

Abstract
Surface erosion has been recognized as a valuable design tool for resorbable biomaterials within the context of drug delivery devices, surface coatings, and when precise control of strength retention is critical. Here we report on high tensile strength, aromatic–aliphatic polycarbonates based on natural phenols, tyrosol (Ty) and homovanillyl alcohol (Hva), that exhibit enzymatic surface erosion by lipase. The Young’s moduli of the polymers for dry and fully hydrated samples are 1.0 to 1.2 GPa and 0.8 to 1.2 GPa, respectively. Typical characteristics of enzymatic surface erosion were confirmed for poly(tyrosol carbonate) films with concomitant mass-loss and thickness-loss at linear rates of 0.14 ± 0.01 mg cm–2 d–1 and 3.0 ± 0.8 μm d–1, respectively. The molecular weight and the mechanical properties of the residual films remained constant. Changing the ratio of Ty and Hva provided control over the glass transition temperature (Tg) and the enzymatic surface erosion: increasing the Hva content in the polymers resulted in higher Tg and lower enzymatic erosion rate. Polymers with more than 50 mol % Hva were stable at 37 °C in enzyme solution. Analysis on thin films using quartz crystal microbalance with dissipation (QCM-D) demonstrated that the onset temperature of the enzymatic erosion was approximately 20 °C lower than the wet Tg for all tested polymers. This new finding demonstrates that relatively high tensile strength polycarbonates can undergo enzymatic surface erosion. Moreover, it also sheds light on the connection between Tg and enzymatic degradation and explains why few of the high strength polymers follow an enzyme-meditated degradation pathway.
Introduction
Surface eroding polymers as compared to bulk eroding polymers have distinct advantages for the design of resorbable medical implants.1 Typically, during bulk erosion a decrease in molecular weight of the polymer occurs before any mass-loss is observed. This leads to unfavorable changes in polymer characteristics such as diminishing mechanical strength and lack of control over long-term drug release. By contrast, surface erosion leads to mass-loss with only negligible molecular weight decrease throughout the bulk of the polymer. Surface erosion is advantageous in applications requiring a controlled retention of mechanical properties during degradation and in drug delivery applications where the rate of drug release can be controlled by the erosion of surface layers of the polymeric matrix. However, hydrolytic surface erosion is only observed when the rate of polymer degradation is faster than the rate of water penetration into the bulk of the polymer.2,3 Hence, for small medical implants, hydrolytic surface erosion is limited to extremely fast degrading polymers such as some polyanhydrides and some poly(ortho esters).4 In the clinic, a surface eroding device (Gliadel) made from polyanhydrides is used to release a chemotherapeutic agent in the brain over 2–3 weeks.5 Most degradable polymers used in the design of medical and drug release devices are materials such as polyesters that invariably undergo bulk erosion.6 Interestingly, Pitt et al. reported that poly(trimethylene carbonate) (PTMC), an aliphatic polycarbonate, showed surface erosion behavior in vivo, while the hydrolytic degradation was slow in vitro.7 It was later demonstrated that PTMC underwent enzymatic degradation by hydrolytic enzymes in vitro, mimicking surface erosion characteristics found in vivo.8 Hence, hydrolytic enzymes are likely to play a significant role in the degradation of PTMC. Further, the involvement of reactive oxygen species in the erosion of aliphatic polycarbonates was recently suggested by Amsden et al., similar to previous findings by Anderson et al. in poly(carbonate urethane)s.9−11 Recent studies have evaluated the suitability of devices from surface eroding, aliphatic polycarbonates for antibiotic delivery, and implantation in a soft tissue environment for vascular and cardiac tissue engineering.12−15 Since the material properties of these aliphatic polycarbonates are characterized as flexible and rubbery (Tg lower than 37 °C), it was previously postulated that enzymatic surface erosion requires a flexible polymer backbone that can comply with the enzyme’s active site.16 Therefore, it is accepted that aromatic polycarbonates and most other currently available biodegradable polymers with Young’s moduli in the GPa range are not susceptible to enzymatic surface erosion, even though amorphous poly(lactic acid) is degradable by Proteinase K.17,18 Likewise, a wide range of tyrosine-derived polycarbonates were extensively studied by Kohn et al., but were not found to degrade by enzyme-mediated processes.19−21 Heretofore, few efforts have been made to discover new polymers of high strength that undergo enzymatic surface erosion.
In this contribution, we report on the preparation and characterization of a series of aromatic–aliphatic polycarbonates based on tyrosol and homovanillyl alcohol. Both monomers are readily available from natural resources such as olive oil mill waste waters and products of fermentation processes.22,23 Tyrosol and hydroxytyrosol are assessed as GRAS (generally recognized as safe) substances by the FDA. Homovanillyl alcohol is a metabolite of hydroxytyrosol and has an LD50 of 3200 mg/kg (oral, rabbit; data from the Material Safety Data Sheet provided by the supplier). As antioxidants they have been credited with benign biological activities.24 The structures of the monomers contain both a phenol and an alcohol group: After polycondensation, polymers with both aromatic and aliphatic carbonate functionalities were obtained. Remarkably, we found enzymatic surface erosion behavior resembling the degradation of soft, aliphatic polycarbonates, while the mechanical properties were strong, similar to aromatic polycarbonates. QCM-D analysis on thin films demonstrated the connection between Tg and enzymatic surface erosion; this finding explains why amorphous poly(lactic acid) and the polycarbonates based on tyrosol and homovanillyl alcohol with Tg below 60 °C can undergo surface erosion, while most of the other high tensile strength polymers with significantly higher Tg cannot.
Experimental Section
Materials
All chemicals used were reagent grade or better. Tyrosol (Ty), homovanillyl alcohol (Hva), bis(trichloromethyl) carbonate (triphosgene), dichloromethane, 2-propanol, hexane, deuterated dimethyl sulfoxide (d6-DMSO), tetrahydrofurane (THF), trifluoroacetic acid (TFA), phosphate buffered saline (PBS) and lipase from Thermomyces lanuginosus (EC3.1.1.3, minimum 105 units g–1) were obtained from Sigma-Aldrich (St. Louis, MO). Pyridine, hexane and N,N-dimethylformamide (DMF) were obtained from Fisher Scientific (Pittsburgh, PA).
Polymer Synthesis and Characterization
CAUTION: Triphosgene used in the following procedure is a hazardous material. Triphosgene can release deadly phosgene, a gas that can be lethal before it can be recognized by its smell. All procedures using triphosgene need to be performed in a closed fume hood and under supervision of an experienced and well-trained operator. A monitor and alarm system for accidental exposure to phosgene is required.
Polycarbonates were synthesized by condensation polymerization using Ty and Hva as monomers in dichloromethane and pyridine with triphosgene as a phosgene source.25 The polymers were purified by repeated precipitation in 2-propanol and dissolution in dichloromethane. The chemical composition was confirmed by analysis of 1H and decoupled 13C nuclear magnetic resonance spectra (500 MHz NMR, Varian, U.S.A.). d6-DMSO was used as solvent and all spectra were referenced to the residual DMSO signal. The number and weight average molecular weights (Mn and Mw) of the copolymers were determined relative to polystyrene standards using gel permeation chromatography (GPC, Waters, Milford, MA) equipped with two PL gel columns of 100 000 and 1000 Å (Polymer Laboratories, Amherst, MA). DMF with 0.1% TFA was applied as eluting solvent. Thermal properties were analyzed using differential scanning calorimetry (DSC 2520, Mettler Toledo, Columbus, OH). The heating rate was 10 °C min–1 and the glass transition temperature (Tg) was calculated using the ASTM midpoint method. The first and second heat scans were used to determine Tg of fully hydrated specimens (Tg(wet)) and dry specimens (Tg(dry)), respectively.
Compression Molding
Polymer films were compression molded at 190 °C using a Carver press (model 4122, Carver, Wabash, IN) with a thickness of 250 μm for water uptake and degradation studies and a thickness of 400 μm thick for mechanical testing, respectively.
Water Uptake
Specimens from compression-molded films, cut to a sample size of approximately 10 mg, were incubated in PBS at 37 °C until the equilibrium water uptake was reached. The specimens were blotted with paper and immediately subjected to thermogravimetric analysis (TGA, Mettler-Toledo, Columbus, OH). Specimens were heated from 25 to 150 °C at a heating rate of 10 °C min–1.
Mechanical Properties
Mechanical properties were characterized using a mechanical tensile testing apparatus equipped with a 10 N submersible load cell (Bose Electroforce, Eden Prairie, MN). Rectangular-shaped specimens were cut to a width of 2 mm from compression-molded films approximately 400 μm thick. The initial grip-to-grip separation was 8 mm and the maximal strain was 150% due to instrument limitation. The crosshead speed was 0.1 mm s–1. Mechanical properties in the dry state were determined at room temperature. To measure the mechanical properties in the wet state, specimens were preconditioned in PBS at 37 °C for 24 h, and then immersed in PBS at 37 °C for testing. To follow the mechanical properties during degradation (see below), specimens were incubated in lipase solution as well as in PBS (as control) and retrieved at respective time points. The tensile tests were performed in the wet state. The tensile modulus was determined from the initial, linear part of the stress–strain curve, using the grip-to-grip distance to measure elongation. The stress and strain at yield (σyield and εyield) values were determined from the upper yield point. The analyses were performed in triplicate.
Spincoating of Polymer Thin Films
Polymer thin films were prepared by spincoating from 1–1.25% (w/v) polymer solution in dry dioxane at 3000 rpm using a spin-coater (Headway Research, Garland, TX) in a humidity-controlled atmosphere (less than 10% relative humidity). For contact angle measurements 15 mm glass coverslips (Fisher Scientific Pittsburgh, PA) and for QCM-D gold-coated quartz crystal QSX100 (Q-sense, Glen Burnie, MD) were used as substrates. The samples were dried in vacuo for at least 12 h.
Static Contact Angle Measurement
To measure the contact angle, a drop of water was applied to the polymer-coated surface and the static contact angle was determined using a goniometer (Rame-Hart, Succasunna, NJ) with at least five independent measurements per composition.
Degradation Experiments
Polymer discs with a diameter of 6 mm and thickness of approximately 250 μm were immersed in 3 mL of lipase solution from Thermomyces lanuginosus as a model enzyme with an activity of 5 kU/ml. For control experiments, specimens were immersed in PBS. All solutions contained 0.02% (w/w) of sodium azide to prevent bacterial growth. The incubation temperature was 37 °C and the solutions were replaced twice per week. Samples in triplicate were removed at respective time points and rinsed with deionized water and 70% (v/v) ethanol. The mass and thickness of the samples were measured after drying in vacuo for 72 h at ambient temperature.
Scanning Electron Microscopy (SEM)
Dried specimens subjected to degradation media or PBS control were sputter coated (SCD 004, Leica Microsystems, Liechtenstein) with gold/palladium, and then the morphology of the specimens was studied by SEM (1830I, Amray, USA, Voltage = 20 kV).
Quartz Crystal Microbalance with Dissipation (QCM-D)
In a Q-sense E4 (Q-sense, Glen Burnie, MD), the surfaces of sensor crystals coated with polymer thin films were equilibrated with PBS buffer at 24 μL min–1. A temperature program was executed between 20 to 49 °C with lipase (or PBS control) preadsorbed at 20 °C. The interval time between temperature steps was 30 min while the frequency was recorded for overtones n = 3, 5, 7, 9. The frequency change (Δfn) and the dissipation change were recorded over time. The change of mass per area was obtained using the Sauerbrey equation with Δm = −CΔfn; (C = 17.7 ng cm–2 s–1 Hz–1) with negligible dissipation changes during erosion.26 The rate of mass-loss for each temperature step was obtained by linear regression after equilibration.
Attachment and Proliferation of Human Mesenchymal Stem Cells (hMSCs) on Polymer Films
Bone marrow derived hMSCs of passage numbers between 2 and 5 (Texas A&M University, College Station, TX) were cultured in MSC basal medium supplemented with SingleQuot’s (Lonza, Walkersville, MD). Compression-molded discs (approximate thickness =250 μm) of (co)polymers from Ty and Hva were cut to fit wells of a 48-well tissue culture polystyrene (TCPS) plate (Corning, Corning, NY). Cells were seeded at a density of 5 × 103 cells cm–2. The hMSCs were cultured at 37 °C in an incubator supplemented with 5% (v/v) of CO2. Cell viability and proliferation were evaluated at time points of 4 h, 4 days, and 7 days. For qualitative fluorescence microscopy imaging, cells were fixed with 4% (w/v) paraformaldehyde for 10 min and then permeabilized with 0.1% (w/v) Triton X-100 for 3 min. Staining was conducted using Alexa Fluor 488 phalloidin for 20 min and Hoechst for 5 min. For quantification of viability, the cell culture medium containing 10% vol. of AlamarBlue reagent (Invitrogen, Carlsbad, CA) was added to wells of live cells after a buffer rinse.27 The fluorescence of the supernatant was measured after 4 h of incubation (λex = 560 nm, λem = 590 nm). Total cell count on polymer substrates was calculated for each time point by comparing fluorescence readouts against a standard curve of known cell numbers. Three independent experiments were carried out (n = 3) with three replicates for each condition.
Results and Discussion
Synthesis and Chemical Structure
A series of polycarbonates from tyrosol (Ty) and homovanillyl alcohol (Hva) (Figure 1A–C) was prepared by condensation polymerization using triphosgene with Ty content of 100, 90, 75, 50, and 0 mol % in the feed. The polymer composition respective to Ty and Hva was confirmed using 1H NMR spectroscopy (Figure S1: 1H NMR spectra, Supporting Information). As illustrated in Figure 1 D, the backbone structure featured sequence isomers with diaryl (head-to-head, h/h), dialkyl (tail-to-tail, t/t) and aryl alkyl (head-to-tail, h/t) carbonates (Table 1: 1H NMR annotations). Chemical shifts of protons in head-to-head, tail-to-tail isomers (aromatic: a1 and b1, aliphatic: d1 and e1) can be distinguished from those in head-to-tail isomers at corresponding positions (aromatic: a2 and b2, aliphatic d2 and e2) for Ty and Hva. Additional protons exclusive to Hva units are annotated accordingly (aromatic: c1 and c2, methoxy: f1 and f2). The ratios of h/h:t/t:h/t shifted from 1:1:1.3 in poly(tyrosol carbonate) to 1:1:2.8 in poly(Hva carbonate) (Table S1: integral ratios in 1H NMR, Supporting Information). We expected dramatic effects of the compositional changes from Ty to Hva and the backbone structure on the properties of these aromatic–aliphatic polycarbonates.
Figure 1.

Chemical structures of (A) tyrosol, (B) homovanillyl alcohol, (C) poly(tyrosol-co-y%homovanillyl carbonate), and (D) polymer sequence isomers at carbonate bond: head-to-head, tail-to-tail, head-to-tail; notations of respective 1H NMR assignments.
Table 1. Chemical Shift Assignment from 1H-NMR Spectra.
| head-to-head (diaryl)/tail-to-tail (dialkyl) |
head-to-tail (aryl
alkyl) |
||
|---|---|---|---|
| proton | chemical shift [ppm] | proton | chemical shift [ppm] |
| a1 (aryl, Ty) | 7.3(m) | a2 (aryl, Ty) | 7.3 (m) |
| a1 (aryl, Hva) | 7.1(m) | a2 (aryl, Hva) | 7.1 (m) |
| b1 (aryl) | 7.3 (m) | b2 (aryl) | 7.1 (m) |
| c1 (aryl, Hva) | 6.9, 6.8 (m) | c2 (aryl, Hva) | 6.7, 6.8 (m) |
| d1 (alkyl) | 2.9 (m) | d2 (alkyl) | 3.0 (m) |
| e1 (alkyl) | 4.3 (m) | e2 (alkyl) | 4.4 (m) |
| f1 (methoxy, Hva) | 3.8 (s, s) | f2 (methoxy, Hva) | 3.7 (s, s) |
s = singlet, m = multiplet.
Physical Properties
The physical properties in the series of polycarbonates from Ty and Hva are listed in Table 2. All polymers were of high molecular weight with Mn values ranging from 117 to 227 × 103 g mol–1. In the dry state, the Tg increased with the content of Hva in the composition from 60 to 74 °C in accordance with the Fox equation.28 This Tg increase may be explained by reduced polymer chain flexibility due to the methoxyl substituent of Hva. As reported elsewhere, additional steric barriers to chain rotations raised Tg in polystyrenes and polymethacrylates.29 In the wet state, the Tg of preconditioned polycarbonates from Ty and Hva was reduced by approximately 10 °C for all compositions; the equilibrium water uptake of the polymer specimens throughout the series was less than 1% (w/w), thus explaining the moderate reduction of the Tg upon hydration. Under physiological conditions all polymers were in the glassy, amorphous state. The polymer surfaces throughout the series were characterized as moderately hydrophobic with water contact angles around 81°.
Table 2. Physical Properties of Polycarbonates from Ty and Hva.
| glass transition temperature (°C) |
|||||
|---|---|---|---|---|---|
| (co)polymer composition Ty/Hva[mol %] | Mn (103 g/mol) | Mw/Mn | dry | wet | static water contact angle(°)a |
| 100/0 | 174 | 1.49 | 60 | 50 | 80 ± 2 |
| 90/10 | 227 | 1.46 | 63 | 54 | 81 ± 1 |
| 75/25 | 212 | 1.42 | 65 | 57 | 82 ± 2 |
| 50/50 | 183 | 1.45 | 69 | 60 | 80 ± 2 |
| 0/100 | 117 | 1.52 | 74 | 63 | 80 ± 2 |
Mean ± standard deviation (SD), n = 5.
To evaluate the applicability of the polycarbonates from Ty and Hva in the fabrication of biodegradable load-bearing devices, the tensile moduli as well as stress and strain at the yield point were determined by tensile testing (see Table 3 and Figure S2, Supporting Information). Changing the ratio of Ty and Hva provided control over the glass transition temperature (Tg) while the mechanical properties remained similar. In the dry state at room temperature, all polymers were characterized as strong and stiff materials with tensile moduli in the range from 1.0 ± 0.1 GPa to 1.2 ± 0.1 GPa and the yield stress (σyield) ranging from 38 ± 2 MPa to 57 ± 3 MPa. The yield strain (εyield) was approximately 6% for all compositions. The polymers were ductile and did not break at 150% strain, which was the maximal elongation that could be measured by our experimental setup. Under simulated physiological conditions at 37 °C, lower tensile moduli (ranging between 0.8 ± 0.2 GPa and 1.2 ± 0.2 GPa) and σyield (27 ± 2 MPa to 43 ± 2 MPa) values of the copolymers were recorded; the εyield values were not affected. The moderate reduction of mechanical performance in the wet state is expected due to hydration and higher temperature during testing. The physical properties and the mechanical performance of the reported polymers were comparable to other resorbable, aromatic polycarbonates. For example, poly(DTE carbonate) (DTE = desaminotyrosyl tyrosine ethyl ester), suitable for biomedical applications, has a modulus of 1.5 GPa in both the dry and the wet states at 22 °C.30 In particular, copolymers of DTE and PEG (5 mol % of PEG, Mn = 5× 103 g mol–1) with 1.2 GPa in the dry state and 0.6 GPa in the wet state match the performance range of polycarbonates from Hva and Ty.31
Table 3. Mechanical properties of polycarbonates from Ty and Hva (mean ± SD, n = 4).
| modulus [GPa] |
yield stress [Mpa] |
yield strainc |
||||
|---|---|---|---|---|---|---|
| copolymer composition Ty/Hva [mol %] | drya | wetb | drya | wetb | drya | wetb |
| 100/0 | 1.1 ± 0.2 | 0.8 ± 0.2 | 38 ± 2 | 27 ± 2 | 6% | 5% |
| 90/10 | 1.0 ± 0.1 | 1.0 ± 0.1 | 42 ± 5 | 27 ± 1 | 6% | 6% |
| 75/25 | 1.0 ± 0.1 | 0.9 ± 0.1 | 42 ± 2 | 32 ± 1 | 6% | 6% |
| 50/50 | 1.2 ± 0.1 | 0.9 ± 0.1 | 57 ± 3 | 34 ± 2 | 8% | 6% |
| 0/100 | 1.2 ± 0.1 | 1.2 ± 0.2 | 54 ± 2 | 43 ± 2 | 7% | 6% |
Tested at room temperature (RT) without preconditioning.
Preconditioned for 24 h and tested in PBS at 37 °C.
SD < 1%.
By contrast, aliphatic polycarbonates, e.g., PTMC, are much more flexible and possess lower moduli in the low megapascal range under physiologically relevant conditions.32
Enzyme-Mediated Surface Erosion in Vitro and Retention of Mechanical Properties
To explore the in vitro degradation behavior, compression-molded specimens of polycarbonates from Hva and Ty were immersed in lipase solution and in PBS as a control. The pH of the degradation medium remained around pH = 7 for all the tested groups measured after 1 week before replacement with fresh solution. At 37 °C in lipase solution, the molecular weight remained unchanged, while linear mass-loss was observed for poly(tyrosol carbonate) and compositions with 90 and 75 mol % Ty (Figure 2A). For poly(tyrosol carbonate) a rate of mass-loss of 0.14 ± 0.01 mg cm–2 d–1 was demonstrated. At this rate, only about half of the mass was retained at a 6-week time point. The last structurally intact specimens were retrieved after 9 weeks with a relative mass-loss of around 80%. The lipase dependent erosion was slower for compositions with 90 and 75 mol % Ty showing rates of 0.07 ± 0.01 mg cm–2 d–1 and 0.03 ± 0.01 mg cm–2 d–1, respectively. However, the mass-loss of compositions with lower than 50 mol % Ty was too slow to be quantified accurately. Poly(homovanillyl carbonate) was stable in lipase solution at 37 °C. In accordance with the mass-loss results, concomitant thickness-loss was observed for poly(tyrosol carbonate) specimens at a rate of 3.0 ± 0.8 μm d–1 (Figure 2B). Likewise, the thickness of specimens containing 90 and 75 mol % Ty decreased over time as well, while no change was observed for compositions with 50 mol % Ty and poly(homovanillyl carbonate). No surface erosion was observed for any composition incubated in PBS, while the long-term stability of poly(tyrosol carbonate) was evaluated for a period of 1 year showing no significant changes in molecular weight.
Figure 2.

(A) Relative mass retention of specimens with compositions of Ty/Hva (mol %) 100/0, 90/10, 75/25, 50/50, and 0/100 incubated in lipase solution at 37 °C (mean ± SD, n = 3). (B) Correlation between relative losses of mass and thickness of poly(tyrosol carbonate) specimens incubated in lipase solution at 37 °C. (mean ± SD, n = 3). The linear regression lines were plotted.
A unique morphology of partially eroded poly(tyrosol carbonate) specimens was revealed in SEM images: while the untreated poly(tyrosol carbonate) specimens showed a smooth surface (Figure 3A), pits and cavities were seen on surfaces when incubated in lipase solution after rinsing with 70% (v/v) ethanol and drying (Figure 3B–E). Over time, pits evolved into regular patterned cavities. The surfaces of specimens were progressively eroded by lipase while control specimens incubated in PBS maintained a smooth surface (Figure 3F).
Figure 3.

SEM morphology of surfaces of compression molded discs of poly(tyrosol carbonate) after incubation in lipase solution: (A) 0 weeks, (B) 1 week, (C) 4 weeks, (D) 6 weeks, (E) 9 weeks, (F) 9 weeks, PBS control; after rinse with 70% (v/v) ethanol. Scale bar =10 μm.
The mechanical properties of poly(tyrosol carbonate), the fastest eroding composition, were evaluated over time in lipase solution and PBS as a control. In both conditions, the modulus, σyield, and εyield values were retained for a period of at least 18 weeks, as shown in Table 4.
Table 4. Mechanical Properties of Poly(tyrosol carbonate) during Erosion (Mean ± SD, n = 4)a.
| modulus [GPa] |
yield stress [Mpa] |
yield strainb |
mass-lossb,c |
|||||
|---|---|---|---|---|---|---|---|---|
| time | lipase | PBS control | lipase | PBS control | lipase | PBS control | lipase | PBS control |
| 24 h | 0.9 ± 0.1 | 0.8 ± 0.2d | 25 ± 2 | 27 ± 2d | 4% | 5%d | 1% | - |
| 1 week | 0.8 ± 0.1 | 0.8 ± 0.1 | 21 ± 2 | 34 ± 5 | 4% | 6% | 7% | - |
| 4 weeks | 0.8 ± 0.1 | 0.9 ± 0.3 | 26 ± 1 | 26 ± 2 | 6% | 5% | 17% | - |
| 18 weeks | 0.9 ± 0.1 | 0.8 ± 0.1 | 22 ± 1 | 27 ± 1 | 4% | 6% | 65% | - |
Specimens were tested while immersed in PBS at 37 °C.
SD < 1%.
Mass loss recorded based on 400 μm thick, rectangular shaped specimens.
Identical to the values shown in Table 3 for preconditioned specimens and included here again for ease of comparison.
When engineering surface eroding devices, however, it has to be considered that due to changes in the specimen’s dimensions the force required to deform the specimens will decrease with time. Nevertheless, the change is predictable, and may be adjusted for by setting the design parameters. In comparison, for bulk-degrading devices, mechanical properties decline in a less controllable manner. For example, poly(dl-lactic acid) specimens are weakened by the possible formation of hollow structures in the bulk with only the retention of an outer shell.33
Glass Transition Temperature and Onset of Enzymatic Erosion
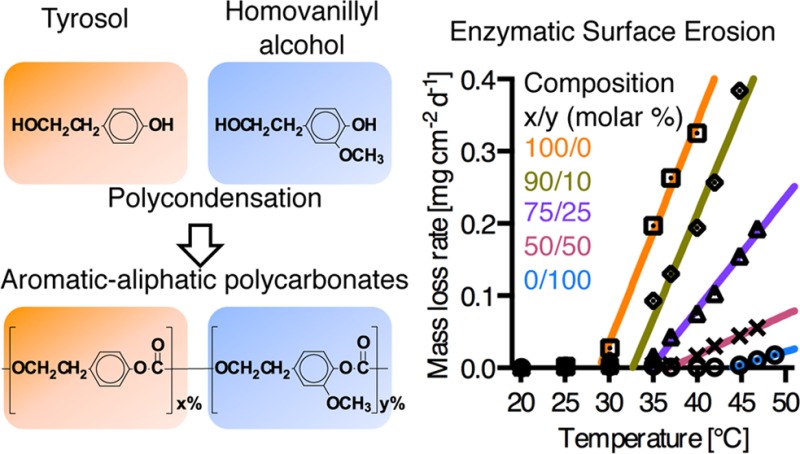
We studied the temperature dependence of enzymatic erosion on thin films of the polycarbonates from Ty and Hva using QCM-D. At the beginning of the experiments, lipase was adsorbed showing comparable adsorption isotherms (Figure S3, Supporting Information) for all compositions with frequency changes (Δf) of −22 Hz and −26 Hz (mass adsorption of 290 to 460 ng cm–2) at 20 °C. At this temperature all thin films were stable against enzymatic degradation. Next, the experiment was continued with stepwise temperature increases in a range between 20 and 49 °C as shown in the QCM-D frequency traces of Figure 4A. At specific, polymer composition-dependent onset temperatures (Tonset), a transition in the frequency traces upon raising the temperature was demonstrated for all the samples, indicating that enzymatic surface erosion started once the temperature of the specimen reached a value that was 20 °C below the wet Tg of fully hydrated samples (see Table 5). We can therefore define an onset temperature for enzymatic surface erosion which can be expected around Tonset = Tg(wet) – 20 °C, where Tg(wet) stands for the glass transition temperature of fully hydrated samples. At each temperature step, a new baseline was recorded after a rapid equilibration of temperature. Once Tonset was reached, frequency increased dramatically even after temperature equilibration, indicating erosion of the polymer films. In control experiments without lipase, no frequency changes beyond baseline equilibration were observed throughout the temperature range.
Figure 4.

(A) QCM-D temperature dependent frequency plots of thin films from compositions of Ty/Hva (mol %) 100/0, 90/10, 75/25, 50/50, and 0/100 with preadsorbed lipase (see also Table 5), and PBS control for 75/25. (B) QCM-D lipase dependent rates of mass-loss dependent on temperature.
Table 5. Temperature-Dependent Erosion Properties from QCM-D and Macroscopic Films.
| (co)polymer composition Ty/Hva [mol %] | QCM-D Tonset [°C] | Δ(Tg/wet – Tonset) [°C] | QCM-D rate of mass-loss at 37 °C [mg cm–2 d–1] | macroscopic rate of mass-loss at 37 °C [mg cm–2 d–1] |
|---|---|---|---|---|
| 100/0 | 29 | 21 | 0.20 ± 0.02 | 0.14 ± 0.01 |
| 90/10 | 33 | 21 | 0.12 ± 0.01 | 0.07 ± 0.01 |
| 75/25 | 34 | 20 | 0.03 ± 0.01 | 0.03 ± 0.01 |
| 50/50 | 37 | 20 | - | - |
| 0/100 | 43 | 20 | - | - |
The rates of mass-loss per unit area derived from the frequency traces are plotted against temperature in Figure 4B. We observed an onset of mass-loss for all compositions. Moreover, when the onset temperature for mass loss was determined by extrapolating the linear section of the curves, the experimentally determined onset temperatures for mass loss were close to Tg(wet) – 20 °C for all compositions. For example, poly(tyrosol carbonate), the polymer with the lowest Tg(wet) in the series (50 °C), has an expected Tonset of 30 °C and was observed to undergo enzymatic surface erosion starting at 29 °C. Most strikingly, even poly(Hva carbonate), which is nonerodible at 37 °C, undergoes enzymatic erosion at 43 °C. As expected, the rates of erosion increase upon heating, reflecting the temperature dependence of enzyme activity. Further differences in the rates of mass-loss among the polymers at a given temperature may be due to structural effects such as steric hindrance at the lipase active site caused by the methoxyaryl groups present in Hva.
Enzyme-mediated degradation of synthetic, degradable polymers is predominantly reported for flexible and rubbery polymers and not for stiff and glassy materials. In this sense, polymer chain flexibility appears to be an important determining factor. It is noteworthy that previous studies suggested increased flexibility of polymer chains in confinement as compared to the bulk: A depression of the bulk Tg value by approximately 20 °C was observed in ultrathin films (thickness below 100 nm) of polystyrene as a model system for confinement.34 We suggest that the polymer chain flexibility at the surface may be increased toward a more rubbery-like behavior compared to a glassy bulk.35 Similarly, the hydrated surface of glassy polymers, such as polycarbonates from Ty and Hva may start to behave in a rubbery manner at about 20 °C below the measured bulk Tg(wet) value. At that temperature, the active site of lipase adsorbed on the polymer surface is able to interact with hydrolyzable carbonate groups to mediate polymer degradation.
Interestingly, the identification of a Tonset for the enzymatic erosion of degradable polymers approximately 20 °C below Tg(wet) may also explain why amorphous poly(l-lactic acid) is susceptible to degradation by Proteinase K at 37 °C, while aromatic polycarbonates such as poly(DTE carbonate) with a Tg of 90 °C21 have a glass transition out of a biologically relevant temperature range and do not exhibit enzymatic erosion under physiological conditions.
Cell Viability and Proliferation
Three polycarbonates from Ty and Hva (100, 90, and 50 mol % Ty) were evaluated as substrates for attachment and proliferation of human mesenchymal stem cells (hMSCs) relative to tissue-culture polystyrene (TCPS). Bone marrow-derived hMSCs were selected for these studies in view of the potential biomedical applications of these high-strength materials in orthopedics. In comparison, all compositions supported cell attachment and proliferation equally with no statistical differences found (Figure 5). After 4 days, hMSCs exhibited spread morphologies on all substrates as observed by confocal microscopy (Figure S5, Supporting Information). Confluence was reached after 7 days in culture on all substrates with cell densities of approximately 6 × 104 cells cm–2.
Figure 5.

Cell viability of hMSCs on polycarbonate substrates of Ty/Hva (mol %) 100/0, 90/10, 50/50 after 4 h, 4 days, and 7 days. Cell density was determined by AlamarBlue assay relative to standards on TCPS (mean ± SE, n = 3).
Conclusions
While the phenomenon of enzymatic surface erosion has been linked to polymer chain flexibility before, it has not been recognized that a rather simple correlation with a polymer’s glass transition temperature can explain why some polymers undergo enzymatic surface erosion while others seem to be unaffected by enzymes. Our results demonstrate that the ability of enzymes to erode a polymer surface is not merely an intrinsic property of the polymer. Instead, it seems that the susceptibility of a polymer to undergo enzymatic surface erosion is determined by the experimental conditions and that a simple correlation (Tonset = Tg(wet) – 20 °C) may allow one to predict if a given polycarbonate will undergo surface erosion under physiological conditions. This new understanding of enzymatic surface erosion can now be used to design innovative polymers that will exhibit enzymatic surface erosion at specific experimental conditions. This is exemplified by the system of new aromatic–aliphatic polycarbonates from the natural phenols tyrosol and homovanillyl alcohol. For selected compositions among these polymers, the hydrated surface layer of the polymer at physiological conditions (37 °C) is flexible enough to allow for enzymatic degradation, while the bulk still maintains the mechanical strength of a glassy material. In future studies, we will explore whether similar correlations can be established for other types of biomedically important polymers such as polyesters and polyamides.
Acknowledgments
This work was supported by Award Number P41EB001046 from the National Institute of Biomedical Imaging and Bioengineering and discretionary funds from the New Jersey Center for Biomaterials at Rutgers University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Biomedical Imaging and Bioengineering or the National Institutes of Health. The authors thank Yangmin Chen for help with degradation studies. Ms. Carole Kantor and Dr. Lauren Macri are acknowledged for assistance editing the manuscript.
Supporting Information Available
Supplementary data include 500 MHz 1H NMR spectra, stress–strain curves, SEM images showing surface morphology, figures showing relative Mw retention, QCM lipase adsorption isotherms, and epifluorescence microscopy images of relevant specimens. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tamada J. A.; Langer R. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpferich A. Biomaterials 1996, 17, 103. [DOI] [PubMed] [Google Scholar]
- von Burkersroda F.; Schedl L.; Göpferich A. Biomaterials 2002, 23, 4221. [DOI] [PubMed] [Google Scholar]
- Heller J. J. Controlled Release 1985, 2, 167. [Google Scholar]
- Brem H.; Tamargo R. J.; Olivi A.; Pinn M.; Weingart J. D.; Wharam M.; Epstein J. I. J. Neurosurg. 1994, 80, 283. [DOI] [PubMed] [Google Scholar]
- Albertsson A. C., Ed. Degradable Aliphatic Polyesters; Advances in Polymer Science Series; Springer: New York, 2002. [Google Scholar]
- Zhu K. J.; Hendren R. W.; Jensen K.; Pitt C. G. Macromolecules 1991, 24, 1736. [Google Scholar]
- Zhang Z.; Kuijer R.; Bulstra S. K.; Grijpma D. W.; Feijen J. Biomaterials 2006, 27, 1741. [DOI] [PubMed] [Google Scholar]
- Chapanian R.; Tse M. Y.; Pang S. C.; Amsden B. G. Biomaterials 2009, 30, 295. [DOI] [PubMed] [Google Scholar]
- Cornacchione L. A.; Qi B.; Bianco J.; Zhou Z.; Amsden B. G. Biomacromolecules 2012, 13, 3099. [DOI] [PubMed] [Google Scholar]
- Christenson E. M.; Anderson J. M.; Hiltner A. J. Biomed. Mater. Res., Part A 2004, 70, 245. [DOI] [PubMed] [Google Scholar]
- Kluin O. S.; van der Mei H. C.; Busscher H. J.; Neut D. Biomaterials 2009, 30, 4738. [DOI] [PubMed] [Google Scholar]
- Dargaville B. L.; Vaquette C.; Peng H.; Rasoul F.; Chau Y. Q.; Cooper-White J. J.; Campbell J. H.; Whittaker A. K. Biomacromolecules 2011, 12, 3856. [DOI] [PubMed] [Google Scholar]
- Bat E.; Harmsen M. C.; Plantinga J. A.; van Luyn M. J.; Feijen J.; Grijpma D. W. J. Controlled Release 2010, 148, e74. [DOI] [PubMed] [Google Scholar]
- Song Y.; Kamphuis M. M.; Zhang Z.; Sterk L. M.; Vermes I.; Poot A. A.; Feijen J.; Grijpma D. W. Acta Biomater. 2010, 6, 1269. [DOI] [PubMed] [Google Scholar]
- Pitt C. G.; Hendren R. W.; Schindler A.; Woodward S. C. J. Controlled Release 1984, 1, 3. [Google Scholar]
- Williams D. F. Eng. Med. 1981, 10, 5. [Google Scholar]
- Yamashita K.; Kikkawa Y.; Kurokawa K.; Doi Y. Biomacromolecules 2005, 6, 850. [DOI] [PubMed] [Google Scholar]
- Tangpasuthadol V.; Pendharkar S. M.; Kohn J. Biomaterials 2000, 21, 2371. [DOI] [PubMed] [Google Scholar]
- Tangpasuthadol V.; Pendharkar S. M.; Peterson R. C.; Kohn J. Biomaterials 2000, 21, 2379. [DOI] [PubMed] [Google Scholar]
- Ertel S. I.; Kohn J. J. Biomed. Mater. Res. 1994, 28, 919. [DOI] [PubMed] [Google Scholar]
- Loscos N.; Hernandez-Orte P.; Cacho J.; Ferreira V. J. Agric. Food Chem. 2007, 55, 6674. [DOI] [PubMed] [Google Scholar]
- Jerman Klen T.; Mozetič Vodopivec B. J. Agric. Food Chem. 2011, 59, 12725. [DOI] [PubMed] [Google Scholar]
- Gris E. F.; Mattivi F.; Ferreira E. A.; Vrhovsek U.; Filho D. W.; Pedrosa R. C.; Bordignon-Luiz M. T. J. Agric. Food Chem. 2011, 59, 7954. [DOI] [PubMed] [Google Scholar]
- Eckert H.; Forster B. Angew. Chem., Int. Ed. Engl. 1987, 26, 894. [Google Scholar]
- Sauerbrey G. Z. Phys. 1959, 155, 206. [Google Scholar]
- Uzunoglu S.; Karaca B.; Atmaca H.; Kisim A.; Sezgin C.; Karabulut B.; Uslu R. Toxicol. Mech. Methods 2010, 20, 482. [DOI] [PubMed] [Google Scholar]
- Fox T. G. Bull. Am. Phys. Soc. 1956, 1, 123. [Google Scholar]
- Mark J. E.Physical Properties of Polymer Handbook; Springer: New York, 2007. [Google Scholar]
- Bedoui F.; Widjaja L. K.; Luk A.; Bolikal D.; Murthy N. S.; Kohn J. Soft Matter 2012, 8, 2230. [Google Scholar]
- Yu C.; Kohn J. Biomaterials 1999, 20, 253. [DOI] [PubMed] [Google Scholar]
- Pego A. P.; Grijpma D. W.; Feijen J. Polymer 2003, 44, 6495. [Google Scholar]
- Grizzi I.; Garreau H.; Li S.; Vert M. Biomaterials 1995, 16, 305. [DOI] [PubMed] [Google Scholar]
- Keddie J. L.; Jones R. A. L.; Cory R. A. Europhys. Lett. 1994, 27, 59. [Google Scholar]
- Ellison C. J.; Torkelson J. M. Nat. Mater. 2003, 2, 695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.