

Abstract
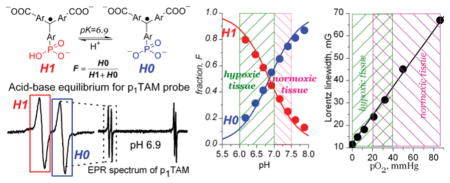
Previously we proposed the concept of dual function pH and oxygen paramagnetic probes based on the incorporation of ionizable groups into the structure of persistent triarylmethyl radicals, TAMs (J. Am. Chem. Soc. 2007, 129, 7240–7241). In this paper, we synthesized an asymmetric monophosphonated TAM probe with the simplest doublet hfs pattern ideally suited for dual function electron paramagnetic resonance (EPR)-based applications. An extraordinary low line width of the synthesized deuterated derivative, p1TAM-D (ΔHpp ≤ 50 mG, Lorentz line width, ≤20 mG) results in high sensitivity to pO2 due to oxygen-induced line broadening (ΔLW/ΔpO2 ≈ 0.5 mG/mmHg or ≈400 mG/mM); accuracy of pO2 measurement, ≈1 mmHg). The presence of a phosphono group in the p1TAM-D structure provides pH sensitivity to its EPR spectra in the physiological range of pH from 5.9 to 8.2 with the ratio of signal intensities of protonated and deprotonated states being a reliable pH marker (accuracy of pH measurements, ± 0.05). The independent character of pH and [O2] effects on the EPR spectra of p1TAM-D provides dual functionality to this probe. The L-band EPR studies performed in breast tumor-bearing mice show a significant difference in extracellular pH and pO2 between tumor and normal mammary gland tissues, as well as the effect of animal breathing with 100% O2 on tissue oxygenation. The developed dual function phosphonated p1TAM-D probe provides a unique tool for in vivo concurrent tissue oxygen and pH monitoring.
INTRODUCTION
Tissue pH and pO2 are among the most important parameters in physiology and pathophysiology of living organisms. For two leading causes of mortality in the United States, cancer and ischemic heart disease, it is well-documented that observed tissue hypoxia1 is associated with changes in glycolysis resulting in tissue acidosis.2,3 Methods that monitor these parameters in vivo are of critical importance for diagnostics and optimization of treatment strategies of these and other diseases.2,4–8
Low-field electron paramagnetic resonance (EPR)-based techniques in combination with paramagnetic probes are powerful tools for in vivo functional biomedical applications. The lack of endogenous EPR signals provides these techniques with an advantage over NMR in signal specificity. Among the paramagnetic probes, triarylmethyl radicals, TAMs, have advantages in extraordinary stability toward tissue redox processes, long relaxation time, and narrow line width, making them particularly attractive for imaging applications.9 In spite of the fact that the synthesis of TAMs has been reported by Gomberg more than century ago,10 the TAM structure with sterically protected trivalent carbon only recently regained attention as the basic fragment for the synthesis of stable organic radicals after innovative development by Nycomed.9,11 The EPR spectra of these TAM derivatives display a very narrow single line of about 100 mG9,12 and long relaxation time13 which makes them attractive for continuous wave (CW)14 and pulsed EPR imaging (EPRI),15–17 proton–electron double-resonance imaging (PEDRI),9,18,19 and hyperpolarized NMR20 and MRI21 applications. TAMs are considered to be the most useful soluble paramagnetic probes for oximetric applications9,14,18,19 with oxygen-induced line broadening effect of about 500 mG/mM of oxygen. The other oxygen-sensitive paramagnetic materials include particulate probes19,22–24 which measure oxygen partial pressure in the place of probe implantation, whereas soluble TAM probes are more suitable for imaging applications.4,23
We proposed the concept of dual function pH and oxygen probes based on TAM structures containing ionizable groups.25,26 It has been realized that TAM derivatives with a single EPR line, such as Oxo63, are not suitable for in vivo pH measurements due to negligible pH-induced spectral shift at low EPR frequencies.25 On the other hand, the hyperfine splitting (hfs) is frequency-independent and can be used as a sensitive pH marker at low EPR fields. Recently, amino26 and triphosphonated27,28 TAM probes with hfs pattern being sensitive to pH in the physiological range were synthesized. However, complex spectral patterns of these TAM derivatives make their applications for concurrent oxygen and pH mapping impractical. Further simplification of spectral properties of dual function TAM probes is desirable ideally providing the simplest doublet hfs pattern.
EXPERIMENTAL METHODS
Synthesis
The route for the synthesis of asymmetric phosphonated TAM derivatives is shown in Scheme 1.
Scheme 1.

Synthesis of Asymmetric Phosponated Trityls. (a) 2.5 M Butyl Lithium in Hexanes, Benzene, [EtO]2CO/[EtO]2P(O)Cl, 2:1 Ratio; (b) NaOH H2O/1,4-Dioxane; (c) TFA; (d) TMSBr, CH2Cl2
(a). One-Step Synthesis of Asymmetric Core Compound
The starting material (1-H or 1-D, 0.53 g, 0.6 mmol) was dissolved in 5 mL of benzene; tetramethylethylenediamine (TMEDA, 6 mmol, 0.723 g, 0.932 mL) was added, and the mixture was placed in an ice bath. Butyl lithium (2.5 M in hexanes, 6 mmol, 2.42 mL) was added slowly via a syringe. The solution was stirred for 30 min, placed at room temperature, and stirred for another hour. This solution was added slowly via syringe to another solution placed in an ice bath of diethyl carbonate (20 mmol, 2.36 g, 2.42 mL) and diethyl chlorophosphate (10 mmol, 1.72 g, 1.44 mL) in 5 mL of benzene. The mixture was stirred over 72 h at room temperature. The reaction mixture was quenched with 20 mL of saturated aqueous ammonium chloride and diluted with 20 mL of ethyl acetate. The organic phase was separated, washed with diluted aqueous solution of hydrochloric acid, dried under anhydrous sodium sulfate, and evaporated under vacuum. The residue was purified by flash chromatography on silica gel eluted with a gradient of hexanes–ethyl acetate. The products were pooled by thin layer chromatography (hexanes–ethyl acetate 3:1). Two fractions, each containing the two products at different ratios, were isolated. The products, 21-H and 22-H synthesized from 1-H, or 21-D and 22-D synthesized from 1-D, were identified using C–OH NMR shift (between 6.5 and 6.8 ppm) as a marker.
(b). General Procedure for the Hydrolysis of the Carboxylic Ester
Each fraction from step (a) (2n-H [n = 1,2] or 2n-D [n = 1,2]) was dissolved in 10 mL of 1,4-dioxane. Aqueous sodium hydroxide solution (2.5 g in 20 mL H2O) was added, and the mixture was heated at reflux over three hours. Upon completion, the reaction was diluted with 20 mL of water and extracted with 20 mL of ethyl acetate. The organic phase was separated and discarded. The aqueous phase was acidified with aqueous hydrochloric acid to pH = 1 and extracted with 20 mL of ethyl acetate. The ethyl acetate phase was separated, dried over anhydrous sodium sulfate, and evaporated under vacuum. The residue was used without further analysis. Purification and identification of specific products by EPR spectroscopy was performed in the next step.
(c). General Procedure for the Synthesis of the Radicals
The residue from step (b) was dissolved in 4 mL of trifluoroacetic acid and stirred overnight. The reaction was evaporated until dry. The residue was purified through a Hypersep C18 cartridge using a gradient of water and methanol. The pure products were isolated and pooled from the mixture of byproducts using the deep green color of the trityl radicals as an indicator. EPR spectral analysis confirmed the presence of trityl radicals, the monophosphate ethyl ester (31-H or 31-D), and the diphosphate ethyl ester (32-H or 32-D).
(d). General Procedure for Deprotecting the Phosphate Ethyl Esters
Each product from step (c) was dissolved in 5 mL of dichloromethane, and 0.6 mL of TMS-Br was added. The solution was stirred overnight, quenched with 2 mL of ethanol, a few drops of water, and evaporated until dry. The product was dissolved in 10 mM aqueous sodium hydroxide solution and purified again through a Hypersep C18 cartridge using water and methanol. The purity of the products was confirmed by EPR spectral and mass spectroscopy analyses.
Overall, approximately 10 mg of pure monohosphonated derivatives, p1TAM-H or p1TAM-D, and 14 mg of pure diphosphonated derivatives, p2TAM-H or p2TAM-D, were obtained. p1TAM-H: MS (ESI, [M + Na]+): 1035.9259 (observed), 1035.9160 (calcd); p1TAM-D: MS (ESI, [M + Na]+): 1072.1402 (observed), 1072.1499 (calcd); p2TAM-H: MS (MALDI, [M+]): 1070.812 (observed), 1070.892 (calcd); p2TAM-D: MS (MALDI, [M + H+]): 1108.993 (observed), 1108.1226 (calcd).
X-Band EPR Studies
Measurements were performed on X-band EMX EPR spectrometer (Bruker, Germany). Temperature and gas composition during EPR measurements were controlled by a temperature and gas controller (Noxygen, Germany). To control gas composition, Teflon tubes with a diameter of 1.14 mm and wall thickness of 60 μm (Zeus, Inc., USA) were used throughout the experiments. The typically used instrument settings were as follows: microwave power, 40 μW; time constant, 20.48 ms; conversion time, 10 ms; sweep time, 81.92 s; modulation frequency, 10 kHz; modulation amplitude, 0.02 G; sweep width, 5 G; number of points, 8192.
L-Band EPR Studies
Measurements were performed on L-band EPR spectrometer (Magnettech, Germany). Temperature was controlled using water bath attached to the thermostat. Gas composition was controlled by bubbling solution with gas delivered from temperature and gas controller (Noxygen, Germany). The typical instrument settings were as follows: attenuation 24 dB; modulation amplitude, 37.5 mG; modulation frequency, 100 kHz; sweep width, 0.9 G; sweep time, 20 s.
pH Titration
Radical solutions were titrated by addition of a small volume of NaOH or HCl with the final dilution of sample less than 1%. pH was controlled by electrode calibrated at 37 °C using pH values for reference solution recommended by National Bureau of Standards (U.S.). Temperature of reference and titrated solutions during pH measurements was controlled using jacketed reaction beaker attached to Lauda Circulator E100. Anoxic conditions were maintained using temperature and gas controller (Noxygen, Germany) for X-band studies or using addition of glucose (10 mM) and glucose oxidase (Sigma, USA, 100 U/mL) to probe solution for L-band studies.
Analysis of the EPR Spectra Line Shape
Lineshape analysis of the EPR spectra was performed as previously described.8 The experimental spectra were fitted by rational approximation of Voigt function given by Hui et al.29 The analysis of fully deprotonated form, p1TAM4– (pH 10), and partially deprotonated form, p1TAM3– (pH 4.5), yields the values of Lorentz and Gaussian line width. The obtained values of Gaussian linewidths which are determined by unresolved hyperfine structure, were used unchanged in the further analysis of the EPR line shape at intermediate pH (when both forms, p1TAM4– and p1TAM3–, were present). In this case line shape fitting yields the line positions and Lorentz linewidths of the both forms. The nonlinear least-squares fittings were performed by Levenberg– Marquardt algorithm using home designed MATLAB code.
pKa Value Calculation
The values of fraction, f, of each ionization state of p1TAM obtained by simulation of experimental spectra were fitted by standard titration equations yielding corresponding values of pKa. Titration of p1TAM is described by the equation:
| (1) |
Animals
Twelve week-old female polyoma middle-T antigen (PyMT+/−) mice with spontaneously formed mammary tumors were used for in vivo evaluation of p1TAM-D probe. For comparison of tissue microenvironments of normal mammary gland and tumor, age-matched littermate females absent of the PyMT oncogene (PyMT−/−) were used. All animal work was performed in accordance with the OSU IACUC approved protocol.
In Vivo L-Band EPR Studies
Mice were narcotized by inhalation of air-isoflurane or oxygen-isoflurane mixture using Ohmeda Fluotec 3 anesthetic machine and then placed into the gap of L-band (1.2 GHz) EPR spectrometer (Magnettech, Germany). The surface coil resonator was placed on to normal mammary gland or mammary tumor and spectrometer was tuned. Solutions of the p1TAM-D probe (0.5–2 mM, 10–30 μL) in saline, pH 7.2 was injected intratissually. Immediately after injection the EPR spectra were acquired for 5–10 min.
RESULTS AND DISCUSSION
Synthesis of Asymmetric Phosphonated Trityl Radicals
The synthesis of asymmetric phosphonated trityls presented challenges due to the harsh deprotonation conditions required in this particular case that are incompatible with either acid labile (tert-butyl) or base labile ester protecting groups. We opted to solve this problem by reacting in one step, upon deprotonation of all three aryl protons of the starting materials 1, with a 2:1 mixture of diethylcarbonate and diethyl chlorophosphate. A mixture of the desired products 2 was produced. Upon further transformations similar to the ones used in our prior syntheses of triphosphonated28 and Finland30 TAMs, the desired monophosphonated, p1TAM (p1TAM-H or p1TAM-D, Scheme 1), and diphosphonated, p2TAM, radicals, were easily separated and identified.
EPR Characterization: Spectral Sensitivity to pH
Figure 1 shows X- and L-band EPR spectra of the deuterated monophosphonated radical, p1TAM-D, at various pH values. The doublet hfs arises from the phosphorus nucleus (SN = 1/2) of single phosphono group of p1TAM-D. First protonation of phosphono group resulted in an increase of the phosphorus hfs constant, ΔaP ≈ 260 mG. The same protonation resulted in the frequency-dependent shift of the EPR spectrum as total toward low magnetic field (37 mG for X- and 4 mG for L-band spectra) which corresponds to an increase of g-factor, Δg ≈ 2 × 10−5. The small pH effect on the g-factor-related spectral shift at low EPR frequencies makes in vivo application of the pH probes with single EPR line difficult. In this respect, the monophosphonated probe provides the simplest frequency-independent doublet hfs pattern, being ideal for in vivo applications. Therefore, we further describe in details the behavior of the p1TAM radical only with the focus on its characterization and in vivo measurements using low-field L-band EPR spectroscopy (see SI for the spectral properties of p2TAM which shows more complex spectral behavior due to the presence of two phosphono groups, and therefore it is less suitable for in vivo applications).
Figure 1.

X-band (A) and L-band (B) EPR spectra of 0.1 mM (A) or 0.2 mM (B) solutions of p1TAM-D probe in 1 mM Na-phosphate buffer, 150 mM NaCl, measured at various pH values under nitrogen atmosphere and temperature of 37 °C.
As seen in Figure 1 at a neutral pH around 7, the EPR spectra of p1TAM-D show superposition of two ionization states, p1TAM4– and p1TAM3–, with fully deprotonated and partially deprotonated phosphonic acid groups, correspondingly (see Scheme SI1 for the structures of all ionization states), in agreement with slow proton exchange between these states on EPR time scale.31 Note that the L-band EPR spectrum is almost symmetrical, while the X-band spectrum shows visible asymmetry due to significant g-factor-induced spectral shift at a higher EPR frequency.
Figure 2 shows the pH dependence of the fraction of the ionization states of p1TAM radicals, calculated from the corresponding EPR spectra. As expected, the pH dependencies are described by standard titration curves with no significant differences between X- and L-band EPR spectra titrations, or between titrations of deuterated and nondeuterated p1TAM derivatives (see also SI). Substitution of the methyl protons with deuterons significantly decreased the individual EPR line width, ΔHpp, from about 90 mG for p1TAM-H to 40 mG for p1TAM-D. The line narrowing results in higher spectral intensity and allows avoiding spectra overlap, therefore simplifying spectra analysis in respect to pH and oxygen sensitivity.
Figure 2.
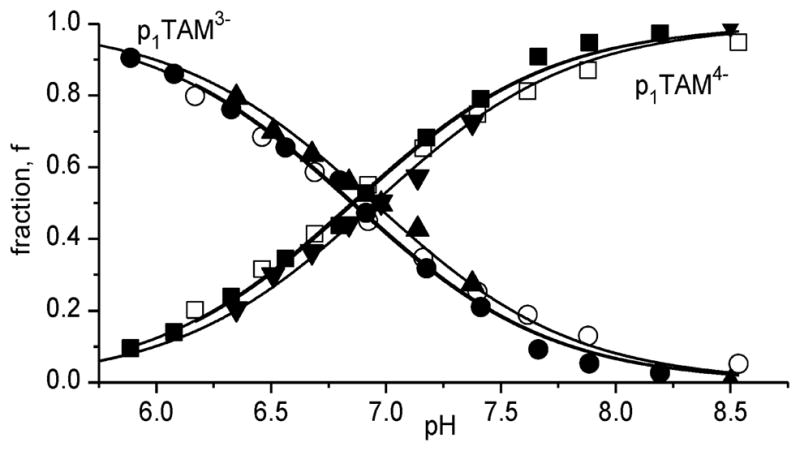
The pH dependencies of fraction of the ionization states of p1TAM probes calculated from the corresponding X-band and L-band EPR spectra. The closed and open symbols denote the data obtained for deuterated and nondeuterated compounds, correspondingly; L-band data are shown by triangle symbols. Solid lines represent the best fits using standard titration equation yielding dissociation constant, pKa2 = 6.90 ± 0.05, for the second dissociation of phosphonic acid group of the probes.
For the second protonation of phosphono group of p1TAM we did not observe superposition of the EPR spectra of the states with a fully protonated, p1TAM0, and a partially protonated, p1TAM1–, phosphono group but only a smooth increase of aP splitting from 3.61 G (pH ≈ 2) to 3.75 G (pH ≈ 0) according to standard titration curve with a pKa1 ≈ 1.3 for the first dissociation of phosphonic acid group. This is in agreement with the fast proton exchange between the ionization states on EPR time scale due to high proton concentration in strong acidic solutions. A similar effect has been previously described in detail for triphosphonated TAMs.28 The protonation of carboxyl groups of p1TAM (pKa ≈ 2.625) precedes the second protonation of the phosphono group but only slightly affects phosphorus hfs (ΔaP ≈ 10–20 mG) because of the long distance between carboxyl group and phosphorus atom of phosphono group. Table 1 summarizes the pKa values of the p1TAM-D probe and spectral parameters of its different ionization states.
Table 1.
The pKa Values of the p1TAM-D Probe and EPR Spectral Parameters of Its Different Ionization States
| state | pKa | aP (mG) | ΔHpp (mG) |
|---|---|---|---|
| p1TAM4- | 6.9 ± 0.05 | 3370 ± 5 | 33 ± 2b; 48 ± 2c |
| p1TAM3- | ≈2.6 ± 0.3a | 3630 ± 5 | 37 ± 2b; 52 ± 2c |
| p1TAM1- | ≈1.3 ± 0.2 | ≈3610 | ≈40 |
| p1TAM0 | ≈3750 | ≈40 |
The pKa value of carboxyl groups.
Measured by X-band EPR.
Measured by L-band EPR.
p1TAM-D Spectral Sensitivity to Oxygen
Figure 3 demonstrates the effect of oxygen-induced line broadening of the L-band EPR spectra of p1TAM-D measured at pH close to pKa2. The narrow individual linewidths allow for the accurate line shape analysis by fitting the experimental spectra with rational approximation of Voigt function.29
Figure 3.

High-field component of the L-band EPR spectra of 0.2 mM p1TAM-D solution in 0.5 mM Na-phosphate buffer, 150 mM NaCl, measured at pH 6.80 under various oxygen partial pressures, temperature of 37 °C. Symbols (○) represent the calculated spectra.
Figure 4 shows dependencies of the Lorentz linewidths on oxygen concentration obtained by line shape analysis of the EPR spectra of p1TAM-D which can be used as calibration curves in further oximetric measurements. The independent character of the two effects, pH on relative signal intensities of p1TAM3– and p1TAM4– states and [O2] on line broadening, provides dual functionality to the p1TAM-D probe allowing for extracting both parameters from a single EPR spectrum.
Figure 4.

Oxygen-induced EPR line broadening of the Lorentz line width of p1TAM3– (●) and p1TAM4– (○) states of p1TAM-D. Solid lines are the linear fits with the slope of 0.48 ± 0.02 mG/mmHg (≈ 400 mG/mM).
Other Factors Influencing EPR Spectra Line Shape
The fast proton exchange between ionization states of the p1TAM-D probe at pH close to pKa1 ≈ 1.3 resulting in coalescence of the corresponding EPR lines is a consequence of high proton concentration. Similar effect can be observed at pH close to pKa2 ≈ 7 upon increasing concentration of the proton-donating buffer molecules with the buffer pKa value in the same pH range. Among the biologically relevant molecules, inorganic phosphate meets these criteria. The phosphate-induced fast proton exchange between p1TAM3– and p1TAM4– ionization states results in coalescence of the corresponding L-band EPR spectra lines of p1TAM-D probe at phosphate concentration about 10 mM (see Figure 5).
Figure 5.

High-field component of the L-band EPR spectra of 200 μM p1TAM-D solution in Na-phosphate buffer, 150 mM NaCl, pH 6.8, measured at different buffer concentrations indicated near the spectra under anoxic conditions and temperature 37 °C. Symbols (○) represent the calculated spectra.
For the most biologically relevant applications, localization of the membrane-impermeable p1TAM-D probe is extracellular where concentration of phosphate buffer is about 1 mM; therefore slow or intermediate frequency exchange between EPR spectral lines of p1TAM-D is expected. Intermediate frequency exchange contributes in the line broadening as shown in Figure 6a masking oxygen-induced line broadening. Fortunately, buffer-induced line broadening, unlike oxygen-induced line broadening, also affects the distance between spectral lines participating in frequency exchange (see Figure 6B).32 Therefore, the buffer-induced line broadening of p1TAM3– and p1TAM4– components can be calculated from the distance between these lines. This allows for easy discrimination between oxygen- and buffer-induced line broadenings.
Figure 6.

(A) Dependencies of the Lorentz linewidths of p1TAM3– (●) and p1TAM4– (○) components of p1TAM-D probe on phosphate buffer concentration. Solid lines represent the best linear fits with the slopes of (5.4 ± 0.8) mG/mM and (8.3 ± 0.5) mG/mM for p1TAM3– and p1TAM4– forms, correspondingly. (B) Dependence of distance between p1TAM3– and p1TAM4– components of p1TAM-D probe on phosphate buffer concentration. The solid line is the best linear fit with the slope of (6.2 ± 0.3) mG/mM.
Concentration-induced line self-broadening is an additional factor which may contribute in the EPR line shape. For the p1TAM-D probe, this line broadening was less or about 10 mG/mM (see Figure SI5), being insignificant at concentrations below 0.3 mM which makes its useful for functional mapping, in vivo.
Aggregation of carboxyl group-containing Finland TAM at low pH or in lipid environments when tendency of carboxyl groups toward protonation is increased has been reported.30 In similar conditions, we did not observe aggregation of p1TAM-D (see Figure SI6) apparently due to low pKa1 for the phosphonic acid dissociation (pKa1 ≈ 1.3), therefore maintaining a negatively charged structure at any reasonable pH. The lower tendency to aggregation and high aqueous solubility of p1TAM-D (lipophilicity coefficient being less than 0.001 at pH = 7, see SI) make it particular attractive for in vivo applications.
Concurrent in Vivo Tissue pO2 and pH Assessment
Figure 7 shows representative in vivo L-band EPR spectra obtained in PyMT mice bearing breast tumors. The spectra clearly demonstrate independent character of the probe sensitivity to pH (cf. Figure 7a and b) and oxygen (cf. Figure 7b and c). For the first time, the use of dual function p1TAM-D probe allows for the concurrent noninvasive in vivo tissue pH and pO2 monitoring, therefore providing complementary information about both parameters independently on probe distribution and time of probe delivery.
Figure 7.
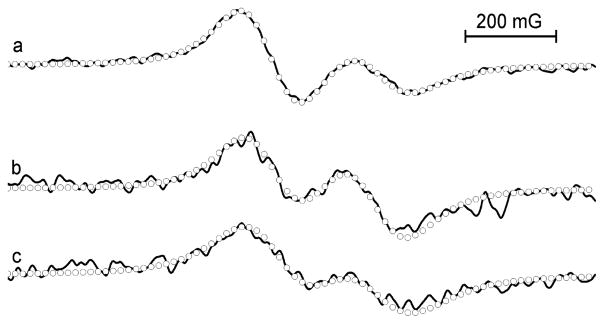
L-band EPR spectra of high-field component of p1TAM-D probe injected into the mammary gland (a) and tumor of the mouse breathing room air (b) or oxygen (c). Circles indicate simulated spectra yielding the pO2 values 54 mmHg (a), 37 mmHg (b), and 66 mmHg (c), and pH values of 7.14 (a), 6.87 (b), and 6.84 (c). See Figure SI7 for the exemplified extraction of pH and pO2 values by fitting experimental EPR spectra.
Figure 8 summarizes the in vivo data obtained using p1TAM-D probe in PyMT tumor-bearing mice. In agreement with the previous reports,2,7 severe acidification of the tumor tissue was observed. This observation confirms expected extracellular probe localization. The EPR-measured average mammary gland tissue pO2 of (52 ± 4) mmHg is a typical value for the breast tissue.33 Lower tumor tissue pO2 of (42 ± 2) mmHg supports the presence of hypoxic areas which is characteristic of tumor pathogenesis.33,34 The sensitivity of the p1TAM-D probe to tissue oxygenation is further supported by the significant increase in tumor tissue pO2 for oxygen-breathing animals.
Figure 8.
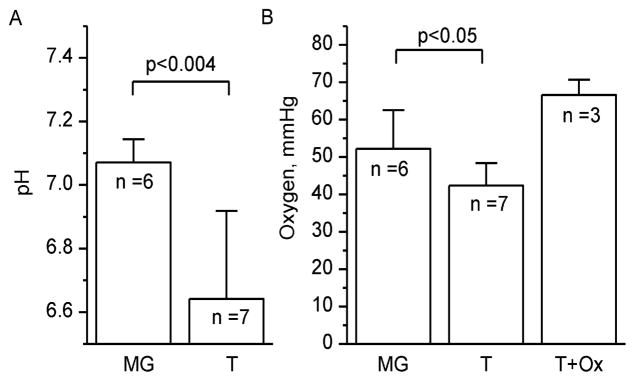
pH (A) and oxygen (B) values measured in mammary gland (MG) and tumor (T) tissues of the PyMT mice. Tumor tissue oxygenation was measured for air- and oxygen-breathing (T+Ox) animals. Average tumor size (0.9 ± 0.4) cm3. Error bars represent SD values.
CONCLUSION
Low-field EPR-based techniques allow for in vivo functional tissue imaging using specific paramagnetic probes. TAM derivatives are emerging superior paramagnetic probes for EPR-based spectroscopic and imaging techniques including CW,14 pulsed EPRI,15,16 and PEDRI.9,18,19 The synthesized monophosphonated probe, p1TAM-D, shows the simplest doublet EPR spectral pattern and enhanced aqueous solubility required for dual function pH and pO2 in vivo measurements. The in vivo L-band EPR studies performed in the mouse model of breast cancer demonstrate the utility of the probe for the dual function assessment from single probe delivery and single spectrum, therefore decreasing method invasiveness and allowing for better correlation of the parameters independently on probe distributions. The demonstrated spectroscopic application has a potential for being extended to imaging modalities taking into account simple EPR spectrum and extraordinary narrow line width of the probe.
Tissue pH and pO2 are among the most important parameters in physiology and pathophysiology of living organisms playing crucial role in pathogenesis of cancer and ischemic heart disease. Therefore, the dual function pH and pO2 TAM probes may significantly advance the area of application of EPR-based magnetic resonance approaches to the biomedicine field.
Supplementary Material
Acknowledgments
This work was partly supported by the grants from NIH (EB014541-01A1 and R00CA131552), RFFI (13-04-1258A) and the Ministry of Education and Science of the Russian Federation (8456).
Footnotes
Author Contributions
I.D. and A.A.B. contributed equally.
Notes
The authors declare no competing financial interest.
Details about synthesis, solubility, and spectral characterization of the phosphonated trityls. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Vaupel P, Thews O, Hoeckel M. In: rhErythropoietin in Cancer Supportive Treatment. Smyth JF, Boogaerts MA, Ehmer BR, editors. Marcel Dekker; New York, Basel: 1996. p. 205. [Google Scholar]
- 2.Gillies RJ, Raghunand N, Garcia-Martin ML, Gatenby RA. IEEE Eng Med Biol Mag. 2004;23:57. doi: 10.1109/memb.2004.1360409. [DOI] [PubMed] [Google Scholar]
- 3.Kumar S, Kasseckert S, Kostin S, Abdallah Y, Schafer C, Kaminski A, Reusch HP, Piper HM, Steinhoff G, Ladilov Y. Cardiovasc Res. 2007;73:172. doi: 10.1016/j.cardiores.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Swartz HM. Antioxid Redox Signal. 2004;6:677. doi: 10.1089/152308604773934440. [DOI] [PubMed] [Google Scholar]
- 5.Mader K, Gallez B, Liu KJ, Swartz HM. Biomaterials. 1996;17:457. doi: 10.1016/0142-9612(96)89664-5. [DOI] [PubMed] [Google Scholar]
- 6.Gallez B, Mader K, Swartz HM. Magn Reson Med. 1996;36:694. doi: 10.1002/mrm.1910360507. [DOI] [PubMed] [Google Scholar]
- 7.Bobko AA, Eubank TD, Voorhees JL, Efimova OV, Kirilyuk IA, Petryakov S, Trofimiov DG, Marsh CB, Zweier JL, Grigor’ev IA, Samouilov A, Khramtsov VV. Magn Reson Med. 2012;67:1827. doi: 10.1002/mrm.23196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komarov DA, Dhimitruka I, Kirilyuk IA, Trofimiov DG, Grigor’ev IA, Zweier JL, Khramtsov VV. Magn Reson Med. 2012;68:649. doi: 10.1002/mrm.23251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ardenkjaer-Larsen JH, Laursen I, Leunbach I, Ehnholm G, Wistrand LG, Petersson JS, Golman K. J Magn Reson. 1998;133:1. doi: 10.1006/jmre.1998.1438. [DOI] [PubMed] [Google Scholar]
- 10.Gomberg M. J Am Chem Soc. 1900;22:757. [Google Scholar]
- 11.Anderson S, Golman K, Rise F, Wikström H, Wistrand L-G. US Patent No. 5,530,140. 1996
- 12.Reddy TJ, Iwama T, Halpern HJ, Rawal VH. J Org Chem. 2002;67:4635. doi: 10.1021/jo011068f. [DOI] [PubMed] [Google Scholar]
- 13.Owenius R, Eaton GR, Eaton SS. J Magn Reson. 2005;172:168. doi: 10.1016/j.jmr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Elas M, Williams BB, Parasca A, Mailer C, Pelizzari CA, Lewis MA, River JN, Karczmar GS, Barth ED, Halpern HJ. Magn Reson Med. 2003;49:682. doi: 10.1002/mrm.10408. [DOI] [PubMed] [Google Scholar]
- 15.Murugesan R, Cook JA, Devasahayam N, Afeworki M, Subramanian S, Tschudin R, Larsen JA, Mitchell JB, Russo A, Krishna MC. Magn Reson Med. 1997;38:409. doi: 10.1002/mrm.1910380309. [DOI] [PubMed] [Google Scholar]
- 16.Yamada K, Murugesan R, Devasahayam N, Cook JA, Mitchell JB, Subramanian S, Krishna MC. J Magn Reson. 2002;154:287. doi: 10.1006/jmre.2001.2487. [DOI] [PubMed] [Google Scholar]
- 17.Mailer C, Subramanian SV, Pelizzari CA, Halpern HJ. Curr Top Biophys. 2005;29:89. [Google Scholar]
- 18.Golman K, Petersson JS, Ardenkjaer-Larsen JH, Leunbach I, Wistrand LG, Ehnholm G, Liu KJ. J Magn Reson Imaging. 2000;12:929. doi: 10.1002/1522-2586(200012)12:6<929::aid-jmri17>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 19.Krishna MC, English S, Yamada K, Yoo J, Murugesan R, Devasahayam N, Cook JA, Golman K, Ardenkjaer-Larsen JH, Subramanian S, Mitchell JB. Proc Natl Acad Sci USA. 2002;99:2216. doi: 10.1073/pnas.042671399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ardenkjaer-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, Servin R, Thaning M, Golman K. Proc Natl Acad Sci USA. 2003;100:10158. doi: 10.1073/pnas.1733835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallagher FA, Kettunen MI, Day SE, Hu DE, Ardenkjaer-Larsen JH, Zandt R, Jensen PR, Karlsson M, Golman K, Lerche MH, Brindle KM. Nature. 2008;453:940. doi: 10.1038/nature07017. [DOI] [PubMed] [Google Scholar]
- 22.Liu KJ, Gast P, Moussavi M, Norby SW, Vahidi N, Walczak T, Wu M, Swartz HM. Proc Natl Acad Sci USA. 1993;90:5438. doi: 10.1073/pnas.90.12.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Presley T, Kuppusamy P, Zweier JL, Ilangovan G. Biophys J. 2006;91:4623. doi: 10.1529/biophysj.106.090175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarkson RB, Odintsov BM, Ceroke PJ, Ardenkjaer-Larsen JH, Fruianu M, Belford RL. Phys Med Biol. 1998;43:1907. doi: 10.1088/0031-9155/43/7/012. [DOI] [PubMed] [Google Scholar]
- 25.Bobko AA, Dhimitruka I, Zweier JL, Khramtsov VV. J Am Chem Soc. 2007;129:7240. doi: 10.1021/ja071515u. [DOI] [PubMed] [Google Scholar]
- 26.Dhimitruka I, Bobko AA, Hadad CM, Zweier JL, Khramtsov VV. J Am Chem Soc. 2008;130:10780. doi: 10.1021/ja803083z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Driesschaert B, Marchand V, Levêque P, Gallez B, Marchand-Brynaert J. Chem Commun. 2012;48:4049. doi: 10.1039/c2cc00025c. [DOI] [PubMed] [Google Scholar]
- 28.Bobko AA, Dhimitruka I, Komarov DA, Khramtsov VV. Anal Chem. 2012;84:6054. doi: 10.1021/ac3008994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hui AK, Armstrong BH, Wray AA. J Quant Spectrosc Radiat Transfer. 1978;19:509. [Google Scholar]
- 30.Dhimitruka I, Velayutham M, Bobko AA, Khramtsov VV, Villamena FA, Hadad CM, Zweier JL. Bioorg Med Chem Lett. 2007;17:6801. doi: 10.1016/j.bmcl.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khramtsov VV, Weiner LM, Eremenko SI, Belchenko OI, Schastnev PV, Grigor’ev IA, Reznikov VA. J Magn Reson. 1985;61:397. [Google Scholar]
- 32.Robinson BH, Mailer C, Reese AW. J Magn Reson. 1999;138:210. doi: 10.1006/jmre.1999.1738. [DOI] [PubMed] [Google Scholar]
- 33.Tatum JL, Kelloff GJ, Gillies RJ, Arbeit JM, Brown JM, Chao KS, Chapman JD, Eckelman WC, Fyles AW, Giaccia AJ, Hill RP, Koch CJ, Krishna MC, Krohn KA, Lewis JS, Mason RP, Melillo G, Padhani AR, Powis G, Rajendran JG, Reba R, Robinson SP, Semenza GL, Swartz HM, Vaupel P, Yang D, Croft B, Hoffman J, Liu G, Stone H, Sullivan D. Int J Radiat Biol. 2006;82:699. doi: 10.1080/09553000601002324. [DOI] [PubMed] [Google Scholar]
- 34.Epel B, Sundramoorthy SV, Barth ED, Mailer C, Halpern HJ. Med Phys. 2011;38:2045. doi: 10.1118/1.3555297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.