

In progeria, a mutant form of lamin A constitutively tethered to the inner nuclear membrane causes disruption of the Ran GTPase system by inducing ROS. However, ROS are also induced by disruption of the Ran system. The data suggest that the nuclear lamina and Ran GTPase system are part of a pathway that contains positive and negative feedback loops.
Abstract
Maintaining the Ran GTPase at a proper concentration in the nucleus is important for nucleocytoplasmic transport. Previously we found that nuclear levels of Ran are reduced in cells from patients with Hutchinson–Gilford progeria syndrome (HGPS), a disease caused by constitutive attachment of a mutant form of lamin A (termed progerin) to the nuclear membrane. Here we explore the relationship between progerin, the Ran GTPase, and oxidative stress. Stable attachment of progerin to the nuclear membrane disrupts the Ran gradient and results in cytoplasmic localization of Ubc9, a Ran-dependent import cargo. Ran and Ubc9 disruption can be induced reversibly with H2O2. CHO cells preadapted to oxidative stress resist the effects of progerin on Ran and Ubc9. Given that HGPS-patient fibroblasts display elevated ROS, these data suggest that progerin inhibits nuclear transport via oxidative stress. A drug that inhibits pre–lamin A cleavage mimics the effects of progerin by disrupting the Ran gradient, but the effects on Ran are observed before a substantial ROS increase. Moreover, reducing the nuclear concentration of Ran is sufficient to induce ROS irrespective of progerin. We speculate that oxidative stress caused by progerin may occur upstream or downstream of Ran, depending on the cell type and physiological setting.
INTRODUCTION
The nuclear lamina is a network of proteins associated with the inner nuclear membrane that plays major roles in defining nuclear structure and function. The most abundant components of the lamina are the A-type and B-type lamins, intermediate filament proteins that assemble into polymers (Goldman et al., 2002). The lamins interact with an expanding group of membrane-associated and nucleoplasmic proteins and chromatin to form macromolecular assemblies that regulate nuclear events, including replication and transcription (Schirmer et al., 2005; Vlcek and Foisner, 2007). Studies have revealed that mutations in genes encoding the lamins, particularly lamin A, are causal to a broad spectrum of human disorders termed laminopathies. These include cardiomyopathy, lipodystrophy, and certain types of muscular dystrophy (Mounkes et al., 2003; Worman, 2012). Remarkably, different mutations within the single gene encoding lamin A (LMNA) give rise to disorders that can be manifest in a cell- and tissue-specific manner, presumably through a combination of changes in nuclear structure and gene expression (Hutchison and Worman, 2004).
One of the most highly studied laminopathies is Hutchinson–Gilford progeria syndrome (HGPS), a premature aging syndrome caused by a de novo mutation in LMNA. The LMNA mutation in HGPS results in utilization of a cryptic pre-mRNA splice site and generation of a transcript that encodes progerin, a form of lamin A missing 50 amino acids near its C-terminus (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). The progerin form of lamin A has a defect in protein processing, which is the basis of its dominant-negative effects on the cell (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). Normally, pre–lamin A is farnesylated on a C-terminal CAAX motif, attached to the nuclear membrane, and subsequently cleaved on the amino-terminal side of Leu-647 by the protease Zmpste24 (Sinensky et al., 1994). Thus fully processed lamin A is not directly attached to the nuclear membrane. By contrast, progerin is farnesylated, but it remains anchored to the nuclear membrane because the Zmpste24 cleavage site is within the 50–amino acid segment that is missing from progerin (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003). Although it is clear that constitutive attachment of progerin drives HGPS phenotypes, the mechanisms that mediate the changes at the cellular and tissue levels are largely undefined. One common phenotype reported for cells overexpressing progerin is aberrant nuclear morphology, including nuclear membrane blebbing (Goldman et al., 2004). A second phenotype of cells expressing progerin is reduced levels of epigenetic marks on histone H3 that are associated with gene repression (Shumaker et al., 2006; Scaffidi and Misteli, 2006). Progerin, which is highly concentrated at the nuclear membrane, elicits epigenetic changes throughout the nucleus (Shumaker et al., 2006; Scaffidi and Misteli, 2006). This observation implies that a dominant effect of nuclear architecture at the nuclear periphery has global effects on the nucleus.
We recently described phenotypic changes in HGPS cells that are associated with alterations in the nuclear transport machinery (Kelley et al., 2011; Snow et al., 2013), in particular, the Ran GTPase. Ran plays a direct role in nucleocytoplasmic transport by regulating transport factor assembly and disassembly, and, in the process, Ran rapidly shuttles between the nucleus and cytoplasm. Under steady-state conditions Ran has a predominantly nuclear distribution, and the concentration of Ran in the nucleus and cytoplasm can be expressed as a ratio (ratio of mean nuclear fluorescence to mean cytoplasmic fluorescence [N/C]) and termed the Ran protein gradient (Bischoff and Ponstingl, 1991; Bischoff et al., 1994; Pemberton and Paschal, 2005; Kelley and Paschal, 2007; Izaurralde et al., 1997; Smith et al., 1998; Görlich et al., 2003). The two nucleotide forms of Ran also generate a gradient between the nucleus and cytoplasm (RanGTP:RanGDP), owing to the subcellular distributions of the nucleotide exchange factor RCC1 (nuclear) and GTPase-activating protein RanGAP (cytoplasmic; Bischoff and Ponstingl, 1991; Bischoff et al., 1994; Paschal and Gerace, 1995; Pemberton and Paschal, 2005; Kelley and Paschal, 2007; Izaurralde et al., 1997; Smith et al., 1998; Görlich et al., 2003). Our laboratory found that the Ran N/C is significantly reduced in HGPS cells and in naive cells transfected with progerin (Kelley et al., 2011). These and other findings led us to suggest that reduced nuclear transport could underpin some of the phenotypic changes in HGPS (Kelley et al., 2011; Snow et al., 2013).
One of the proteins whose nuclear localization is reduced in HGPS is Ubc9, the E2 for SUMOylation (Geiss-Friedlander and Melchior, 2007; Kelley et al., 2011). Ubc9 is imported into the nucleus by importin13, and after its translocation into the nucleus, the Ubc9-importin13 complex is disassembled by RanGTP (Mingot et al., 2001). Working in conjunction with Uba2/Aos1 E1 enzyme, Ubc9 conjugates SUMO to target proteins (Geiss-Friedlander and Melchior, 2007). SUMOylation regulates diverse pathways in the nucleus, including transcription, replication, and DNA repair (Hay, 2005). Ubc9 shuttles between the nucleus and the cytoplasm, and SUMOylation can regulate events in the cytoplasm as well (Geiss-Friedlander and Melchior, 2007). There is also a pool of Ubc9 stably associated with the nucleoporin RanBP2, where it contributes to the SUMOylation of proteins associated with the nuclear pore complex (NPC; Zhang et al., 2002). In a previous study we found that reducing SUMOylation caused changes in the nuclear mobility of RCC1, suggesting that SUMO and possibly Ubc9 might play a role in the Ran protein gradient by modulating GTP-GDP exchange on Ran (Kelley et al., 2011).
Ran distribution and nuclear transport have been shown to be adversely affected by cellular stresses, including hyperosmolarity (Kelley and Paschal, 2007) and oxidative stress (Kodiha et al., 2004; Miyamoto et al., 2004; Yasuda et al., 2006). Oxidative stress caused by reactive oxygen species (ROS)—chemical derivatives of molecular oxygen arising from metabolism—can have profound effects on the cell. ROS in the form of superoxide or hydroxyl radicals are extremely unstable, whereas hydrogen peroxide is freely diffusible and has a longer half-life (Finkel and Holbrook, 2000). ROS levels are kept in check by a collection of enzymes, including superoxide dismutase (SOD) and catalase, that afford protection to the cell by metabolizing ROS (Finkel and Holbrook, 2000).
In the present study we examine the relationship between progerin, Ran, Ubc9, and oxidative stress. ROS levels are elevated in HGPS-patient fibroblasts and in normal fibroblasts treated with a protease inhibitor that prevents pre–lamin A cleavage from the nuclear membrane. Oxidative stress disrupts the Ran gradient and inhibits Ubc9 import, and cells preadapted to oxidative stress are resistant to the effects of progerin. Although these data suggest that progerin effects on the Ran system and protein import are transduced through oxidative stress, we also find that disruption of the Ran gradient is sufficient to induce ROS and inhibit Ubc9 import. We propose that oxidative stress induced by progerin disrupts the Ran gradient and nuclear transport of proteins such as Ubc9 that are highly sensitive to perturbation of Ran distribution. Because Ran gradient disruption is sufficient to increase ROS production irrespective of progerin expression, we suggest that the Ran system and Ubc9 are functionally coupled to the structure of the nuclear lamina and ROS production through feedback loops.
RESULTS
In previous work we showed that Ubc9, the single SUMO E2 in the cell, is mislocalized from the nucleus to the cytoplasm in fibroblasts from HGPS patients expressing the progerin form of lamin A (Kelley et al., 2011). Immunolocalization and biochemical studies from other groups show that there are two pools of Ubc9—a soluble pool that shuttles between the cytoplasm and nucleoplasm, and an insoluble pool associated with RanBP2 at the NPC (Saitoh et al., 1997, 2002). Transient transfection of hemagglutinin (HA)-progerin in HeLa cells caused significant reduction in the nucleoplasmic level of Ubc9 without a major effect on the NPC-associated pool (Figure 1, A–C). Depleting Nup358, the major NPC binding partner for Ubc9, did not appear to affect the nucleoplasmic level of Ubc9 (Supplemental Figure S1, A and B). Progerin therefore affects the pool of Ubc9 undergoing exchange between the nucleus and the cytoplasm, which we set out to investigate using cell biological analyses.
FIGURE 1:

Progerin inhibits nuclear import of Ubc9 through a dominant-negative effect on the nucleus. (A) Endogenous Ubc9 distribution (red) in cells transfected with HA-lamin A and HA-progerin (green). (B) Histogram of Ubc9 N/C (bin size, 0.2) from HA-lamin A (black) and HA-progerin (red). (C) Histogram of Ubc9 fluorescence measured at the nuclear envelope (bin size, 25) in HA-lamin A–transfected (black) and HA-progerin–transfected (red) cells. (D) Heterokaryon cell fusion using HeLa cells transfected with HA-progerin (green) or cyan fluorescent protein (blue). Endogenous Ubc9 (red) was also detected. Of 20 heterokaryons examined, 16 displayed the phenotype shown. (E) Heterokaryon cell fusion using Cos cells transfected with HA-progerin (red). Ran is shown in green. Of 23 heterokaryons examined, 21 displayed the phenotype shown. Scale bars, 20 μm (A) and 10 μm (D, E).
To test whether progerin inhibits Ubc9 import by cytoplasmic retention, we performed heterokaryon assays and analyzed the distribution of Ubc9 by immunofluorescence (IF) microscopy. HeLa cells transfected with HA-progerin were fused with HeLa cells transfected with cyan fluorescent protein, the latter protein (blue) marking the shared cytoplasm within the heterokaryon. Ubc9 was nuclear localized in control nuclei but was not imported into nuclei expressing progerin (Figure 1D). Similarly, a Flag-tagged Ubc9 coexpressed with HA-progerin was imported into control nuclei but was not imported into nuclei expressing progerin (Supplemental Figure S1C). Because Flag-tagged Ubc9 (translated in progerin-expressing cells before fusion) underwent import into control nuclei, we conclude that progerin does not promote cytoplasmic retention or inactivate the import signal on Ubc9. Progerin-mediated disruption of the Ran protein gradient was maintained in progerin-expressing nuclei in heterokaryons, but there was no evidence of Ran defects in control nuclei within heterokaryons (Figure 1E). Our results suggest that progerin disrupts Ubc9 import without affecting the transport competence of Ubc9, and the defect is a feature of the nucleus and not the cytoplasm. Similarly, under the conditions of our assays the dominant-negative mechanism responsible for the Ran distribution defect is not communicated through or rescued by the shared cytoplasm of the heterokaryon.
Nuclear import of Ubc9 is mediated by importin13, a Ran-regulated import receptor that is a member of the importin-β superfamily (Mingot et al., 2001). We reasoned that the Ubc9 import defect induced by progerin is a consequence of Ran gradient disruption, which could reduce import by lowering the efficiency of Ran-dependent import complex disassembly in the nucleoplasm. To test this idea, we used small interfering RNA (siRNA) to deplete the Ran import factor, NTF2. Depletion of NTF2 resulted in a significant reduction in the nuclear concentration of Ran and Ubc9 (Figure 2, A and B). A small reduction in nuclear localization of green fluorescent protein (GFP)–streptavidin (STV)–nuclear localization signal (NLS) was detected in cells in which the Ran gradient was disrupted by NTF2 depletion (Figure 2C), consistent with our recent finding that SV40 NLS–dependent import is relatively efficient even under conditions in which the nuclear concentration of Ran is reduced (Snow et al., 2013). By double-label IF microscopy, we observed correlations between Ubc9 and Ran N/C values in cells treated with control and NTF2 siRNA (Pearson r = 0.4 for siCon and 0.6 for siNTF2; Figure 2D), an indication that subcellular distribution of these proteins is linked. Our data suggest that Ran gradient disruption is sufficient to explain the Ubc9 import defect in cells expressing progerin.
FIGURE 2:

Ubc9 requires the Ran protein gradient for efficient import. (A) Disruption of the Ran gradient by siRNA depletion of NTF2 in GSN2 cells. HeLa cells (stably transfected with GFP-STV-NLS; Black et al., 1999) were cotransfected with siRNA and Flag-Ubc9 and imaged in three channels for endogenous Ran (purple), Flag-Ubc9 (red), and GFP (green) Scale bars, 20 μm. (B) Histogram (bin size, 0.25) showing Flag-Ubc9 N/C from control siRNA (black) and NTF2 siRNA–transfected (red line) cells. (C) Histogram (bin size, 25) showing nuclear fluorescence intensities of GFP-STV-NLS from control (black) and NTF2 siRNA–transfected (red) cells. (D) xy scatter plot of Ubc9 N/C and Ran N/C in control siRNA (black dots) and NTF2 siRNA (red dots) cells.
It was reported that fibroblasts from four HGPS patients (HGADFN167, HGADFN003, AG11513, AG06297) have elevated ROS (Viteri et al., 2010; Pekovic et al., 2011; Richards et al., 2011) and increased protein carbonyl content (Viteri et al., 2010; Richards et al., 2011). Because oxidative stress can inhibit nuclear transport (Stochaj et al., 2000; Miyamoto et al., 2004; Czubryt et al., 2000), we explored whether elevated ROS is potentially related to the Ran distribution changes that we observed in HGPS cells. We measured ROS levels in three commonly used HGPS lines (3199, 1498, 1972) using the fluorescent indicator dichlorofluoroscein diacetate (DCF-DA) and found that all three cell lines showed elevated ROS (Figure 3, A and B). These lines also showed reduced mitochondrial membrane polarization, which could be related to the increase in ROS levels in these cells (Supplemental Figure S2). The fact that seven of seven HGPS lines examined have elevated ROS suggests that oxidative stress might be a common feature of patient-derived HGPS fibroblasts.
FIGURE 3:

Fibroblasts from HGPS patients have elevated ROS. (A) ROS detected in primary fibroblasts from control (8469) and progeria patients (1972, 1498, and 3199) using DCF. (B) Histograms comparing total ROS levels in control (black bars) and progeria patient fibroblasts (red lines). Bin size, 10. Scale bars, 20 μm.
We considered a simple model in which progerin-induced oxidative stress causes disruption of the Ran gradient, which in turn inhibits Ubc9 import. Effects of oxidative stress on nuclear transport have been described and can be induced artificially by exposing cells to H2O2 (Miyamoto et al., 2004; Czubryt et al., 2000; Stochaj et al., 2000). We found that a brief treatment of HeLa cells with H2O2 (50–400 μM, 10 min) was sufficient to markedly reduce the nuclear concentration of Ubc9 and Ran (Figure 4, A–C). The effect of H2O2 on Ubc9 and Ran was reversible, as the nuclear localization of both proteins recovered within 60 min of washout (Figure 4B). The inhibitory effect of H2O2 on nuclear transport was rapid, as a ∼50% reduction in the N/C of both Ran and Ubc9 was observed after a 2-min exposure to H2O2 (Figure 4, D and E). From these data we conclude that localization of Ran and Ubc9 can change in response to oxidative stress.
FIGURE 4:

The Ran gradient and Ubc9 import are sensitive to oxidative stress. (A) Endogenous Ubc9 distribution detected by IF in HeLa cells treated for 10 min with the indicated concentrations of H2O2. (B) Endogenous Ubc9 (red) and Ran (green) in HeLa cells undergo reversible changes in distribution in response to oxidative stress. HeLa cells were treated for 10 min with 200 μM H2O2 and then allowed to recover in fresh media for 30 and 60 min. (C) xy scatter plot of Ubc9 N/C and Ran N/C in untreated (black dots) and H2O2-treated HeLa cells (red dots). (D, E) Ubc9 and Ran N/C levels in HeLa cells treated with 200 μM H2O2 measured as a function of time. (F) Ubc9-Uba2 heterodimer detected by immunoblotting. HeLa cells were treated for 10 min with the indicated concentrations of H2O2 and immunoblotted for Ubc9. Gel samples were prepared without or with 100 mM dithiothreitol, as indicated. Scale bars, 20 μm.
The Melchior group showed that Ubc9 contains a catalytic cysteine that can be oxidized by treating cells with H2O2 (Bossis and Melchior, 2006). In response to oxidative stress, cysteine 93 in Ubc9 forms a disulfide with the catalytic cysteine in the E1 (Bossis and Melchior, 2006). In gel samples prepared without reducing agent, the Ubc9-Uba2 disulfide product can be detected from HeLa cells treated with 50–400 μM H2O2 (Figure 4F). Ubc9 disulfide formation with the E1 is predicted to increase the apparent size of the Ubc9 import cargo from 18 kDa (Ubc9) to ∼130 kDa (Ubc9 + Uba2 + AOS1). Several groups, including ours, have observed that large NLS cargoes require a higher concentration of nuclear Ran for efficient import (Lyman et al., 2002; Ribbeck and Görlich, 2002; Snow et al., 2013). We posited that disulfide formation between Ubc9 and E1 might contribute to the nuclear import defect of Ubc9, either by increasing its size or possibly obscuring the Ubc9 NLS. To test this idea, we used a form of Ubc9 that contains a serine substitution for the catalytic cysteine (C93S), which thereby abrogates disulfide formation with Uba2 (Bossis and Melchior, 2006). We found that Flag-Ubc9-C93S underwent nuclear import similar to Flag-Ubc9 and that both forms of Ubc9 localized to the cytoplasm in cells treated with H2O2 (Figure 5A). In addition, in the presence of cotransfected progerin, both forms of Ubc9 underwent redistribution to the cytoplasm (Figure 5, B–D). These data indicate that Ubc9 localization defects that occur in response to H2O2 and progerin are independent of Ubc9 disulfide formation with Uba2. Consistent with this conclusion, immunoblotting HGPS-patient fibroblast samples under nonreducing conditions gave no evidence for Ubc9 disulfide formation (Figure 5E). The Ubc9 disulfide with Uba2 was clearly inducible by H2O2 treatment of normal fibroblasts (8469), showing that the product can be formed and detected in this cell type (Figure 5E).
FIGURE 5:

Oxidative stress and progerin inhibit Ubc9 import by a mechanism that is independent of Ubc9 cysteine 93 oxidation. (A) Localization of Flag-Ubc9 (WT and C93S mutant; green) in HeLa cells treated with 200 μM H2O2 for 10 min. (B) Localization of Flag-Ubc9 (WT and C93S mutant; green) in HeLa cells cotransfected with HA-lamin A or HA-progerin (red). Scale bars, 10 μm. (C, D) Histograms showing N/C values (bin size, 0.5) of Flag-Ubc9 and Flag-Ubc9-C93S in cells expressing HA-lamin A (black) and HA-progerin (red). (E) Immunoblotting for the Ubc9-Uba2 disulfide in control (8469) and HGPS (1972, 1498, and 3199) fibroblasts. The electrophoretic position of the Ubc9-Uba2 disulfide (arrow) and a nonspecific cross-reaction (asterisk) are indicated. Note that the Ubc9 C93S mutant was expressed at a low level in these experiments to avoid dominant-negative effects on nuclear SUMOylation sufficient to disrupt the Ran gradient (Kelley et al., 2011).
The results obtained with Ubc9-C93S (Figure 5) indicated that the Ubc9 import defect in progerin-expressing cells is not a consequence of Ubc9 oxidation. However, given the elevated ROS in HGPS cells and the rapid, reversible disruption of Ran and Ubc9 distribution by H2O2, the possibility remained that the Ran gradient disruption by progerin is mediated by oxidative stress. To further study the effects of progerin on Ran that might be mediated by oxidative stress, we used an approach based on the drug lopinavir (LPV). LPV is used clinically to inhibit the HIV protease, but it also inhibits Zmpste24, the protease responsible for proteolytic processing of pre–lamin A (Caron et al., 2007; Coffinier et al., 2007; Lefèvre et al., 2010). Treating cultured cells with LPV mimics the effects of progerin expression because it results in the accumulation of unprocessed, nuclear membrane–tethered pre–lamin A and disruption of the Ran gradient (Kelley et al., 2011). Although transient transfection of progerin in HeLa cells appears to recapitulate phenotypes detected in HGPS fibroblasts, with LPV it is possible to test the effect of endogenous levels of membrane-tethered pre–lamin A in the setting of normal fibroblasts as a function of time. Using an antibody specific for pre–lamin A, we found that LPV treatment of normal (8469) human fibroblasts for 72 h resulted in the accumulation of the unprocessed protein (Figure 6A). LPV treatment elevated ROS levels in normal human fibroblasts to about the same extent as 20 μM H2O2 (Supplemental Figure S3, A, B, and D), and the increase in ROS levels was prevented by cotreating the cells with siRNA to lamin A (Figure 6, A and B). By IF microscopy, we observed that LPV treatment resulted in the appearance of pre–lamin A at the nuclear envelope and disruption of the Ran gradient and that both changes were prevented by treating cells with lamin A siRNA (Figure 6, C and D). LPV treatment also reduced the nuclear levels of Ubc9 (Supplemental Figure S3C). These data establish that LPV increases ROS levels and disrupts the Ran gradient by promoting pre–lamin A accumulation.
FIGURE 6:

Blocking pre–lamin A processing with LPV disrupts the Ran gradient and induces ROS. (A) Immunoblotting for pre–lamin A, lamin A, and tubulin (loading control) in control and lamin A siRNA–transfected normal (8469) human fibroblasts treated with and without LPV. (B) Bar graph depicting the mean DCF fluorescence in control and lamin A siRNA–transfected cells with and without LPV treatment. (C) IF detection of pre–lamin A (red) and Ran (green) in control and lamin A siRNA–transfected cells with or without LPV treatment. Scale bars, 20 μm. (D) Bar graph depicting mean Ran N/C in control and lamin A siRNA–transfected cells with and without LPV treatment. Statistical significance as measured by ANOVA with a Tukey posttest: ***p < 0.001.
We performed a time course of LPV treatment to examine the relationship between the appearance of pre–lamin A, ROS levels, and Ran distribution. On immunoblotting of LPV-treated cells, pre–lamin A protein was detected at 12 h, and pre–lamin A levels increased further during the 72-h time course (Figure 7A). ROS levels showed a small but statistically significant elevation at the 12-h time point but returned to control (dimethyl sulfoxide [DMSO]) levels by 18 h and remained low at the 24-, 36-, and 48-h time points (Figure 7A). A relatively large increase in ROS (greater than twofold compared with DMSO control) was observed at the 72-h time point (Figure 7B). Because ROS measurement by DCF-DA staining is a live-cell assay, parallel samples were fixed and processed for IF microscopy to determine Ran localization. Treating cells with LPV for 12 h resulted in a small reduction in the Ran N/C, an effect that became more pronounced at each time point (Figure 7, C and D). Thus LPV induction of pre–lamin A causes a small increase in ROS detected at 12 h and a large increase in ROS detected at 72 h. We also determined that LPV reduces the ratio of reduced to oxidized glutathione (GSH/GSSG; p < 0.0001), which provides an additional assessment of oxidative stress effects of the drug (Figure 7E). LPV has been reported to have off-target inhibitory effects on mitochondria (Deng et al., 2010), which we considered might affect the Ran gradient by reducing ATP levels. We found that ATP levels did not change significantly during a time course of LPV treatment during which the Ran gradient was progressively disrupted (Supplemental Figure S4, A and B).
FIGURE 7:

Time course of LPV-induced pre–lamin A accumulation, disruption of the Ran gradient, and ROS induction. (A) Immunoblot showing the accumulation of prelamin A upon treatment of 8469 fibroblasts with 40 μM LPV at the indicated time points. Ran levels are unchanged. Tubulin represents the loading control. (B) Bar graph comparing mean DCF fluorescence at different time points of LPV treatment. Gray and black bars represent DMSO and LPV samples, respectively. (C) Endogenous Ran distribution (green) at the indicated time points of LPV treatment detected by immunofluorescence. Scale bars, 50 μm. (D) Bar graph representing Ran N/C ratios at the indicated time points of DMSO (gray bars) and LPV (black bars) treatment. Statistical significance as measured by ANOVA with a Tukey posttest: ***p < 0.001. (E) Total glutathione/oxidized glutathione ratio from 8469 fibroblasts treated with DMSO or 40 μM LPV for 24 h.
Although there is a transient ROS increase at 12 h, the major change in ROS appears after the Ran gradient has been disrupted for >48 h (Figure 7). To test the hypothesis that Ran gradient disruption might be upstream of ROS induction, we depleted the Ran import factor NTF2 (Kelley et al., 2011) and measured the effects on Ran distribution and ROS levels. By IF microscopy, NTF2 depletion resulted in a predominantly cytoplasmic localization of Ran, indicating robust disruption of the Ran gradient (Figure 8A). Significantly, disruption of the Ran gradient increased ROS levels as detected with DCF-DA (Figure 8B). Because Ran gradient disruption is sufficient to induce ROS, the large ROS induction observed with 72 h of LPV treatment could be a consequence of Ran gradient disruption. A similar increase in ROS was observed using siRNA to Ran (Figure 8D). Thus ROS induction in these assays is probably due to reduced concentration of Ran in the nucleus and not increased concentration of Ran in the cytoplasm.
FIGURE 8:

Disruption of the Ran protein gradient is sufficient to induce ROS. (A) Disruption of the Ran gradient by siRNA depletion of NTF2 in 8469 cells (5 d), followed by IF microscopy detection of Ran distribution. Scale bars, 20 μm. (B) ROS detection in 8469 fibroblasts treated with control and NTF2 siRNA, showing an increase in levels upon NTF2 knockdown. Scale bars, 100 μm. (C) Histogram depicting an increase in mean DCF fluorescence intensity upon NTF2 knockdown (red lines) in comparison to control siRNA–treated cells (black bars). (D) ROS detection in 8469 fibroblasts treated with control and Ran siRNA. (E) Histogram depicting an increase in mean DCF fluorescence intensity upon Ran knockdown (red lines) in comparison to control (black bars). Bin sizes, 10.
As an additional means to query the relationship between progerin, oxidative stress, and the Ran system, we took advantage of a stress-resistant CHO cell line (OC-14) that was produced by continuous culture in the presence of H2O2 (Spitz et al., 1988a, b, 1990). Comparison of the parental CHO cell line (HA-1) with the resistant line (OC-14) showed that the latter maintains a nuclear distribution of Ubc9 and Ran at 400 μM H2O2 (Figure 9A). The OC-14 line was resistant to formation of the Ubc9-E1 disulfide product, even at the highest concentration of H2O2 tested (Figure 9, B and C). The OC-14 line was also resistant to Ran and Ubc9 localization defects normally caused by progerin expression (Figure 9, D–F). Although the gene expression changes associated with the development of H2O2 resistance in the OC-14 cells are probably complex, the data suggest that progerin disruption of the Ran gradient and Ubc9 distribution in CHO cells involves changes mediated by oxidative stress. These results, together with the other data in this study, suggest that the Ran protein gradient can be disrupted by oxidative stress and that disruption of the Ran gradient can generate oxidative stress (Figure 10).
FIGURE 9:

Nuclear localization of Ubc9 is resistant to the effects of H2O2 and progerin in CHO cells preadapted to oxidative stress. (A) Cells were treated with the indicated concentrations of H2O2 for 10 min, and endogenous Ubc9 and Ran were detected by IF. Scale bars, 20 μm. (B, C) Ubc9-Uba2 disulfide formation in HA-1 and OC14 cells. Cells treated with different concentrations of H2O2 for 10 min were analyzed by immunoblotting under nonreducing conditions. The electrophoretic position of the Ubc9-Uba2 disulfide (arrow) and a nonspecific cross-reaction (asterisk) are indicated. (D) Transfection of HA-lamin and HA-progerin into CHO cell lines, followed by detection of endogenous Ubc9 and Ran by IF. Lamin A and progerin expression was detected with anti-HA antibody. (E, F) Histograms (bin size, 0.5) showing Ran N/C and Ubc9 N/C in the HA-1(parental) and OC-14(resistant) cells transfected with progerin. Scale bars, 20 μm.
FIGURE 10:
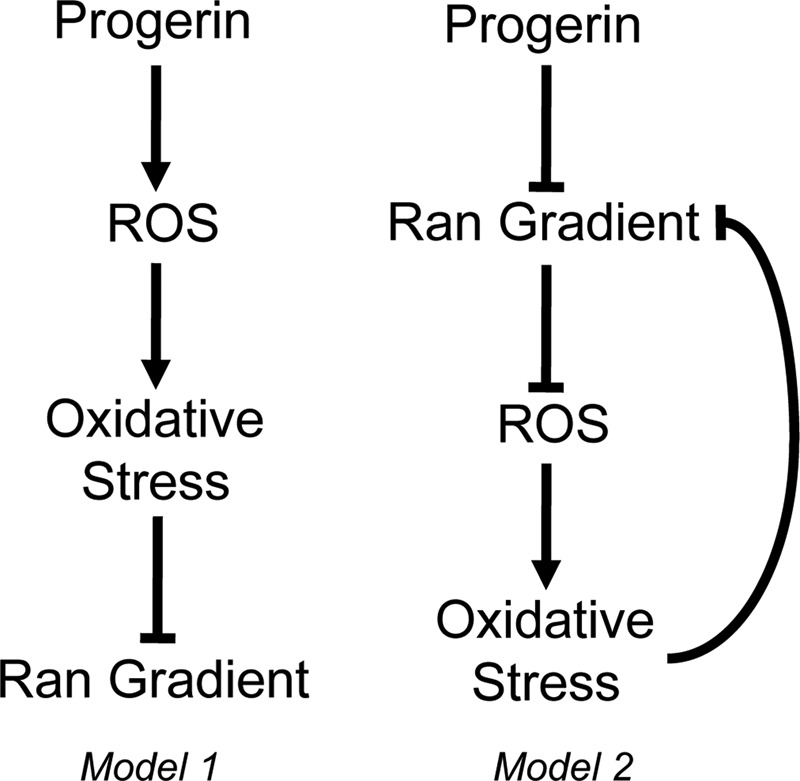
Models for how progerin could affect the Ran gradient in interphase cells. In model 1, progerin induces ROS, which generates oxidative stress in the cell and disrupts the Ran gradient. In model 2, progerin disrupts the N/C distribution of Ran, which induces ROS and oxidative stress. Because oxidative stress is sufficient to disrupt the Ran system, a negative feedback loop is predicted to further reduce the Ran gradient. The specific source of ROS in these pathways has not been identified.
DISCUSSION
SUMOylation plays key roles in cell cycle regulation, transcription, DNA replication and repair, and chromosome dynamics (Wang and Dasso, 2009). Because these are primarily nuclear events, defective import of Ubc9 is expected to affect multiple pathways in the nucleus. Previously we found that Ubc9 is mislocalized from the nucleus to the cytoplasm in HGPS fibroblasts, a change associated with reduced nuclear SUMO2/3 levels detected by IF microscopy (Kelley et al., 2011). In naive cells, progerin expression and forced expression of SENP catalytic domain both reduced the level of nuclear SUMOylation by SUMO2/3 and reduced the nuclear mobility of the RanGEF, RCC1. Because RCC1 nuclear mobility is related to chromatin-dependent nucleotide exchange on Ran, these data, together with disruption of the Ran protein gradient, were taken as evidence that progerin might mediate its effects on the Ran system via changes in SUMOylation. In support of this model, overexpression of Ubc9 fused to the SV40 NLS rescued progerin effects on the Ran gradient, as well as other cellular defects caused by progerin (Kelley et al., 2011). Although these data placed Ubc9 function upstream of Ran, the fact that Ubc9 undergoes nuclear import via a Ran-regulated import receptor (importin13; Mingot et al., 2001) suggests a positive feedback loop that that links the Ran gradient with nuclear Ubc9 activity.
Work published by other groups (Viteri et al., 2010; Pekovic et al., 2011; Richards et al., 2011) and confirmed in this study indicates that HGPS cells have elevated ROS. Given the connections between oxidative stress signaling and the Ran system (Miyamoto et al., 2004; Czubryt et al., 2000; Stochaj et al., 2000), we explored oxidative stress as a mechanism for transducing progerin's effects to Ran and Ubc9. We showed that acute oxidative stress in the form of H2O2 exerts a rapid and reversible effect on Ran and Ubc9 localization, as both proteins delocalized to the cytoplasm within minutes of H2O2 addition. The factor sensing H2O2 in this setting could be a Ran system or a SUMOylation pathway component. Ubc9 is known to be a direct target of oxidation through its catalytic cysteine (Bossis and Melchior, 2006); however, Ubc9 containing a Cys-to-Ser substitution displayed the same responses to H2O2 and progerin as wild-type (WT) Ubc9. Thus the effect of oxidative stress and progerin on Ubc9 is not mediated through its catalytic cysteine. We did obtain evidence that progerin's effects on Ran and Ubc9 can depend on oxidative stress. Both Ran gradient and Ubc9 import were resistant to progerin transfection in a CHO cell line preadapted to oxidative stress (Spitz et al., 1988a). One of the major adaptations of the CHO line OC-14 is overexpression of catalase, which metabolizes H2O2 (Spitz et al., 1988b). It remains possible that the resistance of the OC-14 line to progerin expression involves gene expression changes that are independent of redox regulation. For example, one could imagine that the progerin resistance of OC-14 cells involves the expression of proteins that regulate lamina structure or are involved in sensing the structural changes in the lamina or nuclear membrane induced by membrane tethering of progerin. Finally, it cannot be assumed that progerin affects all cell types in the same way, which is to say, progerin effects might be transduced through redox effects in one cell type and a redox-independent pathway in another cell type. Patient data and animal models indicate that progerin's effects are cell and tissue specific, with clear manifestation in the cardiovascular system (Varga et al., 2006; Olive et al., 2010). Of interest, ROS signaling in the cardiovascular system has both physiological and pathological contributions (Sugamura and Keaney, 2011), but whether there is any relationship to the effects of progerin is unknown.
We used the drug LPV to examine the temporal relationship between progerin expression, oxidative stress, and changes in Ran and Ubc9. Through its inhibition of Zmpste24, LPV treatment results in pre–lamin A tethering to the nuclear membrane (Coffinier et al., 2007). This provides an inducible system for expressing endogenous levels of unprocessed pre–lamin A, which we showed disrupts the Ran gradient (Kelley et al., 2011). The effect on Ran is strictly dependent on WT lamin A expression, since the effects of LPV were abrogated by lamin A siRNA.
LPV-induced expression of membrane-tethered pre–lamin A elevated ROS levels in normal human fibroblasts in what appears to be a biphasic manner. An increase in ROS was observed after 12 h of LPV treatment, which, after returning to near-baseline (DMSO-treated) levels, was followed by second ROS increase at 72 h posttreatment. There are several possible interpretations of how these ROS changes might relate to the Ran protein gradient and nuclear transport. The ROS induction at 12 h could represent an event that initiates disruption of the Ran gradient through a pre–lamin A effect on gene expression (Figure 10, model 1). The ROS induction at 12 h, however, is transient, and ROS levels remained near baseline during a time course in which the Ran gradient was highly disrupted. Thus an alternative interpretation is that the pre–lamin A induction of ROS at 12 h is inconsequential for the Ran gradient, which is instead disrupted by changes in nuclear architecture and chromatin (Figure 10, model 2). According to this view, chronic disruption of the Ran gradient ultimately results in a large increase in ROS at 72 h. Consistent with this explanation is our finding that disruption of the Ran gradient by depletion of the Ran import factor NTF2, or depletion of Ran itself, was sufficient to induce ROS. Oxidative stress generated by Ran disruption could generate a negative feedback loop that exacerbates the phenotype (Figure 10, model 2). Ran depletion in human dermal fibroblasts increases levels of MnSOD (Nagai and Yoneda, 2012), an enzyme known to be induced by oxidative stress. In the same study it was shown that Ran disruption is sufficient to trigger cell senescence (Nagai and Yoneda, 2012). Thus it is plausible that the nuclear concentration of Ran is sensed by a pathway that restrains the induction of ROS and development of cell senescence.
The mechanism linking Ran levels and ROS formation is unknown, but it is fair to speculate that it probably involves nuclear transport that impinges on expression of genes affecting cell redox state, or structural changes in the nuclear envelope that can affect enzymes through scenarios that do not require new protein synthesis. For example, the subset of NADPH oxidases reported to reside in the nuclear membrane (Leto et al., 2009) might respond to changes in the lamina and nuclear Ran, resulting in ROS production. Lamin A encodes several cysteines that can be oxidized in response to stress, implying the lamina itself could sense ROS changes and help transduce the effects to nuclear architecture and chromatin (Pekovic et al., 2011). Defining how Ran controls the activity and localization of factors responsible for ROS production and metabolism should help explain how the nuclear lamina contributes to cell redox state and how Ran system changes might contribute to the pathology in HGPS.
MATERIALS AND METHODS
Cell culture and drug treatment
Primary human fibroblasts from HGPS patients (AGO1972, AG11498, and AGO3199) and a clinically normal father (AGO8469) of an HGPS patient were obtained from the Coriell Cell Repository (Camden, NJ). These are designated HGPS 1972, HGPS 1498, HGPS 3199, and Normal 8469, respectively. Primary fibroblasts were grown at 37°C in 5% CO2 in MEM (Life Technologies, Carlsbad, CA) containing 15% fetal bovine serum (FBS; Hyclone, South Logan, UT), 1% MEM vitamin solution (HyClone), 1% penicillin/streptomycin (Life Technologies), and 1 mM l-glutamine (Life Technologies). The passage number for cells in various experiments was between 8 and 25. HeLa and GSN2 cells (HeLa cells stably transfected with GFP-STV-SV40 NLS; Black et al., 1999) were grown at 37°C in 5% CO2 in DMEM (Life Technologies/Invitrogen) containing 10% FBS (Atlanta Biologicals, Flowery Branch, GA). Cos7 cells were grown in DMEM with 10% FBS. CHO cells (HA-1 and OC-14) were generously provided by Douglas Spitz (University of Iowa; Spitz et al., 1988a). They were grown at 37°C in 5% CO2 in MEM (Life Technologies) supplemented with 15% FBS (Atlanta Biologicals) and 1% penicillin/streptomycin (Life Technologies). H2O2 was purchased from Sigma-Aldrich (St. Louis, MO; catalog no. H-1009) and used at concentrations and time points indicated. Lopinavir was obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (Germantown, MD; catalog no. 9481; a gift from D. Rekosh, University of Virginia) and Selleckchem (Houston, TX; catalog no. S-1380). Normal 8469 fibroblasts were treated with 40 μM lopinavir dissolved in DMSO for 3 d or the indicated time points and processed for immunoblotting, IF microscopy, ROS measurements, or ATP measurements.
Plasmids, siRNA, and transfection
Plasmids encoding the WT and mutant forms of lamin A (pCDNA3-HA-lamin A and pCDNA3-HA-progerin, respectively) were generated as described (Kelley et al., 2011). pCDNA3-HA-progerin C611S mutant was engineered as described (Snow et al., 2013). pCMV5-FLAG-Ubc9 and pCMV5-FLAG-Ubc9-C93S were generously provided by David Wotton, University of Virginia. siRNA against NTF2 was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Stealth siRNA against lamin A (LMNA stealth HSS106094) was purchased from Invitrogen (Carlsbad, CA; catalog no. 1299001), as was siRNA against Nup358/RanBP2 (catalog no. 10620310). siRNA sequence against Ran (5′-GUGAAUUUGAGAAGAAGUATT-3′; Nagai and Yoneda, 2012) was purchased from Invitrogen.
The cells to be transfected with plasmids were plated and grown in six-well plates to a density of ∼60–80% for 24 h, followed by transfection with Transfectin (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. Transfected cells were processed for immunofluorescence microscopy or immunoblotting after 24 h. NTF2 siRNA was transfected by using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions for reverse transfection. GSN2 cells were plated on NTF2 siRNA in 60-mm dishes, split onto glass coverslips at 24 h posttransfection, transfected with pCMV-Flag-Ubc9 24 h postsplitting onto coverslips, and grown for an additional 24 h before processing for IF microscopy. For NTF2 knockdown in normal human (8469) fibroblasts, cells were transfected with NTF2 siRNA by reverse transfection directly on coverslips for immunofluorescence or glass-bottom dishes (MatTek, Ashland, MD) for ROS measurements and allowed to grow for 72 h before processing. For lamin A knockdown in 8469 fibroblasts, cells were transfected with lamin A siRNA by the reverse transfection protocol, followed by LPV treatment after 24 h. The cells were allowed to grow for an additional 72 h before processing for immunoblotting, IF, or ROS measurements.
Heterokaryon cell fusion
For the heterokaryon assays, Cos7 cells were left untransfected or transfected using FuGENE reagent (Promega, Madison, WI) 24 h after plating, as per the requirements of the experiment. After 24 h the donor and acceptor cells were coseeded on coverslips, and after being allowed to grow for an additional 24 h, cells were washed with prewarmed phosphate-buffered saline (PBS) and fused with 50% polyethylene glycol (Roche, Basel, Switzerland). After an additional wash step, cells were incubated in 10 μg/ml cyclohexamide for 30 min or 2 h in a 37°C incubator. The cells were then fixed and processed for IF microscopy. Heterokaryons containing two nuclei per cell were visualized.
ATP measurement
ATP measurements were done using an ATPlite luminescence ATP detection assay system (6016943; PerkinElmer, Waltham, MA) as per manufacturer's protocols. Normal 8469 fibroblasts were plated in 96-well plates in triplicate, and DMSO or LPV was added as indicated in the Figure legends. On the day of processing, simultaneous with ATP measurement, cells from an identical experiment were counted by trypan blue exclusion technique on a hemocytometer, and the luminescence intensities were normalized to the cell numbers so obtained. In the same experiment, IF detection of Ran gradient disruption was performed to verify the efficacy of the LPV treatment.
Antibodies
The following antibodies were used: Ran (mouse monoclonal antibody [mAb], 610341; BD Biosciences, San Jose, CA), lamin A (rabbit polyclonal antibody [pAb], PRB-113c; Covance, Princeton, NJ), Ubc9 (rabbit pAb, ab33044; Abcam), anti-Flag epitope (mouse mAb M2; Sigma-Aldrich), OctA anti-Flag (rabbit pAb; Santa Cruz Biotechnology), RanGAP (mouse MAb 21c7; Zymed, Carlsbad, CA), anti-HA (mouse mAb 16B12 and rabbit pAb Y-11; both from Santa Cruz Biotechnology), pre–lamin A (goat pAb, sc-6214; Santa Cruz Biotechnology), and tubulin (Mouse mAb 1-A2; Sigma-Aldrich).
Immunoblotting
Cells were lysed using standard SDS–PAGE loading buffer. Extracts were sonicated before loading on SDS-polyacrylamide gels. SDS–PAGE and immunoblotting were performed by standard methods using primary antibodies and peroxidase-labeled secondary antibodies and detected by chemiluminescence using ECL reagent. For the immunodetection of Ubc9-Uba2 thioester, cells were lysed in standard SDS–PAGE loading buffer containing 20 mM N-ethylmaleimide, 20 mM iodoacetamide, and protease inhibitors. 100 mM dithiothreitol was added wherever indicated in the figures.
Immunofluorescence microscopy
Cells growing on glass coverslips were washed three times in PBS, fixed with 3.7% formaldehyde for 20 min, washed three times in PBS again, and permeabilized in 0.2% Triton X-100 for 5 min. Coverslips were blocked in IF microscopy blocking buffer (1× PBS, 2% bovine serum albumin, 2% FBS) for 1 h or overnight for Ubc9. Coverslips were then incubated in primary antibody diluted in IF microscopy blocking buffer overnight at 4°C or at room temperature in the case of Ubc9. The secondary antibodies for IF microscopy were diluted in blocking buffer and incubated for 1 h. The antibodies used were fluorescein isothiocyanate (FITC)–labeled donkey anti-mouse, Cy3-labeled donkey anti-mouse, FITC-labeled donkey anti-rabbit, FITC-labeled donkey anti-goat, and Cy3-labeled donkey anti-rabbit antibodies (all from Jackson ImmunoResearch, West Grove, PA). Wide-field microscopy was performed with a Nikon Eclipse E800 upright microscope using a 40×/1.0 numerical aperture (NA) oil immersion objective and recorded with a Hamamatsu C4742-95 charge-coupled device camera using OpenLab software (PerkinElmer). Confocal imaging was done on a Zeiss LSM 510 microscope with 40 and 100×/1.3 NA oil immersion objectives using AxioVision software (Carl Zeiss, Jena, Germany) and on a Zeiss LSM 710 microscope with 20× regular and 63× oil immersion objectives using ZEN software (Carl Zeiss).
ROS measurement
DCF-DA was purchased from Sigma-Aldrich (catalog no. 35845) and a stock solution of 5 mM made in ethanol. Cells growing in glass-bottom dishes (MatTek) were washed three times with serum-free MEM (Life Technologies) to remove traces of serum and incubated with a 1:2000 dilution of stock in serum-free MEM for 30 min at 37°C and 5% CO2. The cells were then washed and mounted in RPMI buffered medium (Life Technologies) followed by visualization of green fluorescence at an excitation wavelength of 488 nm on a Zeiss LSM 710 confocal microscope equipped with a stage warmer with a 10× objective using ZEN software (Carl Zeiss).
Glutathione measurement
GSH/GSSG ratios were determined using a GSH/GSSG-Glo Assay Kit from Promega (Madison, WI). Fibroblasts were plated in 96-well plates (white) at a density of 5000 cells/well. Cells were treated with DMSO or LPV 24 h after plating. Cells were assayed 24 h after treatment according to the manufacturer's instructions, and luminescence was detected using a BioTek (Winooski, VT) Synergy HT Multi-Mode Microplate Reader.
Mitochondrial membrane potential measurement
JC-1 was purchased from Invitrogen (catalog no. T-3168), and a 5 mg/ml stock solution was made in DMSO. Cells growing in glass-bottom dishes (MatTek) were washed three times with regular HGPS medium (see Cell culture and drug treatment) and incubated with 0.3 μg/ml JC-1 in HGPS medium for 1 h at 37°C and 5% CO2. The cells were then washed and mounted in RPMI buffered medium (Life Technologies), followed by visualization on a Zeiss LSM 510-META confocal microscope equipped with a stage warmer with a 40× objective using AxioVision software. JC-1 is a fluorescent dye that accumulates in mitochondria in monomeric form and emits green fluorescence. It uses the mitochondrial potential to form fluorescent J aggregates, which emit red fluorescence (Invitrogen). JC-1 was excited at 488 nm, and both the monomeric and J-aggregate forms of JC-1 were simultaneously visualized using 505- to 530-nm (green) and 585- to 615-nm (red) long-pass filters, respectively. The ratio of red to green fluorescence was quantified as a readout for polarized/total mitochondria.
Image quantification and statistical analyses
Quantitative analysis of IF microscopy images was performed using ImageJ image processing software (National Institutes of Health, Bethesda, MD). For most of the figures >50 cells each from at least three experiments were quantified by circling relevant areas, namely the nucleus and the cytoplasm, measuring the mean fluorescence intensities, and representing the data as mean N/C ratio. For Ubc9 rim quantification (Figure 1C), mean Ubc9 fluorescence intensity was calculated for five uniformly spread selections along the rim, and their average values were plotted. For ROS measurements, cells were outlined and mean DCF fluorescence intensities for the whole cell were plotted. For measurement of mitochondrial potential using JC-1, individual cells were outlined, and the ratio of mean red to green fluorescence intensities for each cell was plotted. For all IF data, a minimum of 50 cells was quantified. Data were binned using Excel (Microsoft) before plotting as histograms, with bin sizes stated in the figure legends, or were represented as bar graphs with error bars using SDs. The “t test: two sample assuming equal variances” function of Excel was used to calculate p values in Figures 1, B and C, 2, B and C, 5, C– E, 8E, and 9, E and F, and Supplemental Figures S1C and S3B, and the type 2 two-tailed p value was used. An analysis of variance (ANOVA) test (Prism; GraphPad) was used to analyze Figures 3B, 6, B and D, and 7, B and D and Supplemental Figures S2 and S4, A and B. To determine statistical significance, Dunnett's multiple comparisons posttest (which compares all conditions to the control condition) was used for Figure 3B and Supplemental Figures S2 and S4B. For the remaining ANOVA tests Tukey's multiple comparison test was used. In all figures, *p < 0.05, **p < 0.01, and ***p < 0.001.
Supplementary Material
Acknowledgments
We thank Douglas Spitz for providing the CHO cell lines and David Wotton for plasmids and valuable input during the study. We also thank Joshua Kelley and Mandovi Chatterjee for helpful discussions during the course of the project and comments on the manuscript. These studies were supported by the National Institutes of Health and a Wagner Fellowship at the University of Virginia.
Abbreviations used:
- DCF-DA
dichlorofluoroscein diacetate
- DMSO
dimethyl sulfoxide
- HGPS
Hutchinson–Gilford progeria syndrome
- LPV
lopinavir
- N/C
ratio of mean nuclear fluorescence to mean cytoplasmic fluorescence
- NPC
nuclear pore complex
- ROS
reactive oxygen species
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-07-0430) on February 12, 2014.
REFERENCES
- Bischoff F, Klebe C, Kretschmer J, Wittinghofer A, Ponstingl H. RanGAP1 induces GTPase activity of nuclear Ras-related Ran. Proc Natl Acad Sci USA. 1994;91:2587–2591. doi: 10.1073/pnas.91.7.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff F, Ponstingl H. Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator RCC 1. Nature. 1991;354:80–82. doi: 10.1038/354080a0. [DOI] [PubMed] [Google Scholar]
- Black BE, Levesque L, Holaska JM, Wood TC, Paschal BM. Identification of an NTF2-related factor that binds Ran-GTP and regulates nuclear protein export. Mol Cell Biol. 1999;19:8616. doi: 10.1128/mcb.19.12.8616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossis G, Melchior F. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol Cell. 2006;21:349–357. doi: 10.1016/j.molcel.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Caron M, Auclair M, Donadille B, Bereziat V, Guerci B, Laville M, Narbonne H, Bodemer C, Lascols O, Capeau J. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007;14:1759–1767. doi: 10.1038/sj.cdd.4402197. [DOI] [PubMed] [Google Scholar]
- Coffinier C, Hudon SE, Farber EA, Chang SY, Hrycyna CA, Young SG, Fong LG. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc Natl Acad Sci USA. 2007;104:13432–13437. doi: 10.1073/pnas.0704212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czubryt MP, Austria JA, Pierce GN. Hydrogen peroxide inhibition of nuclear protein import is mediated by the mitogen-activated protein kinase, ERK2. J Cell Biol. 2000;148:7–16. doi: 10.1083/jcb.148.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Baki L, Yin J, Zhou H, Baumgarten CM. HIV protease inhibitors elicit volume-sensitive Cl− current in cardiac myocytes via mitochondrial ROS. J Mol Cell Cardiol. 2010;49:746–752. doi: 10.1016/j.yjmcc.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055–2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- Eriksson M, Brown W, Gordon L, Glynn M, Singer J, Scott L, Erdos M, Robbins C, Moses T, Berglund P. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Geiss-Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–956. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- Goldman R, Gruenbaum Y, Moir R, Shumaker D, Spann T. Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 2002;16:533–547. doi: 10.1101/gad.960502. [DOI] [PubMed] [Google Scholar]
- Goldman R, Shumaker D, Erdos M, Eriksson M, Goldman A, Gordon L, Gruenbaum Y, Khuon S, Mendez M, Varga R. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görlich D, Seewald M, Ribbeck K. Characterization of Ran-driven cargo transport and the RanGTPase system by kinetic measurements and computer simulation. EMBO J. 2003;22:1088–1100. doi: 10.1093/emboj/cdg113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Hutchison CJ, Worman HJ. A-type lamins: guardians of the soma. Nat Cell Biol. 2004;6:1062–1067. doi: 10.1038/ncb1104-1062. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Kutay U, von Kobbe C, Mattaj IW, Görlich D. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO J. 1997;16:6535–6547. doi: 10.1093/emboj/16.21.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley J, Paschal B. Hyperosmotic stress signaling to the nucleus disrupts the Ran gradient and the production of RanGTP. Mol Biol Cell. 2007;18:4365. doi: 10.1091/mbc.E07-01-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JB, Datta S, Snow CJ, Chatterjee M, Ni L, Spencer A, Yang CS, Cubeñas-Potts C, Matunis MJ, Paschal BM. The defective nuclear lamina in Hutchinson-Gilford progeria syndrome disrupts the nucleocytoplasmic ran gradient and inhibits nuclear localization of Ubc9. Mol Cell Biol. 2011;31:3378–3395. doi: 10.1128/MCB.05087-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodiha M, Chu A, Matusiewicz N, Stochaj U. Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 2004;11:862–874. doi: 10.1038/sj.cdd.4401432. [DOI] [PubMed] [Google Scholar]
- Lefèvre C, Auclair M, Boccara F, Bastard JP, Capeau J, Vigouroux C, Caron-Debarle M. Premature senescence of vascular cells is induced by HIV protease inhibitors implication of prelamin A and reversion by statin. Arterioscler Thromb Vasc Biol. 2010;30:2611–2620. doi: 10.1161/ATVBAHA.110.213603. [DOI] [PubMed] [Google Scholar]
- Leto TL, Morand S, Hurt D, Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal. 2009;11:2607–2619. doi: 10.1089/ars.2009.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman SK, Guan T, Bednenko J, Wodrich H, Gerace L. Influence of cargo size on Ran and energy requirements for nuclear protein import. J Cell Biol. 2002;159:55–67. doi: 10.1083/jcb.200204163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingot J, Kostka S, Kraft R, Hartmann E, Görlich D. Importin 13: a novel mediator of nuclear import and export. EMBO J. 2001;20:3685–3694. doi: 10.1093/emboj/20.14.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto Y, Saiwaki T, Yamashita J, Yasuda Y, Kotera I, Shibata S, Shigeta M, Hiraoka Y, Haraguchi T, Yoneda Y. Cellular stresses induce the nuclear accumulation of importin alpha and cause a conventional nuclear import block. J Cell Biol. 2004;165:617–623. doi: 10.1083/jcb.200312008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounkes L, Kozlov S, Burke B, Stewart CL. The laminopathies: nuclear structure meets disease. Curr Opin Genet Dev. 2003;13:223–230. doi: 10.1016/s0959-437x(03)00058-3. [DOI] [PubMed] [Google Scholar]
- Nagai M, Yoneda Y. Downregulation of the small GTPase Ras-related nuclear protein accelerates cellular aging. Biochim Biophys Acta. 2012;1830:2813–2819. doi: 10.1016/j.bbagen.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Olive M, et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30:2301–2309. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal BM, Gerace L. Identification of NTF2, a cytosolic factor for nuclear import that interacts with nuclear pore complex protein p62. J Cell Biol. 1995;129:925–937. doi: 10.1083/jcb.129.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekovic V, Gibbs-Seymour I, Markiewicz E, Alzoghaibi F, Benham AM, Edwards R, Wenhert M, von Zglinicki T, Hutchison CJ. Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell. 2011;10:1067–1079. doi: 10.1111/j.1474-9726.2011.00750.x. [DOI] [PubMed] [Google Scholar]
- Pemberton L, Paschal B. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic. 2005;6:187–198. doi: 10.1111/j.1600-0854.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- Ribbeck K, Görlich D. The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. EMBO J. 2002;21:2664–2671. doi: 10.1093/emboj/21.11.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards SA, Muter J, Ritchie P, Lattanzi G, Hutchison CJ. The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum Mol Genet. 2011;20:3997–4004. doi: 10.1093/hmg/ddr327. [DOI] [PubMed] [Google Scholar]
- Saitoh H, Pizzi M, Wang J. Perturbation of SUMOlation enzyme Ubc9 by distinct domain within nucleoporin RanBP2/Nup358. J Biol Chem. 2002;277:4755–4763. doi: 10.1074/jbc.M104453200. [DOI] [PubMed] [Google Scholar]
- Saitoh H, Pu R, Cavenagh M, Dasso M. RanBP2 associates with Ubc9p and a modified form of RanGAP1. Proc Natl Acad Sci USA. 1997;94:3736–3741. doi: 10.1073/pnas.94.8.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer E, Florens L, Guan T, Yates J, 3rd, Gerace L. Identification of novel integral membrane proteins of the nuclear envelope with potential disease links using subtractive proteomics. Novartis Found Symp. 2005;264:63–76. [PubMed] [Google Scholar]
- Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci USA. 2006;103:8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. The processing pathway of prelamin A. J Cell Sci. 1994;107:61–67. doi: 10.1242/jcs.107.1.61. [DOI] [PubMed] [Google Scholar]
- Smith A, Brownawell A, Macara IG. Nuclear import of Ran is mediated by the transport factor NTF2. Curr Biol. 1998;8:1403–1406, S1401. doi: 10.1016/s0960-9822(98)00023-2. [DOI] [PubMed] [Google Scholar]
- Snow CJ, Dar A, Dutta A, Kehlenbach RH, Paschal BM. Defective nuclear import of Tpr in progeria reflects the Ran sensitivity of large cargo transport. J Cell Biol. 2013;201:541–557. doi: 10.1083/jcb.201212117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz D, Li G, McCormick M, Sun Y, Oberley L. The isolation and partial characterization of stable H2O2-resistant variants of Chinese hamster fibroblasts. Basic Life Sci. 1988a;49:549. doi: 10.1007/978-1-4684-5568-7_86. [DOI] [PubMed] [Google Scholar]
- Spitz DR, Elwell JH, Sun Y, Oberley LW, Oberley TD, Sullivan SJ, Roberts RJ. Oxygen toxicity in control and H2O2-resistant Chinese hamster fibroblast cell lines. Arch Biochem Biophys. 1990;279:249–260. doi: 10.1016/0003-9861(90)90489-l. [DOI] [PubMed] [Google Scholar]
- Spitz DR, Li GC, McCormick ML, Sun Y, Oberley LW. Stable H2O2-resistant variants of Chinese hamster fibroblasts demonstrate increases in catalase activity. Radiat Res. 1988b;114:114–124. [PubMed] [Google Scholar]
- Stochaj U, Rassadi R, Chiu J. Stress-mediated inhibition of the classical nuclear protein import pathway and nuclear accumulation of the small GTPase Gsp1p. FASEB J. 2000;14:2130–2132. doi: 10.1096/fj.99-0751fje. [DOI] [PubMed] [Google Scholar]
- Sugamura K, Keaney JF., Jr Reactive oxygen species in cardiovascular disease. Free Radical Biol Med. 2011;51:978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga R, et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2006;103:3250–3255. doi: 10.1073/pnas.0600012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viteri G, Chung YW, Stadtman ER. Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech Ageing Dev. 2010;131:2–8. doi: 10.1016/j.mad.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlcek S, Foisner R. A-type lamin networks in light of laminopathic diseases. Biochim Biophys Acta. 2007;1773:661–674. doi: 10.1016/j.bbamcr.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dasso M. SUMOylation and deSUMOylation at a glance. J Cell Sci. 2009;122:4249–4252. doi: 10.1242/jcs.050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman HJ. Nuclear lamins and laminopathies. J Pathol. 2012;226:316–325. doi: 10.1002/path.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Miyamoto Y, Saiwaki T, Yoneda Y. Mechanism of the stress-induced collapse of the Ran distribution. Exp Cell Res. 2006;312:512–520. doi: 10.1016/j.yexcr.2005.11.017. [DOI] [PubMed] [Google Scholar]
- Zhang H, Saitoh H, Matunis M. Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Mol Cell Biol. 2002;22:6498–6508. doi: 10.1128/MCB.22.18.6498-6508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.