

Abstract
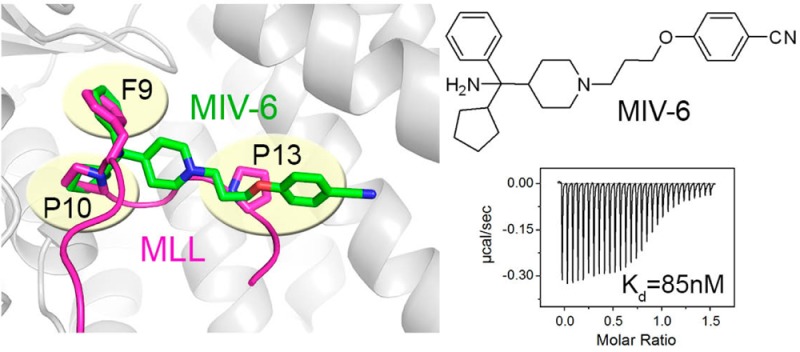
The protein–protein interaction (PPI) between menin and mixed lineage leukemia (MLL) plays a critical role in acute leukemias, and inhibition of this interaction represents a new potential therapeutic strategy for MLL leukemias. We report development of a novel class of small-molecule inhibitors of the menin–MLL interaction, the hydroxy- and aminomethylpiperidine compounds, which originated from HTS of ∼288000 small molecules. We determined menin–inhibitor co-crystal structures and found that these compounds closely mimic all key interactions of MLL with menin. Extensive crystallography studies combined with structure-based design were applied for optimization of these compounds, resulting in MIV-6R, which inhibits the menin–MLL interaction with IC50 = 56 nM. Treatment with MIV-6 demonstrated strong and selective effects in MLL leukemia cells, validating specific mechanism of action. Our studies provide novel and attractive scaffold as a new potential therapeutic approach for MLL leukemias and demonstrate an example of PPI amenable to inhibition by small molecules.
Introduction
Protein–protein interactions (PPIs) play a critical role in many biological processes and in a broad spectrum of human diseases, including cancer,1 and small-molecule modulators of PPIs are highly desired to serve as chemical tools and potential therapeutics. Despite these needs, identification of small-molecule inhibitors of PPIs is considered challenging due to multiple reasons, including large interacting areas, lack of well-defined binding pockets, and flexibility of residues on PPI interfaces,2,3 significantly limiting successful development of PPI modulators. Discovery of cell-permeable small-molecule inhibitors of PPIs provides an additional challenge due to increased molecular weight of PPI inhibitors often required to achieve high potency.4 On the other hand, recent successes with a number of PPI inhibitors3,5−8 demonstrate that some PPIs are amenable to inhibition by small molecules. More importantly, advancing small-molecule inhibitors of PPIs into clinical trials, such as the Bcl-2 protein family inhibitor ABT-2639 and the MDM2 inhibitor RG7112,10 provides an important proof-of-principle that small-molecule inhibitors of PPIs may serve as novel therapeutic agents and justifies the efforts in developing novel PPI inhibitors.
The protein–protein interaction between menin and mixed lineage leukemia (MLL) plays a critical role in acute leukemias with translocations of the MLL gene.11 Fusion of MLL with one out of over 60 partner genes results in expression of chimeric MLL fusion proteins, which enhance proliferation of hematopoietic cells and block hematopoietic differentiation, ultimately leading to acute leukemias.12 The MLL leukemias represent a heterogeneous group of acute myeloid leukemias (AML) and acute lymphoblastic leukemias (ALL), accounting for about 5–10% of acute leukemias in adults13 and ∼70% of acute leukemias in infants.14 Patients with MLL leukemias have very poor prognosis and respond poorly to currently available treatments,12,15 with only about 35% overall five-year survival rate,16 emphasizing the urgent need for development of novel therapies.
The MLL fusion proteins preserve the N-terminal MLL fragment of approximately 1400 amino acids fused with the fusion partner.15,17−19 Importantly, the N-terminal fragment of MLL, retained in all MLL fusion proteins, is involved in the interactions with menin,11,20,21 and this interaction plays a critical role in the MLL–fusion protein mediated leukemogenic transformations.11,22 Menin is a highly specific binding partner of MLL and MLL fusion proteins required for regulation of target genes expression, including HOXA9 and MEIS1 genes, both of which are essential for leukemogenic activity of MLL fusions.11 Therefore, menin represents a critical oncogenic cofactor of MLL fusion proteins in acute leukemias, and disruption of the protein–protein interaction between menin and MLL with small molecules represents a very attractive therapeutic strategy to develop new targeted drugs for the MLL leukemia patients.
Menin interacts with two MLL fragments located within the N-terminal region, with MBM1 (menin-binding motif 1 corresponding to MLL4–15) representing the high affinity menin binding motif.20 We previously reported a high resolution crystal structure of the menin–MBM1 complex, which demonstrated that MLL binds to a very large central cavity on menin.23,24 Furthermore, we developed the thienopyrimidine class of the menin–MLL inhibitors, which represents the first class of small molecules targeting this protein–protein interaction reported to date.23,25 The thienopyrimidine compounds bind to the MLL binding site on menin and mimic a subset of the critical MLL interactions with menin but are incapable of interacting with the P10 pocket,23 which appears to limit their further chemical optimization into more potent, drug-like molecules. Furthermore, the MLL derived peptidomimetics were recently reported as potent in vitro inhibitors of the menin–MLL interaction;26 however, cellular activity of these compounds was not provided, suggesting that optimization of their properties to identify therapeutically useful compounds is required. These limitations, together with the pressing need to develop menin–MLL inhibitors suitable for in vivo studies in animal models of MLL leukemia, emphasize a clear demand for identification of novel menin–MLL inhibitors with distinct chemical scaffolds suitable for optimization of potency and physicochemical properties.
Here, we report development of a novel class of hydroxy- and aminomethylpiperidine inhibitors of the menin–MLL interaction, which we initially discovered by HTS of ∼288000 small molecules. These compounds directly bind to menin at the MLL binding site and specifically block the menin–MLL protein–protein interaction. Crystal structures of menin–inhibitor complexes guided medicinal chemistry optimization, resulting in MIV-6R (IC50 = 56 nM and Kd = 85nM), which demonstrated strong and selective activity in MLL leukemia cells. Overall, this work provides a novel and attractive molecular scaffold together with extensive structural data for menin–ligand complexes, thus paving the way toward further development of these compounds into chemical probes or new potential therapeutics. Importantly, our studies demonstrate the success of HTS in identifying PPI inhibitors and how an effective structure-guided optimization enabled development of nanomolar inhibitors, providing yet another example of PPI interface amenable to inhibition by small molecules.
Chemistry
Synthesis of Hydroxy- and Aminomethylpiperidine Inhibitors of Menin–MLL Interaction
The HTS hit MIV-1 (1), 4-(3-(4-benzhydrylpiperidin-1-yl)propoxy)benzonitrile (compound 3) and 1-(3-phenoxypropyl)piperidin-4-yl)diphenylmethanol, (compound 4, MIV-2), Table 1, were readily prepared starting from either diphenyl(piperidin-4-yl)methanol (compounds 1 and 4) or 4-benzhydrylpiperidine (compound 3) using a one-pot two-step procedure.27 Full experimental details can be found within Supporting Information. Synthesis of the hydroxymethyl piperidine aryl and diaryl analogues 2, 5–10, 12, 18, and 21 (Table 1, Figures 4–5), cyclopentyl analogues 11 and 13–17 (Figure 4), and head group and tail group piperidine analogues 19–20 (Figure 5) were prepared according to Schemes 1–4. Our initial strategy, involving preinstallation of the nitrile tail group prior to Grignard addition, led to final products contaminated with trace amounts of an inseparable side-product presumed to result from Grignard addition to the nitrile (R3 = CN). To circumvent this issue, we performed a selective alkylation of piperidines 22a–b using 1-bromo-3-chloropropane to afford chloride intermediates 23a–b in good yield (Scheme 1). Addition of Grignard reagents to 23a–b provided the crude carbinol chlorides in high conversion. Subsequent alkylation using the appropriate phenol reagent led to final target compounds (Scheme 1).
Table 1. Structures and IC50 Values for Hydroxymethylpiperidine Inhibitors of the Menin–MLL Interaction.
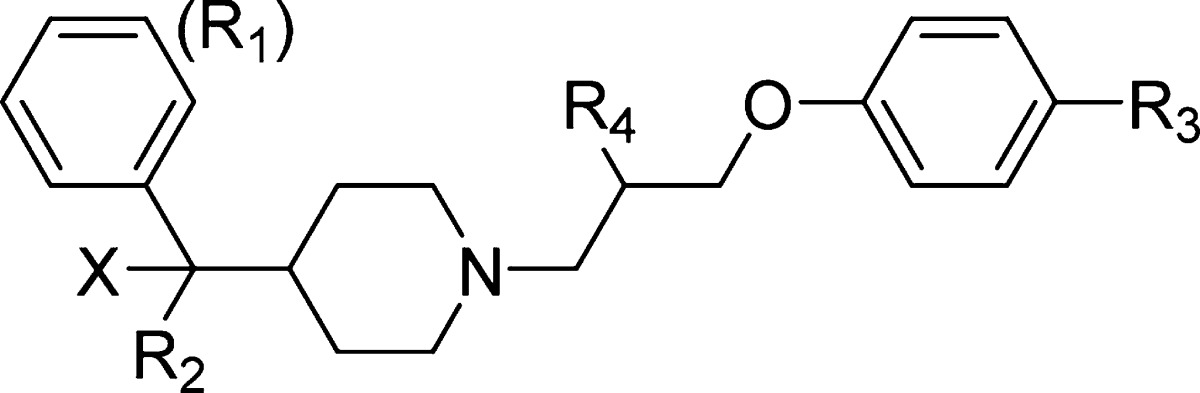
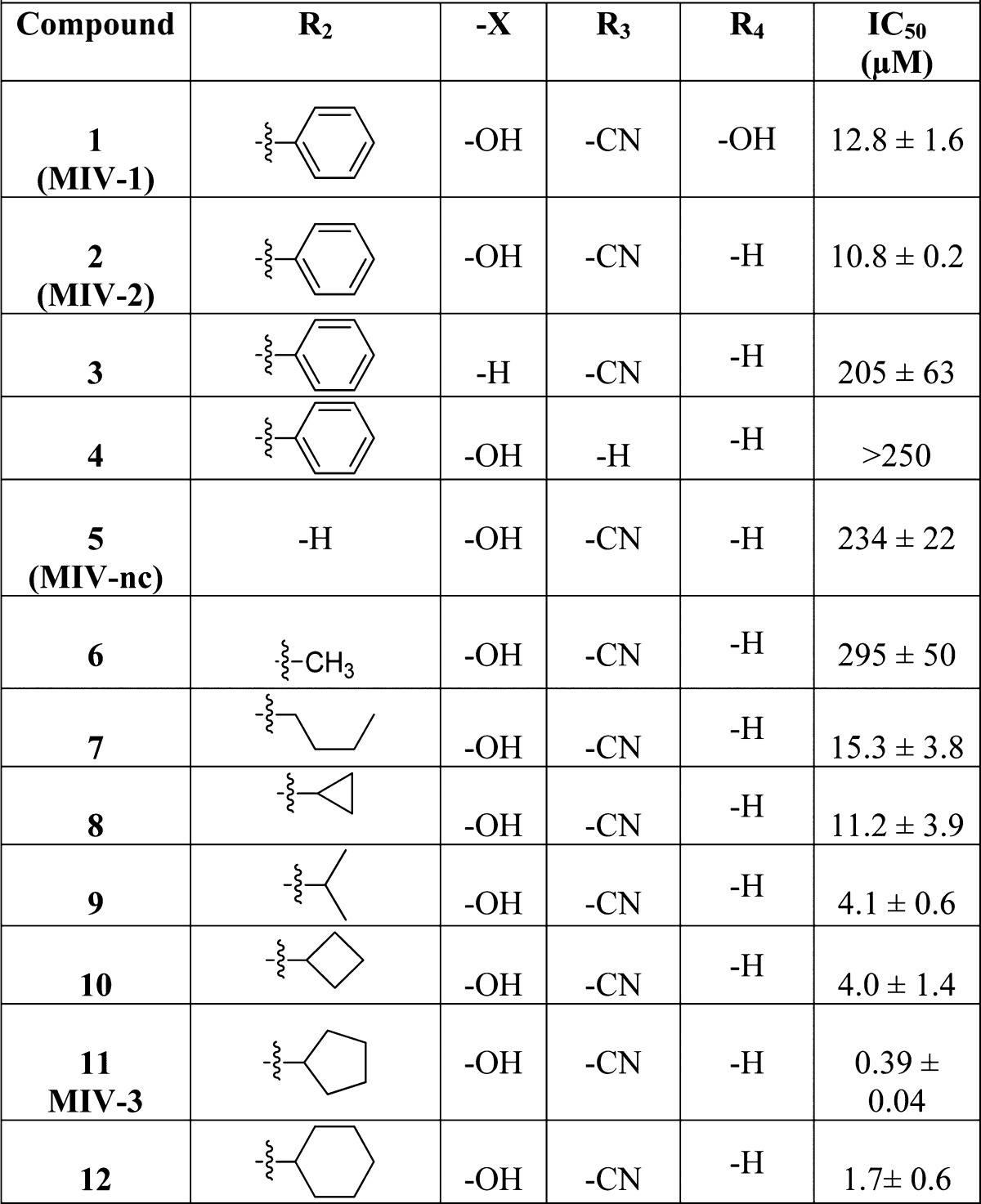
Figure 4.

Structure-based optimization of hydroxymethylpiperidine compounds in the head group region. (a) General structure of the hydroxymethylpiperidine class showing positions of R1 and R2 substituents, which bind to the F9 and P10 pockets, respectively. (b) Structures and inhibitory activities of MIV-3 analogues with modifications introduced at R1. IC50 values are provided for the racemic mixtures. (c) Crystal structure of menin in complex with MIV-4 (green carbon atoms, PDB code 4GO6) showing the interaction of fluorine atom (light blue) in MIV-4 and menin backbone (gray carbon atoms). Close distances involving fluorine from MIV-4 are shown as dashed lines. (d) Structures and activities for analogues with modifications introduced simultaneously at R1 and R2. IC50 values are provided for the racemic mixtures, unless indicated. (e) Crystal structure of menin in complex with MIV-5 (green carbon atoms, PDB code 4GO5) showing water mediated hydrogen bond with Tyr276.
Figure 5.

Structure-based optimization of polar interactions with menin. (a) Structures and activities of analogues with substitution of a hydroxyl (marked as -X) at the head group region. IC50 values are provided for the racemic mixtures, unless indicated. (b) Comparison of the crystal structures of menin in complex with MIV-6R (bottom, PDB code 4GO8) and MIV-3R (top) showing polar interactions between the head group region and menin. Inhibitors are shown in stick representation (green carbon atoms); menin is presented as a gray ribbon. Selected side chains on menin involved in contacts with compounds are shown in stick representation; w represents a water molecule. Hydrogen bonds are shown as dashed lines with distances in angstroms. E359 is present in two alternative conformations. (c) Structures and activities of analogues with substitution of nitrile at the tail group region. IC50 values are provided for the racemic mixtures. (d) Comparison of the crystal structures of menin in complex with MIV-7 (bottom, PDB code 4GO7) and MIV-5 (top) showing the interactions of inhibitor tail groups with menin. Structural representation of the protein and ligand and labeling the same as in (b).
Scheme 1. Synthesis of Aryl Hydroxymethylpiperidines 2, 5–10, 12, 18, and 21.

Scheme 4. Synthesis of Sulfonamide Containing Hydroxymethylpiperidine 20.

Synthesis of cyclopentyl hydroxymethyl piperidines 11 (MIV-3) and 13–17 is outlined in Scheme 2. To circumvent low yields due to competing reduction of the sterically hindered ketones by alkyl Grignard reagents,28 we employed a strategy involving introduction of the saturated cycloalkyl followed by a second aryl or alkyl group (Scheme 2). Thus, initial introduction of the cycloalkyl was accomplished using a known two-step procedure starting from N-benzylpiperidine-4-carbonitrile to give key starting aminoketone 24 (Scheme 2).29 Subsequent one-pot double alkylation using 24 and 1-bromo-3-chloropropane afforded piperidine 25 in 78% yield. Grignard or stabilized aryl lithium additions afforded final target compounds 11 and 13–17 in moderate to good yield. Overaddition of these aryl Grignard and lithium reagents to the nitrile was not evident for substrate 25, and all final compounds displayed excellent purity (>98%).
Scheme 2. Synthesis of Cyclopentyl Hydroxymethylpiperidines 11 and 13–17.

Preparation of tertiary carbinamine, 4-(3-(4-(cyclopentyl(amino)(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile (compound 19, MIV-6), was accomplished via solvolysis of carbinol 11 in the presence of sodium azide in chloroform and sulfuric acid (Scheme 3). The crude azide was reduced using palladium on carbon with atmospheric hydrogen in EtOH to afford amine 19. Despite efforts to screen for alternative conditions to improve solvolysis and formation of the azide intermediate, facile formation of a major elimination side product leading to olefin remained problematic. Resolution of racemic 19 was accomplished using chiral supercritical fluid chromatography (SFC) to afford single enantiomers 19S and 19R for which the stereochemical configuration was subsequently inferred on the basis of the absolute configuration observed in the electron density map of the X-ray structure of the respective menin complex with 19.
Scheme 3. Synthesis of Aminomethylpiperidine 19.

Preparation of 20 began with 2-pyridyl lithium addition to benzyl protected ketone 26 to provide 27 in excellent yield (Scheme 4). Hydrogenolysis of the N-benzyl of 27 using Pearlman’s catalyst overnight in ethanol with heating, followed by alkylation of the crude deprotected piperidine using chloride 28, afforded target sulfonamide 20.
Results and Discussion
Identification of Novel Inhibitors of Protein–Protein Interaction between Menin and MLL
To identify new inhibitors of the menin–MLL interaction, we performed a high throughput screening of ∼288000 small molecules at the NIH Molecular Libraries Probe Production Centers Network (MLPCN, https://mli.nih.gov/mli) using a fluorescence polarization (FP) assay with a fluorescein-labeled MLL-derived peptide MBM1.20 A stepwise procedure, including FP assay for primary screening followed by homogenous time resolved fluorescence (HTRF) assay for secondary screening and NMR saturation transfer difference (STD) experiments to validate direct binding of compounds to menin, was applied to identify menin–MLL inhibitors (detailed description of the HTS screen is provided at PubChem Bioassay, AID 1766: http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1766). The most potent compound identified in the screen was MLS001171971 (4-(2-hydroxy-3-(4-(hydroxydiphenylmethyl)piperidin-1-yl)propoxy)benzonitrile, compound 1, MIV-1), belonging to the hydroxymethylpiperidine class, which exhibited a half maximal inhibitory concentration (IC50) value of 12.8 μM for inhibition of the menin–MLL interaction (Figure 1a). We also identified a compound with a similar structure, MLS001175799 (Supporting Information Figure 1), but with a 6-fold weaker activity (IC50 = 77 μM) than MIV-1. We validated direct binding of MIV-1 to menin using saturation transfer difference (STD) NMR experiments, resulting in a strong STD effect (Figure 1b). Importantly, addition of the MLL peptide strongly decreased the STD effect observed for MIV-1 (Figure 1b), demonstrating that MIV-1 and MLL compete for binding to menin. These results confirm reversible and specific binding of MIV-1 hydroxymethylpiperidine compound to menin.
Figure 1.

Discovery of hydroxymethylpiperidine class of menin–MLL inhibitors by HTS. (a) Structure and activity of the most potent hit from HTS, MIV-1. (b) NMR saturation transfer difference (STD) experiments demonstrating binding of MIV-1 to menin and competition with MLL (competition STD). (c) Structure and activity for MIV-2 representing the essential pharmacophore for the hydroxymethylpiperidine class of menin–MLL inhibitors.
Determination of the Essential Pharmacophore within Hydroxymethylpiperidine Class of Menin–MLL inhibitors
First, we performed synthetic efforts to identify an essential pharmacophore within MIV-1 required for efficient inhibition of the menin–MLL interaction (synthetic procedures used for preparation of hydroxy- and aminomethylpiperidine compounds are provided in Schemes 1–4). We found that removal of the hydroxyl group from MIV-1 in the linker region, resulting in 4-(3-(4-(hydroxydiphenylmethyl)piperidin-1-yl)propoxy)benzonitrile (compound 2, MIV-2, Figure 1c), slightly increased the activity (IC50 = 10.8 μM, Table 1). Thus, to facilitate synthesis and reduce the number of resulting stereoisomers, we eliminated the central hydroxyl group from subsequent analogues. In contrast, removal of the hydroxyl group from the quaternary carbon in the head group region resulted in compound 3 (Table 1) with decreased inhibitory activity by more than 20-fold, emphasizing the importance of this group in binding to menin. Similarly, the binding affinity was strongly decreased by removing the nitrile within the tail group region, which resulted in compound 4 with IC50 > 250 μM (Table 1). On the basis of these data, we concluded that MIV-2 (Figure 1c) represents an essential pharmacophore for the hydroxymethylpiperidine class of menin–MLL inhibitors. These findings remain in a very good agreement with the structural data on the menin–inhibitor complexes (see below), demonstrating that all structural features present in MIV-2 are required for effective interactions with the corresponding binding pockets on menin.
Probing the Hydrophobic Head Group Region to Improve Potency of Menin–MLL Inhibitors
We used MIV-2 as a lead compound for medicinal chemistry optimization to improve potency of hydroxymethylpiperidine class. First, we explored replacement of one of the phenyl rings at the headgroup region of MIV-2 (Figure 1c) with different hydrophobic groups (R2 substituents, Table 1). Interestingly, we found a very pronounced structure–activity relationship (SAR) for modifications at this site (Table 1). The inhibitory activity of 4-(3-(4-(hydroxy(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile (compound 5, MIV-nc), with hydrogen replacing the phenyl group, was very weak (IC50 = 234 μM) and did not improve upon addition of a methyl group at this position (6, Table 1). Introducing more bulky hydrophobic substituents resulted in increased inhibition of the menin–MLL interaction. For example, n-butyl (7) or cyclopropyl (8) substituents yielded about 20-fold increase in activity versus MIV-nc (Table 1). Furthermore, analogues with iso-propyl (9) and cyclobutyl (10) groups had further improved IC50 values (IC50 = 4.1 and 4.0 μM, respectively, Table 1). Finally, cyclopentyl was identified as the most optimal saturated carbocycle at this position, resulting in 4-(3-(4-(cyclopentyl(hydroxy)(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile (compound 11, MIV-3),27 with an IC50 of 390 nM measured for the racemic mixture. Further increase in ring size decreased activity, as cyclohexyl analogue 12 was about 4-fold less active than MIV-3 (Table 1). On the basis of these data, we concluded that cyclopentyl was the preferred hydrophobic group at R2 position.
MIV-3 represents a racemic mixture, and to assess the activity of individual enantiomers, we separated the racemic mixture by chiral SFC. Interestingly, the R enantiomer, MIV-3R (11b, stereochemistry assigned based on the crystal structures, see below), was only about 2-fold more potent than the S enantiomer (11a, MIV-3S) as assessed by FP assay (IC50 = 270 nM and 529 nM for MIV-3R and S, respectively, Figure 2a). We subsequently measured the dissociation constants (Kd) for binding of both enantiomers of MIV-3 to menin using isothermal titration calorimetry (ITC) and found that they both bind to menin with nanomolar affinities (Kd = 285 and 952 nM for R and S isomers, respectively, Figure 2b,c), consistent with the IC50 values, Figure 2a. Overall, development of MIV-3R represents over 40-fold improvement in the activity as compared to the parent compound MIV-1. Interestingly, we found a relatively small difference in inhibition of the menin–MLL interaction by both enantiomers of MIV-3, Figure 2.
Figure 2.

MIV-3 binds to menin with nanomolar affinity. (a) Structure and activity for MIV-3 and two individual enantiomers of this compound. (b,c) Isothermal titration calorimetry experiments demonstrating direct and specific binding of both enantiomers of MIV-3 to menin. N corresponds to the stoichiometry of ligand binding to menin.
Hydroxymethylpiperidine Inhibitors Mimic the Most Critical Interactions of MLL with Menin
To establish the molecular determinants of MIV-3 binding to menin and understand relatively small differences in the binding affinity between the two enantiomers, we determined the crystal structures of menin in complex with both enantiomers of MIV-3 (Figure 3a,b). We found that they both bind in extended conformations to the MLL binding site on menin and occupy three hydrophobic pockets: F9, P10, and P13 (Figure 3). There is a very close overlap of the binding modes of both enantiomers of MIV-3, with the only difference being an alternate positioning of the head group substituents between the F9 and P10 hydrophobic pockets on menin (Figure 3c). The menin–MIV-3 interactions are mostly mediated by the hydrophobic contacts, with only one direct hydrogen bond formed between the nitrile group of both enantiomers of MIV-3 and the indole nitrogen of Trp341 on menin (Figure 3c). Additional water mediated hydrogen bond involves hydroxyl at the head group region of MIV-3R and S and the Asp180 side chain on menin (Figure 3c). The importance of the hydrogen bonds involving head group hydroxyl and tail group nitrile is reflected by more than 20-fold decrease in the activity of the analogues deficient in the corresponding functional groups (compounds 3 and 4, Table 1). Of note, binding of both enantiomers of MIV-3 to menin does not induce conformational changes of the protein.
Figure 3.
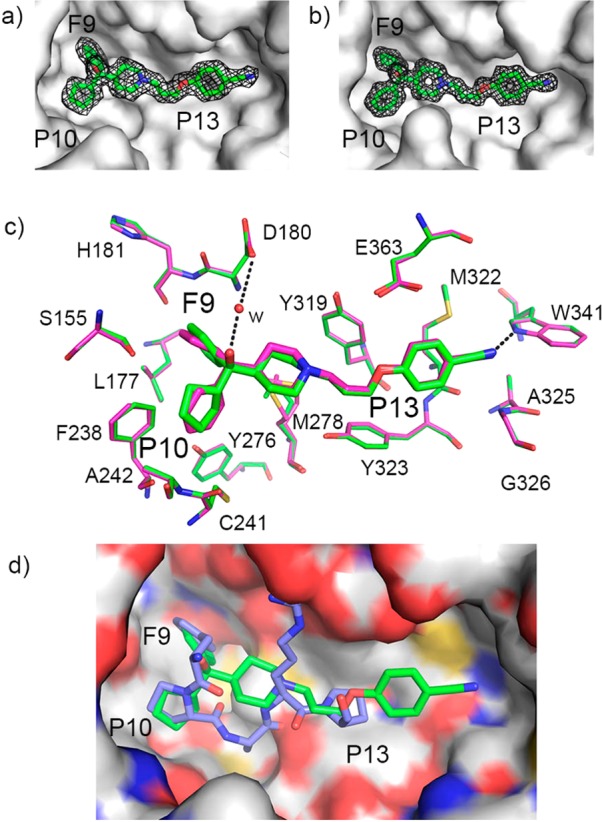
MIV-3 binds to the MLL binding site on menin. (a,b) Crystal structures of menin in complex with MIV-3R (a, PDB code 4GO3) and MIV-3S (b, PDB code 4GO4) with 2Fo – Fc electron density maps for ligands contoured at the 1σ level. Menin is shown in surface representation and location of F9, P10, and P13 pockets is labeled. (c) Superposition of the menin–MIV-3R (carbon atoms in green) and menin–MIV-3S (carbon atoms in magenta) crystal structures showing binding mode of ligands and key protein residues in the binding site. The F9, P10, and P13 pockets are labeled. Dashed lines represent hydrogen bonds between the ligands and protein; w represents a water molecule. (d) Superposition of the crystal structures of menin–MIV-3R (green carbons) and the menin–MLL (blue carbons, PDB code 4GQ6) complexes. Menin is shown in surface representation with atoms colored according to the atom type. The F9, P10, and P13 pockets are labeled.
In the co-crystal structure with more potent enantiomer, MIV-3R, the phenyl ring fits very well into the F9 pocket on menin formed by the side chains of hydrophobic residues (Leu177, Phe238, Ser155, Met278, Figure 3c) and backbone of Asp180 and His181, while the cyclopentyl ring binds to the P10 pocket and interacts with the side chains of Ala242, Cys241, Tyr276, Phe238, and Ser155, Figure 3c. In the case of MIV-3S enantiomer, the positions of cyclopentyl and phenyl rings are swapped between these two pockets on menin (Figure 3c). The piperidine ring and tail groups in both enantiomers of MIV-3 bind to menin in the same manner, extending toward the P13 pocket and Trp341. The alkoxy portion of the linker fits into the P13 pocket formed by Tyr319 and Tyr323 (Figure 3c), while the benzonitrile moiety extends beyond the P13 pocket, where it approaches Met322 and Glu363 side chains to form a hydrogen bond with the side chain of Trp341 (Figure 3c). The interaction of the benzonitrile moiety is of particular interest as this region of the binding site is not occupied by the MLL peptide or our previously identified thienopyrimidine class of menin–MLL inhibitors (Figure 3c,d). Thus this unique and important interaction identified for the new hydoxymethylpiperidine class offers the possibility to introduce additional contacts with menin to further improve affinity through rationally designed modifications. Overall, the binding mode of MIV-3 explains the SAR data (Table 1) and is consistent with the essential pharmacophore identified for this class of menin–MLL inhibitors (Figure 1c).
In our previous studies, we found that three hydrophobic residues in the MLL derived peptide MBM1, namely Phe9, Pro10, and Pro13, have the most critical contributions for binding to menin and their mutations to alanine residues reduce binding affinity by 30–1000-fold.20,23 Superposition of MLL MBM1 peptide and both enantiomers of MIV-3 bound to menin demonstrate a close overlap of these small-molecule inhibitors with the critical residues of MLL required for potent binding to menin (Figure 3d). Specifically, the phenyl ring in the head group of the more potent enantiomer MIV-3R overlaps with the side chain of Phe9 in MLL, while the cyclopentyl ring in MIV-3R adopts a similar position and conformation as the Pro10 side chain in MLL (Figure 3d), thus mimicking very closely the critical MLL interactions with menin by structurally and conformationally similar moieties. Furthermore, the alkoxy portion of the linker in MIV-3R mimics the interactions of MLL Pro13 with menin, while the benzonitrile moiety extends beyond the P13 pocket toward previously unexplored region of the binding site. Overall, the potent activity of MIV-3R results from mimicking key interactions of MLL with menin in F9, P10, and P13 pockets as well as from additional contacts beyond the P13 pocket, which provides further opportunity for optimization of these compounds.
Structure-Based Design of Nanomolar Inhibitors of the Menin–MLL Interaction
We then used the crystal structure of menin in complex with both enantiomers of MIV-3 for rational design of new analogues to further improve potency and physicochemical properties of this class of compounds. Four regions in MIV-3 were explored for modifications (Figures 4 and 5) to rationally design new compounds based upon structural data. First, we explored additional optimization of R1 substituents that bind to the F9 pocket on menin (Figure 4a, Table 1). As demonstrated above, the phenyl ring in MIV-3R closely mimics the interactions of MLL Phe9 with menin, and therefore it was used to design further modifications. In the crystal structure of menin in complex with the MI-2–2 thienopyrimidine inhibitor, we found a very favorable C–F---C=O dipolar interaction between a fluorine atom in MI-2–2 and backbone of His181.23 On the basis of the menin–MIV-3R co-crystal structure, we anticipated that introducing fluorine into position 3 of the phenyl ring should result in similar contacts and in improved activity. Indeed, introduction of a 3-fluoro to give 4-(3-(4-(cyclopentyl(3-fluorophenyl)(hydroxy)methyl)piperidin-1-yl)propoxy)benzonitrile (compound 13, MIV-4) resulted in a 2-fold improvement in potency versus MIV-3 (Figure 4b). We validated the binding mode of MIV-4 to menin by solving the crystal structure of the complex (Figure 4c, Supporting Information Table 1), which confirmed the presence of dipolar interaction between the fluorine atom in MIV-4 (in R enantiomer) and the backbone amide of His181 on menin. Introducing a chloro substituent at the same position (15) or incorporation of additional fluorine to the phenyl ring (14) resulted in a weaker inhibitory activity (Figure 4b). Therefore, structure-based designed 3-fluoro substituted phenyl represents the most preferred substituent identified thus far to interact with the F9 pocket on menin (Figure 4b,c and Table 1).
In the next step, we performed optimization of the R2 substituent that interacts with the P10 pocket. In the complex of menin with more potent MIV-3R enantiomer, this pocket is occupied by the cyclopentyl ring, which is not optimal for further derivatization and contributes significantly to the lipophilic character of the molecule (cLogP = 5.6 for MIV-3; cLogP calculations performed using Molinspiration software, www.molinspiration.com). Because the crystal structure of MIV-3S reveals that the P10 pocket can also accommodate phenyl ring (Figure 3b,c) with only 2-fold decreased IC50 value (Figure 2a), we selected phenyl for further optimization of the R2 substituent as an approach to streamline derivatization and SAR. The analysis of the apo structure of menin and the complex with MIV-3S revealed the presence of a water molecule, which forms a hydrogen bond with Tyr 276, located in a proximity to the ortho position of the phenyl ring in MIV-3S (not shown). Therefore, we introduced a nitrogen atom via replacement of phenyl with a pyridine or thiazole rings to engage in favorable interactions with this structural water molecule. These efforts resulted in 4-(3-(4-(cyclopentyl(hydroxy)(pyridin-2-yl)methyl)piperidin-1-yl)propoxy)benzonitrile (compound 16, MIV-5) and 4-(3-(4-(cyclopentyl(hydroxy)(thiazol-2-yl)methyl)piperidin-1-yl)propoxy)benzonitrile (compound 17), both of which showed improved potency and increased polarity versus MIV-3, Figure 4d. We confirmed the existence of a water-mediated hydrogen bond between MIV-5 and Tyr276 by solving the crystal structure of the complex (Figure 4e). As predicted, despite using the racemic mixture of MIV-5 for co-crystallization with menin, only the S enantiomer was observed in the structure (Figure 4e). This was further confirmed by testing separated enantiomers of MIV-5, showing a ∼4-fold greater inhibitory activity for the S enantiomer (IC50 = 195 nM). Then we combined optimal substituents identified to interact with F9 and P10 pockets and synthesized the hybrid 3-fluorophenyl-2-pyridyl congener 18, with ∼3-fold improved IC50 value (IC50 = 90 nM for R enantiomer of 18, Figure 4d), which represents the most potent hydroxmethylpiperidine compound reported here. Importantly, introduction of heterocyclic versus phenyl ring to occupy the P10 pocket substantially increased polarity (cLogP value reduced by 1 order of magnitude) and may also reduce potential for oxidative metabolism of these compounds.
The crystal structure of MIV-3 enantiomers with menin revealed that the solvent exposed hydroxyl group in the head group region is involved in a water mediated hydrogen bond with Asp180 (Figure 5b). On the basis of structural data, we anticipated that introducing a positively charged group, such as an amino moiety, should result in additional favorable electrostatic interactions with adjacent Asp180 and other acidic residues located in this region of the binding site (e.g., Asp153, Glu 359, Figure 5b). Indeed, MIV-6 (19, Figure 5a,b), which represents the aminomethylpiperidine class, showed almost 6-fold increase in inhibiting the menin–MLL interaction (IC50 = 67nM for racemic mixture of MIV-6) versus MIV-3. The binding mode of MIV-6 was not affected by incorporating the amino group, as validated by the crystal structure of menin in complex with MIV-6R enantiomer (Figure 5b). The MIV-6R represents the most potent inhibitor of the menin–MLL interaction reported here (IC50 = 56 nM), which binds to menin with Kd = 85 nM (Figure 5a and Supporting Information Figure 2). These results demonstrate the importance of considering long-range electrostatic interactions in designing protein ligands to achieve efficient binding.
Finally, we used the crystal structure of menin with MIV-5 to guide replacement of the nitrile, with the overall goal to further improve polarity while retaining the hydrogen bond with Trp341 (Figure 5d). On the basis of the menin–MIV-5 structure, we designed sulfonamide group as the optimal replacement of nitrile. Indeed, the sulfonamide analogue 20 (MIV-7, IC50 = 114 nM) showed ∼3-fold improved inhibitory activity versus the corresponding nitrile analogue MIV-5 (Figure 5c), while 21 demonstrated a modest ∼1.5-fold improvement in IC50 versus the corresponding nitrile derivative 18 (Figures 4d, 5c). The binding mode of MIV-7 to menin was validated by crystallographic studies, confirming the existence of the hydrogen bonds with the side chain of Trp341 and also the presence of an additional water-mediated hydrogen bond with Glu363 (Figure 5d), resulting in increased potency. These data clearly validate the sulfonamide as a viable replacement for the nitrile in this class of menin–MLL inhibitors, which allows for significant increase in polarity relative to the nitrile analogues (cLogP is reduced by an order of magnitude, cLogP = 3.4 for MIV-7 versus 4.4 for MIV-5).
MIV-6 and MIV-3R Disrupt Menin–MLL Fusion Protein Interaction in Cells and Affect Expression of MLL Fusion Protein Downstream Targets
To assess the mechanism of action of this new class of menin–MLL inhibitors, we evaluated their effect on blocking the activity of MLL fusion proteins in leukemia cells. For this purpose, we selected the most potent inhibitor we developed harboring the amino group, MIV-6 (IC50 = 67 nM), and corresponding hydroxyl analogue, MIV-3R (IC50 = 270 nM). First, we performed the co-immunoprecipitation (co-IP) experiment in HEK-293 cells transfected with Flag–MLL-AF9 to evaluate whether the compounds can effectively disrupt the menin–MLL fusion protein interaction in mammalian cells (Figure 6a). Treatment with low micromolar concentrations of both MIV-6 and MIV-3R very effectively disrupts the menin–MLL-AF9 interaction in cells, with more pronounced effect observed for MIV-6 (Figure 6a). Importantly, the expression levels of menin and MLL-AF9 were not affected upon treatment (Figure 6a). These data clearly demonstrate that both MIV-6 and MIV-3R can reach the target protein and effectively disrupt the menin–MLL fusion protein interaction in human cells.
Figure 6.

Biological activity of hydroxy- and aminomethylpiperidine menin–MLL inhibitors. (a) Co-immunoprecipitation experiment in HEK293 cells transfected with MLL-AF9. (b) Quantitative real-time PCR performed in MLL-AF9 transformed murine bone marrow cells after 6 days of treatment with compounds. Expression of Hoxa9 and Meis1 has been normalized to β-actin. Data represent mean values for duplicates ± SD. The experiment was performed two times. (c) Titration curves from MTT cell viability assay performed for MIV-6 and MIV-3R after 7 days of treatment of MLL-AF9 and Hoxa9/Meis1 (HM-2) transformed murine bone marrow cells. Cell growth inhibition (GI50) values are provided for treatment of MLL-AF9 cells. Data represent mean values of quadruplicates ± SD. The experiment was performed three times. (d) Titration curves from MTT cell viability assay performed for MIV-6 and MIV-3R after 7 days of treatment of human MLL leukemia cell lines (MV4;11 and MOLM-13) and control cell line HL-60 (non-MLL leukemia cell line). Cell growth inhibition (GI50) values are provided for MV4;11 and MOLM-13 cell lines. Data represent mean values of quadruplicates ± SD. The experiment was performed three times. (e) Quantification of CD11b expression in MLL-AF9 transformed murine bone marrow cells treated for 6 days with MIV-6 and MIV-3R as detected by flow cytometry. Data represent the mean values for duplicates ± SD. (f) Wright–Giemsa-stained cytospins for MLL-AF9 transformed murine bone marrow cells after 7 days of treatment. Black line represents the scale bar (50 μm).
The menin–MLL fusion protein interaction is required for the maintenance of high expression level of HOXA9 and MEIS1 in MLL leukemia cells and for leukemic transformation by MLL fusions.11 To assess whether MIV-6 and MIV-3R affect the expression level of HOXA9 and MEIS1, we performed real-time quantitative PCR (qRT-PCR) experiments in MLL-AF9 transformed murine bone marrow cells (BMC). Indeed, treatment with both MIV-6 and MIV-3R resulted in a strong and dose dependent reduction in the expression level of Hoxa9 and Meis1 as compared to the DMSO control (Figure 6b), with about 50% decrease in Hoxa9 level upon treatment with 3 μM of MIV-6. The effects on expression level of target genes correlates well with the in vitro inhibition observed for these compounds as MIV-6 showed about 4-fold more pronounced effect on Hoxa9 expression. Importantly, the negative control compound, MIV-nc, which is a very weak menin–MLL inhibitor (IC50 = 234 μM, Table 1), did not show any effect on Hoxa9 and Meis1 expression (Figure 6b). These results demonstrate that MIV-6 and MIV-3R inhibit the menin–MLL fusion protein interaction in cells and block the MLL–fusion protein dependent gene expression, emphasizing on-target effects for these compounds and validating their specific mechanism of action.
MIV-6 and MIV-3R Selectively Block Proliferation and Induce Differentiation in MLL Leukemia Cells
Disruption of the menin–MLL fusion protein interaction is expected to result in growth arrest and differentiation of MLL leukemia cells.11,25 Therefore, we tested activity of MIV-6 and MIV-3R in murine bone marrow cells transformed with either MLL-AF9 or Hoxa9/Meis1 (HM-2), the later served as a negative control cell line. As expected, strong and dose-dependent inhibition of cell proliferation was observed for both compounds in MLL-AF9 transformed BMCs (Figure 6c), with GI50 = 1.1 μM for MIV-6, which showed ∼4-fold more pronounced effect than MIV-3R (GI50 = 4.5 μM), consistent with its higher in vitro activity. In contrast, these two compounds have a very limited effect on proliferation of Hoxa9/Meis1 transformed BMCs (Figure 6c), demonstrating selectivity toward MLL fusion transformed cells. Similar effects were observed in human MLL leukemia cell lines. Treatment of MV4;11 and MOLM-13 cells harboring MLL-AF4, MLL-AF9 fusion proteins, respectively, with MIV-6 and MIV-3R resulted in a strong and dose dependent inhibition of cell proliferation, with a more pronounced effect observed for MIV-6 (Figure 6d). In contrast, marginal effect on cell proliferation was seen upon treatment of HL-60 promyelocytic leukemia cells, which served as a negative control cell line (Figure 6d).
We then assessed the effect of hydroxy- and aminomethylpiperidine compounds on differentiation of MLL fusion protein dependent cells. Indeed, the MLL-AF9 BMCs undergo differentiation upon treatment with MIV-6 and MIV-3R, as assessed by flow cytometry analysis of expression level of CD11b, which serves as a differentiation marker of myeloid cells. Treatment with more potent MIV-6 induces more pronounced increase in CD11b expression (Figure 6e). In addition, Wright–Giemsa staining of MLL-AF9 BMCs treated with low micromolar concentrations of MIV-6 and MIV-3R revealed significant changes in morphology of these cells, such as decreased nuclei to cytoplasm ratio, multilobed nuclei, and highly vacuolated cytoplasm, which clearly demonstrate myeloid differentiation upon treatment with both inhibitors (Figure 6f). Treatment with higher concentration of MIV-6 (6 μM) resulted in terminal differentiation (Figure 6f). All these results further confirm on-target effects and specific mechanism of action for the hydroxy- and aminomethylpiperidine inhibitors.
Conclusions
Menin is a critical oncogenic cofactor of MLL fusion proteins, and the protein–protein interaction between menin and MLL fusion proteins represents a validated and attractive therapeutic target in acute leukemias with translocations of MLL gene.11,21,30 Specific inhibition of this protein–protein interaction with small molecules could lead to development of novel targeted therapy for acute leukemias with MLL translocations.25 The difficulty in developing potent small-molecule inhibitors of the menin–MLL interaction partly arises from a very large size of the binding site on menin (over 5000 Å3),23 as small-molecule inhibitors can occupy only a small portion of this site.
In the present study, we report development of a novel class of the menin–MLL inhibitors, the hydroxy- and aminomethylpiperidine compounds, which we identified by a high throughput screening of over ∼288000 small molecules at the NIH MLPCN. Medicinal chemistry optimization of the HTS hit MIV-1 (IC50 = 10.8 μM) led to a very significant improvement of inhibitory activity, resulting in development of MIV-3 (IC50 = 270 and 530 nM for R and S enantiomers, respectively, Figure 2a). The co-crystal structures of both enantiomers of MIV-3 with menin were used to design new analogues with improved binding affinities. The most pronounced activity increase was obtained after replacing the hydroxyl group with the amino group, leading to electrostatic interactions with Asp180 on menin (Figure 5b). The resulting compound MIV-6R (IC50 = 56 nM, Kd = 85 nM) provides ∼230-fold improvement in the activity versus the initial HTS hit MIV-1. Importantly, despite a relatively low molecular weight (Mw = 416 Da), MIV-6 inhibits the menin–MLL interaction with an affinity similar to the 4-fold larger 12-amino acid MLL-derived peptide. Consequently, the ligand efficiency (LE) index31 for this compound is relatively high (LE = 0.31 for MIV-6R) as compared to 0.24 average value reported for PPI inhibitors.5 Furthermore, MIV-6 has significantly improved polarity (cLogP = 2.9) versus MIV-3 (cLogP = 5.6).
The co-crystal structure of menin in complex with compounds reported here revealed that they mimic the key interactions of MLL with menin by occupying three pockets on menin (F9, P10, and P13), which are the most critical for MLL binding. Importantly, these compounds also extend beyond the P13 pocket toward an additional region on menin, which does not participate in MLL binding, providing a further interaction site for potency optimization and modulation of physicochemical properties of menin–MLL inhibitors. We utilized this site for improvement in compound binding affinity by introducing the benzonitrile group to form a hydrogen bond with Trp341 and for increase in compound polarity by introducing sulfonamide as an efficient replacement of nitrile (Figure 5d). The later example clearly demonstrates the potential for further optimization of physicochemical properties for this class of menin–MLL inhibitors.
The newly developed hydroxy- and aminomethylpiperidine class of compounds have very effective and selective activities in cell-based experiments. The most potent compound, MIV-6, efficiently inhibited proliferation and induced hematopoietic differentiation in MLL leukemia cells and strongly reduced the expression level of key MLL–fusion protein target genes. Overall, these data demonstrate that the novel class of menin–MLL inhibitors reported here has very promising on-target biological activity. On the basis of structural studies, these compounds represent an attractive chemical scaffolds for further optimization as they closely mimic all critical interactions of MLL with menin, including the interactions with the P10 pocket, which remains unoccupied by the previously reported thienopyrimidine class of menin–MLL inhibitors.23 Our studies will pave the way toward further optimization of these compounds into chemical probes for in vivo studies in MLL leukemia models and for potential therapeutic applications. This work provides another example of PPI interface amenable to potent inhibition by small molecules, strongly supporting the efforts in developing inhibitors for novel PPIs as chemical probes and/or potential therapeutic agents.
Experimental Section
General Chemistry Information
The structures of the presented compounds were characterized by 1H, 13C NMR, and mass spectrometry (HRMS). Purity (>95%) was assessed by RP-HPLC. All NMR spectra were recorded on a Bruker 400, 500, or 600 MHz instruments. 1H chemical shifts are reported in δ values relative to residual solvent signals in ppm. Data are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad resonance, m = multiplet), coupling constant (Hz). Low resolution mass spectra were obtained on an Agilent 1200 series 6130 mass spectrometer. High resolution mass spectra were recorded on a Waters Q-TOF API-US plus Acuity system with ES as the ion source. Analytical thin layer chromatography was performed on Sorbent Technologies 250 μm silica plates. Visualization was accomplished via UV light, and/or the use of potassium permanganate solution followed by application of heat. Analytical HPLC was performed on an HP1100 with UV detection at 214 and 254 nm along with ELSD detection, LC/MS (J-Sphere80-C18, 3.0 mm × 50 mm, 4.1 min gradient, 7%[0.1%TFA/H2O]:93%[CH3CN]). Preparative RP-HPLC purification was performed on a custom HP1100 automated purification system with collection triggered by mass detection or using a Gilson Inc. preparative UV-based system using a Phenomenex Luna C18 column (50 × 30 mm ID, 5 μm) with an acetonitrile (unmodified)–water (0.5 mL/L NH4OH) custom gradient. Normal-phase silica gel preparative purification was performed using an automated Combi-flash Companion from ISCO. Semipreparative purifications were carried out via stacked injections on a Waters Investigator SFC using a 10 mm × 250 mm Chiral Technologies Chiralpak ID column heated to 40 °C. Analytical separations were carried out on an Agilent 1260 Infinity SFC using a 4.6 mm × 250 mm Chiral Technologies Chiralpak ID column heated to 40 °C. Solvents for extraction, washing, and chromatography were HPLC grade. All reagents were purchased from Aldrich Chemical Co. and were used without purification. All polymer-supported reagents were purchased from Argonaut Technologies and Biotage.
Procedures for Synthesis of Compounds
Synthesis of presented compounds was performed according to Schemes1–4. Full experimental procedures, analytical data, NMR spectra, and chiral separation of compounds reported here can be found within Supporting Information. Synthesis and analytical data for representative compounds from hydroxy- (MIV-3) and aminomethylpiperidine (MIV-6) classes are provided below.
Synthesis of 4-(3-(4-(Cyclopentyl(hydroxy)(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile, 11 (MIV-3)
4-(3-(4-(Cyclopentanecarbonyl)piperidin-1-yl)propoxy)benzonitrile (30 mg, 0.09 mmol) was dissolved in THF (0.7 mL). Phenylmagnesium bromide (1.0 M in THF) was added dropwise to the solution with stirring. Reaction was then warmed to 50 °C, and stirring was continued for 2 h. Reaction was quenched with satd aqueous NH4Cl and extracted with EtOAc. The combined organic fractions were washed with saturated NaCl and dried over Na2SO4. Concentration in vacuo provided the crude product, which was purified by flash column chromatography (9:1 CH2Cl2/MeOH) to provide 46 mg of the desired products (74% yield). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.54 (2H, d, J = 9 Hz), 7.37 (2H, d, J = 8 Hz), 7.30 (2H, t, J = 8 Hz), 7.22 (1H, t, J = 7 Hz), 6.88 (2H, d, J = 9 Hz), 4.07 (2H, t, J = 6 Hz), 3.21 (2H, m), 2.67 (3H, m), 2.19 (2H, m), 2.05 (2H, m), 1.95 (1H, m), 1.82 (1H, m), 1.72 (1H, m), 1.64 (1H, m), 1.59–1.40 (7H, m), 1.25 (1H, m), 1.07 (1H, m). 13C NMR (100.6 MHz, CDCl3) δ (ppm): 162.4, 143.9, 134.0, 127.7, 126.5, 126.4, 119.4, 115.3, 103.8, 79.6, 68.9, 55.2, 54.5, 54.4, 45.8, 45.3, 27.5, 27.3, 26.7, 26.4, 26.1, 26.0, 25.6. HRMS (ES+, M + H) calcd for C27H35N2O2, 419.2699; found, 419.2698. Chiral separation: Semipreparative purifications were carried out via stacked injections on a Waters Investigator SFC using a 10 mm × 250 mm Chiral Technologies Chiralpak IA column heated to 40 °C. The eluent was 55% EtOH (0.1% DEA) in CO2 at a flow rate of 15 mL/min. Backpressure was maintained at 100 bar. The first eluting peak (11S), retention time = 0.95 min, was inferred as the S stereoisomer based upon the absolute configuration observed in the electron density map of the X-ray structure of the MIV-3S–menin complex. The second eluting peak (11R), retention time = 2.3 min, was inferred as the R stereoisomer based upon the absolute configuration observed in the electron density map of the X-ray structure of the MIV-3R–menin complex.
Synthesis of 4-(3-(4-(Cyclopentyl(amino)(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile, 19 (MIV-6)
A CHCl3 (4.78 mL, 0.25 M) solution of 4-(3-(4-(cyclopentyl(hydroxy)(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile (1.0 g, 2.39 mmol) and sodium azide (1.16 g, 17.9 mmol) was cooled to 0 °C. To the solution H2SO4 was added dropwise (0.28 mL, 9.3 mmol). The mixture was allowed to warm to rt over 4 h with stirring, then cooled to 0 °C and treated with NH4OH until pH was basic. The biphasic solution was extracted with CH2Cl2 (3×) and the organic layers combined and dried over MgSO4. Concentration under reduced pressure and concentration in vacuo afforded a crude oil which was purified by flash column chromatography (9:1 CH2Cl2/MeOH) to afford a colorless oil comprising an inseperable mixture of the desired azide and an elimination byproduct in 30 mg that was carried on to the next step. 4-(3-(4-(Azido(cyclopentyl)(phenyl)methyl)piperidin-1-yl)propoxy)benzonitrile (30 mg, 0.07 mmol) was dissolved in degassed EtOH (0.5 mL) and Pd/C (2.7 mg) added in one portion. Reaction was placed under a balloon of H2 gas and allowed to stir for 4 h at ambient temperature. The reaction was filtered over Celite and rinsed with MeOH. The filtrate was concentrated to afford an oil. RP-HPLC preparative purification afforded the desired product as a TFA salt. The mixture was treated with a StratoSpheres SPE MP-carbonate resin cartridge to give title compound as a free base in 20 mg (2%). 1H NMR (600 MHz, CDCl3) δ 7.54 (2H, d, J = 6.0 Hz), 7.43 (2H, d, J = 7.2 Hz), 7.31 (2H, t, J = 7.2 Hz), 7.22 (1H, t, J = 7.2 Hz), 6.91 (2H, d, J = 6.0 Hz), 4.02 (2H, t, J = 8.4 Hz), 3.05–2.96 (2H, m), 2.62 (1H, m), 2.47 (2H, br s), 1.96–1.89 (4H, m), 1.73 (1H, m), 1.62–1.40 (9H, m), 1.29–1.24 (2H, m), 1.13–1.05 (2H, m). 13C NMR (150.9 MHz, CDCl3) δ (ppm): 162.4, 144.5, 134.1, 127.6, 127.5, 126.1, 119.42, 115.3, 103.9, 66.9, 61.3, 55.2, 54.7, 54.6, 46.4, 45.6, 27.6, 27.1, 26.7, 26.6, 26.5, 25.9, 25.6. HRMS (ES+, M + H) calcd for C28H30N2O2, 418.858; found, 418.2857. Chiral separation: Semipreparative purifications were carried out via stacked injections on a Waters Investigator SFC using a 10 mm × 250 mm Chiral Technologies Chiralpak ID column heated to 40 °C. The eluent was 50% MeOH (0.1% DEA) in CO2 at a flow rate of 15 mL/min. Backpressure was maintained at 100 bar. The first eluting peak (19S), retention time = 3.97 min, was inferred as the S stereoisomer based upon the absolute configuration observed in the electron density map of the X-ray structure of the MIV-6R–menin complex. The second eluting peak (19R), retention time = 5.38 min, was inferred as the R stereoisomer based upon the absolute configuration observed in the electron density map of the X-ray structure of the MIV-6R–menin complex.
Expression and Purification of Human Menin
The expression and purification of menin have been described previously.20,23
High Throughput Screening
The fluorescence polarization assay20,23 was used for HTS of 288000 compounds at NIH MLPCN. Specifically, menin at 200 nM and the 12 amino acid MLL-derived peptide (MLL4–15) labeled with fluorescein or Texas Red, used at 25 and 50 nM, respectively, were utilized in the primary screening with the dual fluorescence polarization read-out at 535 and 632 nm. The primary screening was performed in 1536-well format using 4 μL final volume and four concentrations of each compound (61, 11.7, 2.25, 0.5 μM). Compounds, which showed dose-dependent inhibition in primary screen or at least 20% inhibition at the highest concentration in both FP read-outs were selected for confirmation screen, resulting in 240 compounds representing primary screening hits. The homogenous time resolved FRET (HTRF) assay, with His–menin to bind to anti-His Eu3+ cryptate donor beads and biotinylated MLL4–15 peptide to bind to streptavidin-XL-665 acceptor beads, was applied for confirmation screening performed at the concentration range 383 μM to 0.01 nM using 2-fold dilutions of compounds identified as hits in the primary screen. Donor and acceptor beads for HTRF assay were purchased from Cisbio International. Total 62 compounds were validated to inhibit the menin–MLL interaction in the confirmation HTRF assay and were subsequently tested in NMR STD and competition NMR STD experiments (see below) for direct and specific binding to menin. MLS001171971 (4-(2-hydroxy-3-(4-(hydroxydiphenylmethyl)piperidin-1-yl)propoxy)benzonitrile, compound 1, MIV-1) was the most potent compound identified by HTS to inhibit the menin–MLL interaction and validated to bind to menin by NMR STD. Detailed description of the HTS screen is provided at PubChem Bioassay, AID 1766: http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=1766.
Biochemical Characterization of Menin–MLL Inhibitors
Inhibition of the menin–MLL interaction by small molecules was assessed by fluorescence polarization (FP) assay using the protocol described previously.20,25
NMR Spectroscopy
NMR samples for the saturation transfer difference (STD) experiments contained 2.5 μM menin solution in 50 mM phosphate buffer, 50 mM NaCl, 1 mM DTT, pH = 7.5, with 5% of D2O. The MIV-1 compound was added as stock solutions in DMSO to final 100 μM concentration and 5% DMSO. All NMR experiments were recorded at 25 °C using 600 MHz Bruker Avance III spectrometer. For the STD experiments, we used 2 s irradiation using a train of 50 ms Gaussian pulses centered at 0 ppm with published pulse sequence.32 Samples for the competition STD experiments contained additional MLL1–46 peptide at final concentration of 10 μM, and the measurements were carried out in the same way as described above for STD experiments.
Isothermal Titration Calorimetry
Menin was extensively dialyzed at 4 °C against ITC buffer (50 mM phosphate, pH 7.5, 50 mM NaCl, 1 mM β-mercaptoethanol) and degassed prior to measurement. Compounds were dissolved in DMSO and diluted with the ITC buffer to final concentrations (50–100 μM, 5% DMSO). Protein solution was adjusted to contain 5% DMSO final concentration. The titrations were performed using a VP-ITC titration calorimetric system (MicroCal) at 25 °C. The calorimetric cell, containing menin (concentrations in the range 5–10 μM), was titrated with the compounds (50–100 μM) injected in 10 μL aliquots. Data was analyzed using Origin 7.0 (OriginLab) to obtain Kd and stoichiometry.
Crystallization of Menin Complexes with Small-Molecule Inhibitors
For co-crystallization experiments, 2.5 mg/mL menin was incubated with small-molecule inhibitors (MIV-3R, MIV-3S, MIV-4, MIV-5, MIV-6R, MIV-7) at 1:3 molar ratio. Crystals were obtained using the sitting drop technique at 10 °C by applying the procedure described previously.23 Prior to data collection, crystals were transferred into a cryosolution containing 20% PEG550 MME and flash-frozen in liquid nitrogen.
Crystallographic Data Collection and Structure Determination
Diffraction data for menin and menin complexes were collected at the 21-ID-D and 21-ID-F beamlines at the Life Sciences Collaborative Access Team at the Advanced Photon Source. Data were processed with HKL-2000.33 Structures of the complexes were determined by molecular replacement using MOLREP with the apo-structure of human menin (PDB code: 4GPQ) as a search model in molecular replacement. The model was refined using REFMAC,34 COOT,35 and the CCP4 package.36 In the final stages, refinement was performed with addition of the TLS groups defined by the TLSMD server.37 Validation of the structures was performed using MOLPROBITY38 and ADIT.39 Details of data processing and refinement are summarized in Supporting Information Table 1. Coordinates and structure factors have been deposited in the Protein Data Bank.
Co-immunoprecipitation Experiments
HEK293 cells were transfected with Flag–MLL-AF9 plasmid using Fugene 6 (Roche). Forty-eight hours after transfection, cells were treated with compounds (0.25% final DMSO concentration) or DMSO for 12 h. Whole cell lysates were immunoprecipitated with ANTI-FLAG M-2 magnetic beads (Sigma-Aldrich) and analyzed by SDS-PAGE and Western blotting. For more details, see Grembecka et al.25
Viability Assays
MLL-AF9 transformed mouse bone marrow cells were prepared as described previously.40 MV4;11, KOPN-8, ML-2, and MOLM-13 cells were cultured in RPMI-1640 medium with 10% FBS, 1% penicillin/streptomycin, and NEAA. For viability assay, MOLM-13 (1 × 105/mL), MV4;11 (1 × 105/mL), HL-60 (2 × 105/mL), and MLL-AF9 mouse bone marrow cells (2.5 × 104/mL) were plated (1 mL/well), treated with compounds or 0.25% DMSO, and cultured at 37 °C for 7 days. Media were changed at day four with viable cell number restored to the original concentration, and compounds were resupplied. Then 100 μL of cell suspension were transferred to 96-well plates for each sample in quadruplicates. A Vybrant MTT cell proliferation assay kit (Molecular Probes) was employed. Plates were read for absorbance at 570 nm using a PHERAstar BMG microplate reader. The experiments were performed three times in quadruplicated with calculation of mean and standard deviation for each condition.
Real-Time PCR
Effect of menin–MLL inhibitors on expression level of Hoxa9 and Meis1 was assessed by real-time quantitative PCR (qRT-PCR) using the protocol described previously.23
Expression of CD11b
MLL-AF9 transformed bone marrow cells were plated in 12-well plates at an initial concentration of 3 × 105 cells/mL and treated with compounds or 0.25% DMSO. Media were changed at days three and five with viable cell concentration restored to 3 × 105 cells/mL and compounds resupplied. Seven days after the experiment was setup, the 1.5 × 105 cells were harvested and washed with FACS buffer (PBS, 1% FBS, 0.1% NaN3). Cells were resuspended in 100 μL of FACS buffer and incubated with 1 μL of Pacific Blue rat antimouse CD11b antibody (BD BioLegend) at 4 °C for 30 min. Cells were then washed, resuspended in 100 μL of Annexin V binding buffer, and incubated with 4 μL of Annexin V-FITC (BD Biosciences) and 6 μL of propidium iodide (1 mg/mL, Sigma-Aldrich) at room temperature for 10 min before being analyzed by flow cytometry.
Cytospin/Wright–Giemsa Staining
Mouse bone marrow cells transduced with MLL-AF9 were plated in 12-well plates (1 mL/well) at an initial concentration of 3 × 105/mL cells, treated with compounds or 0.25% DMSO and incubated at 37 °C in a 5% CO2 incubator. Cytospins were prepared as described previously25 at designated time points.
Acknowledgments
This work was funded by the National Institute of Health grants: R03 (1R03MH084875) to J.G. and T.C., R01 (1R01CA160467) to J.G., MLPCN grants (1U54 MH084659) a Leukemia and Lymphoma Society (LLS) TRP grant (6116-12) to J.G., LLS Scholar (1215-14) to J.G., American Cancer Society grants (RSG-11-082-01-DMC to T.C. and RSG-13-130-01-CDD to J.G.), ACS MEDI Predoctoral Fellowship (T.J.S), and the Department of Pathology, University of Michigan. Use of the Advance Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract number DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (grant 085P1000817). We thank Mathew Mulder for HRMS determination and Nathan Kett for preparative chiral SFC. We thank Jay L. Hess and Andrew G. Muntean for cell lines.
Glossary
Abbreviations Used
- FP
fluorescence polarization
- HTRF
homogenous time resolved FRET
- ITC
isothermal titration calorimetry
- MBM1
menin binding motif 1
- MLL
mixed lineage leukemia
- MLPCN
Molecular Libraries Probe Production Centers Network
- STD
saturation transfer difference
- PPI
protein–protein interaction
Supporting Information Available
Crystallography data, experimental data, characterization of compounds by HRMS (ES+, M + H), 1H NMR and 13C NMR, and chiral SFC traces. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinate files have been deposited in the Protein Data Bank with the PDB codes 4GO3, 4GO4, 4GO5, 4GO6, 4GO7, and 4GO8.
Author Contributions
# S.H. and T.J.S. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kar G.; Gursoy A.; Keskin O. Human cancer protein–protein interaction network: a structural perspective. PLoS Comput. Biol. 2009, 5, e1000601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry D. C. Protein–protein interactions as targets for small molecule drug discovery. Biopolymers 2006, 84, 535–552. [DOI] [PubMed] [Google Scholar]
- Buchwald P. Small-molecule protein–protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life 2010, 62, 724–731. [DOI] [PubMed] [Google Scholar]
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. Inhibition of alpha-helix-mediated protein–protein interactions using designed molecules. Nature Chem. 2013, 5, 161–173. [DOI] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 2007, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Morelli X.; Bourgeas R.; Roche P. Chemical and structural lessons from recent successes in protein–protein interaction inhibition (2P2I). Curr. Opin. Chem. Biol. 2011, 15, 475–481. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Whitty A. The road less traveled: modulating signal transduction enzymes by inhibiting their protein–protein interactions. Curr. Opin. Chem. Biol. 2009, 13, 284–290. [DOI] [PubMed] [Google Scholar]
- Smith M. C.; Gestwicki J. E. Features of protein–protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev. Mol. Med. 2012, 14, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C.; Shoemaker A. R.; Adickes J.; Anderson M. G.; Chen J.; Jin S.; Johnson E. F.; Marsh K. C.; Mitten M. J.; Nimmer P.; Roberts L.; Tahir S. K.; Xiao Y.; Yang X.; Zhang H.; Fesik S.; Rosenberg S. H.; Elmore S. W. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [DOI] [PubMed] [Google Scholar]
- Ray-Coquard I.; Blay J. Y.; Italiano A.; Le Cesne A.; Penel N.; Zhi J.; Heil F.; Rueger R.; Graves B.; Ding M.; Geho D.; Middleton S. A.; Vassilev L. T.; Nichols G. L.; Bui B. N. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [DOI] [PubMed] [Google Scholar]
- Yokoyama A.; Somervaille T. C.; Smith K. S.; Rozenblatt-Rosen O.; Meyerson M.; Cleary M. L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [DOI] [PubMed] [Google Scholar]
- Slany R. K. When epigenetics kills: MLL fusion proteins in leukemia. Hematol. Oncol. 2005, 23, 1–9. [DOI] [PubMed] [Google Scholar]
- Marschalek R. Mechanisms of leukemogenesis by MLL fusion proteins. Br. J. Hamaetol. 2011, 152, 141–154. [DOI] [PubMed] [Google Scholar]
- Tomizawa D.; Koh K.; Sato T.; Kinukawa N.; Morimoto A.; Isoyama K.; Kosaka Y.; Oda T.; Oda M.; Hayashi Y.; Eguchi M.; Horibe K.; Nakahata T.; Mizutani S.; Ishii E. Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia 2007, 21, 2258–2263. [DOI] [PubMed] [Google Scholar]
- Popovic R.; Zeleznik-Le N. J. MLL: how complex does it get?. J. Cell Biochem. 2005, 95, 234–242. [DOI] [PubMed] [Google Scholar]
- Dimartino J. F.; Cleary M. L. Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br. J. Hamaetol. 1999, 106, 614–626. [DOI] [PubMed] [Google Scholar]
- Hess J. L. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol. Med. 2004, 10, 500–507. [DOI] [PubMed] [Google Scholar]
- Krivtsov A. V.; Armstrong S. A. MLL translocations, histone modifications and leukaemia stem-cell development. Nature Rev. Cancer 2007, 7, 823–833. [DOI] [PubMed] [Google Scholar]
- Slany R. K. The molecular biology of mixed lineage leukemia. Haematologica 2009, 94, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grembecka J.; Belcher A. M.; Hartley T.; Cierpicki T. Molecular basis of the mixed lineage leukemia–menin interaction: implications for targeting mixed lineage leukemias. J. Biol. Chem. 2010, 285, 40690–40698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caslini C.; Yang Z.; El-Osta M.; Milne T. A.; Slany R. K.; Hess J. L. Interaction of MLL amino terminal sequences with menin is required for transformation. Cancer Res. 2007, 67, 7275–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A.; Cleary M. L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi A.; Murai M. J.; He S.; Lund G.; Hartley T.; Purohit T.; Reddy G.; Chruszcz M.; Grembecka J.; Cierpicki T. Structural insights into inhibition of the bivalent menin–MLL interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai M. J.; Chruszcz M.; Reddy G.; Grembecka J.; Cierpicki T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J. Biol. Chem. 2011, 286, 31742–31748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grembecka J.; He S.; Shi A.; Purohit T.; Muntean A. G.; Sorenson R. J.; Showalter H. D.; Murai M. J.; Belcher A. M.; Hartley T.; Hess J. L.; Cierpicki T. Menin–MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nature Chem. Biol. 2012, 8, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Liu L.; Huang J.; Bernard D.; Karatas H.; Navarro A.; Lei M.; Wang S. Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin–mixed lineage leukemia 1 (MLL1) protein–protein interaction. J. Med. Chem. 2013, 56, 1113–1123. [DOI] [PubMed] [Google Scholar]
- Manka J.; Daniels R. N.; Dawson E.; Daniels J. S.; Southall N.; Jadhav A.; Zheng W.; Austin C.; Grembecka J.; Cierpicki T.; Lindsley C. W.; Stauffer S. R. In Probe Reports from the NIH Molecular Libraries Program; National Institutes of Health: Bethesda, MD, 2013.
- Cowan D. O.; Mosher H. S. Comparison of the Reactions of Grignard Reagents and Dialkylmagnesium Compounds in Addition, Reduction, and Enolization Reactions. J. Org. Chem. 1962, 27, 1–5. [Google Scholar]
- Honkanen E.; Pippuri A.; Kairisalo P.; Nore P.; Karppanen H.; Paakkari I. Synthesis and antihypertensive activity of some new quinazoline derivatives. J. Med. Chem. 1983, 26, 1433–1438. [DOI] [PubMed] [Google Scholar]
- Chen Y. X.; Yan J.; Keeshan K.; Tubbs A. T.; Wang H.; Silva A.; Brown E. J.; Hess J. L.; Pear W. S.; Hua X. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1018–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. In Macromolecular Crystallography, Part A; Methods in Enzymology; Academic Press: New York, 1997; Vol. 276, pp 307–326 [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Painter J.; Merritt E. A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 2006, 39, 109–111. [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. III; Snoeyink J.; Richardson J. S.; Richardson D. C. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Guranovic V.; Dutta S.; Feng Z.; Berman H. M.; Westbrook J. D. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr., Sect. D: Biol .Crystallogr. 2004, 60, 1833–1839. [DOI] [PubMed] [Google Scholar]
- Muntean A. G.; Tan J.; Sitwala K.; Huang Y.; Bronstein J.; Connelly J. A.; Basrur V.; Elenitoba-Johnson K. S.; Hess J. L. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell 2010, 17, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.