

Abstract
Young age at first sexual intercourse is an important risk factor for cervical cancer, but no simple statistical model of its influence has been established. We investigated the relationship between risk of cervical carcinoma and time since first intercourse using data on monogamous women (5,074 cases and 16,137 controls) from the International Collaboration of Epidemiological Studies of Cervical Cancer. Odds ratios (ORs) and 95% confidence intervals (CIs) were estimated from pooled data on 20 studies using conditional logistic regression. The OR for invasive cervical carcinoma is approximately proportional to the square of time since first intercourse (exponent 1.95, 95% CI: 1.76–2.15) up to age 45. First cervical infection with human papillomavirus (HPV) often occurs soon after first sexual intercourse, so early age at first sexual intercourse is a reasonable proxy for early age at first exposure to HPV. In addition, age-specific incidence rates of cervical cancer in unscreened populations remain fairly constant above age 45. Cervical cancer thus resembles other cancers caused by strong early-stage carcinogens, with incidence rates proportional to a power of time since first exposure and also resembles cancers of the breast and other hormone-dependent epithelia where a similar flattening of age-specific incidence rates is seen at the time menopausal changes start. Taken together, these observations suggest that HPV vaccination may prevent the majority of cervical cancers by delaying HPV infection without necessarily providing lifetime protection against HPV.
Keywords: cervical carcinoma, cervical intraepithelial neoplasia, age at first sexual intercourse, multi-stage carcinogenesis
INTRODUCTION
Cervical cancer is caused by sexually transmitted human papillomaviruses (HPVs), 12 of which have been classified as carcinogenic to humans.1 Most HPV infections clear within 1–2 years, but those that persist may progress quite rapidly to cervical intraepithelial neoplasia (CIN) grade 2 or worse.2 Early age at first sexual intercourse (AFI) is an important risk factor for cervical cancer.3 Other risk factors include lifetime number of sexual partners,4 tobacco use5 hormonal contraceptives,6 young age at first full-term pregnancy and number of full-term pregnancies.7
The International Collaboration of Epidemiological Studies of Cervical Cancer collected data on sexual behaviour from 21 national or international epidemiological studies. A collaborative reanalysis of these data found a steadily increasing risk with earlier AFI, with a relative risk of 3.52 (95% confidence interval, (CI): 3.04–4.08) for AFI ≤14 versus ≥25 years, controlling for lifetime number of sexual partners. This effect was reduced, but not eliminated, by further controlling for parity and age at first full-term pregnancy (relative risk=2.05, 95% CI: 1.54–2.73).4
First infection with HPV often occurs soon after first sexual intercourse,2 so early AFI is a reasonable proxy for early age at first exposure to HPV.8 In the current study we revisit the data on AFI to determine whether its association with cervical cancer reflects a progressive increase in risk with the passage of time since first sexual intercourse (TFI).
MATERIALS AND METHODS
Population
Full details of the study design are given elsewhere4 and details of the studies included are given in Appendix A. Briefly, data on women aged 16–89 with invasive cervical carcinoma and controls from 20 epidemiological studies were pooled. One study included in the previous report on sexual factors4 was excluded because it did not collect information on AFI. For cohort studies, a nested case-control sample was selected with up to four age-matched controls per case. All case-control studies were matched by age. Women with CIN3/carcinoma in situ were not included in these analyses, as detection of CIN3 depends on screening coverage and frequency and may sometimes be delayed by many years even in regularly screened populations by the insensitivity of cytology and colposcopy9. There were 9,819 cases of invasive cervical carcinoma and 24,731 controls with complete data on AFI and lifetime number of sexual partners. Confounding by lifetime number of sexual partners was controlled by restricting the analysis to women who reported only 1 sexual partner (5,150 invasive cervical carcinoma cases and 16,686 controls). Women who reported very early (<10 years) or late (>30 years) AFI were excluded. Thus, 5,074 cases and 16,137 controls were included in the analysis. Among cases, 3,837 (75%) were from less developed countries and 1,237 (25%) from more developed countries.
Since the pooled case control data are age matched, they cannot provide information on age-specific effects. The age profile of cervical cancer incidence was estimated using population-based cervical cancer incidence rates from six countries from Asia and South America with no or very little cervical cancer screening.10
Model for incidence rates
The initial model for cervical cancer incidence rates is derived from the multistage model of carcinogenesis,11 which predicts that cancer incidence rates increase as a power of time (See Appendix B for more details). For cervical cancer, the appropriate time scale is TFI since first sexual intercourse determines the first possible exposure to HPV. The basic model is thus that the incidence rate is proportional to (TFI)K, where the exponent K is to be estimated.
This initial model is, however, inconsistent with the age profile of cervical cancer incidence. The model predicts that the incidence rate will continue to increase without limit as age (and hence TFI) increases, whereas cervical cancer incidence rates reach a plateau in middle age.10 We therefore modified the model by fitting TFI truncated at age 45 years. For a woman over 45, truncated TFI takes the value that TFI took when she was 45 years old.
Statistical methods
For the pooled case-control data, odds ratios (ORs) and 95% CIs for invasive cervical carcinoma were estimated using conditional logistic regression stratified by age in 5-year groups and study centre. The exponent of TFI in our model for cervical cancer incidence rates is the coefficient of log(TFI) in this logistic regression model. Other factors considered in a multivariable regression analysis included reproductive age (i.e., number of years since menarche), smoking status (current, never, former, not recorded), oral contraceptive use (any, none, not recorded), parity (0, 1–2, 3–4, ≥5 full-term pregnancies), and delay between first intercourse and first full-term pregnancy (<2, 2–3, 4–5, 6–10, ≥11 years). Reproductive age was calculated by subtracting age at menarche from age at diagnosis or interview, or from age at menopause among post8 menopausal women. For categorical analysis truncated TFI was classified into single-year groups up to 9 years, and 5-year groups thereafter.
Tests for linearity were conducted by comparing the deviance in two models: one using a categorical predictor and one with categories replaced by scores derived from the mean value of the exposure within each category. In Figure 1 “floating” CIs, which are independent of the choice of reference group, are shown.12
Figure 1. Odds ratios (and 95% floating confidence intervals) for cervical cancer by time since first intercourse stratified by age group1.
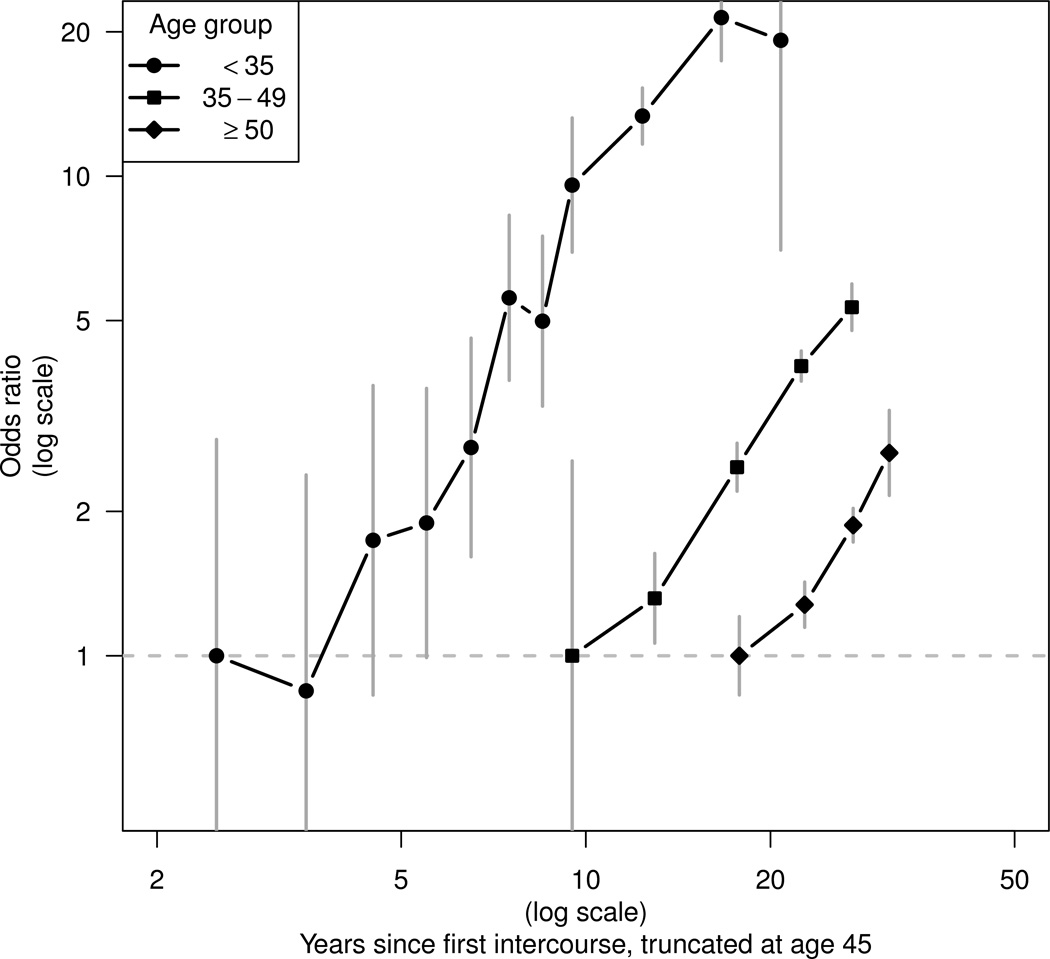
1 Separate reference categories for time since first intercourse are used in each age stratum.
For the population-based cervical cancer incidence data, an age-cohort model was fitted using Poisson regression to the age-specific incidence data in 5-year age- and time-bands for the calendar years 1971–2000.10 The age profile for each country was represented using the fitted values for the 1950–1954 birth cohort. These age profiles were compared with the predicted rates from our model assuming AFI at 18 years, the median AFI in our pooled data. The model predictions were calibrated to reach a maximum of 50 per 100,000 woman years, consistent with the range of incidence rates in middle-age women in these populations.
RESULTS
Figure 1 shows ORs for truncated TFI fitted as a categorical variable separately in women aged <35, 35–49 and ≥50 years. Both axes are shown on a log scale. Tests for linearity in each stratum gave p-values of 0.55, 0.43, and 0.31, respectively. There was no significant difference in slope between the three age strata (χ2 = 2.67 on 2 d.f., p = 0.26). The data are thus consistent with a simplified model in which the OR increases as a power of truncated TFI that is constant at all ages.
Table 1 shows estimates of the coefficient of truncated log(TFI) in a trend model with and without adjustment for other variables. The unadjusted estimate is 1.95 (95% CI: 1.76–2.15), implying that the OR is approximately proportional to the square of truncated TFI. This result is not materially affected by including reproductive age (adjusted estimate=1.93), smoking status (1.95), oral contraceptive use (1.96) or delay between first intercourse and first full-term pregnancy (1.92) in the multiple logistic regression. The estimated coefficient is however attenuated by including parity (adjusted estimate=1.61, 95% CI: 1.40–1.81). When histological types were considered separately, the coefficient was larger for squamous cell carcinoma (1.97 95% CI: 1.77–2.16) than for adenocarcinoma and adenosquamous carcinoma (1.66 95% CI: 1.23–2.08) but there was substantial overlap in the CIs. A case-case comparison between histological types showed no significant difference (p=0.08) (Data not shown). There was also no significant difference in the coefficient between developed and developing countries (p=0.46) (Data not shown).
Table 1.
Conditional logistic regression estimates of the coefficient of the log of time since first intercourse, truncated at age 45.
| Confounders controlled for | Coefficient | 95% CI |
|---|---|---|
| Stratifying variables (age, centre) only | 1.95 | 1.76–2.15 |
| Reproductive age1 | 1.93 | 1.73–2.13 |
| Oral contraceptive use | 1.96 | 1.76–2.16 |
| Delay between first intercourse and first full-term pregnancy | 1.92 | 1.72–2.12 |
| Number of full-term pregnancies | 1.61 | 1.40–1.81 |
Years between menarche and current age or menopause, whichever came first.
CI = confidence interval
Figure 2 shows the age profile of cervical cancer incidence rates in six largely unscreened populations from different continents. The rates increase steeply within each birth cohort up to age 40–44 then flatten abruptly. The same age profile is shown by a model in which the incidence rate is proportional to the square of truncated TFI, assuming first intercourse at 18 years of age.
Figure 2. Age-dependence of cervical cancer incidence in largely unscreened populations in calendar years 1971–2001 from an age-cohort model. The age-specific rates displayed are fitted values for the 1950–1954 birth cohort.
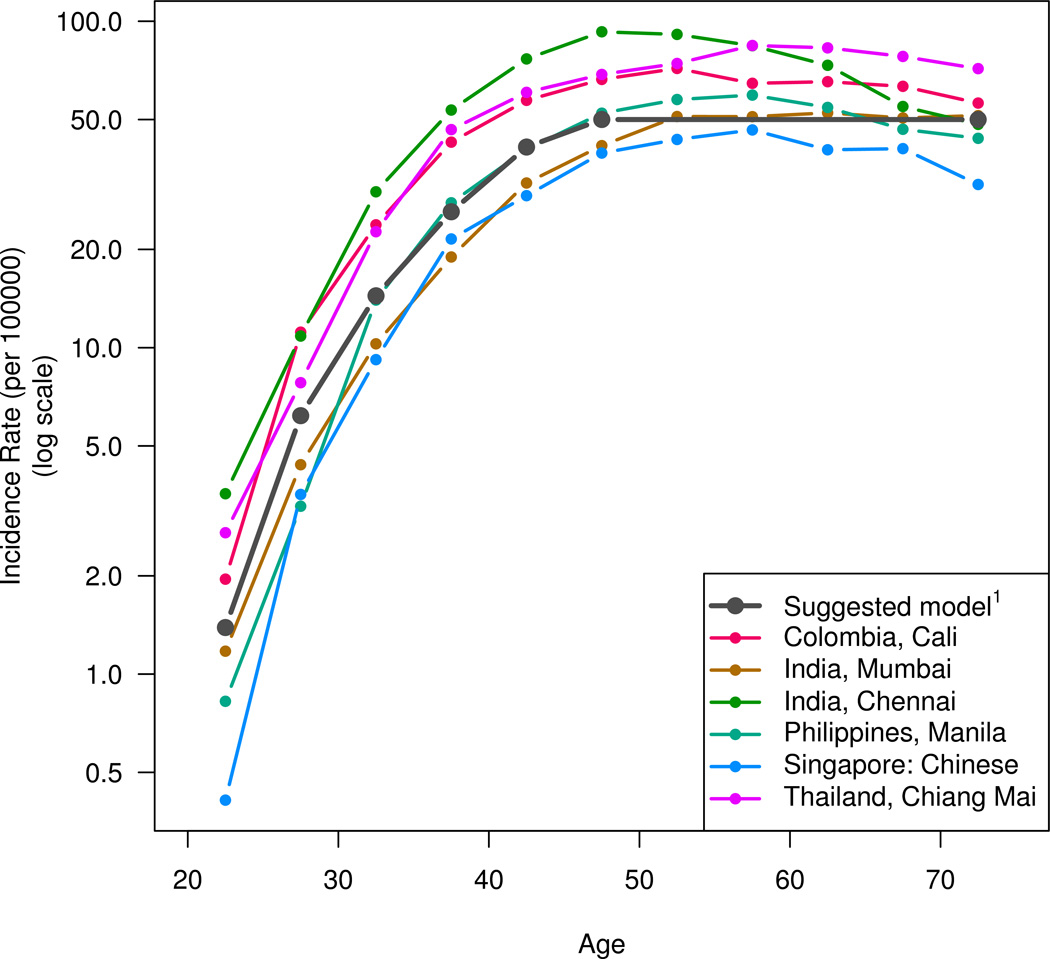
1 The suggested model is that the incidence rate is proportional to the square of time since first intercourse, truncated at age 45.
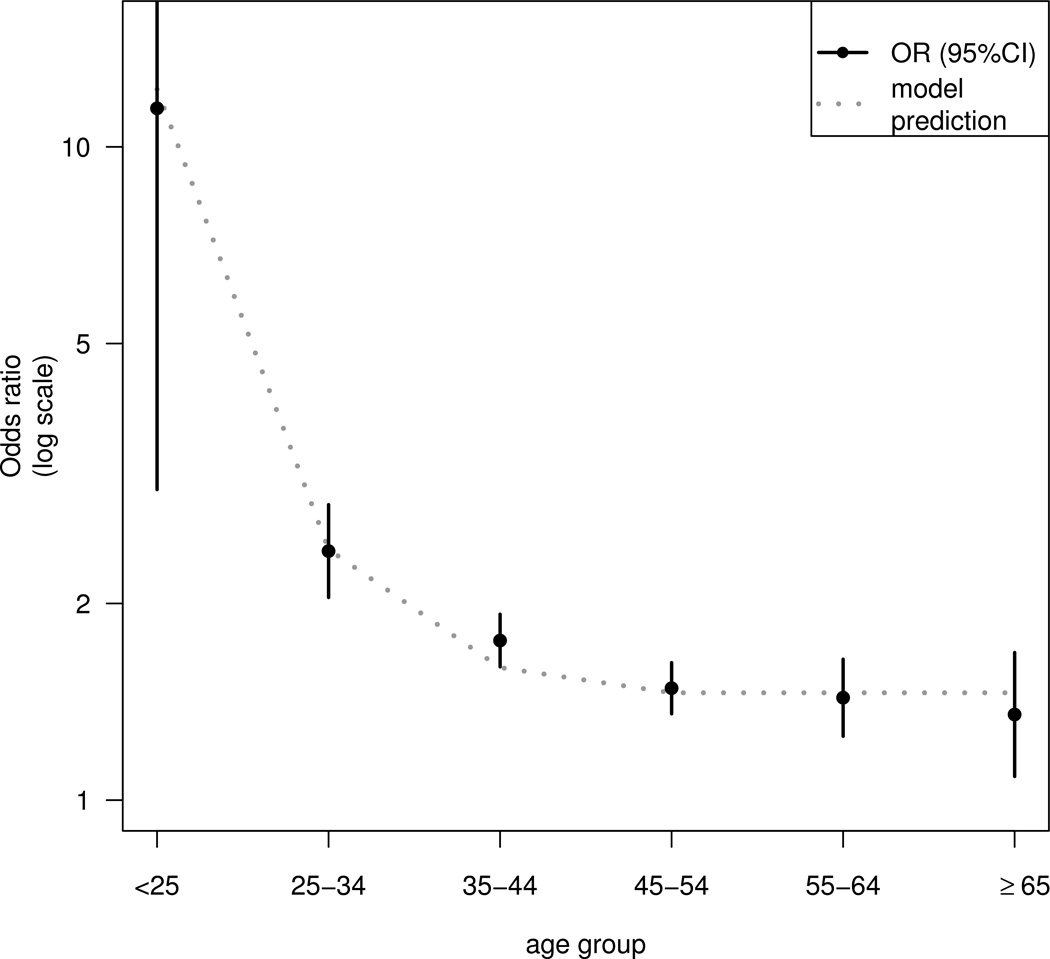
An important prediction of our model is that the effect of earlier age at first intercourse on cervical cancer risk is not homogeneous, but depends on the age of the woman, due to the non-linear relationship between AFI and log(TFI). This is investigated in Figure 3, which shows ORs for 5-year earlier first intercourse in different age groups. The ORs are estimated from a logistic regression model fitted to the pooled case control data in which AFI was included as a continuous variable but TFI was not included. The estimated OR declines sharply with increasing age up to 45 years (11.4, 95% CI: 2.99–43.9 at age <25 years; 2.4, 95% CI: 2.04–2.83 at age 25–34; 1.76, 95% CI: 1.60–1.93 at 35–44; 1.48, 95% CI: 1.36–1.62 at 45–54), but there is no statistically significant reduction at older ages (OR = 1.43, 95% CI: 1.25–1.64 at age 55–64 years; 1.35, 95% CI: 1.09–1.68 at age ≥65). The line in Figure 3 shows the predicted ORs for first intercourse at 16 years versus 21 years, from our model in which the incidence rate is proportional to the square of TFI truncated at age 45. The predictions of this model fit the observed decline in the effect of AFI.
Figure 3. Odds ratio estimates (and 95% confidence intervals) of cervical cancer for age at first intercourse 5 years earlier, and the predictions for first intercourse at age 16 years vs 21 years from the model.
To test the hypothesis that the increased risk of cervical cancer in women with early AFI may be due to an increased vulnerability of the immature cervix to HPV infection, we analysed the effect of the delay between age at menarche and AFI, using women with a delay of 5 years or over as the reference category and controlling for TFI (Table 2). The OR is slightly lower than 1 in the first year after menarche (OR = 0.84, 95% CI: 0.71–0.99) and there is a small excess risk among women with a delay of 4 years (OR = 1.17, 95% CI: 1.03–1.33), but overall there is no significant trend with delay between menarche and first intercourse (p=0.08).
Table 2.
Delay between menarche and first sexual intercourse and risk of invasive cervical carcinoma
| Delay (years) |
Cases | Controls | OR1 | (95% CI) |
|---|---|---|---|---|
| <1 | 379 | 865 | 0.84 | (0.70–0.99) |
| 1 | 417 | 1,046 | 0.96 | (0.82–1.12) |
| 2 | 536 | 1,293 | 1.08 | (0.94–1.24) |
| 3 | 548 | 1,436 | 1.04 | (0.91–1.19) |
| 4 | 599 | 1,514 | 1.17 | (1.03–1.33) |
| ≥5 | 2,535 | 9,550 | 1 |
In strata defined by age and centre and controlling for log(TFI) truncated at age 45.
CI = confidence interval; OR= odds ratio
DISCUSSION
Our pooled case-control data is the largest epidemiological data set on cervical cancer ever assembled. Originally motivated by the study of hormonal contraceptives6 it also allows us to study other risk factors for cervical cancer in much finer detail than has previously been possible. The model suggested by our analysis, together with age-specific incidence rates in unscreened populations, is that the cervical cancer incidence rate increases approximately as the square of TFI up to about age 45 years, then flattens and remains fairly constant at older ages. We interpret this pattern as a result of two effects: increasing risk of cancer following HPV infection, and a strong menopause-related effect on cervical cancer risk.
Duration of HPV infection and the multi-stage model of carcinogenesis
Our analysis is based on age-matched case-control data, so the results of our present pooled analysis provide no direct estimate of the effect of age per se on the incidence rate. Our assumption that the sharp increase with age, up to age 45, is due entirely to the roughly quadratic relationship with TFI is however supported by the similarity of this model and the incidence pattern of cervical cancer in unscreened populations.
Our suggestion that cervical cancer risk increases as a power of time, independent of age, is consistent with findings from other cancers caused by powerful initiating (early-stage) carcinogens. Lung cancer incidence is approximately proportional to the fourth power of time since starting to smoke in continuing smokers,13 and the mesothelioma rate in asbestos workers is proportional to the cube of time since first exposure to asbestos.14 These patterns are explained by mathematical models of carcinogenesis in which a cell must undergo a number of changes before malignant transformation. These changes need not correspond to individual mutations, since many different mutations may lead to the acquisition of the same capability required for malignant transformation.15 The same multi-stage models also predict the increase as a power of age in the incidence rates of many other epithelial cancers that do not have a single well defined cause, including lung cancer in non-smokers.11 Due to the wide applicability of the multi-stage model, it had been hypothesised that the increased risk of early AFI may be an effect of duration of the relevant exposure even before HPV was identified as the cause of cervical cancer. 16,17
The square of time for cervical cancer incidence is lower than the exponent of 4 or more for many epithelial cancers.11,13,14 According to the multi-stage model, the exponent of time is determined by the number of rate limiting cellular changes required for malignant transformation, modified by the dynamics of partially transformed cells. Thus the lower exponent for cervical cancer suggests fewer rate-limiting steps in cervical carcinogenesis or, equivalently, that HPV accelerates the infected cell through some steps leaving relatively few further changes necessary for malignant transformation. This may reflect the functional inactivation by HPV E6 and E7 oncoproteins of the products of both TP53 and RB1,18 two oncosuppressor genes that are commonly mutated or deleted in many human cancers. An in vitro model suggests that the introduction of only one oncogene could generate cervical cancer on the background of expression of HPV16 E6 and E7 oncoproteins in normal human cervical keratinocytes.19 Likewise, incidence rates are greatly increased in individuals with hereditary syndromes caused by inherited or germline mutations in RB1 or APC compared with the general population, but rise as a lower power of age.20
The steps in cervical carcinogenesis discussed here are changes at the molecular level. The correspondence between these changes and lesions detected by cytology and histology is not clear. Inactivation of TP53 and RB1 may account for the rapid progression from HPV infection to CIN3 but other genetic or epigenetic changes may occur both during the development of CIN3 and during the sojourn time from development of CIN3 to invasive cancer, which is usually several decades.
The inflexion in age-specific cancer incidence rates
The sharp inflexion in cervical cancer incidence rates in middle age is also seen in cancers of the breast, ovary and endometrium.21 Pike et al21 explained the incidence pattern in these cancers using the concept of “effective tissue age”, which is proportional to the number of stem cell divisions in these hormone-dependent tissues. Effective tissue age is measured from menarche, not birth, and the rate of change is not constant throughout life, but slows between ages 40 and 50, finally reaching a much lower rate in post-menopausal women.
We hypothesize that cervical cancer rates exhibit a similar inflexion because the cervix is also a hormone-dependent tissue. A strong effect of oestrogen on cervical carcinogenesis in mice was demonstrated experimentally 70 years ago.22 The human cervix is strongly altered by hormonal changes,23 and cervical cancer incidence rates are transiently increased by oral contraceptive use.6 The inflexion in middle age seen in cervical cancer incidence rates may therefore reflect the drop in circulating sex hormone levels during the peri-menopausal period. It is also worth noting that newly incident CIN3 is seldom detected after menopause2.
Whereas Pike et al21 modelled a continous drop in effective tissue ageing rate between ages 40 and 50 years, we have used a model in which effective tissue ageing stops at age 45. This simplification makes the model fitting easier by allowing us to define truncated TFI, but it does not imply that there is an abrupt change at age 45. The menopausal transition lasts a few years and is characterized by gradual increase of anovulatory cycles and fluctuating sex hormone levels24. We note that the final menstrual period, which marks the end of the menopausal transition, typically occurs somewhat later than age 45 but the earlier age that we have used to truncate TFI is around the time that the transition begins.
Despite similarities with other hormone-dependent cancers, there are some differences. Early first full-term pregnancy and high parity are associated with a reduction in risk of breast, ovarian and endometrial cancer,21 but with an increase in cervical cancer risk.7 Furthermore, our model predicts that early menopause is associated with decreased risk of cervical cancer risk, as seen for breast cancer, but the epidemiological evidence on the effect of early menopause on cervical cancer is inconsistent. Among studies contributing this pooled analysis we found statistically significant findings of both increased25 and decreased17 risk associated with earlier final menstrual period.
Biases in the estimated exponent of time
Our results are presented in terms of the empirical relationship between TFI and cervical cancer risk. We assume that AFI has been reported accurately, although we note that AFI may be misreported to conform to social conventions on sexual behaviour. Further interpretation of the effect of TFI in terms of duration of an underlying HPV infection is difficult. Although first HPV infection often occurs soon after first intercourse, the majority of HPV infections are transient, and probably irrelevant to cervical cancer risk. A transforming infection, which leads to cervical cancer, may be acquired much later. If the delay between AFI and this infection is substantial, the fitted exponent of time since infection (if it could be observed) would be lower than our estimated exponent of TFI. A substantial delay between malignant transformation and diagnosis of cervical cancer would have the same effect.
The approximately quadratic effect of TFI on risk of cervical cancer was not modified by controlling for potential confounders with the exception of the number of full term pregnancies, which reduced the exponent from 1.95 to 1.61. This suggests that full term pregnancies may modify “cervical tissue age” as has been shown for breast tissue age.21 The unadjusted exponent of 1.95 is appropriate for population-level predictions when individual data on FTPs are not available but the adjusted value of 1.61 may predict the direct effects of early first intercourse more accurately.
The adolescent cervix
Vulnerability of the adolescent cervix to HPV infection has been cited as a possible explanation for the risk of early age at first sexual intercourse.26 Our findings show that it is not necessary to invoke adolescent vulnerability if TFI is taken into account. We found a non-significant, slightly reduced risk of cancer among women who had first sexual intercourse within a year of menarche (OR = 0.86, 95% CI: 0.72–1.02). A longitudinal study of incident HPV infection among young women27 found a reduced infection rate among those for whom AFI was close to menarche, suggesting that the immature cervix is less easily infected than the mature cervix. This is consistent with the observation that the cervical transformation zone, the tissue target of HPV infection, increases in size after menarche.27 An alternative explanation is that the rate of infection in very young women is low because their partners tend to be young men who may not yet have been infected with HPV.
Implications for vaccination
HPV vaccines are highly effective in preventing HPV infection for up to 7 years,28 but the longer term protection is currently unknown. The cost-effectiveness of short-term vaccine protection has been explored through highly complex Markov models. A consistent feature of these models is their sensitivity to the duration of protection conferred by HPV vaccination.29 It has been suggested that if the duration of protection is not sufficiently long, vaccination of adolescent girls risks being “a costly failed public health experiment in cancer control”.30 In contrast, our model suggests that delaying first exposure to HPV by vaccination would have the same lifelong effect as delaying AFI. For example, a delay of first HPV infection of 12 years from age 18 to 30 years would prevent almost all cervical cancers below age 40 and reduce the risk more than 3-fold above age 45. Conversely, our analysis suggests little advantage of vaccinating older women in the prevention of cervical cancer. Women can be infected by carcinogenic HPV at any age,2 but the lifetime cancer risk caused by a new HPV infection will fall sharply with age at infection and will be very low after about age 40.
Supplementary Material
Novelty and impact.
This study is the first to apply the multistage model of carcinogenesis to cervical cancer as a possible explanation for the well-known association between cervical cancer risk and early age at first intercourse. The model is tested using the largest epidemiological data set ever assembled.
ACKNOWLEDGMENTS
This work was supported by the UNPD/UNFPA/WHO/World Bank Special Program of Research, Development, and Research Training in Human Reproduction, Department of Reproductive Health and Research, World Health Organization, the International Agency for Research on Cancer (IARC), and Cancer Research UK.
Julian Peto was the recipient of an IARC Senior Visiting Scientist Award in 2010.
Abbreviations used
- AFI
age at first sexual intercourse
- CIN
cervical intraepithelial neoplasia
- CI
confidence interval
- HPV
human papillomavirus
- OR
odds ratio
- TFI
time since first sexual intercourse
Collaborators
Cancer Institute (WIA), Chennai, India: T Rajkumar. Cancer Research UK Epidemiology Group, Wolfson Institute of Preventive Medicine, London, UK: J Cuzick. Cancer Research UK Epidemiology Unit, Oxford, UK: P Appleby, V Beral, A Berrington de González, D Bull, K Canfell, B Crossley, J Green, G Reeves, S Sweetland. Danish Cancer Society, Copenhagen, Denmark: S Kjaer. Unit of Health Care Epidemiology, Oxford, UK: R Painter, M Vessey. Fred Hutchinson Cancer Research Center, Seattle, Washington, USA: J Daling, M Madeleine, R Ray, DB Thomas. Guanacaste Epidemiological Project, San José, Costa Rica: R Herrero. Department of Medical Epidemiology and Biostatistics, Karolinska Institut, Stockholm, Sweden: N Ylitalo. Epidemiology and Cancer Registration Unit, IDIBELL, Institut Català d’Oncologia, Barcelona, Spain: FX Bosch, X Castellsagué, S de Sanjosé, K Louie, V Moreno. Institut National de Santé Publique, Algiers, Algeria: D Hammouda. Istituto di Ricerche Farmacologiche ‘Mario Negri’, Milan, Italy: E Negri. Instituto Especializado de Enfermedades Neoplásicas ‘Dr Eduardo Cáceres Graziani’, Lima, Peru: M Alvarez, O Galdos, C Santos, C Velarde. Instituto Nacional de Cancerología, Bogata, Colombia: N Muñoz. International Agency for Research on Cancer, Lyon, France: S Franceschi, M Plummer. London School of Hygiene and Tropical Medicine, London and Institute of Cancer Research, Sutton, UK: J Peto. Lund University, Mälmo, Sweden: J Dillner. Uppsala University Hospital, Uppsala, Sweden: I Silins. Ministère de la Santé Publique et des Affaires Sociales, Bamako, Mali: S Bayo. Ministry of Health, Rabat, Morocco: N Chaouki. Ministry of Health, Asunción, Paraguay: PA Rolón. National Cancer Institute, NIH, Bethesda, Maryland, USA: L Brinton, P Castle, A Hildesheim, J Lacey Jr, M Schiffman. Cancer Epidemiology Research Group, National Health Laboratory Service, Johannesburg, South Africa: L Stein, MI Urban. Royal College of General Practitioners Oral Contraception Study, UK: P Hannaford. Prince of Songkla University, Songkla, Thailand: SB Chichareon. The Cancer Council New South Wales, Sydney, Australia: F Sitas. Universidade de São Paulo, São Paulo, Brazil: J Eluf-Neto. University of Milan, Milan, Italy: C La Vecchia. University of Otago, Dunedin, New Zealand: D Skegg. University of Southern California, Los Angeles, California, USA: R Peters, MC Pike, G Ursin. University of the Philippines, Manila, The Philippines: C Ngelangel. University of Tromsø, Tromsø, Norway: IT Gram. World Health Organization, Geneva, Switzerland: T Farley, O Meirik.
Footnotes
CONFLICT OF INTEREST
None to declare.
REFERENCES
- 1.Schiffman M, Clifford G, Buonaguro FM. Classification of weakly carcinogenic human papillomavirus types: addressing the limits of epidemiology at the borderline. Infect Agent Cancer. 2009;4:8. doi: 10.1186/1750-9378-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodriguez AC, Schiffman M, Herrero R, Hildesheim A, Bratti C, Sherman ME, Solomon D, Guillen D, Alfaro M, Morales J, Hutchinson M, Katki H, et al. Longitudinal study of human papillomavirus persistence and cervical intraepithelial neoplasia grade 2/3: critical role of duration of infection. J Natl Cancer Inst. 2010;102:315–324. doi: 10.1093/jnci/djq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louie KS, de Sanjose S, Diaz M, Castellsague X, Herrero R, Meijer CJ, Shah K, Franceschi S, Munoz N, Bosch FX. Early age at first sexual intercourse and early pregnancy are risk factors for cervical cancer in developing countries. Br J Cancer. 2009;100:1191–1197. doi: 10.1038/sj.bjc.6604974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical carcinoma and sexual behavior: collaborative reanalysis of individual data on 15,461 women with cervical carcinoma and 29,164 women without cervical carcinoma from 21 epidemiological studies. Cancer Epidemiol Biomarkers Prev. 2009;18:1060–1069. doi: 10.1158/1055-9965.EPI-08-1186. [DOI] [PubMed] [Google Scholar]
- 5.International Collaboration of Epidemiological Studies of Cervical Cancer. Carcinoma of the cervix and tobacco smoking: collaborative reanalysis of individual data on 13,541 women with carcinoma of the cervix and 23,017 women without carcinoma of the cervix from 23 epidemiological studies. Int J Cancer. 2006;118:1481–1495. doi: 10.1002/ijc.21493. [DOI] [PubMed] [Google Scholar]
- 6.International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical cancer and hormonal contraceptives: collaborative reanalysis of individual data for 16,573 women with cervical cancer and 35,509 women without cervical cancer from 24 epidemiological studies. Lancet. 2007;370:1609–1621. doi: 10.1016/S0140-6736(07)61684-5. [DOI] [PubMed] [Google Scholar]
- 7.International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical carcinoma and reproductive factors: collaborative reanalysis of individual data on 16,563 women with cervical carcinoma and 33,542 women without cervical carcinoma from 25 epidemiological studies. Int J Cancer. 2006;119:1108–1124. doi: 10.1002/ijc.21953. [DOI] [PubMed] [Google Scholar]
- 8.Edelstein ZR, Madeleine MM, Hughes JP, Johnson LG, Schwartz SM, Galloway DA, Carter JJ, Koutsky LA. Age of diagnosis of squamous cell cervical carcinoma and early sexual experience. Cancer Epidemiol Biomarkers Prev. 2009;18:1070–1076. doi: 10.1158/1055-9965.EPI-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeronimo J, Schiffman M. Colposcopy at a crossroads. Am J Obstet Gynecol. 2006;195:349–353. doi: 10.1016/j.ajog.2006.01.091. [DOI] [PubMed] [Google Scholar]
- 10.Ferlay J, Parkin DM, Curado MP, Bray F, Edwards B, Shin HR, Forman D. Cancer Incidence in Five Continents, Volumes I to IX. 9 ed. Lyon, France: International Agency for Research on Cancer; 2010. [Google Scholar]
- 11.Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plummer M. Improved estimates of floating absolute risk. Stat Med. 2004;23:93–104. doi: 10.1002/sim.1485. [DOI] [PubMed] [Google Scholar]
- 13.Doll R. An epidemiological perspective of the biology of cancer. Cancer Res. 1978;38:3573–3583. [PubMed] [Google Scholar]
- 14.Peto J. Dose-response relationships for asbestos-related disease: implications for hygiene standards. Part II. Mortality. Ann N Y Acad Sci. 1979;330:195–203. doi: 10.1111/j.1749-6632.1979.tb18720.x. [DOI] [PubMed] [Google Scholar]
- 15.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 16.Peto R. Letter: Cervical cancer and early sexual intercourse. Int J Epidemiol. 1976;5:97. [PubMed] [Google Scholar]
- 17.La Vecchia C, Franceschi S, Decarli A, Fasoli M, Gentile A, Parazzini F, Regallo M. Sexual factors, venereal diseases, and the risk of intraepithelial and invasive cervical neoplasia. Cancer. 1986;58:935–941. doi: 10.1002/1097-0142(19860815)58:4<935::aid-cncr2820580422>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 18.IARC. Vol. 90: Human papillomavirus. Lyon: IARC Press; 2007. Monographs on the evaluation of carcinogenic risks to humans. [Google Scholar]
- 19.Narisawa-Saito M, Yoshimatsu Y, Ohno S, Yugawa T, Egawa N, Fujita M, Hirohashi S, Kiyono T. An in vitro multistep carcinogenesis model for human cervical cancer. Cancer Res. 2008;68:5699–5705. doi: 10.1158/0008-5472.CAN-07-6862. [DOI] [PubMed] [Google Scholar]
- 20.Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J. Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. 2004;96:357–363. doi: 10.1093/jnci/djh058. [DOI] [PubMed] [Google Scholar]
- 21.Pike MC, Pearce CL, Wu AH. Prevention of cancers of the breast, endometrium and ovary. Oncogene. 2004;23:6379–6391. doi: 10.1038/sj.onc.1207899. [DOI] [PubMed] [Google Scholar]
- 22.Allen E, Gardner WU. Cancer of the Cervix of the Uterus in Hybrid Mice Following Long-Continued Administration of Estrogen. Cancer Res. 1941;1:359–365. [Google Scholar]
- 23.Chung SH, Franceschi S, Lambert PF. Estrogen and ERalpha: Culprits in cervical cancer? Trends Endocrinol Metab. 2010;21:504–511. doi: 10.1016/j.tem.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burger H. The menopausal transition--endocrinology. J Sex Med. 2008;5:2266–2273. doi: 10.1111/j.1743-6109.2008.00921.x. [DOI] [PubMed] [Google Scholar]
- 25.Franceschi S, Rajkumar T, Vaccarella S, Gajalakshmi V, Sharmila A, Snijders PJ, Munoz N, Meijer CJ, Herrero R. Human papillomavirus and risk factors for cervical cancer in Chennai, India: a case-control study. Int J Cancer. 2003;107:127–133. doi: 10.1002/ijc.11350. [DOI] [PubMed] [Google Scholar]
- 26.Moscicki AB, Winkler B, Irwin CE, Jr, Schachter J. Differences in biologic maturation, sexual behavior, and sexually transmitted disease between adolescents with and without cervical intraepithelial neoplasia. J Pediatr. 1989;115:487–493. doi: 10.1016/s0022-3476(89)80863-7. [DOI] [PubMed] [Google Scholar]
- 27.Collins S, Mazloomzadeh S, Winter H, Rollason T, Blomfield P, Young L, Woodman C. Proximity of first intercourse to menarche and the risk of human papillomavirus infection: a longitudinal study. Int J Cancer. 2005;114:498–500. doi: 10.1002/ijc.20732. [DOI] [PubMed] [Google Scholar]
- 28.De Carvalho N, Teixeira J, Roteli-Martins CM, Naud P, De Borba P, Zahaf T, Sanchez N, Schuind A. Sustained efficacy and immunogenicity of the HPV-16/18 AS04-adjuvanted vaccine up to 7.3 years in young adult women. Vaccine. 2010;28:6247–6255. doi: 10.1016/j.vaccine.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Dasbach EJ, Insinga RP, Elbasha EH. The epidemiological and economic impact of a quadrivalent human papillomavirus vaccine (6/11/16/18) in the UK. BJOG. 2008;115:947–956. doi: 10.1111/j.1471-0528.2008.01743.x. [DOI] [PubMed] [Google Scholar]
- 30.Harper DM. Preliminary HPV vaccine results for women older than 25 years. Lancet. 2009;373:1921–1922. doi: 10.1016/S0140-6736(09)61045-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.