

Abstract
We survey the population genetic basis of social evolution, using a logically consistent set of arguments to cover a wide range of biological scenarios. We start by reconsidering Hamilton's (Hamilton 1964 J. Theoret. Biol. 7, 1–16 (doi:10.1016/0022-5193(64)90038-4)) results for selection on a social trait under the assumptions of additive gene action, weak selection and constant environment and demography. This yields a prediction for the direction of allele frequency change in terms of phenotypic costs and benefits and genealogical concepts of relatedness, which holds for any frequency of the trait in the population, and provides the foundation for further developments and extensions. We then allow for any type of gene interaction within and between individuals, strong selection and fluctuating environments and demography, which may depend on the evolving trait itself. We reach three conclusions pertaining to selection on social behaviours under broad conditions. (i) Selection can be understood by focusing on a one-generation change in mean allele frequency, a computation which underpins the utility of reproductive value weights; (ii) in large populations under the assumptions of additive gene action and weak selection, this change is of constant sign for any allele frequency and is predicted by a phenotypic selection gradient; (iii) under the assumptions of trait substitution sequences, such phenotypic selection gradients suffice to characterize long-term multi-dimensional stochastic evolution, with almost no knowledge about the genetic details underlying the coevolving traits. Having such simple results about the effect of selection regardless of population structure and type of social interactions can help to delineate the common features of distinct biological processes. Finally, we clarify some persistent divergences within social evolution theory, with respect to exactness, synergies, maximization, dynamic sufficiency and the role of genetic arguments.
Keywords: inclusive fitness, game theory, environmental stochasticity, demographic stochasticity, multi-locus models, maximization
1. Introduction
[P]onderous mathematical cortices skimmed my pages like flying saucers and back at their base did not always pronounce favourably on what they saw. Inclusive fitness wasn't ‘well defined’, it was said … [1, p. 95]
Inclusive fitness theory was first described by Hamilton [2] and has delivered insights about the evolution of the biological world, which range from interactions between genes and cells within individuals to the spatial structuring and fighting among groups. Consequently, inclusive fitness theory has become the foundation for social evolution. As illustrated by the above quote, however, Hamilton's results have been controversial, as they became a target in the debate about sociobiology, where issues at stake have not been simply a willingness to understand the biological world [3]. The relationship between social evolution and population genetic theory has consequently been obscured.
Our goal in this paper is to present a mature account of the population genetic basis of social evolution theory. We survey a formulation of this theory that, despite its compactness, has shed light on many of the earlier misunderstandings, and has allowed investigations of many extensions of Hamilton's original analysis. Our aim is not to provide the most general proofs, but to provide an exposition consistent with exacting derivations, rather than simply a rationalization of Hamilton's rule and its extensions. We will progress through a series of examples, and point to potential pitfalls in generalizing from them.
The paper is organized as follows. (i) We present a derivation of Hamilton's [2] first insight in its most attractive form: a description of the result of natural selection on the evolution of a trait that affects its carrier as well as other individuals in the population (i.e. a social behaviour with possible interactions among phenotypes) and that deals as little as possible with unknown details of the genetic basis of the trait considered. (ii) We relax demographic and genetic assumptions. We consider extensions of Hamilton's [2] results to populations with localized dispersal and discuss how complex environmental and population dynamic processes can be handled by the concept of reproductive value. Then, we consider versions of Hamilton's rule including non-additive gene interactions, such as dominance, epistasis and synergies between genotypic effects of different individuals. (iii) We consider the joint evolution of several traits on the longer time scale of the change of trait values in the presence of a recurrent flow of mutations. In this ‘long-term’ perspective [4], coevolving traits can impinge on the ecological and demographic properties of a population so as to result in eco-evolutionary feedback or niche construction. (iv) Finally, we discuss the implications of the results surveyed for long-discussed topics: exact versus approximate results, dynamic sufficiency, maximization of (inclusive) fitness and how behaviours conditional on others' behaviours fit within the general framework.
(a). Fitness and allele frequency change
(i). Stochastic allele frequency change
As a foundation for our later developments, we focus in this section on a description of allele frequency change under natural selection without mutation in a finite population with constant environment. Let p(t) be the frequency of a mutant allele in the population at time t, viewed as the realized value of a random variable P(t), whose change between a parental generation at time t and an offspring generation at time t + 1 is 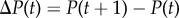 . For simplicity, this change will be denoted
. For simplicity, this change will be denoted 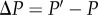 , as throughout this paper a variable without any time index (e.g. P) is by default considered at some parental generation, and we use a prime (′) to denote that variable in the offspring generation (e.g. P′).
, as throughout this paper a variable without any time index (e.g. P) is by default considered at some parental generation, and we use a prime (′) to denote that variable in the offspring generation (e.g. P′).
Our starting point to describe ΔP is gene counting. The allele frequency in the descendent generation is
| 1.1 |
where Ai is a random variable giving the frequency of gene copies in the offspring generation that descend from parent i and 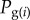 is the frequency of the mutant allele among these gene copies. This expression also applies to diploids if we consider each of the two homologous gene copies of a diploid parent as an individual. Then, i runs over all 2NT gene copies of a diploid population of NT organisms in the parental generation. In a fully assumed gene-centred manner [5], we can envision such gene copies as individuals, to which the following always apply.
is the frequency of the mutant allele among these gene copies. This expression also applies to diploids if we consider each of the two homologous gene copies of a diploid parent as an individual. Then, i runs over all 2NT gene copies of a diploid population of NT organisms in the parental generation. In a fully assumed gene-centred manner [5], we can envision such gene copies as individuals, to which the following always apply.
To express Ai in biological language, we introduce fitness through Hamilton's [2] own words: ‘the number of adult offspring’ of an individual. The general point to be made here out of the word ‘adult’ is that fitness must count the total number of descendants of an individual after one full iteration of the life cycle of the organism (thus including itself through survival, and offspring after density-dependent competition). This can be illustrated by an example. Consider a haploid semelparous population made of many groups of identical and constant size N over generations, where each parent i produces a large but random number Fi of juveniles (Wright's [6] island model). Each juvenile disperses independently with probability m to compete in another randomly chosen group, and density-dependent regulation is assumed to affect each individual independently and equally. Then, a number (1 − m)Fi of the focal's offspring remain in the natal group and compete for settlement in N breeding spots with an average number 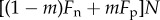 of juveniles, which depends on the average fecundity Fn in the focal group and the average fecundity Fp in the total population. Given these numbers, the expected number of adult offspring of an adult focal individual i is
of juveniles, which depends on the average fecundity Fn in the focal group and the average fecundity Fp in the total population. Given these numbers, the expected number of adult offspring of an adult focal individual i is
| 1.2 |
Thus, when successful offspring are counted after a full iteration of the life cycle (‘adult’ offspring), the fitness of an individual generally depends on the vital rates of others.
Equation (1.2) only provides the expectation of the number Wi of adult offspring of individual i, which itself is a random variable that cannot in general be expressed in terms of average quantities, but unambiguously determines Ai. That is, we have 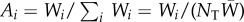 , where
, where 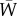 is average fitness. This precise definition of fitness is not consistently followed in the literature. As such, the precise meaning of the variables considered in this paper and the operations made (e.g. various conditional expectations that appear below) may differ among different authors. For the flow of our argument, we will not comment extensively on such similarities and differences, yet we cannot emphasize enough that a consistent adherence to this definition of fitness simplifies all further arguments made to evaluate systematically allele frequency change.
is average fitness. This precise definition of fitness is not consistently followed in the literature. As such, the precise meaning of the variables considered in this paper and the operations made (e.g. various conditional expectations that appear below) may differ among different authors. For the flow of our argument, we will not comment extensively on such similarities and differences, yet we cannot emphasize enough that a consistent adherence to this definition of fitness simplifies all further arguments made to evaluate systematically allele frequency change.
With fitness defined as Wi, we then have
| 1.3 |
[7, eqn (3), first line], where the key element to be retained here from Price's formalism is the use of individual attributes, for example Wi, rather than the older formalism of genotypic attributes. If total population size is constant, then 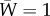 (i.e. one offspring on average for each parent) and
(i.e. one offspring on average for each parent) and 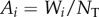 . If total population size is not constant, then
. If total population size is not constant, then 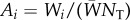 , and
, and 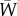 must be retained in equation (1.3). We first assume constant population size and discuss variable population size later. But a case can already be made that expressions in terms of Wi/NT should more generally be understood as expressions in terms of Ai so that the following expressions retain generality.
must be retained in equation (1.3). We first assume constant population size and discuss variable population size later. But a case can already be made that expressions in terms of Wi/NT should more generally be understood as expressions in terms of Ai so that the following expressions retain generality.
(ii). Expected frequency change
Through fitness values, individuals of the parental generation transmit their gene copies to the offspring generation. If reproduction is haploid, then Pg(i) is the allele frequency Pi in individual i. Under diploidy (where the realization of Pi is pi = 0, 1/2 or 1), gene copies in parents are not necessarily transmitted to offspring owing to the randomness of Mendelian segregation. A change Pg(i) − Pi in frequency may then occur for some or all i between the parental and offspring generation. We can avoid this complication by taking each of the two gene copies of a diploid organism as an abstract individual (i.e. gene-centred approach). Alternatively but less generally, since we will focus below on expectations, we can assume that transmission is ‘fair’, i.e. on average the descendant frequency is equivalent to parental frequency [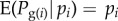 1] a standard assumption in social evolution theory [8,9].
1] a standard assumption in social evolution theory [8,9].
For constant total population size, the conditional expectation of equation (1.3) for any vector p of all realized pi values can be written as
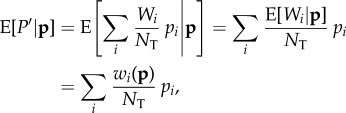 |
1.4 |
where 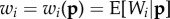 is the expected fitness of individual i conditional on the genotypes of all individuals in the parental population. This yields
is the expected fitness of individual i conditional on the genotypes of all individuals in the parental population. This yields
| 1.5 |
where the covariance is over all individuals in the population (i.e. 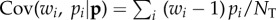 ) and is a form of Price's [7] covariance equation.
) and is a form of Price's [7] covariance equation.
In the example of equation (1.2), a possible expression for fitness wi is
| 1.6 |
in terms of expected fecundities 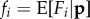 ,
, 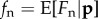 and
and 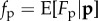 given p. Expressions for expected fitness, for example equation (1.6), dominate the literature [10–15], but potential pitfalls should be noted. The expectation of a ratio of random variables (as shown in equation (1.2)) is in general not a ratio of expectations, so one cannot a priori write the expected Wi in this form. The traditional way to overcome this complication in population genetics and evolutionary biology is an (often implicit) asymptotic argument assuming arbitrarily large fecundities with small variance-to-mean ratios for each genotype (for exceptions see e.g. [16–18]). Otherwise, expected fitness wi may depend on all the moments of the distribution of fecundities of locally interacting individuals as well as those from the whole population [18, eqn (A.6)]. Equation (1.6) can also be reached by assuming finite, Poisson-distributed fecundities [19,20], although this is generally not sufficient to obtain expected number of offspring in age-structured populations, where surviving adults come in competition with youngsters [21,22]. Regardless of the exact underlying assumptions behind wi, they do not affect the conclusions of the next section.
given p. Expressions for expected fitness, for example equation (1.6), dominate the literature [10–15], but potential pitfalls should be noted. The expectation of a ratio of random variables (as shown in equation (1.2)) is in general not a ratio of expectations, so one cannot a priori write the expected Wi in this form. The traditional way to overcome this complication in population genetics and evolutionary biology is an (often implicit) asymptotic argument assuming arbitrarily large fecundities with small variance-to-mean ratios for each genotype (for exceptions see e.g. [16–18]). Otherwise, expected fitness wi may depend on all the moments of the distribution of fecundities of locally interacting individuals as well as those from the whole population [18, eqn (A.6)]. Equation (1.6) can also be reached by assuming finite, Poisson-distributed fecundities [19,20], although this is generally not sufficient to obtain expected number of offspring in age-structured populations, where surviving adults come in competition with youngsters [21,22]. Regardless of the exact underlying assumptions behind wi, they do not affect the conclusions of the next section.
(b). Hamilton's rule
Our aim now is to recover Hamilton's weak-selection result, in the form
| 1.7 |
from the previous expressions, obtaining en route definitions for the fitness cost −c and benefit b and relatedness r. Here, ∼ denotes a first-order approximation with respect to the phenotypic effect of a mutant and p is taken as a deterministic variable.
(i). Phenotypic costs and benefits
We first assume a simple relationship between phenotypes and genotypes: two alleles segregate in the population and the phenotype of individual i is written as zi = z + δpi, where z is the phenotype of individuals carrying the resident allele and δ the phenotypic deviation induced by the expression of the mutant. Fitness wi = wi(z(p)) thus depends on the full phenotypic distribution z in the population, itself a function of the genetic state p. But for a family- or group-structured population, we can simplify the arguments of the fitness function and express wi in terms of the phenotype 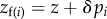 of individual i and of two average phenotypes, the average phenotype
of individual i and of two average phenotypes, the average phenotype 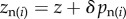 of ‘neighbours’, i.e. family or group mates (excluding the focal individual) and the average phenotype
of ‘neighbours’, i.e. family or group mates (excluding the focal individual) and the average phenotype 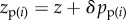 in the population (excluding the focal family or group). Then, assuming that all individuals face the same set of problems (individuals are exchangeable), we can write
in the population (excluding the focal family or group). Then, assuming that all individuals face the same set of problems (individuals are exchangeable), we can write
| 1.8 |
for some function w that is the same for each focal individual and further depends only on the average phenotype of actors on a focal recipient. Fitness can be expressed in this way only to the first order in selection intensity δ (as emphasized by our use of ∼ here), because only in that case can the average of the effects of different individuals of the same class be generally expressed in terms of an average phenotypic value of that class [23, appendix 1; 24, ch. 6].
We can now expand the total derivative of fitness (equation (1.8)) in terms of the partial derivatives of the fitness function w = w(zf, zn, zp). Namely,
| 1.9 |
where the partial derivative ∂w/∂zj represents the effect of the whole set of individuals of category 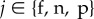 on the fitness of a focal and is evaluated at zj = z for all j. If all parents express the same phenotype, they must all have the same (expected) fitness, w(z) = 1. From this it follows that the sum of partial derivatives of w with respect to elements of z is zero. This means that the last derivative can be expressed in terms of the two others, out of which the traditional fitness costs and benefits −c and b can be defined
on the fitness of a focal and is evaluated at zj = z for all j. If all parents express the same phenotype, they must all have the same (expected) fitness, w(z) = 1. From this it follows that the sum of partial derivatives of w with respect to elements of z is zero. This means that the last derivative can be expressed in terms of the two others, out of which the traditional fitness costs and benefits −c and b can be defined
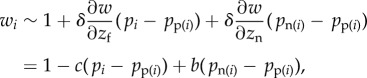 |
1.10 |
where −c = δ∂w/∂zf and b = δ ∂w/∂zn are thus marginal costs and benefits of expressing the mutant allele, respectively, and this expression for fitness holds at all allele frequencies, not only on ‘rare’ mutants.
(ii). Replicates of the evolutionary process
The next step to recover Hamilton's result (equation (1.7)) is to consider expectations of allele frequency change given realized average frequency p in the parental generation. That is, we consider replicates of the evolutionary process starting from an initial p(0) and will evaluate expected allele frequency change from generation t to t + 1 among all replicates that reach a given frequency p(t). From equation (1.5), this expected change can be written as
| 1.11 |
where the expectation is among all realizations with given p. Suppose that over such replicates, variation in wi is to be predicted only from variation in the allele frequency pi in individual i. To each individual, we assign the value 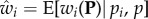 , which is the expected fitness of individuals that bear the same allele as individual i. Then,
, which is the expected fitness of individuals that bear the same allele as individual i. Then,
| 1.12 |
where 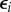 is independent of pi. This basic construct of least-square prediction theory [25,26, ch. 9] shows that
is independent of pi. This basic construct of least-square prediction theory [25,26, ch. 9] shows that 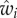 is sufficient to describe selection as
is sufficient to describe selection as
| 1.13 |
(iii). Relatedness as regression
From equation (1.9), 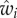 will be a function of the expected value of Pn(i) and Pp(i) among bearers of each allele. We first assume that Pp(i) is independent of Pi, in the sense that the expected value of Pp(i) among bearers of each allele is p and we write the expected value of Pn(i) among bearers of each allele as
will be a function of the expected value of Pn(i) and Pp(i) among bearers of each allele. We first assume that Pp(i) is independent of Pi, in the sense that the expected value of Pp(i) among bearers of each allele is p and we write the expected value of Pn(i) among bearers of each allele as
| 1.14 |
where r is by construction a regression coefficient2. Given that the average Pi value is p, this merely describes two points (either pi = 0 or 1) by a line. This construction is always feasible and implies
| 1.15 |
Then equation (1.13) yields 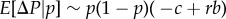 and together with the above assumption that Pp(i) is independent of Pi, equation (1.14) can be seen as the definition of relatedness r that makes Hamilton's rule work (the regression definition [8,27]).
and together with the above assumption that Pp(i) is independent of Pi, equation (1.14) can be seen as the definition of relatedness r that makes Hamilton's rule work (the regression definition [8,27]).
This definition of relatedness can be extended to diploid populations, where one can consider the regression of the focal's mutant allele frequency to transmitted gene copy (see appendix A(a)). A regression coefficient can then be associated to −c, which is simply 1/2 in the absence of inbreeding (the probability that one of the focal's gene copies is the transmitted copy) in a panmictic population, and relatedness can be expressed as a ratio of covariances, that of neighbours' phenotypes to transmitted value and that of focal's phenotype to transmitted value [28].
(iv). Genealogical relatedness
The definition of r as a regression coefficient (equation (1.14)) says little about its biological interpretation: for example, it says nothing about its relationship to pedigrees, and r can a priori be expected to differ for different values of p, and even for different populations with the same p. To obtain more definite results, we need to be more explicit about the underlying biological assumptions, which will allow us to relate the regression definition of r to a genealogical concept of relatedness independent of p.
Consider, for instance, the classical island model of dispersal described above (equation (1.2)), where we define genealogical relatedness to be the probability that two gene lineages have a common ancestor in the same group and we will see that this corresponds precisely to the regression definition under specific assumptions. To that aim, we ignore changes in allele frequency owing to selection or random genetic drift in the total population, and focus on the ancestral lineages of the focal gene's copy and a neighbour's gene copy. In each generation there is a probability (1 − m)2 that none of the two lineages is immigrant, in which case the two lineages can coalesce in a common ancestor. If a coalescence event is the first event back (probability r), it almost certainly occurs over a few recent generations, in which case the common ancestor carries the mutant allele with probability p. If the first event back is the one where at least one lineage is of immigrant origin (probability 1 − r), then the two ancestral gene lineages become independent lineages of the total population, and the probability that the neighbour's allele is the mutant one is p, irrespective of the focal's allele, which is also mutant with probability p (figure 1). This gives the expected allele frequency in the neighbours in the desired form (second term in equation (1.14)) and allele frequency changes according to equation (1.7), with r independent of p, but not necessarily independent of z. The very same argument, where one considers the events back in the ancestry of two gene lineages, underlies the use of genealogical relatedness in the classical family-structured population models [29].
Figure 1.
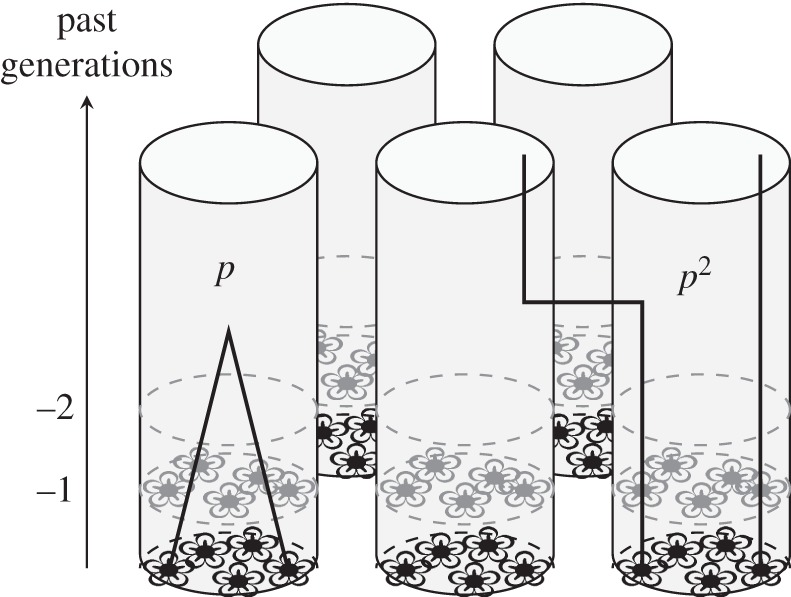
Genealogical events in the ancestry of different pairs of genes and their associated probabilities of identity in the island model of dispersal. This figure shows the position over time of some gene lineages among different groups, each of which is shown as a group of five flowers and the ghosts of some of their ancestors. In the group on the left, the ancestral lineages of two sampled genes coalesce in a recent common ancestor in that group, in which case they are both of the mutant type with probability p. By contrast, the right group illustrates the case where the two sampled lineages have recent ancestors in different groups, in which case they are considered as independent and both are of the mutant type with probability p2.
The defining property of r as a regression coefficient (equation (1.14)) can actually be interpreted in two ways. First, by considering only expectations given p over replicates of the evolutionary process (as done in the previous section), or second, by assuming that the average value of Pn(i)Pi for almost any such replicate is practically equal to the expectation over replicates. As replicates that deviate from the expectation can always be conceived (for example, the configuration where all mutants are in distinct groups cannot be excluded a priori), this then implies that the probability of such replicates is negligible, which is typically obtained by assuming an infinite number of groups. Then, 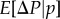 reduces to a deterministic change Δp and this finally yields Hamilton's [2] expression for allele frequency change in its deterministic form (equation (1.7)):
reduces to a deterministic change Δp and this finally yields Hamilton's [2] expression for allele frequency change in its deterministic form (equation (1.7)): 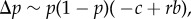 which was really his result.
which was really his result.
(v). Assumptions behind genealogical relatedness
The largely verbal argument subtending the use of genealogical relatedness highlights assumptions that may need to be reconsidered and steps that are to be taken in a formal proof of equation (1.15), for r independent of p. First, we assumed that the allele frequency in the generation of the common ancestor, within a group, is the current allele frequency. Thus, we ignored random drift at the total population level, meaning that the population is large, and we ignored the effect of selection, both on ancestral allele frequency and on probability of coalescence, meaning that we only obtain an expression for allele frequency change to the first order in selection. More precisely, in the context of family groups in a panmictic population, it is assumed that p can be considered constant since the last round of random dispersal. In the island model, it is assumed that p can be considered constant over a few ancestral generations, over a time scale depending on m, meaning that m has to be large relative to the strength of selection. A formal proof of these two cases and others involving selfing rests on the concept of separation of time scales in population genetics [29–31], where certain coalescence events occur at a much faster rate than that of others, which holds in the limit of infinite population size. Then, relatedness can be described conveniently (if somewhat heuristically) as a probability of identity-by-descent or of recent coalescence.
(vi). Frequency independence at the genetic level
One of the most remarkable features of Hamilton's [2] result is that selection appears independent of allele frequency, meaning that (to the first order in selection intensity) allele frequency change is
| 1.16 |
where the phenotypic selection gradient s(z) = ∂w/∂zf + r∂w/∂zn is constant with respect to p. This happens despite fitness being frequency dependent (equation (1.15)) and thus does not imply that social interactions are frequency independent. For instance, consider a social insect population, where parental queens control the phenotype of their offspring, which may be either reproductives or sterile workers that help to raise the reproductive offspring. The latter all disperse randomly over a large number of colony sites. If the survival of juvenile reproductives increases linearly from 1 − ς to 1 as the fraction z of workers in a colony increases from 0 to 1, a first-order approximation for the fitness of a parental gene copy residing in a focal queen (equation (1.8)) can be written as
| 1.17 |
which is the ratio of the focal individual's fecundity to the average in the population. Here, 1 − zf is the fraction of reproductives among juveniles of the focal individual. This yields the selection gradient
| 1.18 |
where both the cost and benefit are seen to depend on the resident investment into workers in the population. Nevertheless, if for some value of self-sacrifice z, s(z) > 0, the allele coding for an increase in self-sacrifice will invade and go to fixation in the population. Hence, game theoretic scenarios are subject to analysis by Hamilton's rule, according to which selection is frequency independent at the genetic level.
(c). Localized dispersal
The simple relationship between genealogical relatedness (probability of common ancestry of pairs of genes), gene identity between actor and recipient, and allele frequency in the total population (equation (1.14)) is essential for the first derivation of Hamilton's rule. But it rests on the assumption that dispersal is homogeneous over the landscape. We now relax this assumption and allow for isolation-by-distance. For example, consider the case where groups of size N are set on a circular array of positions and where juveniles disperse at most to the nearest group on each side, where they compete for settlement as in the classical nearest-neighbour stepping stone model [32,33]. In this case, juveniles from a focal parent compete with juveniles born at most two steps apart. If we further suppose that this parent interacts socially only with its group mates so that her average fecundity depends on her own phenotype and the average phenotype of within-group neighbours, the fitness of a focal individual depends on the average phenotypes of neighbours at most two steps apart, and can be written as 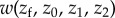 where zk for k = 0, 1 or 2 denotes the average phenotype of parents k steps apart (see appendix A(a) for an explicit example).
where zk for k = 0, 1 or 2 denotes the average phenotype of parents k steps apart (see appendix A(a) for an explicit example).
It is not clear a priori whether the property of frequency-independent selection at the genetic level is retained in this case. However, the previous results turn out to have informative generalizations. In particular, the key result Δp = p(1 − p)δs(z), viewed as p(1 − p) times a constant, can be extended to a form where p(1 − p) is replaced by another non-negative function σ2(p) of the distribution of genetic variation in the total population [34]. Thus, there is still a phenotypic selection gradient that predicts the change of allele frequency at all frequencies under isolation-by-distance. In the above case, where the fitness function is w(zf, z0, z1, z2) and depends only on four arguments, the change of mutant frequency can be written as
| 1.19 |
for some relatedness coefficient Rk describing the similarity of k-neighbours to the focal, relative to the similarity of group neighbours and which can be expressed in terms of probabilities of identity-by-descent (see appendix A(b), equation (B 4) for a derivation). The effect of group neighbours has been cancelled from this equation, which results from the fact that relatedness is no longer measured relative to population average allele frequency (i.e. relatedness is no longer of the form 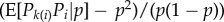 for allele frequency Pk(i) in neighbours at distance k). Although relatedness can still be defined in the latter way (see appendix A(b)), it is then frequency dependent, and thus no longer bears a simple relationship with coalescence probabilities and this conceals the existence of a frequency-independent component of selection. Further attempts at writing a selection gradient as −c + rb potentially involves joint redefinitions of c, r and b, which changes the interpretation of the components of Hamilton's rule [35].
for allele frequency Pk(i) in neighbours at distance k). Although relatedness can still be defined in the latter way (see appendix A(b)), it is then frequency dependent, and thus no longer bears a simple relationship with coalescence probabilities and this conceals the existence of a frequency-independent component of selection. Further attempts at writing a selection gradient as −c + rb potentially involves joint redefinitions of c, r and b, which changes the interpretation of the components of Hamilton's rule [35].
(d). Reproductive value: from sex ratios to environmental and demographic fluctuations
As a result of fluctuations in resource abundances or other biotic and abiotic factors, different individuals may be exposed to different conditions and this will result in the fluctuation of the fitness of several or of all individuals in the population. While within-generation environmental fluctuations are captured by w, since it is an average over all chance effects given a distribution of allele copies in a parental generation (see equation (1.6)), between-generation environmental fluctuation can be analysed in essentially the same manner; in particular, by averaging differentials of expected fitness over environmental states, which again provides a selection gradient independent of allele frequency (see appendix A(c) for an example). More generally, different individuals may also be exposed to different local conditions and this may affect their expected reproductive success compared to that of others. Different individuals may then have different value in transmitting alleles to the next generation, given a distribution of allele copies in a parental generation. A standard way of taking this heterogeneity into account is through the concept of reproductive value [36]. This concept is relevant for scenarios with spatial environmental fluctuations, which extend those of within- and between-generations fluctuations, but classically arises in sex-ratio models where sons and daughters must be given distinct values.
In the following, we consider reproductive values as a vector of weights, which define a weighted average allele frequency. We track the changes of this average through time, whatever the original allele frequencies. The use of reproductive value is often justified in an intuitive manner or through mathematical arguments loosely connected to this computation. In particular, reproductive values appear in an approximation for the growth rate of a rare mutant allele [37–40], but as the asymptotic growth rate of weighted allele frequency is the same whatever the weights, it may not be clear why using reproductive value is necessary in the latter calculation. Further, Fisher [41] is often cited as the origin of the concept, but his original formulation does not exactly match much later usage. In the following, we first recall an intuitive argument for using reproductive value, then reconstruct a more formal argument.
Under biparental inheritance, a son has low reproductive value (i.e. is of little value in transmitting his mother's genes) if the population sex ratio is male-biased, as males will never contribute more than half the genes in the next generation, whatever the sex ratio. In other words, the total reproductive value of all sons and daughters is one-half for each sex, but the individual reproductive value is determined by the sex ratio. In order to describe allele frequency change, one thus expects that the total offspring of one class (say, males) produced by an individual should be weighted by the reproductive values of this class (say, α♂), and thus the fraction of sons that comes from a particular mother (the probability of origin) should likewise be weighted by α♂.
More formally, given parental class c and offspring class 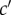 , one can consider probabilities of origin
, one can consider probabilities of origin 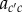 of
of 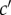 -offspring from c-parents. In an infinite population in a constant environment with unlimited uniform dispersal, the deterministic change in allele frequency vector p(t) gathering the average mutant allele frequency pc(t) in each class c is then given over one generation by
-offspring from c-parents. In an infinite population in a constant environment with unlimited uniform dispersal, the deterministic change in allele frequency vector p(t) gathering the average mutant allele frequency pc(t) in each class c is then given over one generation by
| 1.20 |
where Aδ(t) is the matrix of transition probabilities 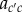 in generation t, depending on allele frequencies among parents in that generation and on mutant effect δ. An average allele frequency
in generation t, depending on allele frequencies among parents in that generation and on mutant effect δ. An average allele frequency 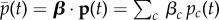 can then be defined through any vector of weights β (normalized such that its elements add up to one). Premultiplying equation (1.20) by this vector, one can write this average as a sum of changes over generations as
can then be defined through any vector of weights β (normalized such that its elements add up to one). Premultiplying equation (1.20) by this vector, one can write this average as a sum of changes over generations as
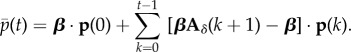 |
1.21 |
Now consider the value A0 of Aδ(k) for mutant effect δ = 0, identical for all generations, and define reproductive value as the normalized left eigenvector α associated with the largest (unit) eigenvalue of A0 (i.e. αA0 = α). In the absence of selection, average allele frequency weighted by β = α does not change over any generation, i.e. 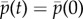 whatever the distribution of the allele among classes in the parental generation (as
whatever the distribution of the allele among classes in the parental generation (as 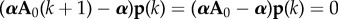 ). In mathematical language, this weighted allele frequency is a martingale, associated with the unit-eigenvalue left eigenvectors of the Markov chain defined by the A0 matrix [26,42]. It allows one to characterize the effect of selection on allele frequency change through a single average
). In mathematical language, this weighted allele frequency is a martingale, associated with the unit-eigenvalue left eigenvectors of the Markov chain defined by the A0 matrix [26,42]. It allows one to characterize the effect of selection on allele frequency change through a single average 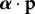 , and to regard the total average allele frequency change over many generations of selection as the sum of the changes of this average only owing to selection in each generation3. It is in this context, which is a standard one for social evolution theory, that a reproductive value weighting is required.
, and to regard the total average allele frequency change over many generations of selection as the sum of the changes of this average only owing to selection in each generation3. It is in this context, which is a standard one for social evolution theory, that a reproductive value weighting is required.
To complete the definition of an inclusive measure of allele frequency change in a social context, we need to express the probabilities of origin 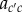 as the function of the genotypes of different actors and to characterize the distribution of allele frequencies in the parental population, as done previously. First, given parent i in class c with mutant allele frequency pc,i one can consider probabilities of origin
as the function of the genotypes of different actors and to characterize the distribution of allele frequencies in the parental population, as done previously. First, given parent i in class c with mutant allele frequency pc,i one can consider probabilities of origin 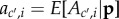 of
of 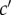 offspring, where
offspring, where 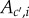 is the frequency of gene copies in class-
is the frequency of gene copies in class-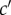 offspring that descend from parent i. This generalizes the Ais from equation (1.1). Then, the conditional expectation of the reproductive value weighted average allele frequency in the descendant generation is
offspring that descend from parent i. This generalizes the Ais from equation (1.1). Then, the conditional expectation of the reproductive value weighted average allele frequency in the descendant generation is
| 1.22 |
where Nc is the total number of individuals in that class.
By the same arguments previously applied to fitness functions, one can further write the probabilities of origin as functions of phenotypes of the different individuals in the parental population for some function 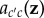 common to all class-c parents (i.e.
common to all class-c parents (i.e. 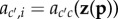 for all i in class c]. Then, applying the same set of arguments that lead to Hamilton's rule, allele frequency change can be written for stationary processes as
for all i in class c]. Then, applying the same set of arguments that lead to Hamilton's rule, allele frequency change can be written for stationary processes as
| 1.23 |
for a frequency-independent selection gradient s(z)4, where fitness effects of actors are weighted by relatedness and different descendant types are weighted by their reproductive values, and where the distribution of classes itself may be affected by the evolving trait [24]. The literature contains many incarnations of these results in a social evolution context, such as age structure [44], sex-ratio evolution [45], environmental and demographic stochasticity [46], host–parasite coevolution [47] or combinations of these factors [48]. The first-order approximation (equation (1.4)) fails if the demographic classes become effectively disjunct populations, this being appropriately quantified in terms of the subdominant eigenvalues of A0 and in particular by the largest of them: the approximation becomes inaccurate if this eigenvalue departs from 1 only by a term of order δ. This potential complication is not apparent in the introductory sex-ratio example, where the subdominant eigenvalue is zero.
(e). Frequency dependence at the genetic level
We now relax assumptions on the genotype–phenotype map of the previous sections and discuss two classical cases where selection will no longer be frequency independent at the genetic level.
(i). Synergies between alleles within individuals: dominance
The first case is genetic dominance. Here, the realized phenotype of individual i can be written as 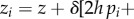
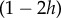 (pi♀pi♂)], where h is the level of dominance and pi♀ (pi♂) is the frequency of the mutant in the maternally (paternally) derived gene (pi = (pi♀+pi♂)/2). In a panmictic population without interactions between relatives, equation (1.10) then becomes
(pi♀pi♂)], where h is the level of dominance and pi♀ (pi♂) is the frequency of the mutant in the maternally (paternally) derived gene (pi = (pi♀+pi♂)/2). In a panmictic population without interactions between relatives, equation (1.10) then becomes
| 1.24 |
where c is defined as previously, only from the phenotype-fitness map and 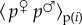 is the average value of the
is the average value of the 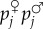 s in the population (excluding the individual i). If the population is of large size, allele frequency change is
s in the population (excluding the individual i). If the population is of large size, allele frequency change is
| 1.25 |
where the frequency-dependent term (1−2h)p represents the effect of the non-additive interaction between the two gene copies on the individual's fecundity [49, eqn 3.29].
Setting this in the more general framework of a situation of social interactions, one sees that the fitness of a focal gene copy depends on the fecundity of competing individuals, which depends on the non-additive effects of gene copies in their genotypes (for h ≠ 1/2). In other words, 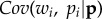 now depends on the covariance between actor's
now depends on the covariance between actor's 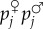 and focal's pi, and this leads to frequency dependence. Under fair Mendelian inheritance, the two gene copies of a focal individual are transmitted independently of their allelic information. Thus, one can consider the covariance between actor's genotype and each of the focal's gene copies, which depends on coalescence probabilities of triplets of genes. Writing Δp as p(1 − p)(−c + rb), with c and b still defined as phenotypic effects, implies that r is in general no longer a probability of recent coalescence of pairs of genes and is itself frequency dependent, a point that has long been understood [8,28].
and focal's pi, and this leads to frequency dependence. Under fair Mendelian inheritance, the two gene copies of a focal individual are transmitted independently of their allelic information. Thus, one can consider the covariance between actor's genotype and each of the focal's gene copies, which depends on coalescence probabilities of triplets of genes. Writing Δp as p(1 − p)(−c + rb), with c and b still defined as phenotypic effects, implies that r is in general no longer a probability of recent coalescence of pairs of genes and is itself frequency dependent, a point that has long been understood [8,28].
Alternatively, the allele frequency change can be expressed in terms of c, b, pairwise coalescence probability, and coalescence probabilities for triplets of genes, by a straightforward extension of the arguments presented in §1b(iv) [29]. Indeed, in a monoecious population the probability that both the maternally and paternally derived copies of an actor and the focal lineage (say 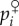 ) carry the mutant can be written as
) carry the mutant can be written as
| 1.26 |
where r3 is the probability that the ancestral lineages of three genes coalesce within their group, and r is the probability that the ancestral lineages of two genes coalesce within their group (e.g. [50], equation (1.5)) and is no longer the relatedness that makes Hamilton's rule work. Nevertheless, it is noteworthy that even with this complication, there are several situations under random dispersal where the direction of selection remains proportional to −c + rb for pairwise r independent of p [29,51], so that dominance does not affect the direction of selection on altruism at allele frequencies under weak selection.
(ii). Synergies between alleles among individuals
Another case where selection can be frequency dependent is in the classical two-person, two-strategies games, such as the hawk–dove or prisoner's dilemma games, here interpreted as scenarios where the fecundity of an individual depends on its pairwise interaction with a randomly chosen partner in the population. Let the relative fecundities be 1 + R, 1 + T, 1 + S and 1 + P, respectively, when both individuals cooperate, the focal cheats on its partner, the focal is cheated by its partner and both partners defect. A synergy occurs when D = R − S + P − T is non-zero, meaning that the pay-off difference of joint defection is not the sum of pay-offs differences of individual defections. Let the phenotype be the probability that an individual acts cooperatively in a pairwise interaction, so that the expected fecundity of focals with phenotype zf interacting with partners with phenotype zn is
| 1.27 |
In a large panmictic population, where the fitness of a focal individual is its fecundity f(zf, zn) relative to the average fecundity in the population, one has
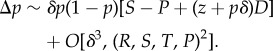 |
1.28 |
Two ways of analysing this model can then be considered. First, R, T, S, P are the given ecological constraints, and we consider the evolution of z. Then, to the first order in δ, equation (1.28) reduces to 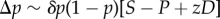 , where selection is independent of allele frequency. This is −cp(1 − p) by definition of the marginal fitness cost and can be extended to games with interaction between relatives. As this example shows, the marginal c takes into account the synergistic interaction (and so would b) so that no additional term is needed to account for them. This result is striking and very useful, as it allows more generally an analysis of modifiers δ of any continuous z affecting signalling or repeated games in terms of only pairwise relatedness [52–55], where the evolving phenotype itself can be a dynamic trait, i.e. a function of the round of the game, as occurs in sequential decision problems [56].
, where selection is independent of allele frequency. This is −cp(1 − p) by definition of the marginal fitness cost and can be extended to games with interaction between relatives. As this example shows, the marginal c takes into account the synergistic interaction (and so would b) so that no additional term is needed to account for them. This result is striking and very useful, as it allows more generally an analysis of modifiers δ of any continuous z affecting signalling or repeated games in terms of only pairwise relatedness [52–55], where the evolving phenotype itself can be a dynamic trait, i.e. a function of the round of the game, as occurs in sequential decision problems [56].
The second and alternative analysis considers an expansion in R, T, S, P, but not in δ. Then the term pδ in the parentheses in equation (1.28) is retained, contributing p2(1 − p)δ2D to the whole expression so that selection is frequency dependent at the genetic level when R − S + P − T is non-zero, i.e. when the acts of each partner non-additively affects the pay-offs. Now, the change of allele frequency Δp depends on associations among three gene positions so that games in pure strategies can be analysed under limited dispersal using coalescence probabilities of triplets of genes [15,57]. From a gene-centred perspective, this case is indeed no different from the case of dominance discussed above.
(f). Multi-locus processes
(i). A simple example with linkage disequilibrium
As illustrated in the last section, one can relax the basic genetic assumptions by changing the genotype–phenotype map, and we now discuss this for multi-locus processes. For example, one may consider that an individual expresses helping only if it harbours a two-locus combination of alleles, in which case the phenotype of individual i can be written 1 + δpiqi, where pi and qi are indicator variables for the realized allelic states at each of the two loci. Equation (1.10) then generalizes as
| 1.29 |
in terms of the average values 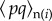 of the product of indicator variables among the within-group neighbours and in the rest of the population
of the product of indicator variables among the within-group neighbours and in the rest of the population 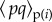 . The deterministic change in mutant allele frequency at the first locus is obtained by taking expectations over replicates of the evolutionary process given the realized average allele frequency p and q at the respective loci in the parental population yielding
. The deterministic change in mutant allele frequency at the first locus is obtained by taking expectations over replicates of the evolutionary process given the realized average allele frequency p and q at the respective loci in the parental population yielding
| 1.30 |
where
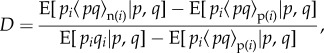 |
1.31 |
and 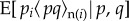 is the expectation of the product of the indicator variable for the transmitted allele pi of the focal individual and of the average value
is the expectation of the product of the indicator variable for the transmitted allele pi of the focal individual and of the average value 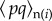 describing the neighbours' acts (as in equation (1.14)), and
describing the neighbours' acts (as in equation (1.14)), and 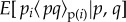 is identically defined but for actors not in the focal group.
is identically defined but for actors not in the focal group.
The similarity of equation (1.30) with Hamilton's rule is superficial, however, as D is a gametic disequilibrium coefficient [58], which is null at neutrality (δ = 0). Allele frequency change is thus at most of second order in δ, in which case a full account of second-order terms on allele frequency changes requires more than expansion (1.29) in terms of the first-order fitness effects c and b.
This example illustrates that multi-locus effects appear as weak forces, which would easily be overcome by any first-order one-locus effect acting on the evolution of the trait. Exceptions can occur, in particular with very strong linkage, because linkage disequilibrium actually depends on the magnitude of selection relative to recombination rather than simply of selection. Another exception occurs, for example, in the case of genetic kin recognition, where helping is conditional on identity between actor and recipient at some recognition locus. Then, the change in allele frequency at one locus can be shown to depend on a so-called identity disequilibrium coefficient, quantifying the dependence between the events that two individuals share alleles at each of different loci [59, fig. 1]. At a genealogical level, it quantifies the fact that the realized genealogical trees at two loci for two group members are not independent of each other. It is indeed non-zero at neutrality in the same conditions (limited dispersal or family-structured interactions) where the one-locus relatedness coefficients are non-zero at neutrality.
(ii). Systematic analysis of allele frequency change
Equation (1.29) is of the same form as equations (1.10) and (1.24), where fitness of a gene position is expanded in terms of selection coefficients weighted by allelic states at homologous or different loci. These expressions illustrate four types of fitness effects on a focal gene copy: those due to homologous genes in the same individual (equation (1.24)), homologous genes in different individuals (equation (1.10)), and different loci in the same and/or different individuals (equation (1.29)). More generally, any number of gene positions within the same or different individuals may affect a focal position, where individuals can further live in different generations and with possible interactions between gene copies. All these situations can be analysed by a logically straightforward extension of the approach delineated in the previous section (although the calculations may be complex), where the dependence of evolution on the genealogical structure is quantified by generalized identity disequilibrium coefficient (e.g. [60] for some general developments and references). Arbitrary levels of ploidy, genomic imprinting, sex linkage, trans-generational effects and cytoplasmic inheritance can all be considered in this unified way and, by systematic perturbation expansion with respect to selection strength, effects on fitness can be evaluated under arbitrary levels of accuracy. For populations without a relatedness structure, this general approach reduces to the quasi-linkage equilibrium (QLE) approach originally formulated by Kimura [61] and more systematically developed in later works [62–64], and the connection between multi-locus processes and social evolution theory extends beyond such approximation frameworks [65].
Although relatedness and linkage disequilibrium may quantify forces of different magnitude, their formal analysis can be based on two similar steps. First, in both cases the effects of selection over several generations are summarized by an expression for one-generation change given the state of the population in the parental generation. Second, the causal chain of events in earlier generations is summarized by its effects on the expected parental states. In the QLE approach, earlier events are summarized by an approximation for expected gametic disequilibrium in the parental population. In the basic social evolution theory approach, earlier events are summarized by relatedness coefficients that quantify the effects of common ancestry on covariances in genotypes among different individuals. In both approaches, concepts of separation of time scale are further used to approximate the relevant parameters: relatedness, or gametic disequilibrium, approach their equilibrium values at faster rates than the rate of changes in allele frequency in the population. These different steps together allow for a systematic analysis of the causes of allele frequency change, rather than simply a statistical description of this change.
(g). Small populations
We have assumed so far that the population was very large (ideally infinite) and emphasized that in this case selection can be understood by focusing on a one-generation change in allele frequency. However, in small populations selection will be frequency dependent in a way not captured by the previous results. Nevertheless, whether the total number of groups is small or not previous expressions for the phenotypic selection gradient s(z) can be reinterpreted so that they are proportional to the effect dπ (z, δ)/dδ of a small phenotypic change δ on the fixation probability 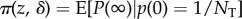 of a single mutant introduced in a resident population. This result captures the cumulative effects on allele frequency change of actor–recipient interactions over generations, until the loss or fixation of the mutant. In this generalization, r can be interpreted as a ratio of average coalescence times of the different pairs of genes compared, rather than as a constant regression coefficient, but the two interpretations coincide when the latter is applicable. Insofar as qualitative features of fixation probabilities determine evolutionary outcomes, all genetic and demographic scenarios considered so far can be analysed in these terms [24,66]. From a quantitative perspective, the approximation
of a single mutant introduced in a resident population. This result captures the cumulative effects on allele frequency change of actor–recipient interactions over generations, until the loss or fixation of the mutant. In this generalization, r can be interpreted as a ratio of average coalescence times of the different pairs of genes compared, rather than as a constant regression coefficient, but the two interpretations coincide when the latter is applicable. Insofar as qualitative features of fixation probabilities determine evolutionary outcomes, all genetic and demographic scenarios considered so far can be analysed in these terms [24,66]. From a quantitative perspective, the approximation 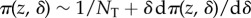 for the fixation probability of a single mutant is accurate only for δ of the order of 1/NT (and this holds in the multi-locus case as well), while more accurate approximations can be obtained for larger δ using diffusion approximations when results for allele frequency change, for example equation (1.7), are available [67].
for the fixation probability of a single mutant is accurate only for δ of the order of 1/NT (and this holds in the multi-locus case as well), while more accurate approximations can be obtained for larger δ using diffusion approximations when results for allele frequency change, for example equation (1.7), are available [67].
2. Evolutionary genetics
(a). Everyone's approximations
Until now, we have considered evolutionary dynamics without mutations and where Hamilton's rule predicts the invasion and substitution of mutant alleles in two-allele systems. Once an allele, say a mutant increasing the level of altruism (see, for example, equation (1.18)), has fixed in the population, a new allele may arise through mutation and may again be selected for. By the repeated invasion of mutant alleles, the trait z then evolves in a step-by-step transformation and may converge to a candidate evolutionary stable point; that is, a phenotypic state where evolution stops. Hence, for such a point to be approached gradually from its neighbourhood phenotypic variation needs to be produced. This entails that whenever a candidate evolutionary stable point is identified as a point where a phenotypic selection gradient vanishes (a singular point), explicit (or implicit) assumptions on the mutation machinery are made.
We now discuss the common assumptions behind models of social interactions in the presence of a constant influx of mutations, which generally deal with continuous phenotypes. Such phenotypic models may come under different labels: ‘adaptive dynamics’, ‘evolutionary game theory’, ‘direct fitness’, ‘kin selection model’ or ‘quantitative trait game theory’. But to a first approximation, everyone makes the same assumptions about the mutation machinery and the effects of mutations. This presumably stems from the fact that the various analyses of evolutionary dynamics face common mathematical difficulties, and so far only common approximations to circumvent these difficulties have been found.
At the risk of oversimplifying, these approximations usually come under two different packages, which delineate two distinct limiting approaches to evaluate long-term evolutionary dynamics. These are the trait substitution sequence (TSS) assumption [68–74] and the quantitative genetics approach to social interactions [75–77], which are now detailed.
(i). Trait substitution sequences
We start with TSSs, where the following set of assumptions has proved useful.
— Small mutation rate and symmetric mutation distribution. The mutation rate is assumed so small that a novel mutation occurs only in a population where polymorphism has previously been eliminated by selection. A single event of phenotypic change can then be analysed by focusing on a mutant–resident system. The distribution of mutant deviations δ is further assumed to have mean zero and individuals of every class have the same mutation rate and produce mutants regardless of their class.
— Selection is weak. Gene action is additive and the mutant deviates phenotypically by a small amount δ from the resident.
— The demographic and/ or environmental processes are stationary Markov processes. Demographic and environmental heterogeneities (as considered in §1d) are assumed to follow an ergodic Markov chain. When a mutant appears in a resident population, the resident demography is further assumed to have reached its stationary state, conditional on non-extinction of the total population.
Under TSSs, whether a mutant is favoured or not by selection is determined by the selection gradient s(z) alone. In general, this should be averaged over the different demographic backgrounds in which the mutant may be introduced (e.g. a population with variable density [20]) and from now on s(z) is thought of in that way. Because the mutation distribution is symmetric, only selection and thus s(z) can determine the direction of expected evolutionary change of the evolving phenotype given current phenotypic value z, which thus necessarily takes the form
| 2.1 |
for some measure v(z) ≥ 0 of genetic variance produced in the resident population. Fluctuations around this expectation will also necessarily and constantly occur through the continuous inflow of mutations, but these effects will average out. Hence, evolution stops (on average) only when
| 2.2 |
which characterizes candidate evolutionary stable (ES) points [4,73,78]. Whether such a point is a local attractor of the mean phenotypic change (equation (2.1)) depends on whether
| 2.3 |
This corresponds precisely to the notion of convergence stable states [4], which is in standard use to determine whether a singular point is a local attractor of the evolutionary dynamics [73,78].
(ii). Approximations to quantitative genetic models
The mean phenotypic change (equation (2.1)) is of the same form as the canonical equation of adaptive dynamics [74,79], which is usually derived under more restrictive assumptions as mutant–mutant interactions are neglected. Regardless of the level of generality of such equations, they are obtained by assuming that only two alleles can segregate in the population. But an equation of the same form also obtains for any number of alleles and any distribution of allelic effects as long as the phenotypic variance in the population is small [75,76,80]. To see this, it suffices to perform a Taylor expansion of the expected fitness wi(z) around the average phenotypic value z in the population. In particular, under additive gene action on phenotypes, and using the covariance equation under the form 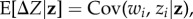 the expected change in average phenotype can then be expressed as
the expected change in average phenotype can then be expressed as
| 2.4 |
for the same c, b and r as in the two-allele model. This stems, in particular, from writing the analogue for phenotypes of the regression definition of relatedness (equation (1.14))
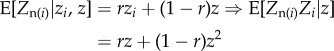 |
2.5 |
[28,81] and additionally showing that the same r applies to all alleles subtending the phenotypes. Equation (2.4) is the actual rationale given by Taylor & Frank [39] (appendix A(b)) for their direct fitness method.
(b). Joint evolution of several traits
(i). Infinite populations
In the one-dimensional case, convergence stable states can be characterized only in terms of fitness costs and benefits and genealogical relatedness. Thus, under well-specified assumptions, the selection gradient s(z) alone predicts long-term evolution. But the situation is likely to differ in at least two ways in the presence of multi-dimensional traits. Here, there may be interactions between traits that affect fitness and genetic variation may be correlated across traits. Can joint adaptive evolution still be characterized by the selection gradient for each trait?
Let us consider a vector z = (z1, … , zn) of n coevolving traits (the realized value of Z = (Z1, … , Zn)) and denote by s(z) the vector of selection gradients. In this case, the conditional average change in phenotype given the resident phenotypic value z satisfies
| 2.6 |
where V(z) is some symmetric variance–covariance mutation matrix. As the net response to selection on a trait may depend on the selection gradient of each other trait, it is a priori not clear whether the evolutionary dynamics starting in a neighbourhood of a singular point, where s(z) = (0, … , 0), can be shown to converge or not to that point, independently of the knowledge of V(z). This raises the question whether the point attractors of equation (2.6) can be predicted from s(z) alone, without further reference to the mutation matrix.
Leimar [82] provides a characterization of multi-dimensional convergence stability and a definite answer to this question. He defines a singular point to be strongly convergence stable if it is an asymptotically stable point of the canonical equation of adaptive dynamics [82, p. 197], which is of the form of the right member of equation (2.6). He further shows that for strong convergence stability of a singular point it is sufficient that the Jacobian matrix of the selection gradient s(z) is negative definite at that point. This owes to the fact that V(z) is a variance–covariance matrix that is necessarily positive definite for a parsimoniously defined model (such that one of the traits is not a linear combination of the others). Hence, for a negative-definite Jacobian matrix pleiotropy does not affect convergence; otherwise, pleiotropy can matter. Although Leimar [82] did not consider interactions between relatives, his result clearly holds in that case, as it rests on the form of the right member of equation (2.6).
(ii). Small populations
Our discussion on long-term evolution under the TSS assumptions also applies to finite populations. In this case, the average evolutionary change of the evolving phenotype is still given by an equation of the form 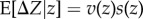 (equation (2.1)), but where s(z) is now interpreted as the average fixation probability perturbation
(equation (2.1)), but where s(z) is now interpreted as the average fixation probability perturbation 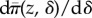 over the different demographic backgrounds in which the mutant may be introduced. Fluctuations around the average change will also occur owing to sampling effects in a small population and the continuous inflow of mutations5. To the leading order, this complicated stochastic adaptive dynamics can be described by a diffusion process (
over the different demographic backgrounds in which the mutant may be introduced. Fluctuations around the average change will also occur owing to sampling effects in a small population and the continuous inflow of mutations5. To the leading order, this complicated stochastic adaptive dynamics can be described by a diffusion process (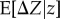 is the infinitesimal mean of the process), which will eventually reach a stationary state describing the phenotypic distribution ψ(z) in the population at a mutation–selection–drift balance. The phenotypic values that dominate this distribution are the most probable outcomes of evolution and, when only one trait evolves and the mutation distribution is independent of z, correspond precisely to the convergence stable states defined previously from the derivative of the selection gradient (i.e.
is the infinitesimal mean of the process), which will eventually reach a stationary state describing the phenotypic distribution ψ(z) in the population at a mutation–selection–drift balance. The phenotypic values that dominate this distribution are the most probable outcomes of evolution and, when only one trait evolves and the mutation distribution is independent of z, correspond precisely to the convergence stable states defined previously from the derivative of the selection gradient (i.e. 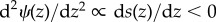 for reflecting boundary conditions [83]). Under multi-dimensional evolution in a finite population, one can also generalize equation (2.6) so that the ith element of s(z) represents the change in the fixation probability of a single mutant when phenotypic component i is varied. This yields a multi-dimensional diffusion equation whose stationary distribution is in general not known, but that can reach the maximum corresponding to the attractor points of the expected dynamics when the variance–covariance matrix does not depend on z [84]. However, the precise conditions under which this occurs are not clear, so further work is needed to establish whether this will be the case for state spaces of interest, for instance when all components of z vary between zero and one, which occurs in many allocation problems.
for reflecting boundary conditions [83]). Under multi-dimensional evolution in a finite population, one can also generalize equation (2.6) so that the ith element of s(z) represents the change in the fixation probability of a single mutant when phenotypic component i is varied. This yields a multi-dimensional diffusion equation whose stationary distribution is in general not known, but that can reach the maximum corresponding to the attractor points of the expected dynamics when the variance–covariance matrix does not depend on z [84]. However, the precise conditions under which this occurs are not clear, so further work is needed to establish whether this will be the case for state spaces of interest, for instance when all components of z vary between zero and one, which occurs in many allocation problems.
(c). The evolutionary stability condition
The selection gradient s(z) predicts whether or not a population will converge to a singular point z* from within a small neighbourhood of that point. But if z* is expressed by most individuals in a population, will it be resistant to the invasion of any alternative mutant phenotype, whenever individuals carrying the mutant phenotype are rare? This is the question of evolutionary stability, and convergence stability does not imply evolutionary stability [4]. For instance, it may be beneficial to consume the most abundant resource among various alternatives when few individuals consume it. But when all individuals in the population consume that resource, individuals consuming a less abundant resource may be favoured by selection owing to the reduction in competition. Hence, by successive allele replacement favouring the consumption of more abundant resources the population may first converge to the state where all the population consumes the most abundant resource. When it is close enough to that point, rare deviant individuals consuming less abundant resources will be favoured and a polymorphism of resource consumption will be maintained, in which case the convergence stable strategy is not evolutionarily stable.
When the population approaches a convergence stable state, the selection gradient vanishes. Second-order terms then become comparatively important. They determine, in particular, whether selection is disruptive on the trait, whereas the weak-selection version of Hamilton's rule is not sufficient to delineate these two cases [78]. The change of allele frequency to the first order of selection (equation (1.16)) can be extended to the second order in the phenotypic deviation δ to give
| 2.7 |
where d(z, p) is a frequency-dependent term that quantifies the intensity of disruptive selection at a phenotypic point z [24, eqn 12.1]. It allows one to check whether a singular point (satisfying s(z*) = 0) is really evolutionarily stable and this will be the case if d(z*, 0) < 0 so that no nearby mutant can invade when rare. ‘Rare’ does not entail that the frequency of mutants is negligible around a single mutant that appears in a group. In the same way as the first-order term depends on local fluctuations in allele frequency even though the mutant originally arises in a single copy in a group, the measure of disruptive selection intensity d(z*, 0) takes mutant interactions into account and involves both second-order partial derivatives and effects of selection on the distribution of mutant number within groups or families.
The full second-order evolutionary stability condition is hardly ever evaluated in models of social evolution under limited dispersal or when interactions occur among family members, except by numerical methods or in cases where there are no non-trivial relatedness coefficients [85,86]. An analytical second-order condition for a one-locus genetic basis in the haploid island model has been given [87] for N = 2 individuals per group [88] for arbitrary N and the methods described in §1f can be used under more general assumptions. If N > 2, the expression for d(z*, 0) involves relatedness coefficients for triplets of genes, already encountered in §1e and it involves first-order effects of selection on relatedness. The complexity of the latter computation, where fitness functions can in general no longer be written in terms of average phenotypes, makes it less attractive, and as a result it is still avoided in recent models [89]. Other features of the computation may also be overlooked, as they are absent from an earlier attempt at defining inclusive fitness criteria of evolutionary stability [85]. Indeed, the very fact that there is such a condition to be computed, distinct from the gradient version of Hamilton's rule, may have been ignored, as it is absent from influential accounts [11].
A further problem is that the biological conclusions to be drawn from the computation are themselves not so clear. For mutations of small effect around a singular point, it can in general be concluded that the change in the phenotypic variance 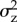 is given by
is given by 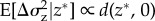 for a rare mutant (p → 0, see appendix A(d)). In particular, when d(z*, 0) > 0 the expected variance in the population will increase as a result of selection. If the mutation rate is high enough and inheritance is clonal, or haploid and uni-locus, two genealogical and phenotypic clusters are formed (‘branching’) and can diverge from each other on both sides of the singular point. It has indeed been shown that an increase in the phenotypic variance can be a very good predictor of the onset of branching and applies to finite populations [90]. However, the response to disruptive selection is sensitive to dominance, polygenic basis and interactions between loci [91] so that there is continuing debate about the biological expectations to be drawn from the models of disruptive selection. Moreover, under TSS assumptions (at most two alleles in the population) branching cannot occur, and therefore long-term evolution can be determined by the condition of convergence stability alone, as implied by results for infinite [82] and finite populations [92].
for a rare mutant (p → 0, see appendix A(d)). In particular, when d(z*, 0) > 0 the expected variance in the population will increase as a result of selection. If the mutation rate is high enough and inheritance is clonal, or haploid and uni-locus, two genealogical and phenotypic clusters are formed (‘branching’) and can diverge from each other on both sides of the singular point. It has indeed been shown that an increase in the phenotypic variance can be a very good predictor of the onset of branching and applies to finite populations [90]. However, the response to disruptive selection is sensitive to dominance, polygenic basis and interactions between loci [91] so that there is continuing debate about the biological expectations to be drawn from the models of disruptive selection. Moreover, under TSS assumptions (at most two alleles in the population) branching cannot occur, and therefore long-term evolution can be determined by the condition of convergence stability alone, as implied by results for infinite [82] and finite populations [92].
3. When the dust settles
(a). Main theoretical messages
There are three main take-home messages behind the approximations to evolutionary dynamics surveyed in this paper.
— The one-generation perspective. Selection on a social behaviour can be understood by focusing on a one-generation change in mean allele frequency. In this perspective, different classes of offspring are weighted by reproductive value, and all multi-generational effects (of selection or of common ancestry) are taken into account by evaluating uni- or multi-locus identity disequilibrium coefficients (generalized relatedness coefficients) quantifying genetic structure in the parental population. The latter is generally done using quasi-equilibrium approximations of different order, which identify in a systematic way forces of different magnitude and often allow the identification of forces common to different biological scenarios.
— Allele frequency change under weak selection. In general, selection is frequency dependent when gene interactions within and between individuals are taken into account. However, under weak selection, the direction of allele frequency change is of constant sign for any allele frequency and is predicted by a phenotypic selection gradient, even in a game theoretic context. This result generally follows from assuming additive gene action and small phenotypic deviation δ. Early studies had reached this conclusion for large panmictic populations, and it has subsequently been extended to spatially structured populations with many groups. This is useful as it provides a description of the direction of microevolution where genetic details are omitted, and that is expressed only in terms of phenotypic costs and benefits and genealogical concepts of relatedness.
— Long-term evolution under weak selection. Multi-dimensional long-term evolution can then be predicted by phenotypic selection gradients on each trait. This is useful as one can obtain a description of long-term evolution and characterize convergence stable states by omitting genetic details (under the more precise assumptions stated in §2b).
(b). Analytical scope of social evolution theory
(i). Common logic versus alternative methods
There has been considerable controversy about social evolution theory, but if the dust is allowed to settle, one can actually see that there is little alternative to this methodology in the literature. By this we mean, for example, that a multilevel selection approach is perfectly feasible, but if developed in a general way it would need the same concepts and analytical tools as described in this paper [93,94]. It is, of course, possible to repeat key arguments, for example, to compute correlations (or higher moments) of allele frequencies based on a separation of time scales in the genealogical structure of the population or to use coalescence probabilities (or times) and reproductive values, without endorsing the language of social evolution theory, but this does not define an alternative methodology.
There is also a common logic between the concepts of social evolution theory and multi-locus selection theory, which shows that the same analytical framework underlies what was previously thought of as different approaches (see §1f). Likewise, there is a common logic between the concepts of inclusive fitness theory and quantitative genetics theory and this is maybe best seen in the light of the exact version of social evolution theory [95]. This exact version can be obtained from the expression for expected fitness 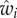 (equation (1.15)) if the partial derivatives are replaced by regression coefficients, which mean here coefficients of a least-square fit to a linear regression model, which can be computed whether the linear regression model is true or not. Then, −c and b are the linear regression coefficients with respect to the predictors pi and pn, and r is a regression coefficient of pn over pi. As the formalism of least-square regression makes no assumptions about the processes controlling the variables considered, writing fitness
(equation (1.15)) if the partial derivatives are replaced by regression coefficients, which mean here coefficients of a least-square fit to a linear regression model, which can be computed whether the linear regression model is true or not. Then, −c and b are the linear regression coefficients with respect to the predictors pi and pn, and r is a regression coefficient of pn over pi. As the formalism of least-square regression makes no assumptions about the processes controlling the variables considered, writing fitness 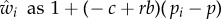 no longer makes any assumption about the strength of selection or genetic architecture. Thus, −c + rb is precisely the average effect of an allele substitution [23] from which the additive genetic variance is evaluated. In this interpretation, Δp = p(1 − p)(−c + rb) holds generally and exactly, not neglecting any effect of order higher than δ, but all components are likely to be frequency dependent [65, p. 219].
no longer makes any assumption about the strength of selection or genetic architecture. Thus, −c + rb is precisely the average effect of an allele substitution [23] from which the additive genetic variance is evaluated. In this interpretation, Δp = p(1 − p)(−c + rb) holds generally and exactly, not neglecting any effect of order higher than δ, but all components are likely to be frequency dependent [65, p. 219].
This general interpretation of Hamilton's rule has been repeatedly emphasized in the literature [65,95–97] and has been useful in conceptual debates, for instance, showing that intergroup selection requires relatedness between group members for altruism to be selected for [98]. However, in most cases of interest in behavioural ecology and even population genetics it is practically impossible to evaluate the different regression coefficients explicitly. As emphasized by Ewens [99, p. 164], the average effects can generally only be expressed implicitly as the unique solution of a gigantic set of simultaneous equations. Thus, it is customary to evaluate approximations that retain only the most important terms, in the presence of social interactions the main such approximation being clearly the weak-selection version of Hamilton's rule and its extensions. This is not to say that alternative approaches cannot be developed but this has not been done in a systematic way to cover classic topics in social evolution theory. For example, an alternative to the one-generation perspective is a multi-generation measure of evolutionary success, for example, the number of successful emigrants descended from an immigrant, summed over all generations since the immigration event [86,100], but simple questions of frequency dependence, dominance or alternative controls of phenotype [10] have not been addressed in this framework.
(ii). Maximization arguments
Hamilton [2] obtained a result for allele frequency change and interpreted it as a maximization result. This interpretation can be formalized as follows. The expected fitness of individual i (equation (1.15)) can be written as
| 3.1 |
where
| 3.2 |
is a value that can be associated to each gene copy and was called ‘inclusive fitness’ by Hamilton [2]. With this, the change in allele frequency owing to selection proceeds as if individuals were changing their behaviour to increase their inclusive fitness
| 3.3 |
The gradient 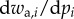 of wa,i points in the direction of the steepest increase in inclusive fitness, which is the path taken by allele frequency change if selection is weak and gene action is additive (as this entails constant −c + rb).
of wa,i points in the direction of the steepest increase in inclusive fitness, which is the path taken by allele frequency change if selection is weak and gene action is additive (as this entails constant −c + rb).
Equation (3.1) shows that the inclusive fitness differential  is equivalent to the fitness differential
is equivalent to the fitness differential 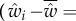
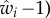 so that both quantities describe the change in allele frequency in exactly the same way. Hence, the mean inclusive fitness increases because the allele frequency changes as if the true fitness values of the alleles were these inclusive fitness values. However, the inclusive fitness values for each allele, as defined by equation (3.2), are not the average fitness (i.e. numbers of adult offspring) for each allele: fitness differs from inclusive fitness by a function of allele frequency and this difference also changes as a result of natural selection.
so that both quantities describe the change in allele frequency in exactly the same way. Hence, the mean inclusive fitness increases because the allele frequency changes as if the true fitness values of the alleles were these inclusive fitness values. However, the inclusive fitness values for each allele, as defined by equation (3.2), are not the average fitness (i.e. numbers of adult offspring) for each allele: fitness differs from inclusive fitness by a function of allele frequency and this difference also changes as a result of natural selection.
Even in the case of additive gene action and weak selection, the inclusive fitness maximization result thus says nothing about adaptation in the usual sense of maximization of fecundity or survival, it says only something about allele frequency changes. Even assuming that all fitness effects are the consequence of effects on fecundity, the average fecundity of the population can actually decrease as a result of selection, which occurs, for instance, in the case of selfish mutants in the prisoner's dilemma game (equation (1.27)). In this sense, Hamilton's results are not generalizations of the classical ‘mean fitness increase’ results (that is, fecundity or survival increase) of the non-social models he took inspiration from [101,102]. Rather, Hamilton's results can be understood as demonstrating a ‘partial increase’ in mean fitness, as in Price's [103] interpretation of Fisher's [41] so-called Fundamental Theorem of Natural Selection [97]. When gene action is no longer additive, inclusive fitness itself does not necessarily increase over generations as a result of selection, as −c + rb will be frequency dependent. Indeed, in the presence of dominance, Hamilton [2] failed to obtain a full proof of total increase in inclusive fitness, which points to a mismatch between his aims for inclusive fitness and his results.
There is thus no univocal relationship between the change in fitness (or inclusive fitness) and allele frequency change under natural selection. Claims to the contrary must be based on other notions of fitness or inclusive fitness than those defined here. In particular, Grafen [104–106] has developed an argument for inclusive fitness maximization based on a different concept of maximization than that implied by equation (3.3) and whose scope does not include all social behaviours discussed in this review. In particular, it applies only when effects on vital rates (fecundity, survival, number of matings) are additive separable; that is, when such effects are the sum of a function of the focal's phenotype and neighbours' phenotypes [107]. Hence, while Hamilton's rule is a general result about allele frequency change, the results on maximization are far more specific and do not have the same breadth.
(iii). Dynamic sufficiency
Analysing the dynamics of a biological scenario requires a closed system of recursions: if the expression for change in allele frequency depends on a frequency of identical pairs of genes, recursions for such a frequency are needed. It is well known that the exact (for any strength of selection) recursions for pairs of genes may depend on triplets of genes, the recursion for triplets may depend on quadruplets and so on, so that a closed system of recursions, said to be dynamically sufficient, may be large (at least of the order of the maximal group size in some cases based on the infinite island model, but much higher in general). The system of recursions needed to obtain the first-order change in allele frequency is much simpler, as, for example, recursions for change in allele frequency depend only on the frequency of identical pairs of genes under neutrality, for which recursions can be written which do not depend on frequency of triplets. A dynamically sufficient system of recursions is then obtained for the approximate solution.
Little biological conclusion can be drawn from an incomplete set of recursions. For example, without a set of equations for brother–sister relatedness there is nothing that prevents one to claim that this relatedness is −2, from which absurd conclusions follow. The Price covariance equation, viewed in isolation, is dynamically insufficient and has thus been criticized but, as emphasized, for example, by Gardner et al. [65], this is not a problem when the covariance equation is only taken as one recursion in a closed set of recursions. The value of such constructs is that they hold as members of many such systems of recursions considered in practice. Nevertheless, dynamically insufficient models have been defended from another perspective. In particular, Grafen [8] viewed Hamilton's original works as showing that an incomplete system of recursions could be more productive than a closed system, with the completely recursive methods following on behind. However, Hamilton's model was actually equivalent to a closed system of recursions for an approximation of the exact process, which was indeed productive because the approximation has an unambiguous and useful meaning, with more exact methods following on behind. This shows that approximations are useful but adds nothing in favour of dynamic insufficiency.
(iv). Interacting phenotypes
Finally, we emphasize that the weak-selection version of Hamilton's inclusive rule applies not only to fixed actions but can also be used to investigate evolution of behavioural rules; that is, the rules for responding to the environment or the actions of others. Behavioural ecologists consider that the actions (behaviours) of an organism can be predicted from knowledge about a set of external stimuli (environmental cues or behaviours expressed by other individuals) and internal states of the organism [108–110]. One can then model behaviour as a function that transforms states (internal and external inputs) to actions or behavioural responses.
As long as the actions expressed by a focal individual during its lifespan can be written as a function of its (continuous) phenotype(s) and that of other individuals (which is not always feasible), the trait(s) expressed by the focal and its neighbours may affect the states of the focal, the transitions between the states and/or the function that maps states into actions. In other words, indirect genetic effects, where genes expressed in one individual affect the phenotype of others [111], or repeated and dynamic games can be analysed in terms of only pairwise relatedness, a point we already mentioned in §1e(ii), but that is repeatedly forgotten and rediscovered. Further, fitness effects of actors may be felt by recipients alive several generations later. This occurs, for instance, under host–parasite coevolution, cultural inheritance or niche construction, which result in processes that can be analysed with the gradient version of social evolution theory, as long as they affect the phenotype-fitness map [11, p. 132; 112–114].
(c). The role of genetics
In order to understand how an organism's behaviour has become adapted to its environment, it may be desirable for an evolutionary biologist to focus on phenotypes, without considering any knowledge of the underlying genetical details. Early evolutionary theory, and thousands of years of artificial selection, was de facto based on such premises, which are therefore the reasonable first start for an evolutionary analysis. They are part of the research strategy known as the phenotypic gambit [115]. The thrust of the phenotypic gambit is that it allows one to build predictions of how behaviours have evolved based only on considerations of trade-offs between various components of fitness, such as survival and fecundity, without incorporating constraints at the genetic level. The gambit was conceived to identify constraints to which different strategies respond equally well and this has led to a rich interplay between data and predictions [116,117].
Phenotypic models have been described as based on the assumption of haploidy [115] but can be more generally said to assume fair transmission of average parental traits to their offspring, which implies additive gene effects on phenotypes and, in particular, semi-dominance in diploid populations. This also suggests that epistasis is absent from the genotype–phenotype map, although this does not prevent epistasis from operating on the genotype-fitness map. Further, this does not exclude genotype–environment interactions. For instance, nothing under the phenotypic gambit excludes the study of evolution of learning rules, considered as constraints on possible alternative strategies defined at the phenotypic level, which themselves are encoded by alternative alleles. Additivity assumptions may seem practically identical to a quantitative genetic formulation in terms of average effects, but in general average effects are not simply fixed properties of alleles, as they depend on the whole population configuration of allele frequencies, so that the quantitative genetic formalism per se does not provide predictions of changes over several generations as definite as those resulting from additivity assumptions.
The phenotypic gambit not only assumes that selection is largely robust to the genetic details, but that any genetic glitch will become negligible in the long term, an argument formalized in models of evolution as TSSs. In its simplest version, the modelling framework we have reviewed is appropriate for the formulation and analysis of such models. In its more general version, it also efficiently deals with complex genotype–phenotype maps. But how far are such complications useful?
There are certainly cases where they are useful. As emphasized in §2c, it has been abundantly documented that the response to diversifying selection depends on genetic details. Further, there are topics in evolution where the concept of the phenotypic gambit has no immediate meaning (such as evolution of reproductive systems in response to inbreeding depression, of recombination, of intragenomic conflicts or genetic kin recognition) and these processes can be viewed as inherently social or can have a social component.
Even leaving these processes aside, how much should one invest in a research strategy, for example the phenotypic gambit, as opposed to more explicit consideration of genetics? This has been much discussed [118] but there is no clear answer. For example, the fact that some presumably optimal mutant is not produced in the course of evolution (experimental or not) can raise questions about the constraints on possible phenotypes, but does not per se invalidate the assumption of additive gene effect on phenotype.
Turning from genetic to environmental constraints, we should finally emphasize that evolutionary arguments based on long time-scales assume that the ecological conditions are constant through time (the environment and demography may fluctuate but they are stationary processes, e.g. [26]). The constant diversification of life forms and repeated occurrence of ecological successions imply that the environment (biotic and abiotic) a gene pool is exposed to is likely to be transient. Long time-scale arguments overlook such unforeseeable changes. Of course, practically none of these considerations are specific to social evolution and all models are approximations. What this actually means is that the relevant way of applying models in disequilibrium conditions may be worth more attention. How far is it useful, for example, to consider joint evolution of different traits in stationary environments to ultimately understand behaviours in non-stationary ones?
All these questions will undoubtedly be dealt with in future research. The answers to these questions will delineate the range of applications of the social evolution theory we surveyed in this paper, which so far has provided the most illuminating and general conceptual machinery for understanding evolution of the sociobiological world.
Acknowledgements
We thank A. Gardner, A. Griffin and S. West for inviting this contribution; L. Étienne, A. Gardner, C. Mullon, D. Roze and two reviewers for helpful comments; and Wilson [119] and Dawkins [120] for suggesting an attractive title.
Appendix A
(a). Relatedness in diploid populations
Here, we make plain the definition of relatedness as a ratio of regression coefficients, in a diploid version of our basic model (see §1b) in a monoecious diploid population of constant size. For simplicity, we assume semi-dominance of gene effects within individuals and that a single fitness function gives both the expected number of successful female and males gametes of individual i. With this, fitness wi can still be written as equation (1.10); that is, 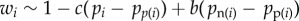 but allele frequencies within individuals take the values 0, 1/2, or 1. In particular,
but allele frequencies within individuals take the values 0, 1/2, or 1. In particular, 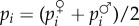 , where
, where 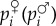 is the frequency of the mutant in the maternally (paternally) derived gene of i. We then have
is the frequency of the mutant in the maternally (paternally) derived gene of i. We then have
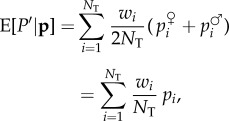 |
A1 |
where, in the first line, 2NT represents the total number of gene copies in the population, and individual i will transmit 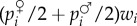 mutant alleles through both male and female gametes, which gives the total number
mutant alleles through both male and female gametes, which gives the total number 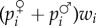 of transmitted mutant alleles.
of transmitted mutant alleles.
As in the haploid case, the linear regression of pn(i) on pi is
| A2 |
which allows us to write predicted fitness as in the main text (equation (1.15)): 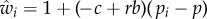 , and where
, and where
| A3 |
The main difference with relatedness r in the haploid case (equation (1.14)) is that r now does not reduce to a probability of recent coalescence (‘probability of identity’) under the separation of time-scale setting presented in §1b(iv), owing to the fact that  is no longer equal to p. Nevertheless, the genealogical interpretation can be extended in a straightforward way by writing
is no longer equal to p. Nevertheless, the genealogical interpretation can be extended in a straightforward way by writing
| A4 |
where both rn and rf are again regression coefficients by construction, whereby
| A5 |
Under the assumptions presented in §1b(iv), rn and rf become, respectively, the probability that two homologous genes randomly sampled in the focal individual coalesce in a recent past and the probability that two homologous genes, one randomly sampled in the focal individual and one in a neighbour, coalesce in a recent past. Then, equation (A 5) is a ratio of probabilities of identity-by-descent, as given in Hamilton [27].
Using equation (A 5) in equation (A 1) and applying the same argument as in §1b gives 
 , where in the absence of inbreeding rf = 1/2.
, where in the absence of inbreeding rf = 1/2.
(b). Allele frequency change under stepping-stone migration
As a concrete example of a fitness function w(zf, z0, z1, z2) for the stepping-stone model, we can assume that each individual can help its group neighbours, giving a relative fecundity benefit B shared among the N − 1 neighbours at a cost C for itself and that the life cycle follows the Moran model [121], where adults are replaced one at a time. Specifically, one of the NT adults taken randomly in the total population dies and is replaced by a juvenile from one parent chosen in proportion to the expected fecundity of the different parents. As the offspring of the focal can compete against a relative number 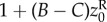 of offspring produced in the focal group (where
of offspring produced in the focal group (where 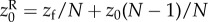 is the average phenotype in this group), a relative number 1 + (B − C)z1 of offspring produced in the group located one step apart from the focal group, and relative number 1 + (B − C)z2 of offspring produced in a group two steps apart, taking into account the various migration events, leads to
is the average phenotype in this group), a relative number 1 + (B − C)z1 of offspring produced in the group located one step apart from the focal group, and relative number 1 + (B − C)z2 of offspring produced in a group two steps apart, taking into account the various migration events, leads to
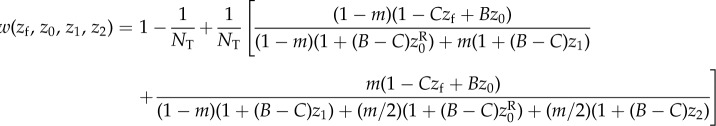 |
B1 |
which generalizes equation (1.6). Then, by the same argument used in Hamilton's rule, we have
| B2 |
where p0(i), p2(i) and p2(i) are, respectively, the average frequency of the mutant allele in neighbours living zero, one and two steps apart from the focal group. As in the derivation of Hamilton's rule, we now consider the expected value  of the focal individual's fitness over replicates of the evolutionary process, for given p. It may be felt that we can evaluate it by applying the regression definition of relatedness (equation (1.14)) for one-step and two-step neighbours, but such regression coefficients will no longer be independent of p and cannot be interpreted as coalescence probabilities. As a result, when relatedness is defined relative to population average allele frequency (i.e. it is of the form
of the focal individual's fitness over replicates of the evolutionary process, for given p. It may be felt that we can evaluate it by applying the regression definition of relatedness (equation (1.14)) for one-step and two-step neighbours, but such regression coefficients will no longer be independent of p and cannot be interpreted as coalescence probabilities. As a result, when relatedness is defined relative to population average allele frequency (i.e. it is of the form 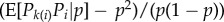 ), the frequency-independent selection gradient is no longer apparent. Hence, we need another argument to go beyond a simple description of allele frequency change.
), the frequency-independent selection gradient is no longer apparent. Hence, we need another argument to go beyond a simple description of allele frequency change.
Using the zero-sum property of partial derivatives and subtracting 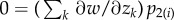 from equation (B 2), we can express the last derivative in terms of the other two, and taking the average of replicates over the evolutionary process and assuming a very large number of groups leads to
from equation (B 2), we can express the last derivative in terms of the other two, and taking the average of replicates over the evolutionary process and assuming a very large number of groups leads to
| B3 |
where 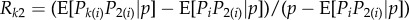 describes the similarity of k-neighbours to the focal, relative to the similarity of 2-neighbours to the focal. Further, Rk2 can be considered as independent of p: for any p, it quantifies a difference in the distributions of coalescence times of the different pairs of genes compared [34]. As for Hamilton's result (equation (1.7)), this conclusion rests on asymptotic results when the strength of selection δ → 0, for non-vanishing dispersal. In this limit, practically nothing is known about
describes the similarity of k-neighbours to the focal, relative to the similarity of 2-neighbours to the focal. Further, Rk2 can be considered as independent of p: for any p, it quantifies a difference in the distributions of coalescence times of the different pairs of genes compared [34]. As for Hamilton's result (equation (1.7)), this conclusion rests on asymptotic results when the strength of selection δ → 0, for non-vanishing dispersal. In this limit, practically nothing is known about 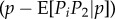 as the function of model parameters, except that it is positive [34].
as the function of model parameters, except that it is positive [34].
Equation (B 3) is perhaps the closest analogue for the localized dispersal of Hamilton's result, displaying a frequency-independent factor. In this equation, the term for the most distant actors plays the role previously played by the average population term: the fitness effects from the most distant actors are absorbed in the other terms, and the ‘relatedness’ Rkl of k-step neighbours is accordingly defined relative to the most distant l-neighbours. There are other ways to exhibit a frequency-independent term, owing to the fact that any of the partial derivatives may be absorbed into the others. These other ways are useful as they may involve simpler relatedness coefficients, but either the phenotypic cost term or the benefit term for within-group neighbours is lost from sight, as we now show. By expressing the second derivative in equation (B 2) in terms of the other two, one obtains
| B4 |
where 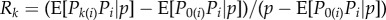 describes the similarity of k-neighbours to the focal, relative to the similarity of group neighbours. Now, the within-group benefits (the b of Hamilton's rule) have been cancelled out, but the Rks have useful compact expressions and can be computed as
describes the similarity of k-neighbours to the focal, relative to the similarity of group neighbours. Now, the within-group benefits (the b of Hamilton's rule) have been cancelled out, but the Rks have useful compact expressions and can be computed as 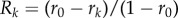 , where ri is the probability of identity-by-descent of two individuals sampled without replacement i steps apart on the lattice. The simplest case is the Moran model assumption leading to equation (B 1), for which
, where ri is the probability of identity-by-descent of two individuals sampled without replacement i steps apart on the lattice. The simplest case is the Moran model assumption leading to equation (B 1), for which 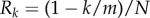 .
.
(c). Between generation fluctuations
In order to illustrate how to analyse how between-generation fluctuations affect allele frequency change, we consider that there may be good and bad years so that the environment determines the survival probability ςg (ςb) of queens in good (bad) years in our social insect colony model (equation (1.17)). In this case, the survival probability in the fitness function (equation (1.17)) becomes a random variable ςd depending on the state  of the environment, and as this fluctuates it may be felt that ‘fitness’ should be measured over several generations in order to evaluate the growth rate of a mutant (i.e. the geometric growth rate). However, allele frequency changes add up over generations and thus it is also correct to describe the change over several generations as an (arithmetic) average of one-generation changes. For example, if there is only one queen per nest so that zn = zf in equation (1.17) and the selection gradient is given by the derivative of fitness with respect to zf, this average change is given as
of the environment, and as this fluctuates it may be felt that ‘fitness’ should be measured over several generations in order to evaluate the growth rate of a mutant (i.e. the geometric growth rate). However, allele frequency changes add up over generations and thus it is also correct to describe the change over several generations as an (arithmetic) average of one-generation changes. For example, if there is only one queen per nest so that zn = zf in equation (1.17) and the selection gradient is given by the derivative of fitness with respect to zf, this average change is given as
| C1 |
where qg (qb) are the probabilities of occurrence of good (bad) years and wd is the fitness in state d ([20], equation (1.23)). Therefore, under temporal fluctuations, the selection gradient is independent of allele frequency, whether environmental change occurs within or between generations.
(d). Disruptive selection and phenotypic variance
Here, we show that the disruptive selection term d(z*, 0) gives the direction of change of the phenotypic variance 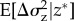 around a singular point when the mutant is rare (p → 0).
around a singular point when the mutant is rare (p → 0).
Given the resident phenotypic value z and mutant phenotype z + δ, the change in phenotypic variance over one generation is
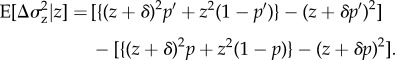 |
D1 |
On substitution of 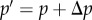 and
and 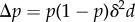
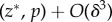 for a singular strategy this produces
for a singular strategy this produces
| D2 |
This shows that when the δ values are small, the remainder can be neglected and the dynamics of the variance is sign equivalent to d(z*, 0) when p → 0.
Endnotes
Throughout the text, we use the shorthand notation E(X | y) for E(X | Y = y).
By definition of the regression coefficient, we have 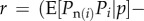
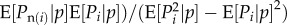 . Since
. Since 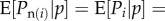
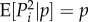 , we have
, we have 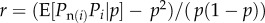 , which gives the right member in equation (1.14) upon rearrangement.
, which gives the right member in equation (1.14) upon rearrangement.
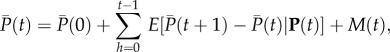 |
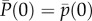 . This representation is unique and obtained by considering (possibly time dependent) weights αc(t) such that
. This representation is unique and obtained by considering (possibly time dependent) weights αc(t) such that 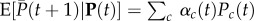 , which entails that in the absence of selection
, which entails that in the absence of selection 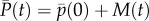 . The argument for using reproductive value makes no further assumptions about the population structure or about the strength of selection.
. The argument for using reproductive value makes no further assumptions about the population structure or about the strength of selection.In a finite population, one further has 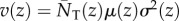 , where
, where 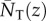 is the average number of gene copies in a population monomorphic for z, μ(z) is the probability that a randomly sampled gene from this population mutates, and σ2(z) is the variance of the mutant step size distribution [83] of genetic variance produced in the resident population.
is the average number of gene copies in a population monomorphic for z, μ(z) is the probability that a randomly sampled gene from this population mutates, and σ2(z) is the variance of the mutant step size distribution [83] of genetic variance produced in the resident population.
References
- 1.Hamilton WD. 2001. Narrow roads to gene land, vol. 2 Oxford, UK: Oxford University Press. [Google Scholar]
- 2.Hamilton WD. 1964. The genetical evolution of social behaviour, 1. J. Theoret. Biol. 7, 1–16. ( 10.1016/0022-5193(64)90038-4) [DOI] [PubMed] [Google Scholar]
- 3.Segerstråle U. 2000. Defenders of the truth. Oxford, UK: Oxford University Press. [Google Scholar]
- 4.Eshel I. 1983. Evolutionary and continuous stability. J. Theoret. Biol. 103, 99–111. ( 10.1016/0022-5193(83)90201-1) [DOI] [Google Scholar]
- 5.Dawkins R. 1978. Replicator selection and the extended phenotype. Z. Tierpsychol. 47, 61–76. ( 10.1111/j.1439-0310.1978.tb01823.x) [DOI] [PubMed] [Google Scholar]
- 6.Wright S. 1931. Evolution in Mendelian populations. Genetics 16, 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price GR. 1970. Selection and covariance. Nature 227, 520–521. ( 10.1038/227520a0) [DOI] [PubMed] [Google Scholar]
- 8.Grafen A. 1985. A geometric view of relatedness. In Oxford surveys in evolutionary biology (eds Dawkins R, Ridley M.), pp. 28–90. Oxford, UK: Oxford University Press. [Google Scholar]
- 9.Grafen A. 2000. Developments of the Price equation and natural selection under uncertainty. Proc. R. Soc. B 267, 1223–1227. ( 10.1098/rspb.2000.1131) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor PD. 1988. An inclusive fitness model for dispersal of offspring. J. Theoret. Biol. 130, 363–378. ( 10.1016/S0022-5193(88)80035-3) [DOI] [Google Scholar]
- 11.Frank SA. 1998. Foundations of social evolution. Princeton, NJ: Princeton University Press. [Google Scholar]
- 12.Gandon S. 1999. Kin competition, the cost of inbreeding and the evolution of dispersal. J. Theoret. Biol. 200, 345–364. ( 10.1006/jtbi.1999.0994) [DOI] [PubMed] [Google Scholar]
- 13.Pen I. 2000. Reproductive effort in viscous populations. Evolution 54, 293–297. [DOI] [PubMed] [Google Scholar]
- 14.Gardner A. 2010. Sex-biased dispersal of adults mediates the evolution of altruism among juveniles. J. Theoret. Biol. 262, 339–345. ( 10.1016/j.jtbi.2009.09.028) [DOI] [PubMed] [Google Scholar]
- 15.Ohtsuki H. 2010. Evolutionary games in Wright's island model: kin selection meets evolutionary game theory. Evolution 64, 3344–3353. ( 10.1111/j.1558-5646.2010.01117.x) [DOI] [PubMed] [Google Scholar]
- 16.Gillespie JH. 1975. Natural selection for within-generation variance in offspring number 2. Discrete haploid models. Genetics 81, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shpak M, Proulx SR. 2007. The role of life cycle and migration in selection for variance in offspring number. Bull. Math. Biol. 69, 837–860. ( 10.1007/s11538-006-9164-y) [DOI] [PubMed] [Google Scholar]
- 18.Lehmann L, Balloux F. 2007. Natural selection on fecundity variance in subdivided populations: kin selection meets bet hedging. Genetics 176, 361–377. ( 10.1534/genetics.106.066910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlin S. 1968. Equilibrium behavior of population genetic models with non-random mating: part II: pedigrees, homozygosity and stochastic models. J. Appl. Probab. 5, 487–566. ( 10.2307/3211920) [DOI] [Google Scholar]
- 20.Rousset F, Ronce O. 2004. Inclusive fitness for traits affecting metapopulation demography. Theoret. Popul. Biol. 65, 127–141. ( 10.1016/j.tpb.2003.09.003) [DOI] [PubMed] [Google Scholar]
- 21.Ronce O, Gandon S, Rousset F. 2000. Kin selection and natal dispersal in an age-structured population. Theoret. Popul. Biol. 58, 143–159. ( 10.1006/tpbi.2000.1476) [DOI] [PubMed] [Google Scholar]
- 22.Vitalis R, Rousset F, Kobayashi Y, Olivieri I, Gandon S. 2013. The joint evolution of dispersal and dormancy in a metapopulation with local extinctions and kin competition. Evolution 67, 1676–1691. ( 10.1111/evo.12069) [DOI] [PubMed] [Google Scholar]
- 23.Lynch M, Walsh B. 1998. Genetics and analysis of quantitative traits. Sunderland, MA: Sinauer. [Google Scholar]
- 24.Rousset F. 2004. Genetic structure and selection in subdivided populations. Princeton, NJ: Princeton University Press. [Google Scholar]
- 25.Davison AC. 2003. Statistical models. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 26.Karlin S, Taylor HM. 1975. A first course in stochastic processes. San Diego, CA: Academic Press. [Google Scholar]
- 27.Hamilton WD. 1970. Selfish and spiteful behavior in an evolutionary model. Nature 228, 1218–1220. ( 10.1038/2281218a0) [DOI] [PubMed] [Google Scholar]
- 28.Michod RE, Hamilton WD. 1980. Coefficients of relatedness in sociobiology. Nature 288, 694–697. ( 10.1038/288694a0) [DOI] [Google Scholar]
- 29.Roze D, Rousset F. 2004. The robustness of Hamilton's rule with inbreeding and dominance: kin selection and fixation probabilities under partial sib mating. Am. Nat. 164, 214–231. ( 10.1086/422202) [DOI] [PubMed] [Google Scholar]
- 30.Ethier SN, Nagylaki T. 1980. Diffusion approximations of Markov chains with two scales and applications to population genetics. Adv. Appl. Probab. 12, 14–49. ( 10.2307/1426492) [DOI] [Google Scholar]
- 31.Lessard S. 2009. Diffusion approximations for one-locus multi-allele kin selection, mutation and random drift in group-structured populations: a unifying approach to selection models in population genetics. J. Math. Biol. 59, 659–696. ( 10.1007/s00285-008-0248-1) [DOI] [PubMed] [Google Scholar]
- 32.Maruyama T. 1970. On the rate of decrease of heterozygosity in circular stepping stone models of population genetics. Theoret. Popul. Biol. 1, 101–119. ( 10.1016/0040-5809(70)90044-4) [DOI] [PubMed] [Google Scholar]
- 33.Malécot G. 1975. Heterozygosity and relationship in regularly subdivided populations. Theoret. Popul. Biol. 8, 212–241. ( 10.1016/0040-5809(75)90033-7) [DOI] [PubMed] [Google Scholar]
- 34.Rousset F. 2006. Separation of time scales, fixation probabilities and convergence to evolutionarily stable states under isolation by distance. Theoret. Popul. Biol. 69, 165–179. ( 10.1016/j.tpb.2005.08.008) [DOI] [PubMed] [Google Scholar]
- 35.Lehmann L, Rousset F. 2010. How life-history and demography promote or inhibit the evolution of helping behaviors. Phil. Trans. R. Soc. B 365, 2599–2617. ( 10.1098/rstb.2010.0138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamilton WD. 1966. The moulding of senescence by natural selection. J. Theoret. Biol. 12, 12–45. ( 10.1016/0022-5193(66)90184-6) [DOI] [PubMed] [Google Scholar]
- 37.Charlesworth B. 1994. Evolution in age-structured populations, 2nd edn Cambridge, UK: Cambridge University Press. [Google Scholar]
- 38.Taylor P. 1990. Allele-frequency change in a class-structured population. Am. Nat. 135, 95–106. ( 10.1086/285034) [DOI] [Google Scholar]
- 39.Taylor PD, Frank SA. 1996. How to make a kin selection model. J. Theoret. Biol. 180, 27–37. ( 10.1006/jtbi.1996.0075) [DOI] [PubMed] [Google Scholar]
- 40.McNamara J. 1997. Optimal life histories for structured populations in fluctuating environments. Theoret. Popul. Biol. 51, 94–108. ( 10.1006/tpbi.1997.1291) [DOI] [Google Scholar]
- 41.Fisher RA. 1930. The genetical theory of natural selection. Oxford, UK: Clarendon Press. [Google Scholar]
- 42.Grimmett G, Stirzaker D. 2001. Probability and random processes. Oxford, UK: Oxford University Press. [Google Scholar]
- 43.Benaïm M, El Karoui N. 2005. Promenade Aléatoire: Chaînes de Markov et Simulations; Martingales et Stratégies. Palaiseau, France: Editions de l'Ecole Polytechnique. [Google Scholar]
- 44.Charlesworth B, Charnov EL. 1981. Kin selection in age-structured populations. J. Theoret. Biol. 88, 103–119. ( 10.1016/0022-5193(81)90330-1) [DOI] [PubMed] [Google Scholar]
- 45.Taylor PD, Getz WM. 1994. An inclusive fitness model for the evolutionary advantage of sibmating. Evol. Ecol. 8, 61–69. ( 10.1007/BF01237666) [DOI] [Google Scholar]
- 46.Lehmann L, Perrin N, Rousset F. 2006. Population demography and the evolution of helping behaviors. Evolution 60, 1137–1151. [PubMed] [Google Scholar]
- 47.Wild G, Gardner A, West SA. 2009. Adaptation and the evolution of parasite virulence in a connected world. Nature 459, 983–986. ( 10.1038/nature08071) [DOI] [PubMed] [Google Scholar]
- 48.Ronce O, Brachet I, Olivieri I, Gouyon PH, Clobert J. 2005. Plastic changes in seed dispersal along ecological succession: theoretical predictions from an evolutionary model. J. Ecol. 93, 431–440. ( 10.1111/j.1365-2745.2005.00972.x) [DOI] [Google Scholar]
- 49.Wright S. 1969. Evolution and the genetics of populations. II. The theory of gene frequencies. Chicago, IL: University of Chicago Press. [Google Scholar]
- 50.Whitlock MC. 2002. Selection, load and inbreeding depression in a large metapopulation. Genetics 160, 1191–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michod RE. 1982. The theory of kin selection. Annu. Rev. Ecol. Systemat. 13, 23–55. ( 10.1146/annurev.es.13.110182.000323) [DOI] [Google Scholar]
- 52.Godfray HCJ. 1995. Signaling of need between parents and young: parent–offspring conflict and sibling rivalry. Am. Nat. 146, 1–24. ( 10.1086/285784) [DOI] [Google Scholar]
- 53.Day T, Taylor PD. 1997. Hamilton's rule meets the Hamiltonian: kin selection on dynamic characters. Proc. R. Soc. Lond. Ser. B 264, 639–644. ( 10.1098/rspb.1997.0090) [DOI] [Google Scholar]
- 54.Johnstone RA. 1999. Signaling of need, sibling competition, and the cost of honesty. Proc. Natl Acad. Sci. USA 26, 12 644–12 649. ( 10.1073/pnas.96.22.12644) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akcay E, Van Cleve J. 2012. Behavioral responses in structured populations pave the way to group optimality. Am. Nat. 179, 257–269. ( 10.1086/663691) [DOI] [PubMed] [Google Scholar]
- 56.Wild G. 2011. Direct fitness for dynamic kin selection. J. Evol. Biol. 24, 1598–1610. ( 10.1111/j.1420-9101.2011.02291.x) [DOI] [PubMed] [Google Scholar]
- 57.Lehmann L, Rousset F, Roze D, Keller L. 2007. Strong reciprocity or strong ferocity? A population genetic view of the evolution of altruistic punishment. Am. Nat. 170, 21–36. ( 10.1086/518568) [DOI] [PubMed] [Google Scholar]
- 58.Crow JF, Kimura M. 1970. An introduction to population genetics theory. New York, NY: Harper and Row. [Google Scholar]
- 59.Rousset F, Roze D. 2007. Constraints on the origin and maintenance of genetic kin recognition. Evolution 61, 2320–2330. ( 10.1111/j.1558-5646.2007.00191.x) [DOI] [PubMed] [Google Scholar]
- 60.Roze D, Rousset F. 2008. Multilocus models in the infinite island model of population structure. Theoret. Popul. Biol. 73, 529–542. ( 10.1016/j.tpb.2008.03.002) [DOI] [PubMed] [Google Scholar]
- 61.Kimura M. 1965. Attainment of quasi-linkage equilibrium when gene frequencies are changing by natural selection. Genetics 52, 875–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagylaki T. 1993. The evolution of multilocus systems under weak selection. Genetics 134, 627–647. ( 10.1534/genetics.105.041301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barton NH, Turelli M. 1991. Natural and sexual selection on many loci. Genetics 127, 229–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kirkpatrick M, Johnson T, Barton N. 2002. General models of multilocus evolution. Genetics 161, 1727–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gardner A, West SA, Barton N. 2007. The relation between multilocus population genetics and social evolution theory. Am. Nat. 169, 207–226. ( 10.1086/510602) [DOI] [PubMed] [Google Scholar]
- 66.Lehmann L, Rousset F. 2009. Perturbation expansions of multilocus fixation probabilities for frequency-dependent selection with applications to the Hill–Robertson effect and to the joint evolution of helping and punishment. Theoret. Popul. Biol. 76, 35–51. ( 10.1016/j.tpb.2009.03.006) [DOI] [PubMed] [Google Scholar]
- 67.Roze D, Rousset F. 2003. Selection and drift in subdivided populations: a straightforward method for deriving diffusion approximations and applications involving dominance, selfing and local extinctions. Genetics 165, 2153–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gillespie JH. 1991. The causes of molecular evolution. Oxford, UK: Oxford University Press. [Google Scholar]
- 69.Eshel I. 1996. On the changing concept of evolutionary population stability as a reflection of a changing point of view in the quantitative theory of evolution. J. Math. Biol. 34, 485–510. ( 10.1007/BF02409747) [DOI] [PubMed] [Google Scholar]
- 70.Hammerstein P. 1996. Darwinian adaptation, population genetics and the streetcar theory of evolution. J. Math. Biol. 34, 511–532. ( 10.1007/BF02409748) [DOI] [PubMed] [Google Scholar]
- 71.Metz JAJ, Geritz SAH, Meszéna G, Jacobs FJA, van Heerwaarden JS. 1996. Adaptive dynamics: a geometrical study of the consequences of nearly faithful reproduction. In Stochastic and spatial structures of dynamical systems (eds van Strien SJ, Verduyn Lunel SM.), pp. 183–231. Amsterdam, The Netherlands: North-Holland. [Google Scholar]
- 72.Eshel I, Feldman M, Bergman A. 1998. Long-term evolution, short-term evolution, and population genetic theory. J. Theoret. Biol. 191, 391–396. ( 10.1006/jtbi.1997.0597) [DOI] [Google Scholar]
- 73.Gertiz SAH, Kisdi E, Meszena G, Metz JAJ. 1998. Evolutionarily singular strategies and the adaptive growth and branching of the evolutionary tree. Evol. Ecol. 12, 35–57. ( 10.1023/A:1006554906681) [DOI] [Google Scholar]
- 74.Champagnat N, Ferrière R, Méléard S. 2006. Unifying evolutionary dynamics: from individual stochastic processes to macroscopic models. Theoret. Popul. Biol. 69, 297–321. ( 10.1016/j.tpb.2005.10.004) [DOI] [PubMed] [Google Scholar]
- 75.Iwasa Y, Pomiankowski A, Nee S. 1991. The evolution of costly mate preferences II. The ‘handicap’ principle. Evolution 45, 1431–1442. ( 10.2307/2409890) [DOI] [PubMed] [Google Scholar]
- 76.Abrams PA, Matsuda H, Harada Y. 1993. Evolutionarily unstable fitness maxima and stable fitness minima of continuous traits. Evol. Ecol. 7, 465–487. ( 10.1007/BF01237642) [DOI] [Google Scholar]
- 77.Day T, Taylor PD. 1996. Evolutionarily stable versus fitness maximizing life histories under frequency-dependent selections. Proc. R. Soc. B 263, 333–338. ( 10.1098/rspb.1996.0051) [DOI] [Google Scholar]
- 78.Taylor PD. 1989. Evolutionary stability in one-parameter models under weak selection. Theoret. Popul. Biol. 36, 125–143. ( 10.1016/0040-5809(89)90025-7) [DOI] [Google Scholar]
- 79.Dieckmann U, Law R. 1996. The dynamical theory of coevolution: a derivation from stochastic ecological processes. J. Math. Biol. 34, 579–612. ( 10.1007/BF02409751) [DOI] [PubMed] [Google Scholar]
- 80.Taper ML, Case TJ. 1992. Models of character displacement and the theoretical robustness of taxon cycles. Evolution 46, 317–333. ( 10.2307/2409853) [DOI] [PubMed] [Google Scholar]
- 81.Orlove MJ, Wood CL. 1978. Coefficients of relationship and coefficients of relatedness in kin selection: a covariance form for the RHO formula. J. Theoret. Biol. 73, 679–686. ( 10.1016/0022-5193(78)90129-7) [DOI] [PubMed] [Google Scholar]
- 82.Leimar O. 2009. Multidimensional convergence stability. Evol. Ecol. Res. 11, 191–208. [Google Scholar]
- 83.Lehmann L. 2012. The stationary distribution of a continuously varying strategy in a class-structured population under mutation-selection-drift balance. J. Evol. Biol. 25, 770–787. ( 10.1111/j.1420-9101.2012.02472.x) [DOI] [PubMed] [Google Scholar]
- 84.Kwon C, Ao P, David JT. 2005. Structure of stochastic dynamics near fixed points. Proc. Natl Acad. Sci. USA 102, 13 029–13 033. ( 10.1073/pnas.0506347102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Day T, Taylor PD. 1998. Unifying genetic and game theoretic models of kin selection for continuous traits. J. Theoret. Biol. 194, 391–407. ( 10.1006/jtbi.1998.0762) [DOI] [PubMed] [Google Scholar]
- 86.Metz J, Gyllenberg M. 2001. How should we define fitness in structured metapopulation models? Including an application to the calculation of evolutionary stable dispersal strategies. Proc. R. Soc. Lond. Ser. B 268, 499–508. ( 10.1098/rspb.2000.1373) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Day T. 2001. Population structure inhibits evolutionary diversification under competition for resources. Genetica 112, 71–86. ( 10.1023/A:1013306914977) [DOI] [PubMed] [Google Scholar]
- 88.Ajar E. 2003. Analysis of disruptive selection in subdivided populations. BMC Evol. Biol. 3, 22 ( 10.1186/1471-2148-3-22) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Biernaskie JM. 2010. The origin of gender dimorphism in animal-dispersed plants: disruptive selection in a model of social evolution. Am. Nat. 175, E134–E148. ( 10.1086/652431) [DOI] [PubMed] [Google Scholar]
- 90.Wakano JY, Iwasa Y. 2013. Evolutionary branching in a finite population: deterministic branching vs. stochastic branching. Genetics 193, 229–241. ( 10.1534/genetics.112.144980) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bürger R, Schneider KA, Willensdorfer M. 2006. The conditions for speciation through intraspecific competition. Evolution 60, 2185–2206. ( 10.1111/j.0014-3820.2006.tb01857.x) [DOI] [PubMed] [Google Scholar]
- 92.Wakano JY, Lehmann L. 2012. Evolutionary and convergence stability for continuous phenotypes in finite populations derived from two-allele models. J. Theoret. Biol. 310, 206–215. ( 10.1016/j.jtbi.2012.06.036) [DOI] [PubMed] [Google Scholar]
- 93.Frank SA. 1986. Hierarchical selection theory and sex ratios I. General solutions for structured populations. Theoret. Popul. Biol. 29, 312–342. ( 10.1016/0040-5809(86)90013-4) [DOI] [PubMed] [Google Scholar]
- 94.Van Dyken JD. 2010. The components of kin competition. Evolution 64, 2840–2854. ( 10.1111/j.1558-5646.2010.01033.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gardner A, West SA, Wild G. 2011. The genetical theory of kin selection. J. Evol. Biol. 24, 1020–1043. ( 10.1111/j.1420-9101.2011.02236.x) [DOI] [PubMed] [Google Scholar]
- 96.Queller DC. 1992. A general model for kin selection. Evolution 46, 376–380. ( 10.2307/2409858) [DOI] [PubMed] [Google Scholar]
- 97.Frank SA. 1997. The Price equation, Fisher's fundamental theorem, kin selection, and causal analysis. Evolution 51, 1712–1729. ( 10.2307/2410995) [DOI] [PubMed] [Google Scholar]
- 98.Nunney L. 1985. Group selection, altruism, and structured-deme models. Am. Nat. 126, 212–230. ( 10.1086/284410) [DOI] [Google Scholar]
- 99.Ewens WJ. 2011. What is the gene trying to do? Br. J. Phil. Sci. 62, 155–176. ( 10.1093/bjps/axq005) [DOI] [Google Scholar]
- 100.Chesson PL. 1984. Persistence of a Markovian population in a patchy environment. Z. Wahr. 66, 97–107. ( 10.1007/BF00532798) [DOI] [Google Scholar]
- 101.Wright S. 1942. Statistical genetics and evolution. Bull. Am. Math. Soc. 48, 223–246. ( 10.1090/S0002-9904-1942-07641-5) [DOI] [Google Scholar]
- 102.Kingman J. 1961. A mathematical problem in population genetics. Math. Proc. Camb. Phil. Soc. 57, 574–582. ( 10.1017/S0305004100035635) [DOI] [Google Scholar]
- 103.Price GR. 1972. Fisher's ‘fundamental theorem’ made clear. Ann. Hum. Genet. 36, 129–140. ( 10.1111/j.1469-1809.1972.tb00764.x) [DOI] [PubMed] [Google Scholar]
- 104.Grafen A. 2006. Optimization of inclusive fitness. J. Theoret. Biol. 238, 541–563. ( 10.1016/j.jtbi.2005.06.009) [DOI] [PubMed] [Google Scholar]
- 105.Grafen A. 2008. The simplest formal argument for fitness optimization. J. Genet. 87, 421–433. ( 10.1007/s12041-008-0064-9) [DOI] [PubMed] [Google Scholar]
- 106.Grafen A. 2013. The formal Darwinism project in outline. Biol. Phil . ( 10.1007/s10539-013-9414-y) [DOI]
- 107.Lehmann L, Rousset F. 2014. Fitness, inclusive fitness, and optimization. Biol. Phil. ( 10.1007/s10539-013-9415-x) [DOI]
- 108.Enquist M, Ghirlanda S. 2005. Neural networks and animal behavior. Princeton, NJ: Princeton University Press. [Google Scholar]
- 109.McFarland D, Houston A. 1981. Quantitative ethology: the state space approach. Boston, MA: Pitman. [Google Scholar]
- 110.Leimar O. 1997. Repeated games: a state space approach. J. Theoret. Biol. 184, 471–498. ( 10.1006/jtbi.1996.0286) [DOI] [Google Scholar]
- 111.Wolf JB, Brodie ED, III, Cheverud A, Moore AJ, Wade MJ. 1998. Evolutionary consequences of indirect genetic effects. Trends Ecol. Evol. 13, 64–69. ( 10.1016/S0169-5347(97)01233-0) [DOI] [PubMed] [Google Scholar]
- 112.Lehmann L. 2008. The adaptive dynamics of niche constructing traits in spatially subdivided populations: evolving posthumous extended phenotypes. Evolution 62, 549–566. ( 10.1111/j.1558-5646.2007.00291.x) [DOI] [PubMed] [Google Scholar]
- 113.Aoki K, Wakano JY, Lehmann L. 2012. Evolutionarily stable learning schedules in discrete generation models. Theoret. Popul. Biol. 81, 300–309. ( 10.1016/j.tpb.2012.01.006) [DOI] [PubMed] [Google Scholar]
- 114.Lion S. 2013. Multiple infections, kin selection and the evolutionary epidemiology of parasite traits. J. Evol. Biol. 26, 2107–2122. ( 10.1111/jeb.12207) [DOI] [PubMed] [Google Scholar]
- 115.Grafen A. 1984. Natural selection, kin selection and group selection. In Behavioural ecology: an evolutionary approach (eds Krebs JR, Davies N.), pp. 62–84. Oxford, UK: Blackwell Scientific Publications. [Google Scholar]
- 116.West SA. 2009. Sex allocation. Princeton, NJ: Princeton University Press. [Google Scholar]
- 117.Bourke A. 2011. Principles of social evolution. Oxford, UK: Oxford University Press. [Google Scholar]
- 118.Bull JJ, Wang IN. 2010. Optimality models in the age of experimental evolution and genomics. J. Evol. Biol. 23, 1820–1838. ( 10.1111/j.1420-9101.2010.02054.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wilson EO. 1975. Sociobiology: the new synthesis. Cambridge, MA: Harvard Press. [Google Scholar]
- 120.Dawkins R. 1976. The selfish gene. Oxford, UK: Oxford University Press. [Google Scholar]
- 121.Ewens WJ. 2004. Mathematical population genetics. New York, NY: Springer. [Google Scholar]