

Abstract
Rapid screening and diagnosis of tuberculosis disease (TB) is still challenging and critically needed for global TB control efforts. In this study, we present a rapid and streamlined technology, using precisely engineered silica nanopore thin films, which are optimized for pore size, structure, capillary force, and film thickness, to isolate Mycobacterium tuberculosis (MTB) antigens in laboratory and clinical samples for rapid TB screening. This technology, referred to here as on-chip fractionation, is integrated with high-throughput matrix-assisted laser desorption/ionization time-of flight mass spectrometry to screen and identify fragments of the MTB antigen, CFP-10, from complex biological samples, without use of immunoaffinity agents. With the use of this comprehensive approach, we were able to clearly distinguish a clinical isolate of MTB from a nonTB species of the genus Mycobacterium avium grown in liquid culture media. This assay can reach a detection limit of 10 fmol and an isolation rate of 90% for the antigen CFP-10. Our strategy has significant potential to fill the conceptual and technical gaps in rapid diagnosis of active TB disease.
Annual worldwide statistics indicate nine million new cases and 1.5 million deaths from tuberculosis (TB), an airborne infectious disease caused by Mycobacterium tuberculosis (MTB), establishing TB as a continued significant public health challenge.1 A major contributing factor against global TB control has been unfavorably influenced by the human immunodeficiency virus (HIV) epidemic and the emergence of tenacious multidrug and extensively drug-resistant TB (M/XDR-TB). To minimize the morbidity and mortality associated with this infectious disease, a rapid and reliable diagnostic and screening test for active TB is needed for early identification and prevention. Current screening methods, including the tuberculin skin test (TST) and interferon gamma releasing assay (IGRAs), suffer from either low sensitivity/specificity or high costs, and importantly, cannot distinguish active TB disease from remote latent TB infection. Addressing specificity, the latest PCR-based GeneXpert test provides relatively increased sensitivity and specificity for TB disease, but it is unable to detect some types of active TB disease (i.e., tuberculous meningitis and other extrapulmonary TB) without invasive procedures (i.e., lumber puncture).2 The MTB culture test (MTCT) remains the standard laboratory diagnosis of active TB disease and identification of drug-resistance strains but requires 10–40 days of turnaround time.3
All attenuated strains of Mycobacterium bovis used for TB vaccination (Bacillus Calmette-Guérin, BCG) and a majority of nonTB mycobacterial species, analyzed to date, lack the CFP-10 (culture filtrate antigen 10 kDa), one of several potent T cell antigens secreted by MTB.4 The detection of CFP-10 in culture supernatants can be used to distinguish MTB from most other mycobacteria.5 Therefore, we advocate an efficient and accurate strategy for the direct identification of CFP-10 in specimens taken from susceptible patient cohorts in order to screen and monitor treatment of active TB disease.6−8
Specific antigen detection methods (immunogenicity) have relied heavily on the use of antibodies, a high-cost commodity. Several commercial CFP-10 antibodies are available for homemade indirect enzyme-linked immunosorbent assay (ELISA), but high sensitive sandwich ELISA is still not commercially available. In addition to immunoassays, matrix-assisted laser desorption/ionization time-of flight-mass spectrometry (MALDI-TOF-MS) has increasingly become a workhorse for profiling analytes in biological samples due to its high degree of molecular mass specificity and sensitivity and its ability to provide peptide sequence information.9−12 Although powerful, MALDI-TOF-MS nevertheless requires sample pretreatment to generate high-quality proteomic profiles, particularly for low abundance proteins/peptides or other analytes, in which MS spectra are overshadowed by more abundant/high-molecular weight (HMW) species. An efficient sample fractionation pathway, therefore, is required for identifying and quantifying low-molecular weight (LMW)/low-abundant CFP-10 from a complex biological sample with MALDI-TOF-MS.
In consideration of all of the above, conceptual and technical obstacles posed by current methods for screening and monitoring TB disease, we explored designs for a robust, antibody-free platform, combining the use of tailorable nanoporous materials (to capture CFP-10 from TB cultures) with MALDI-TOF-MS. The superior properties of nanoporous silica have been demonstrated in various applications, including chemical sensing, filtration, catalysis, and drug-delivery.13−16 Tweaking the architectural, physical, and chemical properties of the materials, by adjusting the processing parameters in evaporation-induced self-assembly (EISA), imparts functionality to these applications. We and others have developed a number of nanoporous materials that exhibit different characteristics (e.g., geometry, size, affinity, etc.) for sequestering the LMW proteins/peptides present in circulation in order to discover the signatures of disease, such as pulmonary metastatic melanoma and breast cancer.16−19 The results reported here, distinctive to those previous studies, demonstrate our ability to capture and identify specific LMW and low abundant TB disease markers, through a comprehensive approach that incorporates precisely designed silica nanopore (NPS or “nanotrap”) thin films with MALDI-TOF-MS, with a remarkably low detection threshold and high isolation rate.
Results
Nanopore-Based Assay to Map the CFP-10 Peptidic “Fingerprints”
An overview of our technology platform is illustrated in Figure 1a. Biological samples, in this case TB or nonTB liquid culture filtrates, were applied to a gasket silicone culture well (3 mm diameter and 1 mm height) array that was placed on top of the NPS film. As described in Materials and Methods, 200 samples can be processed on a 4 in. silicon wafer coated with a nanoporous film. The relatively small size of proteins/peptides such as CFP-10 can be trapped by the silica nanopores. Large proteins cannot enter the nanometer-sized pores and were eliminated by washing. Conducted on fixed silica films, sample capture and washing during on-chip fractionation eliminates the need for tedious and error-prone sedimentation required by particle-based systems. The fabrication of nanoporous film on a 4 in. wafer costs ∼$20. A single wafer can hold 200 samples; hence, the cost of each single test is only $0.1. To generate smaller peptides necessary for achieving high signal-to-noise ratio and resolution in MALDI-TOF-MS, we performed on-chip enzymatic digestion with trypsin prior to establishing a “fingerprint” of recombinant CFP-10 (Figure 1b and Figure S1 of the Supporting Information). This on-chip digestion allows trypsin to be transported into nanopores and catalyze the targeted peptides inside nanopores. The nanopore-based enzymatic digestion has been reported for higher digestion efficiency and better stability of digested peptides.20−22 In addition, the on-chip digestion process eliminates error-prone processes, including buffer exchange and protein extraction required by solution-based digestions. After only 8 h of incubation, we then added an elution buffer to extract the digested proteins/peptides retained inside the “nanotrap”.
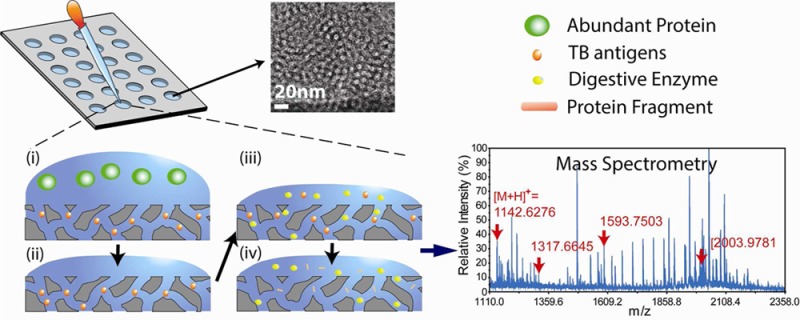
Figure 1.

(a) Schematic representation of the on-chip fractionation and digestion technology. The nanoporous silica films are coated on flat substrates. Above the film lies an adhesive plastic gasket containing multiple sample reservoirs. (i) Small proteins/peptides of interest diffuse into the nanopore while large proteins are excluded. (ii) Extensive washing removes extra proteins but leaves small ones such as the TB antigen CFP-10 within the nanopores. (iii) Trypsin is added to digest CFP-10 into small fragments. (iv) Protein fragments of interest are eluted with buffer in preparation for MALDI-TOF-MS analysis. The inset shows a TEM image of the nanoporous film. (b) MS spectrum shows a “fingerprint” of CFP-10 fragments. (c) The ion mass and sequence identification of the major CFP-10 fragments observed in MALDI-MS.
The utility of NPS is demonstrated in Figure S2 of the Supporting Information. With fractionation, the enriched major CFP-10 signals became more evident in the mass spectrum. The MS spectra of recombinant CFP-10 mapped strong signals for four major fragments (Figure 1, panels b and c, and Figure S2 of the Supporting Information), with the two highest signals observed at [M + H]+ 1142.63 and 1593.75 ([M+H]+: protonated molecule) and the other fragments also displayed significant peaks in MS spectra (Figure 2b). We confirmed by liquid chromatography tandem mass spectrometry (LC–MS/MS) that these fragments originated from CFP-10 (Figure S3 of the Supporting Information). Although these fragments were the part of the trypsin-digested products of CFP-10, we selected the fragments with strong signals in the MS fingerprint as promising signatures of CFP-10 ([M + H]+ 1142.63, 1317.66, 1593.75, and 2003.98).
Figure 2.

(a) BET and ellipsometry were used to measure film characteristics and dimensions. The porosity and film thickness were measured by ellipsometry. The surface area, pore volume, and pore size were determined by N2 adsorption/desorption analysis. The details of nanopore characterization were described in Materials and Methods. The L121 + 25% PPG (thin) is expected to have the same pore morphology as the standard L121 + 25% PPG. (b) The proportion of unfragmented CFP-10 that was retained in the detection well after washing (40 ng of CFP-10 was applied in a 7 mm2 size well, mean ± s.d., n = 6). L121 + 25% PPG can isolate up to 36 ng. (c) MALDI-MS signal intensity of each CFP-10 fragment normalized to its own isotopic fragments. Recombinant CFP-10 was spiked into the culture media, which was then treated through on-chip fractionation and digestion prior to MS analysis (mean ± s.d.; n = 5). (d) Measuring the amount of CFP-10 fragments recovered from sample input. Recombinant CFP-10 (40 ng) was spiked into the culture medium, which was then treated with on-chip fractionation and digestion. The absolute amounts of CFP-10 fragments ([M+H]+ 1142.63 and 1593.75) were quantified by spiking isotopic fragments into eluted samples.
Adapting Nanopore Morphology Influences CFP-10 Enrichment
Different design parameters (e.g., pore size and shape, chemistry, porosity, etc.) dictate the “landscape” and ultimately the peptidic fingerprint of samples processed by on-chip fractionation. To determine the optimal morphology for CFP-10 isolation, we adjusted the pore morphology by using different copolymers and the swelling agent, polypropylene glycol (PPG), which interacts with the hydrophobic domain of polymers to expand the micelle template during NPS film fabrications.16,23 Mixing various compositions of the pluronic triblock copolymers F127, L64, and L121 mixed with PPG at 0, 25, 50, and 100% weight of the copolymers yielded various numbers of film thickness, porosity, surface area, and pore volume and sizes (Figure 2a).
We first investigated the isolation efficiency of recombinant CFP-10 as a function of these NPS configurations. As described in Materials and Methods, 0.05 mg/mL of CFP-10 dissolved in the PBS buffer were incubated in the silicon gasket well (3 mm diameter) placed on the NPS surface. After extensive washes, the amount of CFP-10 remaining in the wash solution was quantified by a homemade indirect ELISA, and the percentages of CFP-10 retained in the morphologically distinct nanopores are reported in Figure 2b. We observed significantly lower isolation efficiency when the highly ordered nanoporous film (F127) for fractionation was compared to the use of nonordered nanoporous films (L121), although the average nanopore sizes of both films are comparable (pore size of F127: 3.7 nm vs L121: 3.9 nm). We have previously shown the F127 film consists of 2-dimensional (2D) hexagonal and closely packed nanopores that are perpendicular to the film’s surface.16 In contrast, L121 or L121 + PPG films are composed of nonordered or wormlike nanoporous structures.16 Our result suggests the nonordered nanopore structure is more conducive to isolating CFP-10.
Pore size also strongly influences the fractionation efficiency. Among the nonordered nanoporous films, the L64 film with 3.2 nm average nanopore diameter displays lower isolation efficiency than other films (L121 and L121 + PPG). Moreover, the films consisting of nanopore size above 3.9 nm show similar CFP-10 isolation efficiencies irrespective of the addition of PPG (L121 and L121 + PPG). This result suggests that the rodlike CFP-10 with dimensions of 1.5 × 6 nm was not significantly excluded by nanopores larger than 3.9 nm.24
Modifying the nanopore film thickness, without interfering with pore morphology also influences the efficiency of sample peptide retention and enrichment. The thickness can be manipulated by diluting the coating sol, which is the silicate sol mixed with polymer (see Materials and Methods) in ethanol or controlling the spin speed of the spin coater. We synthesized L121 + 25% PPG films of two varieties, the 643 nm thick version and the 196 nm thin version, and observed that the thick film captured more CFP-10 peptides (Figure 2b). To further understand this phenomenon, we measured the amount of CFP-10 penetrating into the nanoporous film, using X-ray photoelectron spectroscopy (XPS). As presented in Figure S4 of the Supporting Information, the concentration of CFP-10 declined exponentially as a function of nanoporous film thickness, but the majority of peptide accumulated within the top 100 nm layer. Because the diameter of the nanopore we used here is similar in size to the low molecular weight proteins, the adsorption of proteins inside the nanopore will cause a kinetic trap and minimize the diffusion caused by concentration gradients. We reasoned that thicker films (643 nm vs 196 nm) provide additional reservoirs needed for the capillary-guided water flow. The capillary filling action enhanced the transportation of CFP-10 within nanopores. Of all the nanopore configurations tested, the one structure resulting in an isolation efficiency up to 90% (36 ng of CFP-10) exhibited the following parameters: L121 + 25% PPG, 632 nm thickness, 7 mm2 surface area, and a 30 minute incubation (Figure 2b).
Adjusting the concentration of PPG not only affects pore size but also changes the structure’s porosity, defined as the fraction of void spaces in the film. In Figure 2b, we observed comparable CFP-10 isolation efficiencies when the L121 and L121 + PPG films were used. However, we also considered other parameters that could singly or collectively improve the peptide enrichment and detection procedure, including the likely exclusion of abundant protein species in the sample, the efficiency of trypsin digestion, and sample elution. To test our hypothesis, we spiked recombinant CFP-10 into sterile MTB culture media, processed the samples on nanopore films of distinct characteristics and detected through MS, and then compared the MS signals of CFP-10 fragments (Figure 2c). To minimize the variation caused by the intrinsic fluctuations of MALDI MS,25,26 each extracted sample was spiked with isotopic peptides in known quantities to serve as internal standards. These isotopic peptides were synthesized by digested CFP-10 in 18O-enriched water (H218O), leading to their shift in mass by 4 Da without changing any other physical properties. Each MS signal shown in Figure 2c was normalized by its own isotopic fragments. Although adjusting the pore size and porosity of L121 with PPG did not alter the amount of isolated CFP-10 (Figure 2b), its impact became much more evident when we examined the MS data (Figure 2c). Addition of PPG did have a positive effect on the detection of CFP-10 fragments, with the highest MS signals observed when the sample was processed on the L121 + 25% PPG nanoporous film. This increase tapers down and plateaus with the further addition of PPG (100%). One possible reason for these observations is that the small pore size of L121 without PPG (av pore size: 3.9 nm) hinders the diffusion of globulelike trypsin (4 nm diameter),27 preventing interactions between trypsin and CFP-10 retained inside the nanotraps. With pore sizes beyond 5 nm, the effect of PPG on trypsin digestion is once again minimal to none (compare L121 + 25% PPG and L121 + 50% PPG to L121). Additionally, the larger pores retain more of the abundant proteins present in the sample, leading to a MS signal reduction of CFP-10 fragments. Indeed, we observed a significant decrease in MS signal intensity when the L121 + 100% PPG film (av. pore size: 6.8 nm) was used.
Determining the Amount of CFP-10 from MTB Cultures
To quantify the absolute amount of CFP-10 fragments by their isotopic fragments, we first established a standard curve of the signal ratio of each monoisotopic and 18O-labeled fragment (Figure S6 of the Supporting Information). The isotopic fragments shifted by 4 Da to partially overlap with the monoisotopic fragments. The fragments with [M + H]+ 1142.63 and 1593.75 in MALDI-TOF-MS showed good linear regression between MS signal intensity and fragment quantity below 400 nM (Figure S4 of the Supporting Information, R2 = 1.00 and 0.98, respectively), whereas the fragments of [M + H]+ 1317.66 and 2003.98 demonstrated poor linear regression (R2 = 0.86 and 0.50, respectively). On the basis of standard curves for the fragments with [M + H]+ 1142.63 and 1593.75, we quantified the amount of CFP-10 after on-chip sample processing on different nanopore films. Similar to our earlier results, CFP-10 processed on L121 + 25% PPG resulted in the highest yield at 1.2 pmol (Figure 2d).
To test the sensitivity of our assay and determine its minimum threshold of detection, we titrated recombinant CFP-10 in sterile MTB culture media and processed each sample on the L121 + 25% PPG film and through MS. The MS signals of 4 major fragments, each normalized to its own isotope, are depicted in Figure 3a, plotted as signal intensity versus CFP-10 concentration in the log–log plot. Linear regression ranged from acceptable to good. On the basis of these results, we determined that this assay can detect CFP-10 in culture media at a remarkably low concentration of 14.1 nM. We improved the limit of detection to 1.4 nM by concentrating CFP-10 10-fold by ammonium sulfate precipitation of the culture media (as described in Materials and Methods) before on-chip processing (Figure 3b).
Figure 3.
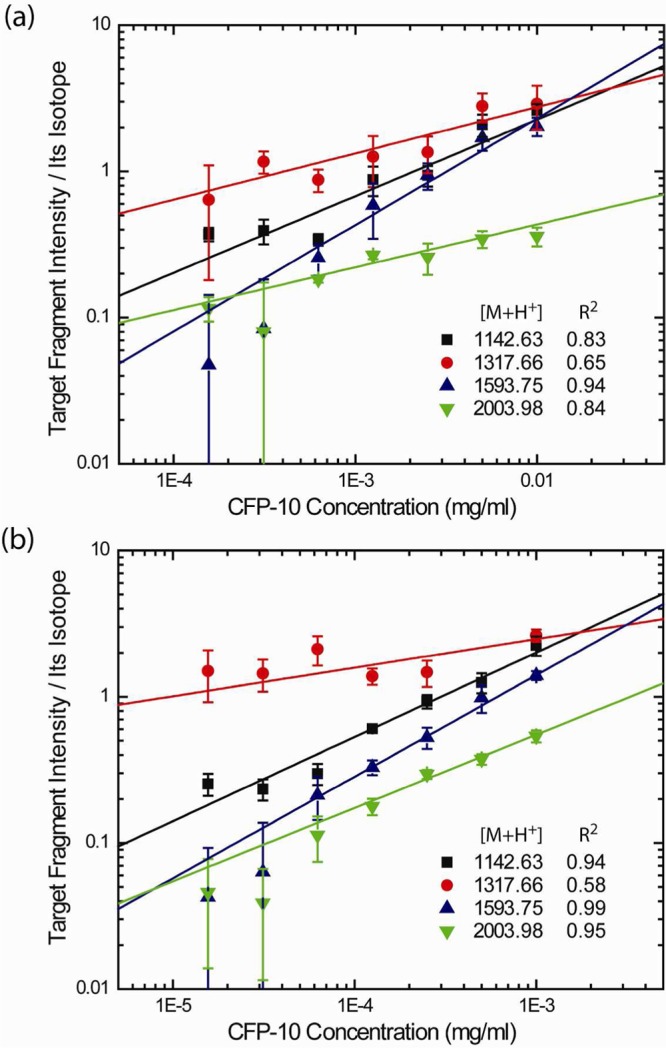
The detection threshold for CFP-10 fragments by MALDI-TOF-MS analysis. Different amounts of recombinant CFP-10 in MTB culture media (mean ± s.d.; n = 5). The signals of each fragment was normalized by its own isotope as an internal standard. (a) Unprecipitated culture medium for each CFP-10 dilution is processed through on-chip fractionation and digestion. The sensitivity plot maintains good linear regression above 14.1 nM in log–log scale. (b) The samples are precipitated 10× by ammonium sulfate prior to on-chip processing. MS analysis shows that the detection limit has been lowered to 1.4 nM, as a result of sample concentration.
To access the interday and intraday variability of our combined on-chip fractionation-MS analysis, we spiked recombinant CFP-10 at three different and defined concentrations, in replicate samples, into sterile culture media. The fragments with [M+H]+ 1142.63 and 1593.75 displayed higher MS signals (Tables 1 & 2), better linear regression with respect to their isotopes, and better quantification accuracy (% RE, relative error) and precision (% coefficient of variation, CV) compared to the fragments with [M + H]+ 1317.66 and 2003.98 (Tables S1 and S2 of the Supporting Information). At 100 nM concentrations, the mean calculated concentrations remained within 10% of the actual values (% RE) and did not exceed 30% of the %CV. At lower concentrations (1.4 nM), the accuracy of quantification decreased. The qualitative identification of CFP-10 remained very precise even at only 1.4 nM. We detected strong MS signals for the fragments with [M+H]+ 1142.63 and 1593.75 in all of the samples (n = 11) and the fragments with [M+H]+ 1317.66 and 2003.98 in 63% and 72% of the samples.
Table 1. Interday Accuracy and Reproducibility of CFP-10 Measurements (1142.628 & 1593.750 Fragments).
| concentration (nM) | N | fragments | mean (μg/mL) | standard deviation | precision (% CV)a | accuracy (% RE)b |
|---|---|---|---|---|---|---|
| 90.3 | 5 | 1142.628 | 1.1057 | 0.2968 | 26.85 | 10.57 |
| 1593.750 | 1.1795 | 0.2810 | 23.82 | 17.95 | ||
| 11.3 | 5 | 1142.628 | 0.0741 | 0.0085 | 11.47 | 40.70 |
| 1593.750 | 0.1309 | 0.0396 | 30.23 | 4.70 | ||
| 1.4 | 5 | 1142.628 | 0.0270 | 0.0100 | 37.01 | 72.83 |
| 1593.750 | 0.0455 | 0.0175 | 38.53 | 191.31 |
The coefficient of variation (CV) = standard deviation/mean.
Relative error (RE) = (measured value – actual value)/actual value.
Table 2. Intraday Accuracy and Reproducibility of CFP-10 Measurements (1142.628 and 1593.750 Fragments).
| concentration (nM) | N | fragments | mean (μg/mL) | standard deviation | precision (% CV) | accuracy (% RE) |
|---|---|---|---|---|---|---|
| 90.3 | 9 | 1142.628 | 0.8914 | 0.1219 | 13.67 | 10.86 |
| 1593.750 | 1.0455 | 0.2717 | 25.99 | 4.55 | ||
| 11.3 | 9 | 1142.628 | 0.0818 | 0.0176 | 23.27 | 34.53 |
| 1593.750 | 0.1465 | 0.0782 | 33.45 | 17.24 | ||
| 1.4 | 9 | 1142.628 | 0.0276 | 0.0072 | 25.99 | 76.43 |
| 1593.750 | 0.0456 | 0.0134 | 29.33 | 191.72 |
Differentiating MTB Based On Its CFP-10 Signatures in Clinical Isolate
To address specificity of our on-chip fractionation-MS technology, we investigated the expression of CFP-10 from MTB grown in culture media (Figure 4). The nonTB Mycobacterium (NTM), Mycobacterium avium lacks the CFP-10 gene and therefore serves as our negative control. To mimic conditions one may find in early disease diagnosis (i.e, low secretion of CFP-10 in the culture supernatant), we performed the same ammonium sulfate concentration protocol prior to on-chip fractionation-MS analysis. Indeed, strong MS signals for all the fragments were observed in the supernatant of the MTB cultures but not in the M. avium culture (Figure 4). These results were corroborated by LC–MS/MS for the fragments of [M+H]+ 1142.63, 1317.66, and 1593.75 (Figure S7 of the Supporting Information).
Figure 4.

Mass spectra of MTB-specific CFP-10 fragments. None of these fragments were observed in the culture of nonTB species of mycobacteria (M. avium).
Discussion
Rapid screening and diagnosis of active TB disease has been a long-standing challenge in global TB control. Current diagnostic tests depend on the actual detection of MTB by bacteriological methods such as AFB smear microscopy and bacterial culture, or by molecular identification of MTB DNA (e.g., PCR-based GeneXpert). Both types of methods require the presence of MTB bacteria in the patient’s specimens (sputum, etc.), often barring or rendering difficult disease detection for pausibacillary TB (e.g., culture-negative TB and extrapulmonary TB). For this study, we hypothesize that all active TB diseases, whether paucibacillary or otherwise, involve the secretion of MTB antigens (CFP10 and ESAT-6) into the host body and further into the circulatory system. As such, we designed a platform that combines the specificity and ease of our nanopore-based on-chip fractionation assay with the powerful MS analysis in terms of sensitivity and specificity. By detecting the patterns of these disease-specific antigens, one can potentially provide a rapid screening method of active TB disease.
One of the first tasks was to determine those design parameters for the nanopore films that are most conducive to high enrichment/isolation of peptides from low sample volumes. We previously established a fabrication protocol to place a thin NPS film onto a flat silicon substrate constructed with highly uniform and textured nanopores.16 The silica nanopore structures are constructed by the self-assembly of structure-directing triblock copolymers consisting of a hydrophobic (PPO) core capped by two hydrophilic (PEO) blocks. Under the cooperative effects of capillary force and size-exclusion, LMW proteins and peptides will diffuse into well-defined and nanometer-sized silica pores, whereas the large and more abundant molecules remain excluded to be washed away during chip processing. We often refer to this procedure as peptide fractionation, isolation, and enrichment, which results in stronger MS signals for LMW or low-abundance species. Here, we adapted this versatile technology to the detection of active TB disease. To our knowledge, this is the first demonstration that employs a convenient and relatively inexpensive, antibody-free nanopore platform coupled to benchtop MALDI-TOF-MS analysis, to isolate and quantify CFP-10 from MTB cultures with a low detection limit.
The detection procedure consists of three major steps: (1) enriching CFP-10 in the nanopore and removing abundant larger proteins; (2) trypsin digesting the isolated CFP-10; and (3) extracting CFP-10 fragments from the nanopore. During the isolation step, absorption of buffer in nanoporous films via capillary action drives CFP-10 into the nanopore of the film, a critical factor for peptide isolation efficiency. As presented above (Figure 2b), using the thicker films with highly interconnected nanopores and additional reservoirs for flow resulted in enhanced peptide isolation.
As we have demonstrated previously and elaborated further in this work, several characteristics such as nanopore size, film architecture, and peptide features (size, shape, charge, etc.) dictate the isolation efficiency,28,29 thus imparting such versatility to the varieties of peptides that can be captured and enriched on this platform. In general, larger pores reduce the diffusion barrier to allow more proteins/peptides into the defined space. On the other hand, because of this capacity to pack into the porous film, abundant proteins/peptides that meet the size-exclusion criteria will also pack into the space and later overwhelm the signals of low-abundance species in MALDI-TOF-MS analysis. In addition, pore size constrains the diffusion of trypsin into the porous film and/or its interaction with enzymatic substrates. Smaller pores (3.9 nm) limit the “wiggle-room” for globular trypsin (4 nm diameter) to maneuver its catalyzing of the rodlike CFP-10 (1.5 × 6 nm). After a comprehensive analysis of nanopore morphology for optimal CFP-10 isolation and MALDI-TOF-MS data quality, we concluded that the best configuration (i.e., L121 + 25% PPG) should exhibit 5 nm, nonordered pores.
Using the L121 + 25% PPG nanotrap, we demonstrated up to 90% isolation of recombinant CFP-10 from a quantified solution. Significant amounts of CFP-10 fragments were recovered from the biological samples (Figure 2d), suggesting that the efficiency of recovery may be improved up to 3 times more, pending further nanopore optimization. As the technology couples on-chip fractionation to MS, the sensitivity of detection logically also relies on MALDI-TOF-MS parameters. The titration curve of CFP-10 fragments shown in Figure S8 of the Supporting Information indicated the detection limit of MALDI-TOF-MS, under current operating conditions, at 0.2 fmol (0.5 μL of a 0.4 nM solution containing CFP-10 spotted on the target plate). Barring no interference from other mycobacteria species in the samples, the detection limit of the current detection protocol is on the order of 1 nM. Hence, in addition to nanopore optimization, adjusting MALDI-TOF-MS parameters (e.g., matrix selection, sample drying, tuning laser energy, etc.) and concentrating samples (e.g., ammonium sulfate precipitation) prior to on-chip fractionation are fine-tuning strategies to further improve sample detection.30−32 Of the latter method, we successfully enhanced detection of CFP-10 by performing a 10-fold concentration procedure. Although this prefractionation procedure simultaneously increased the amount of abundant, and potentially interfering, proteins in the sample, its unintended consequence could easily be minimized or even negated as a result of the on-chip fractionation process through nanoporous films. Combining standard precipitation, where needed, our on-chip fractionation platform remains a feasible alternative and should be especially applicable in situations of extremely low peptide abundance, such as the very early onset of disease.
CFP-10 is an ideal biomarker for MTB because of its absence from the majority of nonTB mycobacteria (NTM). We clearly distinguished the clinical isolate of MTB from M. avium, a NTM organism devoid of CFP-10 secretion. We envision its use as a diagnostic tool, amenable to high-throughput application, to screen patient specimens (e.g., serum, urine, pleural fluid, and cerebrospinal fluids for tuberculous meningitis, etc.) for active TB disease quickly, accurately, and with lead-time for effective treatment.
Materials and Methods
Fabrication of Nanoporous Silica Thin Films
The silicate sol was prepared by adding 14 mL of tetraethyl orthosilicate (TEOS) to a solution of 17 mL of ethanol, 6.5 mL of distilled water, and 0.5 mL of 6 M HCl and then stirred at 80 °C for 2 h to form a clear solution. After the silicate sol cooled down to 25 °C, the silicate sol was added to a mixture of pluronic polymer, ethanol, and different amounts of polypropylene glycol (PPG, labeled as % wt of L121). The recipes for the porous silica films are reported in Table S3 of the Supporting Information. The coating solution was stirred at 25 °C for 2 h. The coating sol was deposited on a Si(100) wafer by spin coating at the spin rate of 2500 rpm for 20 s. To increase the degree of polymerization of the silica framework in the films and to further improve their thermal stability, the deposited films were heated at 80 °C for 12 h. The films were calcinated at 450 °C for 5 hrs to remove the organic compound. The temperature was raised at a rate of 1 °C/min. Pluronic polymers (L121, L64, and F127) were obtained from BASF. All other chemicals were purchased from Sigma-Aldrich.
Characterization of Nanoporous Silica Thin Film
The thickness and porosity of nanoporous silica films were characterized by variable angle spectroscopic ellipsometer (J. A. Woollam Company M-2000DI). The thickness of thin films and their porosities were calculated in Cauchy and effective medium approximation (EMA) models using Complete EASE software (Berlin, Germany, version 4.58). Ellipsometric optical quantities were detected by acquiring spectra at incidence angles of 55, 60, and 65° in wavelengths ranging from 300 to 1800 nm. All fabricated porous silica thin films were characterized by scanning over the entire 4 in. wafer using the ellipsometer. The variations for porosity and thickness were less than 0.5%.
N2 adsorption/desorption analysis was applied in measuring surface area and pore size distribution. Quantachrome Autosorb-3b BET Surface Analyzer was used to record the N2 adsorption/desorption isotherm at 77 K on the full range of relative P/P0 pressures. Nanopore size distributions were calculated from the desorption branch of the isotherms using the Barrett–Joyner–Halenda (BJH) method.
On-Chip Fractionation and Digestion of Proteins/Peptides
To test the detection limit of CFP-10 in complex culture of MTB, different amounts (0–1 μg/mL) of recombinant CFP-10 were mixed in blank culture media (BBL MGIT, purchased Mycobacteria Growth Indicator from BD). Recombinant CFP-10 was purchased from Diagnostics, Inc. (Woburn, MA). Unless otherwise indicated, 8 μL of culture sample was pipetted onto the silica nanoporous film and incubated for 30 min in a humidified chamber at 25 °C. The superfluous solution was then removed, and 10 μL of deionized water was applied onto the silica porous film to remove larger proteins excluded from the nanopores. The washing was then repeated 4 times. For on-chip digestion, 10 μL of 5 μg/mL trypsin (dissolved in 100 mM sodium bicarbonate) was applied onto the silica nanoporous film and incubated overnight at 37 °C. After complete digestion, the nanoporous film was removed from the humidity chamber and allowed to dry. Then, 10 μL of elution buffer [0.1% trifluoracetic acid (TFA) + 50% acetonitrile (ACN) in water] was pipetted to extract the CFP-10 fragments. The elution buffer containing fractionated and digested peptides was then removed and stored in a microcentrifuge tube until MALDI-TOF-MS analysis. In order to detect CFP-10 at extremely low concentrations, the culture medium spiked with recombinant CFP-10 was treated with ammonium sulfate (466 μL of saturated ammonium sulfate solution were mixed with 200 μL samples). The samples were mixed and incubated at 4 °C for 30 min, then centrifuged at 10000g for 10 min to pellet the precipitated peptides. Once dried, the peptide pellets were dissolved in 20 μL of double-deionized water.
18O-Labeling of Trypsin-Catalyzed CFP-10 Fragments
The recombinant CFP-10 was dried and redissolved in 18O-enriched water (97%, Sigma-Aldrich). Trypsin solution (0.5 mg/mL of trypsin and 100 mM of NH4HCO3) was also dissolved in 18O-enriched water. The solution of CFP-10 and trypsin (20:1 weight ratio) was incubated overnight at 37 °C to allow complete digestion of CFP-10.
MALDI-TOF-MS Analysis
A matrix solution of α-cyano-4-hydroxycinnamic acid (CHCA, 4 g/L) was prepared in a mixture of ACN and water (1:1, v/v) containing 0.1% TFA. Each sample was equally mixed with 18O-labeled CFP-10 solution (42 nM). First, 0.5 μL of each CFP-10 mixture was spotted onto the MALDI target plate and allowed to dry at 25 °C. Another 0.5 μL of the matrix solution was spotted onto the dried CFP-10 samples and allowed to dry at 25 °C. Mass spectra were collected using the Applied Biosystems 4700 MALDI TOF/TOF Analyzer (Applied Biosystems). The operating parameters: positive reflection mode in the range of 800–5000 Da, 5000 laser shots, and laser intensity of 4300 instrument units. The spectra were calibrated externally using a peptide calibration standard and processed with Data Explorer (Applied Biosystems).
Quantification of CFP-10 Processed through NPS
To examine the isolation efficiency of CFP-10 on films of different nanopore morphology and configuration, the amount of peptide remaining in the wash buffer was measured using indirect ELISA (enzyme-linked immunosorbent assay). Here, 8 μL of a 5 μg/mL solution containing CFP-10 (dissolved in 1× PBS) were applied in each well of the NPS, allowed to incubate for 30 min, and then washed with 10 μL of 1× PBS 5 times. The wash buffer was collected for protein quantification using indirect ELISA. Mouse monoclonal antibody against CFP-10 was purchased from Abcam, Inc. The indirect ELISA materials, including 96 well plate and TMB (3,3′,5,5′-tetramethylbenzidine) solution, were purchased from Thermo Scientific and eBioscience, respectively. The peptides were incubated at 4 °C overnight to allow coating on the 96 well plate, blocked with 5% serum in 1× PBS buffer, and then probed sequentially with primary followed by secondary antibodies. The chromogenic substrate TMB was used for signal detection.
XPS (X-ray Photoelectron Spectroscopy) Depth Profiling
A 0.01 mg/mL solution of CFP-10 (dissolved in 100 mM NaCl) were incubated on L121 + 25% PPG nanoporous film, washed with deionized water, and then incubated in a vacuum chamber overnight prior to XPS measurement. The PHI Quantera XPS, equipped with an Ar+ ion gun was used to construct the concentration depth profile, sputtering Ar+ ions at 3 kV onto the films within a 2 × 2 mm area. The film thickness determined by an ellipsometer (J.A. Woollam Co, Inc.) was used to calibrate the etching rate on porous silica by sputtering until the oxygen (O1s) signal vanished. A sputtering time interval of 9 s was used to reach a depth spacing of 5.25 nm at 35 nm/min of Ar+ ion etching rate. Nitrogen (N1s) spectra were used to identify the amount of CFP-10 trapped at different depths.
LC–MS/MS Measurement
Two-hundred microliters of MTB culture media were precipitated and processed by on-chip fractionation, as described previously. To avoid remaining impurity, such as polymer damaging the LC column, the processed solutions were further treated with SCX zip-tip (Millipore) before being injected into LC–MS/MS system. Reversed-phase chromatography was performed on Waters Xevo TQ system (Waters corporation, Milford, MA). Gradient solvents used for LC analysis are (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile. Samples were dried in a vacuum centrifuge prior to injection into the LC and resuspended in 1% formic acid and 5 mM NH4OAc. Database search using the PLGS search engine (Waters corporation) was performed to identify the peptide peaks of interest.
Biosafety of MTB Culture
All bacteria cultures were carried out by using BACTEC MGIT 960 system (Becton Dickinson, Sparks, MD). After a 2-week culture, the MTB culture samples were filtered by using a 0.2 μm size Whatman filter (Whatman plc, Kent, U.K.) to remove the bacteria and the culture supernatant was sterilized in a Biosafety level 3 (BL3) facility. To confirm the filtered culture, supernatants were completely disinfected, a 10 μ-filtered sample, s, was inoculated into Lowenstein-Jensen medium for 6-week culture, and no bacterial colonies were found.
Acknowledgments
The work at Houston Methodist Research Institute (HMRI) was primarily supported by research funding provided by Alliance for Nanohealth precenter award, from the Department of Defense (Grant W81XWH-11-2-0168). The authors also acknowledge the support from the National Institute of Health (Grant NIH U54CA151668). The authors also thank Dr. David A. Engler at the Proteomics core of HMRI and Dr. David Hawke in the Proteomic facilities at the University of Texas M.D. Anderson Cancer Center, for their help in operating the mass spectrometer and suggestion in analyzing the resulting data.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- World Health Organization, WHO Library Cataloguing-in-Publication Date: 2011; Vol. 2012, 2012.
- Ioannidis P.; Papaventsis D.; Karabela S.; Nikolaou S.; Panagi M.; Raftopoulou E.; Konstantinidou E.; Marinou I.; Kanavaki S. J. Clin. Microbiol. 2011, 49, 3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention, American Thoracic Society.Am. J. Respir. Crit. Care Med. 2000, 161, 1376. [DOI] [PubMed] [Google Scholar]
- van Ingen J.; de Zwaan R.; Dekhuijzen R.; Boeree M.; van Soolingen D. J. Bacteriol. 2009, 191, 5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T. T.; Shou C. M.; Shen L.; Qian Y.; Wu Z. G.; Fan J.; Zhang Y. Z.; Tang Y. W.; Wu N. P.; Lu H. Z.; Yao H. P. The International Journal of Tuberculosis and Lung Disease 2011, 15, 804. [DOI] [PubMed] [Google Scholar]
- Meher A. K.; Bal N. C.; Chary K. V. R.; Arora A. FEBS J. 2006, 273, 1445. [DOI] [PubMed] [Google Scholar]
- Guinn K. M.; Hickey M. J.; Mathur S. K.; Zakel K. L.; Grotzke J. E.; Lewinsohn D. M.; Smith S.; Sherman D. R. Mol. Microbiol. 2004, 51, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P.; Rosenkrands I.; Andersen P.; Cole S. T.; Brosch R. Trends Microbiol. 2004, 12, 500. [DOI] [PubMed] [Google Scholar]
- Petricoin E. F.; Ornstein D. K.; Paweletz C. P.; Ardekani A.; Hackett P. S.; Hitt B. A.; Velassco A.; Trucco C.; Wiegand L.; Wood K.; Simone C. B.; Levine P. J.; Linehan W. M.; Emmert-Buck M. R.; Steinberg S. M.; Kohn E. C.; Liotta L. A. J. Natl. Cancer Inst. 2002, 94, 1576. [DOI] [PubMed] [Google Scholar]
- Petricoin E. F.; Ardekani A. M.; Hitt B. A.; Levine P. J.; Fusaro V. A.; Steinberg S. M.; Mills G. B.; Simone C.; Fishman D. A.; Kohn E. C.; Liotta L. A. Lancet 2002, 359, 572. [DOI] [PubMed] [Google Scholar]
- Petricoin E. F.; Liotta L. A. Curr. Opin. Biotechnol. 2004, 15, 24. [DOI] [PubMed] [Google Scholar]
- Hortin G. L. Clin. Chem. 2006, 52, 1223. [DOI] [PubMed] [Google Scholar]
- Lan E. H.; Dave B. C.; Fukuto J. M.; Dunn B.; Zink J. I.; Valentine J. S. J. Mater. Chem. 1999, 9, 45. [Google Scholar]
- Stein A.; Melde B. J.; Schroden R. C. Adv. Mater. 2000, 12, 1403. [Google Scholar]
- Lai C. Y.; Trewyn B. G.; Jeftinija D. M.; Jeftinija K.; Xu S.; Jeftinija S.; Lin V. S. Y. J. Am. Chem. Soc. 2003, 125, 4451. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Bouamrani A.; Tasciotti E.; Li L.; Liu X. W.; Ferrari M. ACS Nano 2010, 4, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.; Huang Y.; Finoulst I.; Wu H.-J.; Deng Z.; Xu R.; Xia X.; Ferrari M.; Shen H.; Hu Y. Cancer Lett. 2013, 334, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouamrani A.; Hu Y.; Tasciotti E.; Li L.; Chiappini C.; Liu X. W.; Ferrari M. Proteomics 2010, 10, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J.; Deng X. Y.; Gallagher J. W.; Huang H. Y.; Huang Y.; Wen J. G.; Ferrari M.; Shen H. F.; Hu Y. Philos. Trans. R. Soc., A 2012, 370, 2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Kim B. C.; Lopez-Ferrer D.; Petritis K.; Smith R. D. Proteomics 2010, 10, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L.; Liu Y.; Hudson S. P.; Yang P. Y.; Magner E.; Liu B. H. Chem.—Eur. J. 2008, 14, 151. [DOI] [PubMed] [Google Scholar]
- Savino R.; Casadonte F.; Terracciano R. Molecules 2011, 16, 5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen M. H.; Corkery R. W.; Pedersen J. S.; Rosenholm J.; Alberius P. C. Micropor Mesopor Mater. 2008, 113, 1. [Google Scholar]
- Renshaw P. S.; Lightbody K. L.; Veverka V.; Muskett F. W.; Kelly G.; Frenkiel T. A.; Gordon S. V.; Hewinson R. G.; Burke B.; Norman J.; Williamson R. A.; Carr M. D. EMBO J. 2005, 24, 2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X. D.; Freas A.; Ramirez J.; Demirev P. A.; Fenselau C. Anal. Chem. 2001, 73, 2836. [DOI] [PubMed] [Google Scholar]
- Stewart I. I.; Thomson T.; Figeys D. Rapid Commun. Mass Spectrom. 2001, 15, 2456. [DOI] [PubMed] [Google Scholar]
- Leiros H. K. S.; Brandsdal B. O.; Andersen O. A.; Os V.; Leiros I.; Helland R.; Otlewski J.; Willassen N. P.; Smalas A. O. Protein Sci. 2004, 13, 1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deere J.; Magner E.; Wall J. G.; Hodnett B. K. J. Phys. Chem. B 2002, 106, 7340. [Google Scholar]
- Katiyar A.; Ji L.; Smirniotis P.; Pinto N. G. J. Chromatogr., A 2005, 1069, 119. [DOI] [PubMed] [Google Scholar]
- Dreisewerd K. Chem. Rev. 2003, 103, 395. [DOI] [PubMed] [Google Scholar]
- Karas M.; Gluckmann M.; Schafer J. J. Mass Spectrom. 2000, 35, 1. [DOI] [PubMed] [Google Scholar]
- Gusev A. I.; Wilkinson W. R.; Proctor A.; Hercules D. M. Anal. Chem. 1995, 67, 1034. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.