

Abstract
Background. High mortality and uncertainty about the effectiveness of neuraminidase inhibitors (NAIs) in humans infected with influenza A(H7N9) viruses are public health concerns.
Methods. Susceptibility of N9 viruses to NAIs was determined in a fluorescence-based assay. The NAI oseltamivir (5, 20, or 80 mg/kg/day) was administered to BALB/c mice twice daily starting 24, 48, or 72 hours after A/Anhui/1/2013 (H7N9) virus challenge.
Results. All 12 avian N9 and 3 human H7N9 influenza viruses tested were susceptible to NAIs. Without prior adaptation, A/Anhui/1/2013 (H7N9) caused lethal infection in mice that was restricted to the respiratory tract and resulted in pulmonary edema and acute lung injury with hyaline membrane formation, leading to decreased oxygenation, all characteristics of human acute respiratory distress syndrome. Oseltamivir at 20 and 80 mg/kg protected 80% and 88% of mice when initiated after 24 hours, and the efficacy decreased to 70% and 60%, respectively, when treatment was delayed by 48 hours. Emergence of oseltamivir-resistant variants was not detected.
Conclusions. H7N9 viruses are comparable to currently circulating influenza A viruses in susceptibility to NAIs. Based on these animal studies, early treatment is associated with improved outcomes.
Keywords: human influenza H7N9, oseltamivir, acute respiratory distress syndrome, neuraminidase inhibitor, efficacy, resistance
In February 2013, a patient hospitalized with pneumonia and acute respiratory distress syndrome (ARDS) was confirmed to be infected with a rare subtype of H7N9 avian influenza A virus (A/Shanghai/1/2013) [1]. Since then, a total of 132 laboratory-confirmed human H7N9 infections have been reported in eastern China and Taiwan, resulting in a 28% mortality rate (as of 30 May 2013) [2, 3]. No human H7N9 cases have been reported in other countries. The virus did not spread efficiently among humans, but limited, nonsustained human-to-human transmission could not be excluded in a few family clusters [4]. Viruses of the H7N9 subtype had not been previously isolated from humans, and infections with low-pathogenic avian influenza viruses have not been associated with severe infection or death. This is important because current seasonal influenza vaccines contain only H1N1 and H3N2 influenza A antigens, and all population groups are immunologically naive to both H7 and N9 surface antigens. The presence of H7 surface antigen is also troubling as highly pathogenic viruses of this HA subtype were shown to infect humans and 1 fatal case was reported [5].
Although vaccines are the most effective option for controlling influenza infections, none are available for prevention of H7N9 infections [6]. Human influenza H7N9 viruses are resistant to adamantanes as they harbor an S31N amino acid substitution in the M2; thus, neuraminidase (NA) inhibitors (NAIs) are the only option for control of H7N9 influenza infections [1]. The NAI oseltamivir is recommended for treatment of human H7N9 infections [4, 7]. Challenges in H7N9 antiviral treatment include the development of ARDS as a common complication in patients hospitalized with pneumonia [1, 8] as well as limited clinical experience and knowledge about the effectiveness of oseltamivir on human H7N9 infections. Moreover, human H7N9 viruses with NAI resistance-associated mutations (R152K and R292K, N2 numbering) were reported during treatment, emphasizing that the emergence of drug-resistant viruses should be monitored [1, 9].
Mouse models are well established for evaluating viral pathogenesis and the efficacy of antiviral drugs, as mice and humans develop similar changes in the respiratory tract, with the predominant involvement of the lower airway in mice [10]. Whereas pathological changes caused by human H7N9 viruses in mice have been reported recently [11–13], ARDS was not described, and therapeutic strategies have not been fully explored.
Here we evaluate the susceptibility of a panel of N9 influenza viruses to NAIs in vitro, establish the pathogenicity of A/Anhui/1/2013 (H7N9) in BALB/c mice, and examine the efficacy of oseltamivir treatment against lethal H7N9 virus challenge.
METHODS
Viruses
Human H7N9 influenza viruses were obtained through the World Health Organization surveillance network. Twelve avian influenza viruses of the N9 subtype (Table 1) were obtained from the repository at St Jude Children's Research Hospital (SJCRH). Viruses were propagated in 10-day-old embryonated chicken eggs (eggs) at 35°C for 48 hours. Experiments with human H7N9 influenza viruses were conducted in an animal biosafety level 3+ containment facility approved by the US Department of Agriculture.
Table 1.
Susceptibility of Human H7N9 and Avian N9 Influenza Viruses to Neuraminidase Inhibitors
| Influenza Virus | Subtype | Passage Historya | NA Enzyme Inhibition Assay (Mean IC50 ± SD, nM)b |
||
|---|---|---|---|---|---|
| Oseltamivir Carboxylate | Zanamivir | Peramivir | |||
| Human H7N9 viruses | |||||
| A/Anhui/1/2013 | H7N9 | E3 | 0.33 ± 0.07 | 0.96 ± 0.19 | 0.15 ± 0.03 |
| A/Shanghai/1/2013 | H7N9 | E3 | 0.51 ± 0.01 | 0.82 ± 0.13 | 0.13 ± 0.01 |
| A/Shanghai/1/2013 | H7N9 | E4 | 0.32 ± 0.01 | 0.38 ± 0.30 | 0.20 ± 0.06 |
| A/Shanghai/2/2013 | H7N9 | E4 | 0.34 ± 0.04 | 0.71 ± 0.01 | 0.13 ± 0.03 |
| Avian N9 viruses | |||||
| A/Wild Duck/Shantou/520/2001 | H1N9 | E3 | 0.49 ± 0.02 | 0.89 ± 0.18 | 0.15 ± 0.01 |
| A/Duck/Nanchang/2-0492/2000 | H2N9 | E7 | 0.48 ± 0.09 | 0.86 ± 0.30 | 0.13 ± 0.02 |
| A/Mallard/Alberta/31/2001 | H3N9 | E3 | 0.44 ± 0.05 | 1.41 ± 0.20 | 0.24 ± 0.03 |
| A/Mallard/Alberta/507/2010 | H4N9 | E3 | 0.56 ± 0.12 | 0.87 ± 0.12 | 0.15 ± 0.01 |
| A/Goose/Hong Kong/W217/1997 | H6N9 | E5 | 0.52 ± 0.06 | 0.84 ± 0.07 | 0.15 ± 0.0 |
| A/Turkey/Minnesota/1/1988 | H7N9 | E3 | 0.41 ± 0.04 | 0.87 ± 0.08 | 0.11 ± 0.01 |
| A/Turkey/Minnesota/037767/2009 | H7N9 | E4 | 0.52 ± 0.07 | 0.94 ± 0.08 | 0.14 ± 0.01 |
| A/Duck/Bangladesh/8988/2010 | H10N9 | E2 | 0.35 ± 0.25 | 0.32 ± 0.36 | 0.10 ± 0.05 |
| A/Mallard/Alberta/122/1999 | H11N9 | E4 | 0.49 ± 0.09 | 0.85 ± 0.04 | 0.17 ± 0.01 |
| A/Mallard/Vietnam/66MD/2004 | H12N9 | E3 | 0.57 ± 0.13 | 0.92 ± 0.12 | 0.18 ± 0.01 |
| A/Herring Gull/Delaware/142/2007 | H13N9 | E1(Duck)/E2 | 0.49 ± 0.04 | 1.01 ± 0.09 | 0.17 ± 0.01 |
| A/Australian Shelduck/Western Australia/1762/1983 | H15N9 | E2 | 0.54 ± 0.06 | 1.66 ± 0.24 | 0.23 ± 0.01 |
| Reference viruses | |||||
| A/Fukui/20/2004-WTc | H3N2 | CX/C1 | 0.30 ± 0.15 | 0.52 ± 0.50 | 0.37 ± 0.20 |
| A/Fukui/45/2004-E119Vc | H3N2 | CX/C1 | 35.03 ± 16.32 | 0.76 ± 0.36 | 0.19 ± 0.03 |
Abbreviations: IC50, 50% inhibitory concentration; NA, neuraminidase; WT, wild-type.
a Indicates that virus was propagated in embryonated chicken (or duck) eggs, E; or in Madin-Darby canine kidney cells, C. Number indicates number of passages, X is unknown number.
b Concentration of neuraminidase inhibitor that reduced viral NA activity by 50% relative to NA activity without inhibitor. Values represent the mean ± SD from 2 independent experiments performed in triplicate.
c Oseltamivir-susceptible A/Fukui/20/2004 (H3N2) virus (WT) and oseltamivir-resistant A/Fukui/45/2004 (H3N2) virus carrying an E119V NA mutation were obtained from the Antiviral Group, International Society for Influenza and Other Respiratory Virus Diseases, and were included as internal controls for the standardization of IC50 values.
Neuraminidase Inhibitors
Oseltamivir carboxylate, the prodrug oseltamivir phosphate (oseltamivir), zanamivir, and peramivir were prepared in distillated water and filter-sterilized, and stocks were stored at −20°C.
Susceptibility to NAIs In Vitro
Viral NA activity was determined using the fluorogenic substrate 2′-(4-methylumbelliferyl)-α-D-N-acetylneuraminic acid (MUNANA; Sigma-Aldrich) in 96-well, flat-bottom, black, opaque plates (Corning Costar) [14].
Virus Infectivity
Madin-Darby canine kidney (MDCK) cells were obtained from American Type Culture Collection and maintained as described [14]. The infectivity of H7N9 viruses was determined in MDCK cells (by plaque assay, expressed as log10 plaque-forming units [PFU]/mL, or by 50% tissue culture infectious dose, expressed as log10 TCID50/mL) and in eggs (expressed as log10 EID50/mL).
Pathogenicity of A/Anhui/1/2013 (H7N9) in Mice
All animal experiments were approved by the Animal Care and Use Committee of SJCRH and complied with National Institutes of Health policies and the Animal Welfare Act. The mouse lethal dose that killed 50% of animals (MLD50) was determined in 6-week-old female BALB/c mice (weight, 18–20 g; Jackson Laboratories). Animals that showed severe disease and lost >25% of initial weight were euthanized. Mice were anesthetized with isoflurane and intranasally inoculated with 30 µL of 102–106 PFU/mouse of A/Anhui/1/2013 virus. The mean days to death was calculated by using the log-hazard scale. The weight change was calculated as a percentage of weight on day 0. On days 3, 6, 9, and 12 after inoculation, the lungs, brain, spleen, and small intestine were removed from 3 mice per group, rinsed with sterile phosphate-buffered saline (PBS), homogenized, and resuspended in 1 mL of cold PBS. The suspensions were cleared by centrifugation at 3000g for 20 minutes, and virus yield was determined by TCID50 in MDCK cells.
Assessment of Pulmonary Edema and Vascular Permeability
For the lung-to-body and lung wet-to-dry weight ratios, the blood was drained from excised mouse lungs, and wet lungs were weighed. Lungs were placed into 10% neutral buffered formalin, v/v (NBF; Thermo Scientific) and incubated at 65°C for 48 hours. The NBF was removed, and lungs were dried at 60°C for 96 hours and again weighed. Vascular permeability was measured by the pulmonary extravasation of Evans blue dye (Sigma) [15, 16].
Arterial Blood Gas Analysis
Arterial blood was collected during the terminal bleed from the left ventricle (3 mice/group) on days 0, 3, 6, and 9 postinfection, and arterial blood gases were measured by a portable i-STAT handheld analyzer (Abbott Laboratories) using i-STAT CG8+ cartridges.
Lung Histopathology and Immunohistochemistry
Lung tissues were fixed in NBF, routinely processed, and embedded in paraffin. Hematoxylin-eosin, influenza A nucleoprotein, and Masson trichrome staining were performed by the Veterinary Pathology Core at SJCRH.
Oseltamivir Efficacy in Mice
Groups of 19 mice were anesthetized with isoflurane and inoculated intranasally with 3 MLD50 (102.5 PFU/mouse) of A/Anhui/1/2013 in 50 µL of PBS. Treatment with oseltamivir (5, 20, or 80 mg/kg by oral gavage every 12 hours) was initiated 24, 48, or 72 hours postinfection and continued for 5 days. The mice were observed daily for clinical signs and survival (10 mice/group), and weight changes were monitored. Three mice per group were killed on days 3, 6, and 9 postinfection, and virus lung titers were determined by TCID50 in MDCK cells. Control (inoculated, untreated) mice received sterile water on the same schedule.
Serologic Tests
Sera were collected by retro-orbital bleed, treated with receptor-destroying enzyme, heat-inactivated at 56°C for 1 hour, and tested by hemagglutination inhibition (HI) assay with 0.5% turkey red blood cells (Rockland Immunochemicals).
Sequencing and Clonal Analysis
Viral RNA was isolated from allantoic fluid or lung homogenates using the RNeasy Mini kit (Qiagen). Samples were reverse-transcribed and polymerase chain reaction–amplified using NA gene–specific primers. Sequencing was performed by the Hartwell Center for Bioinformatics and Biotechnology at SJCRH, and DNA sequences were analyzed using the DNASTAR Lasergene analysis package.
Statistical Analysis
Virus infectivity, NAI susceptibility, lung permeability measurements, and mean days to death were compared by analysis of variance using the GraphPad Prism 5.0 software. The probability of survival was estimated by the Kaplan–Meier method and compared between groups using the log-rank test.
RESULTS
Susceptibility of Human H7N9 and Avian N9 Influenza Viruses to NAIs
The 50% inhibitory concentration (IC50) values of the avian N9 influenza viruses ranged from 0.32 nM to 0.52 nM for oseltamivir carboxylate and from 0.32 nM to 1.58 nM for zanamivir (Table 1). The 3 human H7N9 viruses had mean IC50 values of 0.33, 0.68, and 0.16 nM for oseltamivir carboxylate, zanamivir, and peramivir, respectively (A/Shanghai/1/2013 E3 was not used in the calculation). Notably, these values were comparable to those of the NAI-susceptible A/Fukui/20/2004 (H3N2) reference virus. A/Shanghai/1/2013 was reported to have the R292K NA mutation [1], but our tests for this virus showed IC50 values that were within the range of susceptibility. As mixtures of NAI-susceptible and -resistant virus populations can mask phenotypically resistant viruses, we conducted clonal analysis of the virus population after passages in eggs to determine the frequency of the R292K mutation. We determined that 38% and 19% of the virus population contained the mutation after 3 and 4 egg passages, respectively; hence the resistance marker was present within a minor proportion of the viral population. Overall, these analyses demonstrated that the natural baseline NAI susceptibility of human H7N9 and avian N9 influenza viruses was similar to that of NAI-susceptible N2 influenza viruses.
Pathogenicity of A/Anhui/1/2013 (H7N9) in Mice
The 3 human H7N9 viruses (A/Anhui/1/2013, A/Shanghai/1/2013, and A/Shanghai/2/2013) replicated efficiently with similar infectivity both in eggs (8.5–9.75 log10 EID50/mL) and MDCK cells (7.45 to 8.12 log10 PFU/mL) (data not shown). Inoculation of mice with A/Anhui/1/2013 virus resulted in morbidity and death. Mice infected with 104–106 PFU lost weight progressively, and all animals died between days 5 and 6 postinfection (Table 2). Three of 5 and 1 of 5 mice survived after challenge with 102 and 103 PFU, respectively, with a resulting 1 MLD50 of 102.3 PFU. Weight loss as a measure of morbidity correlated with the inoculation dose (Table 2). All doses caused similar levels of replication in mouse lungs on day 3 postinfection (Table 2). No increase in virus titers was observed on day 6 postinfection, suggesting the vast majority of susceptible cells were already infected by day 3 postinfection. Virus was detected in the mouse lungs up to day 9 postinfection. We did not detect virus in the brain; however, low levels of virus were detected in the small intestine (1/3 mice) and spleen (1/3 mice) at day 3 postinfection in mice infected with high virus doses (data not shown). Thus, A/Anhui/1/2013 causes lethal infection in BALB/c mice without prior adaptation and lacks the ability to spread systemically outside the respiratory tract.
Table 2.
Pathogenicity of Influenza A/Anhui/1/2013 (H7N9) Virus in a BALB/c Mouse Model
| Virus Dose (PFU/Mouse) | No. of Mice Survived/ Total No. | Mean Survival Day | Average Percentage of Weight Loss on Indicated Day Postinfection (% Loss Day 0 ± SD) |
Detection of H7N9 Virus at Different Times After Inoculation (Log10 TCID50/mL ± SD) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lungs | ||||||||||
| 3 | 6 | 9 | 12 | 3 | 6 | 9 | 12 | |||
| 102 | 3/5 | 9 ± 15 | 3.9 ± 1.8 | 10.8 ± 5.5 | 18.8 ± 9.2 | 3.4 ± 6.0 | 6.6 ± 0.4 | 6.1 ± 0.8 | 3.7 ± 0.9 | –a |
| 103 | 1/5 | 7 ± 14 | 8.8 ± 3.0 | 21.7 ± 7.0 | 18.4b | 11.6b | 6.9 ± 0.1 | 6.4 ± 0.3 | 4.8b | NA |
| 104 | 0/5 | 6 ± 0 | 12.5 ± 3.2 | 28.6 ± 1.2 | NA | NA | 6.8 ± 0.3 | 6.5 ± 0.4 | NA | NA |
| 105 | 0/5 | 5 ± 1 | 16.1 ± 2.5 | NA | NA | NA | 6.3 ± 0.4 | 5.8 ± 0.5 | NA | NA |
| 106 | 0/5 | 5 ± 0 | 17.1 ± 2.8 | NA | NA | NA | 6.3 ± 0.5 | 5.9 ± 0.3 | NA | NA |
Abbreviations: NA, not applicable (animal deaths occurred between days 5 and 9 postinfection); PFU, plaque-forming units; TCID50, 50% tissue culture infectious dose.
a Data from 2 animals. –, virus not detected by the TCID50 assay (detection limit: 1.5 log10 TCID50/mL).
b Data from 1 animal due to mortality of the other animals.
Effect of A/Anhui/1/2013 (H7N9) Replication on Lung Functions of Mice
To characterize the impact on lung function, we assessed pulmonary edema and vascular permeability in mice infected with 102–106 PFU of H7N9 virus on day 3 postinfection (Figure 1). Differences were observed in lung-to-body weight and lung wet-to-dry weight ratios, showing a virus dose-dependent increase of lung water content (characteristics of pulmonary edema) during H7N9 infection (Figure 1A and 1B). A significant difference was seen between mice infected with 105 or 106 PFU and mock-infected animals (P < .0001). Lungs of H7N9-infected mice showed a virus dose-dependent increase in pulmonary vascular permeability (P < .0001; Figure 1C) as determined by Evans blue dye, which has been previously shown to correlate with the extravasation of radiolabeled albumin of plasma leakage [17]. Histological examinations of the lung tissues revealed interstitial edema, thickening of alveolar walls, peribronchiolar alveolar collapse, and infiltration with inflammatory cells (Figure 2). Necrosis and cell debris in bronchioles suggested that pulmonary cells were destroyed by the virus downstream from the bronchioles (Figure 2A–H). Immunohistochemistry of influenza virus antigen revealed staining in the epithelial cells of bronchioles, terminal bronchioles, and alveolar epithelial cells, indicating viral pneumonia (Figure 2C, D, G, H, K, L, and O). Masson's trichrome stain reveled that alveolar walls and space were filled with collagen, suggesting the formation of hyaline membranes (Figure 2P).
Figure 1.

Infection of BALB/c mice with A/Anhui/1/2013 (H7N9) influenza virus causes acute lung injury and characteristics that closely resemble acute respiratory distress syndrome (ARDS). A, Lung-weight to body-weight ratios. B, Wet-to-dry lung weight ratios. C, Pulmonary vascular permeability index. The lungs were collected 3 days postinfection (p.i.) from mice (n = 2) inoculated with 102–106 plaque-forming units (PFU)/mouse of A/Anhui/1/2013 (H7N9) influenza virus or mock. Increased ratio indicates pulmonary edema (fluid accumulation in lungs). The amount of Evans blue dye measured in pulmonary tissues was normalized to lung tissue weight [15]. D, Oxygenation level. BALB/c mice (n = 3) were infected intranasally with 102.5 PFU/mouse of A/Anhui/1/2013 (H7N9) virus, and arterial blood was drawn 3, 6, or 9 days p.i. Oxygenation level was determined as a ratio of arterial partial pressure of oxygen (PO2) to fraction of inspired oxygen (FIO2) using the i-STAT CG8+ system. Ratios ≤300 and ≤200 indicate mild and moderate ARDS, respectively, as defined by the ARDS working group [18]. *P < .0001, by unpaired Student t test.
Figure 2.

Histopathologic changes in lungs of mice infected with human influenza A/Anhui/1/2013 (H7N9) virus. Mice (n = 3, each day postinfection [p.i.]) were infected with 102–106 plaque-forming units (PFU)/mouse of A/Anhui/1/2013 (H7N9) virus and killed on day 3 (A–L) or day 8 p.i. (M–P). Mouse lungs were fixed in 10% neutral buffered formalin and stained with hematoxylin-eosin (HE) (A, B, E, F, I, J, M, and N) or immunohistochemical staining (IHC) with anti–influenza A nucleoprotein antibody (C, D, G, H, K, L, and O), or Masson trichrome staining was performed to visualize hyaline membrane formation (P). Magnification ×10 (A, C, E, G, I, K, M, and O), ×20 (B, D, F, H, J, L, N, and P). A, B, E, F, I, and J, Necrosis of bronchiole with cell debris in bronchiolar lumen (solid arrow). Alveolar collapse and enlargement of alveolar ducts. Alveoli containing edema (open arrow) and inflammatory cells. C, D, G, H, K, and L, Solid arrow indicates influenza antigen-positive cell debris in bronchiolar lumen and influenza antigen-positive staining in respiratory epithelial cells (a). O, Weak influenza virus antigen-positive staining in bronchiolar (solid arrow) and in respiratory epithelial cells (a), including collapsed areas. P, Masson trichrome stain showing hyaline membrane formation lining alveolar ducts (solid arrow) and alveolar collapse (open arrow).
To assess and quantify the severity of alveolar damage and the resulting changes in oxygenation of infected animals, we determined the ratio of arterial partial pressure of oxygen (PO2) to fraction of inspired oxygen (FIO2) (Figure 1D). On day 3 postinfection, the oxygenation level (PO2/FIO2 ratio) in H7N9-infected mice was 311.1 ± 45.3 mm Hg and lower than mock-infected animals with a normal value of 450 mm Hg. During the course of H7N9 infection, the oxygenation decreased to 246.0 ± 44 mm Hg on day 6 postinfection and 217.5 ± 15.3 mm Hg on day 9 postinfection, levels corresponding those of humans with mild ARDS [18]. None of the infected animals survived past day 9 postinfection, suggesting that severe lung damage and resulting loss of aerated tissues were fatal.
Our findings of pulmonary edema, histopathologic features of acute lung injury with disruption of the alveolar-capillary barrier, hyaline membrane formation, and the loss of aerated lung tissues closely resembles the characteristics of ARDS in humans. Therefore, the pathological process observed in A/Anhui/1/2013-infected mice is a model for the development of human ARDS induced by H7N9 infection.
Efficacy of Oseltamivir Against Lethal Challenge of Mice With A/Anhui/1/2013 (H7N9)
To assess the therapeutic efficacy of oseltamivir, we initiated treatment of mice 24, 48, or 72 hours after virus inoculation. All mice lost weight starting 2 days postinfection (Figure 3A, C, and E), and placebo-treated mice died by day 8 postinfection. When oseltamivir was administered 24 hours postinfection, it protected mice against lethal H7N9 infection, and we observed a dose-dependent survival outcome. The highest survival rate was achieved with 80 mg/kg (88%), followed by 20 mg/kg (80%) and 5 mg/kg (50%) (Figure 3B, D, and F). The survival rate was reduced to 60% and 70% at 80 and 20 mg/kg, respectively; when oseltamivir treatment was delayed until 48 hours postinfection, with no mice surviving from the 5 mg/kg group. None of the oseltamivir doses tested protected mice from lethal infection when treatment was initiated 72 hours postinfection (Figure 3F).
Figure 3.

Effect of delayed oseltamivir treatment in mice infected with lethal dose of A/Anhui/1/2013 (H7N9) influenza virus. BALB/c mice were infected intranasally with 3 MLD50 (102.5 plaque-forming units per mouse) of A/Anhui/1/2013 (H7N9) influenza virus and treated with oseltamivir starting 24, 48, or 72 hours postinfection (p.i.). Oseltamivir was administered by oral gavage twice a day for 5 days at dose of 5, 20, or 80 mg/kg/day. Placebo groups were gavaged twice a day with sterile water on the same schedule. Weight loss (A, C, and E) and survival (B, D, and F) of mice (10 mice per group) treated with oseltamivir at 5 mg/kg (A–D), 20 mg/kg (A–F), or 80 mg/kg (A–F) starting 24 hours (A and B), 48 hours (C and D), or 72 hours (E and F) p.i.
As shown in Figure 4A, C, and E), administration of 20 or 80 mg/kg of oseltamivir 24 hours postinfection resulted in a significant decrease in virus lung titers on day 9 postinfection (P < .005), whereas there was no difference in titers in mice treated with 5 mg/kg 24 hours postinfection or at any dose administered 48 or 72 hours postinfection. The virus lung titers correlated with the loss of aerated lung tissues as determined by the PO2/FIO2 ratio, but a significant decrease in hypoxemia was observed in mice treated with oseltamivir at 20 or 80 mg/kg starting 24 hours postinfection (Figure 4B, D, and F).
Figure 4.

Virus lung replication and development of acute respiratory distress syndrome (ARDS) in oseltamivir-treated mice infected with lethal dose of A/Anhui/1/2013 (H7N9) influenza virus. Virus titers (A, C, and E) were determined 3, 6, and 9 days postinfection (p.i.) in the lungs of BALB/c mice (3 mice per group) by 50% tissue culture infectious dose (TCID50) assay on Madin-Darby canine kidney cells. Levels of hypoxemia (B, D, and F) were determined on day 9 p.i. by arterial blood gas analysis as described in the Figure 1 legend. Ratios ≤300 indicate ARDS [18].
Effect of Oseltamivir Treatment on Emergence of Resistant Variants
To screen for the possible emergence of drug-resistant variants during oseltamivir treatment, we performed genotypic analysis of the NA genes from lung homogenates of mice on days 6 and 9 postinfection. A total of 54 samples (6 from placebo-treated, 12 from 5 mg/kg-treated, 18 from 20 mg/kg-treated, and 18 from 80 mg/kg-treated mice) were analyzed. The consensus NA contigs did not show mixed viral populations in the samples (data not shown). Further analysis of the sequence chromatogram with attention to amino acids 117, 119, 152, 222, 274, and 292 (where changes are associated with resistance or reduced susceptibility to NAIs in group 2 NAs) did not reveal amino acid changes. Thus, oseltamivir-resistant A/Anhui/1/2013 viruses were not selected during or after treatment.
Detection and Persistence of Antibodies in Mouse Survivors Following Recovery
To examine whether the level of immune response was sufficient to protect against H7N9 virus reinfection and whether oseltamivir treatment affected development of anti-HA antibodies, we determined the levels of antibodies in surviving mice and reinfected them with a higher virus dose. All animals reinfected with 18 MLD50 of virus survived and did not lose weight (Figure 5A). Surviving mice from the 24- and 48-hour oseltamivir groups developed HI titers ranging from 1:80 to 1:320, and there was no significant difference in HI titers before or after reinfection in any treatment group (Figure 5B). This confirms that a robust and protective antibody response develops during oseltamivir treatment.
Figure 5.
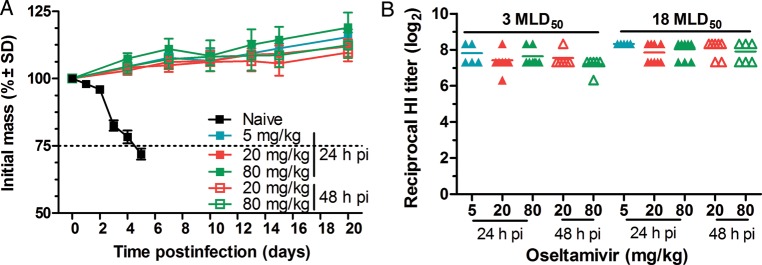
Effect of oseltamivir treatment on development of anti-HA antibody response in mice after infection and reinfection with A/Anhui/1/2013 (H7N9) influenza virus. BALB/c mice were infected intranasally with 3 MLD50 of A/Anhui/1/2013 (H7N9) influenza virus, and 21 days after initial infection, all surviving mice were reinfected with 18 MLD50 of the same virus. A, Weight loss of naive and oseltamivir-treated mice after reinfection with A/Anhui/1/2013 (H7N9) virus. B, Quantification of anti-HA antibody response after oseltamivir treatment. Hemagglutination inhibition (HI) titers were determined for sera collected 20 days postinfection (p.i.) of 3 MLD50 (1 day prior to reinfection) and 20 days p.i. of 18 MLD50.
DISCUSSION
We demonstrated phenotypic and genotypic susceptibility of human H7N9 and avian N9 viruses to NAIs oseltamivir carboxylate, zanamivir, and peramivir, and assessed the effectiveness of oseltamivir against A/Anhui/1/2013 in vivo. We developed a mouse model of H7N9-induced ARDS that closely resembled the pathological changes of infected humans. Importantly, this model is distinct from ARDS development caused by other influenza viruses: non-mouse-adapted H1N1pdm09 virus required high virus dose (106.5 PFU/mouse) [19] and H5N1 infection results in significant induction of inflammatory cytokines and neutrophil recruitment [20, 21]. With our H7N9 mouse model, we demonstrated dose-dependent efficacy of oseltamivir, with high efficacy at 20 and 80 mg/kg when administered within 24 hours postinfection. The efficacy of oseltamivir decreased when administered 48 hours postinfection and was ineffective 72 hours postinfection. Emergence of oseltamivir-resistant variants was not detected.
Antiviral susceptibility has been predominantly examined for human N1 and N2 viruses [22, 23]; however, little is known about the natural baseline IC50 values for N9 viruses. Our analysis showed that the NAI susceptibility of avian N9 viruses and human H7N9 viruses is similar to seasonal human H3N2 viruses. IC50 values for human H7N9 viruses were 550-fold lower than human plasma concentrations achieved with the recommended 75 mg twice daily dose of oseltamivir (Cmax for oseltamivir 330 nM) [24]. This suggests that NAIs should be as effective against NAI-susceptible N9 viruses as against NAI-susceptible N1 and N2 viruses.
One major public health concern with human H7N9 viruses is detection of R292K and R152K NA mutations in NAI-treated patients [1, 9]. The A/Shanghai/1/2013 with the NA-R292K mutation possessed a pan-resistant phenotype to all tested NAIs [25], and this mutation was previously reported in H3N2, H4N2, and H1N9 viruses [26–28]. Our phenotypic analysis of egg-grown A/Shanghai/1/2013 did not show elevated IC50 values for oseltamivir carboxylate, zanamivir, or peramivir. Further clonal analysis of the virus population revealed that prevalence of oseltamivir-resistant variants (with NA-R292K mutation) was reduced to 38% and 19% after 3 and 4 egg passages, respectively, suggesting that human H7N9 viruses with the NA-R292K are not stable upon culture. Our findings are consistent with recent reports [13, 25] and emphasize that virus subpopulations with NAI-resistant mutations can be missed during the routine phenotypic screening of cultured viruses and that susceptibility to NAIs should be based on both phenotypic and genotypic analysis. Notably, NA-R292K mutant virus has less NA activity and binding affinity [25].
A/Anhui/1/2013 caused a lethal infection in BALB/c mice without prior adaptation with 1 MLD50 of 102.3 PFU. Importantly, there was no evidence of sustained systemic virus spread outside of the respiratory tract. Our data are in line with reports by Belser et al [12] and Watanabe et al [13], which demonstrate that human H7N9 viruses of the A/Anhui/1/2013 lineage can cause lethal infections in mice without prior adaptation. Notably, A/Shanghai/1/2013 did not cause mortality in infected mice [11]. The A/Shanghai/1/2013 virus belongs to a different lineage and lacks the HA markers for mammalian adaptation (S138A, G186V, and Q226L; H3 numbering) [29], and binds equally to human-like α2,6- and avian-like α2,3-linked sialic acids whereas A/Anhui/1/2013 virus preferentially binds human-like α2,6-linked sialic acids [13, 30]. Thus, pathogenicity of human H7N9 viruses may be lineage-specific. Clinical reports indicate that patients with H7N9 infections presented with severe pneumonia and ARDS [1, 8]. A/Anhui/1/2013-infected mice exhibited dyspnea starting 2 days postinfection, pulmonary edema due to increased vascular permeability, and severe hypoxemia with the oxygenation level of the virus-infected mice <300 mm Hg. Histologic findings were consistent with acute lung damage. Therefore, we developed a lethal mouse model of ARDS for evaluation of treatment options for human H7N9 infections.
In our study, when administered within 24 hours postinfection, oseltamivir was effective at a dose of 20 mg/kg (the equivalent to the recommended human dose of 75 mg twice daily, based on the interspecies differences in esterase activity required to achieve the same area under the curve of plasma concentrations of oseltamivir carboxylate) [24, 31] in preventing death and development of ARDS in mice infected with a lethal dose of A/Anhui/1/2013 virus. In contrast, only 60%–70% of animals were protected when treatment was initiated 48 hours postinfection. Importantly, increasing the dose to 80 mg/kg did not provide increased protection. There was a dramatic loss in efficacy when oseltamivir therapy was initiated 72 hours postinfection. Survival rates correlated with high virus titers in the lungs and acute lung injury. We detected a decrease in virus lung titers after the 80 mg/kg dose but also protection of lung function (increased PO2/FIO2 ratio) with the 20 and 80 mg/kg doses when initiated 24 hours postinfection. Coinciding with virus replication, increased proinflammatory Th1 and Th2 cytokines were detected in the lungs of infected animals [11, 12]. Along with our data, these reports suggest that treatment strategies for severely ill patients will probably require a complex therapeutic approach targeting the reduction of virus replication, normalizing the aberrant immune response, and improving vascular barrier function. In contrast to our data, Watanabe et al [13] reported a modest effect of oseltamivir in protecting A/Anhui/1/2013-infected BALB/c mice from weight loss when the drug was applied 2 hours postinfection, which is hardly achievable in clinical practice. Importantly, no resistant variants were selected during oseltamivir treatment in our studies, although it was suggested that drug-resistant influenza viruses were difficult to isolate from mice during antiviral treatment [32, 33] with the exception of adamantanes treatment [34, 35] or immunocompromised mouse models [36].
Overall, our data showed that H7N9 viruses were susceptible to NAIs in vitro, and oseltamivir provided protection from lethal infection in a mouse model when administered within 48 hours postinfection. Current use of oseltamivir in humans has been unsuccessful; however, initiation of treatment in these cases began 6–8 days postinfection. Our model shows no benefit to treatment initiated after 72 hours postinfection but suggests that earlier diagnosis and treatment could significantly improve survival outcomes.
Notes
Acknowledgments. The NAIs oseltamivir carboxylate, oseltamivir phosphate, zanamivir, and peramivir were provided by Hoffmann-La Roche, Ltd (Nutley, New Jersey).We are grateful to Dr Lisa Kercher, Kristi Prevost, Patrick Seiler, Beth Little, and David S. Carey for technical support, and David Galloway for editing the manuscript.
Financial support. This work was supported in part by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (contract number HHSN266200700005C) and by the American Lebanese Syrian Associated Charities.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Gao R, Cao B, Hu Y, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368:1888–97. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Number of confirmed human cases of avian influenza A(H7N9) reported to WHO. http://www.who.int/influenza/human_animal_interface/influenza_h7n9/Data_Reports/en/index.html . Accessed 23 July 2013.
- 3.Chang S-Y, Lin P-H, Tsai J-C, Hung C-C, Chang S-C. The first case of H7N9 influenza in Taiwan. Lancet. 2013;381:1621. doi: 10.1016/S0140-6736(13)60943-5. [DOI] [PubMed] [Google Scholar]
- 4.Li Q, Zhou L, Zhou M, et al. Preliminary report: epidemiology of the avian influenza A (H7N9) outbreak in China. N Engl J Med. 0; 0:null. [Google Scholar]
- 5.Fouchier RAM, Schneeberger PM, Rozendaal FW, et al. Avian influenza A virus (H7N7) associated with human conjunctivitis and a fatal case of acute respiratory distress syndrome. Proc Natl Acad Sci U S A. 2004;101:1356–61. doi: 10.1073/pnas.0308352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osterholm MT, Ballering KS, Kelley NS. Major challenges in providing an effective and timely pandemic vaccine for influenza A(H7N9) JAMA. 2013;309:2557–8. doi: 10.1001/jama.2013.6589. [DOI] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. Antiviral agents for treatment of avian influenza A (H7N9) http://www.cdc.gov/flu/avianflu/h7n9-antiviral-treatment.htm . Accessed 26 July 2013.
- 8.Lu S, Zheng Y, Li T, et al. Clinical findings for early human cases of influenza A(H7N9) virus infection, Shanghai, China. Emerg Infect Dis. 2013;19:1142–6. doi: 10.3201/eid1907.130612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Y, Lu S, Song Z, et al. Association between adverse clinical outcome in human disease caused by novel influenza A H7N9 virus and sustained viral shedding and emergence of antiviral resistance. Lancet. 2013;381:2273–9. doi: 10.1016/S0140-6736(13)61125-3. [DOI] [PubMed] [Google Scholar]
- 10.Barnard DL. Animal models for the study of influenza pathogenesis and therapy. Antiviral Res. 2009;82:A110–22. doi: 10.1016/j.antiviral.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mok CKP, Lee HHY, Chan MCW, et al. Pathogenicity of the novel A/H7N9 influenza virus in mice. MBio. 2013;4:e00362–13. doi: 10.1128/mBio.00362-13. http://mbio.asm.org/content/4/4/e00362-13 . Accessed 23 July 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belser JA, Gustin KM, Pearce MB, et al. Pathogenesis and transmission of avian influenza A (H7N9) virus in ferrets and mice. Nature. 2013;501:556–9. doi: 10.1038/nature12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe T, Kiso M, Fukuyama S, et al. Characterization of H7N9 influenza A viruses isolated from humans. Nature. 2013;501:551–5. doi: 10.1038/nature12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baranovich T, Wong S-S, Armstrong J, et al. T-705 (Favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J Virol. 2013;87:3741–51. doi: 10.1128/JVI.02346-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baluk P, Thurston G, Murphy TJ, Bunnett NW, McDonald DM. Neurogenic plasma leakage in mouse airways. Br J Pharmacol. 1999;126:522–8. doi: 10.1038/sj.bjp.0702323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saria A, Lundberg JM. Evans blue fluorescence: quantitative and morphological evaluation of vascular permeability in animal tissues. J Neurosci Methods. 1983;8:41–9. doi: 10.1016/0165-0270(83)90050-x. [DOI] [PubMed] [Google Scholar]
- 17.Patterson CE, Rhoades RA, Garcia JG. Evans blue dye as a marker of albumin clearance in cultured endothelial monolayer and isolated lung. J Appl Physiol (1985) 1992;72:865–73. doi: 10.1152/jappl.1992.72.3.865. [DOI] [PubMed] [Google Scholar]
- 18.Ranieri VM, Rubenfeld GD, et al. ARDS Definition Task Force. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–33. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Sun H, Fan L, et al. Acute respiratory distress syndrome induced by a swine 2009 H1N1 variant in mice. PLoS One. 2012;7:e29347. doi: 10.1371/journal.pone.0029347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu T, Qiao J, Zhao L, et al. Acute respiratory distress syndrome induced by avian influenza A (H5N1) virus in mice. Am J Respir Crit Care Med. 2006;174:1011–7. doi: 10.1164/rccm.200511-1751OC. [DOI] [PubMed] [Google Scholar]
- 21.Garigliany MM, Habyarimana A, Lambrecht B, et al. Influenza A strain-dependent pathogenesis in fatal H1N1 and H5N1 subtype infections of mice. Emerg Infect Dis. 2010;16:595–603. doi: 10.3201/eid1604.091061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitley RJ, Boucher CA, Lina B, et al. Global assessment of resistance to neuraminidase inhibitors, 2008–2011: the Influenza Resistance Information Study (IRIS) Clin Infect Dis. 2013;56:1197–205. doi: 10.1093/cid/cis1220. [DOI] [PubMed] [Google Scholar]
- 23.McKimm-Breschkin JL. Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance. Influenza Other Respir Viruses. 2013;7:25–36. doi: 10.1111/irv.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He G, Massarella J, Ward P. Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64–0802. Clin Pharmacokinet. 1999;37:471–84. doi: 10.2165/00003088-199937060-00003. [DOI] [PubMed] [Google Scholar]
- 25.Yen H-L, McKimm-Breschkin JL, Choy K-T, et al. Resistance to neuraminidase inhibitors conferred by an R292K mutation in a human influenza virus H7N9 isolate can be masked by a mixed R/K viral population. MBio. 2013;4:e00396–13. doi: 10.1128/mBio.00396-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gubareva LV, Robinson MJ, Bethell RC, Webster RG. Catalytic and framework mutations in the neuraminidase active site of influenza viruses that are resistant to 4-guanidino-Neu5Ac2en. J Virol. 1997;71:3385–90. doi: 10.1128/jvi.71.5.3385-3390.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKimm-Breschkin JL, Sahasrabudhe A, Blick TJ, et al. Mutations in a conserved residue in the influenza virus neuraminidase active site decreases sensitivity to Neu5Ac2en-derived inhibitors. J Virol. 1998;72:2456–62. doi: 10.1128/jvi.72.3.2456-2462.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carr J, Ives J, Kelly L, et al. Influenza virus carrying neuraminidase with reduced sensitivity to oseltamivir carboxylate has altered properties in vitro and is compromised for infectivity and replicative ability in vivo. Antiviral Res. 2002;54:79–88. doi: 10.1016/s0166-3542(01)00215-7. [DOI] [PubMed] [Google Scholar]
- 29.Kageyama T, Fujisaki S, Takashita E, et al. Genetic analysis of novel avian A(H7N9) influenza viruses isolated from patients in China, February to April 2013. Euro Surveill. 2013 18. pii:20453. [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J, Wang D, Gao R, et al. Biological features of novel avian influenza A (H7N9) virus. Nature. 2013;499:500–503. doi: 10.1038/nature12379. [DOI] [PubMed] [Google Scholar]
- 31.Ward P, Small I, Smith J, Suter P, Dutkowski R. Oseltamivir (Tamiflu) and its potential for use in the event of an influenza pandemic. J Antimicrob Chemother. 2005;55(suppl 1):i5–21. doi: 10.1093/jac/dki018. [DOI] [PubMed] [Google Scholar]
- 32.Govorkova EA, Ilyushina NA, McClaren JL, Naipospos TSP, Douangngeun B, Webster RG. Susceptibility of highly pathogenic H5N1 influenza viruses to the neuraminidase inhibitor oseltamivir differs in vitro and in a mouse model. Antimicrob Agents Chemother. 2009;53:3088–96. doi: 10.1128/AAC.01667-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yen H-L, Ilyushina NA, Salomon R, Hoffmann E, Webster RG, Govorkova EA. Neuraminidase inhibitor-resistant recombinant A/Vietnam/1203/04 (H5N1) influenza viruses retain their replication efficiency and pathogenicity in vitro and in vivo. J Virol. 2007;81:12418–26. doi: 10.1128/JVI.01067-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goedemans WT, De Bock CA. Inhibition of influenza A viruses by adamantanamines: rate of resistance development in embryonated eggs and in mice. Zentralbl Bakteriol Orig. 1970;213:462–9. [PubMed] [Google Scholar]
- 35.Oxford JS, Logan IS, Potter CW. In vivo selection of an influenza A2 strain resistant to amantadine. Nature. 1970;226:82–3. doi: 10.1038/226082a0. [DOI] [PubMed] [Google Scholar]
- 36.Ison MG, Mishin VP, Braciale TJ, Hayden FG, Gubareva LV. Comparative activities of oseltamivir and A-322278 in immunocompetent and immunocompromised murine models of influenza virus infection. J Infect Dis. 2006;193:765–72. doi: 10.1086/500464. [DOI] [PubMed] [Google Scholar]