3(6):523–531. doi: 10.1007/s11882-003-0065-y. [DOI] [PubMed] [Google Scholar]
- 6.Slavin RG. Sinusitis: viral, bacterial, or fungal and what is the role of Staph? Allergy Asthma Proc. 2006 Nov-Dec;27(6):447–450. doi: 10.2500/aap.2006.27.2890. [DOI] [PubMed] [Google Scholar]
- 7.Zhang N, Holtappels G, Gevaert P, et al. Mucosal tissue polyclonal IgE is functional in response to allergen and SEB. Allergy. 2011 Jan;66(1):141–148. doi: 10.1111/j.1398-9995.2010.02448.x. [DOI] [PubMed] [Google Scholar]
- 8.Lee S, Lane AP. Chronic rhinosinusitis as a multifactorial inflammatory disorder. Curr Infect Dis Rep. 2011 Apr;13(2):159–168. doi: 10.1007/s11908-011-0166-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachert C, Gevaert P, Holtappels G, van Cauwenberge P. Mediators in nasal polyposis. Curr Allergy Asthma Rep. 2002 Nov;2(6):481–487. doi: 10.1007/s11882-002-0088-9. [DOI] [PubMed] [Google Scholar]
- 10.Meltzer EO, Hamilos DL, Hadley JA, et al. Rhinosinusitis: Establishing definitions for clinical research and patient care. Otolaryngol Head Neck Surg. 2004 Dec;131(6 Suppl):S1–62. doi: 10.1016/j.otohns.2004.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mjosberg JM, Trifari S, Crellin NK, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–1062. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 12.van Drunen CM, Mjosberg JM, Segboer CL, Cornet ME, Fokkens WJ. Role of innate immunity in the pathogenesis of chronic rhinosinusitis: progress and new avenues. Curr Allergy Asthma Rep. 2012 Apr;12(2):120–126. doi: 10.1007/s11882-012-0249-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012 Feb 1;188(3):1503–1513. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008 Dec;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prefontaine D, Nadigel J, Chouiali F, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010 Mar;125(3):752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- 16.Pichery M, Mirey E, Mercier P, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol. 2012 Apr 1;188(7):3488–3495. doi: 10.4049/jimmunol.1101977. [DOI] [PubMed] [Google Scholar]
- 17.Kamekura R, Kojima T, Takano K, Go M, Sawada N, Himi T. The role of IL-33 and its receptor ST2 in human nasal epithelium with allergic rhinitis. Clin Exp Allergy. 2011 Feb;42(2):218–228. doi: 10.1111/j.1365-2222.2011.03867.x. [DOI] [PubMed] [Google Scholar]
- 18.Lane AP. The role of innate immunity in the pathogenesis of chronic rhinosinusitis. Curr Allergy Asthma Rep. 2009 May;9(3):205–212. doi: 10.1007/s11882-009-0030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diamond G, Legarda D, Ryan LK. The innate immune response of the respiratory epithelium. Immunol Rev. 2000 Feb;173:27–38. doi: 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- 20.Proud D, Leigh R. Epithelial cells and airway diseases. Immunol Rev. 2011 Jul;242(1):186–204. doi: 10.1111/j.1600-065X.2011.01033.x. [DOI] [PubMed] [Google Scholar]
- 21.Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012 Sep;249(1):158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007 Jan;81(1):1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Yang H, Tracey KJ. Extracellular role of HMGB1 in inflammation and sepsis. J Intern Med. 2004 Mar;255(3):320–331. doi: 10.1111/j.1365-2796.2003.01302.x. [DOI] [PubMed] [Google Scholar]
- 24.Erlandsson Harris H, Andersson U. Mini-review: The nuclear protein HMGB1 as a proinflammatory mediator. Eur J Immunol. 2004 Jun;34(6):1503–1512. doi: 10.1002/eji.200424916. [DOI] [PubMed] [Google Scholar]
- 25.Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007 Dec;220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- 26.Reh DD, Wang Y, Ramanathan M, Jr, Lane AP. Treatment-recalcitrant chronic rhinosinusitis with polyps is associated with altered epithelial cell expression of interleukin-33. Am J Rhinol Allergy. 2010 Mar-Apr;24(2):105–109. doi: 10.2500/ajra.2010.24.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen JE, Wynn TA. Evolution of Th2 immunity: a rapid repair response to tissue destructive pathogens. PLoS Pathog. 2011 May;7(5):e1002003. doi: 10.1371/journal.ppat.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramanathan M, Jr, Lane AP. A comparison of experimental methods in molecular chronic rhinosinusitis research. Am J Rhinol. 2007 May-Jun;21(3):373–377. doi: 10.2500/ajr.2007.21.3034. [DOI] [PubMed] [Google Scholar]
- 29.Borish L, Steinke JW. Interleukin-33 in asthma: how big of a role does it play? Curr Allergy Asthma Rep. 2011 Feb;11(1):7–11. doi: 10.1007/s11882-010-0153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurowska-Stolarska M, Stolarski B, Kewin P, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009 Nov 15;183(10):6469–6477. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 31.Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, Kita H. IL-33-activated dendritic cells induce an atypical TH2-type response. J Allergy Clin Immunol. 2009 May;123(5):1047–1054. doi: 10.1016/j.jaci.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005 Nov;23(5):479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol. 2008 Jun;121(6):1484–1490. doi: 10.1016/j.jaci.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol. 2007 Oct;37(10):2779–2786. doi: 10.1002/eji.200737547. [DOI] [PubMed] [Google Scholar]
- 35.Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010 Feb;10(2):103–110. doi: 10.1038/nri2692. [DOI] [PubMed] [Google Scholar]
- 36.Moffatt MF, Gut IG, Demenais F, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010 Sep 23;363(13):1211–1221. doi: 10.1056/NEJMoa0906312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torgerson DG, Ampleford EJ, Chiu GY, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 2011 Sep;43(9):887–892. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poon AH, Eidelman DH, Martin JG, Laprise C, Hamid Q. Pathogenesis of severe asthma. Clin Exp Allergy. 2012 May;42(5):625–637. doi: 10.1111/j.1365-2222.2012.03983.x. [DOI] [PubMed] [Google Scholar]
- 39.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011 Apr 1;186(7):4375–4387. doi: 10.4049/jimmunol.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price AE, Liang HE, Sullivan BM, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A. 2010 Jun 22;107(25):11489–11494. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koyasu S, Moro K. Innate Th2-type immune responses and the natural helper cell, a newly identified lymphocyte population. Curr Opin Allergy Clin Immunol. 2010 Apr;11(2):109–114. doi: 10.1097/ACI.0b013e3283448808. [DOI] [PubMed] [Google Scholar]
- 42.Lamkanfi M, Dixit VM. IL-33 raises alarm. Immunity. 2009 Jul 17;31(1):5–7. doi: 10.1016/j.immuni.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 43.Lefrancais E, Roga S, Gautier V, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A. 2012 Jan 31;109(5):1673–1678. doi: 10.1073/pnas.1115884109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U S A. 2009 Jun 2;106(22):9021–9026. doi: 10.1073/pnas.0812690106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monticelli LA, Sonnenberg GF, Abt MC, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011 Nov;12(11):1045–1054. doi: 10.1031/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]