

Abstract
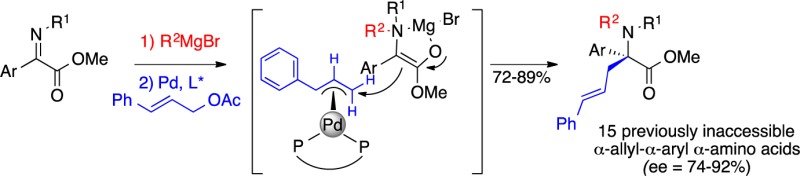
The first asymmetric synthesis of α-allyl-α-aryl α-amino acids by means of a three-component coupling of α-iminoesters, Grignard reagents, and cinnamyl acetate is reported. Notably, the enolate from the tandem process provides a much higher level of reactivity and selectivity than the same enolate generated via direct deprotonation, presumably due to differences in the solvation/aggregation state. A novel method for removal of a homoallylic amine protecting group delivers the free amine congeners. The α-allyl group offers a means to generate further valuable α-amino acid structures as exemplified by ring closing metathesis to generate a higher ring homologue of α-aryl-proline.
Enantiomerically pure α-amino acids and their derivatives are vital synthetic building blocks in organic synthesis and play an integral role in biological research. The α,α-disubstituted α-amino acid structural motif is found in natural products1 and has been utilized by the pharmaceutical industry in numerous antibiotics.2 Peptides with one or more α,α-disubstituted α-amino acid counterparts confer increased stability under physiological conditions and stabilize secondary structure motifs.3 The formation of α,α-disubstituted α-amino acids is difficult and becomes increasingly difficult once structural complexity supersedes analogs of α-Me, α-Et, and α-Bn.4
To date, synthetic access to enantioenriched α-allyl-α-aryl α-amino acids has remained elusive.5 In nucleophilic functionalization, both the Strecker reaction6 and addition to α-iminoesters7 fail. The Strecker reaction succeeds with an α-methyl group when α-aryl groups are employed (Scheme 1, eq 1), but combinations such as α-allyl-α-aryl are difficult due to the relatively unreactive nature of ketoimines8 and difficulty in facial distinction when the two substituents are similar in size.
Scheme 1. General Approaches to α,α-Disubstituted α-Amino Acids.

Electrophilic functionalization has been tremendously successful for generating chiral α-substituted α-amino acids,9 but the enolates required to generate the α,α-disubstituted α-amino acid counterparts are very hindered (Scheme 1, eq 2). As a result, variable yields are obtained when R1 ≠ H,5a,5b,5d and mixed results have been obtained when R1 = Ar.5b,5d Use of cyclic surrogates has improved the outcome of electrophilic functionalization via asymmetric allylic alkylation, but phenylglycine analogs proved sterically hindered and unsuccessful to date (Scheme 1, eq 3).10 To the best of our knowledge, use of achiral acyclic enolates to generate a range of chiral α-allyl-α-aryl α-amino acids has not been successful.
Herein, we report the N-alkylation/asymmetric π-allylation of α-iminoesters (Scheme 2). The tandem process generates an enolate form possessing increased reactivity, which differs from the corresponding enolate generated via direct deprotonation. The in situ generated enolate allows the construction of a diverse group of enantioenriched α-allyl-α-aryl α-amino acids that have not been accessed to date (Scheme 1, eq 4).
Scheme 2. Tandem N-Alkylation/π-Allylation of α-Iminoesters.
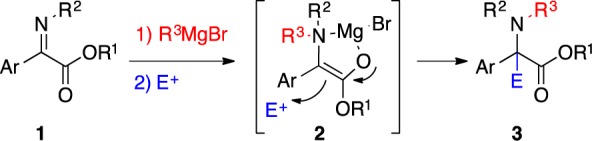
Kagan and Fiaud first reported umpolung N-addition of unstabilized anions to α-iminoesters in 1970.11 Shimizu has expanded upon this work with doubly activated iminomalonates and organoaluminums.12 Recently, we have reported the first tandem reaction to utilize umpolung addition of alkyl Grignard into α-iminoesters to generate racemic α,α-disubstituted α-amino acids.13 In spite of these advances, this umpolung addition has never been combined with an asymmetric process to take advantage of the reactive nucleophile 2 that is produced (Scheme 2).
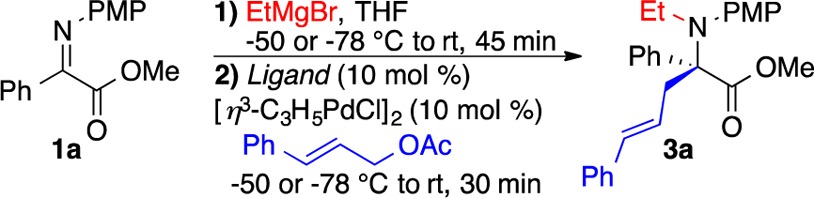
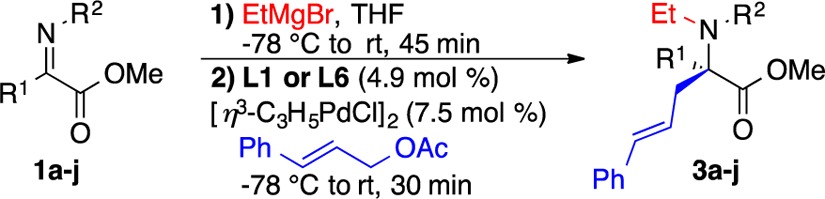
Parallel microscale experimentation (PME), a valuable tool for rapidly screening conditions and drawing out trends,14 was used to optimize the tandem N-alkylation/π-allylation of 1a.15 Table 1 displays results for 11 of >190 enantiopure mono- and bisphosphine ligands conducted at −50 °C utilizing PME.16 Nearly all ligands gave minimal conversion and selectivity, but axial chiral bisphosphines, such as L1, L2, and L5–L9 (Figure 1), were most effective in generating 3a as confirmed in larger scale reactions at −78 °C (Table 1). Trost ligands possessing central chirality and Pfaltz ligands with planar chirality (Table 1, entries 4 and 5) were unsuccessful, showing both poor reactivity and poor selectivity.
Table 1. Results from PME and Scale Up Reactions with Diverse Ligands.
| bench
scaleb |
||||
|---|---|---|---|---|
| entry | ligand (Figure 1) | PMEa ee (%)c | conv (%)d | ee (%)c |
| 1 | BINAP, L1 | 40 | >90 | 80 |
| 2 | BINAP, L1 | >90 | 66e | |
| 3 | H8-BINAP, L2 | 20 | >90 | 48 |
| 4 | DACH Trost, L3 | 0 | <10 | ND |
| 5 | Pfaltz, L4 | 0 | 15 | 14 |
| 6 | SEGPHOS, L5 | 42 | >90 | 40 |
| 7 | DIFLUORPHOS, L6 | 38 | >90 | 88 |
| 8 | TUNEPHOS, L7 | 36 | >90 | 78 |
| 9 | SONIPHOS, L8 | 50 | >90 | 72 |
| 10 | P-PHOS, L9 | 36 | >90 | 80 |
| 11 | WALPHOS, L10 | 36 | >90 | 48 |
| 12 | MANDYPHOS, L11 | 34 | >90 | 66 |
8.6 μmol of 1a at −50 °C.
0.372 mmol of 1a at −78 °C.
Determined by chiral stationary phase (CSP) HPLC.
Conv determined by 1H NMR.
Conducted at −55 °C to simulate PME conditions.
Figure 1.

Ligands in Table 1.
With the optimal conditions, a range of α-iminoesters reacted with satisfactory yields and enantioselectivity (3a–j) (Table 2). Notably, the reaction was equally effective with keto substituents (R1) containing electron-donating and electron-withdrawing aryl groups (Table 2, entries 3–5; see ref (5d)), while ortho-substituted aryls (2-Me, 2-OMe) resulted in lower yields of the desired product.16 Thiophene could also be employed to good effect (Table 2, entry 8), but the indole tandem product was not stable.16 Variation from the PMP activating group on nitrogen to phenyl, p-Me2NC6H4 or 3,4-Me2C6H3 (Table 2, entries 9–11), had a small effect on selectivity, whereas moving from the methyl ester to the ethyl or benzyl ester reduced selectivity further.16
Table 2. Variation of the α-Iminoester in the Tandem N-Alkylation/π-Allylation.
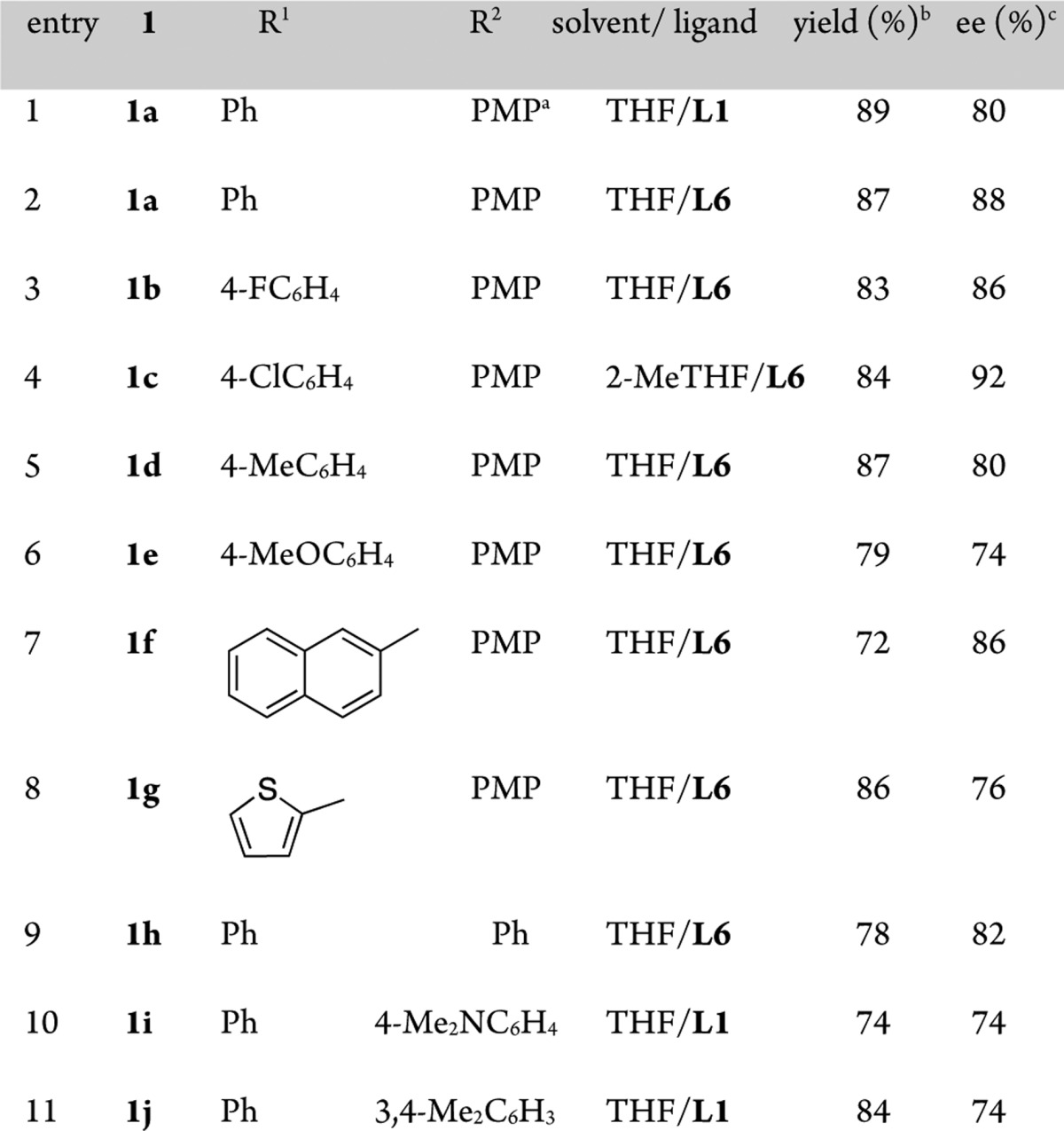
PMP = 4-MeOC6H4.
Isolated yield.
Determined by CSP HPLC.
In contrast to most other reports of N-alkylation of α-iminoesters,17 a range of Grignard reagents could be employed here to provide unique N-alkyl α-aryl-α-allyl α-amino acid derivatives (Table 3, entries 1–5). Notably, more functional Grignards provided terminal silyl and alkenyl derivatives (Table 3, entries 2–5). For alkenyl substrates, the alkene must be distal to the reacting center as allyl, benzyl, and vinyl Grignard reagents were not successful.
Table 3. Variation of the Grignard in the Tandem N-Alkylation/π-Allylation (1a).
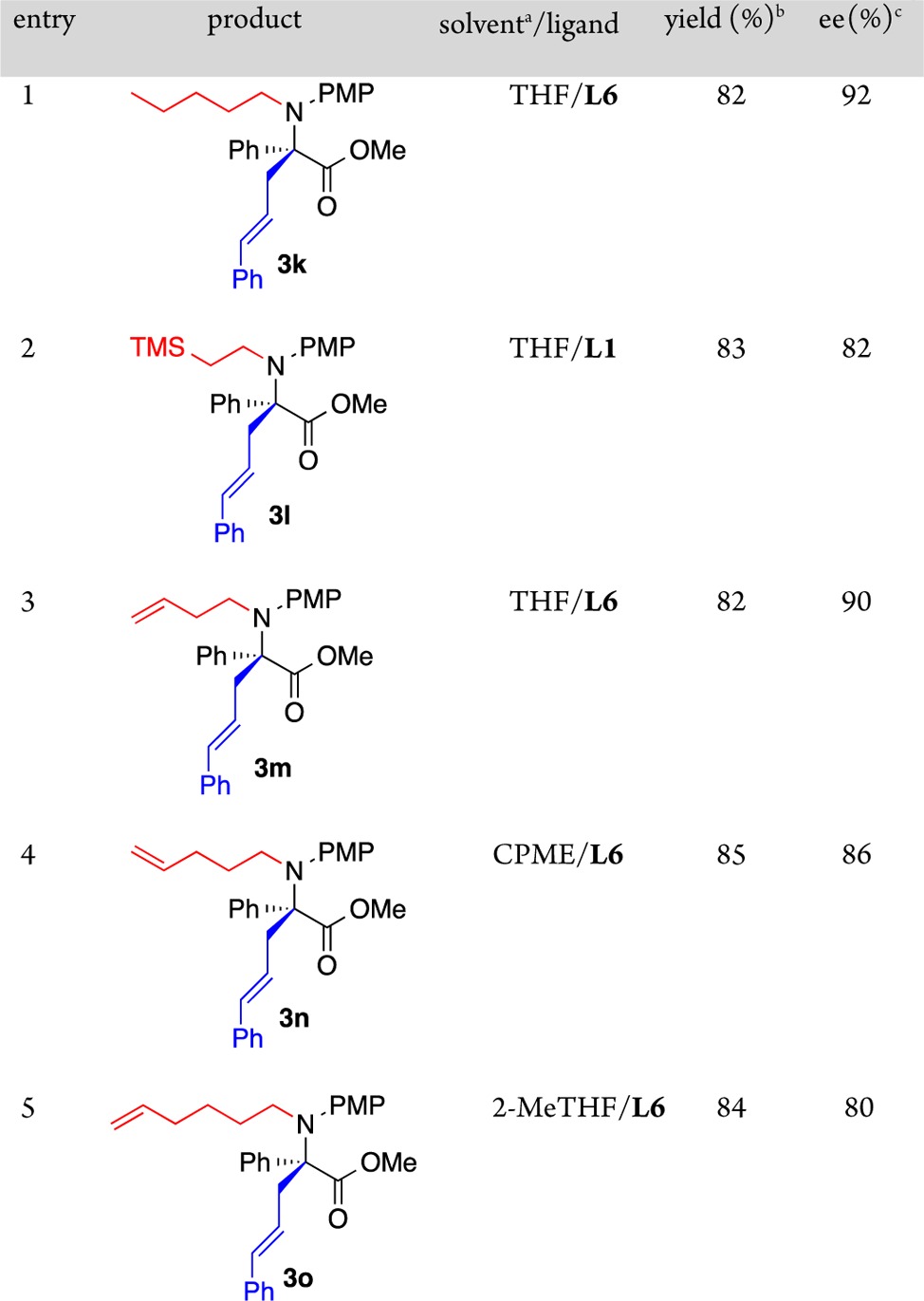
CPME = cyclopentyl methyl ether.
Isolated yield.
Determined by CSP HPLC.
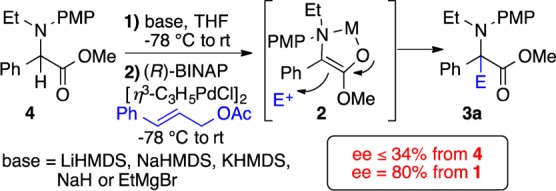
Reasoning that the same products 3 ought to be available from racemic α-phenylgylcine derivatives, the reaction in Scheme 3 was undertaken. A wide range of bases provided good conversion (>85%), but poor enantioselectivity (ee =18–34%), compared to the tandem reaction (Table 2).16 Notably, treatment of 4 with EtMgBr should generate the exact same enolate (2) as in the tandem reaction (2) (Scheme 2). However, this enolate provided low conversion and significantly lower selectivity. We conclude that the exact structural form of the enolate is critical to the outcome. Specifically, the aggregate of 2 obtained from 1 differs from that obtained from 4.18
Scheme 3. Allylation of N-Alkylated Intermediate.
In line with this reasoning, a strong solvent dependence was observed, with ethereal solvents being far superior. Depending on the exact structure of the Grignard reagent, subtle differences were seen among the ethereal solvents THF, 2-MeTHF, Et2O, and CPME (see Tables 2 and 3). On the other hand, amine base additives reduced the selectivity to less than 30% ee.16 Together, these results suggest16 a delicate balance where the most selective species is a solvated dimer, such as B, that lies in between poorly selective forms including a deaggregated form, such as monomer C, and a less solvated form, such as a tetramer or dimer A (Figure 2).19
Figure 2.

Possible enolate forms in solution (sol = solvent).
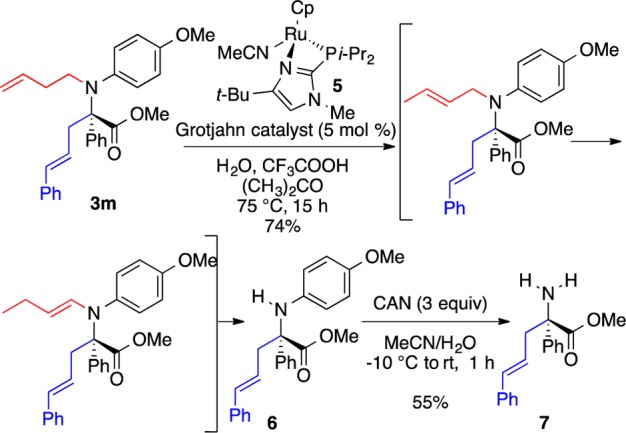
The method provides ready access to N-alkyl α-allyl-α-aryl α-amino acids including a number of N-alkyl substitutions that would be difficult to generate via reductive amination. In addition, routes to the free amine analogs were assessed by means of functionalized Grignard reagents (Table 3), which also offer the option for removal. Surprisingly, the β-silyl group in product 3l could not be removed with fluoride or other nucleophiles under a variety of acidic and basic conditions. Recent reports on the isomerization of alkenes along a longer alkyl chain inspired us to examine whether homoallyl analog 3m could be transformed to the primary amine as shown in Scheme 4. An extensive survey of olefin isomerizing catalysts20 revealed that the Grotjahn catalyst21 was uniquely effective. Elevated temperatures, combined with the addition of water and trifluoroacetic acid, caused the terminal alkene 3m to isomerize by two carbons to the enamine, which underwent hydrolysis in situ. Notably, the sensitive styryl moiety remained intact. Treatment of the product 8 with CAN provided the primary amine 7. Thus, the reaction method described herein can be used to generate both the free amino and the N-alkyl (via PMP deprotection)16 versions of novel α-allyl-α-aryl α-amino ester derivatives (Scheme 4).
Scheme 4. Reduction of the Tandem N-Alkylation/π-Allylation Product to the α-Allyl-α-Aryl α-Amino Acids.
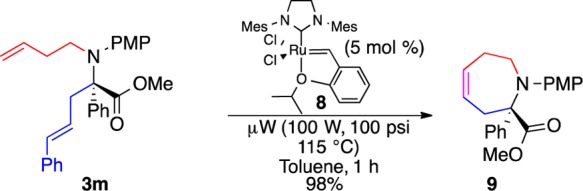
In an effort to access an even greater diversity of the α-allyl-α-aryl α-amino ester derivatives, a preliminary study of metathesis to functionalize the allyl group was undertaken. Microwave irradiation (1 h vs 20–24 h under thermal conditions) was discovered to be critical in the ring closing metathesis of 3m to form cyclic analog 9 efficiently (Scheme 5). Cyclic α-amino acid derivatives are of great synthetic and pharmaceutical interest,22 but asymmetric methods for the synthesis of higher ring homologues of α-substituted prolines23 have not received much attention.24
Scheme 5. Synthesis of Higher Ring Homologue of α-Substituted Proline.
In summary, we have disclosed the first asymmetric tandem N-alkylation/π-allylation of α-iminoesters, which gives rise to complex enantioenriched α-allyl-α-aryl α-amino acids in one step from three commercially available components. This report represents the first enantioselective synthesis of this class of compounds beyond α-allyl-α-phenylglycine. The dramatic effect of enolate aggregation observed herein provides a cautionary tale for other systems. The nature of these effects are the subject of further exploration.
Acknowledgments
We are grateful to the NIH (GM-087605) and the NSF (CHE0911713) for financial support of this research. The NSF provided for funding of the High Throughput Laboratory (GOALI CHE-0848460). Partial instrumentation support was provided by the NIH for MS (1S10RR023444) and NMR (1S10RR022442) and by the NSF for an X-ray diffractometer (CHE 0840438). The invaluable assistance of Dr. D. B. Grotjahn (San Diego State) for providing his catalyst and expertise on alkene isomerization. Our collaboration with Dr. P. Carroll (UPenn) in obtaining the crystal structure is gratefully acknowledged. Also, the contributions of J. Hun (UPenn) and the assistance from Dr. C. Stanciu (Merck) and Dr. S. D. Dreher (Merck) with the PME experiments.
Supporting Information Available
Experimental procedures and characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Xie W.; Zou B.; Pei D.; Ma D. Org. Lett. 2005, 7, 2775. [DOI] [PubMed] [Google Scholar]
- a Savage S. A.; Waltermire R. E.; Campagna S.; Bordawekar S.; Toma J. D. R. Org. Process Res. Dev. 2009, 13, 510. [Google Scholar]; b Washburn W. N.; et al. Bioorg. Med. Chem. Lett. 2004, 14, 3525. [DOI] [PubMed] [Google Scholar]; c Ma D. W.; Tian H. Q.; Zou G. X. J. Org. Chem. 1999, 64, 120. [DOI] [PubMed] [Google Scholar]; d Stilz H. U.; Jablonka B.; Knolle J.; Paulus E. F.; Zoller G. J. Med. Chem. 1996, 39, 2118. [DOI] [PubMed] [Google Scholar]
- a Graver A.; Konig B. Eur. J. Org. Chem. 2009, 30, 5099. [Google Scholar]; b Venkatraman J.; Shankaramma S. C.; Balaram P. Chem. Rev. 2001, 101, 3131. [DOI] [PubMed] [Google Scholar]
- a Liu Z.; Mehta S. J.; Hruby V. J. Organic Preparations and Procedures International: The New Journal for Organic Synthesis 2012, 44, 222. [Google Scholar]; b Cativiela C.; Diaz-De-Villegas M. D. Tetrahedron: Asymmetry 2007, 18, 569. [Google Scholar]; c Vogt H.; Brase S. Org. Biomol. Chem. 2007, 5, 406. [DOI] [PubMed] [Google Scholar]
- a Ooi T.; Takeuchi M.; Kameda M.; Maruoka K. J. Am. Chem. Soc. 2000, 122, 5228. [Google Scholar]; b Ooi T.; Takeuchi M.; Ohara D.; Maruoka K. Synlett 2001, 7, 1185. [Google Scholar]; c Berger R.; Duff K.; Leighton J. L. J. Am. Chem. Soc. 2004, 126, 5686. [DOI] [PubMed] [Google Scholar]; d Application of ref (5b) utilizing an analog of phenylglycine gave a 35% yield and 45% ee; see: Metro T.-X.; Cochi A.; Pardo D. G.; Cossy J. J. Org. Chem. 2011, 76, 2594. [DOI] [PubMed] [Google Scholar]
- Wang J.; Liu X.; Feng X. Chem. Rev. 2011, 111, 6947. [DOI] [PubMed] [Google Scholar]
- a Wieland L. C.; Vieira E. M.; Snapper M. L.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fu P.; Snapper M. L.; Hoveyda A. H. J. Am. Chem. Soc. 2008, 130, 5530. [DOI] [PubMed] [Google Scholar]; c Basra S.; Fennie M. W.; Kozlowski M. C. Org. Lett. 2006, 8, 2659. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zhuang W.; Saaby S.; Jorgensen K. A. Angew. Chem., Int. Ed. 2004, 43, 4476. [DOI] [PubMed] [Google Scholar]
- Riant O.; Hannedouche J. Org. Biomol. Chem. 2007, 5, 873–888. [DOI] [PubMed] [Google Scholar]
- Maruoka K. Org. Process Res. Dev. 2008, 12, 679. [Google Scholar]
- a Trost B. M.; Ariza X. Angew. Chem., Int. Ed. Engl. 1997, 36, 2635. [Google Scholar]; b Chen W.; Hartwig J. F. J. Am. Chem. Soc. 2013, 135, 2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiaud J. C.; Kagan H. Tetrahedron Lett. 1970, 11, 1813. [DOI] [PubMed] [Google Scholar]
- a Mizota I.; Matsuda Y.; Kamimura S.; Tanaka H.; Shimizu M. Org. Lett. 2013, 15, 4206. [DOI] [PubMed] [Google Scholar]; b Shimizu M.; Kurita D.; Mizota I. Asian J. Org. Chem. 2013, 2, 208. [Google Scholar]; c Shimizu M.; Takao Y.; Katsurayama H.; Mizota I. Asian J. Org. Chem. 2013, 2, 130. [Google Scholar]; d Mizota I.; Tanaka K.; Shimizu M. Tetrahedron Lett. 2012, 53, 1847. [Google Scholar]; e Niwa Y.; Shimizu M. J. Am. Chem. Soc. 2003, 125, 3720. [DOI] [PubMed] [Google Scholar]; f Niwa Y.; Takayama K.; Shimizu M. Bull. Chem. Soc. Jpn. 2002, 75, 1819. [Google Scholar]; g Shimizu M.; Niwa Y. Tetrahedron Lett. 2001, 42, 2829. [Google Scholar]
- Dickstein J. S.; Fennie M. W.; Norman A. L.; Paulose B. J.; Kozlowski M. C. J. Am. Chem. Soc. 2008, 130, 15794. [DOI] [PubMed] [Google Scholar]
- a Dreher S. D.; Dormer P. G.; Sandrock D. L.; Molander G. A. J. Am. Chem. Soc. 2008, 130, 9257. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Metz A. E.; Berritt S.; Dreher S. D.; Kozlowski M. C. Org. Lett. 2012, 3, 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For racemic entries to α-allyl compounds from α-iminoesters, see ref (12)d, e.
- See Supporting Information for further details.
- Dickstein J. S.; Kozlowski M. C. Chem. Soc. Rev. 2008, 37, 1166. [DOI] [PubMed] [Google Scholar]
- Meyers A. I.; Knaus G.; Kamata K.; Ford M. E. J. Am. Chem. Soc. 1976, 98, 567. [Google Scholar]
- Magnesium enolates exist as monomers or as dimers bridged through either the halide or the enolate oxygen. The coordination sphere of the magnesium ranges from tetrahedral to octahedral with solvent molecules occupying remaining coordination sites. For the X-ray crystal structure of a dimeric bidentate enolate with Mg, see:; a Di Noto V.; Bandoli G.; Dolmella A.; Zarli B.; Vivani M.; Vidali M. J. Chem. Cryst. 1995, 25, 375. [Google Scholar]; For the X-ray crystal structure of dimeric monodentate enolates with Mg, see:; b Allan J. F.; Clegg W.; Henderson K. W.; Horsburgh L.; Kennedy A. R. J. Organomet. Chem. 1998, 559, 173. [Google Scholar]; c Williard P. G.; Salvino J. M. J. Chem. Soc., Chem. Commun. 1986, 153. [Google Scholar]
- a Arisawa M.; Terada Y.; Takahashi K.; Nakagawa M.; Nishida A. J. Org. Chem. 2006, 71, 4255. [DOI] [PubMed] [Google Scholar]; b Nelson S. G.; Bungard C. J.; Wang K. J. Am. Chem. Soc. 2003, 125, 13000. [DOI] [PubMed] [Google Scholar]
- a Erdogan G.; Grotjahn D. B. J. Am. Chem. Soc. 2009, 131, 10354. [DOI] [PubMed] [Google Scholar]; b Larsen C. R.; Grotjahn D. B. J. Am. Chem. Soc. 2012, 134, 10357. [DOI] [PubMed] [Google Scholar]
- Park K. H.; Kurth M. J. Tetrahedron 2002, 58, 8629. [Google Scholar]
- Seebach D.; Boes M.; Naef R.; Schweizer W. B. J. Am. Chem. Soc. 1983, 105, 5390. [Google Scholar]
- For reviews on the asymmetric synthesis of cyclic α,α-disubstituted α-amino acids, see:; a Cativiela C.; Ordonez M. Tetrahedron: Asymmetry 2009, 20, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cativiela C.; Diaz-De-Villegas M. D. Tetrahedron: Asymmetry 2000, 11, 645. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.